

Abstract
Ribosome profiling and mass spectrometry have revealed thousands of small and alternative open reading frames (sm/alt-ORFs) that are translated into polypeptides variously termed microproteins and alt-proteins in mammalian cells. Some micro-/alt-proteins exhibit stress-, cell type- and/or tissue-specific expression, and understanding this regulated expression will be critical to elucidating their functions. While differential translation has been inferred by ribosome profiling, quantitative mass spectrometry-based proteomics is needed for direct measurement of microprotein and alt-protein expression between samples and conditions. However, while label-free quantitative proteomics has been applied to detect stress-dependent expression of bacterial microproteins, this approach has not yet been demonstrated for analysis of differential expression of unannotated ORFs in the more complex human proteome. Here, we present global micro-/alt-protein quantitation in two human leukemia cell lines, K562 and MOLT4. We identify 12 unannotated proteins that are differentially expressed in these cell lines. The expression of six micro/alt-proteins was validated biochemically, and two were found to localize to the nucleus. Thus, we demonstrate that label-free comparative proteomics enables quantitation of micro-/alt-protein expression between human cell lines. We anticipate that this workflow will enable discovery of regulated sm/alt-ORF products across many biological conditions in human cells.
Keywords: comparative proteomics, microprotein, alternative protein, unannotated protein, proteogenomics
Graphical Abstract
INTRODUCTION
Recent advances in genomic and proteomic technologies have revealed that mammalian genomes harbor thousands of previously unannotated small and alternative open reading frames (sm/alt-ORFs) which are <100 codons and >100 codons in length, respectively1-6. Sm/alt-ORFs encode polypeptide products that have been referred to as smORF-encoded polypeptides (SEPs), micropeptides, or microproteins (<100 amino acids), and alt-proteins (>100 amino acids); we will collectively refer to all unannotated polypeptides, regardless of length, as alt-proteins in this study to comport with recommended nomenclature7. These proteins were previously excluded from genome annotation due to their short size, initiation at non-AUG start codons, and overlap with other coding regions. A rapidly increasing number of sm/alt-ORFs have been shown to play important roles in mammalian biology8. For example, a recent genome-scale Cas9-based knockout screen revealed that 570 out of 2353 smORFs regulate growth of human induced pluripotent stem cells9. A number of sm/alt-ORF-encoded proteins have been characterized at the molecular level, recently including PIGBOS, which regulates the endoplasmic reticulum (ER) stress response10, and BRAWNIN, which is essential for mitochondrial function11. These findings demonstrate that identification and characterization of sm/alt-ORFs can provide new biological insights.
Translation of thousands of mammalian sm/alt-ORFs has been predicted via ribosome profiling12,13. Mass spectrometry offers lower coverage, but provides more direct evidence for the presence of translated alt-proteins in cells2. Differential expression of some functional alt-proteins has been reported to occur under stress conditions, in disease, and during cell division14,15. For example, in humans, CASIMO1 is upregulated in breast tumors, and regulates proliferation of cancer cell lines16. In mouse, AW112010 is induced upon lipopolysaccharide (LPS) treatment, and is required for the innate immune response17. In bacteria, YmcF and YnfQ are small, cold shock-induced alt-proteins18, while GndA is a heat shock-induced membrane alt-protein19. These bacterial stress-response microproteins were identified using a label-free quantitative proteomic approach. Notably, Saghatelian and colleagues have shown that mammalian sm/alt-ORFs exhibit cell- and tissue-specific expression patterns4. Quantitative proteomic profiling of differential sm/alt-ORF expression is therefore needed to further identify stress-induced, and disease- and tissue-specific, functional mammalian alt-proteins. While quantitative proteomics has been applied to a human alt-protein during oxidative stress, differential expression was not observed20, leaving open the question of whether this approach can quantify differences in human alt-proteins between samples.
In this report, we identify 16 and 15 sm/alt-ORF encoded alt-proteins, including microproteins, larger alt-proteins, N-terminal extensions, and an unannotated transcript variant, in K562 and MOLT4 cells, respectively. 28 exhibit differential expression, three are shared by these two cell lines and 12 are reported in this study for the first time. We confirmed differential expression of >30% of these genes with mRNA-seq, performed bioinformatic analysis and tested expression of six alt-proteins from overexpressed, epitope tagged cDNA constructs. Overall, we extend label-free quantitative proteomics to identify differentially expressed, unannotated alt-proteins in human cell lines.
