

Abstract
Motivated by the role that amylin aggregates play in type-II diabetes, we compare the stability of regular amylin fibrils with the stability of fibrils where L-amino acid chains are replaced by D–Retro Inverso (DRI) amylin, i.e., peptides where the sequence of amino acids is reversed, and at the same time, the L–amino acids are replaced by their mirror images. Our molecular dynamics simulations show that despite leading to only marginal difference in fibril structure and stability, aggregating DRI-amylin peptides have different patterns of contacts and hydrogen bonding. Because of these differences does DRI-amylin, when interacting with regular (L) amylin, alters the elongation process and lowers the stability of hybrid amylin fibrils. Our results suggest not only the potential use of DRI-amylin as an inhibitor of amylin fibril-formation but also points to the possibility of using insertion of DRI-proteins in L-assemblies as a way to probe the role of certain kinds of hydrogen bonds in supra-molecular assemblies or aggregates.
Graphical Astract
1. Introduction
While presence of amyloid fibrils is most commonly associated with neurodegenerative diseases such as Alzheimer’s, Parkinson’s, or Huntington’s disease, they are also seen in metabolic and other illnesses. One example is diabetes mellitus type–II, where aggregation of islet amyloid polypeptide (IAPP, also known as amylin) in the pancreas causes endoplasmic reticulum stress, mitochondrial damage, and membrane disruption.1–4 The resulting death of the pancreatic islet β-cells, which in healthy persons modulate the secretion of insulin and glucagon, leads to the onset of type–II diabetes.5–12 Co-localization of amylin and β-amyloid (Aβ) fibrils and computational studies indicate cross-seeding between the two molecules.13–16 Hence, inhibition and degradation of amylin aggregates might be a therapeutic strategy not only against type–II diabetes but also against Alzheimer’s and related disorders.14,17
Available therapies against type–II diabetes include subcutaneous mealtime injections of pramlintide, an amylin analog, that reduces glucose concentrations by regulating the plasma glucagon level, slowing down gastric emptying, and improving satiety.18–21 Unlike amylin itself, pramlintide resists fibril formation. However, because of the short half-life of pramlintide (~ 50 minutes), the drug needs to be administered frequently, leading to undesired side effects such as nausea and hypoglycemia.21,22 Recent studies have also reported that pramlintide possesses the ability to aggregate from mild acidic to neutral conditions 23,24 and can also co-aggregate with amylin fibers. 25 Hence, there is still a need for the development of new drugs with amylin-mimetic properties that improve the life quality of patients.
Short half-life time and the need for frequent administration of the drug are common complications in peptide-based therapies. One way of circumventing these problems is the use of D-Retro Inverso (DRI) peptides. DRI peptides are made up of D-amino acids, stereochemically mirror-images of the L-amino acids, and have at the same time, their sequence of amino acids reversed. As a consequence, DRI peptides resemble the parent peptide and have similar biological activity. However, unlike the parent peptide they are resistant to proteolytic digestion and, therefore, will have longer half-life times. 26,27 Several studies have reported the therapeutic effectiveness of DRI peptides, for instance, for the treatment of tumors.28 Note, however, that the symmetry is not complete and that there are subtle structural variations between DRI-peptides and their L-parents. These differences can impart new chemical properties and might even enhance the potency of the peptides as drugs. For instance, Daggett and co-workers showed that peptides made of alternating L and D amino acids bind preferentially to toxic oligomers instead of monomeric and fibril forms of Aβ, inhibiting Aβ aggregation and reducing cytotoxicity in mouse and C. elegans models of Alzheimer’s disease.29
The above-mentioned studies hint at the possibility that DRI-amylin may provide an alternative to pramlintide as a drug for targeting type–II diabetes. Hence, in this paper, using all-atom molecular dynamics simulations, we first study the ability of DRI-amylin to form amyloids and then explore how the presence of DRI-amylin alters the elongation and the stability of L-amylin fibrils. In the set-up and analysis of these simulations, we use the experience that we gained from a previous study of DRI-Aβ fibril stability.30 We find that DRI-amylin differs from L-amylin by the twist of β-sheets, but has similar or only slightly lower stability. Unlike in our previous work,30 we could now identify the interactions that cause the structural differences and show that they result from a rearrangement of contacts and loss of hydrogen bonds. While DRI-peptides interfere with elongation, their main effect is lowering the stability of hybrid amylin fibrils by easing separation of protofibrils, suggesting a potential for the use of DRI-amylin as an inhibitor of amylin fibril-formation. Our investigations also indicate that the insertion of DRI-proteins in L-assemblies can be used as a tool to probe the role of critical hydrogen bonds in supra-molecular assemblies or aggregates. In the present study, such insertion allowed us to identify the crucial role of Asn ladders and Ser–Ser bifurcated hydrogen bonds for stabilizing amylin fibrils. We speculate that the retention of Asn31 in pramlintide contributes to the observed co-aggregation with amylin and that the effectiveness of the drug could be increased by mutating this residue or replacing it with the D-isomer.
2. Materials and methods
2.1. Amylin fibril polymorphism and choice of model
Various techniques such as atomic force microscopy (AFM),31 electron paramagnetic resonance (EPR),32,33 cryo-electron microscopy (EM),33–35 two-dimensional infrared (2D IR) spectroscopy,36 X-ray crystallography,37 and solid-state nuclear magnetic resonance (ss-NMR)35 have been employed to investigate the structures of fibrils built from amyloidogenic forming segments of amylin. Early models suggested by Kajava et. al (2005)34 consisted of three β-strands, running along the length of the monomer, also known as the “serpentine” model. However, more recent studies have suggested that amylin fibrils are made up of U-shaped (β-turn-β) or β-arch chains, a well-ordered cross-β structure, stacked upon each other to form a parallel β-sheet.33,35,37 Four different models have been presented for self-assembled double-fold amylin fibrils, two ssNMR models, and two crystal models. These models differ slightly in the details of the sidechain packing.35,37 Especially, the topology of the models proposed by Tycko & co-workers35 and Eisenberg & co-workers37 are similar, sharing the same central steric zipper and only differing in the registration of the two inner sheets. For the present investigation, we chose the model proposed by Eisenberg & co-workers.37 This is not only to connect this study with earlier work38 but also because the model interdigitates more closely than the model proposed by Tycko & co-workers,35 due to a zipper-like interaction formed by the sidechains of Ser29 residues extending across the interface.
2.2. Model construction
In order to connect our present study with earlier work investigating the stability of L-amylin aggregates,38 we have first constructed L-amylin decamers from U-shaped chains of a model provided by Eisenberg & co-workers,37 which has been shown to be a reasonable size to investigate the interactions governing the self-assembly of oligomers. Each of the ten chains forms a β-strand-loop-β-strand motif, where the two β-strands are made up of residues 8–17 and 28–37, and the loop region is formed by residues 18–27. The decamer is composed of two pentamers (called by us “protofibrils”), within each of the five “layers” two anti-parallel chains packed together at the C-terminal ends by polar interactions, hydrogen bonds involving Ser29 and Asn31; and hydrophobic interactions between Ala25–Asn35 and Leu27–Gly33. A representation of our model is shown in Figure 1.
Figure 1:
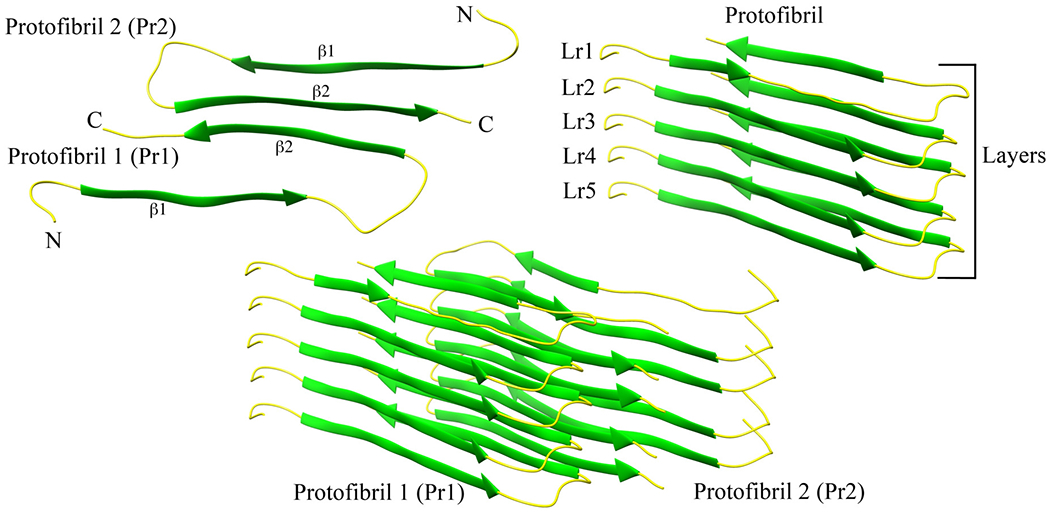
A two-fold human amylin fibril fragment. Chains in each layer interact by the anti-parallel C-terminal ends (a). Each protofibril is a stack of parallel chains (b). The arrangement of the two protofibrils is shown in (c).
Using the protocol described in Xi & Hansmann,30 we have constructed a DRI version of this decamer by replacing the backbone atoms of amylin as such: nitrogen (N) to carbon (C), C to N, hydrogen (H) to oxygen (O), and O to HN (hydrogen bound to nitrogen). The resulting structure is refined by energy minimization and subsequent short molecular dynamics simulation. The resulting model is shown in Suppl. Figure 1 and has by construction the same overall structure as of the L-amylin decamer. However, each of the chains is now made of D-amino acids, with the sequence of residues inverted. Because of this inversion, and in order to compare more easily L and DRI amylin chains, we index the residues in DRI-amylin starting from the C-terminus instead (as in the case of L-amylin), in the usual order starting from the N-terminus.
