

Abstract
Ischemia/reperfusion injury is a complex molecular cascade that causes deleterious cellular damage and organ dysfunction. Stroke, sudden cardiac arrest, and acute myocardial infarction are the most common causes of ischemia/reperfusion injury without effective pharmacologic therapies. Existing preclinical evidence suggests that histone deacetylase inhibitors may be an efficacious, affordable, and clinically feasible therapy that can improve neurologic and cardiac outcomes following ischemia/reperfusion injury. In this review, we discuss the pathophysiology and epigenetic modulations of ischemia/reperfusion injury and focus on the neuroprotective and cardioprotective effects of histone deacetylase inhibitors. We also summarize the protective effects of histone deacetylase inhibitors for other vital organs and highlight the key research priorities for their successful translation to the bedside.
Keywords: epigenetics, histone deacetylase inhibitors, myocardial infarction, stroke, cardiac arrest
Subject Categories: Cardiopulmonary Arrest, Animal Models of Human Disease, Ischemia, Cerebrovascular Disease/Stroke, Myocardial Infarction
Nonstandard Abbreviations and Acronyms
- ARC
apoptosis repressor with caspase recruitment domain
- HDACi
histone deacetylase inhibitors
- HDACs
histone deacetylases
- HSP70
heat shock protein 70
- I/R
ischemia/reperfusion
- MCAO
middle cerebral artery occlusion
- PAI‐1
plasminogen activator inhibitor‐1
- ROS
reactive oxygen species
- SAHA
suberoylanilide hydroxamic acid
- SB
sodium butyrate
- t‐PA
tissue‐type plasminogen activator
- TSA
trichostatin A
- Tub‐A
tubastatin A
- VPA
valproic acid
Ischemia, or insufficient blood supply to an organ or cell, can induce devastating downstream effects. Although ischemia can exert deleterious effects on all tissues,1 the brain and the heart are most susceptible.2, 3 Stroke, sudden cardiac arrest, and acute myocardial infarction are common causes of ischemia and leading causes of morbidity and mortality worldwide.4 The reperfusion injury that follows the restoration of blood flow leads to the activation of complex downstream cellular cascades, which can further worsen organ dysfunction.1, 3, 5, 6, 7, 8 Because both ischemia and reperfusion contribute to significant cellular and organ injuries, the combined processes are frequently referred to as ischemia/reperfusion (I/R) injury.
I/R injury can induce cellular damage through hypoxic and hyperoxic mechanisms (Figure 1). ATP deficiency and subsequent increase in anaerobic metabolism secondary to hypoxic ischemia causes a decrease in cellular pH and intracellular overload of calcium via disruption of ion pumps.1, 3 At the onset of reperfusion, the rapid restoration of intracellular pH and oxygen leads to an increase in the mitochondrial generation of reactive oxygen species (ROS) that further exacerbates cell death via oxidative cellular damage.1, 3, 5 Mitochondrial permeability transition is also facilitated by an increase in ROS and calcium, resulting in necrosis or apoptosis.3, 8
Figure 1. Mechanisms of ischemia‐reperfusion injury. Ischemia induces anerobic glycolysis, intracellular acidosis, and ion pump dysfunction.
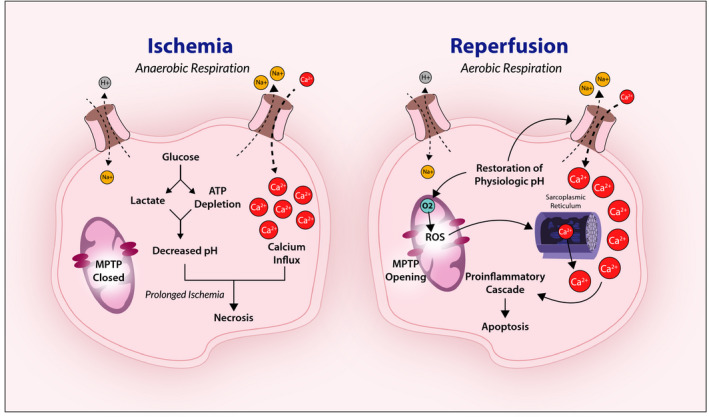
The subsequent calcium influx combined with prolonged ischemia results in cellular necrosis. On reperfusion and restoration of physiologic pH, reactive oxygen species (ROS) generation and intracellular calcium trigger mitochondrial permeability transition pore (MPTP) opening and induce further intracellular calcium overload, proinflammatory cascades, and apoptosis.
Although the duration of ischemia is a critical determinant of subsequent damage, studies have revealed that targeting ischemia with revascularization alone treats only half of the injury.5, 9 Animal models demonstrate that nearly 50% of infarct size is secondary to lethal reperfusion injury regardless of the duration of ischemia.5, 6, 7, 9 Strategies such as cardiopulmonary resuscitation, early defibrillation, thrombolysis, and thrombectomy all aim to minimize ischemic time through the restoration of blood flow. However, they fail to prevent or reverse the progression of the subsequent reperfusion injury. Reperfusion exacerbates cell death through calcium overload, glutamate release, ROS formation, opening of the mitochondrial permeability transition pores, endothelial dysfunction, thrombosis, proteolysis, and activation of inflammatory pathways.1, 3, 6, 7 Mitochondrial dysfunction also contributes significantly to reperfusion injury.10 After prolonged global ischemia from cardiac arrest, microthrombi formation and abnormal leukocyte adhesion in the capillaries can lead to significant secondary ischemia due to the “no‐reflow” phenomenon.11, 12, 13 Hypothermic targeted temperature management is the only clinically available therapy that may attenuate reperfusion injury by upregulating prosurvival pathways after cardiac arrest.14, 15 However, its efficacy is often limited by the inability to achieve target temperature within the therapeutic window,16, 17, 18 side effects,19, 20, 21 and delay in neuroprognostication.22, 23, 24
The paucity of effective treatments for I/R injury demonstrates an urgent need for novel therapeutic approaches. Histone deacetylase inhibitors (HDACi) have emerged as a promising strategy for the treatment of I/R injury (Table). This review highlights the epigenetic modulations and protective effects of HDACi following I/R injury. We focus on the neuroprotective and cardioprotective effects of HDACi after stroke, cardiac arrest, and myocardial infarction. We also discuss the peripheral organ protective effects of HDACi. Special emphasis is placed on valproic acid (VPA), a nonspecific HDACi approved by the US Food and Drug Administration (FDA), with the most preclinical data to support its use following I/R injury.
Table 1.
