

Abstract
Background
Although PAR‐1 (protease‐activated receptor‐1) exerts important functions in the pathophysiology of the cardiovascular system, the role of PAR‐1 signaling in heart failure development remains largely unknown. We tested the hypothesis that PAR‐1 signaling inhibition has protective effects on the progression of cardiac remodeling induced by chronic renin–angiotensin system activation using renin‐overexpressing hypertensive (Ren‐Tg) mice.
Methods and Results
We treated 12‐ to 16‐week‐old male wild‐type (WT) mice and Ren‐Tg mice with continuous subcutaneous infusion of the PAR‐1 antagonist SCH79797 or vehicle for 4 weeks. The thicknesses of interventricular septum and the left ventricular posterior wall were greater in Ren‐Tg mice than in WT mice, and SCH79797 treatment significantly decreased these thicknesses in Ren‐Tg mice. The cardiac fibrosis area and monocyte/macrophage deposition were greater in Ren‐Tg mice than in WT mice, and both conditions were attenuated by SCH79797 treatment. Cardiac mRNA expression levels of PAR‐1, TNF‐α (tumor necrosis factor‐α), TGF‐β1 (transforming growth factor‐β1), and COL3A1 (collagen type 3 α1 chain) and the ratio of β‐myosin heavy chain (β‐MHC) to α‐MHC were all greater in Ren‐Tg mice than in WT mice; SCH79797 treatment attenuated these increases in Ren‐Tg mice. Prothrombin fragment 1+2 concentration and factor Xa in plasma were greater in Ren‐Tg mice than in WT mice, and both conditions were unaffected by SCH79797 treatment. In isolated cardiac fibroblasts, both thrombin and factor Xa enhanced ERK1/2 (extracellular signal‐regulated kinase 1/2) phosphorylation, and SCH79797 pretreatment abolished this enhancement. Furthermore, gene expression of PAR‐1, TGF‐β1, and COL3A1 were enhanced by factor Xa, and all were inhibited by SCH79797.
Conclusions
The results indicate that PAR‐1 signaling is involved in cardiac remodeling induced by renin–angiotensin system activation, which may provide a novel therapeutic target for heart failure.
Keywords: cardiac fibrosis, cardiac hypertrophy, factor Xa, protease‐activated receptor, renin–angiotensin system
Subject Categories: Hypertension, Hypertrophy, Heart Failure, ACE/Angiotension Receptors/Renin Angiotensin System
Nonstandard Abbreviations and Acronyms
- BP
blood pressure
- CF
cardiac fibroblast
- ERK
extracellular signal‐regulated kinase
- FXa
factor Xa
- HF
heart failure
- LV
left ventricular
- MAP
mitogen‐activated protein
- PAR
protease‐activated receptor
- RAS
renin–angiotensin system
- Ren‐Tg
renin‐overexpressing hypertensive
Clinical Perspective
What Is New?
SCH79797, a PAR‐1 (protease‐activated receptor‐1) antagonist, attenuates cardiac hypertrophy, fibrosis, and inflammatory cell infiltration in renin‐overexpressing hypertensive (Ren‐Tg) mice.
SCH79797 inhibits PAR‐1 expression and proinflammatory and profibrotic cytokines in the heart of the Ren‐Tg mice and in isolated cardiac fibroblasts.
SCH79797 may have a protective effect against cardiac hypertrophy and fibrosis induced by enhanced renin–angiotensin system activity, partly through the inhibition of PAR‐1 signaling.
What Are the Clinical Implications?
The cardioprotective effects of PAR‐1 antagonist shown in this study indicate that PAR‐1 signaling may play an important role in cardiac remodeling under the enhanced renin–angiotensin system condition. Our results may provide a novel therapeutic approach for cardiac remodeling and heart failure.
Heart failure (HF) is a major and increasing cause of hospitalization and one of the current serious health and economic issues across the world.1 Despite the advances in medical therapy and devices for treatment, including defibrillator or cardiac resynchronization therapy, the mortality of patients with HF remains significant, even among those who have HF with preserved ejection fraction.2, 3 Therefore, there is an urgent need to identify a new target for the treatment of HF to improve clinical outcomes in patients. The risk factors involved in the development of HF include hypertension, diabetes mellitus, dyslipidemia, smoking, and obesity. In addition to these classic factors, recent evidence demonstrates the involvement of the coagulation cascade in the process of cardiovascular remodeling.4, 5, 6, 7 Among the various signaling pathways affecting coagulation activity, the renin–angiotensin system (RAS) enhances coagulation and decreases fibrinolytic activity, which subsequently contribute to the atherosclerotic process and potentially affect the impact of hypertension on the occurrence of cardiovascular events.8, 9
Thrombin is a principal protease of the coagulation cascade that converts soluble fibrinogen into an insoluble clot, and factor Xa (FXa) is an upstream activator of thrombin.10 In addition to its important role in thrombosis, thrombin and FXa can induce multiple cellular responses through PAR‐1 (protease‐activated receptor‐1).11 PAR‐1 belongs to the G‐protein‐coupled receptor superfamily and is widely expressed in the vascular tissue and the heart, including cardiomyocytes and cardiac fibroblasts (CFs).11, 12, 13 Previous studies have demonstrated that PAR‐1 signaling plays important roles in the regulation of cardiac hypertrophy, ischemic cardiac remodeling,13, 14 and cardiovascular physiology and pathophysiology.5, 11, 15 However, the relationship between PAR‐1 signaling and the development of HF has not yet been elucidated. In this study, we tested the hypothesis that inhibition of PAR‐1 signaling may exert a protective effect on the progression of cardiac hypertrophy and fibrosis induced by continuous RAS activation using renin‐overexpressing hypertensive (Ren‐Tg) mice.
Methods
The data, analytic methods, and study materials will be available to other researchers for purposes of reproducing results or replicating procedures, as described in this article, on reasonable request by contacting the corresponding author.
Animals and Study Protocols
We used wild‐type (WT) mice and Ren‐Tg mice that overexpress renin at a constantly high level, as described previously.16 Briefly, a modified mouse renin transgene driven by a liver‐specific albumin promoter/enhancer was inserted into the genome as a single copy at the liver‐specific locus of ApoA1 (apolipoprotein A1) and ApoC3.16, 17 The resulting transgene expressed renin ectopically in the liver at constantly high levels and resulted in elevated plasma levels of active renin and angiotensin II. We and other investigators have reported that Ren‐Tg mice exhibit elevated systolic blood pressure (BP) at an age as early as 12 weeks.16, 18 Heterozygous (1 copy of the renin transgene) Ren‐Tg mice were backcrossed with C57BL6/N mice for at least 15 generations before the experiments.
Male Ren‐Tg and WT mice aged 12 to 16 weeks were treated with continuous subcutaneous infusion of either the PAR‐1 antagonist SCH79797 (formula, C23H25N5.2HCl; molecular weight, 444.41; 25 μg/kg per day; Abcam)15, 19 or dimethyl sulfoxide (DMSO) as vehicle for 4 weeks using the Alzet osmotic minipump (model 1004; Durect Corp). We dissolved SCH79797 in DMSO at a final concentration of 0.55 mmol/L. Systolic BP was measured, and echocardiography was performed at baseline and after 2 and 4 weeks of the treatment. After the treatment period, mice were euthanized under anesthesia with a mixture of medetomidine (0.75 mg/kg), midazolam (4 mg/kg), and butorphanol tartrate (5 mg/kg) by intraperitoneal injection, and then subsequent experiments were conducted. All procedures were performed according to the Guide for the Care and Use of Laboratory Animals of the US National Institutes of Health and were approved by the institutional animal care and use Committee of Hirosaki University Graduate School of Medicine, Hirosaki, Japan.
