

Abstract
Background
Atrial tissue fibrosis is linked to inflammatory cells, yet is incompletely understood. A growing body of literature associates peripheral blood levels of the antifibrotic hormone BNP (B‐type natriuretic peptide) with atrial fibrillation (AF). We investigated the relationship between pro‐fibrotic tissue M2 macrophage marker Cluster of Differentiation (CD)163+, atrial procollagen expression, and BNP gene expression in patients with and without AF.
Methods and Results
In a cross‐sectional study design, right atrial tissue was procured from 37 consecutive, consenting, stable patients without heart failure or left ventricular systolic dysfunction, of whom 10 had AF and 27 were non‐AF controls. Samples were analyzed for BNP and fibro‐inflammatory gene expression, as well as fibrosis and CD163+. Primary analyses showed strong correlations (all P<0.008) between M2 macrophage CD163+ staining, procollagen gene expression, and myocardial BNP gene expression across the entire cohort. In secondary analyses without multiplicity adjustments, AF patients had greater left atrial volume index, more valve disease, higher serum BNP, and altered collagen turnover markers versus controls (all P<0.05). AF patients also showed higher atrial tissue M2 macrophage CD163+, collagen volume fraction, gene expression of procollagen 1 and 3, as well as reduced expression of the BNP clearance receptor NPRC (all P<0.05). Atrial procollagen 3 gene expression was correlated with fibrosis and BNP gene expression was correlated with serum BNP.
Conclusions
Elevated atrial tissue pro‐fibrotic M2 macrophage CD163+ is associated with increased myocardial gene expression of procollagen and anti‐fibrotic BNP and is higher in patients with AF. More work on modulation of BNP signaling for treatment and prevention of AF may be warranted.
Keywords: atrial fibrillation, fibrosis, gene expression, macrophage, natriuretic peptide
Subject Categories: Fibrosis, Biomarkers, Inflammation, Atrial Fibrillation
Clinical Perspective
What Is New?
This study shows that the pro‐fibrotic macrophage marker CD163+ correlates with gene expression of procollagen and BNP (B‐type natriuretic peptide) in the atrial tissue of patients with undergoing cardiac surgery without heart failure or left ventricular systolic dysfunction.
Furthermore, it shows that the subset of patients with atrial fibrillation had higher tissue CD163+, higher collagen volume fraction, greater gene expression of procollagen 1 and 3, as well as reduced expression of the BNP clearance receptor NPRC.
What Are the Clinical Implications?
These data suggest that BNP may be used clinically to predict atrial fibrosis and that modulation of natriuretic peptide signaling may help in the treatment and prevention of atrial fibrillation.
Nonstandard Abbreviations and Acronyms
- AF
Atrial Fibrillation
- ANP
A‐type natriuretic peptide
- B2M
beta‐2‐microglobulin
- BNP
B‐type natriuretic peptide
- CD
Cluster of Differentiation
- CNP
C‐type natriuretic peptide
- Col I
procollagen 1
- Col III
procollagen 3
- E/e’
ratio between early mitral inflow velocity and mitral annular early diastolic velocity
- GC‐A
Guanyl Cyclase A
- LV
left ventricular
- MMP
matrix metalloproteinases
- NPRC
Natriuretic Peptide Receptor C
- PARABLE
Prospective ARNI in Asymptomatic Patients With Elevated Natriuretic Peptide and Elevated Left Atrial Volume Index eLEvation
- TIMP
tissue inhibitor of matrix metalloproteinase
- ST2
Suppression of Tumorigenicity 2
- STOP‐HF
St Vincent’s Screening TO Prevent Heart Failure
The relationships between anti‐fibrotic hormone BNP (B‐type natriuretic peptide) and atrial fibrosis are incompletely understood. BNP levels measured in peripheral blood are not affected by atrial fibrillation (AF) when patients are matched for left ventricular (LV) function and left atrial volume index.1 However, peripheral concentrations of proBNP, but not pro‐atrial natriuretic peptide, are increased in AF alone.2 The St Vincent's STOP‐HF (Screening To Prevent Heart Failure) study demonstrated that elevated peripheral blood BNP levels can help identify patients at risk of AF, which was also the most common abnormality leading to incident major adverse cardiovascular events requiring hospitalization.3 A greater understanding of BNP myocardial gene expression and atrial fibrosis could explain a growing number of clinical studies, which have shown that BNP is an independent risk factor for the development of new‐onset AF in diverse populations.4, 5, 6, 7, 8, 9
A well‐established part of the pathophysiology of AF is structural remodeling and fibrosis, which result in muscle bundle separation and interference with electrical continuity.10, 11, 12 Therefore, stretch‐ and/or fibrosis‐mediated BNP release may form the basis for elevated expression in the atria of patients with AF, as left atrial volumes expand.13, 14, 15 Inflammatory cells are also implicated in the development and progression of AF16, 17 and we and others have demonstrated that biologically active BNP has antifibrotic effects on cardiac fibroblasts and attenuates monocyte chemotaxis, mediated by activation of its transmembrane receptor, guanyl cyclase A (GC‐A, also known as natriuretic peptide receptor A).14, 15, 18, 19 A recent immunohistochemical analysis of samples from patients undergoing open heart surgery showed an increase in CD68+ expressing pro‐inflammatory monocyte‐derived macrophages in the atrial myocardium of patients with AF compared with those in sinus rhythm.20 These data show that monocyte‐derived macrophages play an important role in the pathophysiology of AF, in addition to well‐known functions as part of the innate immune system, such as removing pathogenic bacteria. Monocyte‐derived macrophages include subtypes that are pro‐inflammatory (M1, “classical”), pro‐fibrotic (M2, “nonclassical”), and intermediate (reviewed in 21). CD68+ investigated is a pan‐macrophage marker expressed by all macrophage subtypes. However, there are no data on CD163+, which identifies more pro‐fibrotic, M2 macrophage subtypes, which may mediate cardiac fibrosis and remodeling in AF. Therefore, in the myocardium of a group of patients undergoing elective cardiac surgery with and without AF, we sought to investigate the relationships between M2 macrophage CD163+ staining, BNP gene expression, and procollagen gene expression.
