

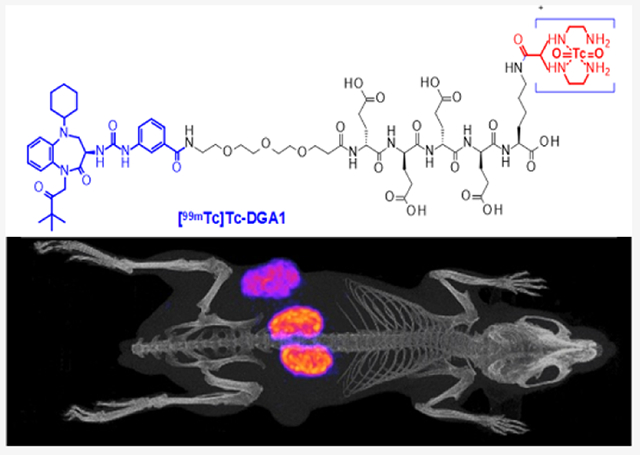
Abstract
Radiolabeled gastrin analogues have been proposed for theranostics of cholecystokinin subtype 2 receptor (CCK2R)-positive cancer. Peptide radioligands based on other receptor antagonists have displayed superior pharmacokinetics and higher biosafety than agonists. Here, we present DGA1, a derivative of the nonpeptidic CCK2R antagonist Z-360 carrying an acyclic tetraamine, for [99mTc]Tc labeling. Preclinical comparison of [99mTc]Tc-DGA1 with [99mTc]Tc-DG2 (CCK2R-agonist reference) was conducted in HEK293-CCK2R/CCK2i4svR cells and mice models, qualifying [99mTc]Tc-DGA1 for further study in patients with CCK2R-positive tumors and single-photon emission computed tomography/CT.
Keywords: molecular imaging, [99mTc]Tc radiotracer, tumor single-photon emission computed tomography imaging, CCK2 receptor, CCK2R antagonist
Graphical Abstract
INTRODUCTION
The cholecystokinin subtype 2 receptor (CCK2R) has been regarded as an attractive molecular target for directing diagnostic and therapeutic radionuclide carriers on tumor sites.1–4 High density and high incidence of CCK2R expression have been reported in medullary thyroid cancer (MTC).5 Overexpression of CCK2R has also been documented in other malignancies, including small-cell lung cancer, tumors of the gastrointestinal system, and stromal ovarian cancer.6–12 A fair number of suitably tailored CCK/gastrin-based radioligands have been developed as candidates in the theranostic management of such CCK2R-expressing cancers, with a few reaching the stage of clinical validation.13–15 Thus far, these agents have been based on peptide CCK2R agonists. As expected, after injection to patients, they bind and activate the CCK2R, eliciting a series of adverse effects that resemble those of the pentagastrin test.16 These effects depend upon the potency and injected dose of each analogue. Side effects may become a major hurdle in the clinical application of agonist-based radioligands, especially in the case of radionuclide therapy, whereby higher peptide amounts need to be administered to patients.
Recently, a shift of paradigm from receptor agonists to receptor antagonists has occurred in the field of radiopeptide development for cancer theranostics.17,18 Radiolabeled receptor antagonists, despite their inability to internalize, have often shown superior pharmacokinetic profiles compared to their agonist counterparts, characterized by higher tumor localization and faster background clearance even from tissues physiologically expressing their cognate receptors.19 Several examples from the somatostatin and bombesin peptide families have confirmed these observations. As a result, a number of theranostic radioligands based on SST2 or GRPR antagonists are currently under clinical evaluation.20–24 However, similar efforts toward the development of radiolabeled CCK2R antagonists seem to be in their infancy so far.
A wide range of CCK2R antagonists reported in the literature could serve as motifs for the development of the respective radioligands, with only very few being peptides. In most cases, diverse classes of small-size organic peptidomimetics with the antagonistic profile at the CCK2R have been proposed for a number of clinical indications.25–28 The two most prominent candidates, currently undergoing clinical testing, were the benzodiazepine derivatives netazepide (YF476)29–31 and nastorazepide (Z-360).32–38 Our interest was particularly triggered by Z-360, allowing the successful conjugation of (i) a [99mTc]Tc-chelate for molecular imaging of CCK2R-expressing tumors with single-photon emission computed tomography (SPECT)/CT,39 (ii) a fluorescent near-infrared dye for guided biopsy and surgery of CCK2R-positive lesions,40 and (iii) delivery of cytotoxic drugs to CCK2R-positive cancer cells with high specificity.41 These findings have verified that the Z-360 motif could be indeed structurally modified to accommodate appropriate entities for a number of theranostic applications and have strongly motivated the present work.
For this purpose, we have attached an acyclic tetraamine chelator, previously shown to facilitate the formation of several classes of peptide radioligands with the eminent diagnostic SPECT radionuclide technetium-99m,42 to Z-360 via a nonmetabolizable PEG3-(DGlu)4 spacer. This spacer was expected to enhance hydrophilicity and background clearance of the end radiotracer without deteriorating receptor affinity. Previous studies on (DGlu)5-gastrin radioligands, as for example, the CCK2R agonist-based [111In]In-CP04,15,43 showed excellent CCK2R affinity, high tumor targeting, and rapid background clearance in preclinical models and in man. Further benefits of selecting a nonpeptidic CCK2R antagonist for clinical use would be higher biosafety and resistance to degrading peptidases, such as neprilysin (neutral endopeptidase; NEP), an ecto-peptidase involved in the rapid in vivo catabolism of CCK and gastrin.44–46 In order to confirm the abovementioned expected advantageous qualities, we conducted head-to-head comparison of DGA1 and DG2 (a CCK2R-agonist reference)13,14 and their end [99mTc]Tc radiotracers, [99mTc]Tc-DGA1 and [99mTc]Tc-DG2, at the cellular level and in mice models. To the best of our knowledge, this is the first direct comparison of a CCK2R antagonist radioligand with an agonist-based reference and is expected to further boost the search for safe and effective CCK2R-directed theranostic agents.
MATERIALS AND METHODS
Chemicals and Instrumentation.
Compounds and Radionuclides.
Fmoc-(9-fluorenylmethoxycarbonyl)-protected amino acid analogues and PEG3 were purchased from Iris Biotech (Marktredwitz, Germany) or Bachem (Bubendorf, Switzerland). 2-Chloro-trityl-chloride polystyrene resin was obtained from Rapp Polymers (Tubingen, Germany). The protected chelator N4(tBoc)4-NHS was sourced from piCHEM (Graz, Austria). Nastorazepide (Z-360) was bought from MedKoo Biosciences, Inc., USA. Solvents and all other organic reagents were purchased from Sigma-Aldrich (Vienna, Austria). Demogastrin 2 (DG2, N4-Gly-DGlu-(Glu)5-Ala-Tyr-Gly-Trp-Met-Asp-Phe-NH2; N4 = 6-carboxy-1,4,8,11-tetraazaundecane) was prepared as previously reported.13 Human [Leu15]gastrin-17 (pGlu-Gly-Pro-Trp-Leu-(Glu)5-Ala-Tyr-Gly-Trp-Leu-Asp-Phe-NH2) used for radioiodination was obtained from Bachem (Bubendorf, Switzerland).
For radiolabeling, [99mTc]TcO4− was eluted in normal saline from a commercial [99Mo]Mo/[99mTc]Tc generator (Ultratechnekow, Tyco Healthcare, Petten, The Netherlands), while [125I]NaI in a 10−5 M NaOH solution (pH 8–10, 185 MBq in ≅ 10 μL) was purchased from Perkin Elmer.
Instrumentation.
Solid-phase peptide synthesis (SPPS) was carried out using a SYRO-II (Biotage Uppsala, Sweden). Analytical reversed-phase high-performance liquid chromatography (RP-HPLC) was performed on an Agilent 1100 low-pressure gradient HPLC system with a UV detector (Agilent, Waldbronn, Germany) using a Nucleosil 100 C18 (5 μm, 150 × 4.0 mm) column (Macherey-Nagel, Duren, Germany). The products were analyzed applying different gradients of 0.1% (v/v) trifluoroacetic acid (TFA) in H2O (solvent A) and 0.1% TFA (v/v) in acetonitrile (solvent B) at a constant flow of 1 mL/min at UV 225 nm (system 1). Preparative RP-HPLC was performed on a high-pressure gradient semi-preparative. HPLC system LC-8A with a Shimadzu UV detector SPD-20A using a Nucleodur C18 HTec (7 μm, 250 × 21 mm) column (Macherey-Nagel, Duren, Germany) at a constant flow of 12 mL/min. The products and intermediates were purified using different gradients of 0.1% (v/v) TFA in H2O (solvent A) and 0.1% TFA (v/v) in acetonitrile (solvent B) at UV 220 nm. For purified DGA1 (Z-360-PEG3-DGlu-DGlu-DGlu-DGu-Lys(N4)-OH; Scheme 1), UV detection at 215 nm was applied, and the abovementioned analytical Nucleosil C18 column was eluted with the following gradient: B 0.1% TFA in MeCN 10% B to 90% B in 50 min. Matrix-assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF-MS) spectra were acquired on an Axima Assurance mass spectrometer (Kratos; Manchester, UK). Further HPLC analysis of DGA1 was conducted on a Waters chromatograph with a 600 solvent delivery system coupled to a 996 photodiode array UV detector; Millennium software was used for data processing and for controlling the HPLC system. A Waters RP-18 XTerra column (5 μm, 3.9 mm × 20 mm) was eluted at a 1.0 mL/min flow rate with the following gradient: 0% B to 50% B in 50 min, where A = 0.1% aq. TFA and B = MeCN (system 2). An automated well-type gamma counter [NaI(Tl) 3” crystal, Canberra Packard Auto-Gamma 5000 series instrument] calibrated for technetium-99m or iodine-125 was used for radioactivity measurements.
Scheme 1.
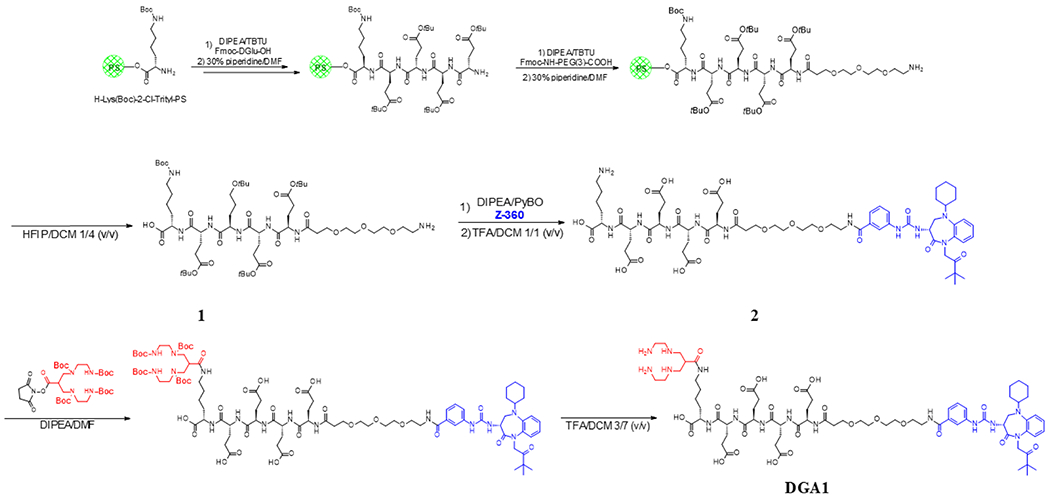
Flow Chart for the Synthesis of DGA1 Adopting a Mixed Mode Strategy of SPPS and LPPS
Synthesis of DGA1.
Synthesis of 1.
