

Abstract
Background
Genetic testing in pediatric primary dilated cardiomyopathy (DCM) patients has identified numerous disease‐causing variants, but few studies have evaluated genetic testing outcomes in this population in the context of patient and familial clinical data or assessed the clinical implications of temporal changes in genetic testing results.
Methods and Results
We performed a retrospective analysis of all patients with primary DCM who presented to our institution between 2008 and 2018. Variants identified by genetic testing were reevaluated for pathogenicity on the basis of current guidelines for variant classification. A total of 73 patients with primary DCM presented to our institution and 63 (86%) were probands that underwent cardiomyopathy‐specific gene testing. A disease‐causing variant was identified in 19 of 63 (30%) of cases, with at least 9/19 (47%) variants occurring de novo. Positive family history was not associated with identification of a causal variant. Reclassification of variants resulted in the downgrading of a large proportion of variants of uncertain significance and did not identify any new disease‐causing variants.
Conclusions
Clinical genetic testing identifies a causal variant in one third of pediatric patients with primary DCM. Variant reevaluation significantly decreased the number of variants of uncertain significance, but a large burden of variants of uncertain significance remain. These results highlight the need for periodic reanalysis of genetic testing results, additional investigation of genotype‐phenotype correlations in DCM through large, multicenter genetic studies, and development of improved tools for functional characterization of variants of uncertain significance.
Keywords: familial dilated cardiomyopathy, genetic testing, idiopathic dilated cardiomyopathy, variant of uncertain significance, variant reanalysis
Subject Categories: Genetics, Cardiomyopathy
Nonstandard Abbreviations and Acronyms
- DCM
dilated cardiomyopathy
- LB
likely benign
- LP
likely pathogenic
- VUS
variant of uncertain significance
Clinical Perspective
What Is New?
Reclassification of previously identified variants results in downgrading of ≈30% of variants of uncertain significance.
Many cases of pediatric primary dilated cardiomyopathy with an identifiable genetic cause arise from de novo mutations.
What Are the Clinical Implications?
The majority of cases of pediatric primary dilated cardiomyopathy have no identifiable genetic etiology on gene panel testing.
Many patients who have previously undergone genetic testing may benefit from reanalysis of testing results or updated testing.
Cascade screening of family members improves variant interpretation and identification of disease‐causing variants.
Dilated cardiomyopathy (DCM) is the prevalent form of pediatric cardiomyopathy and results from intrinsic or extrinsic insults to cardiomyocyte mechanics, calcium signaling, and downstream intracellular signaling pathways.1, 2 In large prospective studies of pediatric cardiomyopathy, 30% to 50% of DCM cases have an identifiable etiology, with neuromuscular disease and myocarditis being the most common causes, and the remaining 50%–70% of cases are categorized as idiopathic.3, 4, 5, 6, 7 More recently, improvement in the ability to determine an etiology in patients with DCM following a formal genetic and metabolic clinical evaluation have been reported8; however, nearly half of all cases remain without a clear cause, even upon evaluation for copy number variants and causal single‐nucleotide variants by whole exome sequencing.9
Pathogenic variants that result in a DCM phenotype have been identified in proteins that affect cardiomyocyte ultrastructure, sarcomeric integrity, force generation, cellular metabolism, and transcriptional regulation.10, 11, 12 A majority of the causal variants reported in the literature have been identified in adult patients, and only a limited number of studies have evaluated the genetic basis of DCM in the pediatric population.13, 14, 15 An estimated 25% to 50% of DCM cases in the adult population are explained by disease‐causing mutations,16 and a similar rate has been reported in small pediatric cohorts.9, 17, 18 Genetic studies that have identified variants in pediatric DCM patients have demonstrated significant overlap in the affected genes with adult DCM studies but have also identified key age‐related differences in the onset of phenotype based on the affected gene.11, 17 Genetic testing is now recommended as part of the evaluation of all patients with cardiomyopathy,19 but the prognostic utility of this information, especially within the pediatric population, remains limited.20, 21
In this study, we review our institutional experience with clinical genetic testing of pediatric probands with primary DCM. By describing the phenotypic characteristics of these patients and evaluating temporal changes in testing results, we aim to better understand the diagnostic utility of genetic testing in this population and the contribution of testing results to overall patient care.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Patient Identification and Diagnostics
Patients were retrospectively identified by querying records between January 1, 2008, and January 1, 2018, for the diagnostic Fyler code for DCM. In general, this code is assigned at our institution on the basis of the presence of structural characteristics of DCM, a dilated left ventricle with systolic dysfunction. Patients with secondary DCM or incomplete clinical documentation were excluded. Three patients with borderline inflammation on endomyocardial biopsy who were subsequently diagnosed with DCM were included (Figure S1). Positive family history was defined as the presence of cardiomyopathy in a first‐degree relative or multiple (≥2) second‐ and third‐degree relatives with cardiomyopathy or sudden cardiac death. All functional cardiac data were calculated from the patient's echocardiogram at the time of initial presentation. Ejection fraction was calculated using the 5/6 area‐length method.
