

Abstract
Despite their notorious adverse effects, glucocorticoids (GC, potent anti-inflammatory drugs) are used extensively in clinical management of rheumatoid arthritis (RA) and other chronic inflammatory diseases. To achieve a sustained therapeutic efficacy and reduced toxicities, macromolecular GC prodrugs have been developed with promising outcomes for the treatment of RA. Fine-tuning the activation kinetics of these prodrugs may further improve their therapeutic efficacy and minimize the off-target adverse effects. To assess the feasibility of this strategy, five different dexamethasone (Dex, a potent GC)-containing monomers with distinctively different linker chemistries were designed, synthesized, and copolymerized with N-(2-hydroxypropyl) methacrylamide (HPMA) to obtain 5 macromolecular Dex prodrugs. Their Dex releasing rates were analyzed in vitro and shown to display a wide spectrum of activation kinetics. Their therapeutic efficacy and preliminary toxicology profiles were assessed and compared in vivo in an adjuvant-induced arthritis (AA) rat model in order to identify the ideal prodrug design for the most effective and safe treatment of inflammatory arthritis. The in vivo data demonstrated that the C3 hydrazone linker-containing prodrug design was the most effective in preserving joint structural integrity. The results from this study suggest that the design and screening of different activation mechanisms may help to identify macromolecular prodrugs with the most potent therapeutic efficacy and safety for the management of inflammatory arthritis.
Keywords: Rheumatoid arthritis, inflammation, dexamethasone, controlled release, HPMA copolymer, prodrug
1. INTRODUCTION
Rheumatoid arthritis (RA) is a chronic autoimmune disease affecting up to 0.8% of general population worldwide [1–3]. It is an inflammatory disease that targets the synovial joint lining, leading to gradual deterioration of articular cartilage, erosion of peri-articular bone and eventual destruction of joint integrity and function [4]. Presently, there is no cure for RA. Current therapies for RA include: nonsteroidal anti-inflammatory drugs (NSAIDs), glucocorticoids (GC), and disease-modifying antirheumatic drugs (DMARDS) and biological-response modifiers [5]. Over one third of RA patients still experience joint inflammation and progressive deterioration in joint structure and function, even though many novel RA therapies have been developed targeting newly identified inflammatory mediators and pathways [6, 7]. NSAIDs have been shown to ease pain and inflammation associated with RA [8, 9] but they do not prevent joint damage and are often associated with significant gastrointestinal, renal [10] and cardiovascular side effects [11]. GC have been used as anti-inflammatory and immunosuppressive agents to regulate the rheumatic activities for more than five decades. Due to their systemic toxicities (e.g., osteoporosis, high blood pressure, obesity, and diabetes, etc.), the utility of GC as long-term RA therapy has been very limited [12, 13]. The lack of organ/tissue specificity after oral or systemic administration often necessitates higher GC dosing levels and/or frequency to achieve effective concentrations at sites of inflammation and pathology, further aggravating the adverse side effects. The ELVIS (Extravasation through Leaky Vasculature and subsequent Inflammatory cell-mediated Sequestration) mechanism-driven, targeted delivery of therapeutic agents to the sites of inflammatory pathology according has been proposed to address this daunting challenge [4]. Accordingly, several nanomedicine formulations of GC have been developed for a variety of different inflammatory conditions, including rheumatoid arthritis [14–17].
Previously, we developed a N-(2-hydroxypropyl)methacrylamide (HPMA) copolymer-based dexamethasone (Dex) prodrug (P-Dex) for the treatment of inflammatory diseases, including inflammatory arthritis, inflammatory bowel disease (IBD), and orthopaedic implant loosening [14–16]. P-Dex was found to passively target to the sites of inflammatory pathology and provided potent and sustained amelioration of the inflammation after a single systemic administration, which was superior to the dose equivalent Dex treatment [16, 18, 19]. Also, different from the Dex treatment, no osteotoxicity was found to be associated with P-Dex therapy [20]. In a head-to-head comparison study of Dex nanomedicine formulations for the treatment of adjuvant-induced arthritis (AA), P-Dex was found to offer superior therapeutic efficacy and safety compared to the other Dex nanomedicine formulations tested. Interestingly, in the same study, we also found that when the P-Dex activation mechanism was changed from a C3 hydrazone to a C21 benzoate ester/hydrazone linker system, a much faster Dex releasing rate was achieved, which resulted in less effective suppression of inflammation and reduced protection from joint damage [21]. Clearly, the mechanism of P-Dex activation has a very significant impact on its pharmacodynamic profile.
In this manuscript, we report the “titration” of the P-Dex activation profiles, in search for the optimal P-Dex structural design that would offer the most potent and safe treatment of RA. A total of 5 different activation chemistries have been designed and incorporated in the P-Dex structure. These macromolecular prodrugs were synthesized, characterized and evaluated in vitro for their different activation profiles. They were then tested in the AA rat model of inflammatory arthritis to compare their therapeutic efficacy and safety profile.
2. MATERIAL AND METHODS
2.1. Materials
N-Methacryloylglycylglycine (MA–Gly–Gly–OH) and N-(2-hydroxypropyl) methacrylamide (HPMA) were prepared according to previously published methods [22–24]. Dexamethasone was purchased from Tianjin Tianyao Pharmaceuticals Co. Ltd. Other chemical reagents and solvents, if not specified, were aquired from either Thermo Fisher Scientific (Waltham, MA USA) or Sigma-Aldrich (St. Louis, MO, USA). All chemicals were reagent-grade or higher and directly used without further purification.
2.2. Instruments
A 500 MHz Varian NMR spectrometer (Varian, Palo Alto, CA, USA) was used to record the 1H and 13C NMR spectra of all chemical compounds. Electrospray Ionization (ESI) Mass Spectrometry (MS) analyses were performed using a Finnigan Mat LCQ Mass Spectrometer System (Thermo Finnigan, San Jose, CA, USA). The copolymers were characterized for their average molecular weights (Mw, Mn) and dispersity (Ð) on an AKTÄ pure FPLC system (GE HealthCare, Chicago, IL), equips with a Superdex 200 Increase 10/300 GL column, a DAWN 8+ multiangle light scattering (MALS) system and an Optilab T-rEX refractive index detector (Wyatt, Santa Barbara, CA). An Agilent HPLC system (1100 series, Agilent Technologies, Inc., Santa Clara, CA) with a Hypersil™ ODS C18 column (4.6 mm × 250 mm, Thermo Fisher Scientific, Waltham, MA) was used for HPLC analysis. A high-resolution micro-CT system (Skyscan 1172, Bruker, Kontich, Belgium) was used for bone quality was analyzed. Histology sections were analyzed on a Leica DM750 Light Microscope with the Leica ICC50 HD Digital Camera.
2.3. Synthesis of HPMA copolymer-based dexamethasone prodrugs with different activation rates
To prepare Dex-containing HPMA copolymer-based prodrugs with different activation rates, five Dex-containing monomers (Monomers A, B, C, D and E) were designed and synthesized (Scheme 1). By adjusting the linker chemistry between the methacryloyl group and Dex, these Dex-containing monomers, once copolymerized with HPMA will produce polymeric prodrugs with a wide spectrum of Dex releasing rates.
Scheme 1.
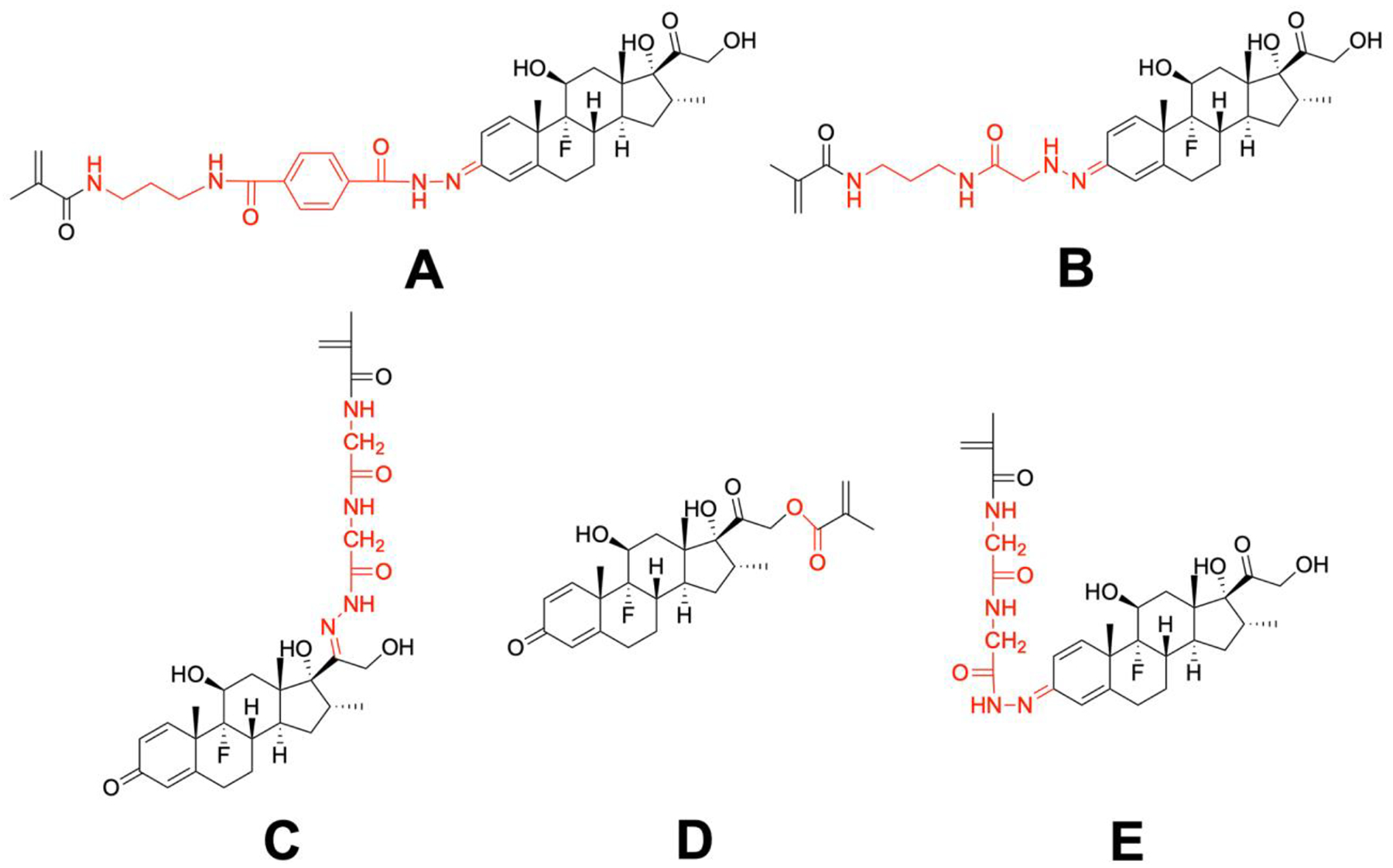
The design of Dex-containing monomers (A, B, C, D, E) for the synthesis of HPMA copolymer-based Dex prodrugs with different releasing rates. Red colored structures represent the different linker chemistries.
2.3.1. Synthesis of Dex-containing monomers A, B, C, D and E
2.3.1.1. Synthesis of monomer A
Synthesis of compound 1 and 2.
Dexamethasone (19.6 g, 50 mmol) and imidazole (6.8 g, 100 mmol) were dissolved in dimethylformamide (DMF, 100 mL, anhydrous) and the solution was cooled to 0 °C by ice bath. tert-Butyldimethylsilyl chloride (TBSCl, 9 g, 60 mmol) was added. Under constant stirring, the solution was maintained at 0 °C for 1 h and then at room temperature for 3 h. After addition of ethyl acetate (150 mL), the solution was washed with saturated brine (100 mL × 4) , separate and dried over Na2SO4. After removal of the solvents, the crude product (TBS-protected Dex, 24.9 g) was obtained.
The TBS-protected Dex was then dissolved in methanol (100 mL). After addition of hydrazine monohydrate (7.5 g, 150 mmol) and acetic acid (1.5 g, 25 mmol), the solution was stirred at room temperature for 3 h. This was followed by the addition of ethyl acetate (200 mL). The resulting solution was washed with brine (150 mL × 2). The organic phase was then separated and dried over Na2SO4. After the solvents’ removal, the crude product was purified by column chromatography (hexanes: ethyl acetate : dichloromethane = 1 : 1 : 0.5) to give recovered TBS-protected Dex (11.12 g, yield: 43.9% ), compound 1 (11.45 g, yield: 44.0%) and compound 2 (0.63 g, yield: 2.4%). The NMR data for Compound 1 has been reported previously [17]. For Compound 2, the NMR data are shown as the following.
1H NMR (500 MHz, DMSO-d6): δ (ppm) = 7.34 (d, J = 10.2 Hz, 1H), 6.25 (s, 2H), 6.19 (dd, J = 10.2 Hz, 1.8Hz, 1H), 5.99 (s, 1H), 5.29 (d, J = 5.8 Hz, 1H), 4.26 (d, J = 11.8 Hz, 1H), 4.20 (d, J = 11.8 Hz, 1H), 4.13 (s, 1H), 4.09 (m, 1H), 3.02 (m, 1H), 2.59 (m, 1H), 2.30 (m, 2H), 2.11 (m, 2H), 1.76 (m, 1H), 1.53 (q, J = 11.8, 1H), 1.48 (s, 3H), 1.41 (d, J = 13.1 Hz, 1H), 1.33 (m, 1H), 1.06 (m, 1H), 0.88 (s, 9H), 0.87 (d, J = 6.2 Hz, 1H), 0.77 (d, J = 7.2 Hz, 1H), 0.11 (s, 6H).
13C NMR (125 MHz, DMSO-d6): δ (ppm) = 185.49, 170.50, 167.51, 153.18, 148.07, 129.03, 124.19, 101.26 (d, J = 174.4 Hz), 86.56, 70.65 (d, J = 36.1 Hz), 59.93, 57.12, 48.28 (d, J = 22.9 Hz), 47.28, 42.32, 36.53, 34.00 (d, J = 19.1 Hz), 33.12, 32.10, 30.59, 27.52, 25.93, 23.16, 23.11, 20.93, 18.02, 17.43, 15.84, 14.26, −5.40, −5.60.
MS (ESI): m/z = 521.5 (M + H+), calculated: 520.3.
