

Abstract
A series of vancomycin C-terminus guanidine modifications are disclosed that improve antimicrobial activity, enhance the durability of antimicrobial action against selection or induction of resistance, and introduce a synergistic mechanism of action independent of D-Ala-D-Ala binding and inhibition of cell wall biosynthesis. The added mechanism of action results in induced bacterial cell permeability, which we show may involve interaction with cell envelope teichoic acid. Significantly, the compounds examined that contain two combined peripheral modifications, a (4-chlorobiphenyl)methyl (CBP) and C-terminus guanidinium modification, offer opportunities for new treatments against not only vancomycin-sensitive, but especially vancomycin-resistant bacteria where they act by two synergistic and now durable mechanisms of action independent of D-Ala-D-Ala/D-Lac binding and display superb antimicrobial potencies (MIC 0.6–0.15 μg/mL, VanA VRE). For the first time, we demonstrate that the synergistic behavior of the peripheral modifications examined requires the presence of both the CBP and guanidine modifications in a single molecule versus their combined use as an equimolar mixture of singly modified compounds. Finally, we show that a prototypical member of the series, G3-CBP-vancomycin (15), exhibits no hemolytic activity, displays no mammalian cell growth inhibition, possesses improved and especially attractive in vivo pharmacokinetic (PK) properties, and displays excellent in vivo efficacy and potency against an especially challenging multidrug-resistant (MRSA) and VanA vancomycin-resistant (VRSA) S. aureus bacterial strain.
Keywords: vancomycin, glycopeptide antibiotics, antibiotic resistance, antimicrobial mechanisms of action, guanidine modification, vancomycin peripheral modification
Graphical abstract
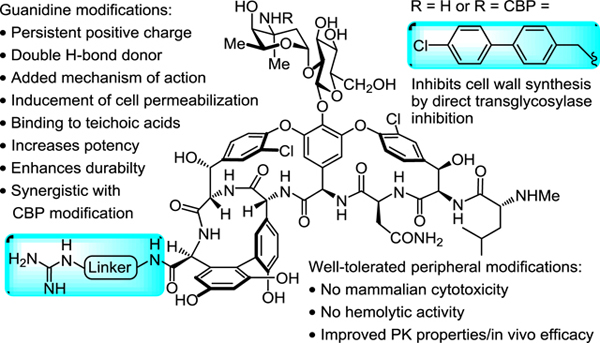
Vancomycin (1) and related glycopeptide antibiotics are one of the most important class of natural product drugs1–5. As the first member of the class, vancomycin has been used in the clinic for over 60 years and most recently as the antibiotic of the last resort for the treatment of infections caused by resistant Gram-positive pathogens, including methicillin-resistant S. aureus (MRSA)6–7. Vancomycin binds to the C-terminus D-Ala-D-Ala moiety of bacterial cell wall precursors and inhibits cell wall biosynthesis8–9. Clinical resistance to vancomycin was initially observed after 30 years of extensive use10–12, first in enterococci (VRE) followed by the recent emergence of vancomycin-resistant S. aureus (VRSA)13–14, which rank 4th and 5th on the WHO global priority list of antibiotic-resistant bacteria treats15. The mechanism of vancomycin clinical resistance found in these pathogens is a late stage remodeling of the bacteria cell wall precursor C-termini from D-Ala-D-Ala to D-Ala-D-Lac, reducing vancomycin binding affinity (1000-fold) and antimicrobial potency (1000-fold)16–18. This widespread (VRE) and emerging (VRSA) resistance presents an urgent need for the development of next generation glycopeptide antibiotics to overcome such resistance. In order to improve potency of the antibiotics against sensitive/resistant strains, peripheral modifications on vancomycin and related glycopeptides have been extensively investigated and some have been shown to enhance antibacterial potency or efficacy19–20, introduce additional mechanisms of action21–24, or improve pharmacokinetic properties25. As a result, three second generation glycopeptide antibiotics, telavancin26, dalbavancin27, and oritavancin28, have been recently approved for clinical use (Figure 1).
Figure 1.

Structure of vancomycin (1) and three clinically approved semisynthetic glycopeptide antibiotics.
We recently reported a new peripheral modification on vancomycin, a C-terminus quaternary trimethylammonium cation, which was found to enhance the antimicrobial potency of vancomycin and its analogues by introducing an additional mechanism of action (bacteria cell envelope permeabilization)29. This trimethylammonium salt modification was combined with a key vancomycin binding pocket modification that conveys dual D-Ala-D-Ala/D-Ala-D-Lac binding and directly overcomes the intrinsic molecular basis of vancomycin resistance30–33, and a well-known peripheral (4-chlorobiphenyl)methyl (CBP) modification that was established to inhibit transglycosylase and cell wall biosynthesis independent of D-Ala-D-Ala/D-Ala-D-Lac binding34–35. This provided antibiotics bearing multiple independent synergistic mechanisms of action that are highly potent against both vancomycin-sensitive and vancomycin-resistant pathogens with an even more enhanced durability. Further investigation into this added trimethylammonium salt resulted in the observation of the structure and site-specific29,36–37 nature of the modification and its conveyance of in vivo efficacy against VRSA and improved pharmacokinetic (PK) properties38. This included a documentation of the improved antibiotic durability, identification of a candidate cell membrane target responsible for the added functional activity, and demonstration that it conveyed improved physical (e.g., solubility) and pharmacological (e.g., PK) properties without introducing acute liabilities. The observed improvement on the properties of vancomycin and its derivatives by a simple trimethylammonium salt modification, which appears to serve as a permanently charged surrogate for a reversibly protonated dimethylamine, inspired us to explore related peripheral modifications. This was addressed by targeting modifications to vancomycin and (4-chlorobiphenyl)methyl-vancomycin (2, CBP-vancomycin) to provide analogues capable of expressing activity through as many as three independent and synergistic mechanisms of action, two of which are not dependent on D-Ala-D-Ala/D-Ala-D-Lac binding (Figure 2).
Figure 2.

A. Structure of CBP-vancomycin (2), C1-vancomycin (3), C1-CBP-vancomycin (4). B. Rationale in the introduction of a guanidine-containing modification.
A protonated guanidinium group (pKa = 13.2) could act as a better, more persistent positive charge under physiological conditions compared with tertiary amine (pKa = 10.6). In contrast to a quaternary trimethylammonium cation, a guanidinium group can also serve as a multiple hydrogen-bond donor, increasing binding affinity to anionic groups of biomolecules. In particular, phosphates found in the phospholipid/teichoic acid39 components of the cell envelope of Gram-positive pathogens implicated in studies with 4 may serve as potential binding sites for positively charged groups where a guanidinium cation would be expected to display improved interactions. Based on this rationale, we examined and herein report structure-activity relationship (SAR) studies and investigations on the mechanism of action of C-terminus guanidine modified vancomycin analogues (Scheme 1). Although these studies were conducted in part to lay the foundation for studies to be conducted on peripherally modified vancomycin analogues that contain key binding pocket modifications, they have provided results highlighting that 5 and related analogues offer opportunities for improved antibiotics active against not only vancomycin-sensitive, but also vancomycin-resistant bacteria even without incorporation of the binding pocket modifications.
Scheme 1.

Structure and synthesis of 5–24.
RESULTS AND DISCUSSION
C-Terminus Guanidine Modifications.