Various hybrid models were generated by applying the above procedure only to a subset of pre-selected chains, leading to fibrils with a mixture of L and DRI peptides. These hybrid models are summarized in Figure 2 and probe possible scenarios by which DRI-amylin chains may interact with L-fibrils. For instance, the fibril model (4L-1D)*2 describes an elongation of a two-layer octamer ((4L)*2) by a DRI-peptide in each protofibril. Note that in the above notation, we abbreviate the ‘DRI’ by a ‘D’. In a similar way describes the model (2L-1D-2L)*2, a case where a DRI-peptide has been incorporated in each of the protofibrils. Cross fibrillization between L and DRI amylin is probed with assembly geometries (L-D-L-D-L)*2, abbreviated as (L/D)*2, and (L-D-L-D-L)*(D-L-D-L-D), abbreviated further as (L/D)*(D/L). Finally, the interaction between a DRI-amylin pentamer with an L-amylin pentamer is probed by the fibril geometry 5D*5L. While various combinations of L and DRI mixtures can be devised, we chose the above models as they are the simplest implementations of the various scenarios by which DRI-amylin and L-amylin can form fibrils (i.e., elongation and cross-seeding).
Figure 2:
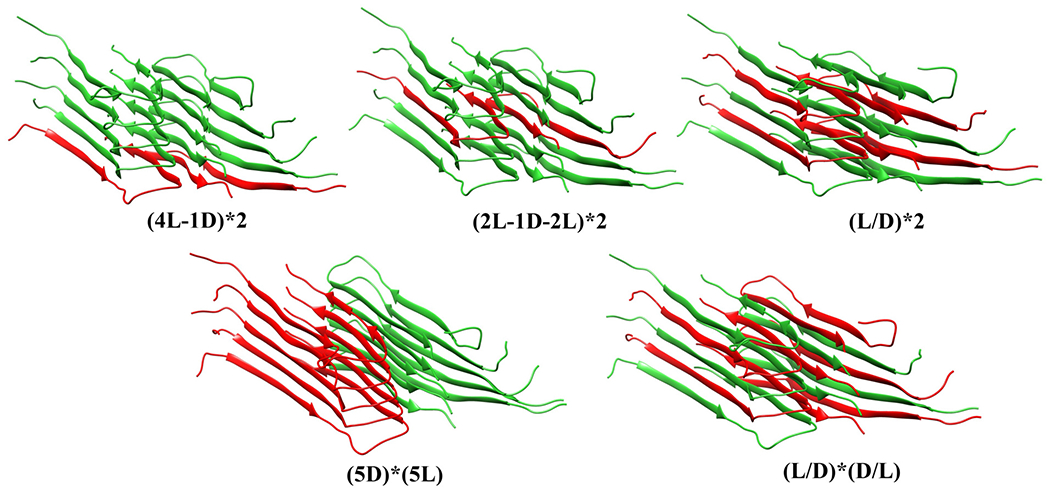
Various models of hybrid fibrils build from mixtures of L and DRI amylin chains.
2.3. Simulation set-up and analysis
We use for our molecular dynamics simulations the software package GROMACS-2018.1,39,40 with the protein interactions approximated by the CHARMM36 force field.41 However, in order to compare our results with our earlier work38 and to gauge any force field dependence of our results, we have also simulated the L-amylin fibril fragment using the AMBER (ff99SB) force field.42 For each run, we place the respective decamer in the center of a cubic box with 12 Å distance to the edges and fill the box with TIP3P water.43 Because of periodic boundary conditions, we evaluate electrostatic interactions by particle-Ewald summation,44,45 and a cut-off of 1.2 nm is used for calculation of vdW-interactions. The resulting systems are energy-minimized by steepest descent, followed by short (500 ps) molecular dynamics in an NVT ensemble, and subsequent 500 ps in an NPT ensemble. Temperature and pressure are controlled by a Parrinello-Danadio-Bussi thermostat46 and Parrinello-Rahman barostat47 and are set to T=310 K and 1 bar. The integration step is 2 f s. For each system, we follow three independent trajectories (starting from different velocity distributions) over 200 ns.
We analyze the resulting trajectories using the tools provided by the GROMACS package. One important quantity that we measure is the root-mean-square deviation of Cα-atoms (RMSD) with respect to the corresponding start configurations, excluding the flexible first seven residues. Monitoring the time evolution of the RMSD for the various models, we find that our simulations converge after 120 ns (Suppl. Figure 2). Therefore, we consider only the last 80 ns of the 200 ns long trajectories for further analysis (i.e., for calculation of average properties). Other quantities that we measure and analyze include the radius of gyration (Rg), solvent-accessible-surface-area (SASA), root-mean-square-fluctuation (RMSF), hydrogen bonds, dihedral angles, twist angle, hydrophobic contacts, and CC–interface contact distance. Hydrogen bonds are defined by a distance cut-off of 3.5 Å between the donor & acceptor atom and an angle cut-off of 30°. Similarly, a hydrophobic contact is defined by the condition that the distance between two residues (i and j, with |i − j| > 3) is less than 4.5 Å. We define the CC–interface contact distance as the distance between the Cα atoms of the residue pairs Leu27–Gly33, Ser29–Asn31, Asn31–Ser29, and Gly33–Leu27. Facing each other, these residues contribute towards fibril packing by forming hydrogen bonds and hydrophobic or polar contacts. We further calculate for the penultimate chains of a protofibril, the vector between the Cα-atom of Gln10 and the Cα-atom of Leu16. We define the twist angle for the β1-strand in a protofibril by the angle between these two vectors. In a similar way, we define the twist angle for the β2-strand by the corresponding vectors that point from the Cα-atom of residue Leu27 to the Cα-atom of Gly33.
3. Results and discussion
3.1. Force field dependence of amylin fibril simulations
A few years ago, we studied in our lab the stability of wild-type and various mutations of amylin fibril fragments.38 The simulation of these decamers (built from L-amino acids) relied on the AMBER (ff99SB) force field. Hence, in order to connect with our earlier work, we have started our investigation by first simulating our L-amylin fibril model (also a decamer) using the AMBER ff99SB force field. As our new results agree within the error bars with previous work,38 see Suppl. Figure 3, we conclude that our present simulation set-up is correct. However, we prefer to replace in the present work the previously used AMBER ff99SB by the newer CHARMM36 force field,41 which in our experience is more suitable for simulations of intrinsically disordered and aggregated proteins.48 Contrasting the ff99SB simulations of the L-amylin fibril with corresponding simulations relying on CHARMM36 allows us also to estimate the force field dependence of our results, and important factors to consider when comparing the stability of L-amylin and DRI-amylin aggregates. For this purpose, we have calculated the distribution of various quantities measured in the two sets of simulations. These distributions are shown in Figure 3. While the distribution of the radius of gyration (Rg), a measure for the compactness of configurations, is similar for the two sets, the situation is different for root-mean-square deviations (RMSD) to the respective start configurations. For this quantity, one derives a much broader distribution from the CHARMM trajectories than from the AMBER trajectories. This broadening goes together with a shift of the solvent-accessible-surface-area (SASA) values toward more solvated configurations in the CHARMM36 simulations. Together with the raised root-mean-square-fluctuations (RMSF) for each residue, suggesting a more diverse ensemble of configurations in the CHARMM36 simulations, these changes illustrate the improvements in force field development in the last years: modern force fields are less focused on the folded states as they aim to describe also correctly the energetics of unfolded configurations. For a more in-depth analysis of the force field dependence of simulations studying amyloid peptide assembly, we refer the reader to the article by Man et. al.48
Figure 3:
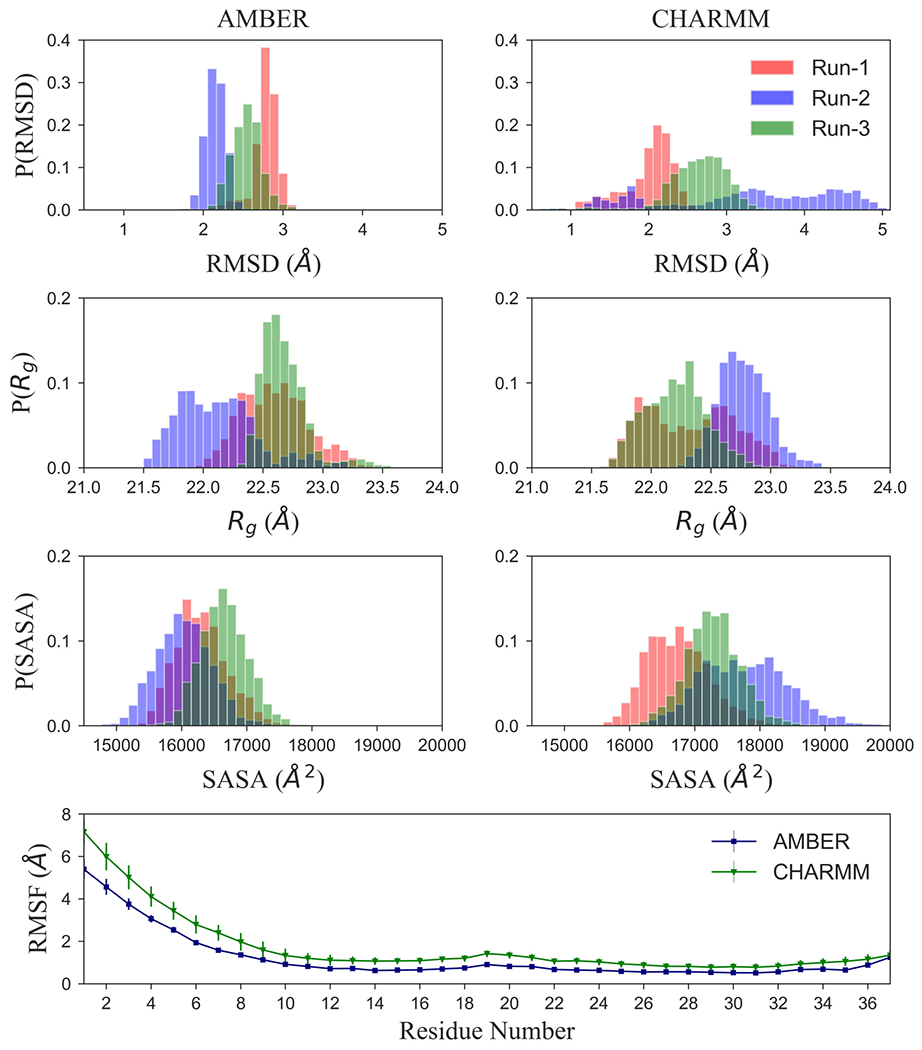
Comparison of molecular dynamics simulations of L-amylin fibril fragment using either AMBER ff99SB or CHARMM36. For this purpose, we show in (a) the normalized distribution of the root-mean-square deviation (RMSD), (b) the radius of Gyration (Rg), and (c) of the solvent-accessible-surface-area (SASA). In (d), we show the root-mean-square-fluctuation (RMSF) of the Cα atoms. For RMSF, values are averaged over all chains in the fibril fragment and all three trajectories.
