

Abstract
Pancreatic cancer is usually advanced and drug resistant at diagnosis. A potential therapeutic approach outlined here uses nanoparticle (NP)-based drug carriers, which have unique properties that enhance intra-tumor drug exposure and reduce systemic toxicity of encapsulated drugs. Here we report that patients whose pancreatic cancers express elevated levels of Death Receptor 5 (DR5) and its downstream regulators/effectors FLIP, Caspase-8, and FADD had particularly poor prognoses. To take advantage of elevated expression of this pathway, we designed drug-loaded NPs with a surface-conjugated αDR5 antibody (AMG 655). Binding and clustering of the DR5 is a prerequisite for efficient apoptosis initiation, and the αDΡ5-NPs were indeed found to activate apoptosis in multiple pancreatic cancer models, whereas the free antibody did not. The extent of apoptosis induced by αDΡ5-NPs was enhanced by downregulating FLIP, a key modulator of death receptor-mediated activation of caspase-8. Moreover, the DNA topoisomerase-1 inhibitor camptothecin (CPT) down-regulated FLIP in pancreatic cancer models and enhanced apoptosis induced by αDΡ5-NPs. CPT-loaded αDΡ5-NPs significantly increased apoptosis and decreased cell viability in vitro in a caspase-8- and FADD-dependent manner consistent with their expected mechanism-of-action. Importantly, CPT-loaded αDΡ5-NPs markedly reduced tumor growth rates in vivo in established pancreatic tumor models, inducing regressions in one model. These proof-of-concept studies indicate that αDΡ5-NPs loaded with agents that downregulate or inhibit FLIP are promising candidate agents for the treatment of pancreatic cancer.
Introduction
Death receptor 5 (DR5) is a member of the TNFα receptor superfamily that is capable of inducing apoptosis in cancer cells when activated. It is agonized endogenously by TRAIL (TNFα-related apoptosis-inducing ligand), which also binds to death receptor 4, osteoprotegrin and the decoy receptors 1 and 2 (1). DR5 has long been of interest as a target for oncology mainly due to its upregulation in many cancers and selective sensitivity of tumour cells to TRAIL (2).
Pancreatic cancer has one of the worst 5 year survival rates and is therefore an area of urgent unmet need (3). The DR5 targeting monoclonal antibody AMG 655 (conatumumab), has previously been investigated in a phase 2 clinical trial for the treatment of pancreatic cancer in combination with the standard-of-care chemotherapeutic agent gemcitabine (4). Ultimately, this study found that AMG 655 did not sufficiently improve survival to pursue further clinical investigation (4,5). AMG 655 and other ‘first-generation’, DR5 targeting antibodies have all been abandoned in clinical evaluations as although well-tolerated, these agents did not generate sufficient survival benefits to warrant their clinical approval (6). This is thought to be due at least in part to the requirement of DR5 crosslinking (“super-clustering”) to activate apoptosis efficiently. It was originally thought that Fcγ receptor engagement from Fcγ receptor-expressing cells would enable sufficient clustering to be achieved at the tumor site; however, this did not prove to be the case (5,7). More recent DR5-targeted therapeutics have been designed as multivalent agents for this reason whereas first-generation DR5 targeting antibodies are only bivalent (8–13).
An alternative approach to enhance multivalency of antibodies is their attachment to the surface of NPs to promote clustering of antibody paratopes, and we have previously shown that efficient DR5 receptor clustering can be achieved through conjugation of AMG 655 to PEGylated poly(lactic-co-glycolide) (PLGA) nanoparticles, leading to activation of apoptosis in colorectal models (14,15). Other groups have also shown that conjugation of monovalent recombinant TRAIL to the surface of multiple nanoparticle types causes an increase in its potency (16–19).
Upregulation of FLIP (Fas-associated death domain (FADD)-like IL-1β-converting enzyme-inhibitory protein) is a potential contributor to the lack of efficacy of DR5 targeting therapies through inhibiting caspase 8 activation (20). FLIP upregulation has been observed in many tumour types and correlates with disease progression (20–28). Many drugs have been shown to downregulate FLIP, which can help increase sensitivity to DR5 therapeutics in the tumour (29).