For the synthesis of 1 (H-PEG3-DGlu(OtBu)-DGlu(OtBu)-DGlu(Ot Bu)-DGu-(OtBu)-Lys(Boc)-OH), H-Lys(Boc)-2Cl-Trityl-PS resin was used with a substitution of>0.52 mmol/g as a starting material. Coupling of the consecutive amino acids was achieved by applying a 10-fold excess of Fmoc amino acid derivatives in relation to the resin loading in DMF in the presence of 10 equiv of 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium tetrafluoroborate (TBTU) and 10 equiv of (N,N-diisopropy-lethylamine) DIPEA for 40 min at ambient temperature. Coupling of Fmoc–NH–PEG3-COOH (3-fold excess) was accomplished with 3 equiv of benzotriazol-1-yloxytris-(pyrrolidino)phosphonium hexafluorophosphate (PyBOP) and 5 equiv of DIPEA in DMF. Fmoc deprotection was carried out by treatment with a solution of 30% piperidine in DMF 3 × 5 min followed by five times DMF wash. Finally, release of 1 from the resin was achieved by treatment with a 1/4 v/v mixture of 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) and dichloromethane (DCM) for 15 min at ambient temperature. After solvent evaporation, crude 1 was purified by preparative RP-HPLC. Pure fractions identified by liquid chromatography-mass spectroscopy (LC-MS) were combined and freeze-dried to furnish 1 (Supporting Information, Figures S1 and S2).
Synthesis of 2.
For the synthesis of 2 (Z-360-PEG3-DGlu-DGlu-DGlu-DGu-Lys-OH), Z-360 was coupled with 2 equiv of 1 by the addition of 2 equiv of PyBOP and 5 equiv of DIPEA in DMF. The conversion was monitored by HPLC and MS (Supporting Information, Figures S3 and S4, respectively). After 4 h of reaction time, the coupling step was completed and the solvent was evaporated, and the residue was redissolved in 30% MeCN and freeze-dried. Finally, all protecting groups were removed by treatment of crude 2 with TFA/DCM in a ratio of 50:50 v/v at ambient temperature. The deprotection step was monitored by LC-MS. The reaction mixture was evaporated to dryness, redissolved in 30% MeCN, and purified by preparative RP-HPLC. Pure fractions (Supporting Information, Figures S5 and S6) were pooled and freeze-dried to yield 2.
Synthesis of DGA1.
For the synthesis of DGA1 (Z-360-PEG3-DGlu-DGlu-DGlu-DGu-Lys(N4)-OH), 2 was dissolved in a mixture of DMF and DIPEA (10 equiv), and a slurry of N4(tBoc)4-NHS (4 equiv) in DMF was added. The reaction mixture was stirred at room temperature for 24 h following the progress of the conversion by LC-MS. After evaporation of the solvents (Supporting Information, Figure S7), the tBoc groups were cleaved by treatment of the residue with a mixture of TFA and DCM in a ratio of 30:70 v/v at ambient temperature with the tBoc removal monitored by LC-MS. The reaction mixture was dried and redissolved in 20% MeCN, and DGA1 was isolated by preparative RP-HPLC (Supporting Information, Figure S8A,B). MALDI-TOF-MS confirmed the formation of DGA1 (calculated MW: 1554.74, found: 1555.1; Supporting Information, Figure S9), and analytical HPLC revealed a purity of the desired conjugate exceeding 96% (Supporting Information, system 1: single peak at tR = 15.3 min/system 2: single peak at tR = 31.0 min; Figure S8B,C, respectively).
Radiolabeling and Quality Control.
Preparation and Analysis of [99mTc]Tc-DGA1 and [99mTc]Tc-DG2.
Lyophilized DGA1 was dissolved in bidistilled water and DG2 in 20 mM NaHCO3, both to a concentration of 2 mg/mL; solutions thereof were stored at −20 °C in 50 μL aliquots. For [99mTc]Tc labeling, the following solutions were added into an Eppendorf tube containing 0.5 M phosphate buffer (pH 11.5; 25 μL): 0.1 M sodium citrate (3 μL), [99mTc]Tc-NaTcO4 (210 μL, 140–280 MBq) generator eluate, DGA1 or DG2 stock solution (7.5 μL, 7.5 nmol), and finally fresh SnCl2 solution in EtOH (5 μg, 5 μL). After 30 min of incubation at ambient temperature, the reaction mixture was neutralized by the addition of 1 M HCl (4 μL), and EtOH was added (25 μL); in the case of DG2, methinone (2 mM) was added to suppress the oxidation of Met15 to the respective sulfoxide(s).13 Quality control comprised radioanalytical HPLC and instant thin-layer chromatography (ITLC). HPLC analyses were performed on a Waters chromatograph coupled to a 2998 photodiode array UV detector (Waters, Vienna, Austria) and a Gabi gamma detector (Raytest RSM Analytische Instrumente GmbH, Germany). A Waters Symmetry Shield RP-18 column (5 μm, 3.9 mm × 20 mm) was eluted at a 1.0 mL/min flow rate with the following gradient: 0% B to 60% B in 20 min, where A = 0.1% aq. TFA B = MeCN (system 3). Under these conditions, [99mTc]TcO4−, [99mTc]Tc-DG2, and [99mTc]Tc-DGA1 eluted at 0.9 min, 12.6 min, and 15.4 min, respectively (Supporting Information, Figure S10). For the detection of reduced hydrolyzed technetium ([99mTc]TcO2-xH2O), ITLC was conducted on ITLC-SG strips (Pall Corporation, NY/USA), as previously described.13
Preparation of [125I][I-Tyr12,Leu15]gastrin-l7.
Radioiodination of [Leu15]gastrin-17 was performed according to the chloramine-T method, and [125I][I-Tyr12,Leu15]gastrin was isolated in high purity by HPLC. Aliquots of the radioligand stock solution in 0.1% BSA-PBS buffer were kept at −20 °C and were used in competition binding experiments (molar activity of 74 GBq/μmol).53,57
All manipulations with gamma and beta emitting radionuclides and their solutions were performed behind sufficient shielding by trained personnel in facilities supervised by the Greek Atomic Energy Commission and in compliance to national and international radiation safety guidelines.
Cells and Cell Culture.
For in vitro assays that monitor ligand binding or internalization and for tumor induction in animals, we used HEK293 cells transfected to stably express the human CCK2R or the CCK2i4svR splice variant, which were a kind gift from Dr. P. Laverman (Randbound University Nijmegen Medical Center, Nijmegen, The Netherlands) and Prof. M. R. Hellmich (University of Texas Medical Branch, Galverston, TX, USA).47–49 Nontransfected HEK293T cells were used as negative controls. Transfected HEK293 cells were grown in Dulbecco’s modified Eagle medium (DMEM) with GlutaMAX-I supplemented with 10% (v/v) fetal bovine serum (FBS), 100 U/mL penicillin, 100 μg/mL streptomycin, and 400 μg/mL G418. Control HEK293T cells were cultured in DMEM with GlutaMAX-I supplemented with 10% (v/v) FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin. All culture media were purchased from Gibco BRL, Life Technologies (Grand Island, NY, USA), and supplements were supplied by Biochrom KG Seromed (Berlin, Germany). Cells were kept in a controlled humidified air containing 5% CO2 at 37 °C. Splitting of cells with a ratio of 1:3 to 1:5 was performed when approaching confluency using a trypsin/EDTA solution (0.05%/0.02% w/v).
For in vitro assays, to determine [Ca2+], HEK293 cells obtained from the American Type Culture Collection (Cat. no. CRL-1573; Manassas, VA) were transfected to transiently express the human CCK2R. The cells were maintained in DMEM media (Thermo Fisher Scientific) containing 25 mM glucose and supplemented with 10% FBS and 1% penicillin-streptomycin. The human CCK2R cDNA (NM_176875) in plasmid pCMV6-Entry was obtained from OriGene Technologies, Inc. (Cat. no. RC200676; Rockville, MD, USA). This plasmid was introduced into HEK293 cells by transfection using Lipofectamine and Plus reagent (Thermo Fisher Sci.). Resultant HEK293-hCCK2R cells were maintained in DMEM cell culture medium. (Thermo Fisher Sci.) containing 25 mM glucose and supplemented with 10% FBS and 1% penicillin— streptomycin.
In Vitro Assays
Receptor Agonism/Antagonism Assays of DGA1 and DG2.
HEK293 cells transfected to transiently express the human CCK2R and grown in monolayers were harvested by trypsinization. Aliquots of the resulting single-cell suspension were plated onto rat tail collagen-coated 96-well Costar 3904 plates for fura-2-based measurements of [Ca2+], using a Flexstation 3 microplate reader in the kinetic mode (Molecular Devices, Sunnyvale, CA).50 The composition of the standard extracellular saline solution (SES) and fura-2 acetoxymethyl ester loading solution (fura-2 AM) were as described previously.50 Test solutions were administered using the automated pipetting feature of the Flexstation under computer control. Spectrofluorimetry was performed using excitation light at 335/9 and 375/9 nm (center/bandpass wavelengths) delivered using a 455 nm dichroic mirror.50 Emitted light was detected at 505/15 nm, and the ratio of emission light intensities due to excitation at 335 and 375 nm was calculated.50 Raw data were exported to Origin v7.5 (Origin Lab., Northhampton, MA) for processing. The percent change in fura-2 ratio (Δ340/380) is illustrated after baseline subtraction so that a value of 0.5 indicates a 50% increase in ratio. The repeatability of findings was confirmed by performing all experiments a minimum of three times.
Competition Binding Assays of DGA1 and DG2 in HEK293-CCK2i4svR Cell Membranes.
Competition binding experiments of DGA1 and DG2 against [125I][I-Tyr12,Leu15]-gastrin were conducted in HEK293-CCK2i4svR cell membranes, harvested as previously described;13 unmodified Z-360 served as the reference compound. Increasing concentrations of the test compound (10−13 to 10−5 M) were mixed with the radioligand (50 pM, ~30,000 cpm) and the membrane homogenate in a total volume of 300 μL binding buffer (pH 7.4, 50 mM HEPES, 1% BSA, 5.5 mM MgCl2, 35 pM bacitracin). Triplicates of each concentration point were incubated for 60 min at 22 °C in an incubator-orbital shaker unit (MPM Instr. SrI, Italy). The incubation was interrupted by adding ice-cold washing buffer (10 mM HEPES pH 7.4, 150 mM NaCl), followed by rapid filtration over glass fiber filters (Whatman GF/B, presoaked in binding buffer) on a Brandel Cell Harvester (Adi Hassel Ingenieur Buro, Munich, Germany). Filters were washed with cold washing buffer and were counted for their radioactivity content in the gamma counter. The 50% inhibitory concentration (IC50) values were calculated by nonlinear regression according to a one-site model applying the PRISM 6.0 program (Graph Pad Software, San Diego, CA) and were expressed as mean ± SD of three experiments performed in triplicate.
Cell Binding and Internalization of [99mTc]Tc-DGA1 and [99mTc]Tc-DG2.
For time-dependent internalization studies with [99mTc]Tc-DGA1 and [99mTc]Tc-DG2, HEK293-CCK2i4svR, HEK293-CCK2R, or HEK293T (negative control) cells were seeded in six-well plates 24 h before the experiment. Cells were rinsed twice with ice-cold internalization medium (DMEM Glutamax-I, supplemented by 1% (v/v) FBS), and then, fresh medium was added (1.2 mL) at 37 °C, followed by test radiopeptide (200 fmol total peptide in 150 μL 0.5% BSA-PBS, 100,000—200,000 cpm). Nonspecific internalization was determined by a parallel triplicate series containing 1 μM DG2. Radiotracer incubation in HEK293-CCK2i4svR or HEK293-CCK2R cells at 37 °C was interrupted at 15, 30, 60, or 120 min by placing the plates on ice, removal of the medium, and washing with 0.5% BSA-PBS; incubation in control HEK293T cells was interrupted only at 1 h. Membrane-bound fractions were collected by incubating the cells 2 × 5 min in acid-wash solution (2 × 0.6 mL, 50 mM glycine buffer pH 2.8, 0.1 M NaCl) at room temperature. Cells were subsequently rinsed with 0.5% BSA—PBS, aspirated and lysed by the addition of 1 M NaOH (2 × 0.6 mL); lysates representing internalized fractions were collected. Samples were measured for radioactivity in the gamma counter, and the percentage of specific internalized and membrane-bound fractions was calculated with Microsoft Excel by subtracting the nonspecific values (obtained in the presence of 1 μM DG2) from totals. Results represent specific internalized±SD of total added radioactivity per well from at least three experiments performed in triplicate.
In Vivo Tests
Metabolic Stability of [99mTc]Tc-DGA1 and [99mTc]Tc-DG2 in Mice.