Genetic Testing
Clinical cardiomyopathy genetic testing was performed at the Partners Healthcare Laboratory for Molecular Medicine (Cambridge, MA) as previously described.11 The number of genes sequenced per patient and sequencing methodology differed over the course of the study period. There was an expansion in the number of genes included in the testing panel from 5 to 62, and 23 patients underwent testing with a next‐generation sequencing–based panel composed of 62 genes associated with cardiomyopathy (Table S1). Variants were classified at the time of initial testing as previously described.11, 22 Given significant changes in cardiomyopathy testing panel content, revisions in guidelines for variant classification, and an increase in the availability of reference population allele databases over the 10‐year study period23 variants were reclassified in accordance with the American College of Medical Genetics and Genomics and the Association for Molecular Pathology Guidelines24 and additional specifications for cardiovascular genes as established by the Clinical Genome inherited cardiomyopathy expert panel.25 Gene‐specific testing was performed in a subset of parents and siblings of probands for segregation analysis.
Statistical Analysis
Continuous variables are presented as medians with ranges, and a Wilcoxon rank sum test was used for comparison. Categorical variables are displayed as frequencies and percentages, and Fisher's exact test was used for statistical comparison. Survival analyses were performed with the Kaplan‐Meier method and were divided according to age (<1 year and >1 year) and the presence or absence of a DCM‐causing genetic variant. For transplant‐free survival analysis, end points of heart transplantation or death were used and subjects were censored at the time of last follow‐up. For transplant‐censored survival analysis, a single end point of death was used, and patients were censored at the time of heart transplantation or time of last follow‐up. Pairwise comparison of survival curves was performed with a log‐rank test. Multivariate Cox proportional hazard modeling was performed to determine the association between survival time and covariates of sex, age, and presence of a DCM‐causing variant. A P<0.05 was considered statistically significant. Statistical analysis was performed in R 3.5.1.
Institutional Approval
All data collection, storage, and analysis was approved by the Institutional Review Board at Boston Children's Hospital and Partners Healthcare. Requirement for informed consent was waived by the Institutional Review Board.
Results
Patient Demographics
We identified 118 patients with DCM who were evaluated at our tertiary care center over a 10‐year period (Figure S1). The etiologies of DCM in this cohort were similar to those described in larger case series,4 with 80 patients having familial (12%) or idiopathic (56%) DCM and the remainder of cases caused by neuromuscular disease (6%), acute myocarditis (8%), metabolic disorder (10%), and genetic syndrome (8%) (Figure S1). Of the 80 patients who presented with primary DCM, 73 were probands with no prior genetic evaluation, and 63 (86%) of these patients underwent cardiomyopathy‐specific gene testing as part of their diagnostic evaluation. The median age at the time of presentation for probands that underwent genetic testing was 6.5 years (range, 0–22) and 37 of 63 (59%) were male (Table 1). There was a bimodal distribution of age at the time of presentation, with 35% of patients presenting before the age of 1 year, and a second group of patients presenting between the ages of 13 and 19 years. Eleven of 60 (18%) probands with available family history data had a positive family history of cardiomyopathy or sudden death, and all patients with a positive family history presented after 1 year of age (P=0.005). The median left ventricular ejection fraction at the time of presentation was 26% (range, 8–65) and tended to be lower in those patients presenting under 1 year of age (23% versus 29.5%; P=0.06). The median left ventricular end‐diastolic volume z score at the time of presentation was +5.85 (range, −0.1 to +21.1) and did not differ between age groups (Table 1). One individual presented with a decreased ejection fraction without ventricular dilation, and another individual with ventricular dilation with a normal ejection fraction, both of whom went on to have DCM.
Table 1.