Here, we explore the use of a PEGylated poly(lactic-co-glycolide) (PLGA) nanoparticle with AMG 655 conjugated to the surface and the DNA topoisomerase 1 inhibitor camptothecin (CPT) entrapped (CPT DR5 NP) in the treatment of pancreatic cancer cell models. We demonstrate that the binding of AMG 655 to the surface of the nanoparticle renders it capable of DR5 agonism, unlike free antibody, suggesting improved avidity. We also show that the addition of CPT leads to enhances the efficacy of the formulation synergistically by causing down regulation of FLIP(L) and FLIP(S). This may sensitise previously resistant tumours to DR5 therapy and therefore provide more clinical options for patients.
Results
High expression of the canonical DR5 pathway is associated with poor prognostic pancreatic cancer.
Canonically, the DR5 (TRAIL-R2) pathway signals via the adaptor protein FADD to activate caspase-8 and initiate apoptosis; however, if expression of the caspase-8 regulator FLIP is high, DR5 activation fails to induce apoptosis and can instead drive tumor growth and metastasis (Figure 1A). Given that the majority of pancreatic cancer patients die from their metastatic disease, we interrogated the GEPIA website, which compares data from The Cancer Genome Atlas and the Genotype-Tissue Expression projects, to assess the impact of expression of the canonical DR5 pathway (DR5, Caspase-8, FADD and FLIP) on pancreatic cancer patient prognosis (30). The results were clear: stratification of patients based on these markers identified two populations with significantly different disease-free survival after the initial 20 months. Those patients with high DR5 pathway transcript expression had significantly worse disease-free survival (Figure 1B). Overall, patients with high expression of FLIP (HR = 1.7, p = 0.026) and FADD (HR = 1.6, p = 0.033) had a significantly worse prognosis. For, caspase-8 and DR5, the same trends were apparent but just failed to reach significance (HR = 1.6, p = 0.052; and HR = 1.5, p = 0.055). Moreover, highly significant correlations were observed between gene expression of all 4 of these DR5 pathway components (Figure 1 and Supplementary Figure 1), indicating that when one component of the pathway is high, the other 3 components are also elevated. It was also notable that high expression of the DR5 ligand, TRAIL, which is normally expressed by immune effector cells, was also more likely to be associated with poorer disease-free survival, although this failed to reach significance (HR = 1.5, p = 0.09; Supplementary Figure 1B). Expression of XIAP, an anti-apoptotic protein that regulates execution of apoptosis downstream of DR5 activation (31), was not associated with poorer prognosis (Supplementary Figure 1C). Overall, these results indicate that pancreatic tumors can be divided into those with high expression of the DR5 pathway, which have a poor prognosis, and those with low expression of the DR5 pathway, which have a better prognosis in those patients surviving beyond 20 months.
Figure 1. The DR5 pathway is upregulated in pancreatic cancer and correlates with poorer prognosis.
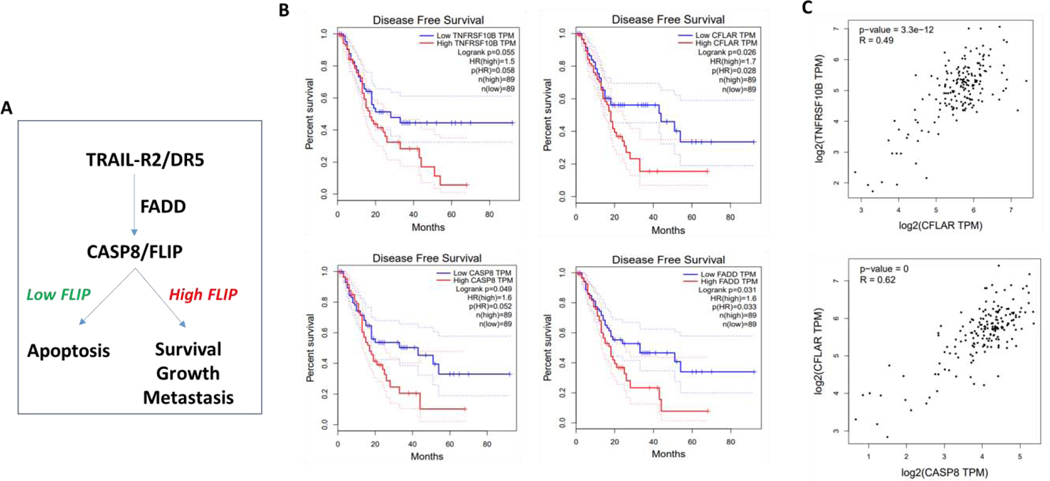
(A) Simplified schematic of the DR5 pathway (B) Disease free survival plots comparing high and low expressers of genes in the DR5 pathway including TNFRSF10B (DR5), CFLAR (FLIP), CASP8 (caspase 8) and FADD (FADD) using RNAseq data from GEPIA.com showing poorer prognosis in high expressors. (C) Correlation plots comparing expression levels of apoptotic pathway genes using RNAseq data from GEPIA.com showing a positive correlation in expression between all genes investigated.
