

Abstract
Background
Alterations of energetic metabolism are suggested to be an important contributor to pressure overload (PO)‐induced heart failure. Our previous study reveals that knockout of endothelial Sirtuin 3 (SIRT3) alters glycolysis and impairs diastolic function. We hypothesize that endothelial SIRT3 regulates glucose utilization of cardiomyocytes and sensitizes PO‐induced heart failure.
Methods and Results
SIRT3 endothelial cell knockout mice and their control SIRT3 LoxP mice were subjected to PO by transverse aortic constriction for 7 weeks. The ratio of heart weight to tibia length was increased in both strains of mice, in which SIRT3 endothelial cell knockout mice+transverse aortic constriction exhibited more severe cardiac hypertrophy. Coronary blood flow and systolic function were significantly decreased in SIRT3 endothelial cell knockout mice+transverse aortic constriction compared with SIRT3 LoxP mice+transverse aortic constriction, as evidenced by lower systolic/diastolic ratio, ejection fraction, and fractional shortening. PO‐induced upregulation of apelin and glucose transporter 4 were significantly reduced in the hearts of SIRT3 endothelial cell knockout mice. In vitro, levels of hypoxia‐inducible factor‐1α and glucose transporter 1 and glucose uptake were significantly reduced in SIRT3 knockout endothelial cells. Furthermore, hypoxia‐induced apelin expression was abolished together with reduced apelin‐mediated glucose uptake in SIRT3 knockout endothelial cells. Exposure of cardiomyocyte with apelin increased expression of glucose transporter 1 and glucose transporter 4. This was accompanied by a significant increase in glycolysis. Supplement of apelin in SIRT3 knockout hypoxic endothelial cell media increased glycolysis in the cardiomyocytes.
Conclusions
Knockout of SIRT3 disrupts glucose transport from endothelial cells to cardiomyocytes, reduces cardiomyocyte glucose utilization via apelin in a paracrine manner, and sensitizes PO‐induced heart failure. Endothelial SIRT3 may regulate cardiomyocyte glucose availability and govern the function of the heart.
Keywords: apelin, endothelial Sirtuin 3, glucose transport, GLUT1, GLUT4, heart failure, transverse aortic constriction
Subject Categories: Heart Failure, Hypertrophy
Nonstandard Abbreviations and Acronyms
- CFR
coronary flow reserve
- CMD
coronary microvascular dysfunction
- EC
endothelial cell
- ECKO
endothelial‐specific knockout
- GLUT1
glucose transporter 1
- GLUT4
glucose transporter 4
- HIF‐1α
hypoxia‐inducible factor‐1α
- LV
left ventricular
- PO
pressure overload
- SIRT3
sirtuin 3
- TAC
transverse aortic constriction
- WT‐ECs
endothelial cells from wild‐type mice
CLINICAL Perspective
What Is New?
An energy sensor sirtuin 3 governs nutrient substrate transport in the endothelium.
Deficiency of sirtuin 3 in endothelial cells reduces hypoxia‐inducible factor‐1α and glucose transporter 1 expression, disrupts glucose uptake and transport in endothelial cells, and thus results in reduction of glucose utilization in cardiomyocytes via apelin paracrine mechanism.
Our study provides the first evidence that endothelial sirtuin 3 governs glucose utilization in cardiomyocytes, which may be a novel target for pressure overload–induced heart failure.
What Are the Clinical Implications?
Modulation of endothelial cell–mediated metabolic substrate transport to cardiomyocytes, thereby governing the function of the supplied heart by regulating nutrient availability to cardiomyocytes, could potentially be a therapeutic target for hypertensive heart failure.
Pressure overload (PO) such as caused by hypertension, advanced aging, and diabetes mellitus is the leading cause of heart failure and death in the United States, but the underlying mechanisms remain poorly understood. Coronary microvascular dysfunction has been shown to be highly prevalent in patients with heart failure. Endothelial dysfunction is considered as the central feature of advanced aging, hypertension, and diabetes mellitus, which has been attributed to the progression of heart failure.1, 2, 3 Endothelial cells have been reported to use glycolysis, instead of oxidative phosphorylation, to provide adjacent cardiomyocytes with the available oxygen.4, 5, 6, 7 Abnormalities in the development of cardiac structure and function are results of disruption of endothelial cell–cardiomyocyte communication and lead to cardiac dysfunction.4, 5, 8, 9, 10 Preexisting coronary microvascular dysfunction (CMD) is usually associated with advanced age, hypertension, and diabetes mellitus.11, 12, 13 CMD has been shown to increase the risk of cardiovascular events, which can be assessed by measuring coronary flow reserve (CFR).13, 14 Reduced CFR is also associated with increased myocardial infarction size, reduced left ventricular (LV) ejection fraction, adverse LV remodeling, and reduced long‐term survival.13 Therefore, novel therapeutic approaches for the treatment of heart failure should consider to target or improve CMD.
Sirtuins are Class III histone deacetylases that have been associated with regulating cellular functions.13 Sirtuin 3 (SIRT3), which is known to be localized in the mitochondria of organs with high metabolic activity, plays a critical role in energy homeostasis, cardiac remodeling, and heart failure.4, 13 The heart has been found to be protected from oxidative stress attributable to SIRT3‐related antioxidative mechanisms.4 Excessive cardiac fibrosis and reduced level of SIRT3 are characteristics of patients with failing hearts who have the cardiovascular disease.15 We have shown that decreased angiogenic capacity and increased reactive oxygen species formation as well as apoptosis in SIRT3‐deficient bone marrow cell–derived endothelial progenitor cells, which limited cardiac repair after myocardial ischemia. Interestingly, apelin, a proangiogenic factor, improves bone marrow cell–mediated post–myocardial infarction cardiac repair and systolic function through a mechanism that potentially involves the upregulation of SIRT3.16 SIRT3 expression was decreased and was associated with reduced capillary density and cardiac dysfunction in diabetic cardiomyopathy.17 In agreement with these findings, apelin gene therapy improves myocardial angiogenesis and alleviates heart failure in diabetic streptozocin, but not in streptozocin‐SIRT3 knockout mice.18 Further, specific knockout of SIRT3 in endothelial cells’ (ECs) impaired angiogenic properties of ECs, including decreased EC tube formation, migration, and aortic sprouting, is associated with an impaired hypoxic signaling pathway involving hypoxia‐inducible factor‐2, angiopoietin‐1, and vascular endothelial growth factor as well as reduced CFR and cardiac function.19 Despite these findings, more studies need to be completed to establish a clearer, comprehensive understanding functional role of endothelial SIRT3 deficiency in the development of CMD and heart failure. In the present study, we tested whether ablation of endothelial SIRT3‐disrupted endothelial glucose transport to cardiomyocytes promoted PO‐induced heart failure. We also investigated the paracrine effects of endothelial apelin in the regulation of cardiomyocyte glucose transporter, glucose uptake, and glycolysis.
Materials and Methods
The authors declare that all supporting data are available within the article.