BP Measurement and Echocardiography
Ren‐Tg and WT mice were maintained in a warm chamber set at 37°C for 10 minutes before measuring their BP and pulse rate. Systolic BP and pulse rate were measured by the tail‐cuff method using BP‐98A (Softron). After discarding the highest and lowest readings, at least 10 readings were averaged, as previously described.20 Echocardiography was performed using an echocardiography system (HD11 XE with L15‐7io Broadband Compact Linear Array; Phillips), and M‐mode tracing was recorded from the short‐axis view at the papillary muscle level, as described earlier.18 Interventricular septum thickness and left ventricular (LV) posterior wall thickness in diastole, LV end‐diastolic dimension, LV end‐systolic dimension, and LV fractional shortening (calculated as the difference of LV end‐diastolic dimension minus LV end‐systolic dimension, divided by LV end‐diastolic dimension) were measured, and measurements obtained from at least 3 cardiac cycles were averaged.
Histological Analysis
Left ventricles were fixed in 10% formalin, embedded in paraffin, and stained with Masson's trichrome to evaluate cardiac interstitial fibrosis. Immunostaining for CD68 was performed to evaluate the infiltration of monocytes or macrophages in the heart. Stained sections were visualized using BZ‐X710 (Keyence), and the fibrotic or immunostaining‐positive area was analyzed using the BZ‐X analyzer (Keyence). The captured images were imported into the software, and the fibrotic or immunostaining‐positive area was automatically extracted from the whole images and calculated.
Mass Spectrometry
Mouse plasma sample preparation was carried out using a solid‐phase extraction kit (Impact; Phenomenex). Briefly, 400 μL of acetonitrile was dispensed to the upper 96‐well plate, and 100 μL of plasma was added directly into the methanol in each well. The sample was vortexed for 2 minutes and stood for 25 minutes. The plate was placed on a collection plate and 5 psi nitrogen gas was applied using a positive‐pressure manifold to filtrate precipitated plasma proteins. The filtrate was dried with nitrogen gas before SCH79797 was extracted using 0.1% formic acid in 50% acetonitrile (200 μL) into the lower 96‐well plate for analysis. Quantification was carried out using external standards with control plasma and a calibration curve. The liquid chromatography–tandem mass spectrometry (LC‐MS/MS) system comprised a high‐performance liquid chromatography system (ExionLC AD; AB Sciex) coupled to a QTRAP6500+ mass spectrometer (AB Sciex) in electrospray ionization mode. SCH79797 was analyzed via LC‐MS/MS in positive mode. Ten microliters of the sample extract were injected onto a high‐performance liquid chromatography C18 column (Zorbax Eclipse XDB‐C18 column, 3×100 mm, 3.5 μm; Agilent) at 40°C using a 10‐minute solvent gradient with 0.1% formic acid in water (solvent A) and 0.1% formic acid in acetonitrile (solvent B). Additional liquid chromatography settings for LC‐MS/MS are as follows: 50% to 100% B in 5 minutes; 100% B in 0.5 minute; 100% to 50% B in 0.5 minute; 50% B in 1 minute at a flow rate of 0.25 mL/min. MS settings for LC‐MS/MS mode are as follows: curtain gas, 30; ion spray voltage, 4500 V; temperature, 400°C; ion source gas 1, 50 psi; ion source gas 2, 70 psi; collision gas, 9 psi; declustering potential, 186 V; entrance potential, 10 V; collision energy, 49 V; collision cell exit potential, 18 V. SCH79797 was identified and quantified using multiple reaction monitoring with quartile 1 (Q1) and Q3 transition of 372.131 and 356.1 m/z, respectively.
Quantitative Reverse Transcriptase Polymerase Chain Reaction
Hearts were rapidly excised, and the atrium was removed and stored in liquid nitrogen. Total RNA was extracted from left ventricles using the RNeasy Fibrous Tissue Mini Kit (Qiagen), according to the manufacturer's protocol. The RNA was converted into cDNA using the Primescript II 1st standard cDNA synthesis kit (Takara Bio). Quantitative polymerase chain reaction was performed using CFX Connect (Applied Biosystems) with TaqMan Universal PCR Master Mix (Applied Biosystems). Specific primers were purchased from Applied Biosystems to detect PAR‐1 (assay ID, Mm00438851_g1), TNF‐α (tumor necrosis factor‐α; assay ID, Mm0043258_m1), TGF‐β1 (transforming growth factor‐β1; assay ID: Mm01178820_m1), α‐myosin heavy chain (α‐MHC; assay ID, Mm00440346_g1), β‐MHC (assay ID, Mm00600532_g1), COL3A1 (collagen type 3 α1; assay ID: Mm01254476_m1), and GAPDH; assay ID: Mm99999915_g1). The mRNA expression levels were normalized by GAPDH.
Biochemical Measurements
After anesthesia with isoflurane, blood was collected from the orbital venous plexus and stored on ice. It was then centrifuged at 1000g for 15 minutes at 4°C, and the supernatant was collected. The concentration of prothrombin fragment 1+2 was measured using the mouse prothrombin fragment 1+2 ELISA kit (MybioSource), and FXa was measured using the mouse coagulation FXa ELISA kit (MybioSource). All procedures were conducted according to the manufacturer's protocol.
Isolation of CFs
Male WT mice aged 12 to 16 weeks were anesthetized by an intraperitoneal injection of a mixture of medetomidine (0.75 mg/kg), midazolam (4 mg/kg), and butorphanol tartrate (5 mg/kg) and then heparinized. The heart was rapidly excised and perfused with a perfusion buffer (118 mmol/L NaCl, 4.7 mmol/L KCl, 1.2 mmol/L MgSO4, 1.2 mmol/L KH2PO4, 5 mmol/L glucose, 25 mmol/L NaHCO3, and 10 mmol/L 2,3‐butanedione 2‐monoxime) through the Langendorff apparatus (Physio‐tech) set at 37°C at a constant flow (3 mL/min). After 3 minutes of perfusion, the buffer was replaced by a digestion buffer containing the perfusion buffer plus 2 μmol/L CaCl2 and 0.24 U/mL of Liberase TH Research Grade (Sigma‐Aldrich) and then perfused for another 20 minutes. Next, the heart was removed from the system, and the left ventricle was minced into small pieces. The minced left ventricle was further digested in the digestion buffer for an additional 10 minutes at 37°C with gentle agitation and filtered through Nitex nylon mesh (Genesee, San Diego, CA, USA) with 100‐μm pore size. The mixture was centrifuged at 25g for 3 minutes to remove cardiomyocytes, and the supernatant was further centrifuged at 500g for 4 minutes to collect CFs. The CFs were grown in DMEM supplemented with 10% fetal bovine serum, penicillin (10 U/mL), and streptomycin (100 μg/mL) on a collagen‐coated dish. For 1 experiment, 1 heart was used for isolation of CFs, and CFs at passage 2 were used.
Western Blot Analysis
The CFs were plated onto 6‐well dishes at a confluence of 70% to 80% and serum‐starved for 4 hours. After pretreatment with the PAR‐1 antagonist SCH79797 (1 μmol/L) or DMSO (final concentration was 0.1%, v/v) as vehicle for 2.5 hours, the CFs were stimulated with thrombin (1 U/mL) for 10 minutes or FXa (20 nmol/L) for 30 minutes. After stimulation, the CFs were immediately lysed with RIPA lysis buffer (Santa Cruz Biotechnology) and centrifuged at 21 600g for 10 minutes. RIPA lysis buffer contains phenylmethylsulfonyl fluoride, protease inhibitor cocktail, and sodium orthovanadate as protease and phosphatase inhibitors, and they were added before use. Protein concentrations were determined by the protein quantification assay (Macherey‐Nagel). The proteins were separated by SDS‐PAGE and then electrophoretically transferred to polyvinylidene fluoride membranes (Bio‐Rad). After blocking with Blocking One‐P (Nacalai Tesque) for phosphorylated ERK1/2 (extracellular signal‐regulated kinases 1 and 2) or 2% skim milk for total ERK, the membranes were incubated overnight with primary antibodies for phosphorylated ERK (1:1000; Cell Signaling) or total ERK (1:2000; Merck Millipore) at 4°C. On the next day, the membranes were incubated with the horseradish peroxidase conjugated secondary antibody goat anti‐rabbit IgG H&L (1:75 000; Abcam) for 1 hour at room temperature. The protein bands were visualized using Amersham ECL Prime Western Blotting Detection Reagents (GE Healthcare), and densitometric analyses were performed with ChemiDoc XRS+ using Image Lab software (Bio‐Rad).