A bolus containing [99mTc]Tc-DGA1 or [99mTc]Tc-DG2 (100 μL, 55.5—111 MBq, 3 nmol of the total peptide in vehicle: saline/EtOH 9/1 v/v) was injected in the tail vein of healthy male Swiss Albino mice (30 ± 5 g, NCSR “Demokritos” Animal House Facility). Animals were euthanized 5 min post injection (pi), and blood was collected in prechilled polypropylene vials containing EDTA and Met placed on ice. Samples were centrifuged at 2000g at 4 °C for 10 min, and plasma was collected and mixed with an equal volume of ice-cold MeCN and centrifuged again for 10 min at 15,000g at 4 °C. The supernatant was collected and concentrated to a small volume (≈0.05 mL) under a gentle N2 flux at 50 °C, diluted with physiological saline (≈400 μL), and filtered through a Millex GV filter (0.22 μm, Ø13 mm). Suitable aliquots of the filtrate were analyzed by RP-HPLC on a Symmetry Shield RP18 (5 μm, 3.9 mm × 20 mm) column (Waters, Germany) eluted at a flow rate of 1 mL/min and adopting gradient system 4: 100% A/0% B to 50% A/50% B in 50 min; A = 0.1% TFA in H2O and B = MeCN. The elution time (tR) of the intact radioligand was determined by coinjection with a sample of the labeling reaction solution. Experiments were repeated three times.
In separate animal groups, a 100 μL bolus containing [99mTc]Tc-DGA1 or [99mTc]Tc-DG2 (100 μL, 55.5–111 MBq, 3 nmol of the total peptide in vehicle: saline/EtOH 9/1 v/v) was injected in the tail vein of healthy male Swiss Albino mice. After 30 min, animals were sacrificed, and the urine was collected from their bladder with a syringe in prechilled polypropylene vials containing EDTA and Met placed on ice. Samples were centrifuged at 35,000 rpm for 10 min, and aliquots of the supernatant (containing >95% of total urine activity) were analyzed by HPLC under the conditions described above.
Biodistribution of [99mTc]Tc-DGAI and [99mTc]Tc-DG2 in Tumor-Bearing Mice.
A ~150 μL bolus containing a suspension of freshly harvested HEK293-CCK2i4svR or HEK293-CCK2R (1.8 × 107) cells in normal saline was subcutaneously inoculated in the left flanks of 6 week-old male SCID mice (NCSR “Demokritos” Animal House, 18 ± 2 g body weight); the respective HEK293T inocula (1.5 × 107 cells in normal saline) were implanted on the right flanks of the animals to serve as negative controls. After approximately 3 weeks, well palpable HEK293-CCK2i4svR or HEK293-CCK2R tumors (200 ± 130 mg) were developed at the inoculation sites, and biodistribution was conducted as follows. Animals were injected in the tail vein with a bolus containing [99mTc]Tc-DGA1 or [99mTc]Tc-DG2 (100 μL, 148–185 kBq, 10 pmol of the total peptide, in vehicle: saline/EtOH 9/1 v/v) and were euthanized in groups of at least four at 1, 4, and 24 h pi; gelofusine (100 μL) was coinjected in additional 4 h pi groups of mice bearing HEK293-CCK2i4svR tumors. Mice were dissected, and blood samples, organs of interest, and tumors were collected, weighted, and counted in the gamma counter. Biodistribution data were calculated as the percent of injected dose per gram tissue (%ID/g) with the aid of suitable standards of the injected dose, using the Microsoft Excel program. Results represent average values ± SD, n ≥ 4.
Statistical Analysis.
For statistical analysis of biodistribution results, the two-way ANOVA with multiple comparisons was used applying Tukey’s post hoc analysis (GraphPad Prism Software, San Diego, CA). P values of <0.05 were considered to be statistically significant.
SPECT/CT Imaging.
For SPECT/CT imaging, four mice bearing twin HEK293-CCK2i4svR and HEK293T tumors were injected in the tail vein with a bolus containing [99mTc]Tc-DGA1 (100 μL, 37 MBq of [99mTc]Tc-DGA1 associated with 1.5 nmol of DGA1, in vehicle: saline/EtOH 9/1 v/v) together with vehicle (100 μL; controls) or with gelofusine (100 μL; Gelo) and were euthanized at 4 h pi. Tomographic SPECT/CT imaging was performed with the y-CUBE/x-CUBE systems (Molecubes, Belgium). The SPECT system is based on monolithic NaI detectors attached to SiPMs, with a 0.6 mm intrinsic resolution. The CT system is based on a structured CMOS detector of CsI with pixels of 75 gm and operates between 35 and 80 kVp and 10–500 μA tube current, with a 33 μm fixed focal spot size. SPECT scans were acquired at 4 h pi, with a 40 min duration protocol based on the injected activity, and each SPECT scan was succeeded by a CT scan, following a general-purpose protocol under 50 kVp, for coregistration purposes. SPECT images were reconstructed using the MLEM reconstruction method with a 250 μm voxel size & 500 iterations. CT images were reconstructed using the ISRA reconstruction method with a 100 μm voxel size.
Images were exported and postprocessed on VivoQuant software, version 4.0 (Invicro, Boston). A smoothing median filter (0.6 mm, spherical window) was applied to the images, and the bladder was removed for consistency purposes. Normalization of images was performed on tumor uptake (i.e., tumors having the same maximum number of counts), to achieve a direct visual comparison between control and experimental groups.
All animal experiments were carried out in compliance with European and national regulations and after approval of protocols by national authorities.
RESULTS
Synthesis of DGA1.
Synthesis was performed by a mixed mode strategy of SPPS and liquid-phase peptide synthesis (LPPS) using the Fmoc-protection methodology, as summarized in Scheme 1. The peptide including the PEG3 spacer (H-PEG3-[DGlu(OtBu)]4-Lys(Boc)-OH) was assembled on a 2-chlor-trityl resin followed by HFIP cleavage of the protected fragment and subsequent RP-HPLC purification. Nonmodified Z-360 was coupled in solution to the N-terminus of the PEG3 spacer followed by TFA deprotection and RP-HPLC purification. Finally, the protected N4 chelator (N,N′,N″,N‴- tetrakis-( t-butoxycarbonyl)-6-( carboxy)-1,4,8,11-tetraazaundecane) was attached to the lysine side chain; after removal of all lateral protecting groups, the DGA1 product was obtained by chromatographic purification and freeze drying. MALDI-TOF-MS data were consistent with the expected formula, whereas analytical HPLC verified a >96% purity (Supporting Information, Figures S1–S9).
Radiochemistry.
Radiolabeling of DGA1 (Scheme 2) and DG2 with technetium-99m was accomplished after 30 min of incubation at room temperature in an alkaline aqueous medium containing citrate anions and SnCl2. A >96% radiometal incorporation into the open-chain tetraamine chelator was verified by RP-HPLC analysis (Supporting Information, Figure S10). Radiopeptides were obtained at molar activities of 18–37 MBq/nmol in >96% radiochemical purity and hence were used as such in all subsequent biological experiments.
Scheme 2.
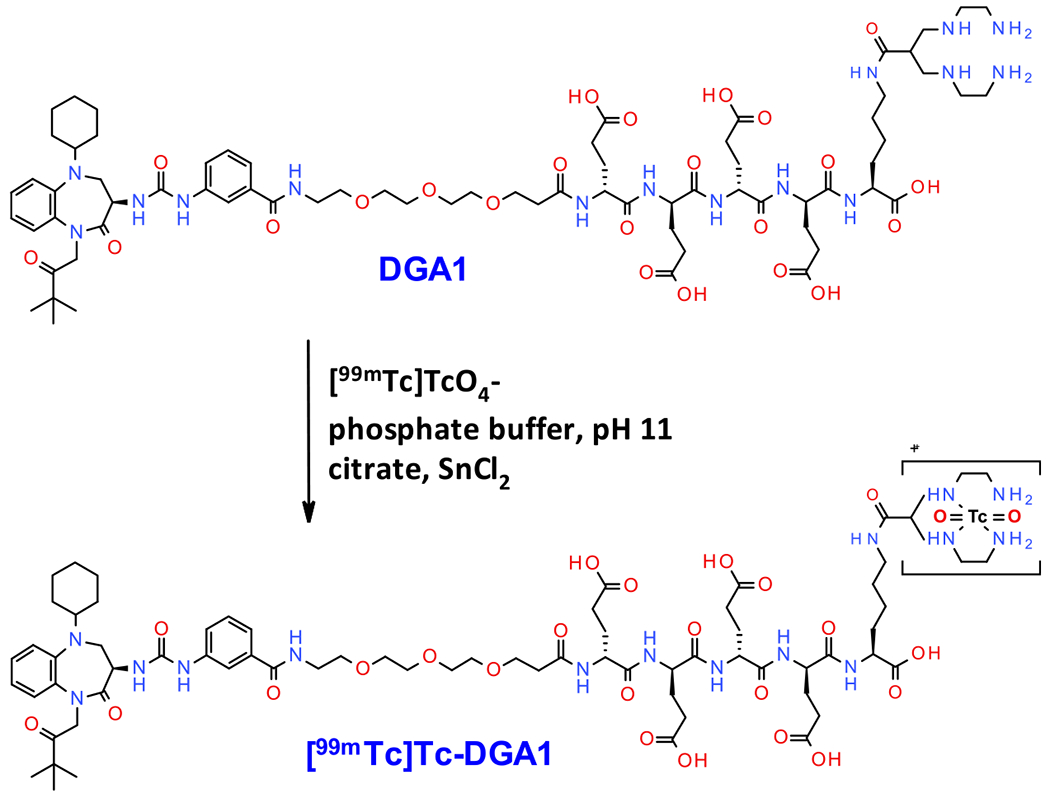
Labeling of DGA1 with Technetium-99m and Formation of [99mTc]Tc-DGA1
In Vitro Assays.
Agonism/Antagonism of DGA1 and DG2 at the hCCK2R.
The CCK2R agonist DG2 (1 pM-100 nM) acted in a dose-dependent manner to stimulate a transient increase in [Ca2+]i in HEK293-hCCK2R cells (Figure 1A). Linear regression analysis of this DG2 dose-response relationship established the EC50 and Hill co-efficient values to be 79 pM and 0.6, respectively (Figure 1B). No such agonist action of DG2 was measured for HEK293 cells not transfected with the CCK2R (Figure 1C), thereby establishing that this cell line does not express endogenous CCK2R for DG2. Still, these untransfected cells did respond to the nucleotide ADP (20 μM) that served as a positive control by virtue of its ability to stimulate endogenous purinergic receptors that are coupled to Ca2+ mobilization in HEK293 cells (Figure 1C).
Figure 1.