Animals
All animals were fed laboratory standard chow and water and housed in individually ventilated cages in the Laboratory Animal Facilities at the University of Mississippi Medical Center. All protocols were approved by the Institutional Animal Care and Use Committee of the University of Mississippi Medical Center (Protocol ID: 1280B) and were in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Pub. No. 85‐23, Revised 1996). The procedures followed were in accordance with institutional guidelines.
The endothelial‐specific SIRT3 knockout (SIRT3 ECKO) mice were generated by using the Cre‐LoxP system as described in the previous study.19 Briefly, SIRT3flox/flox mice (exons 2 and 3 are flanked with LoxP sites, provided by Dr. Eric Verdin at Gladstone Institute of Virology and Immunology, University of California) were crossbred with (B6.FVB‐Tg(Cdh5‐cre)7Mlia/J transgenic mice (Jackson Laboratories) expressing Cre recombinase in vascular endothelial cells. The resulting Cdh5‐Cre/SIRT3flox/− heterozygous mutants were then mated with SIRT3flox/flox to obtain SIRT3 ECKO. Successful knockout was confirmed by tail DNA PCR analysis by using the following primers.
Floxed SIRT3 allele primers: Forward 5′‐TAC TGA ATA TCA GTG GGA ACG‐3′, Reverse 5′‐TGC AAC AAG GCT TTA TCT TCC‐3′; WT SIRT3 primer: Forward 5′‐CTT CTG CGG CTC TAT ACA CAG‐3′.
Cdh5‐Cre transgene primers: Forward 5′‐GCG GTC TGG CAG TAA AAA CTA TC‐3′, Reverse 5′‐GTG AAA CAG CAT TGC TGT CAC TT‐3′; internal positive control Forward 5′‐CTA GGC CAC AGA ATT GAA AGA TCT‐3′ and internal positive control Reverse 5′‐GTA GGT GGA AAT TCT AGC ATC ATC C‐3′.
Mouse Transverse Aortic Constriction Model
SIRT3 ECKO mice and their control mice (SIRT3 LoxP) were subjected to LV PO‐induced by transverse aortic constriction (TAC) for up to 7 weeks, following a previous procedure.20 Briefly, the mice were anesthetized with a single intraperitoneal injection of ketamine (50 mg/kg) and xylazine (10 mg/kg), and successful anesthesia was confirmed by lack of reflex to toe pinching. All hair in the neck and chest area was removed using a topical depilatory agent, and the area was cleaned with betadine and alcohol. The mice were then placed in a supine position on a heating pad, and the temperature was maintained at 37°C. A 2‐ to 3‐mm longitudinal cut was made in the proximal portion of the sternum at the level of the suprasternal notch. After the thyroid was retracted, the aortic arch was visualized under low power magnification. A 6‐0 silk suture was then placed under the aorta between the origin of the right innominate and the left common carotid arteries, followed by 2 loose knots around the transverse aorta. A small piece of a 25‐gauge blunt needle is placed parallel to the transverse aorta. A bent 25‐gauge blunt needle was then placed next to the aortic arch, and the suture was snugly tied around the needle and the aorta. Following ligation, the needle was quickly removed to yield a constriction of 0.51 mm in diameter. The rib cage and skin were closed, and mice were allowed to recover on a warming pad until they were fully awake. The sham procedure was identical except that the aorta was not ligated. Cardiac function and coronary blood flow reserve were evaluated by echocardiography at 1, 4, and 7 weeks after the surgery.
Echocardiography
A Vevo770 High‐Resolution In Vivo Micro‐Imaging System equipped with an RMV 710B scanhead (VisualSonics Inc, Toronto, ON, Canada) was used for the transthoracic echocardiograms on SIRT3 LoxP and SIRT3 ECKO as well as the SIRT3 LoxP mice+TAC and SIRT3 ECKO mice+TAC. Each mouse used in the echocardiogram study was anesthetized by inhalation of 1.5% to 2% isoflurane mixed with 100% medical oxygen administered with a vaporizer in an isolated chamber. Once the mouse was under anesthesia, maintenance of 1% to 1.5% of isoflurane was used and heart rate of ≈400 to 450 beats per minute was monitored. High‐frequency ultrasound imaging software (VisualSonics Inc) was used to analyze M‐mode cine loops for assessment of myocardial parameters and cardiac functions of the left ventricle including ejection fraction and fractional shortening.13
Transmitral inflow Doppler images were obtained in an apical 4‐chamber view using pulsed‐wave Doppler mode to measure the ratio of peak velocity of early to late filling of mitral inflow, isovolumic relaxation time, isovolumic contraction time, and ejection time.21 Myocardial performance index was calculated using the following formula: Myocardial performance index=(isovolumic relaxation time+isovolumic contraction time)/ejection time.
The cine loop of the left proximal coronary artery was visualized in a modified parasternal LV short‐axis view by inhalation of 1% isoflurane for baseline and 2.5% isoflurane for hyperemic conditions for CFR. The ratio of peak blood flow velocity during hyperemia to the peak blood flow velocity at baseline was used to calculate the CFR.19, 22, 23 The systolic/diastolic ratio was calculated by dividing the systolic peak velocity by the diastolic peak velocity at both nonhyperemia (1% isoflurane) and hyperemia (2.5% isoflurane). At the end of the experiments, heart weight/tibia length ratio was measured to determine cardiac hypertrophy.
Immunoblot Analysis
Protein extractions from heart ventricular samples were prepared in lysis buffer with protease inhibitor cocktail. Lysates were separated by SDS‐PAGE under reducing conditions and transferred to polyvinylidene fluoride membranes. The polyvinylidene fluoride membranes were probed with antibodies specific to hypoxia‐inducible factor‐1α (HIF‐1α) (GTX30647, GeneTex, Inc, Irvine, CA), angiopoietin‐1 (A0604, Sigma‐Aldrich, St Louis, MO), apelin (ab56585, Abcam, Cambridge, MA), glucose transporter 1 (GLUT1) and 4 (GLUT4) (2213S, Cell Signaling Technology, Danvers, MA), 6‐phosphofructo‐2‐kinase/fructose‐2,6‐biphosphate 3 and control glyceraldehyde 3‐phosphate dehydrogenase (2118, Cell Signaling Technology). Membranes were washed and incubated with an anti‐rabbit or anti‐mouse secondary antibody conjugated with horseradish peroxidase. Densitometries were analyzed using TINA 2.0 image analysis software.
Cardiomyocyte Cultured With EC‐Conditioned Media and Extracellular Flux Analysis
Rat cardiomyocyte cell line H9C2 (ATCC CRL1446) cells were grown in Dulbecco's Modified Eagle Medium (Sigma‐Aldrich) with the addition of 10% fetal bovine serum (Invitrogen, Carlsbad, CA), 2 mmol/L glutamine, 104×diluted 10 000 U/mL penicillin and 10 mg/mL streptomycin (Sigma‐Aldrich). Microvascular ECs were isolated from SIRT3 LoxP and ECKO mice and maintained in the EGM‐2 medium (Lonza, Walkersville, MD), as previously described.13, 19, 24 In cardiomyocyte cultured with EC‐conditioned media experiments, EC was exposed to hypoxia for 24 hours and culture media were collected, centrifuged, and filtered. Cardiomyocytes were then exposed to hypoxic media for 24 hours, and glycolysis was measured.