Clinical Characteristics of DCM Probands Who Underwent Genetic Testing
| Patient Group | All | Age <1 year | Age >1 year | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Genetic Testing Status | All | + Causal Variant[Link] | − Causal Variant | P Value | All | + Causal Variant[Link] | − Causal Variant | P Value | P Value[Link] | |
| No. of patients | 63 | 23 | 8 | 15 | 40 | 11 | 29 | |||
| Age at presentation, y, median (range) | 6.5 (0–22.3) | 0.2 (0–0.9) | 0.2 (0–0.9) | 0.3 (0–0.8) | 0.86 | 13.4 (1.3–22.3) | 15.4 (5.5–18.3) | 12.8 (1.3–22.3) | 0.03 | |
| Male, n (%) | 37/63 (59) | 13/23 (57) | 3/8 (38) | 10/15 (67) | 0.22 | 24/40 (60) | 6/11 (55) | 18/29 (62) | 1 | 0.80 |
| Positive family history, n (%) | 11/60 (18) | 0/23 (0) | 0/8 (0) | 0/15 (0) | NA | 11/37 (30) | 3/10 (30) | 8/27 (30) | 1 | 0.005 |
| LVEF, median % (range) | 26 (8–65) | 23 (10–48) | 23.5 (15–35.6) | 19.4 (10–48) | 0.45 | 29.5 (8–65) | 30 (8–65) | 29 (10–54) | 0.46 | 0.06 |
| LVEDV z score, median (range) | +5.85 (−0.1 to +21.1) | +5.5 (−0.1 to +21.1 | +4.7 (+2.6 to +9.3) | +6.65 (−0.1 to +21.1) | 0.23 | +6.3 (+1.6 to +24.2 | +6.3 (+1.7 to +13.4) | +6.3 (+1.6 to +24.2) | 0.72 | 0.34 |
| Follow‐up time, y, median (range) | 3.1 (0–9.8) | 3.6 (0–9.7) | 6.1 (0.8–9.7) | 2.1 (0–9.4) | 0.05 | 3.1 (0–9.8) | 2.4 (0.1–9.3) | 4.1 (0–9.8) | 0.74 | 0.98 |
| Death or transplant, n (%) | 29/63 (46) | 6/23 (26) | 1/8 (13) | 5/15 (33) | 0.37 | 23/40 (58) | 10/11 (91) | 13/29 (45) | 0.01 | 0.02 |
LVEDV indicates left ventricular end‐diastolic volume; and LVEF, left ventricular ejection fraction.
Includes likely pathogenic and pathogenic variants as per American College of Medical Genetics and Genomics and the Association for Molecular Pathology classification criteria, as well as those classified as variants of uncertain significance–favor pathogenic where clinical judgment was used to override the initial variant classification following clinical assessment.
P value comparing age <1 year and age >1 year groups.
Genetic Evaluation of Patients With DCM
Cardiomyopathy gene panel testing of 63 probands resulted in the identification of 116 variants that were classified at the initial time of testing as follows: 8 pathogenic, 11 likely pathogenic (LP), 90 variants of unknown significance (VUSs), 3 likely benign (LB), 2 benign, and 2 unclassified on the basis of their position in untranslated regions (Table 2). The burden of VUSs per patient correlated with the size of the testing panel as previously described (Table S2).11, 18 Following variant reevaluation for this study, the 116 variants were classified as 7 pathogenic, 8 LP, 68 VUSs (of which 6 are VUS‐favor pathogenic), 22 LB, and 11 benign (Table 2). The largest shift in variant classification following reevaluation was in the VUS category, where 26 of 90 (29%) of the original VUSs were reclassified as LB or benign (Table 2). The next largest change in classification occurred in variants previously designated as LP, with 4 being downgraded to VUS‐favor pathogenic and 1 being reclassified as pathogenic. No VUS, LB, or benign variants were advanced to a higher‐grade category upon reclassification. These changes in variant classification led to a decrease in the number of “inconclusive” test results with VUS (60%–52%), and an increase in the proportion of “negative” tests that identified only LB or benign variants (14%–24%) (Table S2).
Table 2.
Variant Classifications
| Variants | After Reassessment | ||||||
|---|---|---|---|---|---|---|---|
| P | LP | VUS | LB | B | Not Categorized | ||
| Original | P | 6[Link] | 2 | 0 | 0 | 0 | 0 |
| LP | 1 | 6[Link] | 4[Link] | 0 | 0 | 0 | |
| VUS | 0 | 0 | 64[Link] , [Link] | 18 | 8 | 0 | |
| LB | 0 | 0 | 0 | 2[Link] | 1 | 0 | |
| B | 0 | 0 | 0 | 0 | 2[Link] | 0 | |
| Not categorized | 0 | 0 | 0 | 2 | 0 | 0 | |
Variant classifications at the time of original testing and reclassification after applying current American College of Medical Genetics and Genomics and the Association for Molecular Pathology variant classification criteria. B indicates benign; LB, likely benign; LP, likely pathogenic; P, pathogenic; and VUS, variant of uncertain significance.
Variants that did not change and shaded boxes indicate variant reclassifications.
Four variants reclassified as VUS‐favor pathogenic.
Two VUS‐favor pathogenic.
Familial Testing of Variants
Familial testing for 9 probands with a pathogenic or LP variant revealed that 3 variants were inherited and 6 variants were de novo mutations (Table 3). Additionally, 6 probands with a potentially disease‐causing VUS underwent familial testing to help further clarify the variant's clinical significance. Of these 6 VUSs, 3 occurred de novo, and 3 were inherited from individuals without a cardiac structural phenotype at the time of testing.
Table 3.