HDACi and Their Protective Effects After I/R Injury
| Injury Type | HDAC Class | HDAC Isoform | Nonspecific HDACi | Isoform‐ Specific HDACi | In Vitro Model | Small Animal Model | Mechanistic Outcomes |
|---|---|---|---|---|---|---|---|
| Neuroprotection | |||||||
| Stroke | I, IIa | 1–5, 7–9 | VPA | x | x | Reduced infarct size, neurologic disability score, blood–brain barrier disruption, and neuronal death via hyperacetylation of histones H3 and H4, HSP70 upregulation, and fibrinolysis [#jah35162-bib-0058 #jah35162-bib-0059 #jah35162-bib-0060 #jah35162-bib-0061 #jah35162-bib-0062 #jah35162-bib-0063 #jah35162-bib-0064 #jah35162-bib-0065 #jah35162-bib-0066 #jah35162-bib-0067">58–67, #jah35162-bib-0075 #jah35162-bib-0076 #jah35162-bib-0077">75–77] | |
| I, IIa, IIb, IV | 1–11 | SAHA | x | x | Reduced neuronal death and cerebral inflammation by promoting a protective microglial phenotype [66] | ||
| I, IIa | 1–5, 7–9 | SB | x | x | Reduced neuronal injury and infarct size; increased histone H3 acetylation and HSP70 expression; promoted neuroplasticity via increased BDNF expression [66, 68] | ||
| I, IIb, IV | 1–5, 7–9, 11 | TSA | x | Reduced neuronal injury and infarct size via histone H3 acetylation and HSP70 expression [60] | |||
| IIb | 6 | Tub‐A | x | Reduced neuronal death and infarct size via modulation of Akt/GSK3B and inhibition of mitochondrial apoptosis [29, 69] | |||
| Cardiac arrest | I, IIa | 1–5, 7–9 | VPA | x | Improved survival and neurologic outcome and decreased seizure burden [71, 72, 73] | ||
| Cardioprotection | |||||||
| Myocardial infarction | I, IIa | 1–5, 7–9 | VPA | x | Reduced infarct size, oxidative stress, cell death, and inflammatory response via upregulation of Foxm1 and fibrinolysis [75, 76, 77, 85] | ||
| I, IIa, IIb, IV | 1–11 | SAHA | x | x | Reduced infarct size, cell death, and preserved systolic function; induced autophagy and mitochondrial biogenesis [79, 80] | ||
| I, IIb, IV | 1–5, 7–9, 11 | TSA | xa | Reduced infarct size but failed to preserve contractile function and protect against oxidative stress [83, 84, 86] | |||
| IIb | 6 | Tub‐A | x | xa | In vitro model showed increased cell viability but was not cardioprotective in an ex vivo rat model [82, 86] | ||
| IIb | 3 | Entinostat | xa | Reduced infarct volume and preserved contractility [86] | |||
BDNF indicates brain‐derived neurotrophic factor; GSK3B, glycogen synthase kinase 3β; HDAC, histone deacetylase; HDACi, histone deacetylase inhibitors; HSP70, heat shock protein 70; I/R, ischemia/reperfusion; SAHA, suberoylanilide hydroxamic acid; SB, sodium butyrate; TSA, trichostatin A; Tub‐A, tubastatin A; VPA, valproic acid.
*Ex vivo rat perfusion model of myocardial infarction.
"x" indicates that the type of study for the given column is available for that given row's HDAC Class.
Epigenetic Modulation and HDACi
Epigenetic modulation alters gene expression through the transcriptional regulation of 3 primary mechanisms: DNA methylation, histone modification, and 3‐dimensional chromatin structural modulation.25 In the early studies of epigenetic modulation, I/R injury to the brain and heart were shown to cause a 40% decrease in histone H3 and H4 acetylation.1, 25, 26 The global hypoacetylation induced by I/R injury leads to chromatin condensation, which results in widespread decrease of anti‐inflammatory gene transcriptions, activation of apoptosis, and increase in proinflammatory cytokines such as TNF‐α (tumor necrosis factor α).26, 27 These effects are mediated through histone deacetylases (HDACs) or lysine deacetylases, enzymes that alter the epigenome and gene expression by removing acetyl groups from the tails of histone and nonhistone proteins.27
HDACi can act by targeting multiple classes (nonspecific) or an individual class (isoform‐specific) of HDACs. The latter has greater potential to induce more precise downstream effects.28, 29 HDACi may temporarily alter gene transcription by inhibiting the removal of acetyl groups, thereby promoting global hyperacetylation (Figure 2).28, 30, 31 In turn, this promotes prosurvival pathways in injured cells without disturbing normal cells due to the cell‐state–specific activity of HDACi.32, 33, 34, 35 In addition, HDACi may be acutely effective without inducing histone hyperacetylation by acting directly on lysine residues to regulate fatty acid oxidation and autophagy.36, 37, 38 Over the past 2 decades, research has focused on HDACi as an innovative treatment for various diseases, given the unique organ distribution and distinct physiologic function of HDACs.25, 27, 39 In humans, there are 4 classes of HDACs with total of 18 unique isoforms.27, 30 Because HDACs are overexpressed in many cancers, HDACi was first explored as a novel approach to anticancer therapy through the ability to decrease proliferating gene expression and increase cell‐cycle and apoptotic gene transcriptions.28, 30, 40, 41 To date, 20 distinct HDACi have been assessed in clinical trials for anticancer therapies.28
Figure 2. Epigenetic effects of histone deacetylase (HDAC) inhibition in the brain.
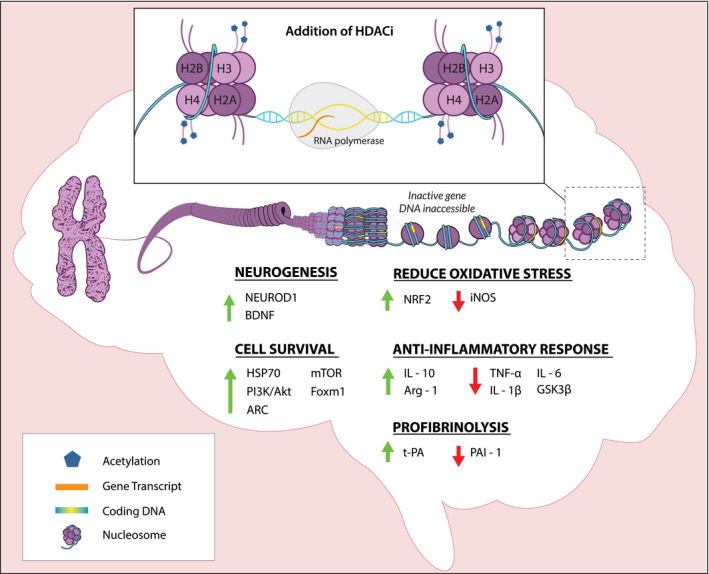
Chromatins are composed of negatively charged DNA wrapped around nucleosomes, each of which consists of 8 positively charged histones (2 sets of H2A, H2B, H3, and H4). HDAC inhibitors (HDACi) inhibit HDACs, resulting in the removal of positive charges on histone H3 and H4 through acetylation. The subsequent unwinding of DNA from the nucleosomes exposes genes to RNA polymerase for transcription. The relevant genes regulated by HDACi for neuroprotection after ischemia/reperfusion injury are highlighted. ARC indicates apoptosis repressor with caspase recruitment domain; BDNF, brain‐derived neurotrophic factor; Foxm1, forkhead box M1; GSK3B, glycogen synthase kinase 3β; HSP70, heat shock protein 70; IL, interleukin; iNOS, inducible nitric oxide synthase; mTOR, mechanistic target of rapamycin kinase; NEUROD1, neuronal differentiation 1; PAI‐1, plasminogen activator inhibitor 1; t‐PA, tissue‐type plasminogen activator.