Glycolysis and mitochondrial function were determined using XFe24 an extracellular flux analyzer from Seahorse Bioscience (Billerica, MA), following the manufacturer's instructions. For the glycolysis test, the cardiomyocytes or ECs were seeded at 2.5×104 cells per well in 24‐well Seahorse XFe24 plates (Agilent Technologies, Santa Clara, CA) 1 day before the assay. The cells were then washed twice with unbuffered XF base medium (Agilent Technologies) and incubated at 37°C without CO2 for 1 hour. Three initial baseline measurements were performed by using the XFe24 Seahorse analyzer followed by injection of glucose (final concentration, 10 mmol/L) and 3 additional measurements. Extracellular acidification rate was calculated in the Wave@ software (Agilent Technologies) and normalized to cell numbers or protein content in each well. For the mitochondrial stress test, the unbuffered assay medium was supplemented with glucose (10 mmol/L), pyruvate (1 mmol/L), and glutamine (2 mmol/L). The baseline oxygen consumption rate was measured, followed by the sequential injection of the following inhibitors with indicated final concentration: oligomycin (1 μmol/L), cyanide p‐trifluoromethoxy‐phenylhydrazone (1 μmol/L) and rotenone/antimycin A (0.5 μmol/L). Basal oxygen consumption rate and ATP production were calculated from the raw data using the Seahorse report generator.
Glucose Uptake Assay
Glucose uptake was measured using a Glucose Uptake Glo‐assay kit from Promega Corporation (Madison, WI) according to the manufacturer's instructions. Briefly, 8×103 cells/well that were seeded in a 96‐well plate were washed with glucose‐free PBS and incubated with 50 μL of 2‐deoxyglucose (1 mmol/L) for 20 minutes at room temperature. The level of intracellular 2‐deoxyglucose‐6‐phosphate converted from 2‐deoxyglucose was detected by incubating with 100 μL of luciferase reagent containing nicotinamide adenine dinucleotide phosphate, glucose‐6‐phosphate dehydrogenase, reductase, and reductase substrate, for 3 hours at room temperature. Luminescence was recorded by using a Synergy H1 microplate reader (Bio‐Tek, Winooski, VT) with a 0.5‐second integration time. The rate of glucose uptake was plotted with a 2‐deoxyglucose‐6‐phosphate standard curve that was run parallel with the samples and calculated by the following formula:
Statistical Analysis
Data are presented as mean±SEM. The assumption of normality in both comparison groups were determined by normality and long‐normality test. Statistical significance was determined using Student unpaired 2‐tailed t test for comparisons between the 2 groups, 1‐way or 2‐way ANOVA followed by Tukey's post hoc test for multiple comparisons using GraphPad Prism 8.1.1 software. P<0.05 was considered as statistically significant.
Results
Ablation of Endothelial SIRT3 Worsens TAC‐Induced Cardiac Dysfunction
A successful TAC surgery was confirmed by echocardiography 1 week after the surgery. As shown in Figure 1A, successful surgical ligation of the transverse aorta led to an elevation in Doppler flow velocity in the right innominate artery but a decrease in flow velocity in the left common carotid artery. We found that the ratio of heart weight to tibia length was increased in both strains of mice following TAC surgery, indicating the development of cardiac hypertrophy, whereas SIRT3 ECKO mice+TAC exhibited more severe cardiac hypertrophy than SIRT3 LoxP mice+TAC (Figure 1B). Also, systolic function decreased after TAC with significant reduction in ejection fraction and fractional shortening in SIRT3 ECKO mice than those of controls at week 7 (Figure 1C and 1D). In contrast, diastolic function decreased after TAC in a similar manner in both strains of mice, despite that preexisting diastolic dysfunction was found in SIRT3 ECKO mice (Figure 1E and 1F).
Figure 1. Effects of pressure overloading induced by TAC on cardiac function.
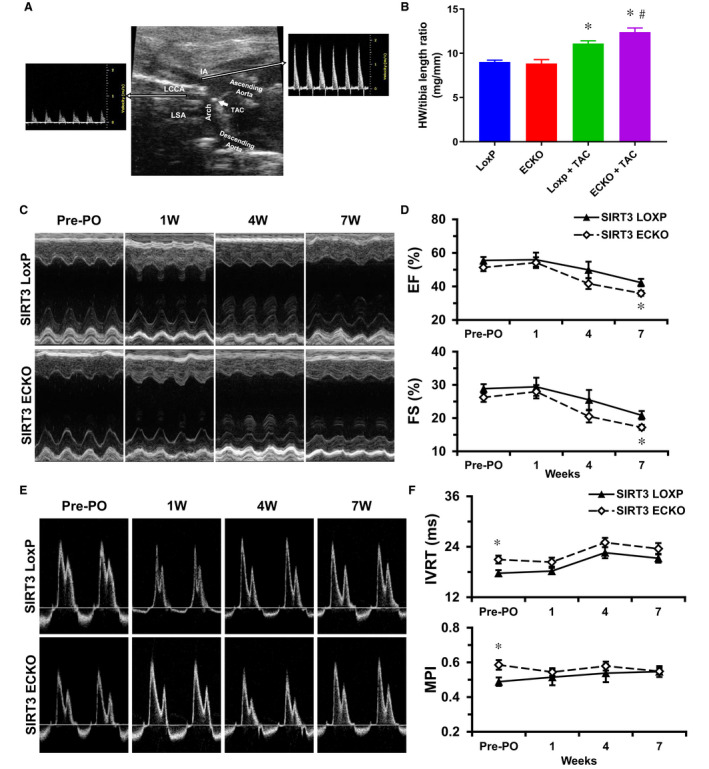
A, Successful surgical ligation of the transverse aorta was confirmed by an elevation in Doppler flow velocity in the right innominate artery but a decrease in flow velocity in the left common carotid artery. B, PO induced by TAC surgery led to increased ratio of heart weight to tibia length in both SIRT3 LoxP mice and SIRT3 ECKO mice. However, SIRT3 ECKO mice+TAC exhibited more severe cardiac hypertrophy than that of SIRT3 LoxP mice+TAC. *P<0.05 vs Sham; # P<0.05 vs SIRT3 LoxP+TAC. n=3 to 4 mice for sham groups, or 9 to 10 mice for TAC groups. C and D, Representative images of M‐mode of echocardiography; PO induced by TAC surgery led to decreased ejection fraction and fractional shortening in both SIRT3 LoxP mice and SIRT3 ECKO mice. However, SIRT3 ECKO+TAC mice exhibited more severe cardiac dysfunction than that of SIRT3 LoxP+TAC mice at 7W. *P<0.05 vs SIRT3 LoxP Baseline; # P<0.05 vs SIRT3 ECKO Baseline; † P<0.05 vs SIRT3 LoxP+TAC. n=9 to 10 mice. E and F, Representative images of transmitral inflow Doppler of left ventricle in an apical 4‐chamber view; PO induced by TAC surgery led to increased IVRT in both SIRT3 LoxP mice and SIRT3 ECKO mice in a similar manner, despite that preexisting diastolic dysfunction was found in SIRT3 ECKO mice. *P<0.05 vs SIRT3 LoxP. n=9 to 10 mice. ECKO indicates endothelial‐specific knockout; IVRT, isovolumic relaxation time; MPI, myocardial performance index; PO, pressure overload; SIRT3, sirtuin 3; and TAC, transverse aortic constriction.