Synthesis of compound 3.
Mono-methyl terephthalate (2.16 g, 12 mmol) and N,N-’dicyclohexylcarbodiimide (DCC, 2.67 g, 13 mmol) were dissolved in dichloromethane (DCM, 20 mL, anhydrous). Under constant stirring, the solution was kept at room temperature for 10 min. N-(3-Aminopropyl) methacrylamide hydrochloride (1.78 g, 10 mmol), hydroxybenzotriazole (HOBt, 3.06 g, 20 mmol) and triethylamine (Et3N, 3.03 g, 30 mmol) were then added. After stirring for 5 h, the solution was filtered, concentrated and purified by flash column chromatography to yield the product (2.42 g, yield: 79.6%).
1H NMR (500 MHz, CDCl3): δ (ppm) = 8.08 (d, J = 8.4 Hz, 2H), 7.94 (d, J = 8.4 Hz, 2H), 7.81 (t, J = 6.0Hz, 1H),, 6.87 (s, 1H), 5.79 (s, 1H), 5.37 (s, 1H), 3.94 (s, 3H), 3.50 (m, 2H), 3.41 (m, 2H), 2.00 (s, 3H), 1.76 (m, 2H), .
13C NMR (125 MHz, CDCl3): δ (ppm) = 169.4, 166.9, 166.3, 139.6, 138.2, 132.5, 129.7, 127.1, 120.1, 52.3, 36.1, 36.0, 29.6, 18.6.
MS (ESI): m/z = 327.1 (M + Na+), calculated: 304.1.
Synthesis of compound 4.
Under constant stirring, a mixture of water (2 mL), methanol (18 mL) and KOH (10 M, 0.4 mL) was used to dissolve compound 3 (1.22 g, 4 mmol) and maintained at room temperature overnight. After evaporation of the solvents, brine (20 mL) and hydrochloride solution (0.6 mL) were added to neutralize the KOH. Ethyl acetate (100 mL × 4) was used to extract the product from the aqueous phase. After removal of the solvent, the residue crude product was purified by flash column chromatography (dichloromethane : methanol = 8 : 1) to produce compound 4 (1.07 g, yield: 92.2%).
1H NMR (500 MHz, DMSO-d6): δ (ppm) = 8.64 (t, J = 5.6 Hz, 1H), 8.01 (d, J = 8.4 Hz, 2H), 7.96 (d, J = 8.4 Hz, 1H), 5.66 (s, 1H), 5.32 (s, 1H), 3.29 (m, 2H), 3.18 (m, 2H), 1.85 (s, 3H), 1.70, (m, 2H).
13C NMR (125 MHz, DMSO-d6): δ (ppm) = 167.65, 167.18, 165.76, 140.21, 138.24, 133.84, 129.37, 127.44, 119.03, 37.25, 36.81, 29.30,18.80.
MS (ESI): m/z = 313.1 (M + Na+), calculated: 290.1.
Synthesis of compound 5.
Compound 1 (312 mg, 0.6 mmol), DCC (206 mg, 1 mmol), HOBt monohydrate (153 mg, 1mmol) and Et3N (150 mg, 1.5 mmol) were dissolved in anhydrous DMF. Compound 4 (145 mg, 0.5 mmol) was then added. Under constant stirring, the solution was maintained at room temperature for 5 h. After addition of the ethyl acetate, the solution was washed with saturated brine, separated and dried over Na2SO4. After solvents’ removal, the crude product was purified by flash column chromatography (dichloromethane: methanol = 8 : 1) to produce compound 5 (335 mg, yield: 84.6%).
1H NMR (500 MHz, DMSO-d6): δ (ppm) = 11.07 (s, 0.38), 11.05 (s, 0.59), 8.63 (s, 1.01 H), 7.98 (t, J = 5.9 Hz, 1.00H), 7.94 (s, br, 3.83H), 7.75 (s, br, 0.28H), 7.03 (d, J = 10.2 Hz, 0.29H), 6.79 (s, 0.57H), 6.70 (d, J = 10.2 Hz, 0.32H), 6.57 (d, J = 10.2 Hz, 0.55H), 6.38 (d, J = 10.2 Hz, 0.59H), 6.12 (s, 0.32H), 5.67 (s, 1.00H), 5.32 (s, 1.01H), 5.20 (s, 0.96H), 4.96 (s, 0.97H), 4.79 (d, J = 19.0 Hz, 0.97H), 4.29 (d, J = 19.0 Hz, 0.97H), 4.15 (m, 1.01H), 3.50 (m, 1.00H), 3.30 (m, 2.00H), 3.19 (m, 2.00H), 2.90 (m, 1.02H), 2.62 (m, 0.89H), 2.28 (m, 1.95H), 2.13 (m, 2.05H), 1.86 (s, 3.12H), 1.71 (m, 3.12H), 1.61 (m, 1.14H), 1.45 (s, 3.00H), 1.35 (s, 2.27H), 1.05 (m, 1.03H), 0.88 (s, 9.15H), 0.87 (d, J = 6.2 Hz, 3.14H), 0.77 (d, J = 7.3 Hz, 3.29H), 0.04 (s, 3H), 0.03 (s, 3.15H).
13C NMR (125 MHz, DMSO-d6): δ (ppm) = 209.24, 167.64, 165.71, 140.19, 136.97, 128.00, 127.11, 119.04, 112.57, 100.87 (d, J = 174.4 Hz), 90.59, 90.52, 79.37, 70.10 (d, J = 36.9 Hz), 68.15, 49.08, 47.62, 47.45, 45.69, 43.75, 43.64, 37.17, 36.77, 36.09, 35.32, 34.09, 33.93, 33.25, 32.07, 30.90, 29.33, 27.48, 27.39, 25.96, 25.47, 24.91, 24.35, 24.31, 23.95, 21.26, 18.81, 18.33, 16.87, 15.41, 9.30, −4.89, −5.04.
MS (ESI): m/z = 815.4 (M + Na+), calculated: 792.4.
Synthesis of monomer A.
After dissolution of compound 5 (300 mg, 0.38 mmol) in tetrahydrofuran (THF), tetrabutylammonium fluoride (TBAF, 1 M in THF, 0.54 mL) was added. Under constant stirring, the solution was maintained at room temperature for 1 h. After the addition of ethyl acetate, the solution was washed with saturated brine, separated and dried over Na2SO4. After the solvents’ removal, the crude product was purified by flash column chromatography (dichloromethane : methanol = 10 : 1) to produce monomer A (236 mg, yield: 93.5%).
1H NMR (500 MHz, DMSO-d6): δ (ppm) = 11.08 (s, br, 0.85H), 8.68 (s, 0.90H), 8.32 (s, 0.45H), 8.02 (s, 1.13H), 7.97 (s, 3.23H), 7.74 (s, 0.28H), 7.04 (d, J = 8.1Hz, 0.29H), 6.86 (s, 0.17H), 6.81 (s, 0.43H), 6.73 (d, J = 8.9 Hz, 0.34H), 6.59 (d, J = 8.0Hz, 0.53H), 6.40 (s, 0.48H), 6.14 (s, 0.28H), 5.67 (s, 0.85H), 5.31 (s, 0.98H), 5.21 (m, 0.87H), 4.95 (m, 1.03H), 4.50 (d, J = 19.1Hz, 1.00H), 4.14 (m, 1H), 4.08 (d, J = 19.1Hz, 1H), 3.30 (d, J = 5.0 Hz, 1.89H), 3.18 (d, J = 5.6 Hz, 2.13H), 2.93 (s, 0.93H), 2.63 (m, 0.55H), 2.27 (m, 1.80H), 2.15 (m, 1.98H), 1.89 (s, 0.60H), 1.86 (s, 2.70H), 1.71 (m, 2.76H), 1.60 (m, 1.24H), 1.45 (s, 3.59H), 1.35 (s, 2.62H), 1.17 (m, 1.02H), 1.06 (m, 1.22H), 0.86 (s, 3.21H), 0.78 (d, J = 5.9Hz).
13C NMR (125 MHz, DMSO-d6): δ (ppm) = 211.41, 172.37, 167.69, 165.74, 140.20, 137.12, 136.47, 128.04, 127.16, 119.11, 112.69, 101.04 (d, J = 174.4 Hz), 90.42, 90.36, 79.40, 70.29, 70.02, 66.48,53.24, 47.63, 43.63, 37.20, 36.79, 36.05, 35.09, 34.56, 34.12, 33.96, 32.19, 30.93, 30.61, 30.22, 29.34, 27.44, 26.03, 24.33, 23.96, 21.50, 21.26, 20.20, 18.84, 16.86, 15.51, 14.09.
MS (ESI): m/z = 701.3 (M + Na+), calculated: 678.3.
2.3.1.2. Synthesis of monomer B
Synthesis of compound 6.
After dissolving compound 2 (1.04g, 2 mmol) and methyl bromoacetate (392 mg, 2.6 mmol) in DMF (10 mL, anhydrous), potassium carbonate (834 mg, 6 mmol) was added to the solution. Under constant stirring, the solution was maintained at room temperature for 2.5 h, followed by the addition of ethyl acetate (100 mL). The solution was then washed with saturated brine (100 mL × 3), separated and dried over Na2SO4. After removal of the solvents, the crude product was purified by flash column chromatography (hexanes :ethyl acetate = 1: 1.5) to produce compound 6 (740 mg, yield: 62.5%).
1H NMR (500 MHz, CDCl3): δ (ppm) = 6.60 (d, J = 10.4 Hz, 0.31H), 6.44 (d, J = 10.4Hz, 0.31H), 6.30 (s, 0.65H), 6.26 (dd, J = 10.2 Hz, 1.6 Hz, 0.66H), 6.13 (d, J = 10.2Hz, 0.66H), 5.94 (s, 0.30H), 5.50 (br, 1H), 4.62 (d, J = 18.3 Hz, 0.31H), 4.60 (d, J = 18.2 Hz, 0.66H), 4.35 (d, J = 18.3 Hz, 0.97H), 4.30 (m, 1.00H), 3.94 (s, 1.46H), 3.92 (s, 0.67H), 3.71(s, 1.93H), 3.70 (s, 0.90H), 3.00 (m, 2H), 2.60 (td, J = 13.6 Hz, 5.5Hz, 0.65H), 2.47 (td, J = 13.6 Hz, 5.5 Hz, 0.31H), 2.20–2.40 (m, 5.46H), 1.68 (m, 1.99H), 1.48 (m, 0.80H), 1.43 (s, 3.18H), 1.36 (d, J = 13.8 Hz, 1H), 1,18 (m, 0.98H), 1.00 (s, 3.00H), 0.89 (s, 9H), 0.87 (d, J = 7.3Hz, 3.21H), 0.08 (s, 3H), 0.07 (s, 3H).
13C NMR (125 MHz, CDCl3): δ (ppm) = 209.47, 209.44, 172.44, 172.34, 154.49, 146.91, 143.57, 143.42, 141.68, 134.76, 127.24, 121.22, 115.57, 110.32, 99.60 (d, J = 173.5Hz), 99.40 (d, J = 173.5Hz), 98.19, 91.16, 71.14 (d, J = 39.2Hz), 71.04 (d, J = 39.2Hz), 69.46, 69.42, 52.22, 51.92, 48.44, 48.40, 47.47 (d, J = 26.7Hz), 43.91, 37.22, 37.16, 36.10, 34.33, 34.18, 32.21, 31.41, 30.27, 27.44, 27.35, 25.81, 24.44, 24.40, 23.99, 23.95, 18.44, 17.23, 14.84, −5.33, −5.47.
MS (ESI): m/z = 615.3 (M + Na+), calculated: 592.3.
Synthesis of compound 7.
Under contant stirring, compound 6 (720 mg, 1.21 mmol) and 1,3-diaminopropane (900 mg, 12.1 mmol) were dissolved in MeOH (10 mL) and maintained at room temperature overnight. After addition of DCM (100 mL), the solution was washed with saturated brine (100 mL × 3), separated and dried over Na2SO4. After removal of the solvents, the crude product was obtained. It was then dissolved in DCM (10 mL, anhydrous). Under constant stirring, Et3N (484 mg, 4.8 mmol), DCC (494 mg, 2.16 mmol), HOBt (555 mg, 3.6 mmol) and methacrylic acid (185 mg, 2.16 mmol) were added and the solution was maintained for 6 h. After the addition of ethyl acetate (100mL), the solution was washed with brine (100 mL × 2), separated and dried over Na2SO4. After the removal of the solvents, the crude product was purified by flash column chromatography (dichloromethane : methanol = 10 : 1) to produce compound 7 (570 mg, yield 67.1% for two steps).
1H NMR (500 MHz, CDCl3): δ (ppm) = 7.67 (s, 0.45H), 7.65 (s, 0.55H), 7.54 (m, 1H), 7.19 (d, J = 10.2 Hz, 1H), 6.97 (t, J = 6.2 Hz, 1H), 6.92 (t, J = 6.2 Hz, 1H), 6.90 (s, 0.56H), 6.79 (t, J = 9.4 Hz, 1H), 6.50 (d, J = 10.2 Hz, 1H), 6.26 (s, 0.44H), 5.78 (s, 1H), 5.33 (s, 1H), 4.64 (d, J = 18.2 Hz, 0.45H), 4.62 (d, J = 18.2 Hz, 0.55H), 4.36 (d, J = 18.2 Hz, 1H), 4.30 (m, 1H), 3.44 (m, 2H), 3.35 (m, 2H), 3.17 (m, 1.19H), 3.02 (m, 1.22H), 2.59 (m, 1H), 2.1–2.4 (m, 4H), 1.98 (s, 1.65H), 1.97 (s, 1.35H), 1.75 (m, 4H), 1.51 (m, 1H), 1.50 (s, 3H), 1.40 (d, J = 13.7 Hz, 1H), 1.24 (m, 2H), 1.03 (s, 3H), 0.90 (s, 9H), 0.87 (d, 3H), 0.09 (s, 3H), 0.08 (s, 3H).