MATERIALS AND METHODS
Cloning
For alt-CCNA2, alt-LEF1, alt-SLC1A5 and alt-SCRIB, a construct comprising the full 5'UTR of the annotated mRNA through the stop codon of each smORF was synthesized by Genscript with a FLAG epitope tag appended to the 3' end of each smORF coding sequence, and was then subcloned into pcDNA3. For ERVK3-1 and alt-C1orf122, a construct comprising the full annotated mRNA sequence was synthesized by Genscript with a FLAG epitope tag appended to the 3' end of each alt-ORF, and was then subcloned into pcDNA3.
Cell Lysis and Protein Size Selection
Cell pellets (2.5 x 106 K562 or MOLT4 cells) were resuspended with 200 μL standard SDS loading buffer, followed by heating for 10 min at 100°C until homogenous. 40 μL of the lysates were loaded into one Tricine gel well, and separated according to established protocol21, followed by Coomassie blue staining and destaining. The gel bands corresponding to 2-5 kDa, 5-10 kDa and 10-15 kDa were excised for further LC-MS/MS analysis. Two biological replicates were performed for MOLT4, and one replicate for K562, which has been extensively profiled in the past3,4,20,22.
Proteomics and Database Searches
Protein-containing gel slices were digested with 13.3 μg/mL trypsin (Promega) at 37°C for 14-16 h. The resulting peptide mixtures were extracted from the gel, dried, extracted with ethyl acetate to remove residual detergent, and then re-suspended in 15 μl of 3:8 70% formic acid:0.1% TFA. A 5 μL aliquot of each sample was injected onto a pre-packed column attached to a nanoAcquity UPLC (Waters) in-line with an LTQ Orbitrap Q Exactive (Thermo Scientific) and a 130-min gradient was used to further separate the peptide mixtures as follows (solvent A: 0.1% formic acid; solvent B: acetonitrile with 0.1% formic acid): Single pump trapping was turned on for 6 min at a flow rate of 2.5 μL/min at 99% A. Isocratic flow was maintained at 0.25 μL/min at 1% B for 40 min, followed by linear gradients from 1% B to 6% B over 2 min, 6% B to 24% B over 58 min, 24% B to 48% B over 5 min, 48% B to 80% B over 5 min. Isocratic flow at 80% B was maintained for 5 min, followed by a gradient from 80% B to 1% B over 5 min, then maintained for 10 min. The full MS was collected over the mass range of 298-1,750 m/z with a resolution of 30,000. MS/MS data was collected using a top 10 high-collisional energy dissociation method in data-dependent mode with a normalized collision energy of 33.0 eV and a 2.0 m/z isolation window. The first mass was 100 m/z in fixed mode. MS/MS resolution was 7,500 and dynamic exclusion was 60 seconds.
For identification of alt- and microproteins, ProteoWizard MS Convert was used for peak picking and files were analyzed using Mascot (version 2.5.1). Oxidation of methionine and N-terminal acetylation were set as variable modifications. A mass deviation of 20 p.p.m. was set for MS1 peaks, with a peptide tolerance of 0.6 Da. A maximum of two missed cleavages were allowed. The false discovery rate (FDR) was set to 1% both on peptide and protein levels. The minimum required peptide length was five amino acids. Protein quantitation was accomplished via spectral counting, followed by comparing the MS1 extracted ion chromatograph (EIC) peak intensity in both cell lines using Xcalibur 4.0 (Thermo). As previously reported3,23, peptide spectra were matched against a 3-frame translation of mRNA-seq from the corresponding cell line, permitting identification of both known and unannotated peptides. Annotated peptides were excluded with a string-matching algorithm via comparison to the human proteome. Because many sm/alt-ORFs are only identified by a single peptide-spectral match, putative unannotated peptide-spectral matches were manually examined for score and sequence coverage, as previously reported18.
PepQuery (version 1.4.1)24 was used to further filter candidate unannotated peptide-spectral matches identified by Mascot. Each spectrum was compared to a database of human proteins (UniProt) and common contaminants using unrestricted post-translational modification searching to exclude the possibility that any annotated, modified peptide would produce a higher-scoring spectrum match. Only peptides which were identified as the top-ranking match by PepQuery were retained.
mRNA-seq and Data Analysis
Whole RNA was isolated from 2.5 x 106 K562 or MOLT4 (n = 3) cells using Qiagen RNeasy Mini Kit spin columns, then treated in solution with DNase I prior to Qiagen column clean-up according to the manufacturer’s protocol. Whole RNA was submitted to the Yale Center for Genomic Analysis for preparation according to the standard Illumina protocol for paired-end sequencing with enrichment of poly-A RNA. Samples were multiplexed and 75 bp fragments were sequenced on the Hiseq2500 sequencer. The reads were mapped to the human genome (hg38) using TopHat (v2.1.1). To identify the genes differentially expressed between these two cell lines, we counted the RNA reads in exons and calculated the reads per kilobase per million reads (RPKM) for each gene as a measure of expression using Cufflinks (v.2.2.2) with the Cuffdiff tool using default parameters and a log2 (fold change) cutoff of 0.5 with p value ≤ 0.05.