Disease‐Causing Variants
| Genes | Transcript | Mutation | Mutation type | AA change | Classification | Age, y | Sex | Additional Clinical Data |
|---|---|---|---|---|---|---|---|---|
| BAG3 | NM_004281.3 | c.1363G>A | Missense | p.Glu455Lys | P | 18.3 | M | No familial data available |
| DES | NM_001927.3 | c.347A>G | Missense | p.Asn116Ser | LP | 11.5 | M | No familial data available |
| DSP | NM_004415.2 | c.1873C>T | Nonsense | p.Gln625X | P | 16.0 | F | Variant paternally inherited. Father (40 y) found to have DCM on cascade screening. |
| LAMP2 | NM_002294.2 | c.294G>A | Nonsense | p.Trp98X | P | 16.7 | M | De novo |
| LMNA | NM_170707.2 | c.1106T>C | Missense | p.Leu369Pro | LP | 12.7 | F | De novo |
| LMNA | NM_170707.2 | c.1621C>T | Missense | p.Arg541Cys | P | 14.8 | M | Mother (no genetic testing) with history of arrhythmia at 16 y and status post ICD placement. Sibling with variant and DCM phenotype at 12 years of age. |
| MYBPC3 | NM_000256.3 | c.2504delG | Frameshift | p.Arg835ProfsX2 | P | 0.9 | M | Paternally inherited. Father (40 y) phenotype negative. Compound heterozygote with additional maternally inherited VUS in MYBPC3 |
| MYH7 | NM_000257.2 | c.1106G>A | Missense | p.Arg369Gln | P | 0.1 | M | De novo |
| MYH7 | NM_000257.2 | c.1922G>C | Missense | p.Gly641Ala | LP | 0.6 | F | De novo |
| MYH7 | NM_000257.2 | c.1798C>T | Missense | p.Pro600Ser | VUS favor pathogenic | 0.3 | F | De novo |
| PKP2 | NM_004572.3 | c.1034+1G>T | Splicing | NA | LP | 16.3 | M | No familial data available |
| TNNI3 | NM_000363.4 | c.544G>A | Missense | p.Glu182Lys | LP | 0.1 | F | De novo |
| TNNT2 | NM_001001430.1 | c.629_631delAGA | Deletion/in‐frame | p.Lys210del | P | 14.4 | F | Sibling with history of DCM and heart transplant at 12 y (genotype unavailable). Parental genotype/phenotype not available. |
| TNNT2 | NM_001001430.1 | c.629_631delAGA | Deletion/in‐frame | p.Lys210del | P | 16.6 | M | De novo |
| TNNT2 | NM_001001430.1 | c.517C>T | Missense | p.Arg173Trp | LP | 17.7 | F | Two siblings genotype and phenotype negative, parental testing not available |
| TNNT2 | NM_001001430.1 | c.391C>T | Missense | p.Arg131Trp | LP | 0.1 | M | No familial data available |
| TNNT2 | NM_001001430.1 | c.264T>G | Missense | p.Asp88Glu | VUS favor pathogenic | 0.3 | F | De novo |
| TPM1 | NM_000366.5 | c.423G>C | Missense | p.Met141Ile | VUS favor pathogenic | 0.1 | F | De novo |
| TTN | NM_133378.4 | c.65683delG | Frameshift | p.Ala21895ProfsX8 | LP | 14.0 | F | Father (35 y) diagnosed with DCM on cascade screening. Genotype unknown. |
Variants identified to be disease causing in the pediatric DCM cohort on the basis of pathogenic or likely pathogenic classification per American College of Medical Genetics and Genomics and the Association for Molecular Pathology classification criteria, as well as variants initially classified as VUS‐favor pathogenic that were determined to be disease causing at the discretion of the clinical geneticist/cardiologist. Listed age is age at presentation.
ICD indicates implantable cardioverter defibrillator; LP, likely pathogenic; P, pathogenic; and VUS, variant of uncertain significance.
Identification of Disease‐Causing Variants
Pathogenic variants, LP, and VUS‐favoring pathogenic, were identified in 12 genes, including those integral to sarcomere function (MYH7, TTN, TNNT2, TNNI3, MYBPC3, TPM1, BAG3), cellular structure (LMNA, DES), cellular junctions (PKP2, DSP), and lysosomal function (LAMP2) (Table 3). Nearly all mutations were heterozygous and thus demonstrated an autosomal dominant pattern. One proband was found to be hemizygous for a truncating mutation on the X chromosome encoded LAMP2. Nine de novo variants were identified in the following genes: LAMP2, MYH7 (n=3), LMNA, TNNI3, TMP1, and TNNT2 (n=2). Three of these variants in MYH7, TNNT2, and TPM1 were classified as VUS‐favor pathogenic, and within the clinical context were interpreted as causal in their respective cases. No individuals had a LP or pathogenic variant or a VUS‐favor pathogenic variant in >1 gene. Collectively, 19 of 63 (30%) patients with idiopathic or familial DCM were determined to have a disease‐causing variant, with at least 9 of 19 (47%) variants occurring de novo (Table 3). The most frequently identified mutations were in the sarcomeric genes TNNT2 (n=5) and MYH7 (n=3). All patients with mutations in MYH7 presented in infancy, while those with TNNT2 mutations presented at 2 discrete time points, either in early infancy (n=2) or adolescence (n=3). Rates at which disease‐causing mutations were identified were similar between patients with a positive family history (3/11; 27%) and those without a positive family history (16/49; 33%). Additionally, the rate of detection of a causal variant did not differ by patient age (Table 1). The percentage of “positive” genetic tests that yielded a pathogenic or LP variant increased by 1.3‐fold with expansion of the gene testing panel to >50 genes (20% for panels <50 genes versus 27% for >50 genes tested) and was largely unaffected by variant reclassification (Tables S2 and S3).