HDACi has also been shown to be promising adjunctive therapy for trauma and sepsis resuscitation in preclinical studies.42 Early studies focused on nonspecific HDACi such as VPA and suberoylanilide hydroxamic acid (SAHA).42 Treatment with a single high‐dose VPA significantly increases survival and attenuates organ dysfunction in large animal models of polytrauma.43, 44, 45, 46 In traumatic brain injury and hemorrhagic shock, high‐dose VPA administered in the early postinjury period has been shown to reduce neurologic injury, expedite recovery, and improve long‐term neurologic outcome.47, 48, 49 In traumatic brain injury, VPA administration induces the activation of master transcription factors such as NEUROD1 (neuronal differentiation 1) and TBR1 (T‐box brain transcription factor 1) to mediate the expression of downstream neurogenic and neuroplastic genes.50 In sepsis, VPA and SAHA treatments improve survival and attenuate organ dysfunction.51, 52, 53 The mechanisms of action of HDACi in trauma and sepsis models have been discussed extensively elsewhere.54
HDACi for Neuroprotection Following I/R Injury
Stroke
Stroke is the second leading cause of death and the leading cause of long‐term disability in the United States.55 Nearly 80% of strokes are caused by ischemia, and the remainder are caused by hemorrhage.4 Stroke occurs by interruption or reduction in the blood supply to the brain, resulting in the deprivation of oxygen, glucose, and electrolytes to neuronal tissues.4 Focal cerebral ischemia is more common in stroke, whereas global ischemia is more prevalent in systemic hypoperfusion such as cardiac arrest and hemorrhagic shock.56
Small animal models of I/R injury have been used to demonstrate the neuroprotective effects of HDACi following stroke.57 In both transient focal and global ischemic rat models of stroke induced by middle cerebral artery occlusion (MCAO) and 4‐vessel occlusion, respectively, intraperitoneal administration of VPA (300 mg/kg) significantly decreased neurologic deficit score, neuronal death, infarct size, and blood–brain barrier disruption.58, 59, 60, 61, 62 Similarly, pre‐ and postinjury treatment with 300 mg/kg of intraperitoneal VPA attenuated infarct size in mouse models of transient MCAO I/R injury. However, only pretreatment with VPA reduced infarct size after permanent MCAO in mice.63 Intraperitoneal VPA injections also reduced hippocampal injury in a gerbil I/R model and decreased CASP1 (caspase 1), IL‐1β (interleukin 1β), and IL‐18.64 Multiple studies have shown that the protective effects of VPA after I/R injury are dependent on the time of drug administration following insult.58, 62, 63 For example, VPA was effective when administered within the first 3 hours following neuronal I/R injury but ineffective when administered after 4 hours in mouse models, making VPA treatment following I/R injury time‐sensitive but clinically feasible.60, 63
The neuroprotective effect of VPA after stroke is thought to be secondary to HDAC inhibition and upregulation of HSP70 (heat shock protein 70). VPA increases the levels of acetylated histones H3 and H4 and HSP70.58, 59, 60 Acetylated histones H3 and H4, in turn, promote prosurvival gene expression and inhibit microglia, GSK3B (glycogen synthase kinase 3β), hippocampal pyroptosis, and cerebral inflammation.58, 59, 63, 64, 65, 66 Furthermore, VPA decreases hippocampal sensitivity to inducible nitric oxide synthase in rat models of focal ischemia, thereby reducing oxidative stress and ROS generation.65 VPA also exhibits antipyroptotic protective effects by increasing the expression of ARC (apoptosis repressor with caspase recruitment domain).63 ARC expression decreases CASP1, IL‐1β, IL‐18, NLRP1 (NLR family pyrin domain containing 1), and NLRP3, which are all part of the pyroptotic neuronal cell injury pathway.64 VPA's protective effect was abolished when ARC was knocked down by small interfering RNA, further confirming ARC‐mediated protection by VPA.64 VPA combined with hypothermia results in synergistic protective effects in hippocampal neurons in vitro.67 Combined treatment significantly increases cell viability and histone H3 acetylation and suppresses the release of lactate dehydrogenase compared with either treatment alone.67 Acetylated histone H3 increases Akt phosphorylation, resulting in the downstream inactivation of GSK3B and expression of antiapoptotic genes.67
The neuroprotective effects of HDACi after stroke are not exclusive to VPA. Postinjury treatment with nonspecific HDACi such as VPA, sodium butyrate (SB), and trichostatin A (TSA) all significantly reduced neuronal injury in rat models of stroke.60 SB also promoted neuroprotective and neurogenic effects in rat models of neonatal postischemic brain injury, suggesting that HDACi could be useful therapy for neonatal I/R injury.68 The anti‐inflammatory and neuroprotective effects of SB are mediated in part through increased expression of oligodendrocyte progenitor cells, which attenuates the infiltration of both microglial cells and macrophage/monocytes.68 SB upregulates the expression of BDNF (brain‐derived neurotrophic factor) mRNA, which is believed to have neurogenic effects.68 Treatment of transient MCAO‐induced I/R brain injury at the onset of ischemia with 50 mg/kg intraperitoneal SAHA also resulted in neuroprotection, attenuation of cerebral inflammation, and promotion of a more protective microglial phenotype.66 Reverse transcriptase polymerase chain reaction confirmed that SAHA significantly reduced the transcription of proinflammatory cytokine IL‐1β, IL‐6, and TNF‐α and increased anti‐inflammatory cytokines IL‐10 and Arg‐1 (arginase 1).66 These mechanisms are thought to reduce microglial activation and monocyte infiltration, resulting in a more protective M2 microglial phenotype overall.66
Studies of isoform‐specific HDACi for neuroprotection after stroke are limited. Tubastatin A (Tub‐A), a class II specific HDAC6 inhibitor, is the most studied isoform‐specific HDACi for I/R injury. Tub‐A protects hippocampal neurons in vitro against injuries from oxygen–glucose deprivation by modulating Akt/GSK3B signaling and inhibiting mitochondria‐mediated apoptosis.69 Tub‐A also significantly reduces neuronal death and infarct size while increasing α‐tubulin and GAP43 (growth‐associated protein 43) acetylation, thus protecting neurons following photothrombotic infarction in mice.29 Further mechanistic and translational studies are needed to validate the efficacy of isoform‐specific HDACi in animal models of I/R injury.
Cardiac Arrest
More than 430 000 people experience cardiac arrest in the United States every year.4 Survival to hospital discharge for out‐of‐hospital cardiac arrest remains low at approximately 11%, and only 9% of survivors have good neurologic outcomes.4, 70 Furthermore, two‐thirds of out‐of‐hospital cardiac arrest and a quarter of in‐hospital cardiac arrest survivors die from neurologic injury.70 Despite increased awareness in bystander cardiopulmonary resuscitation and early defibrillation, there has been minimal improvement in cardiac arrest survival and survival with good neurologic outcome during the past 5 years.4
HDACi has been studied as a neuroprotective therapy following cardiac arrest. Studies of asphyxial cardiac arrest rat models have shown that intravenous high‐dose VPA (300 mg/kg) significantly improves survival and neurologic outcomes when administered immediately after return of spontaneous circulation.71, 72, 73 Specifically, high‐dose VPA combined with hypothermic targeted temperature resulted in significantly greater 72‐hour survival,71, 73 improved survival with good neurologic outcomes,71, 73 and decreased seizure burden compared with normothermia or hypothermic targeted temperature management alone.73 As such, the addition of high‐dose VPA to hypothermic targeted temperature management remains a promising therapy to improve cardiac arrest outcomes.71, 73
The mechanisms of VPA‐mediated neuroprotection after cardiac arrest are likely pleiotropic. High‐dose VPA induces epigenetic modulations through HDAC inhibition, antiepileptic properties,71, 72, 73 and prosurvival effects.54, 74 In addition, high‐dose VPA may also affect neurologic outcome through profibrinolysis. VPA has been shown to induce the release of t‐PA (tissue‐type plasminogen activator) in vitro and in rat models.75, 76 VPA has also been shown to significantly reduce the level of PAI‐1 (plasminogen activator inhibitor 1), thereby altering the t‐PA/PA‐1 ratio in favor of fibrinolysis.77 An observational study of patients with out‐of‐hospital cardiac arrest showed that impairment of fibrinolysis and generation of fibrin were associated with post–cardiac arrest syndrome.78 It is possible that VPA's profibrinolytic effects may result in reduction of microvascular thrombi formation, thereby reducing the no‐reflow phenomenon and end‐organ injury. Future translational research using clinically relevant large animal models of cardiac arrest are needed before HDACi can be adopted as a neuroprotective strategy for cardiac arrest survivors.