Cell Culture and Transfection
All cell lines were purchased from ATCC and early-passage stocks were established in order to ensure cell line identity. Cells were maintained up to only 10 passages. K562 and MOLT4 cells were maintained at a density of 1-10 × 105 cells/mL in RPMI 1640 medium (Gibco, 11875101) with 10% FBS (Sigma, F0392) and 1% penicillin-streptomycin (VWR, 97063-708). HEK 293T cells were cultured in DMEM (Corning, 10-013-CV) with 10% FBS (Sigma, F0392) and 1% penicillin-streptomycin (VWR, 97063-708) in a 5% CO2 atmosphere at 37°C. Plasmid transfection was performed with Lipofectamine 2000 and Opti-MEM (GIBCO, 31985-070) according to the manufacturer’s instructions, or polyethyleneimine (PEI, Polysciences, 23966-1) according to established protocol25.
Data Availability.
The mRNA-seq data have been deposited in the NCBI Gene Expression Omnibus under accession GSE148451.
The mass spectrometry proteomics data have been deposited to the PRIDE Archive (http://www.ebi.ac.uk/pride/archive/) via the PRIDE partner repository with the data set identifier PXD018565 and 10.6019/PXD018565.
RESULTS
Comparative Proteomics Reveals Differential Expression of Unannotated Alt-Protein in Two Human Cell Lines
Previously, we reported a label-free quantitation protocol for comparative profiling of unannotated polypeptides between two conditions in bacteria18,19. As shown in Figure 1A, we aimed to extend this quantitative proteomic workflow to human polypeptides using two human leukemia cell lines, K562 and MOLT4, as a model. We hypothesized that, since both K562 and MOLT4 are leukemia-derived cell lines, they would exhibit similar expression levels of some alt-proteins; however, since they arise from different types of leukemia (chronic myelogenous leukemia and acute lymphoblastic leukemia, respectively) and different progenitor cells (bone marrow and T lymphoblast, respectively)26,27, we reasoned that a subset of alt-proteins would be differentially expressed. Our approach is based on a previously reported method to qualitatively enrich and identify unannotated peptides23. Briefly, after gel-based protein size selection, tryptic digest and LC-MS/MS analysis, all MS/MS spectra are matched to a 3-frame translated RNA-seq database to identify both annotated and unannotated peptides and proteins (Table S1). Subsequently, unannotated peptides are filtered using a string-matching algorithm, and MS/MS spectra are inspected using stringent criteria to remove false positives, as previously reported23. In this work, we additionally utilized PepQuery24 to further analyze any putative smORFs identified by only one peptide-spectral match (Table S2), a common challenge in peptidomics3. The final list of unannotated peptides that passed all filters is provided in Table S3. Finally, differential expression of unannotated peptides identified in only one sample (or non-differential expression of unannotated peptides detected in both) was validated by comparing the MS1 extracted ion chromatograph (EIC) peak intensity in both samples, as previously reported18-20.
Figure 1.

Comparative proteomics reveals differential expression of unannotated alt-proteins in two human cell lines. (A) Schematic overview of the quantitative proteomics protocol. (B) Size distribution of annotated proteins identified in K562 (left) and MOLT4 (right) cells. (C) Venn diagram of alt-proteins identified in K562 (left) or MOLT4 (right) cells. (D) Distribution of locations of the detected alt-proteins relative to the annotated coding sequence (CDS). (E) Frequency of cognate (AUG) vs near-cognate (non-AUG) predicted start codons used by the detected alt-ORFs. (F) Annotation status of the detected alt-proteins: unannotated, predicted in UniProt database without protein level evidence (predicted), and recently published (reported).
Prior to analysis of unannotated sequences, we first validated our workflow and proteomic data quality by analyzing the numbers and sizes of annotated proteins identified from searching the raw spectral data against the UniProt human database. 2,285 and 2,579 annotated proteins were identified in the 2-15 kDa size range from K562 and MOLT4 cells respectively, and a clear enrichment of small proteins was observed (Figure 1B), similar to the number of identifications and size distributions in reported LC-MS/MS proteomics studies of smORFs4,23. Detections of proteins larger than the excised gel band have been previously reported, and may be due to proteolysis during cell lysis20,23.