A G3 unit (a guanidinium-containing amine bearing a C3 linker) was chosen initially for introduction at the vancomycin C-terminus by amide coupling, resulting in the guanidine-modified vancomycin analogue 5 (Scheme 1). It is nearly identical to C1-vancomycin (3) disclosed in our previous reports29 with the only difference being the replacement of the trimethylammonium cation (C1) with a guanidine group at the C-terminus of the glycopeptide. The antimicrobial activity of 5 against vancomycin-resistant organisms (VanA VRE, 4 strains) was evaluated in a standard microdilution assay. We found that the introduction of this small and simple peripheral modification in 5 resulted in potent antimicrobial activity against VanA VRE (MIC = 4 to 16 μg/mL), representing not only a substantial improvement in antimicrobial potency compared with vancomycin (8 to 64-fold), but that it also proved more potent than C1-vancomycin (3, 4-fold) disclosed in our previous reports.
The structure-activity relationships (SAR) of the guanidine modification were explored to establish the source of the outstanding activity of 5. We selected a variety of guanidine-containing modifications with varied linker lengths (6–8) and linker rigidity (9). We also prepared compounds 10–12 that can be viewed as derivatives of compound 7 with an added side chain substituent. In order to demonstrate the effect of the positive charge of the guanidinium group, 13 was also prepared where the positively charged guanidinium group in 10 was replaced by a neutral urea group still capable of hydrogen bonding. Finally, an amine bearing two guanidine groups (14) was attached to vancomycin to examine the influence of an additional positive charge to the properties of the candidate antibiotic. Notably, 5 and the subsequent analogues 6–14 were prepared by direct amide bond coupling of the corresponding guanidine-containing amine with vancomycin without the introduction or removal of intermediate protecting groups. The antimicrobial activity of these analogues against vancomycin-resistant as well as vancomycin-sensitive organisms was determined. The compounds 6–9 maintained the superb antimicrobial potency of 5 against the four VanA VRE strains tested (Table 1), which indicates that the improved activity is insensitive to the linker length and rigidity within the small range examined. The examination of 10 and 12 revealed that the improved activity is not affected by the presence of charge-neutral linker substituents. However, compound 13 that replaced the protonated guanidinium group with a neutral urea group exhibited reduced activity against resistant strains (compared with 10)40. Interestingly and although not initially expected, the presence of an extra negatively charged side chain carboxylic acid led to a significant decrease in antimicrobial potency (8 to 32-fold) of the analogue (11). These observations demonstrate not only the importance of the protonated guanidinium positive charge, but also that the net positive charge change on the C-terminus is essential to the outstanding activity exhibited by these guanidine analogues40. It is also noteworthy that a 2 to 4-fold enhancement in antimicrobial potency against sensitive strains (S. aureus) was also observed for most of the guanidinium-containing vancomycin analogues beyond the already excellent activity of vancomycin itself. Finally, we note that Wender and coworkers independently have recently disclosed compound 12 as a simplification of a vancomycin-octaarginine conjugate (V-r8)41 with incorporation of single arginine (V-r1)42 and that it provided a vancomycin analogue which displayed activity against Gram-negative bacteria and maintained the activity of vancomycin against Gram-positive organisms.
Table 1.
Antimicrobial activity of 1 and 5–14 against four VanA VRE strains, MIC (μg/mL).a
| Compound | VanA E. faecalis | VanA E. faecium | VanA E. faecalis | VanA E. faecium |
|---|---|---|---|---|
| BM 4166 | ATCC BAA-2317 | ATCC BAA-2573 | TX2465 | |
| vancomycin (1) | 250 | 250 | 125 | 250 |
| G3-vancomycin (5) | 16 | 4 | 4 | 16 |
| G2-vancomycin (6) | 16 | 4 | 8 | 16 |
| G4-vancomycin (7) | 16 | 4 | 4 | 16 |
| G6-vancomycin (8) | 16 | 4 | 4 | 16 |
| GBn-vancomycin (9) | 16 | 4 | 4 | 8 |
| Arg(OMe)-vancomycin (10) | 8 | 8 | 4 | 8 |
| Arg-vancomycin (11) | 63 | 63 | 125 | 125 |
| Arg(NH2)-vancomycin (12) | 16 | 4 | 4 | 8 |
| Cit(OMe)-vancomycin (13) | 63 | 31 | 16 | 63 |
| DiG-vancomycin (14) | 8 | 1 | 2 | 4 |
MIC = minimum inhibitory concentration.
The effect of the incorporation of the guanidinium modifications into CBP-vancomycin (2) was subsequently examined. By virtue of the incorporation of the CBP group, CBP-vancomycin (2) already possess one effective mechanism of action independent of D-Ala-D-Ala/D-Ala-D-Lac binding against vancomycin-resistant strains (transglycosylase inhibition)34–35. The guanidinium-containing CBP-vancomycin analogues were prepared from 2 in a single and scalable step by direct amide coupling of the corresponding guanidinium-containing amine without intermediate protection or deprotection (Scheme 1). To our delight, the incorporation of guanidine group at the C-terminus of 2 further enhanced the antimicrobial potency of the analogues (2 to 8-fold) against the four VanA VRE strains tested (Table 2). The net positive charge at the C-terminus was found again to be crucial to the antimicrobial potency, where 21 and 23 exhibited no enhanced antimicrobial potency. Incorporation of two guanidine groups (24) offered no further improvement, and both G3-CBP-vancomycin (15) and GBn-CBP-vancomycin (19) emerged as representative of the most effective compounds in the series. Notable is the superb sub μg/mL activity of the analogues (0.6–0.15 μg/mL) against the small panel of VanA VRE that is derived from two peripheral modifications providing two independent and synergistic mechanisms of action, neither of which directly impact D-Ala-D-Ala/D-Ala-D-Lac binding. Combined, the CBP and guanidine modifications increased the VanA VRE activity as much as 1000-fold relative to vancomycin itself.
Table 2.
Antimicrobial activity of 1, 2 and 15–24 against VanA VRE strains, MIC (μg/mL).
| Compound | VanA E. faecalis | VanA E. faecium | VanA E. faecalis | VanA E. faecium |
|---|---|---|---|---|
| BM 4166 | ATCC BAA-2317 | ATCC BAA-2573 | TX 2465 | |
| vancomycin (1) | 250 | 250 | 125 | 250 |
| CBP-vancomycin (2) | 5 | 2.5 | 0.3 | 5 |
| G3-CBP-vancomycin (15) | 0.6 | 0.3 | 0.15 | 0.6 |
| G2-CBP-vancomycin (16) | 0.6 | 0.3 | 0.15 | 1.2 |
| G4-CBP-vancomycin (17) | 0.6 | 0.3 | 0.15 | 0.6 |
| G6-CBP-vancomycin (18) | 0.6 | 0.3 | 0.3 | 1.2 |
| GBn-CBP-vancomycin (19) | 0.6 | 0.3 | 0.15 | 0.6 |
| Arg(OMe)-CBP-vancomycin (20) | 0.6 | 0.3 | 0.15 | 0.6 |
| Arg-CBP-vancomycin (21) | 1.2 | 1.2 | 0.15 | 2.5 |
| Arg(NH2)-CBP-vancomycin (22) | 0.6 | 0.3 | 0.15 | 0.6 |
| Cit(OMe)-CBP-vancomycin (23) | 1.2 | 2.5 | 0.3 | 5 |
| DiG-CBP-vancomycin (24) | 0.6 | 0.6 | 0.15 | 0.3 |
Just as significantly and as highlighted earlier, the guanidine-containing modifications also increased the antimicrobial activity of both vancomycin or CBP-vancomycin against sensitive bacteria strains by 2 to 4-fold, albeit being less pronounced because of the already superb activity of vancomycin or CBP-vancomycin. This is illustrated with both a sensitive and methicillin-resistant S. aureus strain and an insensitive VanB VRE strain in Table 3.