HDACi for Cardioprotection Following I/R Injury
Cardiovascular disease is the leading cause of death globally and accounts for 1 in 3 deaths in the United States.4 Specifically, myocardial infarction affects ≈8 million people and leads to 115 000 deaths each year in the United States.4, 55 Myocardial infarction causes cardiac ischemia through partial or full occlusion of blood flow to the myocardium.7 The brain is the organ most sensitive to oxygen and glucose deprivation. It sustains irreversible damage in <20 minutes of ischemia because of its high energetic demand and reliance on glucose.1 Although cardiac tissues are also highly sensitive to oxygen deprivation and can suffer irreversible damage after 20 minutes of ischemia, they do not exclusively rely on glucose as a energetic source.1 As such, cardiac tissue may have a longer therapeutic window than the brain, with studies demonstrating therapeutic intervention within the first 2 hours being most optimal and treatment within the first 12 hours of ischemia still associated with better outcomes.1 Given the clear clinical need to improve outcomes after myocardial infarction, HDACi have also been studied for cardioprotection after I/R injury.
Both nonspecific and isoform‐specific HDACi have been evaluated for cardioprotection using in vitro models of I/R injury. Cultured cardiomyocytes pretreated with nonspecific HDACi SAHA demonstrated a 40% reduction in cell death after I/R injury.79, 80 Further investigation suggested that SAHA treatment led to cardioprotection through the reduction of oxidative stress and induction of a protective macrophage phenotype. SAHA pretreatment of mouse cardiomyocytes in I/R injury led to fewer dysfunctional mitochondria, and this protection was lost when ATG7 (autophagy related 7) knockout myocytes were used.79 Moreover, 100 mg/kg of intraperitoneal SAHA administered immediately after myocardial infarction in mice increased the expression of M2 macrophages, thereby improving angiogenesis, wound healing, and left ventricle function.81 A similar study using isoform‐specific class IIb HDAC6 inhibitor Tub‐A on rat cardiomyocytes showed improved cell viability secondary to epigenetic modification.82 Tub‐A increased the expression of acetylated histone H3, PI3K/Akt antiapoptotic pathway, and mTOR (mechanistic target of rapamycin kinase) activation while decreasing autophagy and cellular cytotoxicity on reoxygenation.82
Several small and large animal studies have used the MCAO model to study the protective effects of nonspecific HDACi following myocardial infarction–induced I/R injury.80 Pretreatment with TSA, a nonspecific class I and IIb HDACi, reduced myocardial infarct size by nearly 50%, whereas TSA treatment 12 hours following myocardial infarction had only minimal impact on infarct size, suggesting an optimal therapeutic window for TSA.83, 84 SAHA pretreatment significantly reduced infarct size comparable to TSA in a rabbit model of myocardial infarction.37 High‐dose VPA treatment was able to significantly reduce infarct size and preserve systolic function in a rat model of myocardial infarction. The beneficial cardiac effects of VPA are mediated through the upregulation of Foxm1 (forkhead box M1), a regulator of neonatal cardiomyocyte proliferation and repressor of proinflammatory genes.85 High‐dose VPA may also promote fibrinolysis, thereby mediating a reduction of recurrent myocardial infarction in mice pretreated with intraperitoneal VPA.76
Recent work compared the cardioprotective effects of isoform‐specific HDACi such as entinostat (class I specific) and Tub‐A (class IIb HDAC6 isoform‐specific).86 In a rat model of global cardiac I/R injury, pretreatment with 10 mg/kg intraperitoneal entinostat protected the contractile function following I/R injury by upregulating antioxidant enzymes and decreasing ROS.86 In contrast, pretreatment with 10 mg/kg Tub‐A and 1 mg/kg TSA had no significant protective effect.86 The same group verified the protective effect of entinostat in an ex vivo rat heart I/R injury model.36 Furthermore, treatment with LL‐66, an isoform‐specific inhibitor of HDAC1 found in cardiomyocytes, was equally protective compared with entinostat.36 Such cardioprotection was shown to be mediated by the inhibition of fatty acid oxidation through succinate dehydrogenase.36 Additionally, a meta‐analysis assessing the efficacy of HDACi for organ protection in preclinical injury models of ischemia, trauma, and sepsis concluded that HDACi significantly reduced infarct size compared with untreated animals across injury models.57 These findings highlighted the heterogeneity of preclinical studies and the potential differences in the cardioprotective properties of isoform‐specific HDACi following I/R injury.57, 87
HDACi for Other Organ Protection After I/R injury
In addition to neuroprotection and cardioprotection, HDACi have also been evaluated for protective effects of other peripheral organs after I/R injury.31, 57 Whether I/R injury is a focal primary injury or secondary injury from global ischemia, HDACi may be a protective therapy for organs including the kidneys, lungs, liver, and retinas.
Kidneys
VPA is the major HDACi that has been evaluated as a protective agent after renal I/R injury induced by renal artery occlusions. Multiple studies revealed that VPA attenuates renal dysfunction and expedites renal recovery following I/R injury.88, 89, 90 Although the mechanism remains unclear, a major component appears to be the regulation of inflammation and apoptosis.89, 90 VPA treatment inhibits the proinflammatory response of macrophages and reduces apoptotic regulators such as TNF‐α after renal I/R injuries in rodent models.88, 89, 90
Lungs
HDACi has been evaluated as a pulmonary protective agent in several models of I/R injury. In a rat model of acute lung injury induced by superior mesenteric artery occlusion, VPA has been shown to improve survival and mitigate lung injury through ROS reduction and anti‐inflammatory effects.44 VPA treatment also provides pulmonary protection through the expression of antioxidant HMOX1 (heme oxygenase 1) after I/R injury.91 Blocking HMOX1 with zinc protoporphyrin IX mitigated the pulmonary protective effects of VPA, further supporting VPA's antioxidant mechanisms after pulmonary I/R injury.91
Liver
HDACi may also exhibit protective effects following hepatic I/R injury. Pre‐ and postinjury treatment with nonspecific HDACi SB has been shown to alleviate both partial and total occlusion‐induced hepatic I/R damage.92, 93, 94 SB treatment at the time of injury significantly attenuated hepatic damage, which is associated with the upregulation of histone H3 acetylation and HSP70.92 Despite evidence that supports HDACi as hepatoprotective, 1 study directly contrasted with these results. VPA (300 mg/kg) and SAHA (60 mg/kg) treatments after 90‐minute occlusion of left hepatic lobe failed to provide protection, and VPA treatment exacerbated I/R injury to the liver.95 However, these results might be explained by treatment dose, as VPA is known to be hepatotoxic at high doses and as rapid infusion.96, 97
Retina
HDACi is a potential retinal protective agent following I/R injury. In rat models of retinal I/R injury induced by increasing intraocular pressure, pretreatment with 300 mg/kg subcutaneous VPA reduced retinal damage.98 Subsequent studies demonstrated that VPA attenuates retinal injury by promoting antioxidant effects, antiapoptotic pathways via histone H3 acetylation, and the upregulation of HSP70.99, 100, 101
Future Translation
Most studies of HDACi for organ protection after I/R injury have been in vitro mechanistic studies or proof‐of‐concept studies in small animal models. As such, the logical next step is to use clinically relevant large animal models to validate the protective effects of HDACi. Although large animal studies pose unique challenges, this step is vital before HDACi can be safely translated to treat I/R injury in clinical trials.
Among the HDACi studied, high‐dose VPA has the most promising preclinical evidence supporting its use. A recent phase 1 dose‐escalation trial demonstrated that healthy human participants could tolerate up to 140 mg/kg VPA as a 1‐hour intravenous infusion with minimal side effects.102 Additional early phase clinical studies are necessary to validate VPA's pharmacokinetics, pharmacodynamics, safety, and efficacy in patients with stroke, cardiac arrest, or myocardial infarction. In addition, dose‐finding studies are also needed to determine the therapeutic window and minimal dose of HDACi required to achieve maximal protection. SAHA is another FDA‐approved HDACi with unique promise as a treatment for I/R injury. It is highly potent and has the ability to induce both autophagy and mitochondrial biogenesis.37, 79 Recent advancement in single‐cell sequencing offers a promising strategy to investigate the cell‐ and disease‐specific function of HDACi under different physiologic conditions. To further refine therapeutic interventions using precision medicine, high‐throughput genomic and proteomic studies can help elucidate the pharmacodynamics biomarkers that can best differentiate the treatment responders from nonresponders.