Subsequently analyzing unannotated sequences, we identified peptides mapping to 16 and 15 alt- proteins in K562 and MOLT4 cell lines, respectively. Among alt-proteins identified in this study, 3 are detected in both cell lines, while the rest are detected in only K562 or MOLT4, suggesting differential expression of these sm/alt-ORF candidates (Figure 1C and Tables S1, S2, and S3). Considered together, the 28 sm/alt-ORFs that encode these proteins fall into six categories (Figure 1D): 1) upstream ORFs (uORFs) located in the 5' UTR; 2) downstream ORFs in the 3' UTR; 3) N-terminal extensions of known protein coding sequences from alternative translation initiation sites12; 4) ORFs within “noncoding” RNAs (ncRNAs); 5) ORFs that overlap annotated protein coding sequences in a different reading frame (CDS) and 6) one peptide mapping to an unannotated protein isoform derived from a novel transcript variant, which is supported by reads in our mRNA-seq data (Figure S1). 68% of the 28 alt-proteins presumptively initiate with a non-AUG start codon (Figure 1E). These features are consistent with prior smORF proteomics studies3,4,28, further validating our workflow. Of the 28 detected alt-proteins, 12 (42.9%) are unannotated and reported in this study for the first time; 5 (17.8%) are predicted in the UniProt database without protein-level evidence; and 11 (39.3%) have been detected and/or recently reported3,4,20,28-30 (Figure 1F and Table S3). While only a single replicate of K562 cells was analyzed, the alt-protein identifications in this work were compared to previous studies which have extensively analyzed K562 alt-proteins; 6 out of the 16 alt-proteins detected in this study in K562 cells were reported before in this cell line (Table S3)3,4,29.
Confirmation of Alt-Protein Differential Expression
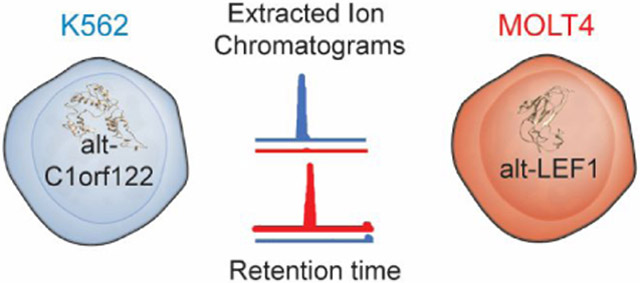
We validated label-free quantitation of six selected alt-proteins detected only in K562, only in MOLT4, or in both cell lines, by comparing the area under the MS1 peak in the extracted ion chromatogram (EIC) for each of these peptides. As shown in Figure 2 and Figure S2, the EIC comparisons revealed that peptides derived from alt-C1orf122 (Figure 2A) were present in K562 cells only, and peptides derived from alt-LEF1 (Figure 2B), alt-CCNA2 (Figure S2A) and alt-SLC1A5 (Figure S2B) were present in MOLT4 cells only, consistent with spectral counting results (Tables S1 and S3). In contrast, peptides derived from ERVK3-1 (Figure 2C) and alt-SCRIB (Figure S2C) were present in both cell lines, again consistent with the spectral counting results (Tables S1 and S3).
Figure 2.

Confirmation of differential expression of three detected alt-proteins with extracted ion chromatograms (EICs). Shown are EICs (left) from MS1 spectra corresponding to MS/MS spectra (right) of three tryptic peptides identified in K562 cells only (A), MOLT4 cells only (B) or both cells (C). The same y-axis scale is used for each matched EIC pair. The EIC intensity at the same retention time was compared for the paired samples.
To orthogonally confirm alt-protein differential expression, we compared the levels of transcripts encoding them in these two cell lines. As shown in Figure 3A, the results of mRNA-seq revealed that, globally, 4,135 genes are upregulated (log2(Fold change) ≥0.5, p-value ≤ 0.05), and 3,664 genes are downregulated (log2(Fold change) ≤ −0.5, p-value ≤ 0.05) in K562 cells compared with MOLT4 cells. We then focused on transcript encoding the alt-proteins of interest. Figure 3B illustrates enrichment of sm/alt-ORF-encoding transcripts in either K562 or MOLT4 cells. For 42% of detected alt-proteins that map to annotated genes, RNA-level quantitation is consistent with the quantitative proteomics analysis, similar to moderate RNA-protein correlation observed in prior studies31. This analysis is therefore partially consistent with our proteomics results, suggesting our proteomics workflow can quantitate differential alt-protein expression.
Figure 3.