Measurement of coronary blood flow, we found that PO resulted in elevated systolic/diastolic ratio at both nonhyperemic (baseline) and hyperemic conditions (Figure 2A through 2C). However, the nonhyperemic systolic/diastolic ratio was significantly higher in SIRT3 LoxP mice than that of SIRT3 ECKO mice after the TAC (Figure 2B).
Figure 2. Effects of pressure overloading induced by TAC on the blood flow of coronary artery.
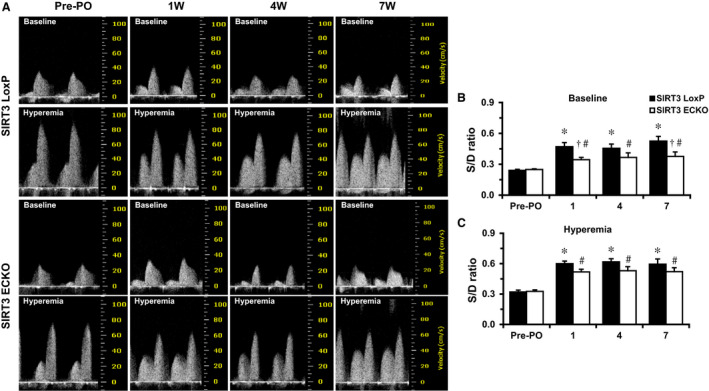
A through C, Representative images of coronary blood flow of left proximal coronary artery in a modified parasternal LV short‐axis view; Nonhyperemic S/D ratio is higher in SIRT3 LoxP mice after the TAC. PO resulted in elevated S/D ratio at both nonhyperemic (B) and hyperemic conditions (C). The nonhyperemic S/D ratio was significantly higher in SIRT3 LoxP mice than that of SIRT3 ECKO mice after the TAC surgeries (B). *P<0.05 vs SIRT3 LoxP baseline; # P<0.05 vs SIRT3 ECKO baseline; † P<0.05 vs SIRT3 LoxP+TAC. n=9 to 10 mice. ECKO indicates endothelial‐specific knockout; LV, left ventricular; O, pressure overload; S/D, systolic/diastolic; SIRT3, sirtuin 3; and TAC, transverse aortic constriction.
SIRT3 ECKO Reduces TAC‐Induced Apelin and GLUT4 Expression
Apelin is mainly produced in endothelial cells which increases proliferation of endothelial cells and the formation of new blood vessels. Angiopoietin‐1 is an important angiogenic growth factor with an important role in vascular maturation and angiogenesis. Thus, apelin and angiopoietin‐1 levels were examined as markers for angiogenesis. SIRT3 LoxP mice subjected to TAC resulted in a significant increase in apelin and Angiopoietin‐1 expression in the heart, but not in the SIRT3 ECKO mice+TAC (Figure 3A and 3B).
Figure 3. Knockout of endothelial SIRT3 impairs PO‐induced apelin and GLUT4 expression.
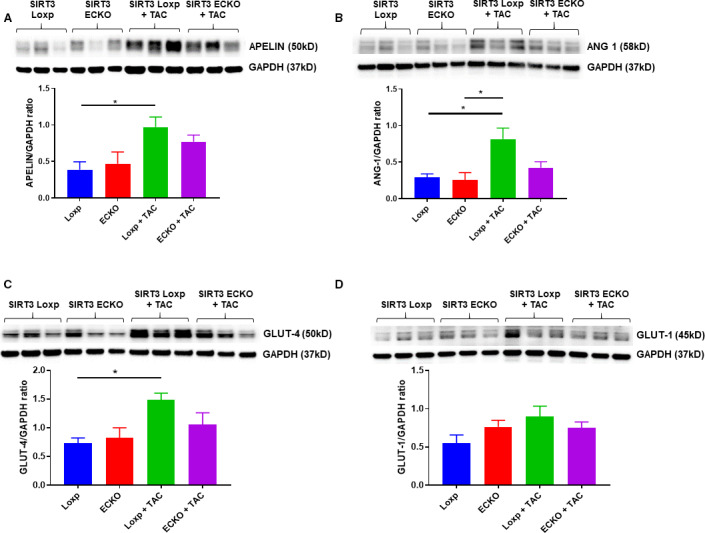
A and B, SIRT3 LoxP mice+TAC had a significant increase in apelin and angiopoietin‐1 expression, but failed to increase in SIRT3 ECKO mice+TAC (n=3 mice; *P<0.05). C and D, SIRT3 LoxP mice+TAC had a significant increase in GLUT4 expression, but failed to increase in SIRT3 ECKO mice (n=3 mice, *P<0.05). GLUT1 expression was increased but did not reach a significant difference. ECKO indicates endothelial‐specific knockout; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; GLUT4, glucose transporter 4; PO, pressure overload; SIRT3, sirtuin 3; and TAC, transverse aortic constriction.
GLUT1 and GLUT4 are 2 major isoforms of glucose transporter responsible for transport exogenous glucose in the blood through the EC and then delivery to the cardiomyocyte.25, 26 Both GLUT1 and GLUT4 levels are increased in human heart failure, which suggests an increase of glucose uptake and glycolysis in the hypertrophic human heart.25 As shown in Figure 3C, SIRT3 LoxP mice subjected to TAC resulted in a significant increase in GLUT4 expression, but not in SIRT3 ECKO mice+TAC. The GLUT1 expression was also upregulated but did not reach a significant difference in the hearts of SIRT3 LoxP mice+TAC (Figure 3D).
Deficiency of SIRT3 Reduces Expression of HIF‐1α and GLUT1 and Impairs Glucose Uptake in Endothelium
Study shows that glucose transport through endothelial cells requires both HIF‐1α and GLUT1.27 To determine whether SIRT3 ablation alters glucose transport in the endothelium, HIF‐1α and GLUT1 expression were examined in the cultured ECs isolated from control SIRT3 LoxP and SIRT3 ECKO mice. The expression of HIF‐1α and GLUT1 was significantly reduced in the ECs of SIRT3 ECKO mice (Figure 4A and 4B). However, GLUT4 expression remained unaltered in the ECs (Figure 4B). This was accompanied by a significant reduction of glucose uptake in SIRT3 knockout ECs (Figure 4C).
Figure 4. The expression of HIF‐1α and GLUT1 and glucose uptake in the cultured endothelial cells.