13C NMR (125 MHz, CDCl3): δ (ppm) = 209.47, 168.77, 164.45, 164.40, 160.46, 160.37, 159.71, 159.13, 150.23, 150.09, 146.83, 145.47, 126.76, 121.12, 120.08, 119.99, 119.32, 114.14, 101.07, 99.68, 91.13, 100.37 (d, J = 174.2 Hz), 91.13, 71.51(d, J = 38.3 Hz), 71.46 (d, J = 38.3 Hz), 69.43, 48.72 (d, J = 22.6 Hz), 48.48, 43.75, 37.06, 37.02, 36.10, 36.02, 35.96, 35.93, 35.87, 34.24 (d, J = 19.4 Hz), 32.26, 31.31, 31.01, 29.61, 29.52, 29.45, 27.43, 23.69 (d, J = 5.3 Hz), 23.39 (d, J = 5.1 Hz), 18.56, 18.42, 17.14, 17.11, 14.82.
MS (ESI): m/z = 703.2 (M + H+), calculated: 702.4.
Synthesis of monomer B.
After dissolving compound 7 (360 mg, 0.51 mmol) in THF (5 mL), TBAF (1 mL, 1 M, 0.26 mmol) was added and the resulting solution was maintained at room temperature for 1 h with constant stirring, followed by the addition of ethyl acetate (50 mL). The resulting solution was washed with brine, separated and dried over Na2SO4. After the removal of the solvents, the crude product was purified by flash column chromatography (dichloromethane : methanol = 7 : 1) to produce monomer B (275 mg, yield 91.7%).
1H NMR (500 MHz, DMSO-d6): δ (ppm)= 7.90 (s, 1H), 7.77 (t, J = 5.3 Hz, 1H), 6.85 (s, 0.35H), 6.82 (s, 0.65H), 6.74 (d, J = 10.5Hz, 0.35H), 6.45 (s, 0.65H), 6.37 (d, J = 10.4 Hz, 0.35H), 6.07 (m, 0.31H), 6.06 (s, 1H), 5.78 (s, 0.34H), 5.64 (s, 1.00H), 5.30 (s, 1.00H), 5.08 (s, 0.35H), 5.06 (s, 0.65H), 4.92 (s, 1.00H), 4.66 (s, 1H), 4.50 (dd, J = 19.3 Hz, 5.7Hz, 1.00H), 4.10 (s, 1H), 4.07 (dd, J = 19.3 Hz, 5.7Hz, 1.00H), 4.06 (dd, J = 19.3 Hz, 5.7Hz, 1H), 3.16 (m, 1H), 3.09 (m, 4.50H), 2.93 (m, 1H), 2.58 (td, J = 13.6 Hz, 5.5 Hz, 0.65H), 2.49 (td, J = 13.6 Hz, 5.5 Hz, 0.52H), 2.10–2.40 (m, 4.2H), 1.84 (s, 3H), 1.75 (s, 3.65H), 1.67 (m, 0.77H), 1.50–1.65 (m, 4.57H), 1.39 (s, 3H), 1.20–1.45 (m, 4H), 1.05 (m, 1H), 0.93 (t, J = 7.3 Hz, 1.53H), 0.84 (s, 3H), 0.78 (d, J = 6.9Hz, 3H).
13C NMR (125 MHz, CDCl3): δ (ppm) = 211.40, 170.98, 167.53, 152.00, 145.03, 140.21, 140.11, 139.98, 139.46, 133.29, 127.15, 121.18, 118.98, 116.83, 111.52, 100.49 (d, J = 172.1 Hz), 100.26 (d, J = 172.1Hz), 90.42, 90.36, 69.85 (d, J = 37.3 Hz), 69.77 (d, J = 37.3 Hz), 67.20, 57.73, 53.71, 47.60, 47.10, 46.91, 46.75, 45.66, 43.71, 36.52, 36.22, 36.10, 36.04, 35.06, 34.18 (d, J = 19.5 Hz), 34.13 (d, J = 19.5 Hz), 32.18, 30.96, 29.85, 29.41, 27.44, 24.91, 24.88, 24.48, 24.44, 23.26, 19.40, 16.84, 15.49, 13.66.
MS (ESI): m/z = 611.1 (M + Na+), calculated: 588.3.
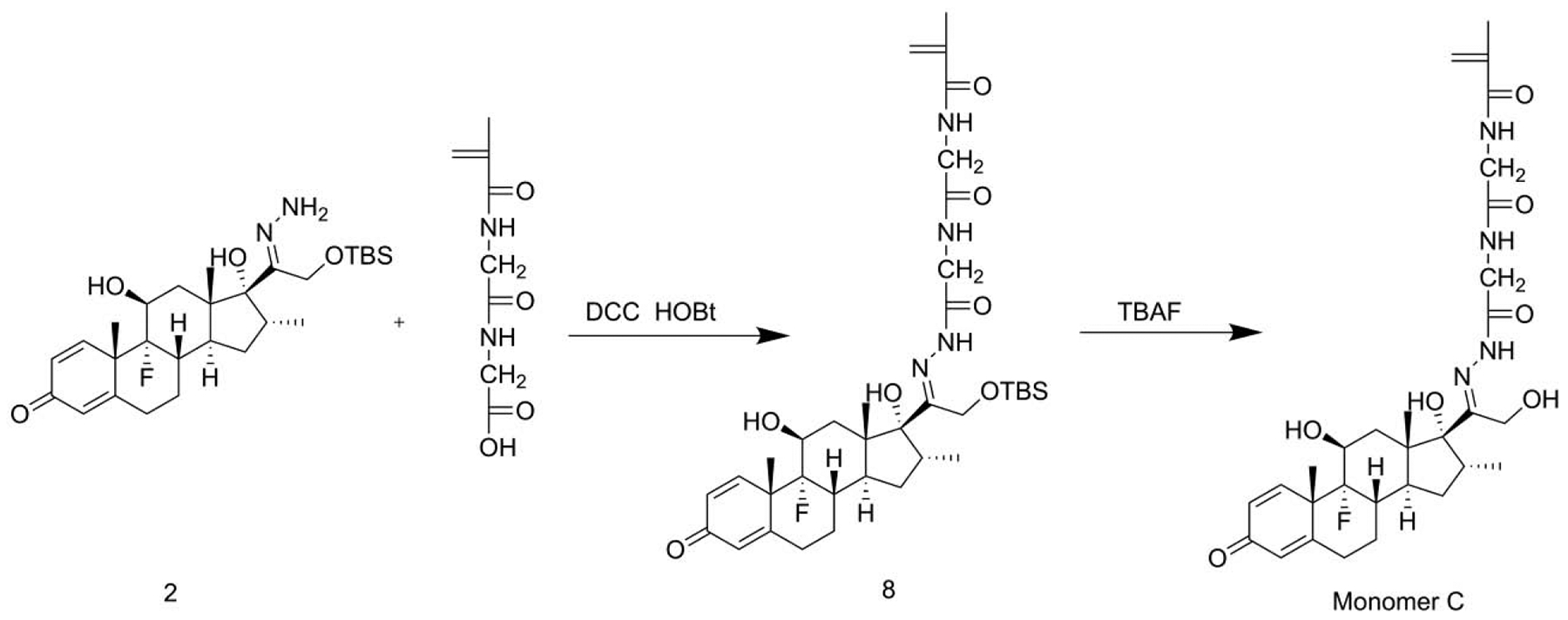
2.3.1.3. Synthesis of monomer C
Synthesis of compound 8.
DCC (154 mg, 0.75 mmol),Et3N (152 mg, 1.5 mmol), HOBt (153 mg, 1 mmol) and compound 2 (260 mg, 0.5 mmol) were dissolved in DMF (5 mL, anhydrous) and maintained at 0 °C with ice-water bath. After the addition of MA-Gly-Gly-OH (120 mg, 0.6 mmol), the solution was allowed to warm up to room temperature and stirred for 5 h, followed by the addition of ethyl acetate (100 mL). The solution was washed with saturated brine, separated and dried over Na2SO4. After the removal of solvents, the crude product was purified by flash column chromatography (dichloromethane : methanol = 10 : 1) to produce compound 8 (293 mg, yield: 83.1%).
1H NMR (500 MHz, CDCl3): δ (ppm) = 10.45 (s, 1H), 7.27 (d, J = 10.1Hz, 1H), 7.03 (s, 1H), 6.87 (s, 1H), 6.31 (d, J = 10.1Hz, 1H), 6.10 (s, 1H), 5.81 (s, 1H), 5.40 (s, 1H), 4.53 (d, J = 14.3 Hz, 1H), 4.37 (d, J = 14.3 Hz, 1H), 4.34 (m, 2H), 4.05 (m, 2H), 3.15 (m, 1H), 2.80 (m, 1H), 2.62 (td, J = 13.4 Hz, 5.4 Hz 1H), 2.38 (m, 3H), 2.30 (m, 1H), 1.99 (s, 3H), 1.60–1.90 (m, 3H), 1.55 (s, 3H), 1.53 (m, 1H), 1.39 (d, J = 13.9 Hz, 1H), 1.23 (m, 1H), 0.97 (s, 3H), 0.94 (d, J = 7.5 Hz, 3H), 0.91 (s, 9H), 0.13 (s, 3H), 0.12 (s, 3H).
13C NMR (125 MHz, CDCl3): δ (ppm) = 186.74, 169.94, 169.10, 168.60, 166.85, 152.84, 152.62, 139.00, 129.54, 124.85, 120.75, 100.45 (d, J = 175.2 Hz), 86.94, 71.86 (d, J = 38.4Hz), 60.36, 60.08, 48.33 (d, J = 22.5 Hz), 43.05, 42.77,40.88, 37.20, 34.33, 34.28, 34.18, 31.79, 31.04, 27.25, 25.62, 22.96 (d, J = 5.6 Hz), 20.99, 18.47, 17.91, 17.55, 14.53, 14.12, −5.58 (2C).
MS (ESI): m/z = 703.3 (M + H+), calculated: 702.4.
Synthesis of monomer C.
After dissolving compound 8 (280 mg, 0.4 mmol) in THF (7 mL), TBAF solution (1 mL, 1 M, THF) was added. After stirring the solution at room temperature for 1 h, ethyl acetate was added. The resulting solution was washed with saturated brine, separated and dried over Na2SO4. After removal of the solvents, the crude product was purified by flash column chromatography to obtain monomer C (210 mg, yield: 89.3%). After removal of the TBS, the hydroxyl group and nitrogen can form a rigid intramolecular hydrogen bond. Due to the different orientations of the atoms, the NMR spectra showed two groups of peaks that can’t be assigned.
1H NMR (500 MHz, DMSO-d6): δ (ppm) = 11.12 (s, 0.25H), 10.51 (s, 0.64H), 8.39 (t, J = 5.8Hz, 0.27H), 8.29 (t, J = 5.7 Hz, 0.28H), 8.21 (t, J = 5.7 Hz, 0.61H), 7.95 (t, J = 5.7 Hz, 0.67H), 7.33 (d, J = 10.2 Hz, 1H), 6.21 (d, J = 10.2 Hz, 1H), 5.99 (s, 1H), 5.86 (br, 0.27H), 5.75 (s, 1H), 5.76 (d, J = 18.1 Hz, 1H), 5.70 (t, J = 4.3 Hz, 1H), 5.39 (s, 0.27), 5.37 (s, 0.73H), 5.31 (m, 1H), 4.58 (d, J = 18.1 Hz, 1H), 4.26 (s, 2H), 4.10 (m, 2.88H), 3.78 (m, 2.64H), 3.35 (m, 3.07H), 3.16 (m, 3.41H), 3.06 (m, 1H), 2.61 (td, J = 13.4 Hz, 5.5Hz,1H), 2.32 (m, 2H), 2.12 (m, 2H), 1.88 (s, 3H), 1.77 (m, 1H), 1.55–1.63 (m, 4H), 1.49 (m, 1H), 1.48 (s, 3H), 1.33 (m, 4H), 1.06 (m, 1H), 0.91 (m, 3H), 0.85 (m, 3H).
13C NMR (125 MHz, DMSO-d6): δ (ppm) = 185.48, 170.05, 169.84, 169.54, 168.00, 167.81, 167.442, 153.74, 153.13, 139.66, 139.51, 129.07, 124.22, 120.16, 119.86, 101.51 (d, J = 174.1 Hz), 86.78, 86.62, 70.77 (d, J = 36.4 Hz), 57.73, 57.26, 54.87, 48.77, 48.17 (d, J = 23.0 Hz), 47.62, 47.55, 42.51, 42.37, 36.41, 34.67, 33.96(d, J = 18.7 Hz), 33.92, 33.51, 32.01, 31.85, 30.53, 27.45, 25.14, 24.19, 23.25, 23.16 (d, J = 5.6 Hz), 19.39, 18.71, 17.41, 17.31, 15.78, 15.51, 13.65.
MS (ESI): m/z = 611.3 (M + Na+), calculated 588.3.
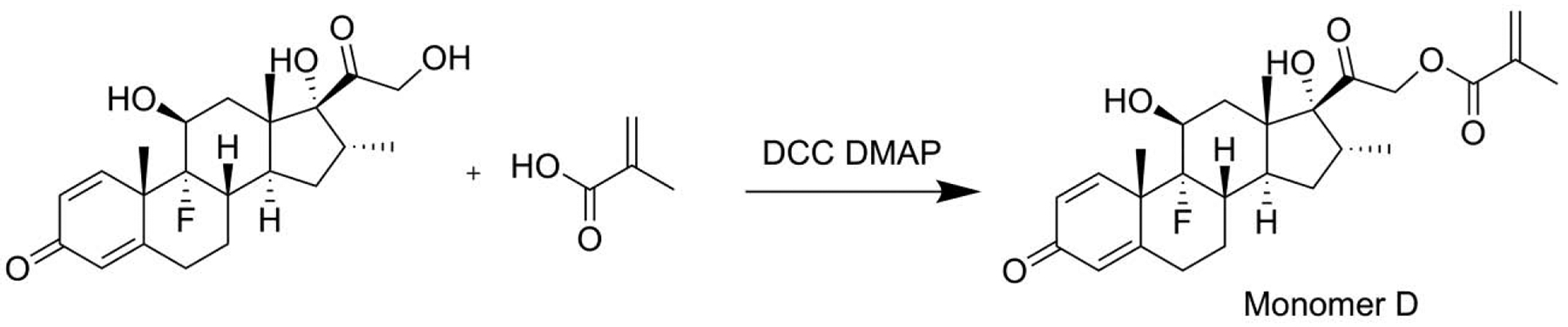
2.3.1.4. Synthesis of monomer D
Dexamethasone (392 mg, 1 mmol), 4-dimethylaminopyridine (DMAP, 24 mg, 0.2 mmol) and methacrylic acid (103 mg, 1.2 mmol) were dissolved in DMF (10 mL, anhydrous) and maintained at 0 °C with ice-water bath. After the addition of DCC (309 mg, 1.5 mmol), the solution was stirred for 3 h, followed by the addition of ethyl acetate (100 mL). The resulting solution was washed with saturated brine, separated and dried over Na2SO4. After removal of the solvents, the crude product was purified by flash column chromatography to produce monomer D (350mg, yield: 76.0%).