The DR5 pathway is maintained in pancreatic cancer cell models of pancreatic cancer and can be activated by DR5-targeted Nanoparticles.
In vitro models of pancreatic cancer retained expression of an intact DR5 pathway at the protein level, albeit with variable levels of expression of DR5 itself and FLIP in particular (Figure 2A). Given the expression and correlation of the DR5 pathway with poor prognostic disease in pancreatic cancer (Figure 1), we next began to assess whether the retention of this pathway could be therapeutically exploited. We therefore set out to generate a DR5-targeted nanoparticle that could present a high density of anti-DR5 paratopes and thereby drive cell surface clustering of DR5 and initiate caspase-8-mediated apoptosis. Importantly for the viability of this approach, the DR5 detected by Western blot analysis was also detected on the cell surface of the pancreatic cancer cells (Figure 2B).
Figure 2. The conjugation of AMG 655 to the surface of the nanoparticle renders it capable of inducing apoptosis and caspase activation in vitro.

(A) Western blots of the relative expression of apoptotic proteins in pancreatic cancer cell lines. (B) Cell surface expression of death receptor 5 on pancreatic cancer cell lines. n=3 ± SD (C) Assessment of cell viability using CellTiter-Glo® reagent following treatment with varying concentrations of BLK DR5 NP and controls. Representative of n = 3
Previously our group generated antibody-conjugated PLGA-based nanoparticles, where conjugation of the antibody was achieved through EDC functionalization of free carboxyl moieties on the surface of the nanoparticles (14). Whilst this approach proved successful, it involves several processing steps that may ultimately limit manufacturing options. In this work, we incorporated an NHS-modified PLGA in the polymer blend, which enables one-step conjugation to the antibody after preparation of the nanoparticles. Using this approach, we conjugated the first generation αDΡ5 therapeutic antibody AMG 655 to the surface of PLGA-NPs.
Notably, compared to free AMG 655, the αDΡ5 NPs activated cell death in each of the 4 pancreatic cancer cell lines (Figure 2C). This was observed even in the PANC-1 model, where expression of DR5 was the lowest, suggesting that the absolute expression level of DR5 is not predictive of the efficacy of this therapeutic approach. Indeed, the most resistant model was AsPC-1, which expresses the highest levels of DR5 (Figure 2A/B); however, this model also expressed the highest levels of FLIP(S). FLIP(S) is an inhibitor that disrupts the formation of procaspase-8 homodimers, a prerequisite for caspase-8 activation (29). Conversely, the long FLIP splice form FLIP(L) can either promote or inhibit DR5-mediated apoptosis depending on its stoichiometry within DISC (32).
FLIP inhibits cell death induced by multivalent TRAIL and anti-DR5 NPs.