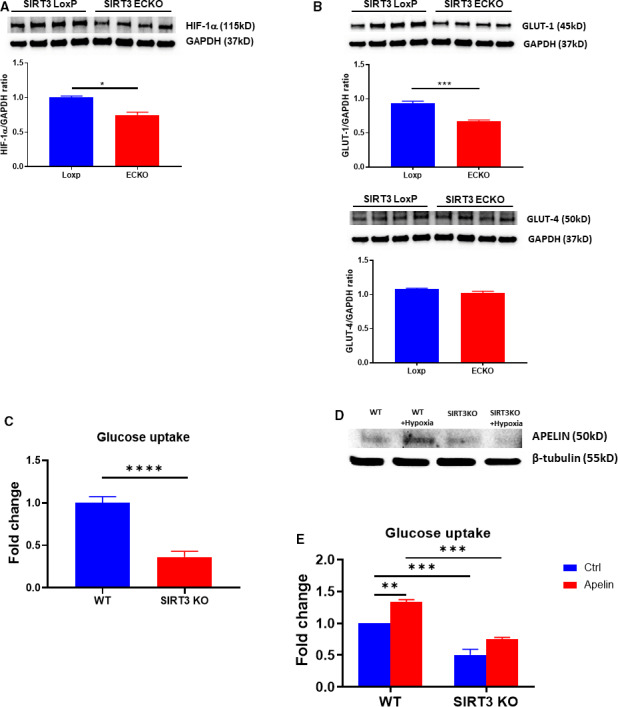
A and B, The expression of HIF‐1α and GLUT1 in cultured ECs. The expression of HIF‐1α and GLUT1 was significantly reduced in the EC isolated from SIRT3 ECKO mice compared with control ECs (*P<0.05 n=4 mice). GLUT4 levels were not significantly altered in EC. C, Glucose uptake in cultured ECs. The glucose uptake was significantly reduced in the ECs isolated from SIRT3 ECKO mice compared with ECs from wild‐type mice (****P<0.0001, n=5 mice). D, Exposure of WT‐ECs to hypoxia for 24 hours resulted in an upregulation of apelin expression. Hypoxia failed to induce apelin expression in the ECs isolated from SIRT3 ECKO mice. The WT‐ECs were isolated from a mouse and used for experiment. E, Treatment of WT‐EC with apelin (1 μmol/L) significantly increased glucose uptake. Apelin‐induced glucose uptake was significantly reduced in the ECs isolated from SIRT3 ECKO mice (n=4 mice, **P<0.01, ***P<0.001). ECKO indicates endothelial‐specific knockout; ECs, endothelial cells; GLUT1, glucose transporter 1; GLUT4, glucose transporter 4; HIF‐1α, hypoxia‐inducible factor‐1α; SIRT3, sirtuin 3; and WT‐ECs, endothelial cells from wild‐type mice.
Deficiency of SIRT3 Abolishes Hypoxia‐Induced Apelin Expression and Reduces Apelin‐Induced Glucose Uptake in Endothelium
Since the expression of apelin was significantly reduced in SIRT3 ECKO+TAC mice, we further examined the expression of apelin in cultured SIRT3 knockout ECs in response to hypoxia for 24 hours. Exposure of ECs from wild‐type mice (WT‐ECs) to hypoxia resulted in an increase in expression of apelin, while hypoxia failed to upregulate apelin expression in SIRT3 knockout ECs (Figure 4D). Exposure of WT‐ECs to apelin (1 μmol/L) resulted in a significant increase in glucose uptake, whereas apelin (1 μmol/L) failed to increase glucose uptake in SIRT3 knockout ECs (Figure 4E).
Apelin Upregulates Glucose Transporter GLUT1 and GLUT4 Expression and Promotes Glycolytic Function in Cardiomyocytes
We further tested whether endothelial derived apelin was attributed to the downregulation of GLUT4 and glycolysis in the cardiomyocytes. As shown in Figure 5A, exposure of cardiomyocyte cell line H9C2 with apelin (1 and 5 μmol/L) resulted in a gradual increase in the expression of GLUT1 and GLUT4. Similarly, the expression of 6‐phosphofructo‐2‐kinase/fructose‐2,6‐biphosphate 3 was upregulated in a time‐dependent manner. Furthermore, exposure of cardiomyocyte cell line with apelin (0.1 and 1 μmol/L) for 24 hours led to a significant increase in the basal glycolysis and glycolytic capacity (Figure 5B). In addition, basal oxygen consumption and ATP production were dose‐dependently increased in the cardiomyocyte cell line following 24 hours apelin (0.1–5 μmol/L) exposure (Figure 5C).
Figure 5. Apelin increases GLUT1 and GLUT4 expression and promotes glycolytic function in cardiomyocytes.
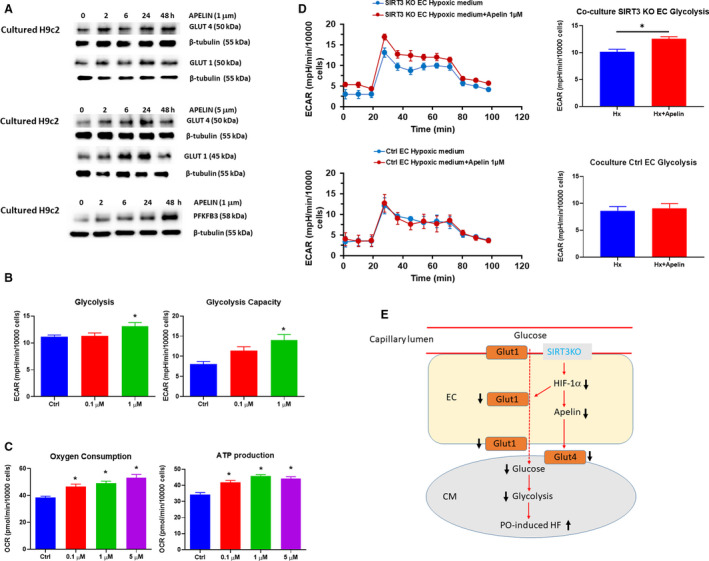
A, Exposure of cardiomyocyte cell line with apelin (1 and 5 μmol/L) for various time up to 48 hours resulted in a gradual increase in the expression of GLUT1, GLUT4, and PFKFB3. B, Exposure of cardiomyocyte cell line with apelin for 24 hours resulted in a significant increase in glycolysis. n=5 independent experiments *P<0.05 vs control vehicle. C, Exposure of cardiomyocyte with apelin (0.1–5 μmol/L) for 24 hours resulted in a dose dependent increase in basal oxygen consumption and ATP production. n=3 independent experiments, *P>0.05 vs control vehicle. D, In cardiomyocytes cultured with control ECs or SIRT3 knockout EC hypoxic‐conditioned media supplement with apelin, the glycolysis was measured in the cardiomyocytes. Supplement with apelin (1 μmol/L) in SIRT3 knockout EC hypoxic‐conditioned media resulted in a significant increase glycolysis in the cardiomyocytes. Supplement with apelin (1 μmol/L) in control EC hypoxic‐conditioned media had little effect on cardiomyocyte glycolytic metabolism. n=5 independent experiments *P<0.05 vs SIRT3 ECKO medium without apelin. E, Proposal working model of endothelial SIRT3 governs nutrient substrate transport in the endothelium and heart failure. Knockout of SIRT3 in ECs leads to downregulation of HIF‐1α and GLUT1 expression, thus impairs glucose uptake and transport to cardiomyocytes, which may reduce glucose availability and utilization of cardiomyocytes. These eventually worsen PO‐induced cardiac hypertrophy and heart failure. Knockout of SIRT3 reduces endothelial apelin expression, which may contribute to controlling cardiomyocyte glucose transport, glycolysis, and angiogenesis via a paracrine mechanism. ECAR indicates extracellular acidification rate; ECKO, endothelial‐specific knockout; ECs, endothelial cells; GLUT1, glucose transporter 1; GLUT4, glucose transporter 4; HF, heart failure; HIF‐1α, hypoxia‐inducible factor‐1α; PFKFB3, 6‐phosphofructo‐2‐kinase/fructose‐2,6‐biphosphate 3; SIRT3, sirtuin 3; and WT‐ECs, endothelial cells from wild‐type mice.