Table 3.
Antimicrobial activity of 1, 2, 5, 9, 15 and 19 against VanB and S. aureus strains, MIC (μg/mL).
| Compound | VanB E. faecalis | Sensitive S. aureus | MRSA S. aureus |
|---|---|---|---|
| ATCC 51299 | ATCC 25923 | ATCC 43300 | |
| vancomycin (1) | 8 | 0.5 | 0.5 |
| G3-vancomycin (5) | 8 | 0.25 | 0.12 |
| GBn-vancomycin (9) | 8 | 0.25 | 0.12 |
| CBP-vancomycin (2) | 0.08 | 0.08 | 0.08 |
| G3-CBP-vancomycin (15) | 0.04 | 0.04 | 0.02 |
| GBn-CBP-vancomycin (19) | 0.04 | 0.04 | 0.02 |
Site Specific Nature of the Guanidine Modification.
Analogous to observations made in our studies with the trimethylammonium salts37, the introduction of the guanidinium group as an A-ring substituent versus at the C-terminus provided less active analogues, highlighting the site selectivity of the impact of the modification (Supporting Information Table S4). Similarly, a C-terminus and A-ring doubly modified guanidinium analogue failed to improve on the activity of the corresponding single C-terminus modification. Finally, and like the C-terminus trend, an A-ring guanidinium modification did prove to be more potent than a corresponding A-ring trimethylammonium salt (C1) analogue (Supporting Information Table S4).
Activity Against Gram-negative Bacteria.
As noted earlier, Wender and coworkers independently disclosed compound 12 as a simplification of a vancomycin-octaarginine conjugate (V-r8)41 with incorporation of single arginine (V-r1)42 and that it provided a vancomycin analogue that displayed activity against Gram-negative bacteria. The generalizability of this observation was investigated with the C-terminus guanidine modified vancomycins (5 and 9) and CBP-vancomycins (15 and 19) and their activity against a small panel of Gram-negative bacteria. Reduced strength broth conditions were required to observe the activity of these analogues, but the C-terminus guanidine modifications in 5 and 9 conveyed potent activity against the small panel of Gram-negative bacteria, increasing activity by 4 to 32-fold and with some MICs as low as 1–2 μg/mL (Supporting Information Table S3). It is noteworthy that although the CBP modification provided an additional mechanism of action and improved activity against Gram-positive bacteria, it was found to reduce this activity of 5 and 9 against Gram-negative bacteria in the small series of compounds examined.
G3-CBP-Vancomycin is a Durable Antibiotic.
We have demonstrated that vancomycin analogues with multiple synergistic mechanisms of action not only exhibit superb potency against vancomycin-resistant organisms, but that they also display an even more important enhanced durability where the rate of emergence of bacterial resistance significantly decreased with each added mechanism of action29. Such durable antibiotics expressing multiple mechanisms of action are expected to suppress the emergence of bacterial resistance even with extensive use. In addition to the importance of demonstrating the enhanced durability, such comparative studies also provide indirect evidence of an added mechanism of action. Thus, in order to establish whether the guanidine-modified vancomycin analogues may express an added antimicrobial mechanism of action, a multi-passage resistance development assay was conducted initially for 25 days, but then extended to 50 days to further illustrate the superb durability. This was conducted with two VanA VRE strains already resistant to vancomycin (MIC = 250 μg/mL), notably no longer susceptible to its cell wall biosynthesis blockage through D-Ala-D-Ala binding and transpeptidase inhibition (Figure 3). Thus, the studies were not only conducted over a longer period than is typical (50 vs 25 days), but were also conducted with vancomycin-resistant organisms that overcome the intrinsic mechanism of action of vancomycin (D-Ala-D-Ala binding) and that are further resistant to many other antibiotic classes43 by virtue of assimilation of additional common mechanisms of resistance. As such, they are on the verge of becoming multidrug and vancomycin-resistant organisms. G3-Vancomycin (5) exhibited a better durability (4 to 8-fold increase in MIC, 1 MOA) than CBP-vancomycin (2, 16 to 32-fold increase in MIC, 1 MOA) over 25 days, and G3-CBP-vancomycin (15), the analogue bearing the two peripheral modifications, exhibited a further enhanced durability (2-fold increase in MIC). Even with extension of the study to 50 days, only a subtle 4-fold increase in MIC was observed with G3-CBP-vancomycin (15), and the antibiotic remains potent (MIC = 1.2–2.5 μg/mL) against the two VanA VRE strains at the end of the study. In contrast, the analogues 2 and 5 with single modifications have lost their activity and display MIC values similar to vancomycin by the end of resistance development assay. Moreover, and with these two VanA VRE strains, the frontline therapies daptomycin, linezolid, and tigecycline raise resistance more rapidly and in a more pronounced manner than either 2 or 529, further highlighting the remarkable behavior of 15 (see Supporting Information Figure S1).
Figure 3.

Resistance acquisition on serial passaging of two strains of VanA VRE in the presence of 0.5 × MIC levels of compound: CBP-vancomycin (2), G3-vancomycin (5) and G3-CBP-vancomycin (15).
These results demonstrate clearly the superb durability of 15, the important consequence of combining the two peripheral modifications as compared with derivatives bearing only a single modification (2 and 5), and illustrate that each peripheral modification and independent mechanism of action are critical in such improvements in durability. Thus, emergence of resistance against one mechanism of action is suppressed by the other when two synergistic mechanisms of action are combined. We observed a similar result in our previous work, where C1-CBP-vancomycin (4) exhibited an enhanced durability compared with CBP-vancomycin (2) due to an added mechanism of action provided by the newly introduced trimethylammonium cation. However, the extended durability of G3-CBP-vancomycin (15) described herein exceeds that of C1-CBP-vancomycin (4), which we attribute to the more potent and robust activity derived from the G3 versus C1 modification. Combined with results of their antimicrobial activity, the studies confirm that an added mechanism of action is introduced by incorporation of the guanidinium group and that G3-CBP-vancomycin (15) and related compounds exhibit their activity through two synergistic and independent mechanisms of action against vancomycin-resistant organisms, both of which are independent of D-Ala-D-Ala/D-Lac binding.
The Guanidine Modification Induces Bacteria Cell Permeabilization: An Added Synergistic Mechanism of Action.
We previously reported that the C-terminus trimethylammonium cation (C1) modification on vancomycin induces bacteria cell permeabilization as well as the enhancement of antimicrobial activity29, as indicated by propidium iodide (PI) influx. While the permeabilization effect was found to be weaker with C1-vancomycin (3) even at high concentration (100 μM), C1-CBP-vancomycin (4), bearing both peripheral modifications, was able to induce pronounced membrane permeabilization. This observation suggests a synergistic nature of the two peripheral modifications, where the presence of one modification (CBP) enhances the effect of the other (C1). We also observed that this inducement of bacterial membrane permeabilization and the resulting enhancement in antimicrobial activity is both structure (C1 vs other trialkylammonium salt) and site (C-terminus of vancomycin vs N-terminus or A-ring) specific36–37. In order to establish whether a similar mechanism of action is introduced by the peripheral guanidine modification, representative members of the guanidine-containing vancomycin analogues were examined in a permeability assay and conducted alongside 1, 2 and 4 (Figure 4).
Figure 4.

Examination of cell membrane permeability induced by compounds 1, 2, 4, 15 and 19 (10 μM added at 5 min) and compound 5 (50 μM added at 5 min) in VanA VRE E. faecalis BM 4166 and E. faecium ATCC BAA-2317.