Apoptosis and Cell Viability Assay
HEK293 cells were cultured in DMEM supplemented with 10% fetal bovine serum, penicillin (10 U/mL), and streptomycin (100 μg/mL), and treated with 0.3 or 1 μmol/L of SCH79797 or 0.1% of DMSO (v/v) as vehicle for 3, 6, or 24 hours. After this period, cells were lysed with RIPA lysis buffer, and the ratio of cleaved Casp3 (caspase‐3; 1:1000; Cell Signaling) to Casp3 (1:1000; Cell Signaling) was evaluated by western blot analysis. As a positive control, we administrated staurosporine 1 μmol/L (Abcam) to cells to induce apoptosis and collected cells 4 hours later. In a cell‐viability assay, HEK293 cells were cultured in DMEM supplemented with 10% fetal bovine serum, penicillin (10 U/mL), and streptomycin (100 μg/mL), and treated with 0.1, 0.3, or 1 μmol/L of SCH79797 or 0.1% of DMSO (v/v) as vehicle for 3 or 24 hours. After this period, cells were detached from the culture dish using Trypsin‐EDTA (Wako), and then trypan blue solution (Wako) was added. The number of whole cells and stained cells were counted with a TC20 automated cell counter (Bio‐Rad), and the survival rate was calculated.
Statistical Analysis
All data were represented as mean±SD. Data were compared using 1‐ or 2‐way ANOVA, followed by the Tukey post hoc test using JMP 12.1.1 software (SAS Institute). Repeated‐measures ANOVA was performed in the serial analysis of systolic BP, interventricular septum thickness, and LV posterior wall thickness evaluated by echocardiography. P<0.05 was considered statistically significant.
Results
PAR‐1 Antagonist SCH79797 Decreases Systolic BP
Our previous study and another report demonstrated that Ren‐Tg mice display elevated systolic BP at an age as early as 12 weeks concomitantly with cardiac hypertrophy and fibrosis.17, 18 Similar to these observations, systolic BP was found to be significantly higher in Ren‐Tg mice than in WT mice at baseline (119±8 versus 99±5 mm Hg) and at 2 weeks (118±6 versus 102±5 mm Hg) and 4 weeks (127±7 versus 105±4 mm Hg) after treatment with vehicle (all P<0.01; Figure 1). Systolic BP measurements at 2 weeks (108±11 mm Hg) and 4 weeks (116±13 mm Hg) after treatment with SCH79797 were significantly lower than the measurements obtained after treatment with vehicle in Ren‐Tg mice (both P<0.05). In contrast, SCH79797 treatment had no effect on pulse rate in both Ren‐Tg and WT mice (Table S1).
Figure 1. Systolic blood pressure in renin‐overexpressing hypertensive (Ren‐Tg) mice and wild‐type (WT) mice at baseline and at 2 and 4 weeks after treatment with vehicle or PAR‐1 (protease‐activated receptor‐1) antagonist SCH79797 (25 μg/kg per day).

*P<0.01 vs WT mice treated with vehicle at the same time point. **P<0.05 vs Ren‐Tg mice treated with vehicle at the same time point.
SCH79797 Attenuates Cardiac Hypertrophy and Fibrosis
The interventricular septum thickness at baseline was significantly greater in Ren‐Tg mice than in WT mice (1.12±0.03 versus 0.92±0.02 mm), and similar trends were observed at 2 weeks (1.17±0.03 versus 0.90±0.03 mm) and 4 weeks (1.22±0.03 versus 0.93±0.01 mm) after treatment with vehicle (all P<0.01; Figure 2A). The interventricular septum thickness at 2 weeks (1.08±0.06 mm) and 4 weeks (1.10±0.04 mm) after treatment with SCH79797 was significantly lower than that measured after treatment with vehicle in Ren‐Tg mice (P<0.05 at 2 weeks and P<0.01 at 4 weeks). Similarly, the LV posterior wall thickness at baseline was significantly greater in Ren‐Tg mice than in WT mice (1.15±0.07 versus 0.97±0.07 mm), and similar trends were observed at 2 weeks (1.20±0.06 versus 0.94±0.05 mm) and 4 weeks (1.24±0.03 versus 0.92±0.01 mm) after treatment with vehicle (all P<0.01; Figure 2B). The LV posterior wall thickness measured at 4 weeks after treatment with SCH79797 (1.15±0.03 mm) was significantly lower than that measured after treatment with vehicle in Ren‐Tg mice (P<0.01). In contrast, SCH79797 treatment had no effect on LV end‐diastolic dimension, LV end‐systolic dimension, or LV fractional shortening in both Ren‐Tg and WT mice. Representative images by echocardiography were shown in Figure 2C. Consistent with these observations, the ratio of heart weight to body weight was greater in Ren‐Tg mice than in WT mice treated with vehicle (6.14±0.42 versus 4.96±0.19 mg/g, P<0.01), and SCH79797 treatment attenuated the increase to 5.29±0.84 mg/g in Ren‐Tg mice (P<0.05) (Figure 2D).
Figure 2. PAR‐1 (protease‐activated receptor‐1) antagonist SCH79797 attenuates cardiac hypertrophy in renin‐overexpressing hypertensive (Ren‐Tg) mice.

A, Interventricular septum thickness and (B) left ventricular posterior wall thickness at baseline and at 2 and 4 weeks after treatment with vehicle or PAR‐1 antagonist SCH79797. C, Representative images by echocardiography at 4 weeks after treatment. D, The ratio of heart weight to body weight. *P<0.01 vs wild‐type (WT) mice treated with vehicle at the same time point. **P<0.05 vs Ren‐Tg mice treated with vehicle at the same time point. ***P<0.01 vs Ren‐Tg mice treated with vehicle at the same time point. † P<0.01 vs baseline in Ren‐Tg mice. †† P<0.05 vs baseline in Ren‐Tg mice.
The representative sections of left ventricle stained with Masson's trichrome in Ren‐Tg and WT mice after treatment with SCH79797 or vehicle at 4 weeks are shown in Figure 3A. The area of cardiac interstitial fibrosis was significantly greater in Ren‐Tg mice than in WT mice (2.5±0.1% versus 1.2±0.2%, P<0.01), and SCH79797 treatment attenuated this area to 1.6±0.5% in Ren‐Tg mice (P<0.01; Figure 3B). Altogether, treatment with the PAR‐1 antagonist SCH79797 inhibited the progression of cardiac hypertrophy and fibrosis induced by the continuous activation of RAS.
Figure 3. PAR‐1 (protease‐activated receptor‐1) antagonist SCH79797 attenuates cardiac interstitial fibrosis.

A, Representative sections of left ventricle stained with Masson's trichrome in renin‐overexpressing hypertensive (Ren‐Tg) mice or wild‐type (WT) mice treated with SCH79797 or vehicle for 4 weeks. B, The area of cardiac interstitial fibrosis was evaluated.
SCH79797 Decreases Monocyte and Macrophage Deposition in the Heart
Figure 4A shows the representative sections of the left ventricle immunostained with CD68. Infiltration of monocyte/macrophage was more frequent in Ren‐Tg mice than in WT mice, and SCH79797 treatment reduced the infiltration of monocytes and macrophages. The area of CD68 infiltration was significantly greater in Ren‐Tg than WT mice (1.7±0.3% versus 0.04±0.1%, P<0.01), and SCH79797 treatment attenuated this area to 0.8±0.6% in Ren‐Tg mice (P<0.05; Figure 4B).
Figure 4. PAR‐1 (protease‐activated receptor‐1) antagonist SCH79797 attenuates cardiac deposition of monocytes or macrophages stained with CD68.