Given more limited side effect profiles, isoform‐specific HDACi could prove to be a superior treatment to nonspecific HDACi.28, 29 However, given that these HDACi are experimental and not already FDA‐approved like VPA, the timeline to translation will be significantly longer. We suspect that HDACi could even be adapted to other I/R injuries, including cardiac bypass surgery and organ transplantation, and as adjunctive therapy during extracorporeal cardiopulmonary resuscitation or aortic balloon occlusion during hemorrhagic shock.
Conclusions
I/R injury remains devastating and can cause irreversible damage to the brain, heart, and other vital organs without effective pharmacologic treatment. HDACi have shown promising protective properties in vitro and in small animal models of I/R injuries. They represent a high‐impact, low cost, and clinically feasible strategy for neuroprotection and cardioprotection. Specifically, high‐dose VPA improves clinical outcomes through pleiotropic effects triggered by HDAC inhibition and is the clear candidate for further clinical translation.
Sources of Funding
Dr Hsu reports research grant K12HL133304‐01 from the National Institutes of Health.
Disclosures
None.
Acknowledgments
The authors acknowledge Katelyn Murphy for her assistance with the figures in this article.
(J Am Heart Assoc. 2020;9:e016349 DOI: 10.1161/JAHA.120.016349.)
For Sources of Funding and Disclosures, see page 8.
References
- 1. Kalogeris T, Baines CP, Krenz M, Korthuis RJ. Cell biology of ischemia/reperfusion injury. Int Rev Cell Mol Biol. 2012;298:229–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ordy JM, Wengenack TM, Bialobok P, Coleman PD, Rodier P, Baggs RB, Dunlap WP, Kates B. Selective vulnerability and early progression of hippocampal CA1 pyramidal cell degeneration and GFAP‐positive astrocyte reactivity in the rat four‐vessel occlusion model of transient global ischemia. Exp Neurol. 1993;119:128–139. [DOI] [PubMed] [Google Scholar]
- 3. Neumar RW. Molecular mechanisms of ischemic neuronal injury. Ann Emerg Med. 2000;36:483–506. [DOI] [PubMed] [Google Scholar]
- 4. Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, de Ferranti SD, Floyd J, Fornage M, Gillespie C, et al. Heart disease and stroke statistics‐2017 update: a report from the American Heart Association. Circulation. 2017;135:e146–e603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bulkley GB. Free radical‐mediated reperfusion injury: a selective review. Br J Cancer Suppl. 1987;8:66–73. [PMC free article] [PubMed] [Google Scholar]
- 6. Sanada S, Komuro I, Kitakaze M. Pathophysiology of myocardial reperfusion injury: preconditioning, postconditioning, and translational aspects of protective measures. Am J Physiol Heart Circ Physiol. 2011;301:H1723–H1741. [DOI] [PubMed] [Google Scholar]
- 7. Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121–1135. [DOI] [PubMed] [Google Scholar]
- 8. White BC, Sullivan JM, DeGracia DJ, O'Neil BJ, Neumar RW, Grossman LI, Rafols JA, Krause GS. Brain ischemia and reperfusion: molecular mechanisms of neuronal injury. J Neurol Sci. 2000;179:1–33. [DOI] [PubMed] [Google Scholar]
- 9. Sekhon MS, Ainslie PN, Griesdale DE. Clinical pathophysiology of hypoxic ischemic brain injury after cardiac arrest: a “two‐hit” model. Crit Care. 2017;21:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kloner RA, Brown DA, Csete M, Dai W, Downey JM, Gottlieb RA, Hale SL, Shi J. New and revisited approaches to preserving the reperfused myocardium. Nat Rev Cardiol. 2017;14:679–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yemisci M, Gursoy‐Ozdemir Y, Vural A, Can A, Topalkara K, Dalkara T. Pericyte contraction induced by oxidative‐nitrative stress impairs capillary reflow despite successful opening of an occluded cerebral artery. Nat Med. 2009;15:1031–1037. [DOI] [PubMed] [Google Scholar]
- 12. Eltzschig HK, Eckle T. Ischemia and reperfusion—from mechanism to translation. Nat Med. 2011;17:1391–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schwartz BG, Kloner RA. Coronary no reflow. J Mol Cell Cardiol. 2012;52:873–882. [DOI] [PubMed] [Google Scholar]
- 14. Hypothermia after Cardiac Arrest Study Group . Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. N Engl J Med. 2002;346:549–556. [DOI] [PubMed] [Google Scholar]
- 15. Bernard SA, Gray TW, Buist MD, Jones BM, Silvester W, Gutteridge G, Smith K. Treatment of comatose survivors of out‐of‐hospital cardiac arrest with induced hypothermia. N Engl J Med. 2002;346:557–563. [DOI] [PubMed] [Google Scholar]
- 16. Kirkegaard H, Soreide E, de Haas I, Pettila V, Taccone FS, Arus U, Storm C, Hassager C, Nielsen JF, Sorensen CA, et al. Targeted temperature management for 48 vs 24 hours and neurologic outcome after out‐of‐hospital cardiac arrest: a randomized clinical trial. JAMA. 2017;318:341–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lascarrou JB, Merdji H, Le Gouge A, Colin G, Grillet G, Girardie P, Coupez E, Dequin PF, Cariou A, Boulain T, et al. Targeted temperature management for cardiac arrest with nonshockable rhythm. N Engl J Med. 2019;381:2327–2337. [DOI] [PubMed] [Google Scholar]
- 18. Nielsen N, Wetterslev J, Cronberg T, Erlinge D, Gasche Y, Hassager C, Horn J, Hovdenes J, Kjaergaard J, Kuiper M, et al. Targeted temperature management at 33 degrees C versus 36 degrees C after cardiac arrest. N Engl J Med. 2013;369:2197–2206. [DOI] [PubMed] [Google Scholar]
- 19. Nolan JP, Neumar RW, Adrie C, Aibiki M, Berg RA, Bottiger BW, Callaway C, Clark RS, Geocadin RG, Jauch EC, et al. Post‐cardiac arrest syndrome: epidemiology, pathophysiology, treatment, and prognostication. A Scientific Statement from the International Liaison Committee on Resuscitation; the American Heart Association Emergency Cardiovascular Care Committee; the Council on Cardiovascular Surgery and Anesthesia; the Council on Cardiopulmonary, Perioperative, and Critical Care; the Council on Clinical Cardiology; the Council on Stroke. Resuscitation. 2008;79:350–379. [DOI] [PubMed] [Google Scholar]
- 20. Polderman KH. Mechanisms of action, physiological effects, and complications of hypothermia. Crit Care Med. 2009;37:S186–S202. [DOI] [PubMed] [Google Scholar]
- 21. Polderman KH. Induced hypothermia and fever control for prevention and treatment of neurological injuries. Lancet. 2008;371:1955–1969. [DOI] [PubMed] [Google Scholar]
- 22. Oddo M, Rossetti AO. Predicting neurological outcome after cardiac arrest. Curr Opin Crit Care. 2011;17:254–259. [DOI] [PubMed] [Google Scholar]
- 23. Grossestreuer AV, Abella BS, Leary M, Perman SM, Fuchs BD, Kolansky DM, Beylin ME, Gaieski DF. Time to awakening and neurologic outcome in therapeutic hypothermia‐treated cardiac arrest patients. Resuscitation. 2013;84:1741–1746. [DOI] [PubMed] [Google Scholar]