While no permeabilization was induced by either vancomycin itself or CBP-vancomycin, weak permeabilization was observed upon addition of G3-vancomycin (5) at an elevated concentration (50 μM), which is accordance with our observations with C1-vancomycin. G3-CBP-vancomycin (15) and GBn-CBP-vancomycin (19) induced strong membrane permeabilization which is comparable or stronger than that observed with C1-CBP-vancomycin (4). A reduced bacteria cell permeabilization was observed with analogues exhibiting weaker antimicrobial potency (21 and 23), where both the initial rate and final extent of the fluorescence signal was reduced (Supporting Information Figure S2). These observations reveal that guanidine-containing vancomycin analogues induce bacterial cell permeabilization, and that the intensity of the signal in the assay directly correlates with the antimicrobial activity of such analogues. Combined with the observations made in the durability assay and similar to the observations made with the trimethylammonium salt vancomycin analogues, the C-terminus incorporation of a guanidine into vancomycin and CBP-vancomycin introduced an additional mechanism of action, resulting in membrane permeabilization independent of D-Ala-D-Ala/D-Lac binding and transglycosylase inhibition.
Exogeneous Lipoteichoic Acid Reduces Antimicrobial Activity and Blocks Induced Bacteria Cell Permeability.
A guanidinium group, serving as both a persistent positive charge under physiological conditions and a multiple hydrogen bond-donor, might well be expected to interact with negatively charged phosphate or carboxylate containing components in the bacteria cell envelope. Therefore, these components are among the possible specific or non-specific binding partners for the guanidinium-containing vancomycin analogues. During the investigation of candidate targets of our earlier trimethylammonium salt modification, we observed that cell envelop teichoic acid (TA) behaved as a potential binding partner38. Teichoic acids are major constituents found in the cell envelop of Gram-positive bacteria either in the form of lipoteichoic acids (LTA) that are anchored in the cell membrane and extend into or through the peptidoglycan cell wall44 or cell wall teichoic acids (WTA) that are covalently linked to N-acetylmuramic acid in the cell wall peptidoglycan45.44 These polyanionic alditol phosphate-containing polymers contribute to cell envelop stability and rigidity, cation (e.g., Mg2+) homeostasis and transport, and are responsible for binding, sequestration, and regulation of autolysins that act to degrade the bacteria cell wall in response to the need for damage repair or replication expansion44–46. In order to establish whether such TAs may also serve as binding partners for the guanidinium-containing vancomycin analogues like cationic peptide antibiotics47,48 that in turn may competitively displace sequestered autolysins, the microdilution antimicrobial assay was conducted in the presence of added LTA (100 and 1000 μg/mL) as a competitive surrogate for both LTA and WTA (Table 4). Whereas the activity of vancomycin (no difference) and CBP-vancomycin (decrease by 2-fold) are insensitive to the added LTA, the antimicrobial activity of both 15 and 19 decreased 8-fold (100 μg/mL LTA). Because of its 10-fold higher MIC and the larger amounts of compound needed for bacterial cell growth inhibition, 5 displayed the same behavior but required a compensating larger amount of added LTA (1000 μg/mL). This impact of LTA concentration on blocking the antimicrobial activity of 5 and 15 was studied with one VanA VRE E. faecium strain (ATCC BAA-2317) and the results are summarized in Supporting Information Table S5. The presence of either NH2CH2CH2CH2NHC(=NH2)NH2+ (G3+, 100 μg/mL) or an inorganic divalent cation (Mg2+, 100 μg/mL) had no effect on this observation, revealing the requirement for covalent attachment of this G3 group to vancomycin for diverted binding. In the case of vancomycin (not shown) and CBP-vancomycin and although not the objective of these studies, the addition of G3 also did not lead to an improvement in the activity, indicating that it must be incorporated into the molecule to have its impact (compare 2 + G3 vs 15). Unlike LTA, inorganic phosphate anion (H2PO4−, 100 μg/mL) or a phospholipid (POPE, 100 μg/mL) found in bacteria cell membranes did not alter the MIC of 15, indicating that the diverted binding observed is selective for TA. Notably, exogeneous LTA only decreases the antimicrobial potency of G3-CBP-vancomycin (15) and GBn-CBP-vancomycin (19) to the level of CBP-vancomycin (2) rather than completely abolishing the activity. These observations are in accordance with competitive binding to exogeneous LTA serving only to disrupt association with the target responsible for the guanidinium-derived mechanism of action. This was further confirmed in the bacteria cell permeabilization assay where the presence of exogenous LTA nearly or completely abolished the permeabilization induced by 15 (Figure 5).
Table 4.
Impact of added lipoteichoic acid (LTA) and other anionic additives on the antimicrobial activity of vancomycin derivatives, MIC (μg/mL).a
| Compound | VanA VRE E. faecalis | VanA VRE E. faecium | ||
|---|---|---|---|---|
| BM 4166 | ATCC BAA-2317 | |||
| Condition | −LTA | +LTA | −LTA | +LTA |
| vancomycin (1) | 250 | 250 | 250 | 250 |
| CBP-vancomycin (2) | 5 | 10 | 2.5 | 5 |
| G3-vancomycin (5) | 16 | 16 | 4 | 8 (63)b |
| G3-CBP-vancomycin (15) | 0.6 | 5 | 0.3 | 2.5 (10)b |
| GBn-vancomycin (9) | 16 | 16 | 4 | 4 |
| GBn-CBP-vancomycin (19) | 0.6 | 5 | 0.3 | 2.5 |
| CBP-vancomycin (2) + G3+ | 5 | 10 | 2.5 | 5 |
| G3-CBP-vancomycin (15) + G3+ | 0.6 | 5 | 0.3 | 2.5 |
| G3-CBP-vancomycin (15) + Mg2+ | 0.6 | 5 | 0.3 | 2.5 |
| Condition | −NaH2PO4 | +NaH2PO4 | −NaH2PO4 | +NaH2PO4 |
| G3-vancomycin (5) | 16 | 16 | 4 | 4 |
| G3-CBP-vancomycin (15) | 0.6 | 0.6 | 0.3 | 0.3 |
| Condition | −POPE | +POPE | −POPE | +POPE |
| G3-vancomycin (5) | 16 | 16 | 4 | 4 |
| G3-CBP-vancomycin (15) | 0.6 | 0.6 | 0.3 | 0.3 |
| G3+ | >800 | >800 | ||
Assays run in the absence or presence of additives (lipoteichoic acid (LTA), NaH2PO4, or POPE, 100 μg/mL) as well as the absence or presence of H2N(CH2)3NHC(=NH2)NH2+ (G3+, 100 μg/mL) or Mg2+ (MgCl2, 100 μg/mL).
With 1000 μg/mL LTA.
Figure 5.

Examination of cell membrane permeability induced by compounds 1, 2, 15 (10 μM added at 5 min) and compound 15 in the presence of 100 μg/mL exogeneous LTA in VanA VRE E. faecalis BM 4166 and E. faecium ATCC BAA-2317.
Synergistic Activity of the Peripheral Modifications Requires Incorporation into Single Molecule.