3.2. Comparison of L-amylin and DRI-amylin fibrils
3.2.1. Structural characteristics
While the structural similarity between DRI-peptides and their L-parents often results in similar biological properties and activity, the existing subtle differences may alter amyloid formation and stability. For instance, in an earlier study, we found noticeable deviations in the stability of L and DRI Aβ40 and Aβ42 peptide fibrils, suggesting that DRI Aβ peptides enhance fibril formation in hybrid systems. However, it is not clear whether these earlier results can be generalized to amylin; and for this reason, we compare here first the stability of L-amylin and DRI-amylin fibrils.
In our earlier work, we could connect some of the stability disparity between L and DRI Ae40-fibrils to the dissimilar twist angles seen in the arrangements of the U-shaped chains. For this reason, we have measured here in both protofibrils the twist angles for the β-strands: β1 and β2. While the Ramachandran plots in Suppl. Figure 4 indicates that the sampling of the backbone ϕ/ψ angles of DRI-amylin mirrors those sampled by L-amylin, and the magnitude of the twist angle varies little between DRI-amylin and L-amylin for the N-terminal β1-strands (L-amylin: −15.1° ± 6.6°, DRI-amylin: 12.8° ± 3.6°), the measured twist angles differ not only by their sign but also in magnitude for the C-terminal β2-strands. Here, the DRI twist angle is more than twice as large than the L-value (L-amylin: −3.3° ± 2.5°, DRI-amylin: 7.6° ± 3.8°) (Suppl. Table 1). Because of these dissimilar twist angles, the residue-residue distances at the CC–interface (dominated by polar residues) between the two protofibrils differ slightly (see Suppl. Table 2), resulting in a tighter packing at the CC–interface in L-amylin fibril fragments than found in the DRI-amylin fibril model. However, the resulting loss in stability is small: the average root-mean-square deviation (RMSD), radius of gyration (Rg), solvent-accessible-surface-area (SASA) and RMSF as shown in Table 1, Suppl. Figure 5 and Suppl. Figure 6 is similar or only slightly higher for DRI-amylin than for L-amylin fibrils, and no obvious differences are seen when comparing individual trajectories (Suppl. Table 3). Snapshots from the simulations show only a marginal difference in the fibrillar structure (Suppl. Figure 7).
Table 1:
Structural changes in L-amylin (L), DRI-amylin (DRI) and Hybrid Amylin models in terms of average root-mean-square deviation (RMSD, in Å), average solvent-accessible-surface-area (SASA, in Å2), number of mainchain hydrogen bonds, sidechain hydrogen bonds, number of hydrogen bonds involved in stacking and packing, and CC–interface distance dCC(Å).
| L | DRI | (4L-1D)*2 | (2L-1D-2L)*2 | (L/D)*2 | 5D*5L | (L/D)*(D/L) | |
|---|---|---|---|---|---|---|---|
| RMSD | 3.0 (0.9) | 3.7 (0.4) | 3.7 (0.5) | 5.0 (0.9) | 5.1 (1.1) | 3.8 (1.1) | 4.3 (0.4) |
| SASA | 17388 (856) | 17720 (587) | 17690 (629) | 17967 (534) | 19172 (1077) | 17428 (698) | 18370 (938) |
| dCC | 6.8 (0.2) | 7.4 (0.7) | 7.9 (0.3) | 10.4 (1.4) | 7.8 (0.4) | 8.7 (1.2) | 8.4 (0.7) |
| Number of Hydrogen Bonds | |||||||
| Backbone | 186 (6) | 175 (9) | 181 (8) | 160 (16) | 161 (13) | 189 (8) | 171 (6) |
| Sidechain | 51 (5) | 43 (6) | 47 (5) | 39 (5) | 31 (6) | 45 (6) | 35 (6) |
| Number of Interchain Hydrogen Bonds | |||||||
| Backbone | 185 (6) | 173 (10) | 177 (9) | 157 (17) | 151 (13) | 187 (9) | 167 (5) |
| Sidechain | 40 (4) | 36 (6) | 38 (5) | 30 (4) | 23 (5) | 38 (6) | 25 (5) |
| Hydrogen Bond Function | |||||||
| Intrachain | 12 (4) | 9 (3) | 13 (4) | 12 (3) | 18 (3) | 9 (3) | 14 (4) |
| Stacking | 217 (7) | 203 (11) | 211 (9) | 184 (19) | 170 (17) | 222 (12) | 188 (6) |
| Packing | 8 (1) | 6 (2) | 4 (2) | 3 (2) | 4 (2) | 3 (2) | 4 (2) |
The CC–interface distance dCC is the average of the Cα distances of residue pairs Ser29–Asn31 and Asn31–Ser29 measured for the middle layer.
As the difference in the magnitude of twist angles is seen solely for the C-terminal β2-strands, the difference likely results from the interaction between chains on opposite protofibrils. The end of the N-terminal β-strand and the loop region connecting the two β-strands in each chain forms a hydrophobic core that contributes to the stability of the β-hairpin (β-strand-loop-β-stand motif) and the stacking of layers. While the total number of these hydrophobic contacts differs little between L-amylin (193 ± 1) and DRI-amylin fibrils (192 ± 7), rearrangements are observed in the inter-strand hydrophobic contacts for the residues Leu12, Ala13, Phe15, Val17, Ala25, Ile26, Leu27, and Val32, with 67 contacts seen only for L-amylin, and 68 only for DRI amylin. For instance, in L-amylin, residue Phe15 on strand i makes a hydrophobic contact with Ala13 on strand i + 1, and Val17 on strand i makes another hydrophobic contact with Phe15 on strand i + 1. In DRI-amylin, the residues switch places, i.e., it is now the Ala13 on strand i that forms a contact with the Phe15 on strand i + 1, while the Phe15 of strand i is now paired with the Val17 of strand i + 1. Similar rearrangements of hydrophobic contacts are observed for residues Leu12, Ala25, Ile26, Leu27, and Val32. Only present in L-amylin is a hydrophobic contact between residue Leu12 on strand i and Ala13 on the neighboring strand i + 1, while DRI-amylin has contact between Ala8–Ala8 on neighboring strands that are not found in L-amylin. As more than 60 % of the hydrophobic contacts are preserved in both forms, and lost contacts are replaced by newly formed, it appears that hydrophobic contacts add in a similar way to the stability of DRI-fibrils and L-fibrils.
3.2.2. Backbone hydrogen bonding
With the exception of an intrachain sidechain hydrogen bond connecting residues Asn35 with Tyr37 and a transiently formed mainchain hydrogen bond connecting N and C-termini, are all hydrogen bonds interchain bonds, and therefore essential for fibril stability. Mainchain hydrogen bonds link β-strands of stacked chains in the same protofibril, while sidechain hydrogen bonds also contribute towards the packing of the two protofibrils. DRI-amylin has 173 ± 10 backbone interchain hydrogen bonds and 36 ± 6 sidechain interchain hydrogen bonds, approximately twelve mainchain and four sidechain hydrogen bonds less than L-amylin (185 ± 6 mainchain hydrogen bonds and 40 ± 4 sidechain hydrogen bonds), see Table 1. While donor and acceptor atoms are switched, DRI-amylin preserves most of the mainchain hydrogen bonds found in L-amylin fibrils. The exception, leading to the unequal number of mainchain hydrogen bonds, are two recurring inter-layer hydrogen bonds which in L-amylin are formed between residues bordering the β-turn region but are in DRI-amylin fibrils only seen with a much lower frequency. The first of these hydrogen bonds connect residue Gly24 of one layer with Ala25 of the layer above and appear in L-amylin with a frequency of 90 ± 10 %, but in DRI-amylin only with a frequency of 13 ± 4 %. Gly24 and Ala25 are part of the large hydrophobic core region of amylin that stabilizes the fibril by hiding the core residues from water. The second L-amylin specific hydrogen bond connects in a similar fashion residue on neighboring layers; however, the two residues, His18 and Ser19, are on the opposite side of the β-turn region. This hydrogen bond is found in L-amylin in 74 ± 28 % of configurations, but only in 20 ± 2 % of DRI-amylin configurations. The difference in the frequency with which these in total 16 inter-layer hydrogen bonds are observed leads to an effective loss of eleven β-turn stabilizing hydrogen bonds in DRI-amylin fibrils, contributing to the slightly lower structural stability of DRI-amylin.