Given the suggestion from AsPC-1 cells that FLIP(S) may inhibit the cell death-inducing capacity of αDΡ5 NPs, we assessed the impact of downregulating FLIP splice forms on sensitivity of pancreatic cancer cells to multivalent DR5 agonists. Using splice form-selective FLIP-targeted siRNAs and dual FLIP(L)/(S)-targeted siRNAs (Figure 3A), we found that downregulating FLIP(S) did indeed enhance the sensitivity of AsPC-1 cells to isoleucine zipper (IZ)-TRAIL, a multivalent recombinant form of TRAIL; however, the greatest degree of enhancement was observed in cells in which both splice forms were downregulated (Figure 3B). A similar effect was observed in MIA PaCa-2 cells. However, in PANC-1 cells, downregulation of FLIP(L) alone had a similar effect to silencing of both splice forms; this may reflect the very low expression of FLIP(S) in the latter model (Figure 1A). In our hands, BxPC-3 cells were highly sensitive to transfection with control siRNAs, so were not included in this analysis.
Figure 3: FLIP downregulation increases response to BLK DR5 NP treatment in siRNA knockdown of FLIP (T) (refers to total, both long and short), FLIP (L) and FLIP (S) in AsPC-1, MIA PaCa-2 and PANC-1 cells:
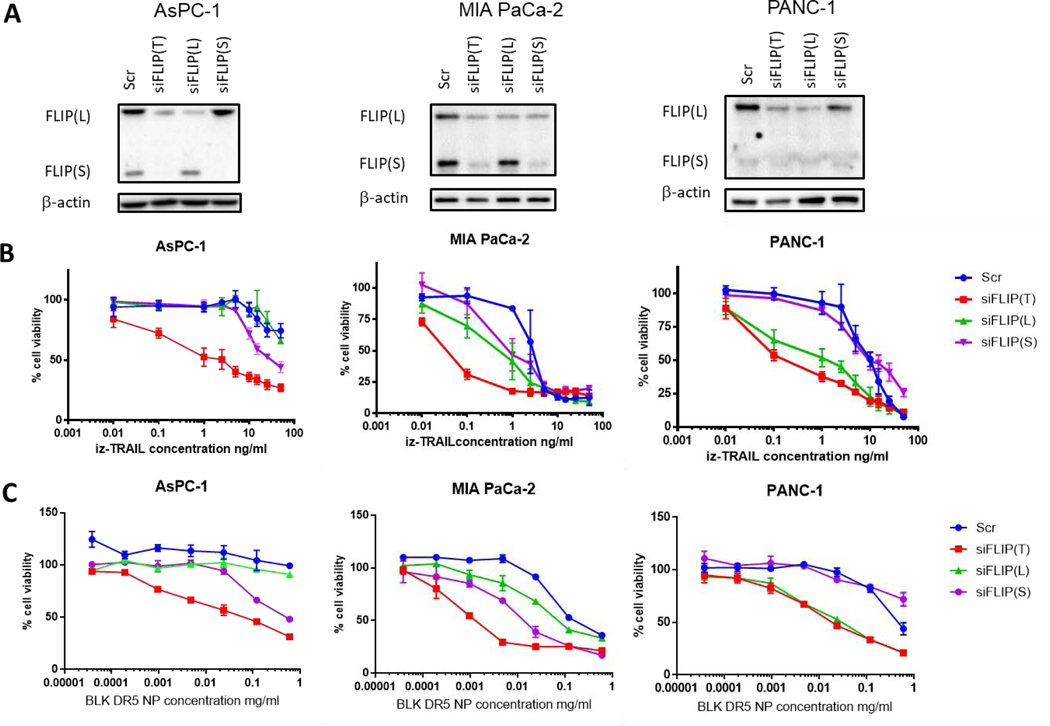
(A) Validation of siFLIP k nock down by western blot. (B) cell viability measured by CellTiter-Glo® after 24 hours treatment with IZ-TRAIL of siFLIP k nock down pancreatic cancer cell lines. Representative of n = 3. (C) cell viability measured by CellTiter-Glo® after 24 hours treatment with BLK DR5 NP of siFLIP k nock down pancreatic cancer cell lines. Representative of n = 3.
Importantly, enhanced sensitivity to αDΡ5 NPs was also observed in FLIP-depleted cells (Figure 3C). Similar to treatment with IZ-TRAIL, the enhancement was greatest in MIA PaCa-2 and AsPC-1 cells when both FLIP(L) and FLIP(S) were simultaneously downregulated. Nonetheless, in PANC-1 cells, silencing FLIP(L) alone was sufficient to drive maximal enhancement of DR5 agonist-induced cell death.