- 24. Zanyk‐McLean K, Sawyer KN, Paternoster R, Shievitz R, Devlin W, Swor R. Time to awakening is often delayed in patients who receive targeted temperature management after cardiac arrest. Ther Hypothermia Temp Manag. 2017;7:95–100. [DOI] [PubMed] [Google Scholar]
- 25. Feinberg AP. The key role of epigenetics in human disease prevention and mitigation. N Engl J Med. 2018;378:1323–1334. [DOI] [PubMed] [Google Scholar]
- 26. Schweizer S, Meisel A, Marschenz S. Epigenetic mechanisms in cerebral ischemia. J Cereb Blood Flow Metab. 2013;33:1335–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10:32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Benedetti R, Conte M, Altucci L. Targeting histone deacetylases in diseases: where are we? Antioxid Redox Signal. 2015;23:99–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Demyanenko S, Berezhnaya E, Neginskaya M, Rodkin S, Dzreyan V, Pitinova M. Cyrilliclass II histone deacetylases in the post‐stroke recovery period‐expression, cellular, and subcellular localization‐promising targets for neuroprotection. J Cell Biochem. 2019. [DOI] [PubMed] [Google Scholar]
- 30. Kim HJ, Bae SC. Histone deacetylase inhibitors: molecular mechanisms of action and clinical trials as anti‐cancer drugs. Am J Transl Res. 2011;3:166–179. [PMC free article] [PubMed] [Google Scholar]
- 31. Halaweish I, Nikolian V, Georgoff P, Li Y, Alam HB. Creating a “Prosurvival Phenotype” through histone deacetylase inhibition: past, present, and future. Shock. 2015;44(suppl 1):6–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cheng X, Liu Z, Liu B, Zhao T, Li Y, Alam HB. Selective histone deacetylase 6 inhibition prolongs survival in a lethal two‐hit model. J Surg Res. 2015;197:39–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Minetti GC, Colussi C, Adami R, Serra C, Mozzetta C, Parente V, Fortuni S, Straino S, Sampaolesi M, Di Padova M, et al. Functional and morphological recovery of dystrophic muscles in mice treated with deacetylase inhibitors. Nat Med. 2006;12:1147–1150. [DOI] [PubMed] [Google Scholar]
- 34. Romagnani P, Lasagni L, Mazzinghi B, Lazzeri E, Romagnani S. Pharmacological modulation of stem cell function. Curr Med Chem. 2007;14:1129–1139. [DOI] [PubMed] [Google Scholar]
- 35. Routy JP. Valproic acid: a potential role in treating latent HIV infection. Lancet. 2005;366:523–524. [DOI] [PubMed] [Google Scholar]
- 36. Herr DJ, Baarine M, Aune SE, Li X, Ball LE, Lemasters JJ, Beeson CC, Chou JC, Menick DR. HDAC1 localizes to the mitochondria of cardiac myocytes and contributes to early cardiac reperfusion injury. J Mol Cell Cardiol. 2018;114:309–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xie M, Kong Y, Tan W, May H, Battiprolu PK, Pedrozo Z, Wang ZV, Morales C, Luo X, Cho G, et al. Histone deacetylase inhibition blunts ischemia/reperfusion injury by inducing cardiomyocyte autophagy. Circulation. 2014;129:1139–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McKinsey TA. Therapeutic potential for HDAC inhibitors in the heart. Annu Rev Pharmacol Toxicol. 2012;52:303–319. [DOI] [PubMed] [Google Scholar]
- 39. Kelly WK, O'Connor OA, Marks PA. Histone deacetylase inhibitors: from target to clinical trials. Expert Opin Investig Drugs. 2002;11:1695–1713. [DOI] [PubMed] [Google Scholar]
- 40. Smith KT, Workman JL. Histone deacetylase inhibitors: anticancer compounds. Int J Biochem Cell Biol. 2009;41:21–25. [DOI] [PubMed] [Google Scholar]
- 41. Zhang J, Zhong Q. Histone deacetylase inhibitors and cell death. Cell Mol Life Sci. 2014;71:3885–3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Williams AM, Bhatti UF, Biesterveld BE, Graham NJ, Chtraklin K, Zhou J, Dennahy IS, Kathawate RG, Vercruysse CA, Russo RM, et al. Valproic acid improves survival and decreases resuscitation requirements in a swine model of prolonged damage control resuscitation. J Trauma Acute Care Surg. 2019;87:393–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bambakidis T, Dekker SE, Liu B, Maxwell J, Chtraklin K, Linzel D, Li Y, Alam HB. Hypothermia and valproic acid activate prosurvival pathways after hemorrhage. J Surg Res. 2015;196:159–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kim K, Li Y, Jin G, Chong W, Liu B, Lu J, Lee K, Demoya M, Velmahos GC, Alam HB. Effect of valproic acid on acute lung injury in a rodent model of intestinal ischemia reperfusion. Resuscitation. 2012;83:243–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Causey MW, Miller S, Hoffer Z, Hempel J, Stallings JD, Jin G, Alam H, Martin M. Beneficial effects of histone deacetylase inhibition with severe hemorrhage and ischemia‐reperfusion injury. J Surg Res. 2013;184:533–540. [DOI] [PubMed] [Google Scholar]
- 46. Alam HB, Shuja F, Butt MU, Duggan M, Li Y, Zacharias N, Fukudome EY, Liu B, Demoya M, Velmahos GC. Surviving blood loss without blood transfusion in a swine poly‐trauma model. Surgery. 2009;146:325–333. [DOI] [PubMed] [Google Scholar]
- 47. Jepsen CH, deMoya MA, Perner A, Sillesen M, Ostrowski SR, Alam HB, Johansson PI. Effect of valproic acid and injury on lesion size and endothelial glycocalyx shedding in a rodent model of isolated traumatic brain injury. J Trauma Acute Care Surg. 2014;77:292–297. [DOI] [PubMed] [Google Scholar]
- 48. Halaweish I, Bambakidis T, Chang Z, Wei H, Liu B, Li Y, Bonthrone T, Srinivasan A, Bonham T, Chtraklin K, et al. Addition of low‐dose valproic acid to saline resuscitation provides neuroprotection and improves long‐term outcomes in a large animal model of combined traumatic brain injury and hemorrhagic shock. J Trauma Acute Care Surg. 2015;79:911–919; discussion 919. [DOI] [PubMed] [Google Scholar]
- 49. Jin G, Duggan M, Imam A, Demoya MA, Sillesen M, Hwabejire J, Jepsen CH, Liu B, Mejaddam AY, Lu J, et al. Pharmacologic resuscitation for hemorrhagic shock combined with traumatic brain injury. J Trauma Acute Care Surg. 2012;73:1461–1470. [DOI] [PubMed] [Google Scholar]
- 50. Higgins GA, Georgoff P, Nikolian V, Allyn‐Feuer A, Pauls B, Higgins R, Athey BD, Alam HE. Network reconstruction reveals that valproic acid activates neurogenic transcriptional programs in adult brain following traumatic injury. Pharm Res. 2017;34:1658–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li Y, Liu B, Zhao H, Sailhamer EA, Fukudome EY, Zhang X, Kheirbek T, Finkelstein RA, Velmahos GC, deMoya M, et al. Protective effect of suberoylanilide hydroxamic acid against LPS‐induced septic shock in rodents. Shock. 2009;32:517–523. [DOI] [PubMed] [Google Scholar]