3.2.3. Sidechain hydrogen bonding
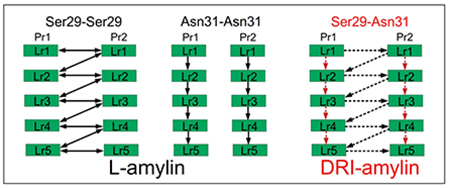
The difference in the number of sidechain hydrogen bonds measured in L and DRI fibrils is surprising as DRI peptides preserve the sidechain geometry of their parent L-peptide. Both fibrils have Asn14–Asn14 hydrogen bonds, and in most cases, Asn22–Asn22 hydrogen bonds (which otherwise are replaced in DRI-amylin fibrils by His18–Asn22 hydrogen bonds), that connects chains on subsequent layers of the same protofibril. However, visual inspection shows that the orientations of the sidechains of Ser29 and Asn31 differ in the two fibrils (Figure 4(d)). In L-amylin, the sidechains of Asn31 residues are aligned on top of each other (homo-stacking), forming Asn31–Asn31 sidechain hydrogen bonds (Figure 4(b)). On the other hand, the Ser29 residues from chains in opposite protofibrils face each other, allowing the formation of Ser29–Ser29 sidechain hydrogen bonds (Figure 4(a)). This is different in DRI-amylin, where Ser29 can now only form hydrogen bonds with Asn31 of the opposite protofibril (either of the same or a neighboring layer) (Figure 4(c)). It is this rearrangement of hydrogen bonds between the sidechains of residues Ser29 and Asn31 in neighboring chains that lead to the lower number of sidechain hydrogen bonds in the DRI-fibril.
Figure 4:
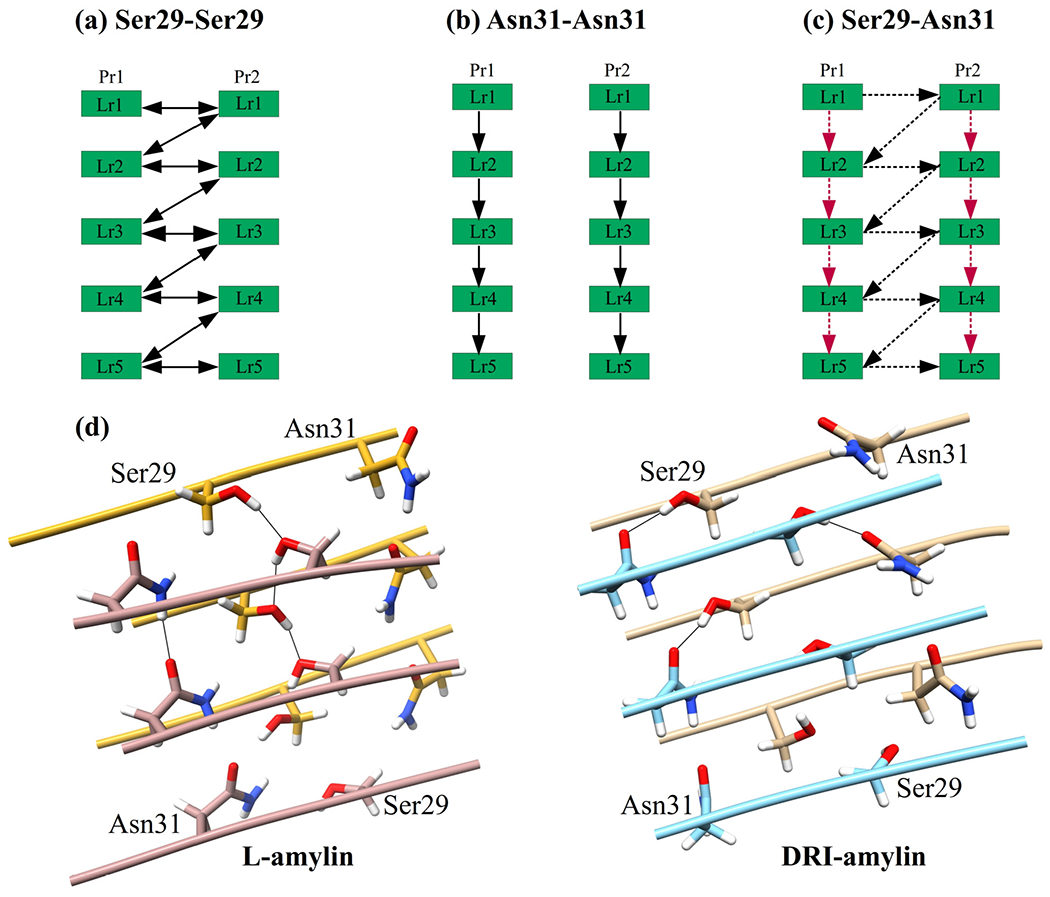
Schematic of the interchain sidechain hydrogen bond observed in L-amylin and DRI-amylin. In L-amylin, Ser29 forms a hydrogen bond with neighboring Ser29 (Ser29@OG1–Ser29@HG1) of the opposite protofibril (a), and Asn31 forms a hydrogen bond with Asn31 (Asn31@OD1–Asn31@2HD2) on subsequent layer of the same protofibril (b) However, in DRI-amylin, Ser29–Ser29 hydrogen bonds do not exist and on average half of the Asn31–Asn31 hydrogen bonds (drawn in red) are replaced by hydrogen bonds between Asn31 and Ser29 (drawn in black) (c). This is because the sidechain of Ser29 and Asn31 are flipped as can be seen in (d). Arrows point from donor to acceptor, and competing hydrogen bonds are drawn with dotted lines.
Since in L-amylin, the interface between the two protofibrils is formed by the C-terminal β-strands of each chain (anti-parallel), the sidechain of residue Ser29 on a given chain can form two hydrogen bonds with the sidechains of Ser29 residues located on chains in the other protofibrils. One hydrogen bond connects Ser29 residues on the same layer and is seen in about 78 ± 23 % of all configurations. The other hydrogen bond connects Ser29 residues on neighboring layers i and i − 1 (but different protofibrils) and is seen in 88 ± 19 % of configurations. The average distance between the donor-acceptor pair in these two hydrogen bonds is comparable: 2.80 ± 0.04 Å (when connecting residues located on the same layer) and 2.79 ± 0.03 Å (when connecting residues on staggered layers). Note that these two hydrogen bonds are bifurcating hydrogen bonds, with the donor and acceptor atoms (OG1 and HG1) switching with a period of 2.2–2.3 ps between the two hydrogen bond forming residues (Data not shown). As a consequence, the Ser29–Ser29 hydrogen bonds support in L-amylin fibrils both the stacking of chains in each protofibril and the packing of the protofibrils. This is not the case in DRI-amylin fibrils, where these bifurcating hydrogen bonds are not observed (the corresponding frequencies are 5 ± 11 % and 5 ± 6 %) for both types.
This effect is partially compensated by alternative hydrogen bonds that Asn31 can form in DRI-fibrils but not in L-fibrils, where Asn31 can only form a single hydrogen bond with an Asn31 on a chain in the above layers of the same protofibril. These hydrogen bonds appear with a frequency of 87 ± 16 % of all configurations and stabilize the stacking of chains in each protofibril. The same kind of hydrogen bond is also seen in DRI-amylin fibril but only with a frequency of 42 ± 27 %, i.e., with half the probability found in L-amylin, and therefore reducing further the cohesion between layers in the protofibrils. However, when in DRI-amylin fibrils not forming an Asn31–Asn31 hydrogen bond, the Asn31 residue on a given chain can form a hydrogen bond with Ser29 instead, on a chain located either on the same layer or on a neighboring layer of the opposite protofibril. These nine possible Asn31–Ser29 hydrogen bonds are observed in 51 ± 35 % of all DRI-fibril conformations, but only in less than 2 % of L-amylin fibrils. While the donor-acceptor distance in these particular hydrogen bonds is within 2.77 ± 0.02 Å, comparable to the Ser29–Ser29 hydrogen bond found in L-amylin, their lifetime is very short (~1.3 ps (Data not shown)), and they are not bifurcating hydrogen bonds as the donor is always Ser29 and acceptor is always Asn31. Nevertheless, replacing in DRI-fibrils the Ser29–Ser29 hydrogen bonds seen in L-amylin, the Asn31–Ser29 bonds stabilize in a similar way the packing of the protofibrils.
However, the loss of all of the nine Ser29–Ser29 hydrogen bonds and, on average, half of the eight Asn31–Asn31 hydrogen bonds is only partially compensated by the formation of, on average, half of the nine Asn31–Ser29 hydrogen bonds possible in the DRI-amylin fibril fragment. While on average in L-amylin, one Asn31–Asn31 sidechain hydrogen bond connects two chains located on layers of the same protofibril, the number is reduced by half in DRI-amylin. Similarly, while in L-amylin, on average, one Ser29–Ser29 hydrogen bond per chains connects the two protofibrils, only half of these are replaced in DRI-amylin by Asn31–Ser29 hydrogen bonds. Hence, the net effect of the diverse hydrogen bond pattern is a reduction of stability in DRI-amylin fibrils by four sidechain hydrogen bonds.