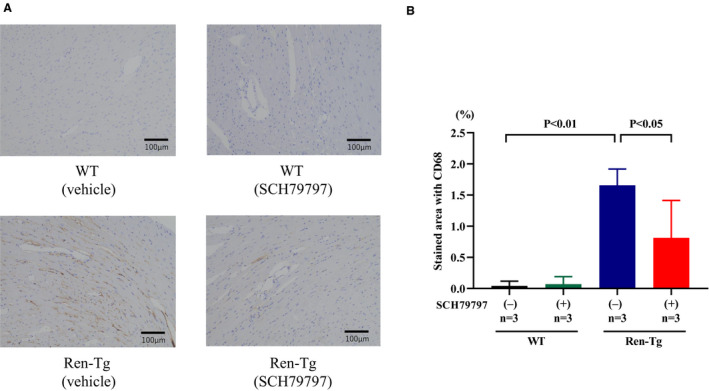
A, Representative sections of left ventricle stained with CD68 in renin‐overexpressing hypertensive (Ren‐Tg) mice or wild‐type (WT) mice treated with SCH79797 or vehicle for 4 weeks. B, The CD68‐positive area was evaluated.
SCH79797 Attenuates Enhancement of Expression Levels of Proinflammatory and Profibrotic Cytokines in the Heart
To investigate the mechanism underlying the protective effects of the PAR‐1 antagonist SCH79797 on cardiac hypertrophy and fibrosis, we measured the mRNA expression levels related to proinflammatory and profibrotic processes in the heart. The expression of PAR‐1 was found to be greater in Ren‐Tg than WT mice (P<0.01), and treatment with SCH79797 significantly inhibited this increase in Ren‐Tg mice (P<0.05; Figure 5A). The expression levels of TNF‐α and TGF‐β1, well‐known cytokines to promote inflammatory and fibrotic processes, were greater in Ren‐Tg than WT mice (P<0.01), and SCH79797 treatment inhibited these increases in Ren‐Tg mice (P<0.05; Figure 5B and 5C). The ratio of the expression of β‐MHC/α‐MHC was greater in Ren‐Tg than WT mice (P<0.01), and treatment with SCH79797 inhibited this increase in Ren‐Tg mice (P<0.01; Figure 5D). The expression of COL3A1 was greater in Ren‐Tg than WT mice (P<0.05), and SCH79797 treatment inhibited this increase in Ren‐Tg mice (P<0.05; Figure 5E). These findings indicate that continuous RAS activation causes the upregulation of PAR‐1 expression in the heart and enhances the production of proinflammatory and profibrotic cytokines, which contribute to the development of cardiac hypertrophy and fibrosis. The PAR‐1 antagonist SCH79797 inhibited cardiac hypertrophy and fibrosis, presumably by attenuating the production of these cytokines.
Figure 5. PAR‐1 (protease‐activated receptor‐1) antagonist SCH79797 attenuates the increase in cardiac mRNA expression levels related to proinflammatory and profibrotic processes.

Expressions of PAR‐1 (A), TNF‐α (tumor necrosis factor‐α) (B), and TGF‐β1 (transforming growth factor‐β1) (C). The ratio of β‐myosin heavy chain (β‐MHC) to α‐MHC (D). Expression of COL3A1 (collagen type 3 α1 chain) (E).
Prothrombin Fragment 1+2 and FXa in Plasma Are Enhanced in Ren‐Tg Mice
Because PAR‐1 is primarily activated by thrombin or FXa, we measured the concentrations of thrombin and FXa in the plasma of Ren‐Tg and WT mice after 4 weeks of treatment with SCH79797 or vehicle. Because fragment 1+2, an activation peptide released from prothrombin during thrombin formation, positively correlates with the concentration of thrombin in plasma,21 we measured fragment 1+2 instead of thrombin. The plasma concentration of fragment 1+2 was found to be higher in Ren‐Tg mice than in WT mice (10.3±3.2 versus 4.7±0.6 ng/mL, P<0.01), and treatment with SCH79797 did not affect this concentration in both Ren‐Tg and WT mice (Figure 6A). Similarly, the FXa in the plasma was greater in Ren‐Tg mice than in WT mice (18.0±3.6 versus 9.9±4.8 ng/mL, P<0.01), and SCH79797 treatment had no effect on this concentration in both Ren‐Tg and WT mice (Figure 6B).
Figure 6. Concentrations of prothrombin fragment 1+2 (A) and factor Xa (B) in plasma. Ren‐Tg indicates renin‐overexpressing hypertensive; WT, wild‐type.

Plasma Concentration of SCH79797
We measured plasma concentration of SCH79797 by LC‐MS/MS. The concentration of SCH79797 in plasma was 1.42±0.98 nmol/L (n=3) in SCH79797‐treated mice but was not detected in DMSO‐treated mice (n=5).
SCH79797 Inhibits ERK Phosphorylation Induced by Thrombin or FXa in Isolated CFs
CFs play a vital role in the production of extracellular matrix and the development of interstitial fibrosis in the heart.22 To further evaluate the inhibitory effect of the PAR‐1 antagonist SCH79797 on cardiac hypertrophy and fibrosis, we examined MAP (mitogen‐activated protein) kinase signaling in response to thrombin or FXa in isolated CFs. We observed that ERK phosphorylation was significantly enhanced in response to thrombin (P<0.01), which was significantly inhibited by pretreatment with SCH79797 (P<0.01; Figure 7A). Similarly, ERK phosphorylation was significantly enhanced in response to FXa (P<0.01), which was significantly inhibited by pretreatment with SCH79797 (P<0.05; Figure 7B).
Figure 7. PAR‐1 (protease‐activated receptor‐1) antagonist SCH79797 (1 μmol/L) inhibits ERK1/2 (extracellular signal‐regulated kinase 1/2) phosphorylation in response to thrombin (1 U/mL) (A) or factor Xa (FXa; 20 nmol/L) (B) in isolated cardiac fibroblasts by western blot analysis. pERK indicates phosphorylated ERK1/2; tERK, total ERK1/2.

SCH79797 Inhibits FXa‐Induced Upregulation of PAR‐1, TGF‐β1, and COL3A1 in Isolated CFs
To further evaluate the cardioprotective effects of SCH79797, we examined the effect of FXa in isolated CFs. FXa significantly enhanced gene expression of PAR‐1, TGF‐β1, and COL3A1 compared with control (P<0.01, P<0.05, and P<0.05, respectively), and pretreatment with SCH79797 abolished these enhancements (all P<0.05; Figure 8A, 8C, and 8D). A similar trend was found in TNF‐α gene expression, albeit not statistically significant (Figure 8B).
Figure 8. PAR‐1 (protease‐activated receptor‐1) antagonist SCH79797 (1 μmol/L) attenuates the increase in proinflammatory and fibrotic‐related gene expression in response to factor Xa (FXa; 20 nmol/L) in isolated cardiac fibroblasts.
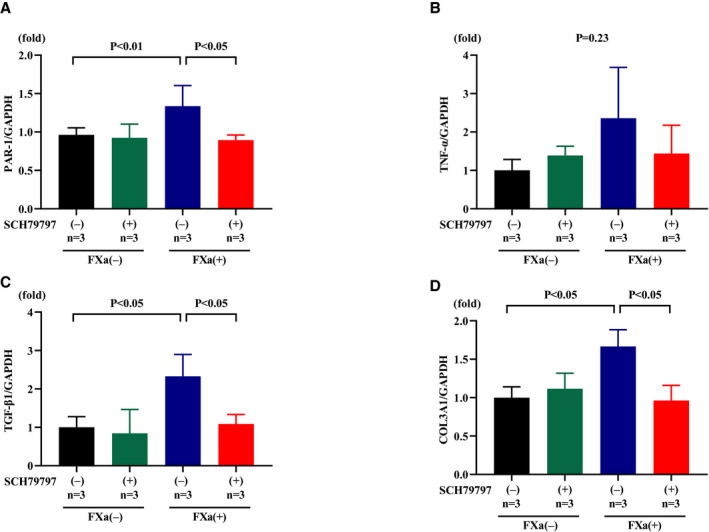
Expressions of PAR‐1 (A), TNF‐α (tumor necrosis factor‐α) (B), TGF‐β1 (transforming growth factor‐β1) (C), and COL3A1 (collagen type 3 α1 chain) (D).