Development and characterization of highly potent CPT-loaded αDR5 NPs.
In order to find a pharmacologically relevant approach to downregulate FLIP expression in pancreatic cancer cell models, we screened a number of agents previously identified as modulators of FLIP expression and/or clinically relevant agents (Supplementary Figure 3). The chemotherapeutic agents, cisplatin and gemcitabine had no effect on FLIP expression. Similarly, inhibitors of MAP kinase and PI3 kinase and PPARγ ligands also had no effect despite studies linking these pathways to FLIP expression (Supp Figure 3B). However, the topoisomerase-I inhibitor camptothecin (CPT) and the HDAC inhibitor SAHA both down-regulated expression of both FLIP splice forms in all 4 pancreatic cancer cell models (Figure 4A & Supplementary Figure 3C). We focused on CPT as this agent has the hydrophobic properties suitable for entrapment in PLGA NPs and initial attempts to encapsulate SAHA failed. CPT was efficiently entrapped, consistent with our previous studies (14,33,34). Notably, there was no significant difference in CPT entrapment between undecorated (“nude”) and αDΡ5-conjugated NPs (Supp Figure 1C), and CPT was released from NPs in a first order manner, with ~50% of total drug released after 24 hours and ~80% total release by 72 hours (Figure 4B). Importantly, in all 4 cell lines, CPT-loaded αDΡ5-NPs had more potent in vitro activity than either free CPT, nude CPT-loaded NPs or non-drug-loaded αDΡ5-NPs (Figure 4C). This increase in potency was found to be synergistic, with combination indices of less than one observed in each cell line (Figure 4D).
Figure 4: CPT downregulates FLIP (L), FLIP (S) and XIAP in a dose dependent manner and increases the efficacy of AMG 655 synergistically:
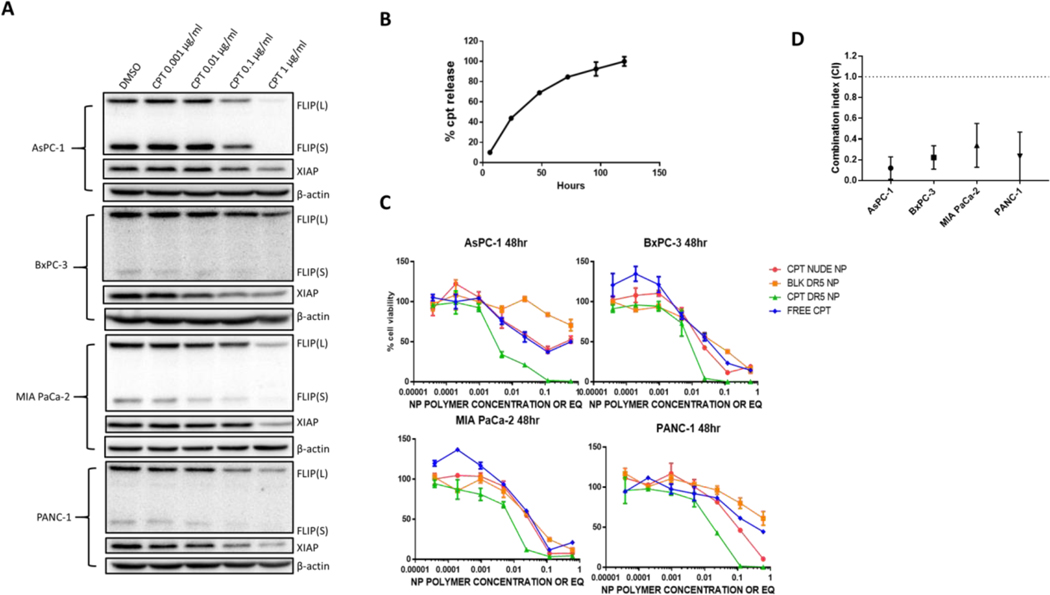
(A) Western blots of FLIP and XIAP post treatment with the concentrations of CPT shown or DMSO controls for 24 hours on AsPC-1, BxPC-3, MIA PaCa-2 and PANC-1. Images representative of n = 3. (B) Camptothecin release from nanoparticles over time. Representative of n = 3 (C) Assessment of cell viability using CellTiter-Glo® reagent following treatment with varying concentrations of CPT DR5 NP and controls at a 48-hour time point. Representative of n = 3 (D) Combination indices for the range of concentrations used in C of CPT DR5 NP compared to BLK DR5 NP and CPT NUDE NP controls at 48 hours. Calculated using CompuSyn. n = 3 ± SD.