Methods
Patient Recruitment
All subjects gave written informed consent to participate in this study. The Ethics Committee at St. Vincent's University Hospital approved all study protocols that conformed to the principles of the Helsinki Declaration. In accordance with Transparency and Openness Promotion (TOP) Guidelines, the data that support the findings of this study are available from the corresponding author upon reasonable request. This study population included 37 consecutive, consenting stable patients without heart failure or LV systolic dysfunction (n=35, preserved ejection fraction, LV ejection fraction ≥50%; n=2 midrange ejection fraction, LV ejection fraction 40–49%) and classified as having a documented history of AF (either persistent or chronic AF). Patients were stable, non–intensive care unit patients undergoing elective/nonemergency surgeries for coronary artery bypass grafting or valve replacement under the care of the cardiology staff at St Vincent's University Hospital and the Cardiology Department of the Blackrock Clinic in Dublin, Ireland. Right atrial appendage tissue and peripheral venous blood were procured from consenting patients following screening with echocardiography before surgery.
Immunohistochemical Analysis of CD163+ Macrophage Marker
Assessment of CD68+ and CD163+ macrophage markers in atrial tissue samples was performed with the use of the EnVision Detection System Kit (Dako). Tissue sections (6 μm) underwent deparaffinization, rehydration, and antigen retrieval using Trilogy solution (Cell Marque) in a pressure cooker for 15 minutes. Slides were washed in distilled water and blocked in 1% hydrogen peroxide in the dark for 10 minutes. Nonspecific binding was blocked by incubation in PBS/5% bovine serum albumin/1% goat serum for 30 minutes. A mouse anti‐human CD68+ (Dako) or CD163+ (Novocastra) monoclonal antibodies (1:100 dilution in PBS) were applied for 1 hour. Following washes in PBS/0.05% Tween, the Dako EnVision Kit was used to complete the staining. This included an incubation in prediluted HRP‐conjugated secondary antibody for 25 minutes, followed by washes and incubation with diaminobenzidine colorimetric substrate for 5 minutes. Slides were counterstained with hematoxylin for nuclear visualization. Following dehydration and coverslip addition, slides were scanned with Aperio ScanScopeXT Slide Scanner (20‐fold magnification) and image analysis was performed with ImageScope software. A positive pixel count algorithm was used to quantify brown‐colored CD68+ or CD163+ within each scanned image.
Myocardial Tissue Gene Expression Analysis
For analysis of myocardial tissue gene expression, the tissue was individually disrupted and homogenized using an Ultra Turrax T25 Dispersing Instrument (IKA). RNA was extracted using the AllPrep DNA/RNA extraction kit (Qiagen) according to the manufacturer's instructions. First strand cDNA synthesis was carried out using SuperScript II RT (Invitrogen). Quantitative real‐time polymerase chain reaction primers were designed for procollagen 1 (Col I), procollagen 3 (Col III), BNP, GC‐A, and NPRC. Primer sequences used were are follows: Collagen 1 a1 (COL1A1): GAACGCGTGTCATCCCTTGT (forward, F), GAACGAGGTAGTCTTTCAGCAACA (reverse, R); Collagen 3 a1 (COL3A1): AACACGCAAGGCTGTGAGCT (F), GAACGAGGTAGTCTTTCAGCAACA (R); BNP: ACCGCAAAATGGTCCTCTAC (F), CGCCTCAGCACTTTGCAG (R); GC‐A: CGCAAAAGGCCGAGTTATCTA (F), AACGTAGTCCTCCCCACACA (R); NPRC: CCCAGGAGGTTATTGGTGATTATTT (F), ACATTCGGCCGCATTTCAA (R); quantitative real‐time polymerase chain reaction reactions were normalized to the housekeeping gene beta‐2‐microglobulin (B2M): AGGCTATCCAGCGTACTCCA (F), CCAGTCCTTGCTGAAAGACA (R). Quantitative real‐time polymerase chain reaction was performed with Platinum SYBR Green qPCR SuperMix‐UDG mix (Invitrogen) using the Mx3000P System (Stratagene). The quantitative real‐time polymerase chain reaction cycling program consisted of 40 3‐step cycles of 15 s/95°C, 30 s/TA (annealing temperature), and 30 s/72°C.
Assessment of Tissue Fibrosis
Fibrosis assessment was performed using Picrosirius red and Masson's trichrome tissue staining and subsequent image analysis. For picrosirius red staining, tissue sections (5 μm thick) were rehydrated and incubated with 0.2% phosphomolybdic acid for 2 minutes. Following a rinse in distilled water, the slides were stained with picrosirius red (Direct Red 80 in picric acid, Sigma) for 90 minutes. The slides were then placed in 0.4% HCl for 2 minutes, 70% ethanol for 45 s, dehydrated, and cover‐slipped for analysis. The degree of collagen deposition was quantified by automated digital image analysis (Aperio ScanScope XT Slide Scanner, Aperio Technologies) at 20‐fold magnification. Automated image analysis was performed using ImageScope (Aperio). A positive pixel count algorithm was used to quantify dark pink‐stained collagen within each image. Required analysis input parameters for each stain were based on the hue, saturation, and intensity color model. To detect collagen with picrosirius red, a hue value of 0.8 was specified, and a hue width of 0.5 was used to include the moderate range of color shades.
Masson's trichrome staining kit (Dako) was used for analysis of interstitial collagen within the myocardial tissue. Tissue sections (8 μm) were rehydrated and incubated overnight in Bouin's solution. Sections were then incubated with Weigert's hematoxylin, Biebrich Scarlet‐acid Fuchsin solution, phosphotungstic/phosphomolybdic acid solution, and Aniline Blue solution. Following this, tissue sections were incubated in acetic acid and subsequently dehydrated and cover‐slipped for analysis. Image analysis of Masson's trichrome staining was performed by automated analysis using the Aperio ScanScopeXTSlide Scanner system at 20‐fold magnification, and ImageScope software. A positive pixel count algorithm was used to quantify blue‐colored collagen within each scanned image. A hue value of 0.66 was specified. The default hue width value of 0.5 was used to allow inclusion of a moderate range of color shades. A collagen volume fraction was calculated based on the percent of blue collagen staining quantified within a tissue section.
Doppler‐Echocardiography
Doppler‐echocardiography was blinded to clinical, histological, and biochemical assessment. A Philips IE33 ultrasound scanner with standard adult probe was used for data acquisition. All studies were performed in accordance with the American Society of Echocardiography recommendations using the following procedure sequence:
Conventional long axis view, 2 chamber and 4 chamber view of 2D.
M‐mode study.
Doppler and tissue evaluation and hemodynamic study.
LV systolic dysfunction was assessed using the Teichholz method. LV mass was calculated using the Devereux formula. Assessment of LV diastolic dysfunction involved evaluation of left atrial volume index and the ratio of mitral inflow velocities and tissue Doppler analysis of early diastolic mitral annular velocity at the lateral wall (lateral E/e’).