Outcomes of Patients With DCM
Patients with DCM who underwent genetic testing had a median clinical follow‐up of 37 months (range, 0–118), and 29 of 63 (46%) patients underwent heart transplantation or died during follow‐up. Echocardiographic measurements of left ventricular size and systolic function did not differ on the basis of the presence or absence of a causal variant in age‐stratified patients (Table 1). Patients presenting after 1 year of age with a disease‐causing variant had decreased transplant‐free survival, with 10 of 11 (91%) patients having a composite outcome of death or transplant versus 13 of 29 (45%) patients >1 year of age without an identified variant (P=0.01) (Table 1 and Figure A).On proportional hazards modeling there was an increased risk of death or transplantation in patients presenting after the first year of life (hazard ratio [HR], 1.2; 95% CI, 1.05–1.3; P=0.007) (Figure B),consistent with previously reported age‐related risk of death or transplantation.4 The presence of a disease‐causing variant was not significantly associated with risk of transplantation or death in the overall cohort (HR, 1.9; 95% CI, 0.87–4.2; P=0.11) (Figure B). Transplant‐censored survival was not different between groups, and the observed differences were largely driven by rates of heart transplantation (Figure C).
Figure 1. Kaplan–Meier analysis of transplant‐free and transplant‐censored Survival.
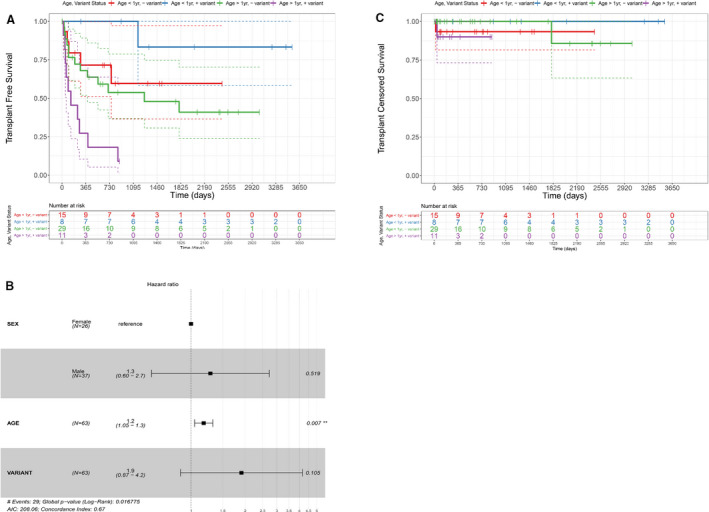
A, Kaplan‐Meier survival analysis of transplant‐free survival stratified by age and genetic testing result. “Variant” denotes the presence of a DCM‐causing variant on genetic testing. Patients who presented with DCM at >1 year of age and who had an identified causal genetic variant have significantly worse transplant‐free survival when compared with those of a similar age without an identified genetic variant (P=0.0139, log‐rank test). Dashed lines denote the 95% confidence interval. B, Kaplan‐Meier survival analysis of transplant‐censored survival demonstrates that differences in (A) are largely driven by the rate of transplantation. C, Forest plot of multivariate Cox proportional hazards modeling for transplant‐free survival and selected covariates. Age but not DCM variant status is an independent risk factor for death or transplant. DCM indicates dilated cardiomyopathy.
Discussion
In this study, we performed a retrospective analysis of cardiomyopathy genetic testing in one of the largest reported pediatric cohorts with primary DCM to undergo genetic evaluation, and identified a disease‐causing variant in 30% of patients. This detection rate for causal variants in probands was similar to results from adult DCM studies and other pediatric cohorts.9, 15, 17, 18
In probands presenting after 1 year of age, we observed lower transplant‐free survival in those with a positive genetic testing result compared with those without an identified causal variant. Previous studies have similarly reported an association between genetic testing results and transplant‐free survival.17 We are unable to report whether the natural history of patients with DCM with a positive genetic testing result differs significantly from those without an identified causal variant because of the high rates of transplantation in our pediatric DCM population. As a result, the observed difference in transplant‐free survival could be the result of confounding by indication.