1H NMR (500 MHz, DMSO-d6): δ (ppm) = 7.23 (d, J = 10.1Hz, 1H), 6.34 (d, J = 10.1Hz, 1H), 6.21 (s, 1H), 6.11 (s, 1H), 5.66 (s, 1H), 4.99 (d, J = 17.5 Hz, 1H), 4.95 (d, J = 17.5Hz, 1H), 4.37 (d, J = 8.9 Hz, 1H), 3.12 (m, 1H), 2.53(td, J = 13.4 Hz, 5.4Hz 1H), 2.38 (m, 3H), 2.18 (m, 1H), 1.99 (s, 3H), 1.95 (m, 1H), 1.60–1.90 (m, 3H), 1.56 (s, 3H), 1.55 (m, 1H), 1.23 (m, 1H), 1.06 (s, 3H), 0.94 (d, J = 7.5Hz, 3H).
13C NMR (125 MHz, CDCl3): δ (ppm) = 204.73, 186.67, 167.25, 166.20, 152.24, 135.45, 129.75, 126.76, 125.06, 100.19 (d, J = 175.1 Hz), 91.14, 72.08 (d, J = 38.7 Hz), 68.6, 48.41, 48.26 (d, J = 22.7 Hz), 44.00, 36.55, 35.93, 34.17 (d, J = 19.3 Hz), 32.23, 31.00, 27.35, 22.90 (d, J = 5.6 Hz), 18.25, 16.54, 14.63.
MS (ESI): m/z = 483.2 (M + Na+), calculated: 460.2.
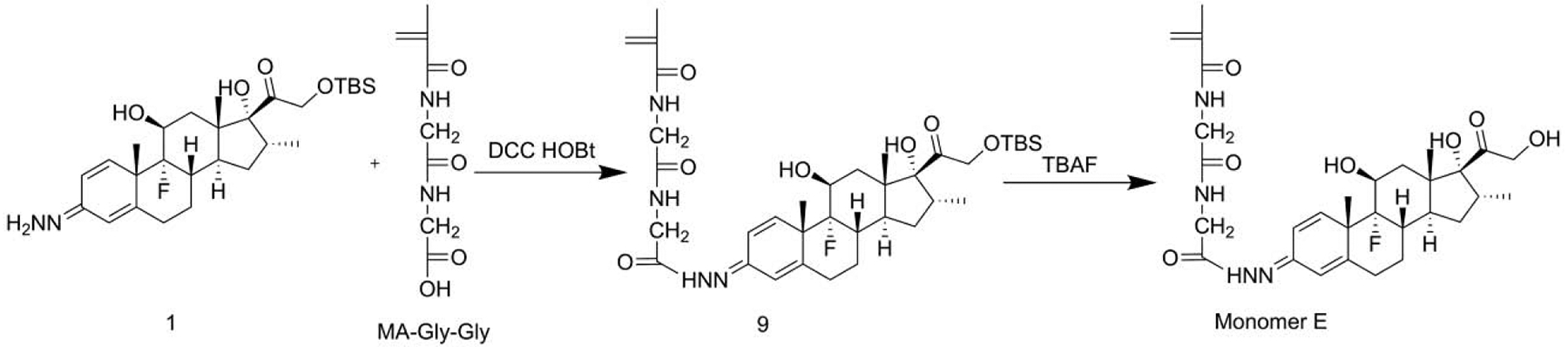
2.3.1.5. Synthesis of monomer E
Synthesis of compound 9.
Under constant stirring, DCC (82.4 mg, 0.4 mmol), MA-Gly-Gly-OH (60 mg, 0.3 mmol) and Et3N (60 mg, 0.6 mmol) were dissolved in DMF (3 mL, anhydrous) at 0 °C and maintained for 15 min before the addition of compound 1 (130 mg, 0.25 mmol). After warming up to room temperature, the solution was stirred overnight, followed by the addition of ethyl acetate (50 mL). The resulting solution was washed with saturated brine (50 mL × 4), separated and dried over Na2SO4. After the removal of the solvents, the crude product was purified by flash column chromatography (ethyl acetate : MeOH = 15:1) to produce compound 9 (106 mg). Yield 60.4%.
1H NMR (500 MHz, DMSO-d6): δ (ppm) = 10.93 (s, 0.23H), 10.85 (s, 0.30H), 10.50 (s, 0.12H), 10.49 (s, 0.23H), 8.21 (t, J = 4.2 Hz, 0.35H), 8.19 (t, J = 5.7 Hz, 0.65H), 8.10 (t, J = 4.9Hz, 0.37H), 7.89 (t, J = 5.4 Hz, 0.57H), 7.02 (d, J = 10.6 Hz, 0.24H), 6.93 (d, J = 10.2 Hz, 0.13H), 6.79 (s, 0.32H), 6.68 (s, 0.31H), 6.62 (t, J = 10.5 Hz, 0.25H), 6.46 (d, J = 11.1 Hz, 0.23H), 6.43 (d, J = 11.2 Hz, 0.36H), 6.28 (t, J = 10.2 Hz, 0.23H), 6.22 (d, J = 10.1 Hz, 0.32H), 6.00 (s, 0.15H), 5.96 (s, 0.25H), 5.74 (s, 1H), 5.38 (s, 1H), 5.16 (m, 1H), 4.95 (s, 1H), 4.77 (d, J = 19.0 Hz, 1H), 4.13 (m, 2.29H), 3.86 (m, 0.78H), 3.78 (dd, J = 14.7 Hz, 5.8Hz, 2H), 2.90 (m, 1H), 2.60 (m, 0.62H), 2.25 (m, 1.78H), 2.11 (m, 2.47H), 1.87 (s, 3.2H), 1.71 (m, 1.08H), 1.58 (m, 1.09H), 1.41 (m, 4.20H), 1.31 (m, 1.14H), 1.06 (m, 1.10H), 0.88 (s, 9H), 0.84 (s, 3H), 0.78 (d, J = 7.1 Hz, 3.26H).
13C NMR (125 MHz, DMSO-d6): δ (ppm) = 209.27, 170.60, 170.57, 170.50, 169.63, 169.43, 167.93, 167.82, 165.62, 165.53, 157.41, 157.02, 152.24, 151.46, 146.77, 146.31, 144.12, 143.95, 143.17, 139.69, 139.62, 126.60, 120.58, 120.00, 119.90, 117.07, 116.63, 111.61, 111.44, 101.47 (J = 173 Hz), 101.42 (J = 173 Hz), 100.08 (J = 173 Hz), 100.03 (J = 173 Hz), 90.63, 90.56, 79.37, 70.08 (J = 36.9 Hz), 68.19, 59.95, 47.43 (J = 22.8 Hz), 47.27 (J = 22.8 Hz), 43.68, 42.64, 42.50, 41.56, 40.70, 36.15, 36.08, 35.36, 34.10 (J = 21.4 Hz), 34.03 (J = 19.4 Hz), 32.10, 31.06, 31.01, 30.13, 29.97, 27.50, 27.44, 24.42 (J = 4.9 Hz), 24.06 (J = 4.5 Hz), 23.98(J = 4.7 Hz), 20.94, 18.76, 18.73, 18.36, 16.90, 15.42, 15.40, 14.28, −4.86, −5.01.
MS (ESI): m/z = 703.3 (M + H+), calculated: 702.4.
Synthesis of monomer E.
Under constant stirring, compound 9 (70 mg, 0.1 mmol) was dissolved in THF (5 mL), followed by the addition of TBAF (0.2 mL, 1 M). The resulting solution was maintained at room temperature for 15 min, followed by the addition of ethyl acetate (50 mL). The resulting solution was washed with saturated brine (50 mL × 4), separated and dried over Na2SO4. After removing the solvents, the crude product was purified by flash column chromatography (MeOH : ethyl acetate = 1 : 10) to produce monomer E (48 mg), Yield: 81.6%.
1H NMR (500 MHz, DMSO-d6): δ (ppm) = 10.92 (s, 0.21H), 10.84 (s, 0.32H), 10.50 (s, 0.13H), 10.50 (s, 0.23H), 8.21 (t, J = 4.2 Hz, 0.31H), 8.18 (t, J = 5.7 Hz, 0.67H), 8.10 (t, J = 4.9Hz, 0.40H), 7.88 (t, J = 5.4 Hz, 0.57H), 7.02 (d, J = 10.6 Hz, 0.22H), 6.93 (d, J = 10.2 Hz, 0.12H), 6.78 (s, 0.34H), 6.68 (s, 0.38H), 6.62 (d, J = 10.4 Hz, 0.23H), 6.47 (d, J = 11.1 Hz, 0.25H), 6.43 (d, J = 11.2 Hz, 0.36H), 6.27 (t, J = 10.2 Hz, 0.25H), 6.22 (d, J = 10.1 Hz, 0.35H), 6.00 (s, 0.12H), 5.96 (s, 0.22H), 5.74 (s, 1H), 5.37 (s, 1H), 5.14 (m, 1H), 4.93 (s, 1H), 4.67 (br, 1H), 4.49 (d, J = 19.2 Hz, 1H), 4.14 (m, 2.05H), 4.07 (d, J = 19.2 Hz, 1H), 3.87 (m, 0.90H), 3.79 (dd, J = 14.9 Hz, 5.7Hz, 2H), 2.93 (m, 1H), 2.60 (m, 0.60H), 2.50 (m, 1.12H), 2.25 (m, 1.80H), 2.13 (m, 2.25H), 1.87 (s, 3.2H), 1.71 (m, 1.04H), 1.59 (m, 1.11H), 1.42 (m, 4.37H), 1.31 (m, 1.79H), 1.06 (m, 1.15H), 0.88 (s, 9H), 0.84 (s, 3.61H), 0.787 (d, J = 7.0 Hz, 3.34H).
13C NMR (125 MHz, DMSO-d6): δ (ppm) = 211.47, 170.63, 170.60, 169.71, 169.52, 168.02, 167.91, 165.74, 165.66, 162.62, 157.56, 157.15, 152.37, 151.56, 146.89, 146.44, 144.26, 144.06, 143.25, 139.77, 139.72, 139.66, 139.12, 117.11, 116.67, 111.69, 111.49, 100.93 (J = 172.9 Hz), 100.88 (J = 172.9 Hz), 100.75 (J = 174,7 Hz), 100.72 (J = 173.9 Hz), 90.48, 90.42, 70.18 (J = 35.9 Hz), 66.55, 60.02, 53.07, 47,69 47.47(J = 22.8 Hz), 47.33 (J = 22.8 Hz), 43.70, 42.71, 42.56, 41.64, 40.76, 36.16, 36.09, 35.16, 34.12 (J = 20.5 Hz), 34.08 (J = 20.5 Hz), 32.24, 31.08 (br), 30.18, 30.03, 28.18, 27.55, 24.45 (J = 5.0 Hz), 24.09 (J = 5.9 Hz), 21.34, 20.99, 20.14, 18.79, 18.77, 16.91, 15.54, 15.52, 14.32, 14.07.
MS (ESI): m/z = 589.1 (M + H+), calculated 588.3.
2.3.2. Synthesis of HPMA copolymer-based Dex prodrugs
Five HPMA copolymer-based Dex prodrugs were synthesized by RAFT (reversible addition-fragmentation chain transfer) copolymerization of HPMA and the five Dex-containing monomers (Scheme 1), respectively. HPMA (2.79 mmol), a Dex-containing monomer (0.20 mmol), 2,2″-azobis(isobutyronitrile) (AIBN, 0.026 mmol) and S,S′-bis(α,α′-dimethyl-α″-acetic acid) trithiocarbonate (RAFT agent, 0.15 mmol) were dissolved in methanol (3.8 mL, anhydrous) in an ampule. After purging the solution with Argon for 2 min, the ampule was flame-sealed and immersed in an oil bath (50 °C). After polymerization for 48 h, the resulting solution was purified by size exclusion chromatography (LH-20 column). After removing the solvent, the resulting polymer was dissolved in water and lyophilized to produce the polymeric Dex prodrugs as white powder.
2.4. Characterization of the P-Dex copolymers
The molecular weights (Mn and Mw) and dispersity (Ð) of polymeric prodrugs were determined using a AKTÄ pure FPLC system, equips with a Superdex 200 Increase 10/300 GL column, a DAWN 8+ multiangle light scattering (MALS) system and an Optilab T-rEX refractive index detector. To quantify Dex content in the polymeric prodrugs, the prodrugs (1 mg/ mL) were hydrolyzed in HCl (0.1 N) overnight. Before analyzing on an Agilent 1100 HPLC system with a Hypersil™ ODS C18 column, the solution was neutralized. Conditions for the HPLC analysis are listed as the following. Mobile phase: water : acetonitrile = 60 : 40; UV detection wavelength: 240 nm; injection volume: 10 μL; flow rate: 1 mL/min. All HPLC analyses were performed in triplicate. The mean value and standard deviation were calculated using Microsoft® Office Excel.
2.5. In vitro release of Dex from the polymeric prodrugs
In vitro releasing profiles of the copolymers (P-Dex-A, P-Dex-B, P-Dex-C, P-Dex-D and PDex-E) were assessed in different buffered solutions (i.e., pH 5.0 acetate buffer, pH 6.0 acetate buffer, pH 7.0 and pH 7.4 phosphate buffer). The polymeric prodrugs were dissolved into different buffers at the concentration of 4 mg/mL (polymer/solution) with Pluronic F127 (1 w/v %) added to ensure the “sink” condition [25]. The solutions were then agitated in a shaking incubator (60 r/min) at 37 °C. At pre-designated time points, the releasing solutions were withdrawn, neutralized and then extracted with METB 9 times. The 1/3 volume of combined METB solution was then withdrawn and evaporated using a vacuum evaporator. The residues were reconstituted into 100 uL H2O/MeOH solution (H2O/MeOH=1:9) resulting in samples for HPLC analyses. The HPLC analyses were performed based on the Dex calibration under the same HPLC conditions used in the Dex loading analysis. The METB extraction recovery efficiency was analyzed using the same method as described above with the Dex concentration ranging 5–500 μg/mL.