Differential expression of the detected alt-ORFs at the mRNA level. (A) Global volcano plot of mRNA-seq from K562 and MOLT4 cells. Transcripts corresponding to the six alt-ORFs selected for further confirmation are indicated in red and gene names are labeled. (B) Differential expression of transcripts encoding the detected alt-ORFs in K562 and MOLT4 cells. Genes corresponding to MOLT4- and K562-specific alt-proteins are represented in red, and blue, respectively. Genes corresponding to alt-proteins detected in both cell lines are shown in purple. Gene names are indicated to the right.
Overexpression Analysis of Alt-Proteins
The six alt-proteins for which MS1 quantitation was performed above represent a mix of K562-specific, MOLT4-specific, and ubiquitous expression. While alt-CCNA2, alt-SCRIB, ERVK3-1 and alt-C1orf122 have been previously reported in proteomics studies3,20,29, alt-LEF1 and alt-SLC1A5 are reported for the first time in this work, and none, except alt-CCNA23, have been previously validated to express at the molecular level. Therefore, we generated constructs containing the full-length cDNA (or cDNA sequence from the 5'UTR through the stop codon of the alt-ORF) with a FLAG tag appended to the C-terminus of each putative alt-ORF. These constructs were overexpressed in HEK 293T cells and visualized with immunofluorescence. As shown in Figure 4, immunostaining revealed that all six constructs produce FLAG signal, indicating all six alt-ORFs can be translated from cDNA in cells. Alt-C1orf122 and alt-LEF1 are primarily nuclear. We note that confirmation that the endogenous alt-ORFs are expressed requires genomic tagging or specific antibodies, so these identifications must be regarded as putative.
Figure 4.

Confirmation of the expression of six detected alt-proteins with immunostaining. HEK 293T cells were transfected with a construct containing the full 5'UTR or transcript and the indicated alt-protein coding sequence with a FLAG tag appended to the C-terminus, followed by immunostaining with anti-FLAG (left) and DAPI (center). Scale bar 10 μm. (A-B) show two proteins, ERVK3-1 and alt-SCRIB, detected in both K562 and MOLT4 cells, (C) alt-C1orf122, detected in K562 only, and (D-F) three alt-proteins, alt-LEF1, alt-CCNA2, and alt-SLC1A5, which were detected in MOLT4 only.
To confirm the immunostaining results, we performed Western blotting. As shown in Figure 5, ERVK3-1, alt-LEF1, alt-C1orf122 and alt-SCRIB were robustly detected. We did not see clear Western blot bands for alt-CCNA2 and alt-SLC1A5, although they were detected via immunostaining, suggesting their expression levels may be low. ERVK3-1 produces two bands, which may be caused by phosphorylation32, proteolytic processing or other post-translational modifications.
Figure 5.

Confirmation of alt-protein expression with Western blotting. HEK 293T cells were transfected with a construct containing the full 5'UTR or transcript and the indicated alt-protein coding sequence with a FLAG tag appended to the C-terminus, followed by Western blotting with anti-FLAG antibody. The bands indicated by a red asterisk correspond to ERVK3-1, alt-LEF1, alt-C1orf122 and alt-SCRIB.
Alt-ORF Start Codon Prediction
Next, we identified the start codon used by the six alt-ORFs by searching the upstream, in-frame DNA sequences relative to the detected tryptic peptides for ATG or near-cognate start codons in strong Kozak consensus sequences. As shown in Figure S3, we found that ERVK3-1 and alt-SCRIB contain an upstream, in-frame ATG start codon, A205TG and A326TG, respectively (numbered relative to the first nucleotide of the cDNA), and are predicted to encode 109 amino acid (aa) and 120 aa proteins, respectively. Alt-C1orf122, alt-LEF1, alt-CCNA2 and alt-SLC1A5 contain near-cognate start codons in a strong Kozak sequence context: A138CG, A40GG, C135TG and A301AG, respectively, and may respectively encode 317 aa, 141 aa, 115 aa and 61 aa proteins. The predicted protein sizes are generally consistent with the observed molecular weights (MW) in Western blot analysis (Figure 5), supporting these start codon assignments. The observed MW of alt-LEF1 is 20 kDa, while the predicted MW is 15 kDa, which we hypothesize may be explained by the basic amino acid composition of alt-LEF1 (PI = 10.61); other similarly basic alt-proteins such as NBDY (PI = 9.51) and MRI-2 (PI = 9.30) also show aberrantly low mobility in SDS-PAGE gels33,34. Deletion analysis of the alt-LEF1 predicted start codon will be required to confirm its assignment.