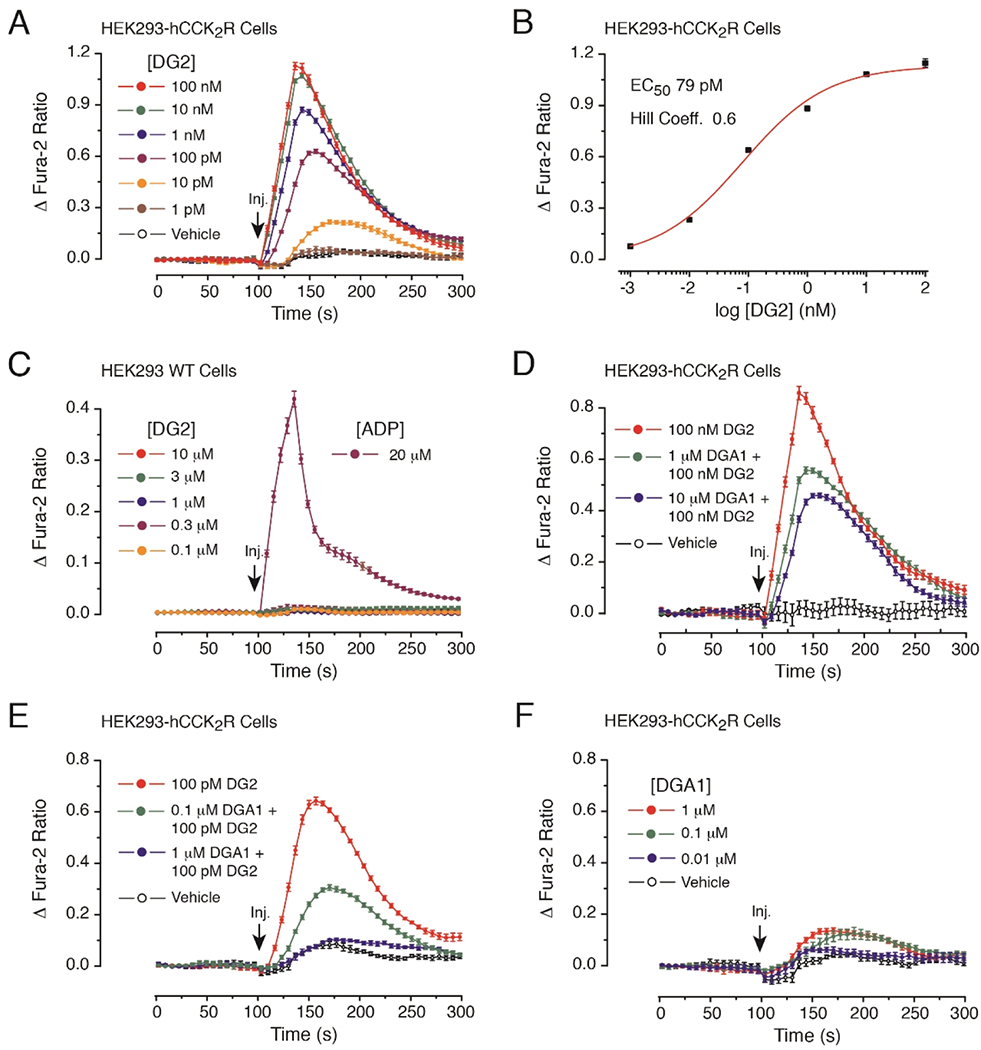
Antagonist properties of DGA1 at the CCK2R monitored in fura-2 assays of [Ca2+]i. (A) Dose-dependent action of the CCK2R agonist DG2 (1 pM-100 nM) to increase levels of [Ca2+]i in HEK293-hCCK2R cells. (B) Linear regression analysis of the dose response data illustrated in the panel (A). (C) DG2 (0.1–10 μM) fails to elevate levels of [Ca2+]i in untransfected HEK293 cells, whereas ADP (20 μM) does exert an agonist action. (D) Partial antagonism of 100 nM DG2 agonist action by 1 or 10 μM DGA1. (E) Full antagonism of 100 pM DG2 agonist action by 1 μM DGA1. (F) Little or no agonist action of DGA1 administered alone when tested at 0.01–1 μM. For panels (A-F), test solutions were administered at the 100 s time point. Each data point was sampled at 6 s intervals so that the time course of the change in fura-2 ratio could be measured over a 300 s time frame. Standard deviation (SD) error bars indicate the average of eight wells for each time point in a single experiment. Illustrations are representative examples obtained in n = 3 separate experiments.
We next demonstrated that DGA1 counteracted the agonist action of DG2, as measured in HEK293-hCCK2R cells. For this analysis, cells were pretreated for 20 min with DGA1 and then stimulated with a test solution that contained DG2 in combination with DGA1. It was established that the action of 100 nM DG2 was reduced by ca. 50% by pretreatment with 10 μM DGA1 (Figure 1D). Additional dose-response analysis was then performed using a lower concentration of DG2 (100 pM) that approximates its EC50 value in this assay. This revealed that the agonist action of 100 pM DG2 was partially inhibited (ca. 54%) by 0.1 μM DGA1, whereas DG2 agonist activity was fully suppressed by 1 μM DGA1 (Figure 1E). Importantly, when DGA1 (0.01–1 μM) was administered alone in the absence of DG2, the basal [Ca2+]; was barely altered (Figure 1F). It may be concluded that DGA1 exhibits antagonist properties to block DG2 agonist action at the CCK2R.
Affinities of DGA1 and DG2 for the HEK293-CCK2i4svR.
As shown in Figure 2, DGA1 and DG2 displaced [125I][I-Tyr12,Leu15]gastrin from CCK2i4svR sites on HEK293-CCK2i4svR cell membranes in a monophasic and dose-dependent manner. The antagonist DGA1 displayed equally high affinity for the CCK2i4svR (IC50 = 1.62 ± 0.17 nM) compared to the unmodified Z-360 reference (IC50 = 1.57 ± 0.14 nM), indicating that all structural modifications undertaken were well tolerated by the receptor. In comparison, the agonist DG2 showed less CCK2i4svR affinity (IC50 = 5.04 ± 0.29 nM) compared to either DGA1 or Z-360.
Figure 2.
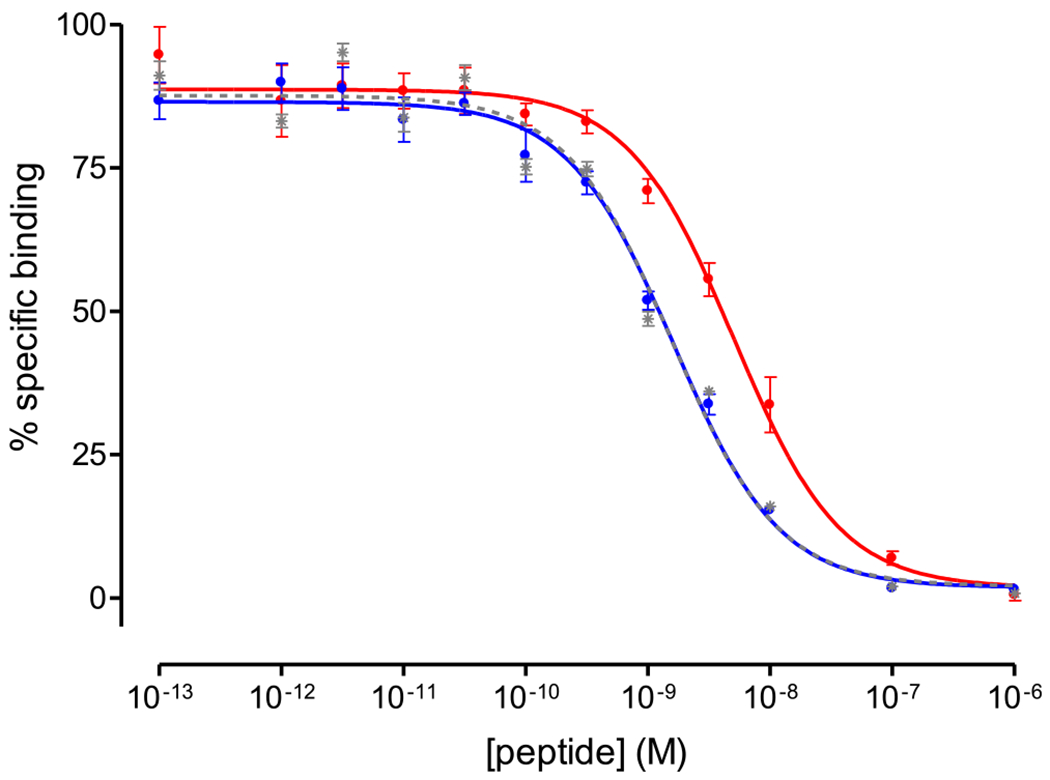
Displacement curves of [125I][I-Tyr12,Leu15]gastrin from CCK2i4svR sites on HEK293-CCK2i4svR cell membranes by increasing concentrations of Z-360 (gray; IC50 = 1.57 ± 0.14 nM), DGA1 (blue; IC50 = 1.62 ± 0.17 nM), and DG2 (red; IC50 = 5.04 ± 0.29 nM); results are given as mean IC50 values ± SD, n = 3.
Cell Binding—Internalization of [99mTc]Tc-DGA1 and [99mTc]Tc-DG2.
Time-dependent cell association and internalization rate curves for [99mTc]Tc-DGA1 and [99mTc]Tc-DG2 in HEK293-CCK2i4svR cells are shown in Figure 3A,B, respectively. As shown in the figure, the overall CCK2i4svR-specific uptake of the antagonist [99mTc]Tc-DGA1 was significantly lower than that of the agonist [99mTc]Tc-DG2 in all time points. For example, at 1 h of incubation at 37 °C, the total specific cell uptake for [99mTc]Tc-DGA1 reaches 20.8 ± 2.0% of total added radioactivity and is almost equally distributed in the membrane-bound (10.0 ± 1.1%) and the internalized (10.0 ± 1.1%; n = 3) fractions. The respective value for [99mTc]Tc-DG2 is 9 ± 0.4% of total added radioactivity. Notably, the major portion of the cell-associated activity is found within the cell (42.8 ± 0.7% internalized and 10.1 ± 0.3% membrane-bound; n = 3), as consistent with a radioagonist profile. Specific cell binding and internalization rate curves for [99mTc]Tc-DGA1 and [99mTc]-Tc-DG2 in HEK293-CCK2R cells are additionally presented in Figure S11A,B, respectively (Supporting Information), leading to similar observations. In all the abovementioned cases, nonspecific values did not exceed 1% of total added activity (representative graphs for [99mTc]Tc-DGA1 and [99mTc]Tc-DG2 in HEK293-CCK2i4svR cells at 1 h are shown in the Supporting Information, Figure S12). It should be noted that the specific cell uptake of [99mTc]Tc-DGA1 in wild-type HEK293T cells, devoid of CCK2R/CCK2i4svR expression, was negligible at 1 h of incubation at 37 °C (Supporting Information, Figure S12).
Figure 3.
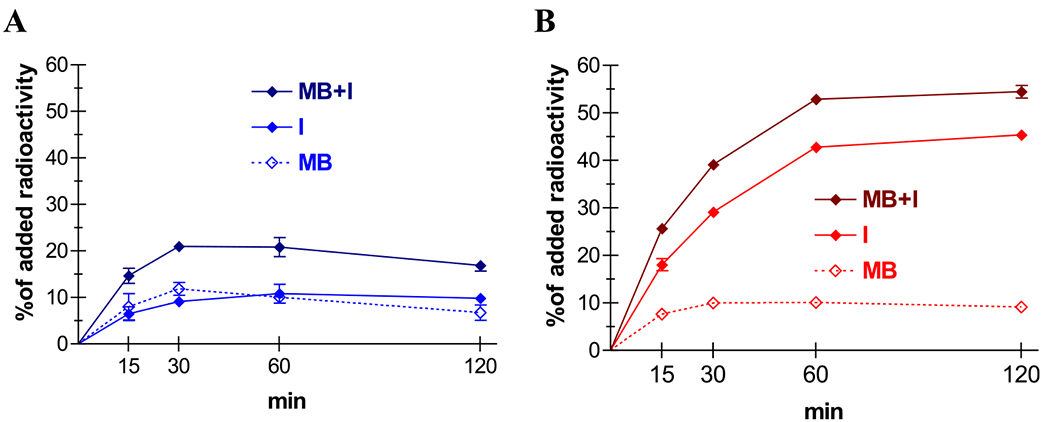
Specific cell association of (A) [99mTc]Tc-DGA1 (blue) and (B) [99mTc]Tc-DG2 (red) in HEK293-CCK2i4SVR cells at 37 °C; solid lines represent the internalized fractions (I), and scattered lines represent the membrane bound fractions (MB), whereas the darker solid lines represent specific total cell uptake (MB + I); results represent mean values ± SD, n = 3.
In Vivo Results
Metabolic Stability of [99mTc]Tc-DGA1 and [99mTc]Tc-DG2 in Mice.
[99mTc]Tc-DGA1 remained practically intact (97.0 ± 0.5% intact; n = 3) in peripheral mouse blood at 5 min pi, as opposed to [99mTc]Tc-DG2 that showed noticeable degradation within the same period in mouse circulation (54.4 ± 5.7% intact; n = 4). Representative radiochromatograms for the metabolic fate of [99mTc]Tc-DGA1 and [99mTc]Tc-DG2 in mouse circulation at 5 min pi are displayed in Figure 4A,B, respectively. Interestingly, radioactivity excreted in the urine at 30 min pi contained a considerable portion of intact [99mTc]Tc-DGA1 (>55% intact; Figure 4C).
Figure 4.