Apelin Increases Glycolysis in Cardiomyocyte Cultured With SIRT3 knockout EC–Conditioned Media
To further determine the paracrine action of endothelial derived apelin on cardiomyocyte glycolysis, we mimic in vivo conditions by culture cardiomyocyte cell line with WT‐ECs and SIRT3 knockout EC hypoxic conditioned media supplement with or without apelin. The glycolysis was measured in the cardiomyocyte cell line. As shown in Figure 5D, supplement with apelin (1 μmol/L) in SIRT3 ECKO hypoxic conditioned media resulted in a significant increase glycolysis in the cardiomyocytes. Surprisingly, supplement with apelin in control EC hypoxic conditioned media had little effect on cardiomyocyte glycolytic metabolism.
Discussion
Hypertension elevates blood vessel resistance and leads to PO in the heart, which plays a key role in the development of heart failure, similar to the PO‐induced heart failure in our study. In this study, PO induced by TAC resulted in cardiac hypertrophy and systolic and diastolic dysfunction over time. PO also increases genes associated with glucose uptake and angiogenesis but is subsequently attenuated in SIRT3 ECKO mice. Our results indicate that SIRT3 could potentially be a therapeutic target for patients with hypertensive heart failure.
Mice deficient in SIRT3 have been characterized by an accelerated aging process as well as the development of cancer, metabolic, and cardiovascular disease.13, 28, 29, 30, 31 Decline of SIRT3 and alterations in the coronary vascular homeostasis and myocardial blood flow predispose the heart to be more susceptible to pathological conditions, including myocardial infarction.13 In addition, if the coronary flow is impaired that will result in insufficient perfusion to meet the myocardial demand, it will lead to possible myocardial hypoxia and hypertrophy.13, 32 Our results obtained with the echocardiography and Doppler images showed a reduced coronary blood flow and more severe cardiac hypertrophy in SIRT3 ECKO mice+TAC than that of SIRT3 LoxP mice+TAC at 7 weeks with significantly lower ejection fraction and fractional shortening. Consistent with our previous studies, diastolic dysfunction exhibited in SIRT3 ECKO mice with the elevation of isovolumic relaxation time and myocardial performance index. However, SIRT3 ECKO mice+TAC had similar diastolic dysfunction as compared with SIRT3 LoxP mice+TAC over the 7 weeks of study. Our study indicates a critical role of SIRT3 in endothelium on cardiac systolic dysfunction but not the diastolic function under PO.
During myocardial ischemia or PO, mitochondrial oxidative phosphorylation is no longer a reliable source of ATP generation when oxygen supply is limited and depleted. In response to low oxygen partial pressure, increasing glucose uptake, upregulation of glucose transport, and glycolysis are achieved by activation of many signaling pathways of glucose metabolism and to maintain energy homeostasis.13 The endothelium plays a critical role in the regulation of tissue glucose metabolism. GLUT1 is widely expressed in endothelial cells and is the major transporter of exogenous glucose across the endothelial cells.27 GLUT4 is found primarily in cardiomyocyte and skeletal muscle.27 Both GLUT1 and GLUT4 were found to be expressed in the heart. Exogenous glucose in the circulation was first transported into the endothelial cell via GLUT1,27, 33, 34 where it was further transported to cardiomyocytes by GLUT4.27, 33, 34 Studies have shown that cardiac‐specific knockout of GLUT4 resulted in more severe heart failure in response to pressure overload,35, 36 whereas cardiac‐specific overexpression of GLUT1 protected the heart against pressure overload‐induced LV hypertrophy.35, 37, 38, 39, 40 Our data showed that GLUT4 protein expression was significantly increased in the mouse heart subjected to TAC; in line with that, there is a metabolic shift from the use of fatty acids for oxidative phosphorylation to the use of glucose for glycolysis to produce ATP in cardiomyocytes during PO.41 We reasoned that GLUT4 expression was significantly declined in the SIRT3 ECKO mice+TAC, which may contribute to, at least in part, exacerbation of LV hypertrophy and heart failure. Previously, we have shown that knockout of SIRT3 in ECs increased glycolytic enzyme 6‐phosphofructo‐2‐kinase/fructose‐2,6‐biphosphate 3 acetylation and disrupted glycolytic metabolism, and thus led to CMD in the mouse heart.19 Both endothelial HIF‐1α and GLUT1 have been shown to be necessary for glucose transport through endothelial cells. GLUT1 expression was mainly upregulated by endothelial HIF‐1α.27 The specific knockout of endothelial HIF‐1α reduced GLUT1 expression and inhibited glucose uptake in the brain and heart.27 HIF‐1α‐GLUT1 axis is considered an important pathway by which the endothelium regulates glucose delivery to tissues such as heart and brain. To better understand the role of SIRT3 on EC glucose transport and glycolytic metabolism, we specifically investigated the expression of HIF‐1α and GLUT1 in the cultured ECs. We found that the expression of HIF‐1α and GLUT1 was dramatically reduced in SIRT3 knockout ECs. Furthermore, EC glucose uptake was reduced about 70% as compared with normal ECs. Similarly, apelin‐induced glucose uptake was significantly reduced in ECs isolated from SIRT3 knockout mice. These results further support our hypothesis that SIRT3 deficiency impairs EC glucose uptake and glucose transport from ECs to cardiomyocytes. Because of the unique localization of ECs that is between the glucose in the circulation and cardiomyocytes, disruption of glucose uptake such as reduced GLUT1 within the endothelium may lower the rate of exogenous glucose being transported to the cardiomyocytes, thereby reducing glucose availability and utilization of cardiomyocytes. Eventually, this may cause CFR reduction and cardiac dysfunction in SIRT3 ECKO mice under PO. Although our data revealed that both GLUT1 and GLUT4 are impaired, which are crucial for glucose transport through ECs or cardiomyocytes, the underlying mechanisms by which SIRT3 regulates glucose transport have not been elucidated. Further studies are warranted to investigate protein acetylation in glucose transport.