With the establishment of this newly introduced mechanism of action by the guanidine modification, we conducted a study to determine whether the synergistic behavior of the two mechanisms of action attributable to the CBP and guanidine modifications requires the two to be located on the same glycopeptide molecule. The antimicrobial potency of a 1:1 mixture of CBP-vancomycin (2) and G3-vancomycin (5) or GBn-vancomycin (9) was tested alongside G3-CBP-vancomycin (15) and GBn-CBP-vancomycin (19) (Table 5). Unlike the synergistic activity observed with 15 and 19, the equimolar mixtures of the singly modified vancomycins did not display this enhanced potency, exhibiting antimicrobial activity only at the level of the most potent compound in the mixture (CBP-vancomycin, 2). Thus, the expression of the synergistic activity observed with 15 and 19 requires that both peripheral modifications be incorporated in a single molecule. A study was also conducted with our earlier C1 trimethylammonium cation modification and provided analogous results (Table 5). Although we were uncertain what to expect before conducting the study, the results with the singly modified antibiotic combinations are consistent with simple additive effects of the two antibiotics and observation of only the potency of the most active compound in the mixture. This typical behavior stands in contrast to the synergistic, and we would suggest special, activity observed when both peripheral modifications are found in a single molecule.
Table 5.
Antimicrobial activity of equimolar mixtures of CBP-vancomycin (2) and C-terminus vancomycin analogues (3, 5 and 9) against VanA E. faecium, presented alongside the individual activity of compounds 2, 3, 4, 5, 9, 15, 19, MIC (μg/mL).
| Compound | VanA E. faecium |
|---|---|
| ATCC BAA-2317 | |
| CBP-vancomycin (2) | 2.5 |
| G3-vancomycin (5) | 4 |
| G3-CBP-vancomycin (15) | 0.3 |
| CBP-vancomycin (2) + G3-vancomycin (5) | 2.5 + 2.5 |
| GBn-vancomycin (9) | 4 |
| GBn-CBP-vancomycin (19) | 0.3 |
| CBP-vancomycin (2) + GBn-vancomycin (9) | 2.5 + 2.5 |
| C1-vancomycin (3) | 31 |
| C1-CBP-vancomycin (4) | 0.3 |
| CBP-vancomycin (2) + C1-vancomycin (3) | 2.5 + 2.5 |
Additional Key Properties.
Neither 5 or 15 exhibited red blood cell hemolytic activity derived from cell membrane lysis or disruption even at concentrations >100-fold above their MICs (Supporting Information Figure S3). Similarly, neither 5 or 15 displayed mammalian cell toxicity (IC50 >20 μM) when assessed for growth inhibition against the NIH/3T3 (mouse embryonic fibroblasts), HepG2 (human liver cancer cell line) and HCT116 (human colon cancer) cell lines.
Although a maximum tolerated dose (MTD) for 15 was not established, we did find that it is tolerated at doses up to and including 50 mg/kg in the following PK studies in mice. In earlier studies, we reported a MTD of 75 mg/kg for CBP-vancomycin (2) and 50 mg/kg for C1-CBP-vancomycin (4). Not surprisingly given the safety and tolerability of oritavancin that bears a peripheral CBP substituent, the CBP-vancomycin (2) MTD in these studies was found to be only 4-fold lower than vancomycin (MTD = 300 mg/kg). The further addition of the trimethylammonium cation in 4 did not significantly alter this tolerability and the addition of the guanidine modification in 15 detailed herein proved to be at least as well tolerated if not better (MTD ≥50 mg/kg). Notably, CBP-vancomycin (2, ca. 100-fold), C1-CBP-vancomycin (4, ca. 1000-fold) and G3-CBP-vancomycin (15, ca.1000-fold) are progressively more potent than vancomycin, and would be administered at accordingly much lower doses, making the small MTD distinctions even more impressive. These studies establish that there is no significant additional acute toxicity associated with the added guanidinium (or trimethylammonium cation) modification, a key question initial studies were designed to answer.
The in vivo PK properties of 15 in mice (iv, n = 3/time point @ 50 and 10 mg/kg) were established and conducted side-by-side with 4, which we characterized earlier38 (Table 6). These earlier studies revealed that 4 (t1/2 = 5 h), bearing the CBP group and trimethylammonium cation, improved the short terminal half-life (0.5–1.3 h, t1/2), lower exposure (AUC), consistent volume of distribution (Vd), and rapid clearance (CL) of vancomycin and mitigated the poor dose proportionality49 and extended terminal half-life (t1/2) of CBP-vancomycin (2) that makes clinical administration of the structurally related drug oritavancin challenging (Supporting Information Table S6). Like vancomycin and 4, 15 displayed well behaved dose proportional PK across all parameters, whereas CBP-vancomycin (2) does not, exhibiting a relatively lower plasma exposure at the higher dose (Supporting Information Table S6). Compound 15 improved the short terminal half-life, lower exposure, and rapid clearance of vancomycin like 4. It displayed overall PK parameters similar to 4, but was found to exhibit an improved Cmax, now approaching that of vancomycin itself, and improved AUC while maintaining the excellent terminal half-life in mice (t1/2 4.3–4.4 h). Notably, the improved terminal half-life relative to vancomycin along with other properties illustrate that 15 and its guanidine modification are not subject to rapid metabolism. These preliminary studies establish that 15, bearing both the peripheral CBP group and guanidine modification, displays substantial in vivo PK improvements over both vancomycin (1) and CBP-vancomycin (2).
Table 6.
Comparison PK properties of 4 and 15.a
| Parameter | CI-CBP-vancomycin (4) | G3-CBP-vancomycin (15) | |
|---|---|---|---|
| 10 mg/kg | 10 mg/kg | 50 mg/kg | |
| Cmax (Mg/mL) | 14.5 | 35.9 | 152 |
| tmax (h) | 0.08 | 0.14 | 0.28 |
| AUC (Mg-h/mL) | 48.4 | 66.0 | 312 |
| Vss (L/kg) | 0.64 | 0.41 | 0.35 |
| CL (L/h/kg) | 0.12 | 0.09 | 0.09 |
| MRTINF obs (h) | 5.6 | 3.9 | 4.6 |
| t1/2 (h) | 5.1 | 4.4 | 4.3 |
Compounds administered iv @ 50 or 10 mg/kg in mice (n = 3/time point, measured at 0.083, 0.25, 0.5, 1, 2, 4, 6, 8 and 24 h).
In vivo Efficacy.
Finally, an in vivo efficacy study of 15 was conducted in neutropenic mice (n = 5/dosed group) using a well-established thigh infection model with an especially challenging VanA VRSA strain (VRS250). This S. aureus strain is representative of an especially challenging class of antibiotic-resistant pathogens, being both multidrug-resistant (MRSA) and vancomycin-resistant (VRSA). Therefore, the results are remarkable especially with this challenging pathogen. Following the experimental protocol of our previous in vivo efficacy study with C1-CBP-vancomycin (4)38, the right thigh of each mouse was injected intramuscularly (im) with the pathogen. After 2 h, control vehicle, linezolid (po, 50 mg/kg × 2) and 15 (sc, 12.5–100 mg/kg) were administered. After 26 h, the bacterial CFU/g of harvested thighs was established and compared with baseline bacterial count at 2 h and vehicle control at 26 h. While vancomycin was found to be inactive in this model50, a nearly 3-log10 reduction in bacterial count was observed after treatment with 15 (50 or 100 mg/kg), outperforming linezolid (administered po twice at 2 and 12 h) (Figure 6). Notably, a >2-log10 reduction was observed even at the lower doses of 15 (12.5 and 25 mg/kg), outperforming 2 and 4 at the same dose (Figure S4).38
Figure 6.

Efficacy of G3-CBP-vancomycin (15) and linezolid against the multidrug resistant and vancomycin-resistant S. aureus (VRSA) strain VRS-2 in the mouse thigh infection model (n = 5/dose). Compound 15 and control vehicle were administered sc once at 2 h at the doses indicated. The reference standard linezolid was administered orally (po) twice at 2 and 12 h at 50 mg/kg. Left: dose-dependent reduction in bacterial load. Right: dose-dependent bactericidal effect (relative to 2 h baseline).