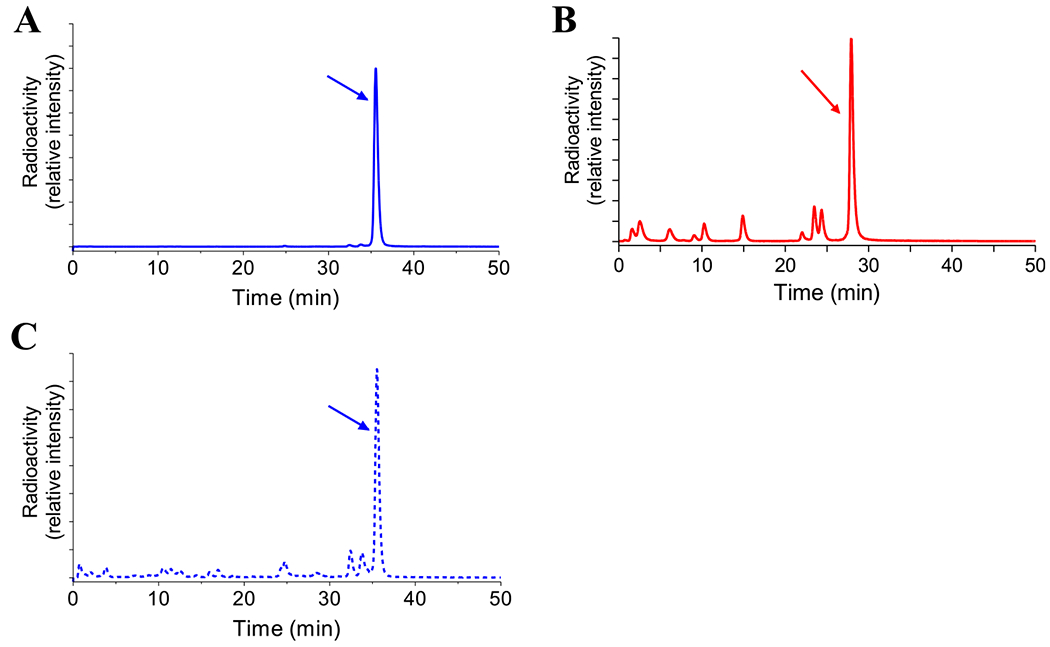
Representative radiochromatograms of metabolic patterns in peripheral mouse blood 5 min after injection of (A) [99mTc]Tc-DGA1 (blue line; 96.8% intact) and (B) [99mTc]Tc-DG2 (red line; 55.0% intact), revealing the superior metabolic stability of the receptor antagonist; (C) representative radiochromatogram of a mouse urine sample collected 30 min after [99mTc]Tc-DGA1, showing the major part of excreted radioactivity in the form of the intact radioligand (blue scattered line).
Biodistribution of [99mTc]Tc-DGA1 and [99mTc]Tc-DG2 in Tumor-Bearing Mice.
Biodistribution data for [99mTc]Tc-DGA1 and [99mTc]Tc-DG2 at 1, 4, and 24 h pi in animals bearing double HEK293-CCK2i4svR-positive and HEK293-CCK2i4svR-negative tumors are summarized in Table 1; results are expressed as %ID/g and represent mean values ± SD, n = 4. We observe a very rapid washout of both radiotracers from the body of mice predominantly via the kidneys and the urinary tract. [99mTc]Tc-DGA1 displayed higher values in blood, liver, and intestines compared to [99mTc]Tc-DG2, especially in the initial time intervals. For example, blood values for [99mTc]Tc-DGA1 were significantly higher than those for [99mTc]Tc-DG2 at 1 h pi (7.46 ± 1.38%ID/g vs 0.33 ± 0.03%ID/g; P < 0.001). Likewise, uptake in the kidneys was significantly higher for [99mTc]Tc-DGA1 (96.09 ± 12.01%ID/g at 4 h pi) compared to [99mTc]Tc-DG2 (51.73 ± 8.91%ID/g at 4 h pi; P < 0.0001), but renal values were found similar at 24 h pi (16.69 ± 3.30%ID/g vs 11.92 ± 1.81%ID/g; P > 0.05). Interestingly, coinjection of gelofusine resulted in a drastic reduction of renal accumulation for both radiotracers.51 For example, renal values after gelofusine coinjection dropped to 33.61 ± 2.67%ID/g at 4 h pi for [99mTc]Tc-DGA1 (in controls: 96.09 ± 12.01%ID/g; P < 0.0001).
Table 1.
Biodistribution of [99mTc]Tc-DGA1 and [99mTc]Tc-DG2 in SCID Mice Bearing Twin HEK293-CCK2i4svR-Positive and HEK293-CCK2i4svR-Negative Tumors at 1, 4, and 24 h pi, Expressed as %ID/g and Representing Mean Values ± SD, n = 4
| % ID/g ± SD, n = 4 |
||||||||
|---|---|---|---|---|---|---|---|---|
| [99mTc]Tc-DGA1 |
[99mTc]Tc-DG2 |
|||||||
| organs | 1 h | 4 h | 4 ha | 24 h | 1h | 4 h | 4 ha | 24 h |
| blood | 7.46 ± 1.38 | 0.94 ± 0.31 | 0.91 ± 0.12 | 0.09 ± 0.03 | 0.33 ± 0.03 | 0.09 ± 0.01 | 0.10 ± 0.03 | 0.04 ± 0.01 |
| liver | 4.59 ± 0.23 | 1.80 ± 0.62 | 1.65 ± 0.24 | 0.33 ± 0.04 | 0.37 ± 0.02 | 0.22 ± 0.03 | 0.29 ± 0.18 | 0.09 ± 0.03 |
| heart | 3.12 ± 0.35 | 0.64 ± 0.13 | 0.50 ± 0.07 | 0.12 ± 0.03 | 0.28 ± 0.03 | 0.08 ± 0.03 | 0.11 ± 0.07 | 0.08 ± 0.02 |
| kidneys | 113.53 ± 15.26 | 96.09 ± 12.01 | 33.61 ± 2.67 | 16.69 ± 3.30 | 99.97 ± 8.2 | 51.73 ± 8.91 | 9.47 ± 0.85 | 11.92 ± 1.81 |
| stomach | 1.68 ± 0.38 | 0.75 ± 0.21 | 0.79 ± 0.24 | 0.13 ± 0.05 | 2.00 ± 0.5 | 1.26 ± 0.23 | 1.95 ± 0.58 | 0.61 ± 0.02 |
| intestines | 1.60 ± 0.33 | 1.32 ± 0.39 | 1.15 ± 0.37 | 0.23 ± 0.05 | 0.55 ± 0.15 | 0.29 ± 0.09 | 0.51 ± 0.15 | 0.11 ± 0.03 |
| spleen | 2.31 ± 0.53 | 0.88 ± 0.18 | 0.82 ± 0.08 | 0.43 ± 0.01 | 0.38 ± 0.06 | 0.20 ± 0.02 | 0.32 ± 0.12 | 0.33 ± 0.07 |
| muscle | 1.03 ± 0.12 | 0.19 ± 0.04 | 0.19 ± 0.03 | 0.06 ± 0.02 | 0.12 ± 0.03 | 0.03 ± 0.01 | 0.05 ± 0.03 | 0.04 ± 0.01 |
| lungs | 4.67 ± 0.45 | 1.02 ± 0.20 | 1.10 ± 0.18 | 0.21 ± 0.01 | 0.34 ± 0.03 | 0.06 ± 0.01 | 0.13 ± 0.05 | 0.05 ± 0.01 |
| pancreas | 2.06 ± 0.24 | 0.38 ± 0.09 | 0.39 ± 0.07 | 0.07 ± 0.02 | 0.25 ± 0.01 | 0.12 ± 0.01 | 0.25 ± 0.15 | 0.09 ± 0.03 |
| CCK2i4svR(+) tumor | 31.92 ± 3.66 | 31.63 ± 4.59 | 32.13 ± 3.53 | 11.85 ± 1.02 | 16.67 ± 0.71 | 16.39 ± 3.18 | 11.56 ± 1.31 | 9.11 ± 0.65 |
| CCK2i4svR(-) tumor | 1.87 ± 0.11 | 0.60 ± 0.11 | 0.65 ± 0.15 | 0.16 ± 0.01 | 0.43 ± 0.03 | 0.24 ± 0.05 | 0.26 ± 0.06 | 0.15 ± 0.01 |
Coinjection of 100 μL of gelofusine.
Both tracers displayed considerably higher uptake in the HEK293-CCK2i4svR-positive tumors compared to the tumors devoid of CCK2i4svR expression in all time intervals (P < 0.00001), consistent with a receptor-specific process. Notably, [99mTc]Tc-DGA1 displayed about twice as high uptake in the HEK293-CCK2i4svR-expressing tumors (e.g., 31.63 ± 4.59%ID/ g at 4 h pi) compared to [99mTc]Tc-DG2 (e.g., 16.39 ± 3.18% ID/g at 4 h pi; P < 0.0001), and this uptake was not affected by gelofusine coinjection (e.g., 32.13 ± 3.53%ID/g at 4 h pi; P > 0.05). At the 24 h time intervals, the uptake of both radiotracers declined in the CCK2i4svR-expressing xenografts to comparable levels ([99mTc]Tc-DGA1: 11.85 ± 1.02%ID/g and [99mTc]Tc-DG2: 9.11 ± 0.65%ID/g; P > 0.05).
Similar observations were made for the biodistribution of [99mTc]Tc-DGA1 and [99mTc]Tc-DG2 in animals bearing double HEK293-CCK2R-positive and HEK293-CCK2R-negative tumors for the 1, 4, and 24 h pi intervals (Supporting Information, Table S1).
SPECT/CT Imaging.
Mice SPECT/CT images obtained 4 h after injection of [99mTc]Tc-DGA1 are presented in Figure 5. Significant accumulation was achieved in the CCK2i4svR-expressing xenografts, but no uptake was evident in the HEK293T tumors devoid of CCK2i4svR expression. Clear differences in renal accumulation could be observed between control (Figure 5A,B) and gelofusine-treated animals (Figure 5C,D). Normalization on the tumor uptake (i.e., tumors having the same maximum number of counts) has been performed to provide a clear visualization of the reduced accumulation on kidneys, when gelofusine is administered, confirming biodistribution results.
Figure 5.
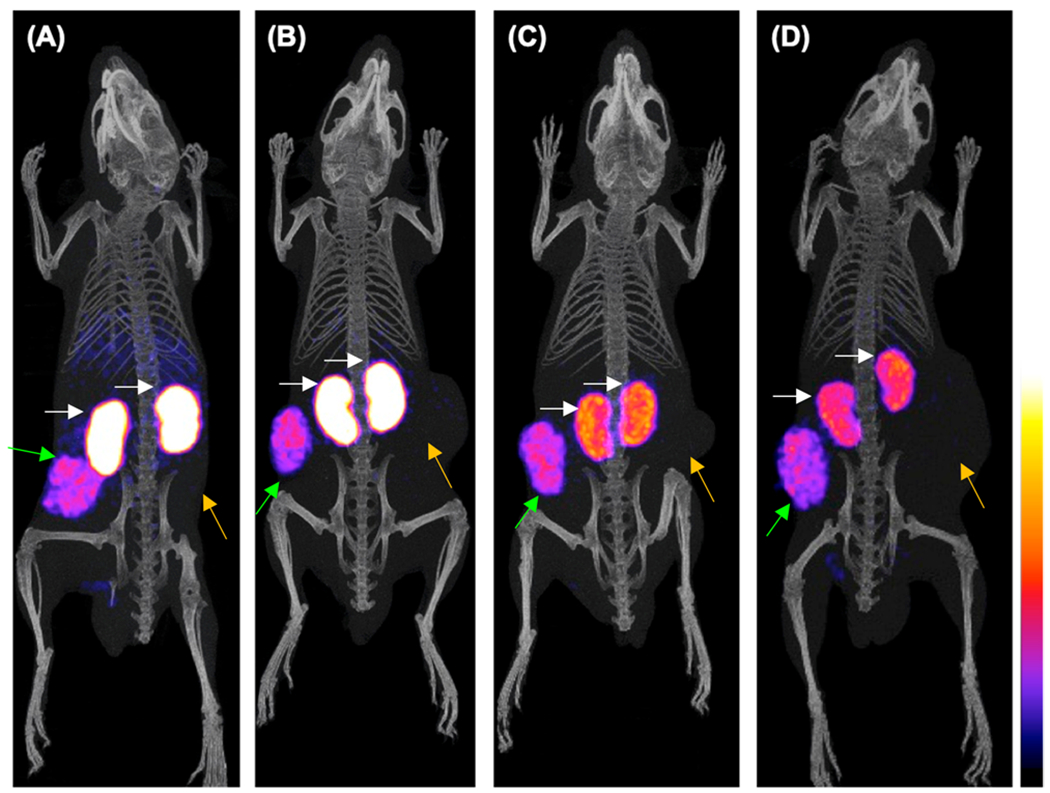
Static whole body SPECT/CT images of mice 4 h after injection of [99mTc]Tc-DGA1 (A,B) alone or (C,D) with gelofusine coinjection; green arrows are showing the radiotracer uptake in the HEK293-CCK2i4svR xenografts, orange arrows are pointing to the HEK293T tumors, and white arrows are indicating the kidneys. Intense uptake is observed in the CCK2i4svR-expressing tumors, but no uptake is evident in the tumors devoid of CCK2i4svR expression; when gelofusine is administered, the renal uptake has been significantly reduced. The color bar indicates the difference in accumulated activity (purple being the lowest and white being the highest level of accumulation).