3.2.4. Stability differences
Our discussion of the differences in hydrogen bonding also explains the rather small differences in stability between L-amylin fibrils and DRI-fibrils. In L-amylin fibrils are neighboring layers hold together by, on average, 23 mainchain hydrogen bonds and four sidechain hydrogen bonds per pair of chains, leading to a total of 217 (7) hydrogen bonds that stabilize stacking of chains. In DRI-amylin are the corresponding numbers 21 mainchain hydrogen bonds and three sidechain hydrogen bonds per pair of chains, leading to a total of 203 (11) hydrogen bonds that stabilize the stacking of chains. Hence, the stacking of chains is less stable in DRI-amylin fibrils by about two hydrogen bonds per pair of chains, a total reduction in stacking of about 7 %. On the other hand, the two protofibrils are in the L-amylin fibril hold together by, on average, eight sidechain hydrogen bonds, but only by six sidechain hydrogen bonds in the DRI-amylin fibril, reducing the packing stability by about 25 %.
While the loss of hydrogen bonds in DRI-amylin fibrils reduces only slightly the overall stability, and individual β-strands keep their structure, it still leads to notable structural differences. The looser packing of the individual protofibrils, resulting from the smaller number of sidechain hydrogen bonds, leads to a more twisted conformation, with the DRI twist angle for the C-terminal β2-strands twice as large as the corresponding L-value. In L-amylin, the twisting of the C-terminal β2-strands is restrained by the hydrogen bonds formed between the side chains of the Ser29 residues on the two protofibrils (Ser29–Ser29), leading to twist angles of −2.9°±0.5° and −3.6°±0.4° (see Suppl. Table 1). The effective loss of these hydrogen bonds in DRI-amylin is only partially compensated by the newly formed sidechain hydrogen bonds between Ser29 and Asn31, but not sufficient to counterbalance the intrinsic propensity of the protofibrils to twist. As a consequence, we observe a higher twist angle in the β2-strands of the protofibrils of DRI-amylin (7.2° ± 2.9° and 8.0° ± 4.5°); see Suppl. Table 1. The consequence of this larger twist is the two protofibrils less close than in L-amylin. For instance, we measure the CC–interface distance value of 7.4 ± 0.7 Å for DRI-amylin, compared to 6.8 ± 0.2 Å for L-amylin fibrils.
3.3. Stability of Hybrid Assemblies
Despite the only marginal structural differences, there are distinct differences in the hydrogen bond pattern between L-amylin fibrils and such made of DRI-amylin, with the effect most pronounced for sidechain hydrogen bonds that can alternate (or oscillate) between two residue pairs sharing one residue as either donor or acceptor (in our case Ser29). This suggests that the insertion of DRI-proteins in L-assemblies may be an alternative to mutations for probing the role of such hydrogen bonds in supra-molecular assemblies or aggregates. Another possible application would be to use these differences to modulate the formation and growth of fibrils.
Fibril formation is a nucleation process where rapid fibril growth follows the formation of a critical nucleus. The long lag-phase required for nucleation can be shortened by orders of magnitude through seeding, i.e., the addition of preformed fibrils as seeds. These seeds do not have to be of the same protein, as amyloids can also be formed by cross-seeding with other proteins.17 The latter process is not fully understood and is especially interesting for mixtures of L and DRI peptides where the differences in chirality impart subtle differences in the hydrogen bond and hydrophobic interactions (as seen in the previous section), which can either promote or inhibit seeding. Hence, in order to understand how the presence of DRI-amylin affects the growth and stability of L-amylin fibrils, we have studied the stability of various hybrid constructs made up of L and DRI amylin.
3.3.1. Fibril elongation by DRI-peptides
In the first set of simulations, we have looked into the effect of DRI-peptides on the elongation of amylin fibrils. For this purpose, we considered the case of DRI-peptides attached at the end of both layers of a double-layer L-amylin fibril, i.e., the fibril construct (4L-1D)*2. This decamer assembly models the elongation of a double-layer L-amylin octamer ((4L)*2) by a DRI-peptide at each layer. As can be seen from Table 1, the (4L-1D)*2 fibril has 0.7 A higher average RMSD (3.7 ± 0.5 Å), 0.3 Å higher Rg (22.7 ± 0.4 Å) and 302 Å2 more SASA (17690 ± 629 Å2) values than the corresponding L-amylin decamer, and therefore is only slightly less stable (Suppl. Figure 6 and Suppl. Figure 7). Since hydrogen bonds across the chains are the major player in the elongation process of the fibril assembly, we have calculated the number of mainchain and sidechain hydrogen bonds, and observe that most of the mainchain (181 ± 8) and sidechain hydrogen bonds (47 ± 5) are conserved in (4L-1D)*2 hybrid fibril, with discrepancies only at the L and DRI interface. In a pure L-fibril or a pure DRI-fibril, each i residue in one chain forms two mainchain hydrogen bonds with the residues in the subsequent layer: one with i − 1 residue with the backbone amide hydrogen and the other with i + 1 residue with the backbone carbonyl oxygen (Figure 5). However, at the L–DRI interface, the i residue from L-chain forms both the mainchain hydrogen bonds with the i − 1 residue from DRI-chain, i.e., the DRI strand is shifted in relation to the rest of the fibril by one residue towards the terminus. As a consequence, the loop region and C-terminus end of the DRI-chain are no longer in close proximity to the loop region and the C-terminus end of the underlying L-chain, hindering the formation of mainchain hydrogen bonds in this region, specifically between the residue pairs Asn22–Phe23 (Pr2Lr4–Pr2Lr5), Ala25–Ile26 (Pr1Lr4–Pr1Lr5) & Gly24–Ala25 (Pr1Lr4–Pr1Lr5) (beta-turn region) and Ser34–Asn35 (Pr2Lr4–Pr2Lr5) & Thr36–Tyr37 (Pr2Lr4–Pr2Lr5) (terminal regions). The hydrogen bonds between the residue pairs Asn22–Phe23, Gly24–Ala25 & Ala25–Ile26 occur with a frequency of 91 ± 19%, 95 ± 5% and 98 ± 1 % at the interacting L–L interface, while the corresponding values for the L-DRI interface are within the error bars consistent with zero. The loss of four backbone hydrogen bonds leads to a weakening of the inter-layer stacking at the interface between L and DRI chains, causing rearrangement of hydrophobic contacts at this interface residues Leu12, Ala25, Ile26, Leu27, and Val32.
Figure 5:
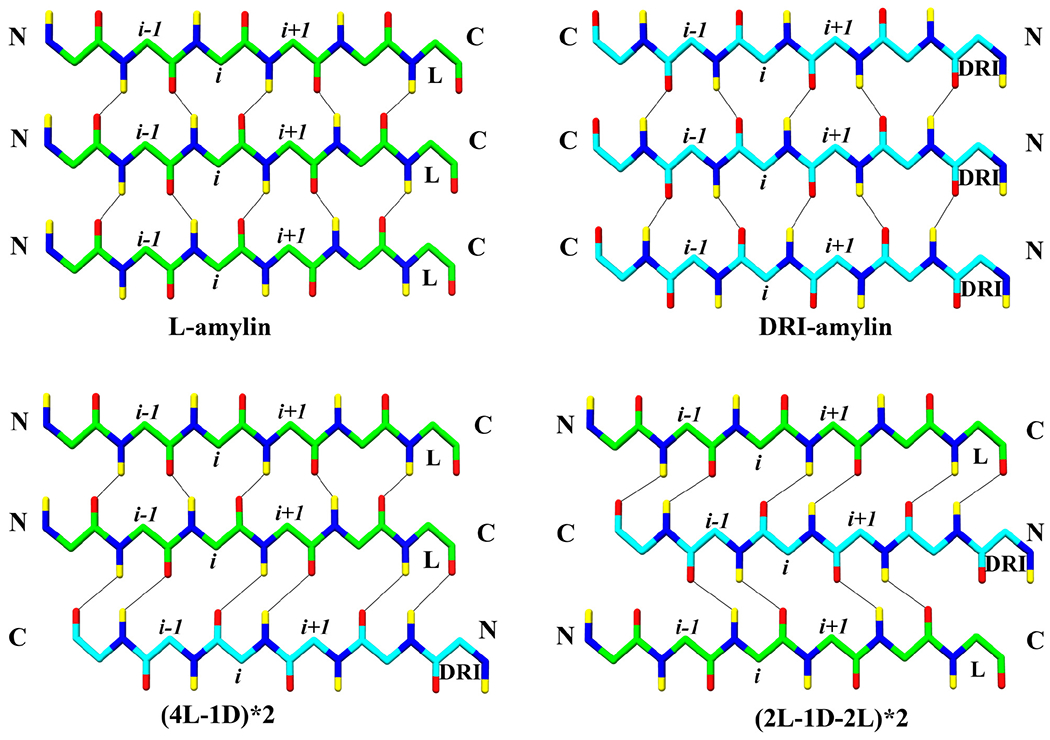
Interchain hydrogen bonds formed in L-amylin, DRI-amylin and hybrid fibrils. Between L–L and DRI–DRI strands, interchain mainchain hydrogen bonds are formed connecting the i residue from one strand to the i — 1 and i + 1 residue from another strand. In case of a L–DRI interface, the i residue from one strand forms interchain mainchain hydrogen bonds with the i − 1 residue or with the i + 1 residue from another strand. Only backbone atoms are shown for clarity. Oxygen atoms are coloured in red, nitrogen in blue, hydrogens in yellow and Cα and C atom of L-chain is coloured in light green, while that of DRI-chain is coloured in cyan. Note that residues are counted from the C-terminal in DRI chains.