Alt-Protein Bioinformatic Analysis
Previous studies have suggested that many sm/alt-ORF-encoded proteins contain signal peptides and/or localize to organelles3,11. To determine whether any of our 28 alt-proteins contain signal peptides, mitochondrial transfer peptides, or subcellular localization motifs like nuclear localization sequences (NLS), we performed bioinformatics analyses with TargetP 2.035 and cNLS-mapper36,37. These analyses revealed that CHTOP (N-terminal extension) is predicted to contain a mitochondrial transfer peptide (Figure S4A), which we hypothesize may change the localization of the annotated CHTOP protein from nuclear to mitochondrial. Previous reports of localization signal-encoding N-terminal extensions, for example FGF2 initiation at either an upstream CUG start codon or a downstream AUG to produce two protein isoforms with distinct localizations and interactions38,39, provide precedent for this observation. Additionally, SF3A1 N-terminal extension is predicted to contain a signal peptide (Figures S4B and Table S4). Furthermore, three alt-proteins (alt-C1orf122, alt-LEF1 and A0A499FIZ0) are predicted to encode NLS sequences (Figure S5), consistent with nuclear immunofluorescence for two of these candidates upon overexpression (Figures 4C and 4D). These results suggest that differentially expressed alt-proteins may be imported into the secretory pathway or organelles.
Establishing conservation of small open reading frames throughout evolution remains a challenge due to poor annotation of orthologs across species and limited sequence lengths40. To determine whether any of the six selected alt-proteins are conserved, CCNA2, LEF1, SLC1A5, C1ORF122 and SCRIB mRNA sequences from different species were obtained from NCBI nucleotide database, then translated in the +1, +2 and +3 reading frames using the ExPASy translate tool. ATG or near-cognate start codons in strong Kozak consensus sequences in frame with sequences homologous to human alt-CCNA2, alt-LEF1, alt-SLC1A5, alt-C1orf122 or alt-SCRIB were identified in the 5'UTR of each transcript in order to predict the full-length sequence of hypothetical homologs. ClustalW alignment of these hypothetical homologs against human alt-CCNA2 (Figure S6A), alt-LEF1 (Figure S6B), alt-SLC1A5 (Figure S6C), alt-C1orf122 (Figure S6D), and alt-SCRIB (Figure S6E), revealed significant sequence similarity, though protein-level existence for these hypothetical homologs does not currently exist. Finally, both alt-C1orf122 and alt-LEF1 are predicted to be structured using I-TASSER41 (Figure S7). The observations that alt-proteins contain localization signals, exhibit conservation and may be structured permit hypothesis generation about their possible functions.
DISCUSSION
In this study, we applied a workflow for comparative proteomics to quantify expression of 28 unannotated alt-proteins in two human leukemia cell lines. The observed differential expression can be partially confirmed by mRNA-seq. It is worth noting that (1) mRNA levels are poorly correlated with protein levels in general due to post-transcriptional regulation31, and (2) mRNA-seq quantifies the annotated genes taking all transcript variants into account, while alt-proteins may be encoded by a specific transcript isoform34, so mRNA-seq is not expected to perfectly correlate with alt-protein expression levels. For six selected alt-proteins, we confirmed via MS1 quantitation that two are expressed in both cell lines, while the other four are specifically expressed in either K562 or MOLT4 cell lines, suggesting that leukemia-derived cells can have substantially different alt-protein expression profiles. Taken together, these results show that this workflow enables comparison of the expression levels of alt-proteins in human cell lines.
Several of the alt-proteins examined in this study are conserved in mammals or primates. While this degree of conservation is limited, similar clade-specific alt-proteins have previously been shown to be functional. For example, alt-proteins specific to mammals, such as NBDY and Minion, have been shown to impact biological processes such as RNA decapping and cellular fusion33,42, and Storz and colleagues have noted similarly limited conservation of functional smORFs in bacteria40.
Interestingly, alt-C1orf122, a K562-specific alt-protein, and alt-LEF1, a MOLT4-specific alt-protein, both localize primarily to the nucleus and may fold into tertiary structures. Therefore, the differentially expressed proteins identified in this study, especially those exhibiting specific subcellular localization, may be candidates for follow-up functional characterization. In the future, we anticipate that this method can be extended to profiling of unannotated human alt-proteins expressed under different conditions, including stress or disease states.
Supplementary Material
Figure S1. mRNA-Seq support for novel SCRIB transcript variant.
Figure S2. Confirmation of differential expression of three additional detected alt-proteins with extracted ion chromatograms (EICs).
Figure S3. Predicted start codons used by the six selected alt-ORFs.
Figure S4. Results of TargetP bioinformatic analysis.
Figure S5. Nuclear localization signals predicted using cNLS-mapper.