- 52. Zhao T, Li Y, Liu B, Liu Z, Chong W, Duan X, Deperalta DK, Velmahos GC, Alam HB. Novel pharmacologic treatment attenuates septic shock and improves long‐term survival. Surgery. 2013;154:206–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Finkelstein RA, Li Y, Liu B, Shuja F, Fukudome E, Velmahos GC, deMoya M, Alam HB. Treatment with histone deacetylase inhibitor attenuates MAP kinase mediated liver injury in a lethal model of septic shock. J Surg Res. 2010;163:146–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Williams AM, Dennahy IS, Bhatti UF, Biesterveld BE, Graham N, Li Y, Alam HB. Histone deacetylase inhibitors: a novel strategy in trauma and sepsis. Shock. 2019;52:300–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Levine GN, Bates ER, Bittl JA, Brindis RG, Fihn SD, Fleisher LA, Granger CB, Lange RA, Mack MJ, Mauri L, et al. 2016 ACC/AHA Guideline Focused Update on Duration of Dual Antiplatelet Therapy in Patients With Coronary Artery Disease: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines: An Update of the 2011 ACCF/AHA/SCAI Guideline for Percutaneous Coronary Intervention, 2011 ACCF/AHA Guideline for Coronary Artery Bypass Graft Surgery, 2012 ACC/AHA/ACP/AATS/PCNA/SCAI/STS Guideline for the Diagnosis and Management of Patients With Stable Ischemic Heart Disease, 2013 ACCF/AHA Guideline for the Management of ST‐Elevation Myocardial Infarction, 2014 AHA/ACC Guideline for the Management of Patients With Non‐ST‐Elevation Acute Coronary Syndromes, and 2014 ACC/AHA Guideline on Perioperative Cardiovascular Evaluation and Management of Patients Undergoing Noncardiac Surgery. Circulation. 2016;134:e123–e155. [DOI] [PubMed] [Google Scholar]
- 56. Traystman RJ. Animal models of focal and global cerebral ischemia. ILAR J. 2003;44:85–95. [DOI] [PubMed] [Google Scholar]
- 57. Yusoff SI, Roman M, Lai FY, Eagle‐Hemming B, Murphy GJ, Kumar T, Wozniak M. Systematic review and meta‐analysis of experimental studies evaluating the organ protective effects of histone deacetylase inhibitors. Transl Res. 2019;205:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ren M, Leng Y, Jeong M, Leeds PR, Chuang DM. Valproic acid reduces brain damage induced by transient focal cerebral ischemia in rats: potential roles of histone deacetylase inhibition and heat shock protein induction. J Neurochem. 2004;89:1358–1367. [DOI] [PubMed] [Google Scholar]
- 59. Xuan A, Long D, Li J, Ji W, Hong L, Zhang M, Zhang W. Neuroprotective effects of valproic acid following transient global ischemia in rats. Life Sci. 2012;90:463–468. [DOI] [PubMed] [Google Scholar]
- 60. Kim HJ, Rowe M, Ren M, Hong JS, Chen PS, Chuang DM. Histone deacetylase inhibitors exhibit anti‐inflammatory and neuroprotective effects in a rat permanent ischemic model of stroke: multiple mechanisms of action. J Pharmacol Exp Ther. 2007;321:892–901. [DOI] [PubMed] [Google Scholar]
- 61. Wang Z, Leng Y, Tsai LK, Leeds P, Chuang DM. Valproic acid attenuates blood‐brain barrier disruption in a rat model of transient focal cerebral ischemia: the roles of HDAC and MMP‐9 inhibition. J Cereb Blood Flow Metab. 2011;31:52–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lv L, Tang YP, Han X, Wang X, Dong Q. Therapeutic application of histone deacetylase inhibitors for stroke. Cent Nerv Syst Agents Med Chem. 2011;11:138–149. [DOI] [PubMed] [Google Scholar]
- 63. Qian YR, Lee MJ, Hwang S, Kook JH, Kim JK, Bae CS. Neuroprotection by valproic acid in mouse models of permanent and transient focal cerebral ischemia. Korean J Physiol Pharmacol. 2010;14:435–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhu S, Zhang Z, Jia LQ, Zhan KX, Wang LJ, Song N, Liu Y, Cheng YY, Yang YJ, Guan L, et al. Valproic acid attenuates global cerebral ischemia/reperfusion injury in gerbils via anti‐pyroptosis pathways. Neurochem Int. 2019;124:141–151. [DOI] [PubMed] [Google Scholar]
- 65. Silva MR, Correia AO, Dos Santos GCA, Parente LLT, de Siqueira KP, Lima DGS, Moura JA, da Silva Ribeiro AE, Costa RO, Lucetti DL, et al. Neuroprotective effects of valproic acid on brain ischemia are related to its HDAC and GSK3 inhibitions. Pharmacol Biochem Behav. 2018;167:17–28. [DOI] [PubMed] [Google Scholar]
- 66. Li S, Lu X, Shao Q, Chen Z, Huang Q, Jiao Z, Huang X, Yue M, Peng J, Zhou X, et al. Early histone deacetylase inhibition mitigates ischemia/reperfusion brain injury by reducing microglia activation and modulating their phenotype. Front Neurol. 2019;10:893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Jin G, Liu B, You Z, Bambakidis T, Dekker SE, Maxwell J, Halaweish I, Linzel D, Alam HB. Development of a novel neuroprotective strategy: combined treatment with hypothermia and valproic acid improves survival in hypoxic hippocampal cells. Surgery. 2014;156:221–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ziemka‐Nalecz M, Jaworska J, Sypecka J, Polowy R, Filipkowski RK, Zalewska T. Sodium butyrate, a histone deacetylase inhibitor, exhibits neuroprotective/neurogenic effects in a rat model of neonatal hypoxia‐ischemia. Mol Neurobiol. 2017;54:5300–5318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Chang P, Tian Y, Williams AM, Bhatti UF, Liu B, Li Y, Alam HB. Inhibition of histone deacetylase 6 protects hippocampal cells against mitochondria‐mediated apoptosis in a model of severe oxygen‐glucose deprivation. Curr Mol Med. 2019;19:673–682. [DOI] [PubMed] [Google Scholar]
- 70. Laver S, Farrow C, Turner D, Nolan J. Mode of death after admission to an intensive care unit following cardiac arrest. Intensive Care Med. 2004;30:2126–2128. [DOI] [PubMed] [Google Scholar]
- 71. Lee JH, Kim K, Jo YH, Lee MJ, Hwang JE, Kim MA. Effect of valproic acid combined with therapeutic hypothermia on neurologic outcome in asphyxial cardiac arrest model of rats. Am J Emerg Med. 2015;33:1773–1779. [DOI] [PubMed] [Google Scholar]
- 72. Lee JH, Kim K, Jo YH, Lee SH, Kang C, Kim J, Park CJ, Kim MA, Lee MJ, Rhee JE. Effect of valproic acid on survival and neurologic outcomes in an asphyxial cardiac arrest model of rats. Resuscitation. 2013;84:1443–1449. [DOI] [PubMed] [Google Scholar]
- 73. Oh JS, Tulasi J, Xiaodan R, Stacey WC, Neumar RW. Valproic acid combined with postcardiac arrest hypothermic‐targeted temperature management prevents delayed seizures and improves survival in a rat cardiac arrest model. Crit Care Med. 2017;45:e1149–e1156. [DOI] [PubMed] [Google Scholar]
- 74. Soria‐Castro R, Schcolnik‐Cabrera A, Rodriguez‐Lopez G, Campillo‐Navarro M, Puebla‐Osorio N, Estrada‐Parra S, Estrada‐Garcia I, Chacon‐Salinas R, Chavez‐Blanco AD. Exploring the drug repurposing versatility of valproic acid as a multifunctional regulator of innate and adaptive immune cells. J Immunol Res. 2019;2019:9678098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Larsson P, Ulfhammer E, Magnusson M, Bergh N, Lunke S, El‐Osta A, Medcalf RL, Svensson PA, Karlsson L, Jern S. Role of histone acetylation in the stimulatory effect of valproic acid on vascular endothelial tissue‐type plasminogen activator expression. PLoS ONE. 2012;7:e31573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Larsson P, Alwis I, Niego B, Sashindranath M, Fogelstrand P, Wu MC, Glise L, Magnusson M, Daglas M, Bergh N, et al. Valproic acid selectively increases vascular endothelial tissue‐type plasminogen activator production and reduces thrombus formation in the mouse. J Thromb Haemost. 2016;14:2496–2508. [DOI] [PubMed] [Google Scholar]