Since the DRI-amylin chain is shifted by one residue in relation to the L-amylin chains, we expect to see also differences in the sidechain hydrogen bond pattern at the L–DRI interface. For instance, the Asn31–Asn31 hydrogen bond is lost at the interface between L and DRI chains (Pr1Lr4–Pr1Lr5 and Pr2Lr4–Pr2Lr5). Interestingly, the attachment of DRI at the end of the L-amylin fibril also affects the network of Ser29–Ser29 hydrogen bonds beyond the L and DRI interface, where these hydrogen bonds now appear only with a frequency of 20 ± 20 %. In pure DRI fibrils, the loss of Ser29–Ser29 hydrogen bonds is partially compensated by the formation of hydrogen bonds between Ser29 and Asn31, but this is not the case in the (4L-1D)*2 model. Since the Asn31–Asn31 sidechain hydrogen bonds are still retained in the L–L layers, though appearing with a lower frequency of 45 ± 36 %, the hydrogen bond forming atoms of Asn31 are not available to form hydrogen bonds with Ser29 to make up for the loss of Ser29–Ser29 hydrogen bonds. As a consequence, at the L–DRI interfaces between layers 4 and 5 in the two protofibrils, three sidechain hydrogen bonds are less involved in stacking, with approximately one hydrogen bond at each L–DRI interface. On the other hand, the two L–DRI interfaces between chains located on opposite protofibrils, lead to the loss of four sidechain hydrogen bonds for packing, two for each L–DRI interface. The loss of packing-supporting hydrogen bonds is due to the larger twist of the C-terminal β2-strands in the DRI-amylin chains, which also affects the corresponding strands of the L-amylin chains in the other layers of the fibril which have about three times larger twist angles than seen in pure L-amylin fibrils. Hence, addition of the DRI-fibril leads to the loss of the sidechain hydrogen bonds that stabilize the packing of the two protofibrils: only four hydrogen bonds stabilize the packing. In contrast, in L-amylin fibrils, the packing is stabilized by eight hydrogen bonds, and by six hydrogen bonds in DRI-amylin fibrils. Hence, elongation of an L-fibril by DRI peptide is energetically less favorable than elongation by an L peptide, since the packing of layers is reduced at the interface by four mainchain hydrogen bonds, and the packing between protofibrils by four hydrogen bonds, reducing the interaction between the protofibrils even beyond the interfacing layer (see Table 1). As a consequence is the CC–interface distance with 7.9 Å substantially larger than in either L- or DRI- amylin fibrils.
3.3.2. DRI-peptides as defects in L-amylin fibrils
While the simulations of the fibril construct (4L-1D)*2 allows us to probe the elongation of existing L-fibrils by DRI-amylin and the nucleation of L-amylin fibrils by DRI-amylin seeds, they describe settings where there is only one interface between L and DRI peptide. Hence, these simulations cannot tell us about the stability of hybrid fibrils where DRI peptides are incorporated within L-layers, i.e., whether DRI-amylin as a fibril breaker or enhances the stability of amylin fibrils. The simplest system to study this question is an arrangement (2L-1D-2L)*2, where in each protofibril, the central chain is a DRI-amylin, surrounded by two L-amylin chains on each side. We find that this hybrid construct is even less stable than the (4L-1D)*2 construct, with a higher RMSD (5.0 ± 0.6 Å), and more solvated structures seen at the end of the trajectories (see Table 1). As a consequence, this model has 199 (17) hydrogen bonds, about 30–40 hydrogen bonds less than the L-amylin model (or even the (4L-1D)*2 model), and about 20 less than DRI-amylin fibril (see Table 1). All of the lost hydrogen bonds are interchain. The differences in hydrogen bonding and hydrophobic contacts are again concentrated at the interfaces between L and DRI peptides. Since in (2L-1D-2L)*2, the DRI-chain interfaces with two L-amylin chains, the i residue from the DRI-chain forms both backbone hydrogen bonds either with the i + 1 residue of the L-chain in the next layer or with the i + 1 residue of the L-chain on the preceding layer (see Figure 5). However, at both interfaces are hydrogen bonds lost in the beta-turn region (Asn22–Phe23 (47 ± 46 %), Gly24-Ala25 (8 ± 23 %) & Ala25-Ile26 (36 ± 44 %)) and terminal region (Ser34-Asn35 (0 %) & Thr36-Tyr37 (0 %)). In total, there are 157 (17) interchain backbone hydrogen bonds stabilizing the stacking of chains, 28 less than L-amylin. Of these are 14 lost at the four L–DRI interfaces, i.e., around four backbone hydrogen bonds per L–DRI interface, the same value as seen in the (4L-1D)*2 model. Note that while 14 hydrogen bonds are lost at the four L–L interfaces, this loss results from increased fluctuation in the loop region and C-terminus due to the inclusion of the DRI-chain, not from a relative shift of chains as in the case of L–DRI interfaces. This can be clearly seen from the increased RMSD of the L-chains in the (2L-1D-2L)*2 model (Suppl. Figure 6).
Similar to the (4L-1D)*2 case, in the (2L-1D-2L)*2 model the sidechain hydrogen bonds are lost between Ser29–Ser29 and Asn31–Asn31 (14 ± 17 %), and only 3 (2) sidechain hydrogen bonds (less than one per pair of chains) contribute to the packing of the two protofibrils. However, in addition, the sidechain hydrogen bonds between Asn22–Asn22 (29 ± 35 %) are lost, not compensated by the formation of His18–Asn22 (3 ± 8 %) sidechain hydrogen bonds. As a consequence, the stability of stacking in these fibrils is reduced by an additional five sidechain hydrogen bonds when compared to pure L fibrils. Four of these hydrogen bonds are lost at the four L–DRI interlayer interfaces, while three are lost at the two interlayer L–DRI interfaces in the (4L-1D)*2 model. Contributing to this loss of stacking stability is the lack of 28 hydrophobic contacts (when compared to L-amylin) involving residues Ala25, Ile26, Leu27, and Val32. Especially, the hydrophobic contacts between Leu27 & Val32 were lost across all the layers, while the contacts between residues Ala25 & Ile26 and Ile26 & Leu27 were lost only at the interfaces between L and DRI strands. The net effect of this loss of hydrophobic contacts and hydrogen bonds is a much larger reduction of stability than observed for pure DRI-amylin fibrils or (4L-1D)*2 construct, as seen, for instance, by the time evolution of RMSD (Suppl. Figure 2 and Suppl. Figure 6) or the much larger distance between the two protofibrils at the CC–interface (10.4 ± 1.4 Å), more than 4 Å larger than seen in the L-amylin fibril.
3.3.3. The effect of DRI-peptides on fibril packing and stacking
The fibril fragment (2L-1D-2L)*2 serves as a model for a defect in the fibril resulting from incorporating a DRI-chain into an L-fibril, a situation that may occur if the concentration of DRI-chains is small compared to that of L-chains. Once the concentration of DRI-chains is sufficiently large, alternating assemblies of L and DRI chains could appear instead, if they were energetically favorable. We have considered three such assemblies. The first one is the model (L/D)*2, where in a given layer, the chains are either both L or both DRI peptides. In the second model, 5D*5L, one protofibril is made of L-amylin chains and the other of DRI-amylin. In the third model, (L/D)*(D/L), the chains differ not only in the two protofibrils but also in each layer. In neither case, do we see a stabilizing effect caused by the alternating L and DRI chains. In the (L/D)*2 model, designed by us for probing the effect of alternating L and DRI amylin on the stacking of chains, we find that in comparison to the L-amylin fibril the average RMSD increases by two Å (5.1 ± 1.1 Å) and the average solvent accessible surface area (SASA) by 1784 Å2. Both the number of the interchain backbone (151 ± 13) and sidechain hydrogen bonds (23 ± 5) is lower than in all cases, and only partially compensated by an increase in intrachain hydrogen bonds (18 ± 13). The effect of the alternating L and DRI amylin chains is most visible on the stacking of chains, which is now stabilized by only 170 ± 16 hydrogen bonds, 151 backbone hydrogen bonds, and 19 sidechain hydrogen bonds, connecting L and DRI chains. Hence, as in the previous models, the stability of stacking is reduced by about four backbone hydrogen bonds and one sidechain hydrogen bond per L–DRI interface. Note that the number of hydrogen bonds stabilizing the packing of the two protofibrils is with 4 (2) hydrogen bonds stronger than in the (2L-1D-2L)*2 model, leading to a separation of CC–interfaces (7.8 ± 0.2 Å) by only one Å larger than L-amylin fibril, and is comparable with the pure DRI fibril. However, these small differences suffice to ease the separation of protofibrils and dissociation of the fibril, see the increased RMSD of individual chains (Suppl. Figure 6) and the structural snapshot obtained after 200 ns simulation (Suppl. Figure 7).
On the other hand, in the second model, 5D*5L, designed by us to probe the effect of alternating L and DRI amylin chains on packing of the two protofibrils, we find a much smaller disturbance of the fibril structure as can be seen from the RMSD and SASA values in Table 1. Here, the two protofibrils preserved most of the interactions seen in their corresponding pure fibril forms, with the number of hydrogen bonds stabilizing the stacking of chains (222 ± 12) comparable to those found in the L-amylin fibril. However, the number of sidechains stabilizing the packing is again strongly reduced as the loss of Ser29–Ser29 sidechain hydrogen bonds are again only partially compensated by newly formed Ser29–Asn31 hydrogen bonds, which are observed only with a frequency of about 30 % (compared to about 50 % in pure DRI-amylin fibrils). The number of remaining hydrogen bonds (3 ± 2) that stabilize packing of the protofibrils is comparable to that seen in the (2L-1D-2L)*2 model, but the packing is less weakened and the distance between the CC-interfaces with 8.7 ± 1.2 Å, less increased than in the (2L-1D-2L)*2 model. Additional stability comes from π – π interaction observed between the Phe23 on the DRI protofibril and Tyr37 of the L protofibril. Although these interactions are weaker than hydrogen bonds, they add up and partially compensate for the loss of hydrogen bonds that stabilize in L-amylin fibril the packing of the two protofibrils.