SCH79797 Is Not Associated With Either Apoptosis or Cell Death
Although SCH79797 at various doses (30 nmol/L to 10 μmol/L) was shown to inhibit PAR‐1 signaling,23, 24 there may be concern regarding the toxicity of SCH79797, even at a low concentration (150 nmol/L).25 To examine the possibility of toxicity of SCH79797, we performed apoptosis and a cell‐viability assay. The ratio of cleaved Casp3/Casp3 was not altered in either 0.3 or 1 μmol/L of SCH79797 or DMSO as vehicle at any time points in HEK293 cells (Figure 9A). The survival rates of HEK293 cells incubated with 0.1, 0.3, or 1 μmol/L of SCH79797 for 3 or 24 hours were similar to cells incubated with DMSO (Figure 9B and 9C). These results indicate that SCH79797, even at 1 μmol/L, is not associated with either apoptosis or cell death.
Figure 9. SCH79797 is not associated with either apoptosis or cell death.
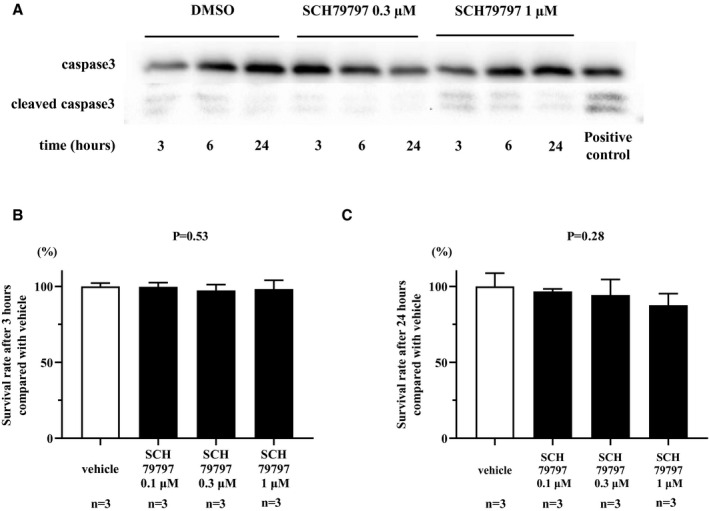
A, The ratio of cleaved Casp3 (caspase‐3) to Casp3 was examined in either 0.3 or 1 μmol/L of SCH79797 or dimethyl sulfoxide (DMSO) as vehicle at 3, 6, and 24 hours in HEK293 cells. B and C, The survival rate of HEK293 cells incubated with 0.1, 0.3, or 1 μmol/L of SCH79797 for 3 or 24 hours assessed by trypan blue staining.
Discussion
In this study, we examined the effects of the PAR‐1 antagonist SCH79797 on cardiac hypertrophy and fibrosis induced by the continuous enhancement of RAS activity. We observed that SCH79797 attenuates cardiac hypertrophy and fibrosis by reducing the expression levels of proinflammatory and profibrotic cytokines and monocyte/macrophage deposition in the heart. Moreover, continuous enhancement of RAS activity increased thrombin generation and FXa in plasma; both act as agonists for PAR‐1. In isolated CFs, SCH79797 inhibited ERK phosphorylation in response to thrombin or FXa. Furthermore, SCH79797 inhibited PAR‐1 expression and proinflammatory and profibrotic cytokines induced by FXa in isolated CFs. These findings indicate that the PAR‐1 antagonist SCH79797 may have a protective effect against cardiac hypertrophy and fibrosis induced by RAS activity partly through the inhibition of PAR‐1 signaling.
Recent studies demonstrate the involvement of the coagulation cascade in pathologic cardiac remodeling and the relationship between RAS and coagulation activities.6, 8 In fact, plasma renin activity was found to have a positive correlation with concentrations of fibrinogen, D‐dimer, and plasminogen activator inhibitor‐1 (PAI‐1) in hypertensive patients,9 and fragment 1+2 concentration was found to be higher in hypertensive patients than in normotensive controls and positively correlated with BP.8 Furthermore, our recent study demonstrated that FXa in plasma was enhanced in Ren‐Tg mice compared with WT mice.20 In recent years, thrombin and FXa have been viewed as important mediators of the inflammatory process and cardiac remodeling because of the widespread use of direct oral anticoagulants. These medications are commonly used worldwide for patients with atrial fibrillation or venous thrombosis to prevent thromboembolic events, and their cardioprotective effect has been appreciated. Studies have also reported that the direct thrombin inhibitor dabigatran attenuated cardiac fibrosis induced by pressure overload26 or myocardial infarction27 and improved cardiac systolic dysfunction in a dilated cardiomyopathy mouse model.7, 28 Furthermore, the direct FXa inhibitor rivaroxaban was found to attenuate cardiac fibrosis induced by pressure overload29 and the progression of cardiac systolic dysfunction in a myocardial infarction model.30 These results indicate that thrombin and FXa are involved in the pathogenesis of cardiac remodeling, and their inhibition appears to be protective.
In the present study, we showed that continuous RAS activation results in the enhancement of thrombin generation and FXa in plasma, both of which function as agonists for PAR‐1. Several studies have demonstrated that PAR‐1 activates multiple intracellular signaling pathways and exerts important functions in regulating the cardiovascular physiology and pathophysiology5, 11, 13, 15; a possible involvement of PAR‐1 signaling in cardiac remodeling has also been reported. Other research has demonstrated that knocking out PAR‐1 or using the PAR‐1 antagonist SCH79797 attenuated the progression of cardiac systolic dysfunction and fibrosis induced by ischemia/reperfusion injury15 and cardiomyocyte‐specific PAR‐1 overexpression induced LV enlargement and systolic dysfunction.14 However, the detailed mechanism and the effects of PAR‐1 inhibition on cardiac hypertrophy and fibrosis induced by RAS activation remain largely unknown.
In the present study, PAR‐1 expression in the heart was enhanced in Ren‐Tg mice compared with WT mice, and SCH79797 attenuated the increase. Expression of PAR‐1 and PAR‐2 was shown to be increased in an ischemic heart,31 and cardiac PAR‐2 expression was increased by pressure overload.29 Furthermore, our previous study showed that PAR‐2 expression was enhanced in the kidneys of Ren‐Tg mice.20 Previous reports also showed the involvement of inflammatory cytokines in regulation of PAR expression. TNF and IL‐1β (interleukin‐1β) upregulate PAR2 and PAR‐4 expression in human endothelial cells,32 and TNF induces PAR‐2 upregulation in human lung fibroblasts.33 Furthermore, IL‐1β has been shown to upregulate PAR‐2 expression in human airway smooth muscle cells.34 In the present study, TNF‐α expression in the heart was enhanced in Ren‐Tg mice and was downregulated by SCH79797. In addition, PAR‐1 expression in isolated CFs was enhanced by FXa and was suppressed by SCH79797 pretreatment. These findings indicate that enhancement of inflammatory cytokines including TNF‐α upregulate PAR‐1 expression in the heart, and SCH79797 inhibits expression of inflammatory cytokines and, consequently, may decrease PAR‐1 expression.
A hypertrophied myocardium provides multiple stimuli for monocyte recruitment to the heart, which is further amplified by the presence of tissue hypoxia and ischemia. Excessive monocyte and macrophage recruitment leads to enhanced inflammation of the heart, resulting in myocardial damage and remodeling.35 In fact, TNF‐α released from monocytes and macrophages triggers production of the inducible type of nitric oxide synthase, uncontrolled oxidative stress, apoptosis, and tissue necrosis.36 Cardiac fibrosis seems to be a consequence of an activated monocyte or macrophage cascade of HF.37 In the present study, monocyte or macrophage deposition in the heart was increased in Ren‐Tg mice, and SCH79797 inhibited this increase. Thus, PAR‐1 inhibition may attenuate cardiac hypertrophy and fibrosis, partly through inhibition of monocyte or macrophage infiltration in the heart.