Surprisingly, we did not find a difference in rates of positive cardiomyopathy genetic testing in patients with (27%) and without (33%) a family history of cardiomyopathy or sudden death. In our study, this is likely the result of a high proportion of de novo variants identified in patients with a negative family history. Few published case series have comprehensively evaluated the mechanism of inheritance of variants in pediatric DCM, but small studies of pediatric DCM have shown a high fraction of de novo variants in established DCM genes.26 In the 73% of patients with DCM and positive family history who did not have a causal variant identified, incomplete testing for the full spectrum of potential cardiomyopathy‐associated genes, oligogenic or polygenic causes of disease, or undetected types of genomic variation such as structural variants could contribute to inability to identify a genetic cause. The lack of association between family history and genotype in our study supports the current guideline recommendation for clinical genetic testing in all cardiomyopathy probands, regardless of family history19 and the need for additional genetic studies in large cohorts of pediatric patients with DCM.
Our results highlight the importance of genetic and phenotypic evaluation in parents and first‐degree relatives of probands for the interpretation of genetic testing results in DCM probands. Familial testing of healthy parents provided supporting evidence of variant pathogenicity in 3 probands by identifying the variant as de novo. In our small cohort, the use of this familial data aided in the identification of 3 of 19 (16%) of the disease‐causing variants. Given the large number of patients with VUSs and inconclusive test results in our data set, additional information ascertained from familial genetic testing and cardiac screening, particularly in the parents, could provide evidence to help interpret the clinical significance of these variants. Beyond the value of familial screening to interpretation of variants identified in probands, data from screening help inform risk stratification of siblings and family planning.
We report that variant reclassification was able to reduce the overall burden of VUSs by nearly 30%. This is primarily attributable to the availability of reference databases such as gnomAD,23 which allow the use of population allele frequencies to aid in the classification variants.23, 27 Initial comparison of clinical genetic testing results in DCM with allele frequencies in the ExAC reference database demonstrated a large number of rare variants in purported DCM genes, leading to likely overestimation of pathogenicity in some cases.28 Our results are in line with these findings and others,11 with a large proportion of variants being downgraded on reclassification. Despite a reduction in the number of VUSs after reclassification, a large number of VUSs remain in our cohort. Additional population‐level allele data that include diverse ethnic subgroups, increased data sharing among testing laboratories, and efforts to functionally classify VUSs will likely aid in further decreasing this burden. These results highlight the need for periodic reevaluation of variants, either by the testing laboratory, a geneticist, or cardiologist versed in variant interpretation. While general guidelines for variant reevaluation by testing laboratories have been put forth by the ACMG29, there are no guidelines to inform when a clinician should initiate this process. A reasonable approach might be to perform a review of genetic testing if there is an inherited VUS or LP variant, more than a year has passed, and the parents are planning to have more children; when pediatric patients transition care to adult providers; or when discussing family planning with patients with primary DCM.
The disease‐causing variants identified in our study are located in 12 genes that have been previously implicated in pediatric DCM. Variants in MYH7 and TNNT2 represent 25% of the disease‐causing variants in our cohort and have been frequently identified in other pediatric DCM cohorts.8, 9, 11, 17, 30 All patients with a pathogenic MYH7 variant presented in infancy, while the patients with TNNT2 variants presented at 2 disparate time points, with 2 patients presenting in early infancy and 3 patients during adolescence. Three patients were found to have pathogenic variants in genes that are associated with arrhythmogenic cardiomyopathy (DSP, PKP2, and DES).31 Eight probands who presented in adolescence and in whom a pathogenic or LP variant was not identified were not tested for variants in TTN, the most frequently identified gene harboring pathogenic variants in DCM in both adolescent and adult patients.10 Given the importance of TTN variants in the pathogenesis of DCM and the increase in diagnostic yield observed with expansion of testing panels11 patients evaluated early in the era of genetic testing for DCM are likely to benefit from additional testing. As genetic testing costs decrease, a comprehensive whole genome sequencing approach may afford the best opportunity to identify pathogenic single‐nucleotide variants, copy number variants, and/or polygenic causes of DCM.
There are several important limitations to our study. First, the use of diagnostic codes to identify DCM patients may not have identified all cases and may select for a subset with a more severe phenotype. All genetic testing was performed in the same laboratory, but there were significant changes in the number of cardiomyopathy‐associated genes tested over the study period. As such, our study may underestimate the number of pathogenic or LP variants in this cohort, and those determined to have negative testing results early in the study period could harbor a pathogenic variant in untested genes. Finally, our data collection was retrospective, and some patients were lost to follow‐up, which could underestimate the number of important clinical events.