Our third model, (L/D)*(D/L), is designed to allow us to observe the combined effect of alternating L and DRI-amylin chains on stacking and packing. This arrangement where chain type (L or DRI amylin) changes both between layers and protofibrils leads in terms of RMSD (4.3 ± 0.4 Å), solvent accessible surface area (18370.6 ± 938.2 Å2), and distance between the two protofibrils (8.4 ± 0.7 Å) to values that lay in between the 5D*5L and the (L/D)*2 model and shares with both of these models, a comparable number of hydrogen bonds (4 ± 6) stabilizing the packing of the two protofibrils. Note that the number of hydrogen bonds stabilizing the stacking of chains is (188 ± 6) much higher than in the (L/D)*2 model (170 ± 17), i.e., the staggering of chains increases the stacking stability but does not affect the packing of protofibrils. This is because unlike in the previous models, the β2-strands of the two chains in each layer are now parallel instead of anti-parallel, adding not only to the stability of the protofibril packing but by stabilizing the U-shaped geometry of the chains also eases their alignment and stacking.
Hence, in cases where L and DRI strands appear in alternating fashion, i.e., the fibrillar assemblies (L/D)*2 and (L/D)*(D/L), the stacking is reduced by at least four backbone hydrogen bonds and one sidechain hydrogen bond per L–DRI interface, and packing is reduced by four sidechain hydrogen bonds to at most one hydrogen bond connecting two chains on opposite protofibrils (see Table 1). As the latter is a 50 % reduction in the number of hydrogen bonds stabilizing the packing of protofibrils, but the former only a 15–20 % reduction in stacking-stabilizing hydrogen bonds, the main effect is a separation of double fold assemblies into two single fold fibrils. We conjecture that the addition of DRI-amylin discourages elongation of amylin fibrils and causes separation into single-fold fibrils.
4. Conclusion
The present study is motivated by the question of whether DRI-amylin could provide an alternative to the often used pramlintide whose utility as a drug for targeting type–II diabetes is limited by its short lifetime. DRI-amylin has a long biological lifetime that is characteristic for peptides built from D-amino acids,26,27 but in order to function as a drug, it also needs to reproduce pramlintide’s ability to inhibit or dis-aggregate amylin aggregates. We have therefore studied by means of molecular dynamics simulations the stability of DRI-amylin fibrils and how the insertion of DRI-amylin alters the aggregation of regular L-amylin. Our research builds on a previous study of DRI-Aβ fibril stability30 but extends it to a system with a large number of polar and charged residues. We find that fibrils made of DRI-amylin are only marginally less stable than the L-amylin fibrils. Going beyond our previous work,30 we can now present a mechanism for the observed stability differences relating them to the loss of about 20 hydrogen bonds (about two per chain), both backbone and sidechain hydrogen bonds. The loss of Ser29–Ser29 sidechain hydrogen bonds in DRI-fibrils and their partial replacement by Ser29–Asn31 sidechain hydrogen bonds leads to a net-reduction of packing stabilizing hydrogen bonds by about 25 %, that changes the twist angle of the β2-strands forming the interface between the two protofibrils.
The presence of DRI-amylin affects the elongation and the stability of L-amylin fibrils by shifting the backbone hydrogen bonds at the L–DRI interface by one residue toward the C-terminus. Unlike in pure DRI-amylin fibrils are the Ser29–Ser29 sidechain hydrogen bonds completely lost and not partially replaced by Ser29–Asn31 hydrogen bonds. This perturbation of hydrogen bonding spreads from the L–DRI interface to successive layers, easing separation of the two protofibrils. Hence, while the incorporation of DRI-chains weakens the stacking of chains in each protofibril, the main effect is the reduction of the packing stability between the two protofibrils. While we see at low concentrations cross-seeding, with increasing concentration and growing number of L-DRI interfaces, DRI-amylin peptides start to inhibit fibril formation. Such a concentration dependence was also seen in recent work by Ghosh et. al49 for the inhibitory effect of TK9 (a nine-residue peptide of the extra membrane C-terminal tail of the SARS corona-virus envelope) on amylin aggregation.
While our simulations indicate that the presence of DRI-amylin will inhibit elongation of (L) amylin fibrils and reduce their stability, further studies are needed to explore the mechanical stability of the fibril models 50 and the kinetics of this process and whether it leads indeed to inhibition of fibril formation. Besides such direct applications, our investigation also suggests that the insertion of DRI-proteins in L-assemblies may be an alternative way to mutations to probe the role of such hydrogen bonds in supra-molecular assemblies or aggregates. Despite only marginal structural differences, there are distinct differences in the hydrogen bond pattern in aggregating DRI-peptides, the fact that we used in the present study to point out some key interactions that stabilize amylin fibril geometries. In particular, Asn ladders (Asn14, Asn22, and Asn31) and Ser–Ser bifurcated hydrogen bonds seem to play an important role in stabilizing L-amylin fibrils. We speculate that the retention of Asn31 in pramlintide contributes to the observed co-aggregation with amylin and that the effectiveness of the drug could be increased by mutating this residue or replacing it with the D-isomer.
Supplementary Material
Acknowledgement
We thank Elliott K. Vanderford for help at early stages of this project. The simulations in this work were done using the SCHOONER cluster of the University of Oklahoma, the Blue Waters sustained - petascale computing project, and XSEDE resources allocated under grant MCB160005 (National Science Foundation). We acknowledge financial support from the National Institutes of Health under grants GM120578 and GM120634.
Footnotes
Supporting Information Available
The following tables and figures are provided in a supplementary document, available free of charge at http://pubs.ac.org:
Supplementary Tables: Twist angle for L-amylin and DRI-amylin (Suppl. Table 1); Face-to-face contact distance of L-amylin and DRI-amylin (Suppl. Table 2); Structural changes in terms of RMSD, Rg, and SASA (Suppl. Table 3);
Supplementary Figures: Generating DRI-peptides (Suppl. Figure 1); Time evolution of the root-mean-square deviation (RMSD) (Suppl. Figure 2); Comparison of root-mean-square fluctuations (RMSF) for AMBER results with Bernhardt et. al (2013) (Suppl. Figure 3); Ramachandran plot for L-amylin and DRI-amylin (Suppl. Figure 4); Root-mean-square fluctuations (RMSF) of the Cα atoms for L-amylin and DRI-amylin (Suppl. Figure 5); Time evolution of root-mean-square deviation (RMSD) of individual monomers (Suppl. Figure 6); Representative snapshots (Suppl. Figure 7).