Accumulating evidence demonstrates that cardiac function is maintained by the critical EC and cardiomyocyte interactions.13, 42, 43 By increasing angiogenesis and releasing regulatory proteins, such as apelin and angiopoietins, ECs support cardiomyocyte function.4, 5, 8, 43 Although the roles of EC‐mediated angiogenesis on cardiomyocyte growth have been well documented, few studies investigated the crosstalk of ECs and cardiomyocytes on cardiomyocyte energetic metabolisms. For the first time, we demonstrated a paracrine action of endothelial SIRT3‐apelin axis in the regulation of cardiomyocyte glucose transporter and glycolysis. Our data showed that PO‐induced apelin was significantly reduced in SIRT3 ECKO mice. Furthermore, endothelial‐derived apelin was completely abolished in SIRT3 knockout EC exposure to hypoxia. In contrast, treatment of cardiomyocytes with apelin led to a gradual upregulation of GLUT1, GLUT4, and glycolytic enzyme 6‐phosphofructo‐2‐kinase/fructose‐2,6‐biphosphate 3 together with increased glycolysis. So far, the underlying mechanism of apelin‐regulated glucose transport remains unknown. Our previous study showed that apelin increased SIRT3 expression in diabetic hearts.17 Whether apelin increases glucose transport via SIRT3 warranted further investigation. In the present study, apelin treatment also significantly improved mitochondrial oxygen consumption and ATP production in cardiomyocytes. Most intriguingly, cardiomyocytes cultured with SIRT3 knockout EC hypoxic‐conditioned media supplement with apelin mimicked in vivo conditions and significantly improved cardiomyocyte glycolysis, but this was not seen in the cardiomyocytes cultured with WT‐EC hypoxic‐conditioned media. These novel findings indicated an essential role of endothelial apelin paracrine action and EC‐cardiomyocyte interactions on glucose utilization and energetic metabolism of cardiomyocytes in SIRT3 ECKO mice.
Our previous study demonstrated that the expression of the two key angiogenic growth factors, angiopoietin‐1 and vascular endothelial growth factor R2, was dramatically reduced in SIRT3 knockout mice, indicating that there was impairment of angiogenic capabilities.13, 17 Similarly, results from the present study using SIRT3 ECKO mice showed an increase in angiogenesis in the TAC mice as evidenced by an increase in apelin and angiopoietin‐1 protein expression levels, whereas SIRT3 ECKO mice failed to do so. Taken together, these alterations may be attributed to worsening PO‐induced cardiac hypertrophy and heart failure seen in the SIRT3 ECKO mice.
In conclusion, our study provides more of an understanding of the energy sensor SIRT3 controlling substrate transport in the endothelium. Our study provides, to our knowledge, the first evidence that endothelial SIRT3 controls glucose uptake and transport to cardiomyocytes, which may regulate glucose availability and utilization of cardiomyocytes. Also, ablation of endothelial SIRT3 downregulates apelin, which may contribute to reduction of cardiomyocyte glucose transport, glycolysis, and angiogenesis via a paracrine mechanism; these eventually worsen PO‐induced cardiac hypertrophy and heart failure (Figure 5E). Therefore, modulation of endothelial‐mediated metabolic substrate transport to cardiomyocytes, thereby governing the function of the supplied heart by regulating nutrient availability to cardiomyocytes, could potentially be a therapeutic target for heart failure.
Sources of Funding
This work was supported by the National Heart, Lung, and Blood Institute 2R01HL102042‐7 and University of Mississippi Medical Center Intramural Research Support Program to Dr Chen.
Disclosures
None.
(J Am Heart Assoc. 2020;9:e015895 DOI: 10.1161/JAHA.120.015895.)
For Sources of Funding and Disclosures, see page 11.
References
- 1. Hadi HA, Suwaidi JA. Endothelial dysfunction in diabetes mellitus. Vasc Health Risk Manag. 2007;853–876. [PMC free article] [PubMed] [Google Scholar]
- 2. Park KH, Park WJ. Endothelial dysfunction: clinical implications in cardiovascular disease and therapeutic approaches. J Korean Med Sci. 2015;1213–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tang EH, Vanhoutte PM. Endothelial dysfunction: a strategic target in the treatment of hypertension? Pflugers Arch. 2010;995–1004. [DOI] [PubMed] [Google Scholar]
- 4. He X, Zeng H, Chen JX. Emerging role of SIRT3 in endothelial metabolism, angiogenesis, and cardiovascular disease. J Cell Physiol. 2019;2252–2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wan A, Rodrigues B. Endothelial cell‐cardiomyocyte crosstalk in diabetic cardiomyopathy. Cardiovasc Res. 2016;172–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong BW, Cantelmo AR, Quaegebeur A, Ghesquiere B, Cauwenberghs S, Eelen G, et al. Role of PFKFB3‐driven glycolysis in vessel sprouting. Cell. 2013;651–663. [DOI] [PubMed] [Google Scholar]
- 7. Vallerie SN, Bornfeldt KE. Metabolic flexibility and dysfunction in cardiovascular cells. Arterioscler Thromb Vasc Biol. 2015;e37–e42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lim SL, Lam CS, Segers VF, Brutsaert DL, De Keulenaer GW. Cardiac endothelium‐myocyte interaction: clinical opportunities for new heart failure therapies regardless of ejection fraction. Eur Heart J. 2015;2050–2060. [DOI] [PubMed] [Google Scholar]
- 9. Wang Y, Zhang D, Chiu AP, Wan A, Neumaier K, Vlodavsky I, Rodrigues B. Endothelial heparanase regulates heart metabolism by stimulating lipoprotein lipase secretion from cardiomyocytes. Arterioscler Thromb Vasc Biol. 2013;894–902. [DOI] [PubMed] [Google Scholar]
- 10. Wang F, Zhang D, Wan A, Rodrigues B. Endothelial cell regulation of cardiac metabolism following diabetes. Cardiovasc Hematol Disord Drug Targets. 2014;121–125. [DOI] [PubMed] [Google Scholar]
- 11. Niccoli G, Scalone G, Lerman A, Crea F. Coronary microvascular obstruction in acute myocardial infarction. Eur Heart J. 2016;1024–1033. [DOI] [PubMed] [Google Scholar]
- 12. Camici PG, Crea F. Coronary microvascular dysfunction. N Engl J Med. 2007;830–840. [DOI] [PubMed] [Google Scholar]
- 13. He X, Zeng H, Chen JX. Ablation of SIRT3 causes coronary microvascular dysfunction and impairs cardiac recovery post myocardial ischemia. Int J Cardiol. 2016;349–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fordyce CB, Gersh BJ, Stone GW, Granger CB. Novel therapeutics in myocardial infarction: targeting microvascular dysfunction and reperfusion injury. Trends Pharmacol Sci. 2015;605–616. [DOI] [PubMed] [Google Scholar]