- 77. Saluveer O, Larsson P, Ridderstrale W, Hrafnkelsdottir TJ, Jern S, Bergh N. Profibrinolytic effect of the epigenetic modifier valproic acid in man. PLoS ONE. 2014;9:e107582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wada T. Coagulofibrinolytic changes in patients with post‐cardiac arrest syndrome. Front Med (Lausanne). 2017;4:156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Yang J, He J, Ismail M, Tweeten S, Zeng F, Gao L, Ballinger S, Young M, Prabhu SD, Rowe GC, et al. HDAC inhibition induces autophagy and mitochondrial biogenesis to maintain mitochondrial homeostasis during cardiac ischemia/reperfusion injury. J Mol Cell Cardiol. 2019;130:36–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Xie M, Tang Y, Hill JA. HDAC inhibition as a therapeutic strategy in myocardial ischemia/reperfusion injury. J Mol Cell Cardiol. 2019;129:188–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kimbrough D, Wang SH, Wright LH, Mani SK, Kasiganesan H, LaRue AC, Cheng Q, Nadig SN, Atkinson C, Menick DR. HDAC inhibition helps post‐MI healing by modulating macrophage polarization. J Mol Cell Cardiol. 2018;119:51–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Williams AM, He W, Li Y, Bhatti UF, Nikolian VC, Chang P, Chang Z, Halaweish I, Liu B, Cheng X, et al. Histone deacetylase inhibition attenuates cardiomyocyte hypoxia‐reoxygenation injury. Curr Mol Med. 2018;18:711–718. [DOI] [PubMed] [Google Scholar]
- 83. Zhao TC, Cheng G, Zhang LX, Tseng YT, Padbury JF. Inhibition of histone deacetylases triggers pharmacologic preconditioning effects against myocardial ischemic injury. Cardiovasc Res. 2007;76:473–481. [DOI] [PubMed] [Google Scholar]
- 84. Granger A, Abdullah I, Huebner F, Stout A, Wang T, Huebner T, Epstein JA, Gruber PJ. Histone deacetylase inhibition reduces myocardial ischemia‐reperfusion injury in mice. FASEB J. 2008;22:3549–3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Tian S, Lei I, Gao W, Liu L, Guo Y, Creech J, Herron TJ, Xian S, Ma PX, Eugene Chen Y, et al. HDAC inhibitor valproic acid protects heart function through Foxm1 pathway after acute myocardial infarction. EBioMedicine. 2019;39:83–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Aune SE, Herr DJ, Mani SK, Menick DR. Selective inhibition of class I but not class IIb histone deacetylases exerts cardiac protection from ischemia reperfusion. J Mol Cell Cardiol. 2014;72:138–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Wang Z, Pagani FD, Chen Y, Alam H, Li Y. Valproic acid for treatment of myocardial ischemia. 2018.
- 88. Amirzargar MA, Yaghubi F, Hosseinipanah M, Jafari M, Pourjafar M, Rezaeepoor M, Rezaei H, Roshanaei G, Hajilooi M, Solgi G. Anti‐inflammatory effects of valproic acid in a rat model of renal ischemia/reperfusion injury: alteration in cytokine profile. Inflammation. 2017;40:1310–1318. [DOI] [PubMed] [Google Scholar]
- 89. Costalonga EC, Silva FM, Noronha IL. Valproic acid prevents renal dysfunction and inflammation in the ischemia‐reperfusion injury model. Biomed Res Int. 2016;2016:5985903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Speir RW, Stallings JD, Andrews JM, Gelnett MS, Brand TC, Salgar SK. Effects of valproic acid and dexamethasone administration on early bio‐markers and gene expression profile in acute kidney ischemia‐reperfusion injury in the rat. PLoS ONE. 2015;10:e0126622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Wu SY, Tang SE, Ko FC, Wu GC, Huang KL, Chu SJ. Valproic acid attenuates acute lung injury induced by ischemia‐reperfusion in rats. Anesthesiology. 2015;122:1327–1337. [DOI] [PubMed] [Google Scholar]
- 92. Sun J, Wu Q, Sun H, Qiao Y. Inhibition of histone deacetylase by butyrate protects rat liver from ischemic reperfusion injury. Int J Mol Sci. 2014;15:21069–21079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Liu B, Qian J, Wang Q, Wang F, Ma Z, Qiao Y. Butyrate protects rat liver against total hepatic ischemia reperfusion injury with bowel congestion. PLoS ONE. 2014;9:e106184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Qiao YL, Qian JM, Wang FR, Ma ZY, Wang QW. Butyrate protects liver against ischemia reperfusion injury by inhibiting nuclear factor kappa B activation in Kupffer cells. J Surg Res. 2014;187:653–659. [DOI] [PubMed] [Google Scholar]
- 95. Ruess DA, Probst M, Marjanovic G, Wittel UA, Hopt UT, Keck T, Bausch D. HDACi Valproic Acid (VPA) and Suberoylanilide Hydroxamic Acid (SAHA) delay but fail to protect against warm hepatic ischemia‐reperfusion injury. PLoS ONE. 2016;11:e0161233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Gopaul S, Farrell K, Abbott F. Effects of age and polytherapy, risk factors of valproic acid (VPA) hepatotoxicity, on the excretion of thiol conjugates of (E)‐2,4‐diene VPA in people with epilepsy taking VPA. Epilepsia. 2003;44:322–328. [DOI] [PubMed] [Google Scholar]
- 97. Kondo T, Ishida M, Kaneko S, Hirano T, Otani K, Fukushima Y, Muranaka H, Koide N, Yokoyama M, Nakata S, et al. Is 2‐propyl‐4‐pentenoic acid, a hepatotoxic metabolite of valproate, responsible for valproate‐induced hyperammonemia? Epilepsia. 1992;33:550–554. [DOI] [PubMed] [Google Scholar]
- 98. Zhang Z, Tong N, Gong Y, Qiu Q, Yin L, Lv X, Wu X. Valproate protects the retina from endoplasmic reticulum stress‐induced apoptosis after ischemia‐reperfusion injury. Neurosci Lett. 2011;504:88–92. [DOI] [PubMed] [Google Scholar]
- 99. Zhang Z, Qin X, Tong N, Zhao X, Gong Y, Shi Y, Wu X. Valproic acid‐mediated neuroprotection in retinal ischemia injury via histone deacetylase inhibition and transcriptional activation. Exp Eye Res. 2012;94:98–108. [DOI] [PubMed] [Google Scholar]
- 100. Zhang Z, Qin X, Zhao X, Tong N, Gong Y, Zhang W, Wu X. Valproic acid regulates antioxidant enzymes and prevents ischemia/reperfusion injury in the rat retina. Curr Eye Res. 2012;37:429–437. [DOI] [PubMed] [Google Scholar]
- 101. Zhang ZZ, Wu XW, Gong YY, Zhang W, Yin LL. [Protective effect of valproic acid on ischemia‐reperfusion induced injury in retina of rat]. Zhonghua Yan Ke Za Zhi. 2012;48:739–743. [PubMed] [Google Scholar]
- 102. Georgoff PE, Nikolian VC, Bonham T, Pai MP, Tafatia C, Halaweish I, To K, Watcharotone K, Parameswaran A, Luo R, et al. Safety and tolerability of intravenous valproic acid in healthy subjects: a phase I dose‐escalation trial. Clin Pharmacokinet. 2018;57:209–219. [DOI] [PMC free article] [PubMed] [Google Scholar]