2.6. Therapeutic efficacy of polymeric Dex prodrugs on adjuvant-induced arthritis rats
The adjuvant-induced arthritis (AA) rat model was established using Lewis rats (male, body weight 175–200 g, Charles River Laboratories) as described previously [16]. Seven groups were randomly assigned as the following: P-Dex-A, P-Dex-C, P-Dex-D and P-Dex-E treatment (7 rats per group, dose equivalent of Dex = 10 mg/kg, the prodrugs were given via tail vein injection14 days after arthritis induction), dexamethasone sodium phosphate treatment (7/group, equivalent of Dex = 10 mg/kg, divided into 4 equal aliquots which were given i.v. on days 14, 15, 16, 17), saline control (4/group) and healthy group (4/group). Due to its low Dex content, P-Dex-B was not included in this in vivo study. Joint edema of the rats was monitored daily from day 11 post arthritis induction. At euthanasia on day 44, the animals’ hind paws were isolated, fixed in buffered formalin and then paraffin embedded. Joints tissue sections (8 μm) were collected and processed for H&E and safranin O staining. The blinded histological evaluation was performed by a pathologist (Dr. Subodh M. Lele). All animal studies were performed according to a protocol approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Nebraska Medical Center.
2.7. Observational assessment of the arthritic joints
During the arthritis development, articular index (AI) scores were recorded consistently by the same observers. The AI scoring system is based on a 0–4 numeric system as followed: 0 = no signs of swelling or erythema; 1 = slight swelling and/or erythema; 2 = low-to-moderate edema and signs involving the tarsals; 3 = pronounced edema with limited use of the joint and signs extending to the metatarsals; 4 = excessive edema with joint rigidity and severe signs involving the entire hind paw. The sum of the two hind limb scores for each animal was recorded. Ankle diameter (medial to lateral) was measured using a digital caliper as verification of inflammation-associated edema/hyperplasia [16].
2.8. Micro-CT analysis of articular bone quality
The bone quality of ankle joint was evaluated using a Bruker Skyscan 1172 micro-CT system as described previously [26]. The following scanning parameters were kept consistent during the analysis of the entire set of ankle joints: 70 kV (voltage), 142 μA (current), 3650 ms (exposure time), 13.1 μm (resolution), 0.5 mm (aluminum filter), 0.4° (rotation step), 6 (frame averaging), 10 (random movement), 360° (rotation scanning). The data were processed with the software (i.e., NRecon and CTvox) supplied by the Skyscan. The entire calcaneus and the selected region of interest (ROI) of the trabecular bone within the calcaneus were analyzed to assess the impacts of different treatments. Using DataViewer software, the calcaneus bone was oriented and aligned along the sagittal plane, with 3D-registeration. Specifically, the ROI was defined as the region starting at the 0.98 mm away from the epiphyseal plate and continued for 1.00 mm. The diameter of the cylindrical ROI was set at 1.00 mm. The morphometric parameters including bone volume fraction (BV/TV), bone surface density (BS/TV), trabecular number (Tb.N), trabecular thickness (Tb.Th), trabecular separation (Tb.Sp) and bone mineral density (BMD) and were calculated.
2.9. Histological analysis of the arthritic joints
The limbs, after micro-CT analysis, were decalcified with 5% formic acid. They were paraffin-embedded, sectioned (8 μm) approximately 200 μm apart. After hematoxylin and eosin (H&E) and safranin O staining, the sections were reviewed and graded by a professional pathologist (Dr. Subodh M. Lele) in a blinded fashion. The histopathologic features were graded according to the following standard: synovial cell lining hyperplasia (0 to 2); pannus formation (0 to 3); mononuclear cell infiltration (0 to 3); polymorphonuclear leukocytes infiltration in periarticular soft tissue (0 to 3); cellular infiltration and bone erosion at the distal tibia (0 to 3); and cellular infiltration of cartilage (0 to 2). The score for each histological feature was then added up for individual animals.
2.10. The impact of prodrug treatments on the systemic bone quality
Lumbar vertebral bodies were isolated at euthanasia and fixed in formalin. The quality of the bone was evaluated using the same Bruker Skyscan 1172 micro-CT system. Micro-CT scanning parameters were set the same as the ankle scanning parameters. The 5th lumbar vertebral body was aligned along the sagittal plane using DataViewer software, with 3D-registeration. For quantitative analysis, the ROI of the 5th lumbar vertebra was selected beginning from secondary spongiosa above the bottom endplate for 0.52 mm and the trabecular bone within the vertebra were used for analysis. The morphometric parameters, such as bone volume fraction (BV/TV), bone surface density (BS/TV), trabecular separation (Tb.Sp), trabecular number (Tb.N), trabecular thickness (Tb.Th) and bone mineral density (BMD) were calculated.
2.11. Statistical methods
Two-way analysis of variance (ANOVA), followed by Tukey’s post hoc test to account for multiple comparisons, was used for data analysis using GraphPad Prism Software. P-values ≤ 0.05 were considered as statistically significant.
3. RESULTS
3.1. Characterization of HPMA copolymer-based Dex prodrugs
The characterization of Dex-containing HPMA copolymer prodrugs are presented in Table 1. The Mw of the polymeric prodrugs were ~35 kDa, the Ð values were between 1.1 to 1.5. The Dex content of the prodrugs were determined to be ~300 μmol/g with the exception of P-Dex-B which was about 100 μmol/g. Due to its low Dex content, P-Dex-B was excluded from the in vivo therapeutic evaluation [27].
Table 1.
The characterization of Dex-containing polymeric prodrugs.
| Dex-containing polymeric prodrugs | Mw (×103 g/mol) | Ð | Dex Content (μmol/g) |
|---|---|---|---|
| P-Dex-A | 35.8 | 1.32 | 329.76 ± 8.80 |
| P-Dex-B | 36.7 | 1.36 | 113.61 ± 13.19 |
| P-Dex-C | 35.4 | 1.13 | 418.27 ± 12.63 |
| P-Dex-D | 31.9 | 1.48 | 303.66 ± 9.22 |
| P-Dex-E | 39.1 | 1.23 | 258.11 ± 6.40 |
Mw: weight-average molecular weight; Ð: dispersity.
3.2. In vitro releasing profiles of the P-Dex prodrugs in various buffers
The in vitro Dex release was evaluated by measuring the Dex concentration in the solution after incubating P-Dex-A, P-Dex-B, P-Dex-C, P-Dex-D and P-Dex-E in buffered solutions with pH = 5.0, 6.0, 7.0 and 7.4, human serum and rat serum at 37°C. The Dex in the releasing buffer was extracted as illustrated in the methods section. The recovery rate was determined to be 97.18 ± 2%. In Figure 1, the results indicated that the polymeric prodrugs showed different releasing rates in each of the tested buffers. Under acidic condition (e.g. pH = 5.0 and 6.0), the releasing rate trend (from fast to slow) was RP-Dex-B > RP-Dex-C > RP-Dex-E > RP-Dex-A > RP-Dex-D. Under physiological pH (pH 7.4) the releasing rate trend was RP-Dex-B > RP-Dex-D > RP-Dex-C > RP-Dex-E > RP-Dex-A. All P-Dex prodrugs demonstrated good stability in rat and human serum except P-Dex-B, with ~50% activated in rat serum within 500 h,
Figure 1.
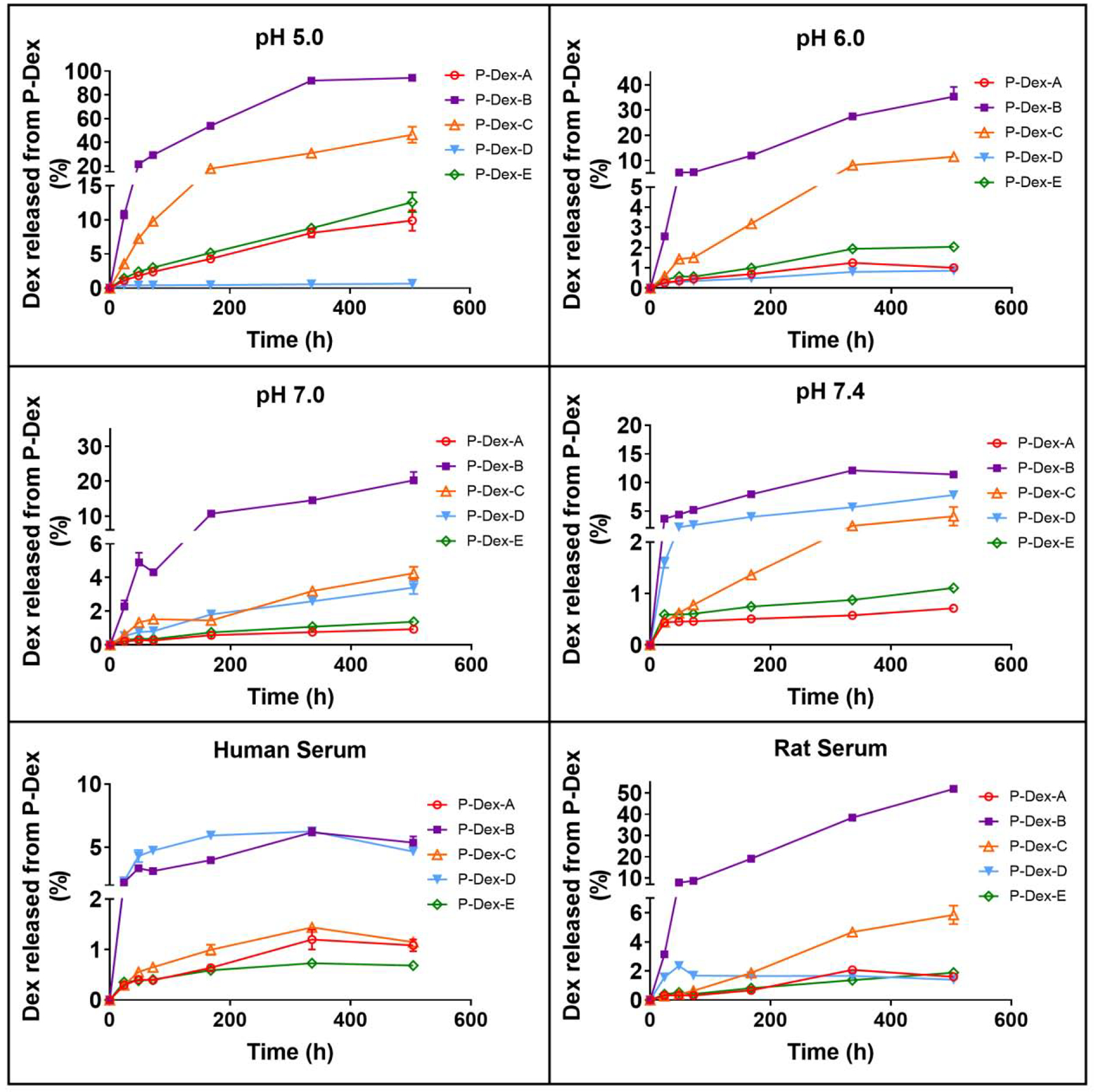
In vitro Dex release kinetics of P-Dex-A, P-Dex-B, P-Dex-C, P-Dex-D and P-Dex-E in the different releasing buffers. Each sample was measured three times. The mean values and standard deviation were calculated using GraphPad Prism 7.
3.3. Therapeutic evaluation of P-Dex prodrugs treatment of AA rats
After induction, the rat ankle joints began to swell on day 8 and progressed to a plateau by day 13. On day 14 post-induction, the treatments were initiated. The disease progression was monitored to compare the therapeutic effect in each group.
3.3.1. The P-Dex prodrugs demonstrated different patterns of sustained amelioration of joint inflammation in AA rats
The therapeutic outcomes associated with the different treatments were characterized by assessment of the ankle diameters and AI scores (Figure 2). The Dex-treated group showed an immediate reduction of the swelling, which flared on day 19 upon cessation of the treatment. A single injection of the polymeric prodrugs (dose equivalent to that of Dex treatment) resulted in different levels of reduction in ankle swelling and AI score from day 15 to day 44. As shown in Figure 2A, the average ankle diameter of P-Dex-C treatment rats showed a sharp reduction the day after the treatment, lasted about 8 days and then flared. Comparatively, the average ankle diameters of P-Dex-E and P-Dex-A treated rats experienced a gradual reduction until the end of the study. Results indicate that the differences in therapeutic efficacy correlated with the in vitro releasing profiles of the prodrugs.
Figure 2.