- 15. Sundaresan NR, Bindu S, Pillai VB, Samant S, Pan Y, Huang JY, Gupta M, Nagalingam RS, Wolfgeher D, Verdin E, et al. SIRT3 blocks aging‐associated tissue fibrosis in mice by deacetylating and activating glycogen synthase kinase 3beta. Mol Cell Biol. 2015;678–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li L, Zeng H, Hou X, He X, Chen JX. Myocardial injection of apelin‐overexpressing bone marrow cells improves cardiac repair via upregulation of Sirt3 after myocardial infarction. PLoS One. 2013;e71041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zeng H, He X, Hou X, Li L, Chen JX. Apelin gene therapy increases myocardial vascular density and ameliorates diabetic cardiomyopathy via upregulation of sirtuin 3. Am J Physiol Heart Circ Physiol. 2014;H585–H597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hou X, Zeng H, He X, Chen JX. Sirt3 is essential for apelin‐induced angiogenesis in post‐myocardial infarction of diabetes. J Cell Mol Med. 2015;53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. He X, Zeng H, Chen ST, Roman RJ, Aschner JL, Didion S, Chen JX. Endothelial specific SIRT3 deletion impairs glycolysis and angiogenesis and causes diastolic dysfunction. J Mol Cell Cardiol. 2017;104–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. deAlmeida AC, van Oort RJ, Wehrens XH. Transverse aortic constriction in mice. J Vis Exp. 2010;38:1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gao S, Ho D, Vatner DE, Vatner SF. Echocardiography in mice. Curr Protoc Mouse Biol. 2011;71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. You J, Wu J, Ge J, Zou Y. Comparison between adenosine and isoflurane for assessing the coronary flow reserve in mouse models of left ventricular pressure and volume overload. Am J Physiol Heart Circ Physiol. 2012;H1199–H1207. [DOI] [PubMed] [Google Scholar]
- 23. Chang WT, Fisch S, Chen M, Qiu Y, Cheng S, Liao R. Ultrasound based assessment of coronary artery flow and coronary flow reserve using the pressure overload model in mice. J Vis Exp. 2015;e52598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. He X, Zeng H, Roman RJ, Chen JX. Inhibition of prolyl hydroxylases alters cell metabolism and reverses pre‐existing diastolic dysfunction in mice. Int J Cardiol. 2018;281–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Geraets IME, Glatz JFC, Luiken J, Nabben M. Pivotal role of membrane substrate transporters on the metabolic alterations in the pressure‐overloaded heart. Cardiovasc Res. 2019;1000–1012. [DOI] [PubMed] [Google Scholar]
- 26. Kolwicz SC Jr, Tian R. Glucose metabolism and cardiac hypertrophy. Cardiovasc Res. 2011;194–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huang Y, Lei L, Liu D, Jovin I, Russell R, Johnson RS, Di Lorenzo A, Giordano FJ. Normal glucose uptake in the brain and heart requires an endothelial cell‐specific HIF‐1alpha‐dependent function. Proc Natl Acad Sci USA. 2012;17478–17483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ahn BH, Kim HS, Song S, Lee IH, Liu J, Vassilopoulos A, Deng CX, Finkel T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci USA. 2008;14447–14452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, Grueter CA, Harris C, Biddinger S, Ilkayeva OR, et al. SIRT3 regulates mitochondrial fatty‐acid oxidation by reversible enzyme deacetylation. Nature. 2010;121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McDonnell E, Peterson BS, Bomze HM, Hirschey MD. SIRT3 regulates progression and development of diseases of aging. Trends Endocrinol Metab. 2015;486–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kim HS, Patel K, Muldoon‐Jacobs K, Bisht KS, Aykin‐Burns N, Pennington JD, van der Meer R, Nguyen P, Savage J, Owens KM, et al. SIRT3 is a mitochondria‐localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell. 2010;41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. van de Hoef TP, Echavarria‐Pinto M, van Lavieren MA, Meuwissen M, Serruys PW, Tijssen JG, Pocock SJ, Escaned J, Piek JJ. Diagnostic and prognostic implications of coronary flow capacity: a comprehensive cross‐modality physiological concept in ischemic heart disease. JACC Cardiovasc Interv. 2015;1670–1680. [DOI] [PubMed] [Google Scholar]
- 33. Abel ED. Glucose transport in the heart. Front Biosci. 2004;201–215. [DOI] [PubMed] [Google Scholar]
- 34. Szablewski L. Glucose transporters in healthy heart and in cardiac disease. Int J Cardiol. 2017;70–75. [DOI] [PubMed] [Google Scholar]
- 35. Shao D, Tian R. Glucose transporters in cardiac metabolism and hypertrophy. Compr Physiol. 2015;331–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wende AR, Kim J, Holland WL, Wayment BE, O'Neill BT, Tuinei J, Brahma MK, Pepin ME, McCrory MA, Luptak I, et al. Glucose transporter 4‐deficient hearts develop maladaptive hypertrophy in response to physiological or pathological stresses. Am J Physiol Heart Circ Physiol. 2017;H1098–H1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liao R, Jain M, Cui L, D'Agostino J, Aiello F, Luptak I, Ngoy S, Mortensen RM, Tian R. Cardiac‐specific overexpression of GLUT1 prevents the development of heart failure attributable to pressure overload in mice. Circulation. 2002;2125–2131. [DOI] [PubMed] [Google Scholar]
- 38. Luptak I, Yan J, Cui L, Jain M, Liao R, Tian R. Long‐term effects of increased glucose entry on mouse hearts during normal aging and ischemic stress. Circulation. 2007;901–909. [DOI] [PubMed] [Google Scholar]
- 39. Pereira RO, Wende AR, Olsen C, Soto J, Rawlings T, Zhu Y, Anderson SM, Abel ED. Inducible overexpression of GLUT1 prevents mitochondrial dysfunction and attenuates structural remodeling in pressure overload but does not prevent left ventricular dysfunction. J Am Heart Assoc. 2013;e000301 DOI: 10.1161/JAHA.113.000301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yan J, Young ME, Cui L, Lopaschuk GD, Liao R, Tian R. Increased glucose uptake and oxidation in mouse hearts prevent high fatty acid oxidation but cause cardiac dysfunction in diet‐induced obesity. Circulation. 2009;2818–2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhabyeyev P, Gandhi M, Mori J, Basu R, Kassiri Z, Clanachan A, Lopaschuk GD, Oudit GY. Pressure‐overload‐induced heart failure induces a selective reduction in glucose oxidation at physiological afterload. Cardiovasc Res. 2013;676–685. [DOI] [PubMed] [Google Scholar]
- 42. Leucker TM, Bienengraeber M, Muravyeva M, Baotic I, Weihrauch D, Brzezinska AK, Warltier DC, Kersten JR, Pratt PF Jr. Endothelial‐cardiomyocyte crosstalk enhances pharmacological cardioprotection. J Mol Cell Cardiol. 2011;803–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zeng H, Chen JX. Microvascular rarefaction and heart failure with preserved ejection fraction. Front Cardiovasc Med. 2019;15. [DOI] [PMC free article] [PubMed] [Google Scholar]