Our work highlights that genetic testing of probands, especially when coupled with familial testing of first‐degree relatives, is a useful diagnostic tool for identifying disease‐causing variants and determining an etiology in patients with primary DCM. Genetic testing results are dynamic, and changes in variant classification over time significantly reduced the burden of VUSs; however, a significant number of probands with inconclusive test results remain. Additional genetic studies in this patient subgroup, continuous improvement of tools and data sharing mechanisms that aid in the clarification of the clinical significance of VUSs, and functional assays to better understand the effects of genetic variants on cardiomyocyte function will provide further clarification of VUSs. Additionally, a whole‐genome sequencing approach to allow for the simultaneous evaluation of copy number variants and comprehensive gene sequencing in a significantly larger number of interrogated genes is likely to be the next frontier in the genetic evaluation of pediatric DCM.
Sources of Funding
DQ is supported by T32 HL 7572‐33 from the National Heart, Lung, and Blood Institute—NIH. This research was funded in part by a generous donation from the Joseph Middlemiss Big Heart Foundation, Inc.
Disclosures
None.
Supporting information
Tables S1–S3 Figure S1
Acknowledgments
The authors thank Christopher Koch for technical assistance in streamlining analysis of patient genetic testing data.
(J Am Heart Assoc. 2020;9:e016195 DOI: 10.1161/JAHA.120.016195.)
Supplementary Materials for this article are available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.120.016195
For Sources of Funding and Disclosures, see page 8.
See Editorial by Towbin
References
- 1. Colan SD. 2005. Ventricular Function in Volume Overload Lesions. In: Fogel MA, ed. Ventricular Function and Blood Flow in Congenital Heart Disease, ; 2007. DOI: 10.1002/9780470994849.ch12. [DOI] [PubMed] [Google Scholar]
- 2. Davis J, Davis LC, Correll RN, Makarewich CA, Schwanekamp JA, Moussavi‐Harami F, Wang D, York AJ, Wu H, Houser SR, et al. A tension‐based model distinguishes hypertrophic versus dilated cardiomyopathy. Cell. 2016;165:1147–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wilkinson JD, Westphal JA, Bansal N, Czachor JD, Razoky H, Lipshultz SE. Lessons learned from the pediatric cardiomyopathy registry (PCMR) study group. Cardiol Young. 2015;25(suppl 2):140–153. [DOI] [PubMed] [Google Scholar]
- 4. Towbin JA, Lowe AM, Colan SD, Sleeper LA, Orav EJ, Clunie S, Messere J, Cox GF, Lurie PR, Hsu D, et al. Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA. 2006;296:1867–1876. [DOI] [PubMed] [Google Scholar]
- 5. Lipshultz SE, Sleeper LA, Towbin JA, Lowe AM, Orav EJ, Cox GF, Lurie PR, McCoy KL, McDonald MA, Messere JE, et al. The incidence of pediatric cardiomyopathy in two regions of the United States. N Engl J Med. 2003;348:1647–1655. [DOI] [PubMed] [Google Scholar]
- 6. Nugent AW, Daubeney PE, Chondros P, Carlin JB, Cheung M, Wilkinson LC, Davis AM, Kahler SG, Chow CW, Wilkinson JL, et al. The epidemiology of childhood cardiomyopathy in Australia. N Engl J Med. 2003;348:1639–1646. [DOI] [PubMed] [Google Scholar]
- 7. Lipshultz SE, Law YM, Asante‐Korang A, Austin ED, Dipchand AI, Everitt MD, Hsu DT, Lin KY, Price JF, Wilkinson JD, et al. Cardiomyopathy in children: classification and diagnosis: a scientific statement from the American Heart Association. Circulation. 2019;140:e9–e68. [DOI] [PubMed] [Google Scholar]
- 8. Kindel SJ, Miller EM, Gupta R, Cripe LH, Hinton RB, Spicer RL, Towbin JA, Ware SM. Pediatric cardiomyopathy: importance of genetic and metabolic evaluation. J Card Fail. 2012;18:396–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Herkert JC, Abbott KM, Birnie E, Meems‐Veldhuis MT, Boven LG, Benjamins M, du Marchie Sarvaas GJ, Barge‐Schaapveld D, van Tintelen JP, van der Zwaag PA, et al. Toward an effective exome‐based genetic testing strategy in pediatric dilated cardiomyopathy. Genet Med. 2018;20:1374–1386. [DOI] [PubMed] [Google Scholar]
- 10. Burke MA, Cook SA, Seidman JG, Seidman CE. Clinical and mechanistic insights into the genetics of cardiomyopathy. J Am Coll Cardiol. 2016;68:2871–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pugh TJ, Kelly MA, Gowrisankar S, Hynes E, Seidman MA, Baxter SM, Bowser M, Harrison B, Aaron D, Mahanta LM, et al. The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet Med. 2014;16:601–608. [DOI] [PubMed] [Google Scholar]