To further evaluate the mechanism underlying the protective effects of the PAR‐1 antagonist SCH79797 on cardiac hypertrophy and fibrosis induced by chronic RAS activation, we examined the role of CFs. CFs represent the largest population of interstitial cells in the heart and are responsible for producing the extracellular matrix.22 Furthermore, CFs function as a regulator of cardiac hypertrophy and contractility.38, 39 Notably, thrombin and FXa induce MAP kinase activation and the conversion into myofibroblasts and subsequently enhance TGF‐β expression and collagen production in CFs.15, 40, 41 These findings indicate that thrombin and FXa contribute to the development of interstitial fibrosis in the heart. In the present study, either thrombin or FXa significantly enhanced ERK phosphorylation in CFs, and treatment with SCH79797 inhibited this enhancement. Furthermore, SCH79797 inhibited enhancement of TGF‐β1 and COL3A1 expressions induced by FXa in CFs. Because ERK activation has been shown to be closely associated with extracellular matrix production and tissue fibrosis,42, 43 our results indicate that enhanced thrombin generation and FXa via chronic RAS activation contribute to cardiac fibrosis through PAR‐1 signaling.
In terms of vascular physiology, studies have reported that PAR‐1 agonist induces rapid and transient hypotension followed by sustained hypertension,44 and PAR‐1 agonist–induced relaxation was completely abolished and contraction was observed in its place in coronary arteries without intimal endothelium or with severe atherosclerotic lesions.45, 46 These results indicate that the PAR‐1 agonist–induced vasodilation depends on the existence of a healthy endothelium, and vasoconstriction is caused in the arteries with dysfunctional endothelium. In the present study, continuous infusion of the PAR‐1 antagonist SCH79797 decreased systolic BP in Ren‐Tg mice. Endothelial dysfunction has been commonly observed in the arteries of hypertensive animals47 and humans48; therefore, in our hypertensive mouse model, PAR‐1 inhibition may cause vasodilation and decrease systolic BP. An earlier study examined the effect of PAR‐1 knockout on cardiac remodeling induced by continuous angiotensin II infusion and demonstrated that the progression of cardiac systolic dysfunction and perivascular fibrosis was attenuated by PAR‐1 knockout, whereas no effect was observed on cardiac hypertrophy.49 It should be noted that angiotensin II infusion for 4 weeks had no effect on BP and cardiac hypertrophy in that study. The differences observed in our results may be explained by the differences in angiotensin II stimulation or the mouse model used in our study. Furthermore, the arteries of Ren‐Tg mice were continuously exposed by excessive angiotensin II soon after birth, possibly resulting in more endothelial dysfunction compared with the model of angiotensin II infusion for 4 weeks. Further studies are clearly required.
In contrast, no obvious change in baseline BP was observed in PAR‐1‐deficient mice,50 and SCH79797 treatment did not affect systolic BP in WT mice in the present study, suggesting that PAR‐1 is not necessary for the maintenance of the normal vascular tone. A possibility exists that decreased systolic BP reduced the afterload, which potentially affected the progression of cardiac hypertrophy and fibrosis in the present study. However, our previous study and another report demonstrated that RAS‐induced cardiac hypertrophy was not attenuated by lowering BP.18, 49 In addition, treatment with the PAR‐1 antagonist did not affect the thrombin concentration or the FXa in plasma, both of which enhanced the phosphorylation of ERK in CFs in the present study. A recent report showed that FXa led to neonatal cardiomyocyte hypertrophy and that SCH79797 abolished it. Furthermore, SCH79797 inhibited migration and proliferation of neonatal CFs induced by FXa.51 These findings indicate that the inhibition of PAR‐1 signaling contributes to protective effects on the development of cardiac hypertrophy and fibrosis, at least in part through a BP‐independent mechanism. Further experimental studies are required to elucidate the mechanism underlying the BP‐lowering effect of SCH79797. Although we used SCH79797 (25 μg/kg per day) according to the previous reports,15, 19 its mean plasma concentration was 1.42 nmol/L in SCH79797‐treated mice, which was quite lower than expected.52 Because we and other researchers have reported significant effects of SCH79797 on animal studies,15, 19 further pharmacokinetic studies may be needed to resolve this important issue.
Recent studies have appreciated the relationship between coagulation activity and cardiovascular events in humans.7, 8 In fact, rivaroxaban was found to reduce cardiovascular death in patients with a recent acute coronary syndrome,53, 54 and addition of rivaroxaban to aspirin was found to possibly improve the outcome in patients with coronary artery disease.55 Strikingly, PAR‐1 expression was enhanced in the hearts of patients with ischemic or idiopathic dilated cardiomyopathy, and treatment with the PAR1 inhibitor vorapaxar reduced ischemic events in patients with cardiovascular disease.56, 57 These results clearly indicate the clinical significance of PAR‐1 signaling in the pathogenesis of cardiovascular diseases. Furthermore, cardiac hypertrophy and fibrosis increase the stiffness of the left ventricle, leading to the development of HF with preserved ejection fraction characterized by cardiac diastolic dysfunction. PAR‐1‐dependent profibrotic responses result in increased cardiac stiffness and subsequent diastolic dysfunction of the heart. Thus, our results indicate that PAR‐1 antagonist might be a potential candidate for the treatment of HF with preserved ejection fraction.
In conclusion, treatment with the PAR‐1 antagonist attenuates cardiac hypertrophy and fibrosis induced by chronic RAS activation. Inhibition of PAR‐1 signaling might be a novel strategy for treating HF induced by chronic RAS activation.
Sources of Funding
None.
Disclosures
Dr Tomita received research funding from Boehringer Ingelheim, Bayer, Daiichi‐Sankyo, and Pfizer and speakers’ bureau/honoraria from Boehringer Ingelheim, Bayer, Daiichi‐Sankyo, and Bristol‐Myers Squibb. The remaining authors have no disclosures to report.
Supporting information
Table S1
Acknowledgments
We thank Nahomi Miyamoto, Ritsuko Kasai, and Chuya Murakami in the Department of Cardiology for their excellent technical support and Miyu Miyazaki in the Center for Scientific Equipment Management, Hirosaki University Graduate School of Medicine, for help with mass spectrometry.
(J Am Heart Assoc. 2020;9:e015616 DOI: 10.1161/JAHA.119.015616.)
For Sources of Funding and Disclosures, see page 13.