References
- (1).Kahn SE; D’Alessio DA; Schwartz MW; Fujimoto WY; Ensinck JW; Taborsky GJ; Porte D Evidence of Cosecretion of Islet Amyloid Polypeptide and Insulin by β-Cells. Diabetes 1990, 39, 634–638. [DOI] [PubMed] [Google Scholar]
- (2).Kahn SE; Andrikopoulos S; Verchere CB Islet amyloid: a long-recognized but underappreciated pathological feature of type 2 diabetes. Diabetes 1999, 48, 241–253. [DOI] [PubMed] [Google Scholar]
- (3).Goldsbury C; Goldie K; Pellaud J; Seelig J; Frey P; Müller S; Kistler J; Cooper G; Aebi U Amyloid Fibril Formation from Full-Length and Fragments of Amylin. Journal of Structural Biology 2000, 130, 352–362. [DOI] [PubMed] [Google Scholar]
- (4).Pillay K; Govender P Amylin Uncovered: A Review on the Polypeptide Responsible for Type II Diabetes. BioMed Research International 2013, 2013, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Hiddinga HJ; Eberhardt NL Intracellular Amyloidogenesis by Human Islet Amyloid Polypeptide Induces Apoptosis in COS-1 Cells. The American Journal of Pathology 1999, 154, 1077–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Lorenzo A; Razzaboni B; Weir GC; Yankner BA Pancreatic islet cell toxicity of amylin associated with type-2 diabetes mellitus. Nature 1994, 368, 756–760. [DOI] [PubMed] [Google Scholar]
- (7).Verchere CB; D’Alessio DA; Palmiter RD; Weir GC; Bonner-Weir S; Baskin DG; Kahn SE Islet amyloid formation associated with hyperglycemia in transgenic mice with pancreatic beta cell expression of human islet amyloid polypeptide. Proceedings of the National Academy of Sciences 1996, 93, 3492–3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Mirzabekov TA; Lin M.-c.; Kagan BL Pore Formation by the Cytotoxic Islet Amyloid Peptide Amylin. Journal of Biological Chemistry 1996, 271, 1988–1992. [DOI] [PubMed] [Google Scholar]
- (9).Anguiano M; Nowak RJ; Lansbury PT Protofibrillar Islet Amyloid Polypeptide Permeabilizes Synthetic Vesicles by a Pore-like Mechanism that May Be Relevant to Type II Diabetes. Biochemistry 2002, 41, 11338–11343. [DOI] [PubMed] [Google Scholar]
- (10).Cernea S; Dobreanu M Diabetes and beta cell function: from mechanisms to evaluation and clinical implications. Biochemia Medica 2013, 23, 266–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Kaufman RJ; Back SH; Song B; Han J; Hassler J The unfolded protein response is required to maintain the integrity of the endoplasmic reticulum, prevent oxidative stress and preserve differentiation in β-cells. Diabetes, Obesity and Metabolism 2010, 12, 99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Leibowitz G; Bachar E; Shaked M; Sinai A; Ketzinel-Gilad M; Cerasi E; Kaiser N Glucose regulation of β-cell stress in type 2 diabetes. Diabetes, Obesity and Metabolism 2010, 12, 66–75. [DOI] [PubMed] [Google Scholar]
- (13).Jackson K; Barisone GA; Diaz E; Jin L.-w.; DeCarli C; Despa F Amylin deposition in the brain: A second amyloid in Alzheimer disease? Annals of Neurology 2013, 74, 517–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Lutz TA; Meyer U Amylin at the interface between metabolic and neurodegenerative disorders. Frontiers in Neuroscience 2015, 9, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Baram M; Atsmon-Raz Y; Ma B; Nussinov R; Miller Y Amylin–Aβ oligomers at atomic resolution using molecular dynamics simulations: a link between Type 2 diabetes and Alzheimer’s disease. Physical Chemistry Chemical Physics 2016, 18, 2330–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Raimundo AF; Ferreira S; Martins IC; Menezes R Islet Amyloid Polypeptide: A Partner in Crime With Aβ in the Pathology of Alzheimer’s Disease. Frontiers in Molecular Neuroscience 2020, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Berhanu WM; Hansmann UHE Inter-Species Cross-Seeding: Stability and Assembly of Rat - Human Amylin Aggregates. PLoS ONE 2014, 9, e97051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Pullman J; Darsow T; Frias JP Pramlintide in the management of insulin-using patients with type 2 and type 1 diabetes. Vascular health and risk management 2006, 2, 203–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Alrefai HA; Latif KA; Hieronymus LB; Weakley CR; Moss RJ Pramlintide: Clinical Strategies for Success. Diabetes Spectrum 2010, 23, 124–130. [Google Scholar]
- (20).Akter R; Cao P; Noor H; Ridgway Z; Tu L-H; Wang H; Wong AG; Zhang X; Abedini A; Schmidt AM; Raleigh DP Islet Amyloid Polypeptide: Structure, Function, and Pathophysiology. Journal of Diabetes Research 2016, 2016, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Chaudhury A; Duvoor C; Reddy Dendi VS; Kraleti S; Chada A; Ravilla R; Marco A; Shekhawat NS; Montales MT; Kuriakose K; Sasapu A; Beebe A; Patil N; Musham CK; Lohani GP; Mirza W Clinical Review of Antidiabetic Drugs: Implications for Type 2 Diabetes Mellitus Management. Frontiers in Endocrinology 2017, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).McQueen J Pramlintide acetate. American Journal of Health-System Pharmacy 2005, 62, 2363–2372. [DOI] [PubMed] [Google Scholar]
- (23).Nonoyama A; Laurence JS; Garriques L; Qi H; Le T; Middaugh C A Biophysical Characterization of the Peptide Drug Pramlintide (AC137) Using Empirical Phase Diagrams. Journal of Pharmaceutical Sciences 2008, 97, 2552–2567. [DOI] [PubMed] [Google Scholar]
- (24).da Silva DC; Fontes GN; Erthal LC; Lima LMT Amyloidogenesis of the amylin analogue pramlintide. Biophysical Chemistry 2016, 219, 1–8. [DOI] [PubMed] [Google Scholar]
- (25).Wang H; Ridgway Z; Cao P; Ruzsicska B; Raleigh DP Analysis of the ability of pramlintide to inhibit amyloid formation by human islet amyloid polypeptide reveals a balance between optimal recognition and reduced amyloidogenicity. Biochemistry 2015, 54, 6704–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Cardoso MH; Candido ES; Oshiro KGN; Rezende SB; Franco OL In Peptide Applications in Biomedicine, Biotechnology and Bioengineering; Koutsopoulos S, Ed.; Woodhead Publishing, 2018; pp 131–155. [Google Scholar]
- (27).Weinstock MT; Francis JN; Redman JS; Kay MS Protease-resistant peptide design—empowering nature’s fragile warriors against HIV. Peptide Science 2012, 98, 431–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Warso MA; Richards JM; Mehta D; Christov K; Schaeffer C; Rae Bressler L; Yamada T; Majumdar D; Kennedy SA; Beattie CW; Das Gupta TK A first-in-class, first-in-human, phase I trial of p28, a non-HDM2-mediated peptide inhibitor of p53 ubiquitination in patients with advanced solid tumours. British journal of cancer 2013, 108, 1061–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Shea D; Hsu C-C; Bi TM; Paranjapye N; Childers MC; Cochran J; Tomberlin CP; Wang L; Paris D; Zonderman J; Varani G; Link CD; Mullan M; Daggett V α-Sheet secondary structure in amyloid β-peptide drives aggregation and toxicity in Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America 2019, 116, 8895–8900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Xi W; Hansmann UHE The effect of retro-inverse D-amino acid Aβ-peptides on Aβ-fibril formation. The Journal of Chemical Physics 2019, 150, 095101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Wei L; Jiang P; Xu W; Li H; Zhang H; Yan L; Chan-Park MB; Liu X-W; Tang K; Mu Y; Pervushin K The Molecular Basis of Distinct Aggregation Pathways of Islet Amyloid Polypeptide. Journal of Biological Chemistry 2011, 286, 6291–6300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Jayasinghe SA; Langen R Identifying Structural Features of Fibrillar Islet Amyloid Polypeptide Using Site-directed Spin Labeling. Journal of Biological Chemistry 2004, 279, 48420–48425. [DOI] [PubMed] [Google Scholar]
- (33).Bedrood S; Li Y; Isas JM; Hegde BG; Baxa U; Haworth IS; Langen R Fibril Structure of Human Islet Amyloid Polypeptide. Journal of Biological Chemistry 2012, 287, 5235–5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Kajava AV; Aebi U; Steven AC The Parallel Superpleated Beta-structure as a Model for Amyloid Fibrils of Human Amylin. Journal of Molecular Biology 2005, 348, 247–252. [DOI] [PubMed] [Google Scholar]
- (35).Luca S; Yau W-M; Leapman R; Tycko R Peptide conformation and supramolecular organization in amylin fibrils: constraints from solid-state NMR. Biochemistry 2007, 46, 13505–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Wang L; Middleton CT; Singh S; Reddy AS; Woys AM; Strasfeld DB; Marek P; Raleigh DP; de Pablo JJ; Zanni MT; Skinner JL 2DIR Spectroscopy of Human Amylin Fibrils Reflects Stable β-Sheet Structure. Journal of the American Chemical Society 2011, 133, 16062–16071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Wiltzius JJ; Sievers SA; Sawaya MR; Cascio D; Popov D; Riekel C; Eisenberg D Atomic structure of the cross-β spine of islet amyloid polypeptide (amylin). Protein Science 2008, 17, 1467–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Bernhardt NA; Berhanu WM; Hansmann UHE Mutations and Seeding of Amylin Fibril-Like Oligomers. The Journal of Physical Chemistry B 2013, 117, 16076–16085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Abraham MJ; Murtola T; Schulz R; Páll S; Smith JC; Hess B; Lindahl E GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1-2, 19–25. [Google Scholar]
- (40).Páll S; Abraham MJ; Kutzner C; Hess B; Lindahl E In Solving Software Challenges for Exascale; Markidis S, Laure E, Eds.; Springer International Publishing: Cham, 2015; pp 3–27. [Google Scholar]
- (41).Huang J; MacKerell AD CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. Journal of Computational Chemistry 2013, 34, 2135–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Lindorff-Larsen K; Piana S; Palmo K; Maragakis P; Klepeis JL; Dror RO; Shaw DE Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins: Structure, Function, and Bioinformatics 2010. , 78, NA–NA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML Comparison of simple potential functions for simulating liquid water. The Journal of Chemical Physics 1983, 79, 926–935. [Google Scholar]
- (44).Darden T; York D; Pedersen L Particle mesh Ewald: An N . log( N ) method for Ewald sums in large systems. The Journal of Chemical Physics 1993, 98, 10089–10092. [Google Scholar]
- (45).Essmann U; Perera L; Berkowitz ML; Darden T; Lee H; Pedersen LG A smooth particle mesh Ewald method. The Journal of Chemical Physics 1995, 103, 8577–8593. [Google Scholar]
- (46).Bussi G; Donadio D; Parrinello M Canonical sampling through velocity rescaling. The Journal of Chemical Physics 2007, 126, 014101. [DOI] [PubMed] [Google Scholar]
- (47).Parrinello M; Rahman A Polymorphic transitions in single crystals: A new molecular dynamics method. Journal of Applied Physics 1981, 52, 7182–7190. [Google Scholar]
- (48).Man VH; He X; Derreumaux P; Ji B; Xie X-Q; Nguyen PH; Wang J Effects of All-Atom Molecular Mechanics Force Fields on Amyloid Peptide Assembly: The Case of Aβ 16–22 Dimer. Journal of Chemical Theory and Computation 2019, 15, 1440–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Ghosh A; Pithadia AS; Bhat J; Bera S; Midya A; Fierke CA; Ramamoorthy A; Bhunia A Self-Assembly of a Nine-Residue Amyloid-Forming Peptide Fragment of SARS Corona Virus E-Protein: Mechanism of Self Aggregation and Amyloid-Inhibition of hIAPP. Biochemistry 2015, 54, 2249–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Kouza M; Co NT; Li MS; Kmiecik S; Kolinski A; Kloczkowski A; Buhimschi IA Kinetics and mechanical stability of the fibril state control fibril formation time of polypeptide chains: A computational study. The Journal of Chemical Physics 2018, 148, 215106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.