Peripheral Blood Sampling
Peripheral venous blood was obtained at the time of clinical assessment/surgery. BNP was measured using the Alere Triage Point‐of‐care BNP assay with a sensitivity limit <5 pg/mL (Alere Inc.). Although the Alere Triage BNP assay used in our study is not specific for active BNP 1‐32, which is virtually undetectable in peripheral blood, it cross‐reacts with proteolyzed BNP fragments (eg, BNP3‐32) and proBNP, especially the glycosylated form, which, in turn, are elevated in LV dysfunction and heart failure.22, 23
Serum samples were obtained following centrifugation of peripheral blood samples at 2500g for 10 minutes at 4°C. Serum was aliquoted and stored at −80°C until required. Each serum sample underwent no more than 3 freeze–thaw cycles before its use in enzyme‐linked immunosorbent assays and radioimmunoassays.
Serum Biomarker Analysis
Serum levels of carboxy‐terminal pro‐peptide of Collagen 1 were quantified using an ELISA assay from Takara Biochemicals (Osaka, Japan) with assay sensitivity of 2.0 ng/mL according to the manufacturer's instructions. Serum levels of amino‐terminal pro‐peptide of Collagen 1, Collagen 3, and carboxy‐terminal telo‐peptide of Collagen 1 were measured using radioimmunoassays from Orion Diagnostica (Espoo, Finland). Assay sensitivity for each of these markers was 13.0, 1.9, and 0.5 ng/mL, respectively. Serum levels of matrix metalloproteinases MMP2/9 and tissue inhibitor of matrix metalloproteinase 1 (TIMP1) were measured using immunoassays with colorimetric detection (GE Healthcare) and were analyzed using SpectraMax 190 microplate reader (Molecular devices). Assay sensitivity was 0.4, 0.6, and 1.3 ng/mL, respectively. Cytokine quantification of the inflammatory markers tumor necrosis factor alpha, interleukin 6, interleukin 8, high‐sensitivity C‐reactive protein, and monocyte chemoattractant protein 1 was performed with custom‐made multispot ultrasensitive immunoassay with electrochemiluminescence detection (Meso Scale Discovery) and analyzed with SECTOR Imager 2400 (MSD). Assay sensitivity for each of these cytokines was ≤0.7 pg/mL. Suppression of Tumorigenicity 2 (ST2) serum levels were quantified with Presage ST2 assay with colorimetric detection (Critical Diagnostics) and assay sensitivity of 1.8 ng/mL.
Statistical Analysis
The primary objective of this study was to assess the relationships between M2 macrophage CD163+ staining, myocardial BNP gene expression, and procollagen 1 and 3 gene expression using Pearson correlation on normally distributed raw data. Log transformed values or using Spearman rank order correlations were used for non‐normally distributed variables. Nominal statistical significance was set at P<0.05 with Bonferroni correction for the 6 pairwise correlations, meaning a P value of 0.05/6=0.0083 was considered statistically significant.
Descriptive data are presented as either mean±SD or median (25th:75th percentile) for normally and non‐normally distributed continuous variables, respectively. Frequencies and percentages (in parentheses) summarize categorical variables. Comparisons between the AF and control groups were conducted with an independent t test or Mann–Whitney rank sum test for normal and non‐normal distributions, respectively. Categorical variables were compared using χ2 analysis. Statistical analysis was performed using the statistical software package GraphPad Prism v6 and R v3.6.2.
Results
Clinical, Cardiovascular, and Imaging Characteristics
The descriptive characteristics of the total patient population, including a comparison of those with AF (n=10) or not (n=27) are described in Table. In terms of demographics, clinical characteristics, cardiovascular history, cardiometabolic phenotype, AF and non‐AF patients were similar apart from a higher prevalence of valvular disease in the AF group. Cardiovascular medications were similar. Doppler echocardiography showed that AF patients had higher left atrial volume index, but significantly lower posterior wall thickness and LV mass index measurements.
Table 1.
Demographic, Clinical, Medication, Biochemistry, and Doppler Echocardiographic Characteristics of the Study Population
| Measure (Mean/SD, Median [IQR], n [%]) | Total Population (n=37) | AF (n=10) | Non‐AF Controls (n=27) | P Valuea |
|---|---|---|---|---|
| Age, y | 67.7±9.9 | 70.7±9.4 | 66.6±10.1 | NS |
| Male, n (%) | 26 (70) | 8 (80) | 18 (67) | NS |
| Cardiovascular history | ||||
| Vascular disease, n (%) | 25 (68) | 5 (50) | 20 (74) | NS |
| Valvular heart disease, n (%) | 19 (51) | 9 (90) | 10 (37) | 0.003 |
| Diabetes mellitus, n (%) | 6 (16) | 2 (20) | 4 (15) | NS |
| Hypertension, n (%) | 30 (70) | 7 (70) | 23 (85) | NS |
| Dyslipidemia, n (%) | 23 (61) | 4 (40) | 17 (63) | NS |
| CHA2DS2VASC Score | 2.9±1.3 | 2.9±1.4 | 2.9±1.3 | NS |
| Cardiometabolic phenotype | ||||
| Body mass index, kg/m2 | 27.2±3.2 | 27.4±3.3 | 27.2±3.2 | NS |
| Systolic blood pressure, mm Hg | 135.5±7.0 | 136.0±3.7 | 135.1±8.0 | NS |
| Diastolic blood pressure, mm Hg | 79.8±6.7 | 79.8±6.8 | 79.9±6.8 | NS |