References
- 1. Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr, Colvin MM, Drazner MH, Filippatos GS, Fonarow GC, Givertz MM, et al. 2017 ACC/AHA/HFSA focused update of the 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology/American Heart Association Task Force on clinical practice guidelines and the Heart Failure Society of America. Circulation. 2017;136:e137–e161. [DOI] [PubMed] [Google Scholar]
- 2. Mosterd A, Cost B, Hoes AW, de Bruijne MC, Deckers JW, Hofman A, Grobbee DE. The prognosis of heart failure in the general population: the Rotterdam Study. Eur Heart J. 2001;22:1318–1327. [DOI] [PubMed] [Google Scholar]
- 3. Vasan RS, Larson MG, Benjamin EJ, Evans JC, Reiss CK, Levy D. Congestive heart failure in subjects with normal versus reduced left ventricular ejection fraction: prevalence and mortality in a population‐based cohort. J Am Coll Cardiol. 1999;33:1948–1955. [DOI] [PubMed] [Google Scholar]
- 4. Steffel J, Luscher TF, Tanner FC. Tissue factor in cardiovascular diseases: molecular mechanisms and clinical implications. Circulation. 2006;113:722–731. [DOI] [PubMed] [Google Scholar]
- 5. Takada M, Tanaka H, Yamada T, Ito O, Kogushi M, Yanagimachi M, Kawamura T, Musha T, Yoshida F, Ito M, et al. Antibody to thrombin receptor inhibits neointimal smooth muscle cell accumulation without causing inhibition of platelet aggregation or altering hemostatic parameters after angioplasty in rat. Circ Res. 1998;82:980–987. [DOI] [PubMed] [Google Scholar]
- 6. Zhao JV, Schooling CM. Coagulation factors and the risk of ischemic heart disease: a Mendelian randomization study. Circ Genom Precis Med. 2018;11:e001956. [DOI] [PubMed] [Google Scholar]
- 7. Ito K, Hongo K, Date T, Ikegami M, Hano H, Owada M, Morimoto S, Kashiwagi Y, Katoh D, Yoshino T, et al. Tissue thrombin is associated with the pathogenesis of dilated cardiomyopathy. Int J Cardiol. 2017;228:821–827. [DOI] [PubMed] [Google Scholar]
- 8. Sechi LA, Zingaro L, Catena C, Casaccio D, De Marchi S. Relationship of fibrinogen levels and hemostatic abnormalities with organ damage in hypertension. Hypertension. 2000;36:978–985. [DOI] [PubMed] [Google Scholar]
- 9. Sechi LA, Novello M, Colussi G, Di Fabio A, Chiuch A, Nadalini E, Casanova‐Borca A, Uzzau A, Catena C. Relationship of plasma renin with a prothrombotic state in hypertension: relevance for organ damage. Am J Hypertens. 2008;21:1347–1353. [DOI] [PubMed] [Google Scholar]
- 10. Coughlin SR. Thrombin signalling and protease‐activated receptors. Nature. 2000;407:258–264. [DOI] [PubMed] [Google Scholar]
- 11. Leger AJ, Covic L, Kuliopulos A. Protease‐activated receptors in cardiovascular diseases. Circulation. 2006;114:1070–1077. [DOI] [PubMed] [Google Scholar]
- 12. Sabri A, Short J, Guo J, Steinberg SF. Protease‐activated receptor‐1‐mediated DNA synthesis in cardiac fibroblast is via epidermal growth factor receptor transactivation: distinct PAR‐1 signaling pathways in cardiac fibroblasts and cardiomyocytes. Circ Res. 2002;91:532–539. [DOI] [PubMed] [Google Scholar]
- 13. Sabri A, Muske G, Zhang H, Pak E, Darrow A, Andrade‐Gordon P, Steinberg SF. Signaling properties and functions of two distinct cardiomyocyte protease‐activated receptors. Circ Res. 2000;86:1054–1061. [DOI] [PubMed] [Google Scholar]
- 14. Pawlinski R, Tencati M, Hampton CR, Shishido T, Bullard TA, Casey LM, Andrade‐Gordon P, Kotzsch M, Spring D, Luther T, et al. Protease‐activated receptor‐1 contributes to cardiac remodeling and hypertrophy. Circulation. 2007;116:2298–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sonin DL, Wakatsuki T, Routhu KV, Harmann LM, Petersen M, Meyer J, Strande JL. Protease‐activated receptor 1 inhibition by SCH79797 attenuates left ventricular remodeling and profibrotic activities of cardiac fibroblasts. J Cardiovasc Pharmacol Ther. 2013;18:460–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Caron KM, James LR, Kim HS, Morham SG, Sequeira Lopez ML, Gomez RA, Reudelhuber TL, Smithies O. A genetically clamped renin transgene for the induction of hypertension. Proc Natl Acad Sci USA. 2002;99:8248–8252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Caron KM, James LR, Kim HS, Knowles J, Uhlir R, Mao L, Hagaman JR, Cascio W, Rockman H, Smithies O. Cardiac hypertrophy and sudden death in mice with a genetically clamped renin transgene. Proc Natl Acad Sci USA. 2004;101:3106–3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tanno T, Tomita H, Narita I, Kinjo T, Nishizaki K, Ichikawa H, Kimura Y, Tanaka M, Osanai T, Okumura K. Olmesartan inhibits cardiac hypertrophy in mice overexpressing renin independently of blood pressure: its beneficial effects on ACE2/An (1‐7)/Mas axis and NADPH oxidase expression. J Cardiovasc Pharmacol. 2016;67:503–509. [DOI] [PubMed] [Google Scholar]
- 19. Strande JL, Hsu A, Su J, Fu X, Gross GJ, Baker JE. SCH 79797, a selective PAR1 antagonist, limits myocardial ischemia/reperfusion injury in rat hearts. Basic Res Cardiol. 2007;102:350–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ichikawa H, Shimada M, Narita M, Narita I, Kimura Y, Tanaka M, Osanai T, Okumura K, Tomita H. Rivaroxaban, a direct factor Xa inhibitor, ameliorates hypertensive renal damage through inhibition of the inflammatory response mediated by protease‐activated receptor pathway. J Am Heart Assoc. 2019;8:e012195 DOI: 10.1161/JAHA.119.012195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Aronson DL, Stevan L, Ball AP, Franza BR Jr, Finlayson JS. Generation of the combined prothrombin activation peptide (F1‐2) during the clotting of blood and plasma. J Clin Invest. 1977;60:1410–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brown RD, Ambler SK, Mitchell MD, Long CS. The cardiac fibroblast: therapeutic target in myocardial remodeling and failure. Annu Rev Pharmacol Toxicol. 2005;45:657–687. [DOI] [PubMed] [Google Scholar]
- 23. Yue R, Li H, Liu H, Li Y, Wei B, Gao G, Jin Y, Liu T, Wei L, Du J, et al. Thrombin receptor regulates hematopoiesis and endothelial‐to‐hematopoietic transition. Dev Cell. 2012;22:1092–1100. [DOI] [PubMed] [Google Scholar]
- 24. Bukowska A, Zacharias I, Weinert S, Skopp K, Hartmann C, Huth C, Goette A. Coagulation factor Xa induces an inflammatory signalling by activation of protease‐activated receptors in human atrial tissue. Eur J Pharmacol. 2013;718:114–123. [DOI] [PubMed] [Google Scholar]
- 25. Di Serio C, Pellerito S, Duarte M, Massi D, Naldini A, Cirino G, Prudovsky I, Santucci M, Geppetti P, Marchionni N, et al. Protease‐activated receptor 1‐selective antagonist SCH79797 inhibits cell proliferation and induces apoptosis by a protease‐activated receptor 1‐independent mechanism. Basic Clin Pharmacol Toxicol. 2007;101:63–69. [DOI] [PubMed] [Google Scholar]
- 26. Dong A, Mueller P, Yang F, Yang L, Morris A, Smyth SS. Direct thrombin inhibition with dabigatran attenuates pressure overload‐induced cardiac fibrosis and dysfunction in mice. Thromb Res. 2017;159:58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jumeau C, Rupin A, Chieng‐Yane P, Mougenot N, Zahr N, David‐Dufilho M, Hatem SN. Direct thrombin inhibitors prevent left atrial remodeling associated with heart failure in rats. JACC Basic Transl Sci. 2016;1:328–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bulani Y, Srinivasan K, Sharma SS. Attenuation of type‐1 diabetes‐induced cardiovascular dysfunctions by direct thrombin inhibitor in rats: a mechanistic study. Mol Cell Biochem. 2019;451:69–78. [DOI] [PubMed] [Google Scholar]