CONCLUSIONS
Inspired by the discovery of the productive impact of a vancomycin C-terminus trimethylammonium cation modification and its underlying mechanism, we examined an analogous and further improved guanidine modification. The rationale for the studies, the evolution of the improvements, and the impact of this new peripheral modification on antimicrobial potency are summarized in Figure 7. This C-terminus guanidine modification improves antimicrobial activity, enhances the durability of action against selection of resistance, and introduces a synergistic mechanism of action independent of D-Ala-D-Ala/D-Lac binding and inhibition of cell wall biosynthesis. The added mechanism of action results in induced bacterial cell permeability, which we suggest involves interaction with teichoic acid in the cell envelope. For the first time, we show that the synergistic behavior of the combined peripheral modifications we identified requires the presence of both the CBP and guanidinium modifications (or C1 modification) in a single molecule versus their combined use as an equimolar mixture of singly modified compounds. Finally, we show that a prototypical member of the series, G3-CBP-vancomycin (15), exhibits no hemolytic activity, displays no mammalian cell growth inhibition, possesses especially attractive in vivo PK properties, and exhibits exciting in vivo efficacy and potency against an especially challenging multidrug-resistant (MRSA) and VanA vancomycin-resistant (VRSA) S. aureus bacterial strain (VRS-2). Although the inspiration for the studies has been the incorporation of such combined peripheral modifications into pocket modified vancomycin analogues29–33,51–55, their incorporation into vancomycin (1) or CBP-vancomycin (2) provide attractive new semisynthetic vancomycin analogues that act by two or three independent and synergistic mechanisms of action, only one of which is dependent on D-Ala-D-Ala binding. Significantly, those that contain the two combined peripheral modifications offer attractive opportunities for new treatments against not only vancomycin-sensitive, but especially for vancomycin-resistant bacteria even without incorporation of binding pocket modifications where they still can act by two synergistic and now durable mechanisms of action and display superb antimicrobial potencies (MIC 0.6–0.15 μg/mL, VanA VRE). As such, the C-terminus guanidine modifications found in G3-vancomycin (5), G3-CBP-vancomycin (15) and related analogues herein constitute new prototypical representatives of an exciting new class of vancomycin analogues that include not only our earlier C1-vancomycin (3) and C1-CBP-vancomycin (4), but also other basic or cationic C-terminus modifications disclosed in the recent studies of Cooper56, Halder24 and most notably Wender41–42. The latter recently disclosed that simplification of a vancomycin-octaarginine conjugate (V-r8)41 with incorporation of single arginine (V-r1)42 provided a vancomycin analogue that displayed activity against Gram-negative bacteria and maintained the activity of vancomycin against Gram-positive organisms.
Figure 7.

Evolution and comparison of the C-terminus cationic modifications examined.
METHODS
Reagents and solvents were purchased from commercial sources and used as received unless otherwise noted. 1H and 13C NMR spectra were obtained using a Bruker Avance III HD 600 MHz spectrometer equipped with either a 5 mm QCI or 5 mm CPDCH probe or a Bruker Avance III 500 MHz spectrometer equipped with a 5 mm BBFO probe at 298 K. Residual solvent peaks were used as an internal reference. Coupling constants (J) (H, H) are given in Hz. Coupling patterns are designated as singlet (s), doublet (d), triplet (t), quadruplet (q), multiplet (m), or broad signal (br). High resolution mass spectra were measured with a TOF mass spectrometer. Analytical and preparative reverse-phase HPLC was performed using a Waters HPLC and an Agilent HPLC-MS. In vitro antimicrobial activity was determined on samples established to be ≥95% pure (HPLC: Nacalai Tesque, Inc., ARII-C18, 5 μm, 10 × 150 mm, 1–40% (analogues 5-14) or 20–80% (analogues 15-24) MeCN/H2O–0.07% TFA gradient over 40 min, 3 mL/min) through a standard microdilution assay. UV-Vis signal of hemolysis assay and cytotoxicity assay and flourescence signal of bacteria cell permeability assay was recorded with a TECAN plate reader.
Supplementary Material
ACKNOWLEDGEMENTS
We gratefully acknowledge financial support from the NIH (CA041101 to D.L.B.) and a JITRI-Scripps Fellowship (Z.-C.W.). We thank Jelena Momirov, Nilanjana Chakraborty, and Rosa Al-Herbawi for conducting the cell growth inhibition (cytotoxic activity) assays. We especially thank Eurofins Pharmacology Discovery Services Taiwan, Ltd., a partner lab of Eurofins Pharma Discovery Services, for conduct of the in vivo efficacy study.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://doi.org/10.1021/acsinfecdis.0c00258.
Full experimental details for synthesis of 5–24, in vitro antimicrobial assay, site specificity of guanidine modified analogues, resistance development assay, permeability assay, assays for lipoteichoic acid impact on antimicrobial activity and membrane permeability, hemolysis assay, PK studies, and in vivo efficacy studies (PDF)
The authors declare no competing financial interest.
REFERENCES
- 1.McCormick MH (1956) Vancomycin, a new antibiotic. I. Chemical and biologic properties. Antibiot. Annu 3, 606–611. [PubMed] [Google Scholar]
- 2.Perkins HR (1982) Vancomycin and related antibiotics. Pharmacol. Ther 16, 181–197 DOI 10.1016/0163-7258(82)90053-5. [DOI] [PubMed] [Google Scholar]
- 3.Van Bambeke F, Van Laethem Y, Courvalin P, Tulkens PM (2004) Glycopeptide antibiotics. Drugs 64, 913–936 DOI 10.2165/00003495-200464090-00001. [DOI] [PubMed] [Google Scholar]
- 4.Levine D. (2006) Vancomycin: a history. Clin. Infect. Dis 42, S5–S12 10.1086/491709. [DOI] [PubMed] [Google Scholar]
- 5.Kahne D, Leimkuhler C, Lu W, Walsh C. (2005) Glycopeptide and lipoglycopeptide antibiotics. Chem. Rev 105, 425–448 DOI 10.1021/cr030103a. [DOI] [PubMed] [Google Scholar]
- 6.Barna JCJ, Williams DH (1984) The structure and mode of action of glycopeptide antibiotics of the vancomycin group. Annu. Rev. Microbiol 38, 339–357 DOI 10.1146/annurev.mi.38.100184.002011. [DOI] [PubMed] [Google Scholar]
- 7.Hubbard BK, Walsh CT (2003) Vancomycin assembly: nature’s way. Angew. Chem., Int. Ed 42, 730–765 DOI 10.1002/anie.200390202. [DOI] [PubMed] [Google Scholar]
- 8.Williams DH, Bardsley B. (1999) The vancomycin group of antibiotics and the fight against resistant bacteria. Angew. Chem., Int. Ed 38, 1172–1193 DOI 10.1002/(SICI)1521-3773(19990503)38:9<1172::AID-ANIE1172>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 9.Perkins HR (1982) Vancomycin and related antibiotics. Pharmacol. Ther 16, 181–197 DOI 10.1016/0163-7258(82)90053-5. [DOI] [PubMed] [Google Scholar]
- 10.Leclercq R, Derlot E, Duval J, Courvalin P. (1988) Plasmid-mediated resistance to vancomycin and teicoplanin in Enterococcus faecium. N. Engl. J. Med 319, 157–161 DOI 10.1056/NEJM198807213190307. [DOI] [PubMed] [Google Scholar]
- 11.Pootoolal J, Neu J, Wright GD (2002) Glycopeptide antibiotic resistance. Annu. Rev. Pharmacol. Toxicol 42, 381–408 DOI 10.1146/annurev.pharmtox.42.091601.142813. [DOI] [PubMed] [Google Scholar]