Figure S6. Amino acid-level conservation of alt-proteins in other species.
Figure S7. Structure prediction of two nuclear alt-proteins.
Figure S8. Uncropped Western blots for confirmation of alt-protein expression (related to Figure 5).
Figure S9. K562 MS/MS spectra.
Figure S10. MOLT4 MS/MS spectra.
Figure S11. MS/MS spectra of peptides found in both cell lines.
Table S1. Full list of peptides and proteins detected in each sample. (XLSX)
Table S2. PepQuery results. (XLSX)
Table S3. Full list of peptides detected from unannotated alt-proteins. (XLSX)
Table S4. TargetP 2.0 prediction results. (XLSX)
ACKNOWLEDGMENT
This work was supported by the Leukemia Research Foundation, the Searle Scholars Program, and Yale University West Campus start-up funds (to S.A.S.). X.C. was supported in part by a Rudolph J. Anderson postdoctoral fellowship from Yale University. A.K. was in part supported by an NIH Predoctoral Training Grant (5T32GM06754 3-12). D.G.D. was in part supported by an NIH Predoctoral Training Grant (2T32GM06754 3-16).
Footnotes
SUPPORTING INFORMATION
The following supporting information is available free of charge at ACS website.
REFERENCES
- 1.Ingolia NT, Ghaemmaghami S, Newman JR & Weissman JS Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324, 218–23 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martinez TF et al. Accurate annotation of human protein-coding small open reading frames. Nat Chem Biol (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Slavoff SA et al. Peptidomic discovery of short open reading frame-encoded peptides in human cells. Nat Chem Biol 9, 59–64 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ma J et al. Discovery of human sORF-encoded polypeptides (SEPs) in cell lines and tissue. J Proteome Res 13, 1757–65 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brunet MA et al. OpenProt: a more comprehensive guide to explore eukaryotic coding potential and proteomes. Nucleic Acids Res 47, D403–D410 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Samandi S et al. Deep transcriptome annotation enables the discovery and functional characterization of cryptic small proteins. Elife 6(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Orr MW, Mao Y, Storz G & Qian SB Alternative ORFs and small ORFs: shedding light on the dark proteome. Nucleic Acids Res 48, 1029–1042 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saghatelian A & Couso JP Discovery and characterization of smORF-encoded bioactive polypeptides. Nat Chem Biol 11, 909–16 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen J et al. Pervasive functional translation of noncanonical human open reading frames. Science 367, 1140–1146 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chu Q et al. Regulation of the ER stress response by a mitochondrial microprotein. Nat Commun 10, 4883 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang S et al. Mitochondrial peptide BRAWNIN is essential for vertebrate respiratory complex III assembly. Nat Commun 11, 1312 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee S et al. Global mapping of translation initiation sites in mammalian cells at single-nucleotide resolution. Proc Natl Acad Sci U S A 109, E2424–32 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ingolia NT, Lareau LF & Weissman JS Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 147, 789–802 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khitun A, Ness TJ & Slavoff SA Small open reading frames and cellular stress responses. Mol Omics 15, 108–116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brar GA et al. High-resolution view of the yeast meiotic program revealed by ribosome profiling. Science 335, 552–7 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Polycarpou-Schwarz M et al. The cancer-associated microprotein CASIMO1 controls cell proliferation and interacts with squalene epoxidase modulating lipid droplet formation. Oncogene 37, 4750–4768 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Jackson R et al. The translation of non-canonical open reading frames controls mucosal immunity. Nature 564, 434–438 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.D'Lima NG et al. Comparative Proteomics Enables Identification of Nonannotated Cold Shock Proteins in E. coli. J Proteome Res 16, 3722–3731 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yuan P, D'Lima NG & Slavoff SA Comparative Membrane Proteomics Reveals a Nonannotated E. coli Heat Shock Protein. Biochemistry 57, 56–60 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma J et al. Improved Identification and Analysis of Small Open Reading Frame Encoded Polypeptides. Anal Chem 88, 3967–75 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schagger H Tricine-SDS-PAGE. Nat Protoc 1, 16–22 (2006). [DOI] [PubMed] [Google Scholar]