| Creatinine, mmol/L | 91±11 | 95±20 | 90±4 | NS |
| Cardiovascular medications | ||||
| RAAS modifying therapy, n (%) | 15 (41) | 3 (30) | 12 (44) | NS |
| Beta‐blocker, n (%) | 23 (62) | 5 (50) | 18 (67) | NS |
| Diuretic, n (%) | 13 (35) | 5 (50) | 8 (30) | NS |
| Calcium channel blocker, n (%) | 4 (11) | 1 (10) | 3 (11) | NS |
| Statin, n (%) | 21 (57) | 4 (40) | 17 (63) | NS |
| Doppler echocardiography | ||||
| EF, % | 58±5 | 56±8 | 58±4 | NS |
| LVIDD, mm | 53±6 | 52±5 | 53±6 | NS |
| IVS, mm | 9.7±1.4 | 9.6±1.3 | 9.7±1.5 | NS |
| PW, mm | 10.9±1.7 | 9.9±1 | 11.2±1.8 | 0.035 |
| E/e’ | 9.5±2.8 | 10.0±2.6 | 9.4±2.9 | NS |
| LAVI, mL/m2 | 28.9±4.3 | 32.3±5.4 | 27.7±3.2 | 0.003 |
| LVMI, g/m2 | 108.5±19.7 | 97.0±18.5 | 112.8±18.7 | 0.028 |
| Blood biochemistry | ||||
| PICP, ng/Ml | 300±114 | 253±42 | 317±127 | NS |
| CITP, ng/mL | 5.21±1.65 | 6.11±2.31 | 4.87±1.22 | 0.042 |
| PINP, ng/mL | 51.2±34.2 | 32.3±12.3 | 58.2±37.1 | 0.016 |
| PIIINP, ng/mL | 5.46±2.72 | 6.66±3.94 | 5.01±2.04 | NS |
| MCP1, pg/mL | 292±122 | 277±120 | 297±125 | NS |
| IL6, pg/mL | 9.13±9.15 | 10.54±9.75 | 8.61±9.06 | NS |
| IL8, pg/mL | 11.34±9.72 | 10.21±7.20 | 11.76±10.60 | NS |
| TNFα, pg/mL | 4.82±3.08 | 4.82±2.94 | 4.82±3.18 | NS |
| TIMP1, ng/mL | 1015±364 | 1146±576 | 966±244 | NS |
| MMP2, ng/mL | 907±217 | 951±238 | 890±211 | NS |
| MMP9, ng/mL | 302±361 | 432±469 | 253±308 | NS |
CHA2DS2VASC Score is a clinical prediction score summing: 1 for congestive heart failure (or left ventricular systolic dysfunction); 1 for hypertension; 2 for age ≥75 years; 1 for diabetes mellitus; 2 for prior stroke, transient ischemic attack, or thromboembolism; 1 for vascular disease; 1 for age 65 to 74 years; 1 for female sex. AF indicates atrial fibrillation; CITP, carboxy‐terminal telo‐peptide of Collagen 1; E/E’, Tissue Doppler early diastolic mitral annular velocity; EF, ejection fraction; IL6, interleukin 6; IL8, interleukin 8; IVS, intraventricular septal thickness; LAVI, left atrial volume index; LVIDD, diastolic left ventricular internal diameter; LVMI, left ventricular mass index; MCP1, monocyte chemoattractant protein 1; MMP2, matrix metalloproteinase 2; MMP9, matrix metalloproteinase 9; NS, Not Significant; PICP, carboxy terminal pro‐peptide of Collagen 1; PIIINP, amino‐terminal pro‐peptide of Collagen 3; PINP, amino‐terminal pro‐peptide of Collagen 1; PW, posterior wall thickness; RAAS, renin‐angiotensin‐aldosterone system; TIMP1, tissue inhibitor of matrix metalloproteinase 1; and TNFα, tumor necrosis factor alpha.
All pairwise comparisons P<0.05 are included.
Tissue Studies
The main finding of this study is that atrial tissue CD163+ staining was highly correlated with BNP and procollagen 1 gene expression (Figure 1A and 1B) across the entire cohort of patients undergoing elective cardiac surgery. Furthermore, BNP gene expression was highly correlated with procollagen 1 and 3 gene expression (Figure 1C and 1D). These primary analyses met the prespecified, Bonferroni‐corrected threshold for statistical significance. There was also a positive correlation between CD163+ and procollagen 3 gene expression, but this did not reach the adjusted threshold for significance (R=0.38, P=0.02).
Figure 1.
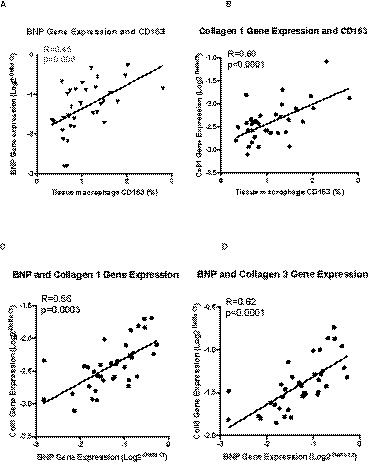
Correlations between cardiac atrial tissue staining of M2‐macrophage marker CD163 and gene expression of BNP (A) and Collagen 1 (B). Furthermore, BNP gene expression in the atrial tissue correlates positively with Collagen 1 (C) and Collagen 3 (D) gene expression. BNP indicates B‐type natriuretic peptide; CD163, Cluster of differentiation 163; Col1, Collagen 1; and Col3, Collagen 3.
Secondary analyses reported herein are subsidiary, require confirmation in larger studies, and were not subjected to correction for multiple testing. With the caveat that numbers are small and the patient populations were not matched for valve disease, we observed increased CD163+ M2 macrophage marker positivity in AF patients compared with controls (Figure 2A) and also a higher collagen volume fraction (Figure 2B) evidenced by picrosirius red staining. CD163+ M2‐macrophage staining and picrosirius red collagen staining were evident in both the perivascular and interstitial regions (illustrative scans, Figure 2C and 2D, respectively). In accordance with this, there was increased atrial tissue gene expression of procollagen 1 and 3 in AF patients compared with controls (Figure 3A and 3B) and tissue collagen volume fraction was correlated with procollagen 3 gene expression (R=0.38, 95% CI [0.04, 0.64], P=0.029).
Figure 2.
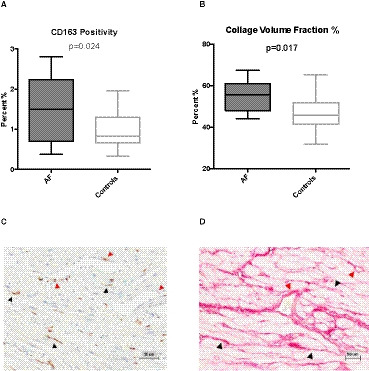
Cardiac atrial tissue staining for CD163 positivity (A) and collagen volume fraction (CVF, picrosirius red) (B).
Also shown is typical staining of CD163‐positive macrophages (C) and collagen accumulation (D, picrosirius red) in cardiac interstitial (black arrowheads) and perivascular (red arrowheads) regions. AF indicates atrial fibrillation; CD163, cluster of differentiation 163; and CVF, collagen volume fraction.
Figure 3.