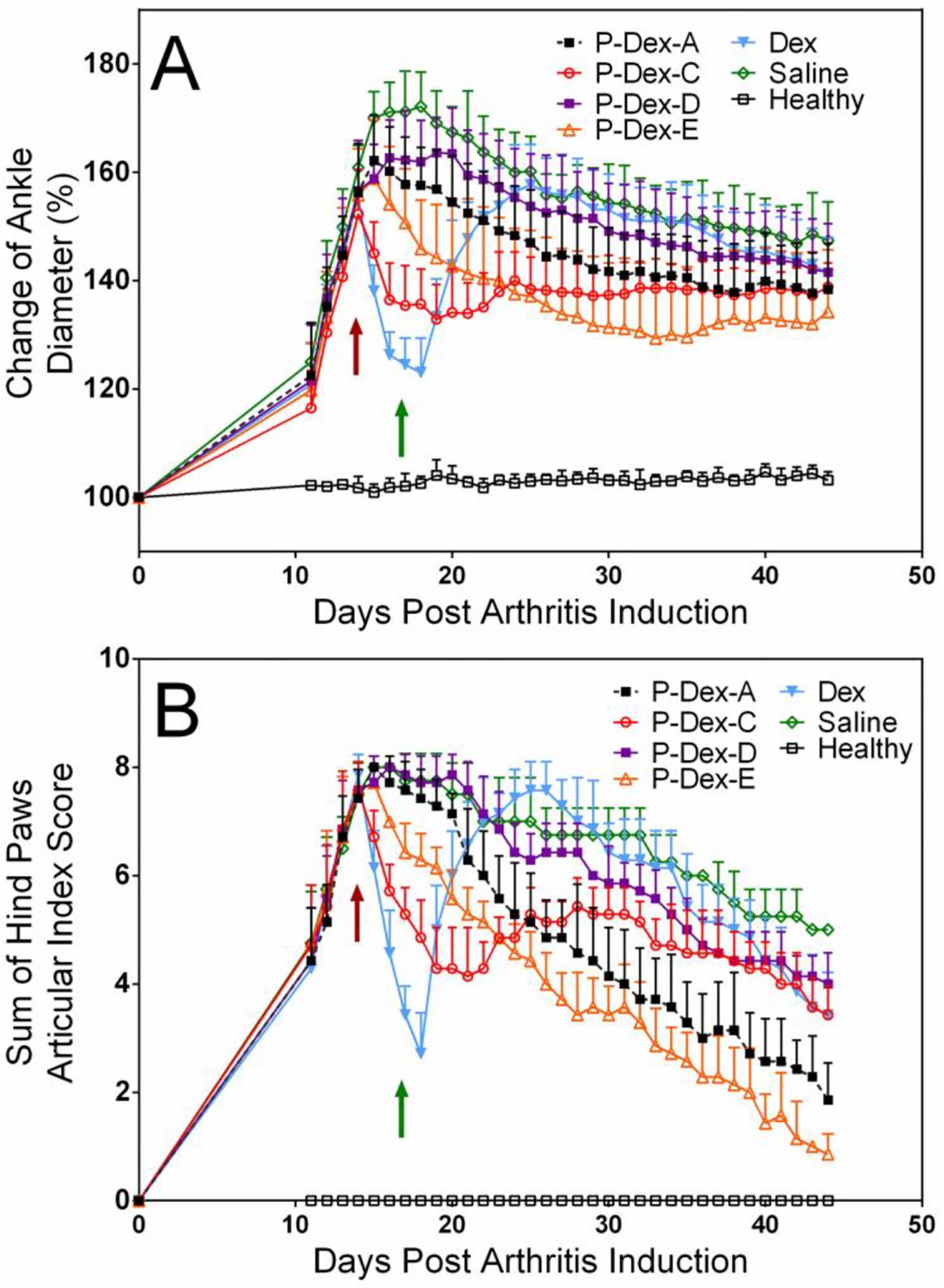
HPMA copolymer-based Dex prodrugs with different releasing mechanisms demonstrated different therapeutic effects on amelioration of joint inflammation in an adjuvant-induced arthritis (AA) rat model. (A) The changes in the left ankle joint diameter of AA rats treated with different Dex-containing polymeric prodrugs during the entire experiment; (B) The changes in the articular index scores of AA rats treated with different Dex-containing polymeric prodrugs during the entire experiment. The red arrow indicated the day when rats received the single polymeric prodrug injection or the first injection of the four daily free Dex treatments. The green arrow indicated the day when daily Dex-treated rats received their last injection. The sustained amelioration of arthritic ankle swelling by single injection of P-Dex-A and P-Dex-E lasted for about 1 month from day 15 to day 44.
3.3.2. The P-Dex prodrugs demonstrated different levels of articular bone protection in AA rats
As shown in Figure 3, the ankle joints of the rats in the saline group were associated with the most severe joint bone damage. P-Dex-A, P-Dex-C, P-Dex-D, and P-Dex-E treated rats demonstrated various levels of preservation of ankle bone when compared to the rats in saline group. One month following the single dose prodrug administration, only minor bone erosion was found in P-Dex-A, P-Dex-C and P-Dex-E treated groups. According to Figure 3C, P-Dex-E treated rats demonstrated the best overall ankle joint bone preservation. The quantitative micro-CT analysis of the hind paw calcaneus trabecular bone showed that P-Dex-A, P-Dex-C and P-Dex-E treatment preserved joint bone at different levels (different P value compared with Saline control group) (Figure 3A), as evident in the morphometric parameters, such as BV/TV (bone volume fraction), Tb.N (trabecular number), Tb.Th (trabecular thickness), and BMD (bone mineral density), with their values similar to those observed from healthy controls, and significantly better than those of the free Dex-treated rats. When the entire calcaneus bone was analyzed (Figure 3B), the P-Dex-D, Dex and saline groups were found with significantly higher calcaneus tissue volume (TV), calcaneus total porosity (Po(tot)), calcaneus bone surface (BS), and significantly lower calcaneus bone volume fraction (BV/TV), when compared with the healthy and P-Dex-A, P-Dex-C and P-Dex-E treated groups.
Figure 3.
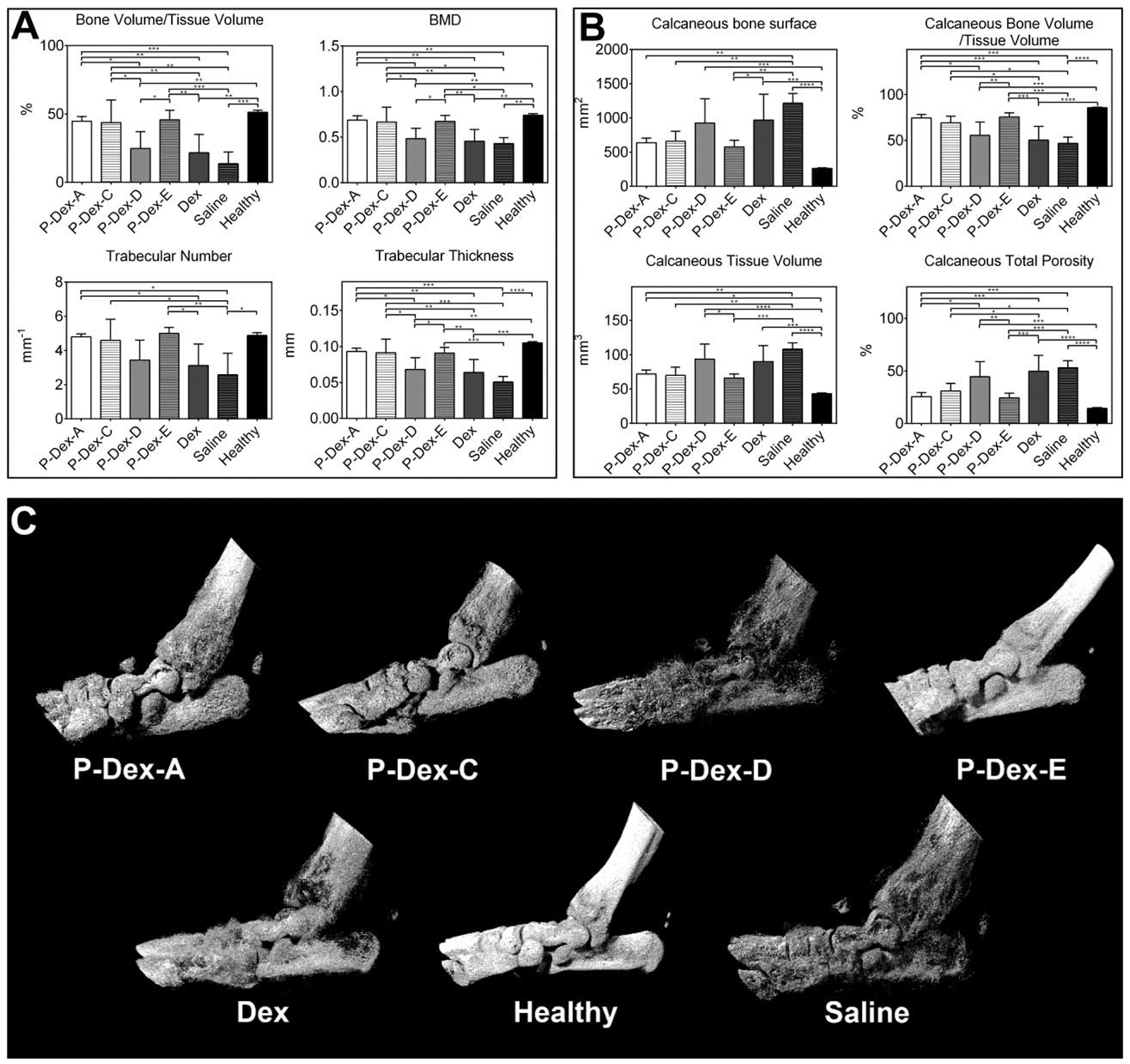
Micro-CT analyses of the hind paw of the rats from different treatment groups. (A) Bone morphometric parameters of the trabecular bone ROI within calcaneus bone. (B) Bone morphometric parameters of the entire calcaneus bone. (C) Representative images of 3-D rendering of the arthritic ankle joints. (*, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001).
3.3.3. Histological analysis of different Dex-containing prodrug treated AA rats
Analysis of the bone histological morphology confirmed that the P-Dex-A and P-Dex-E treatment preserved bone morphology when compared to the healthy controls. The obvious bone and cartilage destruction of the distal tibia, periosteal expansion and inflammatory cell infiltration were found in the P-Dex-C, P-Dex-D, Dex, and saline treated groups. The histology score evaluated by the pathologist for each animal was summed up and shown in Figure 4. Significant difference was found between healthy vs P-Dex-C, P-Dex-D, saline and Dex; saline vs P-Dex-A, P-Dex-D and P-Dex-E groups.
Figure 4.
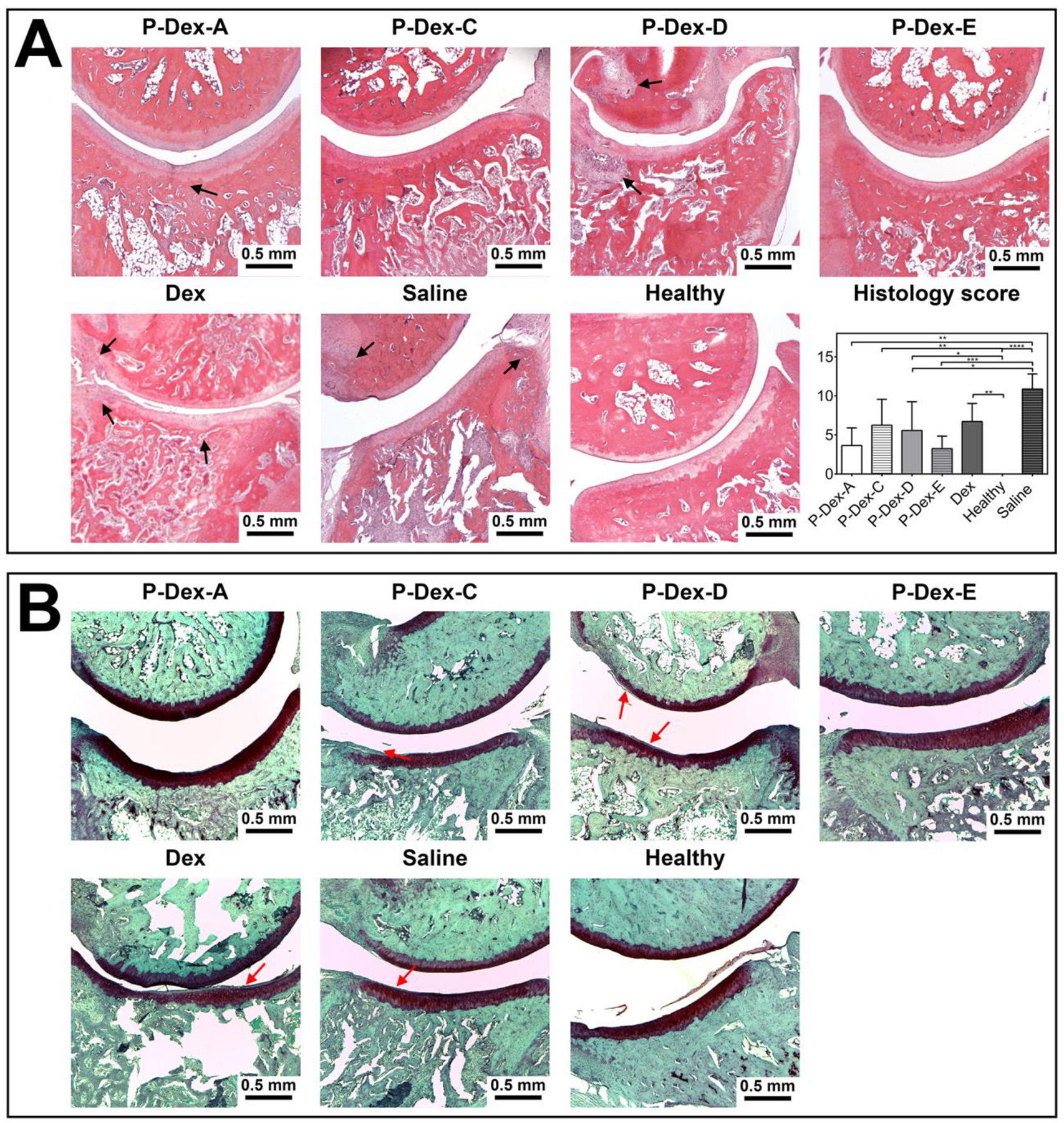
Histological evaluation of ankle joints from different treatment groups. (A) Representative images of the H&E-stained joint sections (400×) and semiquantitative comparisons of histology scores of all treatment groups (*, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001) were presented. (B) Representative images of the Safranin O-stained joint sections (400×). Scale bar = 0.5 mm. Black arrow indicated bone destruction. Red arrow indicated cellular infiltration in cartilage.
3.3.4. The impact of the polymeric Dex prodrug treatments on systemic bone quality of the AA rats.
To evaluate the impacts of the different prodrug treatments on systemic bone quality, the 5th lumbar vertebrae were collected and scanned using micro-CT. The micro-architectures of the trabecular bone and cortical bone are shown in the Figure 5. All the treatment groups showed significantly lower lumber spine trabecular bone volume fraction compared to the healthy group, indicating that the skeleton was negatively affected by the systemic inflammation associated with the arthritis [28, 29]. The results also indicate that the healthy group had significant thicker cortical bone compared to the P-Dex-C, P-Dex-D, Dex and saline treated groups, but not significantly thicker than P-Dex-A or P-Dex-E treatment groups. The cortical bone volume fraction from all the groups were similar (no statistic significant difference found).
Figure 5.