DISCUSSION
Aiming to explore the potential of antagonist-based radiotracers in the diagnostic imaging of CCK2R-expressing tumors in man, we herein introduced DGA1. In this peptidomimetic, the small-size CCK2R-antagonist Z-36032,33,35,36,3S was combined with an acyclic tetraamine chelator for technetium-99m labeling,42 and the two entities were linked together by a PEG3-(DGlu)4 chain (Schemes 1 and 2). Z-360 displayed high affinity and antagonistic profile at the CCK2R and was shown to elicit antiproliferative effects in gastrointestinal cancer models. As a result, it has been selected for clinical validation in the treatment of pancreatic cancer in combination with gemcitabine.38 Recently, Z-360 has been used as a motif for the development of antagonist-based probes in the molecular targeting of CCK2R-positive lesions with near-infrared dyes (for fluorescence-guided surgery applications)40 and technetium-99m (for diagnostic imaging with SPECT)39 and for delivering cytotoxic payloads to cancer cells.41 First, results from these studies have demonstrated the feasibility of structurally modifying Z-360 to obtain conjugates retaining good affinity to the CCK2R and showing promising qualities for a number of clinical applications. However, important parameters in the biological profile of the abovementioned technetium-99m-based CCK2R-antagonists at the cellular level (cell binding and internalization experiments; functional assays) and in animals (metabolic stability) have not been investigated. Moreover, comparison with established radiolabeled CCK2R agonists have not been made.
In the present work, we aimed to acquire the abovementioned missing information for further assessing the applicability and efficacy of radiolabeled CCK2R antagonists in the theranostic management of oncological patients. For this purpose, we have designed our first CCK2R antagonist, DGA1. In this new analogue, an acyclic tetraamine was selected for technetium-99m labeling, instead of the peptide-based chelator used in the initial reports on Z-360 which forms square pyramidal mono-oxo Tc-complexes. Tetraamines have been shown to form hydrophilic monocationic and in vivo robust octahedral frans-dioxo Tc-complexes in high purity and high molar activity, most suitable for labeling peptide ligands with technetium-99m.42 In addition, this modification offered us the advantage of head-to-head comparisons with [99mTc]Tc-DG2, carrying the same tetraamine moiety via a Gly-linker to [DGlu1]minigastrin for labeling with technetium-99m. In previous studies, [99mTc]Tc-DG2 displayed an attractive profile in preclinical models and in MTC patients,13,14 qualifying as a suitable CCK2R-agonist reference during the preclinical evaluation of [99mTc]Tc-DGA1.
Finally, the PEG3-(DGlu)4 linker was selected mainly for improving pharmacokinetics. Data on Z-360 conjugates have hinted at the problem of high lipophilicity and ensuing high background levels after radioligand administration.39 Hence, the introduction of hydrophilic spacers serving as pharmacokinetic modifiers was proposed. For this goal, we decided to use the PEG3-(DGlu)4 linker. First, the (DGlu)4 chain, although nonsusceptible to protease degradation, could provide four negatively charged carboxylate groups which, in addition to the pendant carboxylate group of Lys, were expected to enhance hydrophilicity and promote rapid clearance of [99mTc]Tc-DGA1 via the kidneys and the urinary tract. Radiolabeled gastrin analogues containing the (Glu)5 sequence of the natural CCK2R ligand have been notorious for their high kidney uptake and retention.51 However, recent studies in animal models have shown that such renal retention n could be drastically cut down by simply replacing the original (Glu)5 chain with (DGlu)5, without, at the same time, compromising tumor uptake.15,43,52 Moreover, either (Glu)5/ (DGlu)5 group was reported to favor affinity to the CCK2R and to be associated with higher metabolic stability.45,53,56
Notably, the abovementioned structural interventions did not deteriorate CCK2R affinity, with DGA1 and Z-360 displaying identical affinities during competition assays (Figure 2). Furthermore, DGA1 displayed an antagonistic profile at the CCK2R during a [Ca2+], mobilization assay, by barely altering basal [Ca2+], when administered alone and by suppressing the potent agonist activity of DG2 (EC50 = 79 pM), newly established herein, under the same experimental settings (Figure 1). To our knowledge, this is the first study confirming the antagonistic properties of a Z-360 bioconjugate at the CCK2R at the cellular level. Furthermore, the cell uptake and internalization rates of the end radiotracer [99mTc]Tc-DGA1 were directly compared with those of the CCK2R-agonist reference [99mTc]Tc-DG2. Comparisons were performed in two cell lines transfected to stably express the human CCK2R and its splice variant CCIK2i4svR, characterized by a 64 amino acid insertion in the third cytoplasmic domain.47 The CCK2i4svR was found to be more tumor-specific and often coexpressed with the CCK2R in cancer cells. As shown in Figure 3 (and Figure S11, Supporting Information), [99mTc]-Tc-DGA1 reached the highest cell uptake at 60 min of incubation with the radioactivity equally distributed between the cell membrane and the intracellular compartment. This behavior albeit intriguing for a receptor antagonist is not unique. Previous studies at the cellular level have shown that well-characterized CCK2R antagonists may induce receptor internalization while others do not.54,55 In contrast, [99mTc]Tc-DG2 displayed considerably higher internalization as consistent with a receptor agonist profile.
Of particular interest was the superior metabolic stability of [99mTc]Tc-DGA1 versus the peptide-based [99mTc]Tc-DG2 in peripheral mouse blood (Figure 4A,B, respectively). The metabolic fate of radiolabeled gastrin analogues, carrying the radiometal chelate at the N-terminus, was shown to depend on two major proteases, ACE and NEP.45,53 Several studies have demonstrated that the (Glu)5 chain of native minigastrin increased the resistance of related radioligands to proteolytic enzymes, whereas truncated analogues lacking this sequence were rapidly degraded in mice and human.14,45,56 However, even the (Glu)5/(DGlu)5 chain containing radioligands, such as [99mTc]Tc-DG2 or [111In]In-CP04, displayed a considerable degree of in vivo degradation.53,57 In contrast, [99mTc]Tc-DGA1 was detected intact, confirming that the organic Z-360 moiety coupled to the PEG3-(DGlu)4 linker could resist hydrolyzing proteases and other enzymes as well. Moreover, the radioactivity excreted in urine contained a significant amount of intact [99mTc]Tc-DGA1, whereas [99mTc]Tc-DG2 was excreted totally degraded.13
Of great interest are the comparative biodistribution results of [99mTc]Tc-DGA1 and [99mTc]Tc-DG2 in mice bearing CCK2i4svR-(Table 1) or CCK2R-expressing xenografts (Table S1, Supporting Information). In all cases, [99mTc]Tc-DGA1 showed notably superior uptake compared with [99mTc]Tc-DG2 at 1 h and 4 h pi, reflecting the higher receptor affinity and metabolic stability of the receptor antagonist. Elevated values of [99mTc]Tc-DGA1 in blood and the liver at 1 h pi rapidly declined with time, resulting in lower tumor-to-blood and tumor-to-liver ratios for the receptor antagonist. It should be noted that these ratios were superior for [99mTc]Tc-DGA1 at all time points compared with the previously reported Z-360 radiotracer [99mTc]Tc-CRL-339 (tumor-to-blood, [99mTc]Tc-DGA1/[99mTc]Tc-CRL-3: 33.6/7.0 at 4 h pi, 131.7/63 at 24 h pi; tumor-to-liver, [99mTc]Tc-DGA1/[99mTc]Tc-CRL-3: 17.6/ 2.9 at 4 h pi, 35.9/4.5 at 24 h pi).
On the other hand, the renal uptake of [99mTc]Tc-DGA1 was unfavorably high, higher than [99mTc]Tc-DG2 at both 1 and 4 h pi. It remains to be seen, if this elevated renal uptake is clinically relevant. During the evaluation of the structurally related [99mTc]Tc-DG2 agonist in MTC patients, renal clearance turned out to be very fast and kidney uptake in human was found to be very low as opposed to the corresponding values observed in mice.13,14 The renal uptake of [99mTc]Tc-DGA1 was clearly unexpected, in view of the fact that the previously reported [99mTc]Tc-CRL-3 displayed remarkably lower kidney uptake (13.4 ± 2.1%ID/g at 4 h pi and 2.6 ± 1.6%ID/g at 24 h pi),39 whereas the (DGlu)x chain (x ≤ 6) was previously shown not to induce renal accumulation in the case of minigastrin-based radiotracers.43,52 Each of the metal chelate, linker, or vector parts of [99mTc]Tc-DGA1 was not individually associated with elevated kidney uptake with their combination, however, leading to this unfavorable result. Interestingly, treatment of mice with gelofusine was successful to strongly reduce kidney values of [99mTc]Tc-DGA1, hinting at the involvement of the cubilin— megalin system in the observed retention.51 Following gelofusine injection, the tumor-to-kidney ratios of [99mTc]Tc-DGA1 were reduced, matching those of [99mTc]Tc-CRL-3 at 4 h pi (≈1). However, comparison of biodistribution data, including kidney uptake values, between [99mTc]Tc-DGA1 and [99mTc]Tc-CRL-3, should be taken with caution because different animal species were used in each case (male SCID versus female nu/nu mice) and markedly different radioactivity and peptide amounts (185 kBq associated with 10 pmol DGA1 versus 5.55 MBq associated with 10 nmol CRL-3)39 were applied.
CONCLUSIONS
A new technetium-99m radiotracer based on a CCK2R antagonist has been developed and evaluated for its applicability in the diagnostic imaging of human tumors with SPECT in a series of cell and animal models. To our knowledge, this is a first study whereby a CCK2R antagonist was characterized at the cellular level in a head-to-head comparison with the corresponding CCK2R agonist. Moreover, clear advantages of the new [99mTc]Tc-DGA1 versus the established [99mTc]Tc-DG2 CCK2R-agonist reference could be shown, including higher metabolic stability and superior localization in CCK2R-positive tumors in mice, although there is still space for improvement of pharmacokinetics. These findings strongly support previous data on the feasibility of developing a new generation of radioligands, based on nonpeptidic CCK2R antagonists and associated with higher safety, for use in theranostic management of cancer patients. Systematic structure—activity relationship investigations in search of further clinically effective and safe radioligands seem therefore warranted.