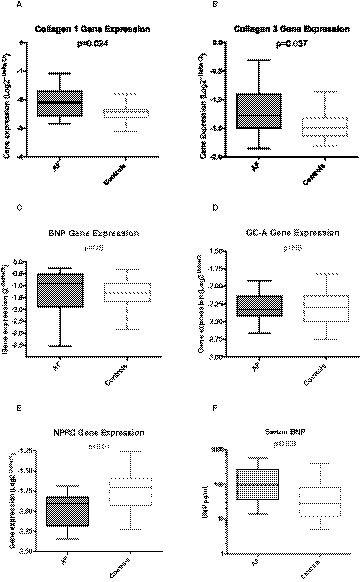
Cardiac atrial tissue gene expression of Collagen 1 (A), Collagen 3 (B), BNP (C), GC‐A (D), and NPRC (E) with significant differences observed between AF and Controls in all except BNP and GC‐A.
Higher serum BNP levels were observed in patients with AF compared with Controls (F). Serum BNP data are not normally distributed and are presented as medians (line), interquartile ranges (box) and ranges (whiskers). AF indicates atrial fibrillation; BNP, B‐type natriuretic peptide; GG‐A, Guanyl‐cyclase‐A, also known as natriuretic peptide receptor A; and NPRC, natriuretic peptide receptor C.
Interestingly, in these exploratory analyses, we did not observe differences between AF and controls in gene expression of BNP (−1.30±0.321 versus −1.372±0.117, P=NS) or GC‐A (−2.297±0.071 versus −2.314±0.0448, P=NS, Figure 3C and 3D, respectively). However, we observed reduced expression of the NPRC clearance receptor (AF −1.969±0.06578) versus controls (−1.74±0.23, P<0.01 versus AF, Figure 3E) and this may help explain elevated levels of blood BNP in these patients (Figure 3F).
Furthermore, in a post‐hoc analysis we found that GC‐A mRNA expression correlated with CD163+ (R=0.51, P=0.0015), collagen 1 gene expression (R=0.51, P=0.0013), and collagen 3 gene expression (R=0.41, P=0.01). Albeit these unplanned analyses are exploratory and were not subjected to multiplicity adjustments, they are similar in pattern to the BNP mRNA expression in the primary analyses (Figure 1A through 1D).
Peripheral Markers, BNP, and Natriuretic Peptide Receptors
Secondary biochemistry analyses showed higher circulating BNP, lower levels of the collagen synthesis marker amino‐terminal pro‐peptide of collagen 1, and higher levels of the collagen degradation marker carboxy‐terminal telo‐peptide of collagen 1 in the AF group (Table).
Blood levels of BNP were elevated in AF patients (Figure 3F) and were positively correlated with cardiac atrial tissue BNP gene expression across the entire patient cohort (R=0.37, P=0.02). Median circulating BNP was 95 pg/mL [interquartile range 36, 264] in the AF patients and 27 pg/mL [interquartile range 12, 79] in controls (P=0.023),
Subpopulation With Valvular Heart Disease
Since most of the AF patients also had valvular heart disease, we also evaluated this subset dependent on whether they had AF (n=9) or not (n=10) in an exploratory, post‐hoc analysis without correction for multiple testing. Although the numbers are small, there were no differences in these subgroups in terms of age, sex, cardiovascular history, body mass index, blood pressure, creatinine, ejection fraction, and E/e’, similar to the main population. Median circulating BNP was 131 pg/mL [interquartile range 41, 282] in the AF patients and 55 pg/mL [interquartile range 21, 156] in controls, although this did not reach statistical significance. Differences in left atrial volume index did not reach statistical significance (32.9±5.2 versus 30.7±1.9). However, in accordance with the overall population, LV mass index was lower (97.1±19.6 versus 109.5±25.5 g/m2, P=0.04), collagen volume fraction was higher (52.9±7.5% versus 47.0±8.4%, P=0.03) and CD163+ positivity was higher (1.6±8.6% versus 0.87±3.4%, P=0.02) in patients with AF versus controls in the subset with valvular heart disease.
Furthermore, in this post‐hoc analysis without multiplicity adjustments, CD163+ staining was also positively correlated with procollagen 1 gene expression (R=0.62, P=0.008), and procollagen 3 gene expression (R=0.54, P=0.026). CD163+ staining was also negatively correlated with expression of NPRC in this subset (R=−0.56, P=0.019). Finally, pan‐macrophage marker CD68+ staining in a subset of the population showed a nonsignificant trend to increased levels in AF patient tissue versus controls (1.47±0.21% versus 0.94±0.18%, P=0.08) and was significantly different between AF and controls within the valvular heart disease subset (1.47±0.57% versus 0.65±0.44%, P=0.02).
Discussion
The STOP‐HF and other clinical studies demonstrate that peripheral blood levels of BNP can help identify patients at high risk of AF progression.3, 4, 5, 6, 7, 8, 9 BNP has anti‐inflammatory effects in cardiac tissue14, 15, 18 and recent studies have highlighted excess inflammatory cell infiltration in the myocardium of patients with AF.16, 20 However, BNP also has anti‐fibrotic effects,14, 19 and the present study extends our understanding of the relationship between BNP and the pathophysiology of AF by evaluating pro‐fibrotic M2 (nonclassical) macrophage marker CD163+, myocardial fibrosis, and gene expression profiles in the myocardium of patients with and without AF.
Among our small cohort of patients without heart failure or LV systolic dysfunction, we demonstrate that CD163+ staining was strongly correlated with gene expression of procollagen 1 and also with BNP gene expression in right atrial appendage tissue (Figure 1A and 1B), suggesting a relationship between pro‐fibrotic M2 macrophages, BNP, and atrial fibrosis. Similarly, we observed correlations between BNP gene expression and procollagen expression across the entire cohort, suggesting a continuous relationship between these markers among people without AF as well as those with established AF (Figure 1C and 1D). Furthermore, in secondary analyses not subjected to multiplicity testing, AF patients had higher CD163+ staining, higher median serum BNP, higher atrial tissue collagen volume fraction, and increased gene expression of collagen 1 and 3, which may extend our understanding of alternative inflammatory cell involvement in AF. Myocardial BNP gene expression significantly correlated with the peripheral blood levels of BNP protein across the entire cohort, supporting the emerging evidence for BNP as a marker of AF and progression of AF. Given the known anti‐fibrotic effects of BNP on cardiac fibroblasts and anti‐inflammatory effects on monocytes,14, 18 these data may point to an endogenous, protective BNP response in the myocardium. They may help provide a pathophysiological basis, involving pro‐fibrotic monocyte‐derived M2 macrophages and atrial fibrosis, for the observed relationship between BNP and AF. Finally, they may open new opportunities for investigation of therapies for modulation of BNP in the prevention and treatment of AF.