- 29. Kondo H, Abe I, Fukui A, Saito S, Miyoshi M, Aoki K, Shinohara T, Teshima Y, Yufu K, Takahashi N. Possible role of rivaroxaban in attenuating pressure‐overload‐induced atrial fibrosis and fibrillation. J Cardiol. 2018;71:310–319. [DOI] [PubMed] [Google Scholar]
- 30. Bode MF, Auriemma AC, Grover SP, Hisada Y, Rennie A, Bode WD, Vora R, Subramaniam S, Cooley B, Andrade‐Gordon P, et al. The factor Xa inhibitor rivaroxaban reduces cardiac dysfunction in a mouse model of myocardial infarction. Thromb Res. 2018;167:128–134. [DOI] [PubMed] [Google Scholar]
- 31. Liu J, Nishida M, Inui H, Chang J, Zhu Y, Kanno K, Matsuda H, Sairyo M, Okada T, Nakaoka H, et al. Rivaroxaban suppresses the progression of ischemic cardiomyopathy in a murine model of diet‐induced myocardial infarction. J Atheroscler Thromb. 2019;26:915–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ritchie E, Saka M, Mackenzie C, Drummond R, Wheeler‐Jones C, Kanke T, Plevin R. Cytokine upregulation of proteinase‐activated‐receptors 2 and 4 expression mediated by p38 MAP kinase and inhibitory kappa B kinase beta in human endothelial cells. Br J Pharmacol. 2007;150:1044–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ramachandran R, Sadofsky LR, Xiao Y, Botham A, Cowen M, Morice AH, Compton SJ. Inflammatory mediators modulate thrombin and cathepsin‐G signaling in human bronchial fibroblasts by inducing expression of proteinase‐activated receptor‐4. Am J Physiol Lung Cell Mol Physiol. 2007;292:L788–L798. [DOI] [PubMed] [Google Scholar]
- 34. Freund‐Michel V, Frossard N. Inflammatory conditions increase expression of protease‐activated receptor‐2 by human airway smooth muscle cells in culture. Fundam Clin Pharmacol. 2006;20:351–357. [DOI] [PubMed] [Google Scholar]
- 35. Shahid F, Lip GYH, Shantsila E. Role of monocytes in heart failure and atrial fibrillation. J Am Heart Assoc. 2018;7:e007849 DOI: 10.1161/JAHA.117.007849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bradham WS, Moe G, Wendt KA, Scott AA, Konig A, Romanova M, Naik G, Spinale FG. TNF‐alpha and myocardial matrix metalloproteinases in heart failure: relationship to LV remodeling. Am J Physiol Heart Circ Physiol. 2002;282:H1288–H1295. [DOI] [PubMed] [Google Scholar]
- 37. Frangogiannis NG. Regulation of the inflammatory response in cardiac repair. Circ Res. 2012;110:159–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Xiang FL, Fang M, Yutzey KE. Loss of beta‐catenin in resident cardiac fibroblasts attenuates fibrosis induced by pressure overload in mice. Nat Commun. 2017;8:712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Woodall MC, Woodall BP, Gao E, Yuan A, Koch WJ. Cardiac fibroblast GRK2 deletion enhances contractility and remodeling following ischemia/reperfusion injury. Circ Res. 2016;119:1116–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Blanc‐Brude OP, Archer F, Leoni P, Derian C, Bolsover S, Laurent GJ, Chambers RC. Factor Xa stimulates fibroblast procollagen production, proliferation, and calcium signaling via PAR1 activation. Exp Cell Res. 2005;304:16–27. [DOI] [PubMed] [Google Scholar]
- 41. Altieri P, Bertolotto M, Fabbi P, Sportelli E, Balbi M, Santini F, Brunelli C, Canepa M, Montecucco F, Ameri P. Thrombin induces protease‐activated receptor 1 signaling and activation of human atrial fibroblasts and dabigatran prevents these effects. Int J Cardiol. 2018;271:219–227. [DOI] [PubMed] [Google Scholar]
- 42. Hayashida T, Decaestecker M, Schnaper HW. Cross‐talk between ERK MAP kinase and Smad signaling pathways enhances TGF‐beta‐dependent responses in human mesangial cells. FASEB J. 2003;17:1576–1578. [DOI] [PubMed] [Google Scholar]
- 43. Liu X, Hubchak SC, Browne JA, Schnaper HW. Epidermal growth factor inhibits transforming growth factor‐beta‐induced fibrogenic differentiation marker expression through ERK activation. Cell Signal. 2014;26:2276–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cheung WM, Andrade‐Gordon P, Derian CK, Damiano BP. Receptor‐activating peptides distinguish thrombin receptor (PAR‐1) and protease activated receptor 2 (PAR‐2) mediated hemodynamic responses in vivo. Can J Physiol Pharmacol. 1998;76:16–25. [DOI] [PubMed] [Google Scholar]
- 45. Ku DD, Zaleski JK. Receptor mechanism of thrombin‐induced endothelium‐dependent and endothelium‐independent coronary vascular effects in dogs. J Cardiovasc Pharmacol. 1993;22:609–616. [DOI] [PubMed] [Google Scholar]
- 46. Ku DD, Dai J. Expression of thrombin receptors in human atherosclerotic coronary arteries leads to an exaggerated vasoconstrictory response in vitro. J Cardiovasc Pharmacol. 1997;30:649–657. [DOI] [PubMed] [Google Scholar]
- 47. Brandes RP. Endothelial dysfunction and hypertension. Hypertension. 2014;64:924–928. [DOI] [PubMed] [Google Scholar]
- 48. Mordi I, Mordi N, Delles C, Tzemos N. Endothelial dysfunction in human essential hypertension. J Hypertens. 2016;34:1464–1472. [DOI] [PubMed] [Google Scholar]
- 49. Antoniak S, Cardenas JC, Buczek LJ, Church FC, Mackman N, Pawlinski R. Protease‐activated receptor 1 contributes to angiotensin II‐induced cardiovascular remodeling and inflammation. Cardiology. 2017;136:258–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Darrow AL, Fung‐Leung WP, Ye RD, Santulli RJ, Cheung WM, Derian CK, Burns CL, Damiano BP, Zhou L, Keenan CM, et al. Biological consequences of thrombin receptor deficiency in mice. Thromb Haemost. 1996;76:860–866. [PubMed] [Google Scholar]
- 51. Guo X, Kolpakov MA, Hooshdaran B, Schappell W, Wang T, Eguchi S, Elliott KJ, Tilley DG, Rao AK, Andrade‐Gordon P, et al. Cardiac expression of factor X mediates cardiac hypertrophy and fibrosis in pressure overload. JACC Basic Transl Sci. 2020;5:69–83. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 52. Ahn HS, Foster C, Boykow G, Stamford A, Manna M, Graziano M. Inhibition of cellular action of thrombin by N3‐cyclopropyl‐7‐[[4‐(1‐methylethyl)phenyl]methyl]‐7H‐pyrrolo[3, 2‐f]quinazoline‐1,3‐diamine (SCH 79797), a nonpeptide thrombin receptor antagonist. Biochem Pharmacol. 2000;60:1425–1434. [DOI] [PubMed] [Google Scholar]
- 53. Mega JL, Braunwald E, Wiviott SD, Bassand JP, Bhatt DL, Bode C, Burton P, Cohen M, Cook‐Bruns N, Fox KA, et al. Rivaroxaban in patients with a recent acute coronary syndrome. N Engl J Med. 2012;366:9–19. [DOI] [PubMed] [Google Scholar]
- 54. Mega JL, Braunwald E, Wiviott SD, Murphy SA, Plotnikov A, Gotcheva N, Ruda M, Gibson CM. Comparison of the efficacy and safety of two rivaroxaban doses in acute coronary syndrome (from ATLAS ACS 2‐TIMI 51). Am J Cardiol. 2013;112:472–478. [DOI] [PubMed] [Google Scholar]
- 55. Connolly SJ, Eikelboom JW, Bosch J, Dagenais G, Dyal L, Lanas F, Metsarinne K, O'Donnell M, Dans AL, Ha JW, et al. Rivaroxaban with or without aspirin in patients with stable coronary artery disease: an international, randomised, double‐blind, placebo‐controlled trial. Lancet. 2018;391:205–218. [DOI] [PubMed] [Google Scholar]
- 56. Capodanno D, Bhatt DL, Goto S, O'Donoghue ML, Moliterno DJ, Tamburino C, Angiolillo DJ. Safety and efficacy of protease‐activated receptor‐1 antagonists in patients with coronary artery disease: a meta‐analysis of randomized clinical trials. J Thromb Haemost. 2012;10:2006–2015. [DOI] [PubMed] [Google Scholar]
- 57. Morrow DA, Braunwald E, Bonaca MP, Ameriso SF, Dalby AJ, Fish MP, Fox KA, Lipka LJ, Liu X, Nicolau JC, et al. Vorapaxar in the secondary prevention of atherothrombotic events. N Engl J Med. 2012;366:1404–1413. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1