Supplementary Material
HPLC analysis of H-PEG3-[DGlu(OtBu)]4-Lys(Boc)-OH before and after purification, MALDI-TOF-MS spectrum of H-PEG3-[DGlu(OtBu)]4-Lys(Boc)-OH) HPLC analysis of Z-360-PEG3-[DGlu(OtBu)]4-Lys-(Boc)-OH before and after purification, MALDI-TOF-MS spectrum of Z-360-PEG3-[DGlu(OtBu)]4-Lys(Boc)-OH, HPLC analysis of Z-360-PEG3-(DGlu)4-Lys-OH, MALDI-TOF-MS spectrum of Z-360-PEG3-(DGlu)4-Lys-OH, HPLC analysis of Z-360-PEG3-(DGlu)4-Lys-[N4(Boc)4]-OH before and after purification, Figure S8: HPLC analysis of DGA1 before and after purification, MALDI-TOF-MS spectrum of DGA1, HPLC radiochromatogram of [99m]Tc-DGA1, specific cell association/internalization rates of [99mTc]Tc-DGA1 and [99mTc]Tc-DG2 in HEK293-CCK2R cells at 37 °C; Cell association/internalization of [99mTc]Tc-DGA1 and [99mTc]Tc-DG2 in HEK293-CCK2i4svR cells and of [99mTc]Tc-DGA1 in HEK293T cells at 37 °C at 1 h, including total, nonspecific, and specific values; and biodistribution of [99mTc]Tc-DGA1 and [99mTc]Tc-DG2 in SCID mice bearing twin HEK293-CCK2R-positive and HEK293-CCK2R-negative tumors at 1, 4, and 24 h pi (PDF)
ACKNOWLEDGMENTS
This study has been partially supported by the project “NCSRD—INRASTES research activities within National RIS3 (MIS5002559)” and the Operational Program “Competitiveness, Entrepreneurship and Innovation” (NSRF 2014— 2020), cofinanced by Greece and the European Union (European Regional Development Fund). The IKY scholarship to A.K., funded from the Action “Reinforcement of Postdoctoral Researchers” with the resources of the Operational Program “Human Resources Development, Education and Lifelong Learning” with priority axes 6,8,9, cofinanced by the European Social Fund (ESF) and the Greek State, is gratefully acknowledged. G.G.H. acknowledges the support of NIH R01 DK069575.
ABBREVIATIONS
- SPECT
single-photon emission computed tomography
- CT
computed tomography
- CCK2R
cholecystokinin subtype 2 receptor
- N4
6-(carboxyl)-1,4,8,11-tetraazaundecane
- Z-360
Nastorazepide, 3-[[[[(3R)-5-cyclohexyl-1-(3,3-dimethyl-2-ox-obutyl)-2,3,4,5-tetrahydro-2-oxo-1H-1,5-benzodiazepin-3-yl]-amino]carbonyl]amino]benzoic acid
- YF476
netazepide, 1-[(3R)-1-(3,3-dimethyl-2-oxobutyl)-2-oxo-5-pyridin-2-yl-3H-1,4-benzodiazepin-3-yl]-3-[3-(methylamino)phenyl]urea CP04, DOTA-(DGlu) 6-Ala-Tyr-Gly-Trp-Met-Asp-Phe-NH2
- NEP
neprilysin, neutral endopeptidase
- [Leu15]gastrin
pGlu-Gly-Pro-Trp-Leu-(Glu)5-Ala-Tyr-Gly-Trp-Leu-Asp-Phe-NH2
- [125I][I-Tyr12,Leu15]gastrin
pGlu-Gly-Pro-Trp-Leu-(Glu)5-Ala-[125I]I-Tyr-Gly-Trp-Leu-Asp-Phe-NH2
- HPLC
high-performance liquid chromatography
- ITLC
instant thin-layer chromatography
- MALDI-TOF-MS
matrix-assisted laser desorption/ionization time of flight mass spectrometry
- LC— MS
liquid chromatography—mass spectroscopy
- SPPS
solid-phase peptide synthesis
- LPPS
liquid-phase peptide synthesis
- MTC
medullary thyroid cancer
- SAR
structure—activity relationships
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.molpharmaceut.0c00605.
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.molpharmaceut.0c00605
The authors declare no competing financial interest.
Contributor Information
Aikaterini Kaloudi, Molecular Radiopharmacy, INRASTES, NCSR “Demokritos”, 15310 Athens, Greece.
Panagiotis Kanellopoulos, Molecular Radiopharmacy, INRASTES, NCSR “Demokritos”, 15310 Athens, Greece.
Thorsten Radolf, piCHEM Forschungs-und Entwicklungs GmbH, 8074 Grambach, Austria.
Oleg G. Chepurny, SUNY Upstate Medical University, Syracuse, New York 13210, United States
Maritina Rouchota, BIOEMTECH, Lefkippos Attica Technology Park NCSR “Demokritos”, 15310 Athens, Greece.
George Loudos, BIOEMTECH, Lefkippos Attica Technology Park NCSR “Demokritos”, 15310 Athens, Greece.
Fritz Andreae, piCHEM Forschungs-und Entwicklungs GmbH, 8074 Grambach, Austria.
George G. Holz, SUNY Upstate Medical University, Syracuse, New York 13210, United States
Berthold Artur Nock, Molecular Radiopharmacy, INRASTES, NCSR “Demokritos”, 15310 Athens, Greece.
Theodosia Maina, Molecular Radiopharmacy, INRASTES, NCSR “Demokritos”, 15310 Athens, Greece.
REFERENCES
- (1).Noble F; Wank SA; Crawley JN; Bradwejn J; Seroogy KB; Hamon M; Roques BP International Union of Pharmacology. XXI. Structure, distribution, and functions of cholecystokinin receptors. Pharmacol. Rev 1999, 51, 745–781. [PubMed] [Google Scholar]
- (2).Noble F; Roques BP CCK-B receptor: chemistry, molecular biology, biochemistry and pharmacology. Prog. Neurobiol 1999, 58, 349–379. [DOI] [PubMed] [Google Scholar]
- (3).Behr TM; Béhé M; Angerstein C; Gratz S; Mach R; Hagemann L; Jenner N; Stiehler M; Frank-Raue K; Raue F; Becker W Cholecystokinin-B/gastrin receptor binding peptides: preclinical development and evaluation of their diagnostic and therapeutic potential. Clin. Cancer Res 1999, 5, 3124s–3138s. [PubMed] [Google Scholar]
- (4).Behr TM; Jenner N; Behe M; Angerstein C; Gratz S; Raue F; Becker W Radiolabeled peptides for targeting cholecysto-kinin-B/gastrin receptor-expressing tumors. J. Nucl. Med 1999, 40, 1029–1044. [PubMed] [Google Scholar]
- (5).Reubi JC; Waser B Unexpected high incidence of cholecystokinin-B/gastrin receptors in human medullary thyroid carcinomas. Int. J. Cancer 1996, 67, 644–647. [DOI] [PubMed] [Google Scholar]
- (6).Sethi T; Herget T; Wu SV; Walsh JH; Rozengurt E CCKA and CCKB receptors are expressed in small cell lung cancer lines and mediate Ca2+ mobilization and clonal growth. Cancer Res. 1993, 53, 5208–5213. [PubMed] [Google Scholar]
- (7).Reubi JC; Waser B; Gugger M; Friess H; Kleeff J; Kayed H; BUchler MW; Laissue JA Distribution of CCK1 and CCK2 receptors in normal and diseased human pancreatic tissue. Gastroenterology 2003, 125, 98–106. [DOI] [PubMed] [Google Scholar]
- (8).Reubi JC; Waser B; Schaer JC; Laederach U; Erion J; Srinivasan A; Schmidt MA; Bugaj JE Unsulfated DTPA-and DOTA-CCK analogs as specific high-affinity ligands for CCK-B receptor-expressing human and rat tissues in vitro and in vivo. Eur. J. Nucl. Med 1998, 25, 481–490. [DOI] [PubMed] [Google Scholar]
- (9).Reubi JC; Schaer JC; Waser B Cholecystokinin(CCK)-A and CCK-B/gastrin receptors in human tumors. Cancer Res. 1997, 57, 1377–1386. [PubMed] [Google Scholar]
- (10).Hur K; Kwak MK; Lee H-J; Park DJ; Lee HK; Lee HS; Kim WH; Michaeli D; Yang H-K Expression of gastrin and its receptor in human gastric cancer tissues. J. Cancer Res. Clin. Oncol 2006, 132, 85–91. [DOI] [PubMed] [Google Scholar]
- (11).Aly A; Shulkes A; Baldwin GS Gastrins, cholecystokinins and gastrointestinal cancer. Biochim. Biophys. Acta 2004, 1704, 1–10. [DOI] [PubMed] [Google Scholar]
- (12).Roosenburg S; Laverman P; van Delft FL; Boerman OC Radiolabeled CCK/gastrin peptides for imaging and therapy of CCK2 receptor-expressing tumors. Amino Acids 2011, 41, 1049–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Nock BA; Maina T; Behe M; Nikolopoulou A; Gotthardt M; Schmitt JS; Behr TM; Macke HR CCK-2/gastrin receptor-targeted tumor imaging with 99mTc-labeled minigastrin analogs. J. Nucl. Med 2005, 46, 1727–1736. [PubMed] [Google Scholar]
- (14).Froberg AC; de Jong M; Nock BA; Breeman WAP; Erion JL; Maina T; Verdijsseldonck M; de Herder WW; van der Lugt A; Kooij PPM; Krenning EP Comparison of three radiolabelled peptide analogues for CCK-2 receptor scintigraphy in medullary thyroid carcinoma. Eur. J. Nucl. Med. Mol. Imaging 2009, 36, 1265–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Erba PA; Maecke H; Mikolajczak R; Decristoforo C; Zaletel K; Maina-Nock T; Peitl PK; Garnuszek P; Froberg A; Goebel G; de Jong M; Jabrocka-Hybel A; Konijnenberg M; Virgolini I; Nock B; Lenda-Tracz W; Pawlak D; Rangger C; Trofimiuk-Muldner M; Sowa-Staszczak A; Tomaszuk M; von Guggenberg E; Scarpa L; Hubalewska-Dydejczyk A A novel CCK2/gastrin receptor-localizing radiolabeled peptide probe for personalized diagnosis and therapy of patients with progressive or metastatic medullary thyroid carcinoma: a multicenter phase I GRAN-T-MTC study. Pol. Arch. Intern. Med 2018, 128, 791–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Ubl P; Gincu T; Keilani M; Ponhold L; Crevenna R; Niederle B; Hacker M; Li S Comparison of side effects of pentagastrin test and calcium stimulation test in patients with increased basal calcitonin concentration: the gender-specific differences. Endocrine 2014, 46, 549–553. [DOI] [PubMed] [Google Scholar]
- (17).Ginj M; Zhang H; Waser B; Cescato R; Wild D; Wang X; Erchegyi J; Rivier J; Macke HR; Reubi JC Radiolabeled somatostatin receptor antagonists are preferable to agonists for in vivo peptide receptor targeting of tumors. Proc. Natl. Acad. Sci. U.S.A 2006, 103, 16436–16441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Cescato R; Maina T; Nock B; Nikolopoulou A; Charalambidis D; Piccand V; Reubi JC Bombesin receptor antagonists may be preferable to agonists for tumor targeting. J. Nucl. Med 2008, 49, 318–326. [DOI] [PubMed] [Google Scholar]
- (19).Maina T; Nock BA; Kulkarni H; Singh A; Baum R P. Theranostic prospects of gastrin-releasing peptide receptor-radio-antagonists in oncology. PET Clin. 2017, 12, 297–309. [DOI] [PubMed] [Google Scholar]
- (20).Fani M; Nicolas GP; Wild D Somatostatin receptor antagonists for imaging and therapy. J. Nucl. Med 2017, 58, 61S–66S. [DOI] [PubMed] [Google Scholar]
- (21).Nock BA; Kaloudi A; Lymperis E; Giarika A; Kulkarni HR; Klette I; Singh A; Krenning EP; de Jong M; Maina T; Baum RP Theranostic perspectives in prostate cancer with the gastrin-releasing peptide receptor antagonist NeoBOMB1: preclinical and first clinical results. J. Nucl. Med 2017, 58, 75–80. [DOI] [PubMed] [Google Scholar]
- (22).Maina T; Bergsma H; Kulkarni HR; Mueller D; Charalambidis D; Krenning EP; Nock BA; de Jong M; Baum RP Preclinical and first clinical experience with the gastrin-releasing peptide receptor-antagonist [68Ga]SB3 and PET/CT. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 964–973. [DOI] [PubMed] [Google Scholar]
- (23).Minamimoto R; Hancock S; Schneider B; Chin FT; Jamali M; Loening A; Vasanawala S; Gambhir SS; Iagaru A Pilot comparison of 68Ga-RM2 PET and 68Ga-PSMA-11 PET in patients with biochemically recurrent prostate cancer. J. Nucl. Med 2016, 57, 557–562. [DOI] [PubMed] [Google Scholar]