The main strength of this study is the characterization of the pro‐fibrotic M2 macrophage marker CD163+ and markers of fibrosis in the atrial tissue of patients with and without AF. The pathophysiology of AF is widely understood to involve electrical, contractile, and structural remodeling. Structural remodeling involves reactive interstitial fibrosis, which separates muscle bundles and reparative fibrosis replacing dead cardiomyocytes, both interfering with electrical continuity and slowing conduction.10, 11, 12 It is also known that inflammatory cells migrate into the right and left atria of patients with AF,16, 20 and the present study extends our understanding of the pathophysiology of structural remodeling in AF and the potential role of M2‐type, nonclassical (CD163+) monocyte‐derived macrophages. The data build on a recent immune‐histochemical analysis of samples from patients undergoing open heart surgery, which identified monocyte‐derived macrophages as the most abundant inflammatory cell in both atria of patients with AF.20 Similar to our study, using the pan‐macrophage marker (CD68+), they found evidence of inflammatory cells in all patients undergoing open heart surgery, regardless of the presence of AF, but overall with higher tissue levels in AF.
We extend these observations by showing for the first time an excess of pro‐fibrotic M2 macrophages, identified by an increase in CD163+ expression, in atrial tissue of patients with AF. While these observations should be confirmed in larger studies and there is some debate about the precise classification of monocyte/macrophage subsets because of their plasticity depending on the pathophysiological environment, M2 macrophages (denoted by CD163+) are of particular interest in AF because of their pro‐fibrotic phenotype.21 We further show novel data in support of the relationship between monocyte‐derived M2 macrophages and myocardial fibrosis (picosirius red), as well as myocardial gene expression of collagen.
Taken together, our data may support the hypothesis that circulating peripheral blood monocytes recruited by the inflamed myocardium give rise to increased proportion of M2 (CD163+) tissue macrophages and that these macrophages are strongly associated with collagen formation, deposition, and myocardial fibrosis, which contribute to AF. Similar to previous work on inflammatory cell infiltration,16, 20 the correlations between CD163+, myocardial fibrosis, and collagen expression across the entire cohort with and without AF suggest a continuum between patients with and without AF. This may have implications for potential diagnostic and even therapeutic approaches to preventing deterioration of pre‐existing AF, but also to prevention of new‐onset AF.
The association between natriuretic peptides and AF is not new, because they have been studied as regulators of cardiac electrophysiology via L‐type calcium channels24 and have been genetically linked to the development of AF.25 Our work shows that cardiac tissue M2 macrophages were strongly associated with the gene expression and regulation of BNP. BNP activation of GC‐A results in guanylyl cyclase–mediated increases in cyclic guanosine monophosphate, which attenuates monocyte inflammation, chemotaxis, and inflammation in macrophages.18, 19, 26 This is counterbalanced by activation of NPRC, a clearance receptor, which also activates inhibitory G‐proteins that cause a reduction in cAMP production. Importantly, our secondary analyses suggest that increased BNP in patients with AF is associated with reduced expression of the clearance receptor NPRC. Increased myocardial fibrosis also altered levels of serum markers of collagen turnover.
Our AF patients demonstrate significantly elevated left atrial volume indices compared with sinus rhythm controls, a well‐established feature of AF progression.1, 15, 27 Pathological expansion of the atrium involves both myocardial fibrosis and stretch, which mediates increased BNP expression as atrial volumes expand.14, 15, 28 We have previously shown that gene expression of BNP and GC‐A is increased significantly by stretch of cardiac tissue 14 and it is well established that deletion of the BNP or GC‐A genes results in cardiac fibrosis.29, 30 We did not observe differential gene expression of BNP and GC‐A when comparing AF and control patients (Figure 3C and 3D). Rather, we observed a significant reduction in NPRC gene expression between AF and control patients, associated with increased collagen volume fraction, atrial fibrosis, and CD163+. NPRC has also been associated with antifibrotic effects in the atria31, 32 and may be responsible for increased circulating levels of BNP. Indeed, NPRC expression levels are higher in atrial versus ventricular tissues, and co‐treating wild‐type mice with angiotensin II and an NPRC agonist reduced atrial fibrosis and AF inducibility.32 We could speculate that antifibrotic effects of natriuretic peptide signaling may involve different balances of BNP, GC‐A, and NPRC expression at different stages of atrial fibrosis and AF, but further work is required to fully clarify these data. However, our study supports previous observations and the hypothesis that NPRC, as well as BNP and GC‐A, may be targets for prevention of atrial fibrosis and structural remodeling, which contribute to AF.
Limitations
There are few opportunities in clinical practice for obtaining cardiac tissue from patients with and without AF in order to explore the pathophysiology and correlates of tissue fibrosis. However, there are a number of important limitations of our study: first, it is a small, selected cohort and the comparisons between populations with and without AF were secondary, exploratory in nature, and statistical significance thresholds were not subjected to adjustment for multiple testing; it is difficult to match groups perfectly and there is limited scope for confounder adjustment using multivariable analyses, including adjusting for difference between the groups in terms of valvular disease and LV mass index; although the numbers are small, a post‐hoc subanalysis of 19 patients with valvular heart disease supports the hypothesis that pro‐fibrotic M2 macrophage marker CD163+ is associated with AF, procollagen gene expression, and natriuretic peptide signaling; second, we obtained atrial tissue from the right appendage, which may not exactly reflect the structure, physiology, and expression profile of left atrial tissue, even though some parameters, such as cardiomyocyte dimensions16 and number of infiltrating inflammatory cells (including macrophages),16, 20 were recently shown to be similar in the left and right atria of patients with AF16, 20; third, duration of AF was not available and patients were classified as AF only if a documented history of persistent/chronic AF was present. Finally, we did not measure other natriuretic peptides (ANP, CNP), angiotensin II, and aldosterone levels in our patients and our BNP assay captured proBNP and BNP3‐32. We cannot exclude differences in these unmeasured natriuretic peptides or other unmeasured cardiovascular hormones, which interact with natriuretic peptide signaling, in our population.