- 12.Courvalin P. (2006) Vancomycin resistance in Gram-positive cocci. Clin. Infect. Dis 42, S25–S34 DOI 10.1086/491711. [DOI] [PubMed] [Google Scholar]
- 13.Weigel LM, Clewell DB, Gill SR, Clark NC, McDougal LK, Flannagan SE, Kolonay JF, Shetty J, Killgore GE, Tenover FC (2003) Genetic analysis of a high-level vancomycin-resistant isolate of Staphylococcus aureus. Science 302, 1569−1571 DOI 10.1126/science.1090956. [DOI] [PubMed] [Google Scholar]
- 14.Walsh TR, Howe RA (2002) The prevalence and mechanisms of vancomycin resistance in Staphylococcus aureus. Annu. Rev. Microbiol 56, 657–675 DOI 10.1146/annurev.micro.56.012302.160806. [DOI] [PubMed] [Google Scholar]
- 15.Willyard C. (2017) The drug-resistant bacteria that pose the greatest health threats. Nature 543, 15 DOI 10.1038/nature.2017.21550. [DOI] [PubMed] [Google Scholar]
- 16.Walsh CT (1993) Vancomycin resistance: decoding the molecular logic. Science 261, 308–310 DOI 10.1126/science.8392747. [DOI] [PubMed] [Google Scholar]
- 17.Healy VL, Lessard IA, Roper DI, Knox JR, Walsh CT (2000) Vancomycin resistance in enterococci: reprogramming of the D-Ala-D-Ala ligases in bacterial peptidoglycan biosynthesis. Chem. Biol 7, R109–R119 DOI 10.1016/S1074-5521(00)00116-2. [DOI] [PubMed] [Google Scholar]
- 18.McComas CC, Crowley BM, Boger DL (2003) Partitioning the loss in vancomycin binding affinity for D-Ala-D-Lac into lost H-bond and repulsive lone pair contributions. J. Am. Chem. Soc 125, 9314–9315 DOI 10.1021/ja035901x. [DOI] [PubMed] [Google Scholar]
- 19.Guan D, Chen F, Xiong L, Tang F, Qiu Y, Zhang N, Gong L, Li J, Lan L, Huang W. (2018) Extra sugar on vancomycin: new analogues for combating multidrug-resistant Staphylococcus aureus and vancomycin-resistant enterococci. J. Med. Chem 61, 286–304 DOI 10.1021/acs.jmedchem.7b01345. [DOI] [PubMed] [Google Scholar]
- 20.Yarlagadda V, Konai MM, Manjunath GB, Ghosh C, Haldar J, (2015) Tackling vancomycin-resistant bacteria with ‘lipophilic–vancomycin–carbohydrate conjugates’. J. Antibiot 68, 302–312 DOI 10.1038/ja.2014.144. [DOI] [PubMed] [Google Scholar]
- 21.Yarlagadda V, Sarkar P, Samaddar S, Haldar J. (2016) A vancomycin derivative with a pyrophosphate‐binding group: a strategy to combat vancomycin‐resistant bacteria. Angew. Chem., Int. Ed 55, 7836–7840 DOI 10.1002/anie.201601621. [DOI] [PubMed] [Google Scholar]
- 22.Yarlagadda V, Samaddar S, Paramanandham K, Shome BR, Haldar J. (2015) Membrane disruption and enhanced inhibition of cell‐wall biosynthesis: a synergistic approach to tackle vancomycin‐resistant bacteria. Angew. Chem., Int. Ed 54, 13644–13649 DOI 10.1002/anie.201507567. [DOI] [PubMed] [Google Scholar]
- 23.Long DD, Aggen JB, Chinn J, Choi SK, Christensen BG, Fatheree PR, Green D, Hegde SS, Judice JK, Kaniga K, Krause KM (2008) Exploring the positional attachment of glycopeptide/β-lactam heterodimers. J. Antibiot 61, 603–614 DOI 10.1038/ja.2008.80. [DOI] [PubMed] [Google Scholar]
- 24.Yarlagadda V, Akkapeddi P, Manjunath GB, Haldar J. (2014) Membrane active vancomycin analogues: a strategy to combat bacterial resistance. J. Med. Chem 57, 4558–4568 DOI 10.1021/jm500270w. [DOI] [PubMed] [Google Scholar]
- 25.Leadbetter MR, Adams SM, Bazzini B, Fatheree PR, Karr DE, Krause KM, Lam BM, Linsell MS, Nodwell MB, Pace JL, Quast K. (2004) Hydrophobic vancomycin derivatives with improved ADME properties. J. Antibiot 57, 326–336 DOI 10.7164/antibiotics.57.326. [DOI] [PubMed] [Google Scholar]
- 26.Corey GR, Stryjewski ME, Weyenberg W, Yasothan U, Kirkpatrick P. (2009) Telavancin. Nat. Rev. Drug Discov 8, 929–930 DOI 10.1038/nrd3051. [DOI] [PubMed] [Google Scholar]
- 27.Anderson VR, Keating GM (2008) Dalbavancin. Drugs 68, 639–648 DOI 10.2165/00003495-200868050-00006. [DOI] [PubMed] [Google Scholar]
- 28.Markham A. (2014) Oritavancin: first global approval. Drugs 74, 1823–1828. DOI 10.1007/s40265-014-0295-4. [DOI] [PubMed] [Google Scholar]
- 29.Okano A, Isley NA, Boger DL (2017) Peripheral modifications of [Ψ [CH2NH]Tpg4]vancomycin with added synergistic mechanisms of action provide durable and potent antibiotics. Proc. Natl. Acad. Sci. U. S. A 114, E5052–E5061 DOI 10.1073/pnas.1704125114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Okano A, Nakayama A, Wu K, Lindsey EA, Schammel AW, Feng Y, Collins KC, Boger DL (2015) Total synthesis and initial examination of [Ψ[C(=S)NH]Tpg4]-vancomycin, [Ψ[C(=NH)NH]Tpg4]vancomycin, [Ψ[CH2NH]Tpg4]vancomycin and their (4-chlorobiphenyl)methyl derivatives: synergistic binding pocket and peripheral modifications for the glycopeptide Antibiotics. J. Am. Chem. Soc 137, 3693–3704 DOI 10.1021/jacs.5b01008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Okano A, Nakayama A, Schammel AW, Boger DL (2014) Total synthesis of [Ψ[C(=NH)NH]Tpg4]vancomycin and its (4-chlorobiphenyl)methyl derivative: impact of peripheral modifications on vancomycin analogs redesigned for dual D-Ala-D-Ala and D-Ala-D-Lac binding, J. Am. Chem. Soc 136, 13522–13525 DOI 10.1021/ja507009a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Crowley BM, Boger DL (2006) Total synthesis and evaluation of [Ψ (CH2NH)Tpg4]vancomycin aglycon: reengineering vancomycin for dual D-Ala-D-Ala and D-Ala-D-Lac binding. J. Am. Chem. Soc 128, 2885–2892 DOI 10.1021/ja0572912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boger DL (2017) The difference a single atom can make synthesis and design at the chemistry−biology interface. J. Org. Chem 82, 11961–11980 DOI 10.1021/acs.joc.7b02088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ge M, Chen Z, Russell H, Kohler J, Silver LL, Kerns R, Fukuzawa S, Thompson C, Kahne D. (1999) Vancomycin derivatives that inhibit peptidoglycan biosynthesis without binding D-Ala-D-Ala. Science 284, 507–511 DOI 10.1126/science.284.5413.507. [DOI] [PubMed] [Google Scholar]
- 35.Kerns R, Dong SD, Fukuzawa S, Carbeck J, Kohler J, Silver L, Kahne D. (2000) The role of hydrophobic substituents in the biological activity of glycopeptide antibiotics. J. Am. Chem. Soc 122, 12608–12609 DOI 10.1021/ja0027665. [DOI] [Google Scholar]
- 36.Wu Z−C, Isley NA, Boger DL (2018) N-terminus alkylation of vancomycin: ligand binding affinity, antimicrobial activity, and site-specific nature of quaternary trimethylammonium salt modification. ACS Infect. Dis 4, 1468–1474 DOI 10.1021/acsinfecdis.8b00152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu Z−C, Boger DL (2019) Exploration of the site-specific nature and generalizability of a trimethylammonium salt modification on vancomycin: A-ring derivatives. Tetrahedron 75, 3160–3165 DOI 10.1016/j.tet.2019.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu Z−C, Isley NA, Okano A, Weiss WJ, Boger DL (2020) C1-CBP-Vancomycin: impact of a vancomycin C-terminus trimethylammonium cation on pharmacological properties and insights into its newly introduced mechanism of action. J. Org. Chem 85, 1365–1375 DOI 10.1021/acs.joc.9b02314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rajagopal M, Walker S. (2015) Envelope structures of Gram-positive bacteria In Protein and sugar export and assembly in Gram-positive bacteria, pp 1−44, Springer, Cham: DOI 10.1007/82_2015_5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Unpublished studies suggest vancomycin C-terminus amide formation, neutralizing the carboxylic acid negative charge, may modestly enhance activity against vancomycin-resistant organisms.