- (24).Mansi R; Fleischmann A; Macke HR; Reubi JC Targeting GRPR in urological cancers-from basic research to clinical application. Nat. Rev. Urol 2013, 10, 235–244. [DOI] [PubMed] [Google Scholar]
- (25).de Tullio P; Delarge J; Pirotte B Therapeutic and chemical developments of cholecystokinin receptor ligands. Expert Opin. Invest. Drugs 2000, 9, 129–146. [DOI] [PubMed] [Google Scholar]
- (26).Kalindjian SB; McDonald IM Strategies for the design of non-peptide CCK2 receptor agonist and antagonist ligand. Curr. Top. Med. Chem 2007, 7, 1195–1204. [DOI] [PubMed] [Google Scholar]
- (27).Herranz R Cholecystokinin antagonists: pharmacological and therapeutic potential. Med. Res. Rev 2003, 23, 559–605. [DOI] [PubMed] [Google Scholar]
- (28).Berna MJ; Tapia JA; Sancho V; Jensen RT Progress in developing cholecystokinin (CCK)/gastrin receptor ligands that have therapeutic potential. Curr. Opin. Pharmacol 2007, 7, 583–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Takinami Y; Yuki H; Nishida A; Akuzawa S; Uchida A; Takemoto Y; Ohta M; Satoh M; Semple G; Miyata K YF476 is a new potent and selective gastrin/cholecystokinin-B receptor antagonist in vitro and in vivo. Aliment. Pharmacol. Ther 1997, 11, 113–120. [DOI] [PubMed] [Google Scholar]
- (30).Moore AR; Boyce M; Steele IA; Campbell F; Varro A; Pritchard DM Netazepide, a gastrin receptor antagonist, normalises tumour biomarkers and causes regression of type 1 gastric neuroendocrine tumours in a nonrandomised trial of patients with chronic atrophic gastritis. PLoS One 2013, 8, No. e76462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Boyce M; Moore AR; Sagatun L; Parsons BN; Varro A; Campbell F; Fossmark R; Waldum HL; Pritchard DM Netazepide, a gastrin/cholecystokinin-2 receptor antagonist, can eradicate gastric neuroendocrine tumours in patients with auto-immune chronic atrophic gastritis. Br. J. Clin. Pharmacol 2017, 83, 466–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Morita H; Miura N; Hori Y; Matsunaga Y; Ukawa H; Suda H; Yoneta T; Kurimoto T; Itoh Z Effects of Z-360, a novel CCKB/gastrin (CCK2) receptor antagonist, on meal-induced acid secretion and experimental ulcer models in dogs and rats. Gastroenterology 2001, 120, A311. [Google Scholar]
- (33).Miura N; Yoneta T; Ukawa H; Fukuda Y; Eta R; Mera Y; Omata T; Kinomoto T; Kurimoto T; Itoh Z Pharmacological profiles of Z-360, a novel CCKB/gastrin (CCK2) receptor antagonist with excellent oral potency. Gastroenterology 2001, 120, A311. [Google Scholar]
- (34).Ukawa H; Miura N; Morita H; Hori Y; Ueki S; Yoneta T; Kurimoto T; Itoh Z Effect of Z-360, a selective CCKB/gastrin receptor antagonist, on chronic acid reflux esophagitis in rats. Gastroenterology 2002, 122, A194. [Google Scholar]
- (35).Grabowska AM; Morris TM; McKenzie AJ; Kumari R; Hamano H; Emori Y; Yoshinaga K; Watson SA Pre-clinical evaluation of a new orally-active CCK-2R antagonist, Z-360, in gastrointestinal cancer models. Regul Pept. 2008, 146, 46–57. [DOI] [PubMed] [Google Scholar]
- (36).Kawasaki D; Emori Y; Eta R; Iino Y; Hamano H; Yoshinaga K; Tanaka T; Takei M; Watson SA Effect of Z-360, a novel orally active CCK-2/gastrin receptor antagonist on tumor growth in human pancreatic adenocarcinoma cell lines in vivo and mode of action determinations in vitro. Cancer Chemother. Pharmacol 2008, 61, 883–892. [DOI] [PubMed] [Google Scholar]
- (37).Meyer T; Caplin ME; Palmer DH; Valle JW; Larvin M; Waters JS; Coxon F; Borbath I; Peeters M; Nagano E; Kato H A phase Ib/IIa trial to evaluate the CCK2 receptor antagonist Z-360 in combination with gemcitabine in patients with advanced pancreatic cancer. Eur. J. Cancer 2010, 46, 526–533. [DOI] [PubMed] [Google Scholar]
- (38).Ueno M; Li CP; Ikeda M; Ishii H; Mizuno N; Yamaguchi T; Ioka T; Oh DY; Ichikawa W; Okusaka T; Matsuyama Y; Arai D; Chen LT; Park YS; Furuse J A randomized phase II study of gemcitabine plus Z-360, a CCK2 receptor-selective antagonist, in patients with metastatic pancreatic cancer as compared with gemcitabine plus placebo. Cancer Chemother. Pharmacol 2017, 80, 307–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Wayua C; Low PS Evaluation of a nonpeptidic ligand for imaging of cholecystokinin 2 receptor-expressing cancers. J. Nucl Med 2015, 56, 113–119. [DOI] [PubMed] [Google Scholar]
- (40).Wayua C; Low PS Evaluation of a cholecystokinin 2 receptor-targeted near-infrared dye for fluorescence-guided surgery of cancer. Mol. Pharm 2014, H, 468–476. [DOI] [PubMed] [Google Scholar]
- (41).Wayua C; Roy J; Putt KS; Low PS Selective tumor targeting of desacetyl vinblastine hydrazide and tubulysin B via conjugation to a cholecystokinin 2 receptor (CCK2R) ligand. Mol. Pharm 2015, 12, 2477–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Nock BA; Maina T Tetraamine-coupled peptides and resulting 99mTc-radioligands: an effective route for receptor-targeted diagnostic imaging of human tumors. Curr. Top. Med. Chem 2012, 12, 2655–2667. [DOI] [PubMed] [Google Scholar]
- (43).Maina T; Konijnenberg MW; KolencPeitl P; Garnuszek P; Nock BA; Kaloudi A; Kroselj M; Zaletel K; Maecke H; Mansi R; Erba P; von Guggenberg E; Hubalewska-Dydejczyk A; Mikolajczak R; Decristoforo C Preclinical pharmacokinetics, biodistribution, radiation dosimetry and toxicity studies required for regulatory approval of a phase I clinical trial with 111In-CP04 in medullary thyroid carcinoma patients. Eur. J. Pharm. Sci 2016, 91, 236–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Deschodt-Lanckman M; Pauwels S; Najdovski T; Dimaline R; Dockray GJ In vitro and in vivo degradation ofhuman gastrin by endopeptidase 24.11. Gastroenterology 1988, 94, 712–721. [DOI] [PubMed] [Google Scholar]
- (45).Kaloudi A; Nock BA; Krenning EP; Maina T; De Jong M Radiolabeled gastrin/CCK analogs in tumor diagnosis: towards higher stability and improved tumor targeting. Q. J. Nucl. Med. Mol. Imaging 2015, 59, 287–302. [PubMed] [Google Scholar]
- (46).Nock BA; Maina T; Krenning EP; de Jong M “To serve and protect”: enzyme inhibitors as radiopeptide escorts promote tumor targeting. J. Nucl. Med 2014, 55, 121–127. [DOI] [PubMed] [Google Scholar]
- (47).Korner M; Waser B; Reubi JC; Miller LJ CCK(2) receptor splice variant with intron 4 retention in human gastrointestinal and lung tumours. J. Cell. Mol. Med 2010, 14, 933–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Chao C; Han X; Ives K; Park J; Kolokoltsov AA; Davey RA; Moyer MP; Hellmich MR CCK2 receptor expression transforms non-tumorigenic human NCM356 colonic epithelial cells into tumor forming cells. Int. J. Cancer 2010, 126, 864–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Laverman P; Roosenburg S; Gotthardt M; Park J; Oyen WJG; deJong M; Hellmich MR; Rutjes FPJT; van Delft FL ; Boerman OC Targeting of a CCK(2) receptor splice variant with 111In-labelled cholecystokinin-8 (CCK8) and 111In-labelled minigastrin. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 386–392. [DOI] [PubMed] [Google Scholar]
- (50).Chepurny OG; Holz GG; Roe MW; Leech CA GPR119 agonist AS1269574 activates TRPA1 cation channels to stimulate GLP-1 secretion. Mol. Endocrinol 2016, 30, 614–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Gotthardt M; van Eerd-Vismale J; Oyen WJG; de Jong M ; Zhang H; Rolleman E; Maecke HR; Behe M; Boerman O Indication for different mechanisms of kidney uptake of radiolabeled peptides. J. Nucl. Med 2007, 48, 596–601. [DOI] [PubMed] [Google Scholar]
- (52).Sauter AW; Mansi R; Hassiepen U; Muller L; Panigada T; Wiehr S; Wild A-M; Geistlich S; Behe M; Rottenburger C; Wild D; Fani M Targeting of the cholecystokinin-2 receptor with the minigastrin analog Lu-177-DOTA-PP-F11N: does the use of protease inhibitors further improve in vivo distribution? J. Nucl. Med 2019, 60, 393–399. [DOI] [PubMed] [Google Scholar]
- (53).Kaloudi A; Nock BA; Lymperis E; Krenning EP; de Jong M; Maina T 99mTc-labeled gastrins of varying peptide chain length: Distinct impact ofNEP/ACE-inhibition on stability and tumor uptake in mice. Nucl. Med. Biol 2016, 43, 347–354. [DOI] [PubMed] [Google Scholar]
- (54).Roettger BF; Ghanekar D; Rao R; Toledo C; Yingling J; Pinon D; Miller LJ Antagonist-stimulated internalization of the G protein-coupled cholecystokinin receptor. Mol. Pharmacol 1997, 51, 357–362. [PubMed] [Google Scholar]
- (55).Akgun E; Korner M; Gao F; Harikumar KG; Waser B; Reubi JC; Portoghese PS; Miller LJ Synthesis and in vitro characterization of radioiodinatable benzodiazepines selective for type 1 and type 2 cholecystokinin receptors. J. Med. Chem 2009, 52, 2138–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Breeman WAP; Froberg AC; de Blois E; van Gameren A; Melis M; de Jong M; Maina T; Nock BA; Erion JL; Macke HR; Krenning EP Optimised labeling, preclinical and initial clinical aspects of CCK-2 receptor-targeting with 3 radiolabeled peptides. Nucl. Med. Biol 2008, 35, 839–849. [DOI] [PubMed] [Google Scholar]
- (57).Kaloudi A; Nock BA; Lymperis E; Krenning EP; de Jong M; Maina T Improving the in vivo profile of minigastrin radiotracers: a comparative study involving the neutral endopeptidase inhibitor phosphoramidon. Cancer Biother. Radiopharm 2016, 31, 20–28 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
HPLC analysis of H-PEG3-[DGlu(OtBu)]4-Lys(Boc)-OH before and after purification, MALDI-TOF-MS spectrum of H-PEG3-[DGlu(OtBu)]4-Lys(Boc)-OH) HPLC analysis of Z-360-PEG3-[DGlu(OtBu)]4-Lys-(Boc)-OH before and after purification, MALDI-TOF-MS spectrum of Z-360-PEG3-[DGlu(OtBu)]4-Lys(Boc)-OH, HPLC analysis of Z-360-PEG3-(DGlu)4-Lys-OH, MALDI-TOF-MS spectrum of Z-360-PEG3-(DGlu)4-Lys-OH, HPLC analysis of Z-360-PEG3-(DGlu)4-Lys-[N4(Boc)4]-OH before and after purification, Figure S8: HPLC analysis of DGA1 before and after purification, MALDI-TOF-MS spectrum of DGA1, HPLC radiochromatogram of [99m]Tc-DGA1, specific cell association/internalization rates of [99mTc]Tc-DGA1 and [99mTc]Tc-DG2 in HEK293-CCK2R cells at 37 °C; Cell association/internalization of [99mTc]Tc-DGA1 and [99mTc]Tc-DG2 in HEK293-CCK2i4svR cells and of [99mTc]Tc-DGA1 in HEK293T cells at 37 °C at 1 h, including total, nonspecific, and specific values; and biodistribution of [99mTc]Tc-DGA1 and [99mTc]Tc-DG2 in SCID mice bearing twin HEK293-CCK2R-positive and HEK293-CCK2R-negative tumors at 1, 4, and 24 h pi (PDF)