- 22.Oyama M et al. Diversity of translation start sites may define increased complexity of the human short ORFeome. Mol Cell Proteomics 6, 1000–6 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Khitun A & Slavoff SA Proteomic Detection and Validation of Translated Small Open Reading Frames. Curr Protoc Chem Biol 11, e77 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wen B, Wang X & Zhang B PepQuery enables fast, accurate, and convenient proteomic validation of novel genomic alterations. Genome Res 29, 485–493 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Longo PA, Kavran JM, Kim MS & Leahy DJ Transient mammalian cell transfection with polyethylenimine (PEI). Methods Enzymol 529, 227–40 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lozzio CB & Lozzio BB Human chronic myelogenous leukemia cell-line with positive Philadelphia chromosome. Blood 45, 321–34 (1975). [PubMed] [Google Scholar]
- 27.Minowada J, Onuma T & Moore GE Rosette-forming human lymphoid cell lines. I. Establishment and evidence for origin of thymus-derived lymphocytes. J Natl Cancer Inst 49, 891–5 (1972). [PubMed] [Google Scholar]
- 28.Na CH et al. Discovery of noncanonical translation initiation sites through mass spectrometric analysis of protein N termini. Genome Res 28, 25–36 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu Y et al. Discovery of coding regions in the human genome by integrated proteogenomics analysis workflow. Nat Commun 9, 903 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu L et al. Interaction between p12CDK2AP1 and a novel unnamed protein product inhibits cell proliferation by regulating the cell cycle. Mol Med Rep 9, 156–62 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Cheng Z et al. Pervasive, Coordinated Protein-Level Changes Driven by Transcript Isoform Switching during Meiosis. Cell 172, 910–923 e16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wegener AD & Jones LR Phosphorylation-induced mobility shift in phospholamban in sodium dodecyl sulfate-polyacrylamide gels. Evidence for a protein structure consisting of multiple identical phosphorylatable subunits. J Biol Chem 259, 1834–41 (1984). [PubMed] [Google Scholar]
- 33.D'Lima NG et al. A human microprotein that interacts with the mRNA decapping complex. Nat Chem Biol 13, 174–180 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Slavoff SA, Heo J, Budnik BA, Hanakahi LA & Saghatelian A A human short open reading frame (sORF)-encoded polypeptide that stimulates DNA end joining. J Biol Chem 289, 10950–7 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Almagro Armenteros JJ et al. Detecting sequence signals in targeting peptides using deep learning. Life Sci Alliance 2(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kosugi S et al. Six classes of nuclear localization signals specific to different binding grooves of importin alpha. J Biol Chem 284, 478–85 (2009). [DOI] [PubMed] [Google Scholar]
- 37.Kosugi S, Hasebe M, Tomita M & Yanagawa H Systematic identification of cell cycle-dependent yeast nucleocytoplasmic shuttling proteins by prediction of composite motifs. Proc Natl Acad Sci U S A 106, 10171–6 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Quarto N, Finger FP & Rifkin DB The NH2-terminal extension of high molecular weight bFGF is a nuclear targeting signal. J Cell Physiol 147, 311–8 (1991). [DOI] [PubMed] [Google Scholar]
- 39.Bugler B, Amalric F & Prats H Alternative initiation of translation determines cytoplasmic or nuclear localization of basic fibroblast growth factor. Mol Cell Biol 11, 573–7 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Storz G, Wolf YI & Ramamurthi KS Small proteins can no longer be ignored. Annu Rev Biochem 83, 753–77 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roy A, Kucukural A & Zhang Y I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 5, 725–38 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Q et al. The microprotein Minion controls cell fusion and muscle formation. Nat Commun 8, 15664 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. mRNA-Seq support for novel SCRIB transcript variant.
Figure S2. Confirmation of differential expression of three additional detected alt-proteins with extracted ion chromatograms (EICs).
Figure S3. Predicted start codons used by the six selected alt-ORFs.
Figure S4. Results of TargetP bioinformatic analysis.
Figure S5. Nuclear localization signals predicted using cNLS-mapper.
Figure S6. Amino acid-level conservation of alt-proteins in other species.
Figure S7. Structure prediction of two nuclear alt-proteins.
Figure S8. Uncropped Western blots for confirmation of alt-protein expression (related to Figure 5).
Figure S9. K562 MS/MS spectra.
Figure S10. MOLT4 MS/MS spectra.
Figure S11. MS/MS spectra of peptides found in both cell lines.
Table S1. Full list of peptides and proteins detected in each sample. (XLSX)
Table S2. PepQuery results. (XLSX)
Table S3. Full list of peptides detected from unannotated alt-proteins. (XLSX)
Table S4. TargetP 2.0 prediction results. (XLSX)
Data Availability Statement
The mRNA-seq data have been deposited in the NCBI Gene Expression Omnibus under accession GSE148451.
The mass spectrometry proteomics data have been deposited to the PRIDE Archive (http://www.ebi.ac.uk/pride/archive/) via the PRIDE partner repository with the data set identifier PXD018565 and 10.6019/PXD018565.