Conclusions
We observed strong correlations between atrial tissue M2 macrophage CD163+, gene expression of BNP, procollagen expression, and atrial fibrosis in a cohort of patients with and without AF. Higher levels were evident in those patients with AF in secondary analyses. Further work is needed to understand the generalizability of these observations from a small, selected cohort undergoing cardiac surgery. However, the wider implications of these data may extend beyond the pathophysiology of AF and the role of BNP as a blood biomarker of AF. Subcutaneous BNP and a dual GC‐A/GC‐B agonist can attenuate cardiac fibrosis,19 and our study may support evaluation of further pharmacological modulation of the natriuretic peptide system, via inhibition of NPRC or neutral endopeptidase, as a therapeutic approach in AF and prevention of AF. In this regard, it is noteworthy that clinical studies have demonstrated that augmenting endogenous, active natriuretic peptide levels using sacubitril/valsartan resulted in a significant reduction in left atrial volume index in a relatively short time‐frame in populations with a substantial baseline prevalence of AF.33, 34 The ongoing investigator‐led PARABLE study (Prospective ARNI in Asymptomatic Patients With Elevated Natriuretic Peptide and Elevated Left Atrial Volume Index Elevation, NCT02682719) will provide more information in this regard among patients at risk of AF who have elevated left atrial volume and BNP levels.
Sources of Funding
This work was supported by the Health Research Board of Ireland (Grant numbers CSA‐2012‐36 and HRB POR 2012/119) and European Commission Framework Programme 7 Metabolic Road to Diastolic Heart Failure (MEDIA) Grant.
Disclosures
Professors Ledwidge and McDonald are Co‐Principal Investigators of the ongoing, investigator‐led PARABLE randomized controlled trial (NCT02682719), which is funded by an unrestricted research grant from Novartis. The remaining authors have no disclosures to report.
(J Am Heart Assoc. 2020;9:e013416 DOI: 10.1161/JAHA.119.013416.)
For Sources of Funding and Disclosures, see page 11.
References
- 1. Rossi A, Enruquez‐Sarano M, Burnett JC, Lerman A, Abel MD, Seward JB. Natriuretic peptide levels in atrial fibrillation: a prospective hormonal and Doppler‐echocardiographic study. J Am Coll Cardiol. 2000;35:1256–1262. [DOI] [PubMed] [Google Scholar]
- 2. Ellinor PT, Low AF, Patton KK, Shea MA, Macrae CA. Discordant atrial natriuretic peptide and brain natriuretic peptide levels in lone atrial fibrillation. J Am Coll Cardiol. 2005;45:82–86. [DOI] [PubMed] [Google Scholar]
- 3. Ledwidge M, Gallagher J, Conlon C, Tallon E, O'Connell E, Dawkins I, Watson C, O'Hanlon R, Bermingham M, Patle A, et al. Natriuretic peptide‐based screening and collaborative care for heart failure: the STOP‐HF randomized trial. JAMA. 2013;310:66–74. [DOI] [PubMed] [Google Scholar]
- 4. Wang TJ, Larson MG, Levy D, Benjamin EJ, Leip EP, Wolf PA, Vasan R. Plasma natriuretic peptide levels and the risk of cardiovascular events and death. N Engl J Med. 2004;350:655–663. [DOI] [PubMed] [Google Scholar]
- 5. Patton KK, Ellinor PT, Heckbert SR, Christenson RH, DeFilippi C, Gottdiener JS, Kronmal RA. N‐terminal pro‐B‐type natriuretic peptide is a major predictor of the development of atrial fibrillation: the Cardiovascular Health Study. Circulation. 2009;120:1768–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yamauchi T, Sakata Y, Miura M, Onose T, Tsuji K, Abe R, Oikawa T, Kasahara S, Sato M, Nochioka K, et al. Prognostic impact of atrial fibrillation and new risk score of its onset in patients at high risk of heart failure—a report from the CHART‐2 Study. Circ J. 2017;81:185–194. [DOI] [PubMed] [Google Scholar]
- 7. Kara K, Geisel MH, Möhlenkamp S, Lehmann N, Kälsch H, Bauer M, Neumann T, Dragano N, Moebus S, Jöckel KH, et al. B‐type natriuretic peptide for incident atrial fibrillation‐The Heinz Nixdorf Recall Study. J Cardiol. 2015;65:453–458. [DOI] [PubMed] [Google Scholar]
- 8. Svennberg E, Henriksson P, Engdahl J, Hijazi Z, Al‐Khalili F, Friberg L, Frykman V. N‐terminal pro B‐type natriuretic peptide in systematic screening for atrial fibrillation. Heart. 2017;103:1271–1277. [DOI] [PubMed] [Google Scholar]
- 9. Inohara T, Kim S, Pieper K, Blanco RG, Allen LA, Fonarow GC, Gersh BJ, Ezekowitz MD, Kowey PR, Reiffel JA, et al. B‐type natriuretic peptide, disease progression and clinical outcomes in atrial fibrillation. Heart. 2018;05:370–377. [DOI] [PubMed] [Google Scholar]
- 10. Burstein B, Comtois P, Michael G, Nishida K, Villeneuve L, Yeh YH, Nattel S. Changes in connexin expression and the atrial fibrillation substrate in congestive heart failure. Circ Res. 2009;105:1213–1222. [DOI] [PubMed] [Google Scholar]
- 11. Burstein B, Nattel S. Atrial fibrosis: mechanisms and clinical relevance in atrial fibrillation. J Am Coll Cardiol. 2008;51:802–809. [DOI] [PubMed] [Google Scholar]
- 12. Yue L, Xie J, Nattel S. Molecular determinants of cardiac fibroblast electrical function and therapeutic implications for atrial fibrillation. Cardiovasc Res. 2011;89:744–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. De Jong AM, Maass AH, Oberdorf‐Maass SU, De Boer RA, Van Gilst WH, Van Gelder IC. Cyclical stretch induces structural changes in atrial myocytes. J Cell Mol Med. 2013;17:743–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Watson CJ, Phelan D, Xu M, Collier P, Neary R, Smolenski A, Ledwidge M, McDonald K, Baugh J. Mechanical stretch up‐regulates the B‐type natriuretic peptide system in human cardiac fibroblasts: a possible defense against transforming growth factor‐β mediated fibrosis. Fibrogenesis Tissue Repair. 2012;5:9, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Phelan D, Watson C, Martos R, Collier P, Patle A, Donnelly S, Ledwidge M, Baugh J, McDonald K. Modest elevation in BNP in asymptomatic hypertensive patients reflects sub‐clinical cardiac remodeling, inflammation and extracellular matrix changes. PLoS One. 2012;7:e49259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen MC, Chang JP, Liu WH, Yang CH, Chen YL, Tsai TH, Wang YH, Pan KL. Increased inflammatory cell infiltration in the atrial myocardium of patients with atrial fibrillation. Am J Cardiol. 2008;102:861–865. [DOI] [PubMed] [Google Scholar]