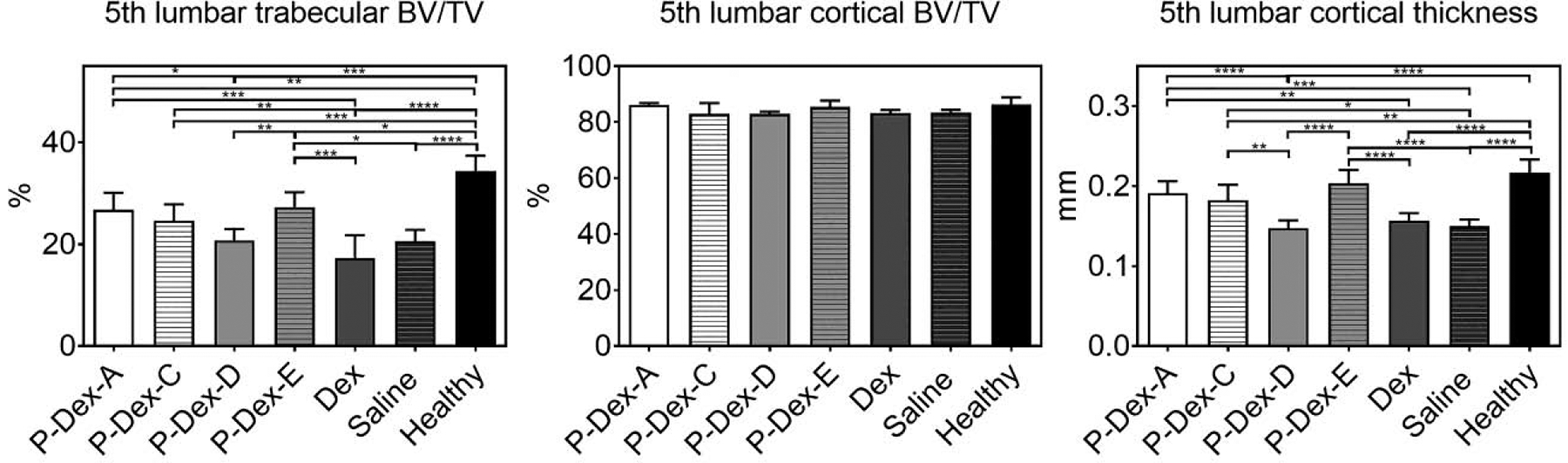
The quantitative analysis of the 5th lumbar vertebrate using micro-CT. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001.
4. DISCUSSION
During the past 2 decades, our research team has focused on the development of multiple macromolecular GC prodrugs for the treatment of inflammatory/autoimmune diseases (e.g., rheumatoid arthritis, IBD, lupus nephritis, etc.) [14–16]. We found that when administered systematically in relevant animal models, the prodrugs passively target to the inflammatory lesions via a mechanism that we have termed as “Extravasation through Leaky Vasculature and Inflammatory cell-mediated Sequestration (ELVIS)”, which provides sustained anti-inflammatory and disease modifying effects with greatly reduced toxicities. Modifications to the structural parameters of the macromolecular prodrugs (e.g., molecular weight, drug contents) have been found to affect their pharmacokinetics and biodistribution in murine models of inflammatory arthritis and orthopaedic implant loosening [27]. Furthermore, we hypothesized that the fine-tuning of the macromolecular prodrugs’ activation mechanism would directly affect their therapeutic efficacy and safety by regulating the free Dex levels in different organs/tissues. To validate this hypothesis, we initiated this comprehensive in vitro and in vivo comparison study of five macromolecular prodrugs of Dex.
In the initial design of P-Dex, hydrazone, an acid-cleavable bond, was employed as the activation mechanism [16]. The rationale for this design was intended to utilize the unique pathophysiological features of the tissue acidosis associated with inflammatory arthritis and the reduced pH of the lysosomes where the prodrugs were sequestered after cellular internalization, which would allow the prodrug activation to be more specific to the joint tissue pathology [30, 31]. Upregulated enzyme levels (e.g., cathepsin K) associated with inflammatory arthritis [32] were also considered as prodrug activation triggers. Given the structural variations of the enzymes between human and laboratory animal models [33], however, the utility of a hydrazone bond as the prodrug activation trigger was deemed to be more appropriate for clinical translation.
Given the chemical structure of Dex, the acid-cleavable hydrazone bond may be formed at either C3 (monomer E) or C20 (monomer C) positions. Due to its poor solubility in common chromatography organic solvents, minute amount and similar polarity, the minor product monomer C was not completely separated at the time of our initial monomer E synthesis [16]. As the major product, the monomer E has a hydrazone bond with a conjugation system involving 9 atoms and 4 double bonds, while monomer C only has a conjugation involving 5 atoms and 2 double bonds. Compared to monomer C, the larger electron delocalization in monomer E stabilized the hydrazone structure, resulting in it being the major product. To obtain pure monomers C and E, we developed a new synthetic strategy. Dex was first reacted with TBSCl to generate a TBS-protected Dex which is much more soluble in common organic solvents to facilitate purification. Both C3 and C20 carbonyl groups in TBS-protected Dex can react with hydrazine to form isomers that can be easily separated by column chromatography. The two purified products can then react with MA-Gly-Gly-OH to afford TBS-protected monomers E and C. After removal of TBS, pure monomers E and C were obtained with high yield. Due to the enhanced stability of the monomer E and the steric hindrance of TBS to hydrazine’s nucleophilic attack on C20, hydrazone formation at C3 position was the predominant product (monomer E).
P-Dex-C and P-Dex-E showed different impacts on amelioration of joint inflammation (Figure 2), though animals in both groups outperformed those in the Saline group. P-Dex-C treatment showed a faster joint swelling amelioration compared to the Saline group at day 17 post induction (P < 0.0001). But it was not as sustained as P-Dex-E, which may be attributed to the faster Dex releasing kinetics of P-Dex-C. This faster activation of P-Dex-C may have led to its reduced protection of the ankle bone structure when compared to P-Dex-E, as evident in the micro-CT data (Figure 3).
To allow further manipulation of the P-Dex activation rate, we decided to modify the hydrazone bond-associated electron conjugation system. Mechanistically, the delocalization of the electron cloud is a major factor affecting the chemical stability of organic molecules. By introducing double bonds, triple bonds, aromatic structures, the local electron conjugation may vary significantly, leading to a wide spectrum of prodrug activation rates.
As shown in Scheme 1, the addition of a benzene ring and an amide to the monomer E linker chemistry results in monomer A with a stronger conjugation system, which encompasses 18 atoms, 5 double bonds and 1 aromatic ring. Because the lone electron pair on nitrogen atoms can form p-π conjugation with double bonds and the aromatic ring, the resulting large delocalized electron system further improved the stability of the hydrazone. As shown in Figure 1, under acidic conditions, P-Dex-A was activated at a slower rate than P-Dex-E. According to the in vivo assessment of the therapeutic efficacy (Figure 2), the P-Dex-A group showed delayed resolution of ankle joint inflammation, suggesting the slower Dex releasing kinetics of P-Dex-A did not provide an immediately effective Dex concentration in the arthritic joint when compared to free Dex and P-Dex-C. Compared to P-Dex-E, the slower activation rate of P-Dex-A further moderated the resolution of joint inflammation (Figure 2) and the preservation of ankle joint integrity (Figure 3C).
To prepare a monomer with a faster activation rate, we decided to interrupt the conjugation system of monomer E. As shown in Scheme 1, a methylene group was inserted between the carbonyl group and the hydrazone bond, resulting in monomer B with a smaller conjugation system, including 3 double bonds and 7 atoms. Its relative instability was reflected in the reduced Dex loading in P-Dex-B and its much faster in vitro activation rate under acidic conditions as compared to P-Dex-A and P-Dex-E (Figure 1). It is important to recognize, however, that the electron delocalization is not the only factor that affects the hydrazone stability. If that is the case, then monomer B (with a conjugation system encompassing 7 atoms and 3 double bonds) should have a slower activation rate than monomer C (with a conjugation system encompassing 5 atoms and two double bonds). The in vitro releasing study, on the contrary, showed that P-Dex-B activated faster than P-Dex-C under acidic pH. We believe this observation may be attributed to the formation of a 6-membered ring via the hydrogen bond between the C21 hydroxyl oxygen and the hydrogen connected with the nitrogen of hydrazone bond (Scheme 1). It is well-recognized that when 6 atoms form a ring structure, they can achieve a chair-like conformation in order to minimize the electron cloud impulsion and release the strain tension, resulting in the arrangement of the atoms and bonds at a much lower energy level. Clearly, the formation of the 6-membered ring in monomer C via hydrogen bond improved its stability and reduced the P-Dex-C’s activation rate to be slower than that of P-Dex-B. Furthermore, we speculate the instability of monomer B may have resulted in its methanol-mediated partial cleavage and Dex release during copolymerization, which has led to the significantly lower Dex content in P-Dex-B after RAFT polymerization (Table 1). Since the Dex content may directly affect the P-Dex’ PK/BD profile [27], P-Dex-B was therefore excluded from the in vivo evaluation.
Different from the other four monomers, the linker in monomer D was an ester bond, which showed the slowest releasing rate under the acidic conditions (Figure 1). Initially, we did not select the C21 -OH for Dex conjugation to HPMA copolymer, fearing that the well-known high level of serum esterase may lead to the prodrug’s premature activation before distributing to the arthritic joints. According to the work by Timofeevski et al. [34], however, we recognized that the C21 ester may have reasonable stability due to the potential formation of intramolecular micelles, which may have sterically prohibited the access of C21 ester to the serum esterase, resulting in its unusual stability in a macromolecular prodrug. P-Dex-D was synthesized and evaluated in vitro and in vivo. As shown in Figure 1, P-Dex-D was found to be relatively stable in rat and human serum. In vivo data suggest that P-Dex-D treatment only presented marginal improvement in reducing ankle joint edema when compared to the Saline control group (Figure 2), with no apparent impact on joint protection as evident in micro-CT analyses (Figure 3). This suggest that the insufficient activation of P-Dex-D in vivo may have limited its therapeutic efficacy.
In addition to their impact on the local arthritic joint protection, the prodrug activation rates may also exert different impact on systemic skeletal quality. As shown in Figure 5, P-Dex-A and P-Dex-E effectively preserved the bone quality of the 5th lumber vertebral body. The interpretation of the extra-articular skeletal quality data from animals treated with different polymeric Dex prodrugs can be complex, as it may reflect the combined detrimental impact of the activated Dex on bone and the dampening of the systemic inflammation associated with arthritis during the polymeric Dex prodrug treatments. Clearly, to fully assess the osteotoxicity and other aspects of the systemic toxicities of these prodrugs, more thorough investigations are still needed.
Overall, the in vivo assessment of these polymeric Dex prodrugs in the AA rat model clearly established a direct correlation between their therapeutic efficacy and their activation kinetics. While faster prodrug activation provided a powerful immediate therapeutic response, the prodrugs with slower releasing kinetics were found to provide more sustained joint protection. Therefore, for further improvement of these prodrugs’ efficacy and safety of the prodrugs, one may argue for co-formulation of both the fast and slow activated prodrugs.
5. CONCLUSIONS
In this study, we have successfully designed and synthesized five N-(2-hydroxypropyl) methacrylamide (HPMA) copolymer-based dexamethasone (Dex) prodrugs (i.e., P-Dex-A, P-Dex-B, P-Dex-C, P-Dex-D, P-Dex-E) by systematically adjusting the linker chemistry between Dex and the methacryloyl group. These polymeric prodrugs demonstrated a wide spectrum of activation kinetics under different conditions. When evaluated in an adjuvant-induced arthritis (AA) rat model, these polymeric Dex prodrugs exhibit different therapeutic efficacy profiles, which directly correlated with their activation kinetics. P-Dex-E was identified as the most promising prodrug for further development and clinical translation. The results from this systematic structure-activity relationship (SAR) study suggest that the rational design of prodrug activation mechanisms is a critical element in successful development of macromolecular prodrugs.
Scheme 2.
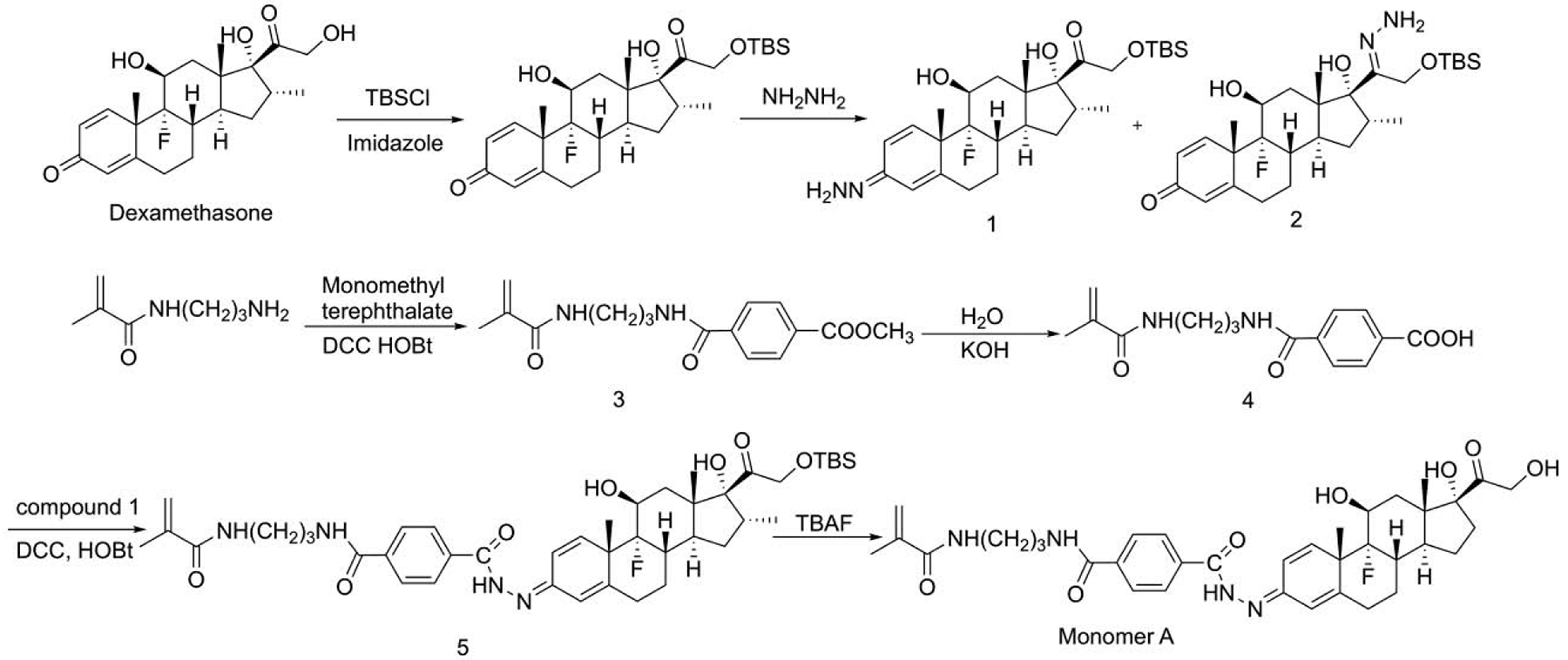
The synthesis of monomer A
Scheme 3.