- 41.Antonoplis A, Zang X, Huttner MA, Chong KK, Lee YB, Co JY, Amieva MR, Kline KA, Wender PA, Cegelski L, (2018) A dual-function antibiotic-transporter conjugate exhibits superior activity in sterilizing MRSA biofilms and killing persister cells. J. Am. Chem. Soc 140, 16140–16151 DOI 10.1021/jacs.8b08711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Antonoplis A, Zang X, Wegner T, Wender PA, Cegelski L. (2019) Vancomycin–arginine conjugate inhibits growth of carbapenem-resistant E. coli and targets cell wall synthesis. ACS Chem. Biol 14, 2065–2070 DOI 10.1021/acschembio.9b00565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.For VanA E. faecalis (VanA VRE, BM 4166): resistant to erythromycin, gentamicin, chloramphenicol, and ciprofloxacin as well as vancomycin and teicoplanin, sensitive to daptomycin. For VanA E. faecium (VanA VRE, ATCC BAA-2317): resistant to ampicillin, benzylpenicillin, ciprofloxacin, erythromycin, levofloxacin, nitrofurantoin, and tetracycline as well as vancomycin and teicoplanin, insensitive to linezolid, sensitive to tigecycline and dalfopristine.
- 44.Percy MG, Gründling A. (2014) Lipoteichoic acid synthesis and function in Gram-positive bacteria. Annu. Rev. Microbiol 68, 81–100 DOI 10.1146/annurev-micro-091213-112949. [DOI] [PubMed] [Google Scholar]
- 45.Brown S, Santa Maria JP Jr., Walker S. (2013) Wall teichoic acids of Gram-positive bacteria. Annu. Rev. Microbiol 67, 313–336 DOI 10.1146/annurev-micro-092412-155620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Höltje J−V, Tomasz A. (1975) Lipoteichoic acid: a specific inhibitor of autolysin activity in pneumococcus. Proc. Natl. Acad. Sci. U. S. A 72, 1690–1694 DOI 10.1073/pnas.72.5.1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bierbaum G, Sahl H−G (1985) Induction of autolysis of staphylococci by the basic peptide antibiotics Pep 5 and nisin and their influence on the activity of autolytic enzymes. Arch. Microbiol 141, 249–254 DOI 10.1007/BF00408067. [DOI] [PubMed] [Google Scholar]
- 48.Bierbaum G, Sahl H−G (1987) Autolytic system of Staphylococcus simulans 22: influence of cationic peptides on activity of N-acetylmuramoyl-L-alanine amidase. J. Bacteriol 169, 5452–5458 DOI 10.1128/jb.169.12.5452-5458.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fetterly GJ, Ong CM, Bhavnani SM, Loutit JS, Porter SB, Morello LG, Ambrose PG, Nicolau DP (2005) Pharmacokinetics of oritavancin in plasma and skin blister fluid following administration of a 200-milligram dose for 3 days or a single 800-milligram dose. Antimicrob. Agents Chemother 49, 148–152 DOI 10.1128/AAC.49.1.148-152.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.For VanA S. aureus (VRSA, VRS2): resistant to methicillin, oxacillin, gentamicin, erythromycin, clindamycin, levofloxacin, trimethoprim-sulfamethoxazole, ceftriaxone, amoxicillin-clavulanic acid, piperacillin-tazobactam, cefepime, and imipenem as well as vancomycin, sensitive to linezolid, daptomycin, mupirocin and tigecycline.
- 51.Xie J, Okano A, Pierce JG, James RC, Stamm S, Crane CM, Boger DL (2012) Total synthesis of [Ψ[C(=S)NH]Tpg4]vancomycin aglycon, [Ψ[C(=NH)NH]Tpg4]-vancomycin aglycon and related key Compounds: reengineering vancomycin for dual D-Ala-D-Ala and D-Ala-D-Lac binding. J. Am. Chem. Soc 134, 1284–1297 DOI 10.1021/ja209937s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xie J, Pierce JG, James RC, Okano A, Boger DL (2011) A redesigned vancomycin engineered for Dual D-Ala-D-Ala and D-Ala-D-Lac binding exhibits potent antimicrobial activity against vancomycin-resistant bacteria. J. Am. Chem. Soc 133, 13946–13949 DOI 10.1021/ja207142h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Okano A, Isley NA, Boger DL (2017) Total synthesis of vancomycin related glycopeptide antibiotics and key analogues. Chem. Rev 117, 11952–11993 DOI 10.1021/acs.chemrev.6b00820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nakayama A, Okano A, Feng Y, Collins JC, Collins KC, Walsh CT, Boger DL (2014) Enzymatic glycosylation of vancomycin aglycon: completion of a total synthesis of vancomycin and N- and C-terminus substituent effects of the aglycon substrate. Org. Lett 16, 3572–3575 DOI 10.1021/ol501568t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.James RC, Pierce JG, Okano A, Xie J, Boger DL (2012) Redesign of glycopeptide antibiotics: back to the future. ACS Chem. Biol 7, 797–804 DOI 10.1021/cb300007j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Blaskovich MA, Hansford KA, Gong Y, Butler MS, Muldoon C, Huang JX, Ramu S, Silva AB, Cheng M, Kavanagh AM, Ziora Z, Premraj R, Lindahl F, Bradford TA, Lee JC, Karoli T, Pelingon R, Edwards DJ, Amado M, Elliott AG, Phetsang W, Daud NH, Deecke JE, Sidjabat HE, Ramaologa S, Zuegg J, Betley JR, Beevers APG, Smith RAG, Roberts JA, Paterson DL, Cooper MA (2018) Protein-inspired antibiotics active against vancomycin-and daptomycin-resistant bacteria. Nat. Commun 9, 1−17 DOI 10.1038/s41467-017-02123-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.