- 12. Ware SM. Genetic diagnosis in pediatric cardiomyopathy: clinical application and research perspectives. Prog Pediatr Cardiol. 2011;31:99–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ware SM. Genetics of paediatric cardiomyopathies. Curr Opin Pediatr. 2017;29:534–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Khan R, Pahl E, Dellefave‐Castillo L, Rychlik K, McNally E, Webster G. Genetic testing outcomes in pediatric dilated cardiomyopathy. J Heart Lung Transplant. 2019;38:S201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vasilescu C, Ojala TH, Brilhante V, Ojanen S, Hinterding HM, Palin E, Alastalo TP, Koskenvuo J, Hiippala A, Jokinen E, et al. Genetic basis of severe childhood‐onset cardiomyopathies. J Am Coll Cardiol. 2018;72:2324–2338. [DOI] [PubMed] [Google Scholar]
- 16. Haas J, Frese KS, Peil B, Kloos W, Keller A, Nietsch R, Feng Z, Muller S, Kayvanpour E, Vogel B, et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur Heart J. 2015;36:1123–1135a. [DOI] [PubMed] [Google Scholar]
- 17. Ellepola CD, Knight LM, Fischbach P, Deshpande SR. Genetic testing in pediatric cardiomyopathy. Pediatr Cardiol. 2018;39:491–500. [DOI] [PubMed] [Google Scholar]
- 18. Ouellette AC, Mathew J, Manickaraj AK, Manase G, Zahavich L, Wilson J, George K, Benson L, Bowdin S, Mital S. Clinical genetic testing in pediatric cardiomyopathy: is bigger better? Clin Genet. 2018;93:33–40. [DOI] [PubMed] [Google Scholar]
- 19. Hershberger RE, Givertz MM, Ho CY, Judge DP, Kantor PF, McBride KL, Morales A, Taylor MRG, Vatta M, Ware SM, et al. Genetic evaluation of cardiomyopathy: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2018;20:899–909. [DOI] [PubMed] [Google Scholar]
- 20. Lakdawala NK. Using genetic testing to guide therapeutic decisions in cardiomyopathy. Curr Treat Options Cardiovasc Med. 2013;15:387–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kayvanpour E, Sedaghat‐Hamedani F, Amr A, Lai A, Haas J, Holzer DB, Frese KS, Keller A, Jensen K, Katus HA, et al. Genotype‐phenotype associations in dilated cardiomyopathy: meta‐analysis on more than 8000 individuals. Clin Res Cardiol. 2017;106:127–139. [DOI] [PubMed] [Google Scholar]
- 22. Duzkale H, Shen J, McLaughlin H, Alfares A, Kelly MA, Pugh TJ, Funke BH, Rehm HL, Lebo MS. A systematic approach to assessing the clinical significance of genetic variants. Clin Genet. 2013;84:453–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss‐of‐function intolerance across human protein‐coding genes. bioRxiv. 2019;531210. [Google Scholar]
- 24. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier‐Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kelly MA, Caleshu C, Morales A, Buchan J, Wolf Z, Harrison SM, Cook S, Dillon MW, Garcia J, Haverfield E, et al. Adaptation and validation of the ACMG/AMP variant classification framework for MYH7‐associated inherited cardiomyopathies: recommendations by Clingen's inherited cardiomyopathy expert panel. Genet Med. 2018;20:351–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Long PA, Evans JM, Olson TM. Diagnostic yield of whole exome sequencing in pediatric dilated cardiomyopathy. J Cardiovasc Dev Dis. 2017;4:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell‐Luria AH, Ware JS, Hill AJ, Cummings BB, et al. Analysis of protein‐coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Walsh R, Thomson KL, Ware JS, Funke BH, Woodley J, McGuire KJ, Mazzarotto F, Blair E, Seller A, Taylor JC, et al. Reassessment of mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet Med. 2017;19:192–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Deignan JL, Chung WK, Kearney HM, Monaghan KG, Rehder CW, Chao EC, ACMG Laboratory Quality Assurance Committee. Points to consider in the reevaluation and reanalysis of genomic test results: a statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2019;21:1267–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rampersaud E, Siegfried JD, Norton N, Li D, Martin E, Hershberger RE. Rare variant mutations identified in pediatric patients with dilated cardiomyopathy. Prog Pediatr Cardiol. 2011;31:39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. DeWitt ES, Chandler SF, Hylind RJ, Beausejour Ladouceur V, Blume ED, VanderPluym C, Powell AJ, Fynn‐Thompson F, Roberts AE, Sanders SP, et al. Phenotypic manifestations of arrhythmogenic cardiomyopathy in children and adolescents. J Am Coll Cardiol. 2019;74:346–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S3 Figure S1