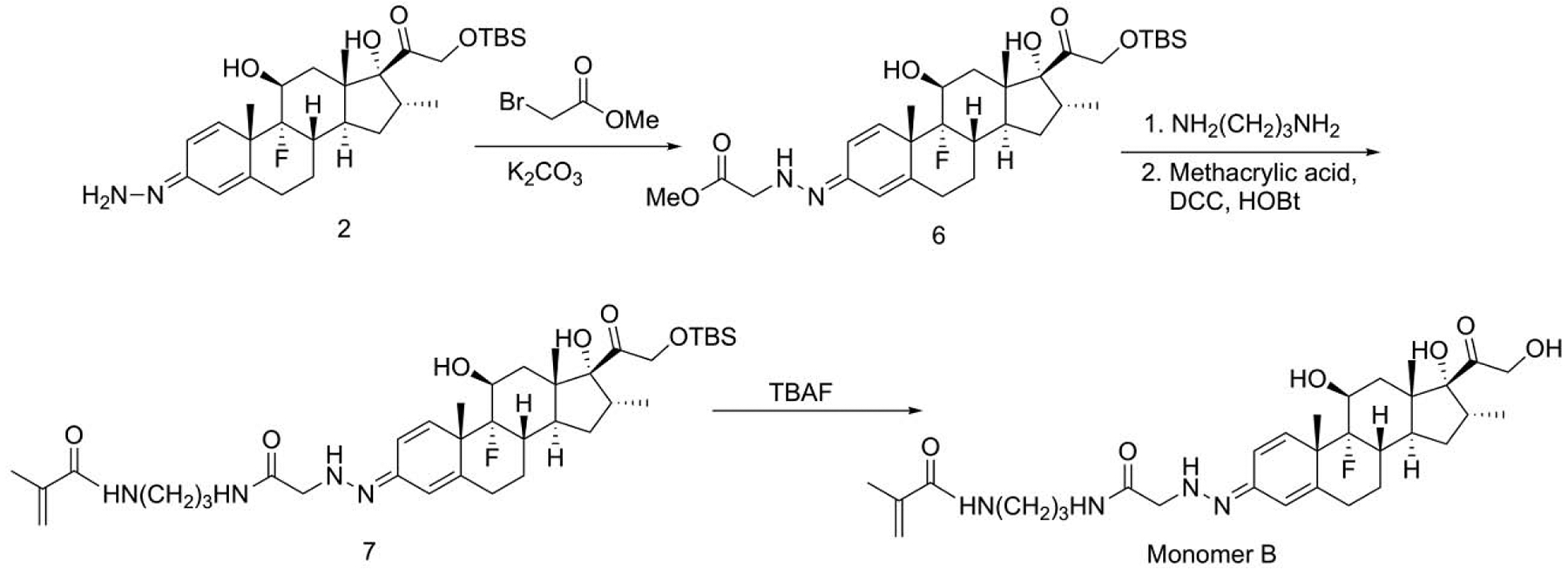
Synthesis of monomer B
Scheme 4.
Synthesis of monomer C
Scheme 5.
Synthesis of monomer D
Scheme 6.
Synthesis of monomer E
Synthesis of polymeric dexamethasone prodrugs with different linker chemistry.
The prodrugs demonstrated a wide spectrum of activation kinetics in vitro.
The prodrugs exhibit different efficacy in preserving arthritic joint in vivo.
The prodrug activation mechanism is critical for its therapeutic efficacy.
ACKNOWLEDGEMENTS
This study was supported in part by grants from National Institute of Health (R01AR062680, R01 AI119090), China Scholarship Council (XW, GZ, DK, YZ), and UNMC College of Pharmacy. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
REFERENCES
- [1].Gabriel SE, The epidemiology of rheumatoid arthritis, Rheumatic Disease Clinics of North America 27(2) (2001) 269–281. [DOI] [PubMed] [Google Scholar]
- [2].Zhang W, Anis AH, The economic burden of rheumatoid arthritis: beyond health care costs, Clin Rheumatol 30 Suppl 1 (2011) S25–32. [DOI] [PubMed] [Google Scholar]
- [3].Control C.f.D., Prevention, National and state medical expenditures and lost earnings attributable to arthritis and other rheumatic conditions--United States, 2003, MMWR. Morbidity and mortality weekly report 56(1) (2007) 4. [PubMed] [Google Scholar]
- [4].Yuan F, Quan LD, Cui L, Goldring SR, Wang D, Development of macromolecular prodrug for rheumatoid arthritis, Adv Drug Deliv Rev 64(12) (2012) 1205–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].van den Hoven JM, Van Tomme SR, Metselaar JM, Nuijen B, Beijnen JH, Storm G, Liposomal drug formulations in the treatment of rheumatoid arthritis, Mol Pharm 8(4) (2011) 1002–15. [DOI] [PubMed] [Google Scholar]
- [6].Quan LD, Thiele GM, Tian J, Wang D, The Development of Novel Therapies for Rheumatoid Arthritis, Expert Opin Ther Pat 18(7) (2008) 723–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Smolen JS, Aletaha D, Barton A, Burmester GR, Emery P, Firestein GS, Kavanaugh A, McInnes IB, Solomon DH, Strand V, Yamamoto K, Rheumatoid arthritis, Nature Reviews Disease Primers 4(1) (2018) 18001. [DOI] [PubMed] [Google Scholar]
- [8].Longo UG, Petrillo S, Denaro V, Current concepts in the management of rheumatoid hand, International journal of rheumatology 2015 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].De Cock D, Van der Elst K, Meyfroidt S, Verschueren P, Westhovens R, The optimal combination therapy for the treatment of early rheumatoid arthritis, Expert opinion on pharmacotherapy 16(11) (2015) 1615–1625. [DOI] [PubMed] [Google Scholar]
- [10].El Desoky ES, Pharmacotherapy of rheumatoid arthritis: an overview, Current therapeutic research 62(2) (2001) 92–112. [Google Scholar]
- [11].Danelich IM, Wright SS, Lose JM, Tefft BJ, Cicci JD, Reed BN, Safety of nonsteroidal antiinflammatory drugs in patients with cardiovascular disease, Pharmacotherapy: The Journal of Human Pharmacology and Drug Therapy 35(5) (2015) 520–535. [DOI] [PubMed] [Google Scholar]
- [12].Ferreira JF, Mohamed AAA, Emery P, Glucocorticoids and rheumatoid arthritis, Rheumatic Disease Clinics 42(1) (2016) 33–46. [DOI] [PubMed] [Google Scholar]
- [13].Rasch LA, Bultink IE, van Tuyl LH, Lems WF, Glucocorticoid safety for treating rheumatoid arthritis, Expert opinion on drug safety 14(6) (2015) 839–844. [DOI] [PubMed] [Google Scholar]
- [14].Yuan F, Tabor DE, Nelson RK, Yuan H, Zhang Y, Nuxoll J, Bynoté KK, Lele SM, Wang D, Gould KA, A Dexamethasone Prodrug Reduces the Renal Macrophage Response and Provides Enhanced Resolution of Established Murine Lupus Nephritis, PLOS ONE 8(11) (2013) e81483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ren K, Yuan H, Zhang Y, Wei X, Wang D, Macromolecular glucocorticoid prodrug improves the treatment of dextran sulfate sodium-induced mice ulcerative colitis, Clinical Immunology 160(1) (2015) 71–81. [DOI] [PubMed] [Google Scholar]
- [16].Liu XM, Quan LD, Tian J, Alnouti Y, Fu K, Thiele GM, Wang D, Synthesis and evaluation of a well-defined HPMA copolymer-dexamethasone conjugate for effective treatment of rheumatoid arthritis, Pharm Res 25(12) (2008) 2910–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Jia Z, Wang X, Wei X, Zhao G, Foster KW, Qiu F, Gao Y, Yuan F, Yu F, Thiele GM, Bronich TK, O’Dell JR, Wang D, Micelle-Forming Dexamethasone Prodrug Attenuates Nephritis in Lupus-Prone Mice without Apparent Glucocorticoid Side Effects, ACS Nano 12(8) (2018) 7663–7681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Quan L, Zhang Y, Dusad A, Ren K, Purdue PE, Goldring SR, Wang D, The Evaluation of the Therapeutic Efficacy and Side Effects of a Macromolecular Dexamethasone Prodrug in the Collagen-Induced Arthritis Mouse Model, Pharm Res 33(1) (2016) 186–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Quan LD, Purdue PE, Liu XM, Boska MD, Lele SM, Thiele GM, Mikuls TR, Dou H, Goldring SR, Wang D, Development of a macromolecular prodrug for the treatment of inflammatory arthritis: mechanisms involved in arthrotropism and sustained therapeutic efficacy, Arthritis Res Ther 12(5) (2010) R170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wang D, Miller SC, Liu X-M, Anderson B, Wang XS, Goldring SR, Novel dexamethasone-HPMA copolymer conjugate and its potential application in treatment of rheumatoid arthritis, Arthritis Research & Therapy 9(1) (2007) R2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Quan L, Zhang Y, Crielaard BJ, Dusad A, Lele SM, Rijcken CJF, Metselaar JM, Kostková H, Etrych T, Ulbrich K, Kiessling F, Mikuls TR, Hennink WE, Storm G, Lammers T, Wang D, Nanomedicines for Inflammatory Arthritis: Head-to-Head Comparison of Glucocorticoid-Containing Polymers, Micelles, and Liposomes, ACS Nano 8(1) (2014) 458–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kopeček J, Baẑilová H, Poly [N-(2-hydroxypropyl) methacrylamide]—I. Radical polymerization and copolymerization, European Polymer Journal 9(1) (1973) 7–14. [Google Scholar]
- [23].Rejmanová P, Labský J, Kopeček J, Aminolyses of monomeric and polymeric 4-nitrophenyl esters of Nmethacryloylamino acids, Die Makromolekulare Chemie: Macromolecular Chemistry and Physics 178(8) (1977) 2159–2168. [Google Scholar]
- [24].Strohalm J, Kopeček J, Poly [N(2hydroxypropyl) methacrylamide]. IV. Heterogeneous polymerization, Die Angewandte Makromolekulare Chemie: Applied Macromolecular Chemistry and Physics 70(1) (1978) 109–118. [Google Scholar]
- [25].Zheng H, Rao Y, Yin Y, Xiong X, Xu P, Lu B, Preparation, characterization, and in vitro drug release behavior of 6-mercaptopurine-carboxymethyl chitosan, Carbohydrate polymers. 83(4) (2011) 1952–1958. [Google Scholar]
- [26].Wei X, Wu J, Zhao G, Galdamez J, Lele SM, Wang X, Liu Y, Soni DM, Purdue PE, Mikuls TR, Goldring SR, Wang D, Development of a Janus Kinase Inhibitor Prodrug for the Treatment of Rheumatoid Arthritis, Molecular Pharmaceutics 15(8) (2018) 3456–3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wei X, Li F, Zhao G, Chhonker YS, Averill C, Galdamez J, Purdue PE, Wang X, Fehringer EV, Garvin KL, Goldring SR, Alnouti Y, Wang D, Pharmacokinetic and Biodistribution Studies of HPMA Copolymer Conjugates in an Aseptic Implant Loosening Mouse Model, Molecular Pharmaceutics 14(5) (2017) 1418–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Bao L, Zhu Y, Elhassan AM, Wu Q, Xiao B, Zhu J, Lindgren JU, Adjuvant-induced arthritis: IL-1β, IL-6 and TNF-α are up-regulated in the spinal cord, NeuroReport 12(18) (2001) 3905–3908. [DOI] [PubMed] [Google Scholar]
- [29].Stolina M, Bolon B, Middleton S, Dwyer D, Brown H, Duryea D, Zhu L, Rohner A, Pretorius J, Kostenuik P, Feige U, Zack D, The Evolving Systemic and Local Biomarker Milieu at Different Stages of Disease Progression in Rat Adjuvant-Induced Arthritis, Journal of Clinical Immunology 29(2) (2008) 158. [DOI] [PubMed] [Google Scholar]
- [30].Geborek P, Saxne T, Pettersson H, Wollheim F, Synovial fluid acidosis correlates with radiological joint destruction in rheumatoid arthritis knee joints, The Journal of rheumatology 16(4) (1989) 468–472. [PubMed] [Google Scholar]
- [31].Yu M, Wu X, Lin B, Han J, Yang L, Han S, Lysosomal pH Decrease in Inflammatory Cells Used To Enable Activatable Imaging of Inflammation with a Sialic Acid Conjugated Profluorophore, Analytical Chemistry 87(13) (2015) 6688–6695. [DOI] [PubMed] [Google Scholar]
- [32].Hou W-S, Li W, Keyszer G, Weber E, Levy R, Klein MJ, Gravallese EM, Goldring SR, Brömme D, Comparison of cathepsins K and S expression within the rheumatoid and osteoarthritic synovium, Arthritis & Rheumatism 46(3) (2002) 663–674. [DOI] [PubMed] [Google Scholar]
- [33].Law S, Andrault P-M, Aguda AH, Nguyen NT, Kruglyak N, Brayer GD, Brömme D, Identification of mouse cathepsin K structural elements that regulate the potency of odanacatib, Biochemical Journal 474(5) (2017) 851–864. [DOI] [PubMed] [Google Scholar]
- [34].Timofeevski SL, Panarin EF, Vinogradov OL, Nezhentsev MV, Anti-inflammatory and antishock water-soluble polyesters of glucocorticoids with low level systemic toxicity, Pharmaceutical research 13(3) (1996) 476–480. [DOI] [PubMed] [Google Scholar]