- 17. Hu YF, Chen YJ, Lin YJ, Chen SA. Inflammation and the pathogenesis of atrial fibrillation. Nat Rev Cardiol. 2015;12:230–243. [DOI] [PubMed] [Google Scholar]
- 18. Glezeva N, Collier P, Voon V, Ledwidge M, McDonald K, Watson C, Baugh J. Attenuation of monocyte chemotaxis‐a novel anti‐inflammatory mechanism of action for the cardio‐protective hormone B‐type natriuretic peptide. J Cardiovasc Transl Res. 2013;6:545–557. [DOI] [PubMed] [Google Scholar]
- 19. Ichiki T, Schirger JA, Huntley BK, Brozovich FV, Maleszewski JJ, Sandberg SM, Sangaralingham SJ, Park SJ, Burnett JC Jr. Cardiac fibrosis in end‐stage human heart failure and the cardiac natriuretic peptide guanylyl cyclase system: regulation and therapeutic implications. J Mol Cell Cardiol. 2014;75:199–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Smorodinova N, Blaha M, Melenovsky V, Rozsivalova K, Pridal J, Durisova M, Pirk J, Kautzner J, Kučera T. Analysis of immune cell populations in atrial myocardium of patients with atrial fibrillation or sinus rhythm. PLoS One. 2017;12:e0172691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shahid F, Lip GYH, Shantsila E. Role of monocytes in heart failure and atrial fibrillation. J Am Heart Assoc. 2018;7:e007849 DOI: 10.1161/JAHA.117.007849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Saenger A, Rodriguez‐Fraga O, Ler R, Ordonez‐Llanos J, Jaffe AS, Goetze JP, Apple FS. Specificity of B‐type natriuretic peptide assays: cross‐reactivity with different BNP, NT‐proBNP, and proBNP peptides. Clin Chem. 2017;63:351–358. [DOI] [PubMed] [Google Scholar]
- 23. Lam CSP, Burnett JRJC, Costello‐Boerrigter L, Rodeheffer RJ, Margaret M, Redfield MR. Alternate circulating Pro‐B‐type natriuretic peptide and B‐type natriuretic peptide forms in the general population. J Am Coll Cardiol. 2007;49:1193–1202. [DOI] [PubMed] [Google Scholar]
- 24. Springer J, Azer J, Hua R, Robbins C, Adamczyk A, McBoyle S, Bissell MB, Rose RA. The natriuretic peptides BNP and CNP increase heart rate and electrical conduction by stimulating ionic currents in the sinoatrial node and atrial myocardium following activation of guanylyl cyclase‐linked natriuretic peptide receptors. J Mol Cell Cardiol. 2012;52:1122–1134. [DOI] [PubMed] [Google Scholar]
- 25. Hodgson‐Zingman DM, Karst ML, Zingman LV, Heublein DM, Darbar D, Herron KJ, Ballew JD, de Andrade M, Burnett JC Jr, Olson TM. Atrial natriuretic peptide frameshift mutation in familial atrial fibrillation. N Engl J Med. 2008;359:158–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chiurchiu V, Izzi V, D'Aquilio F, Carotenuto F, Di Nardo P, Baldini PM. Brain natriuretic peptide (BNP) regulates the production of inflammatory mediators in human THP‐1 macrophages. Regul Pept. 2008;148:26–32. [DOI] [PubMed] [Google Scholar]
- 27. Jang AY, Yu J, Park YM, Shin MS, Chung WJ, Moon J. Cardiac structural or functional changes associated with CHA2DS2‐VASc scores in non‐valvular atrial fibrillation: a cross‐sectional study using echocardiography. J Cardiovasc Imaging. 2018;26:135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Patel JB, Valencik ML, Pritchett AM, Burnett JC Jr, McDonald JA, Redfield MM. Cardiac‐specific attenuation of natriuretic peptide A receptor activity accentuates adverse cardiac remodeling and mortality in response to pressure overload. Am J Physiol Heart Circ Physiol. 2005;289:H777–H784. [DOI] [PubMed] [Google Scholar]
- 29. Tamura N, Ogawa Y, Chusho H, Nakamura K, Nakao K, Suda M, Kasahara M, Hashimoto R, Katsuura G, Mukoyama M, et al. Cardiac fibrosis in mice lacking brain natriuretic peptide. Proc Natl Acad Sci USA. 2000;97:4239–4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Holtwick R, van Eickels M, Skryabin BV, Baba HA, Bubikat A, Begrow F, Schneider MD, Garbers DL, Kuhn M. Pressure‐independent cardiac hypertrophy in mice with cardiomyocyte‐restricted inactivation of the atrial natriuretic peptide receptor guanylyl cyclase‐A. J Clin Invest. 2003;111:1399–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rahmutula D, Zhang H, Wilson EE, Olgin JE. Absence of NPR‐C attenuates TGF‐ß1 induced selective atrial fibrosis and atrial fibrillation. Cardiovasc Res. 2019;115:357–372. [DOI] [PubMed] [Google Scholar]
- 32. Jansen HJ, Mackasey M, Moghtadaei M, Liu Y, Kaur J, Egom EE, Tuomi JM, Rafferty SA, Kirkby AW, Rose RA. NPR‐C (natriuretic peptide receptor‐C) modulates the progression of angiotensin II‐mediated atrial fibrillation and atrial remodeling in mice. Circ Arrhythm Electrophysiol. 2019;12:e006863. [DOI] [PubMed] [Google Scholar]
- 33. Solomon SD, Zile M, Pieske B, Voors A, Shah A, Kraigher‐Krainer E, Shi V, Bransford T, Takeuchi M, Gong J, et al. The angiotensin receptor neprilysin inhibitor LCZ696 in heart failure with preserved ejection fraction: a phase 2 double‐blind randomised controlled trial. Lancet. 2012;380:1387–1395. [DOI] [PubMed] [Google Scholar]
- 34. Januzzi JL Jr, Prescott MF, Butler J, Felker GM, Maisel AS, McCague K, Camacho A, Piña IL, Rocha RA, Shah AM, et al. Association of change in N‐terminal pro–B‐type natriuretic peptide following initiation of sacubitril‐valsartan treatment with cardiac structure and function in patients with heart failure with reduced ejection fraction. JAMA. 2019;322:1085–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]