In vitro confirmation of the mode of action of CPT-loaded αDR5-NPs.
To examine the mechanism-of-action of the CPT-loaded αDΡ5-NPs in more detail, we focused on the MIA PaCa-2 and PANC-1 models. Consistent with the effects of free drug, both nude and αDΡ5-conjugated CPT-loaded NPs down-regulated expression of both FLIP(L) and FLIP(S) (Figure 5A). Notably, a 43 kDa version of FLIP (p43-FLIP(L)) was observed from BLK DR5 NP and CPT DR5 NP and is evidence of DISC formation as p43-FLIP(L) is an intermediate form of FLIP(L) only formed through cleavage with procaspase 8 at the DISC (Figure 5A). Moreover, the enhanced efficacy of the αDΡ5-conjugated, CPT-loaded NPs was found to correlate with enhanced apoptosis induction (Figure 5B), which itself correlated with enhanced mitochondrial outer membrane permeabilization (MOMP, Figure 5C), a critical point in the commitment of cells to undergo apoptosis (35). Moreover, enhancement of caspase-8 and caspase-3 processing (Figure 5D) and activity (Figure 5E) were observed in response to αDΡ5-conjugated CPT-loaded NPs compared to nude, CPT-loaded NPs and αDΡ5-conjugated blank.
Figure 5: In vitro confirmation of the mode of action of CPT DR5 NP.

(A) Western blots of FLIP splice forms post treatment of 0.2 mg/ml CPT DR5 NP and appropriate controls for 24 hours on MIA PaCa-2 and PANC-1 cells (B) Apoptosis was identified using Annexin V/PI staining analyzed by flow cyt ometry after 24- or 48-hour treatment with CPT DR5 NP and appropriate controls. n = 3 ± SD (C) CPT DR5 NP treatment causes mitochondrial membrane depolarization. Post 24-hour treatment with CPT DR5 NP cells were incubated with 25 μM tetra-methyl-rhodamine ethyl ester (TMRE). n = 3 ± SD. (D) CPT DR5 NP treatment (0.2 mg/ml) induces cleavage of procaspase 8 and procaspase 3 as shown through western blot and validated by (E) Caspase-Glo® assays. Black dotted lines = crop
To probe the mechanism of cell death induction activated by αDΡ5-conjugated CPT-loaded NPs further, we developed pancreatic cancer cell models in which either the critical adaptor protein FADD or effector enzyme caspase-8 of the DR5 pathway were deleted (Figure 6A). The CRISPR knockout cells retained expression of the other pathway components, although there were alterations in the levels of expression of some proteins compared to the control (EV) cell line, most notably a reduction in FLIP(L) expression in both caspase-8 knockouts; this suggests a role for procaspase-8 in regulating FLIP(L) expression. In MIA PaCa-2 or PANC-1 cells lacking either FADD or caspase-8, the effectiveness of the αDΡ5-conjugated CPT-loaded NPs was significantly reduced (Figure 6B). In fact, in the FADD and caspase-8 KO models, the efficacy of the αDΡ5-conjugated CPT-loaded NPs was identical to that of free CPT and nude CPT-loaded NPs. This is consistent with the mechanism-of-action of the αDΡ5-conjugated CPT-loaded NPs operating by enhanced activation of DR5-induced, FADD-and caspase-8-mediated apoptosis as a result of FLIP downregulation.
Figure 6: Knockout of FADD and caspase 8 stops conjugated AMG 655 efficacy.
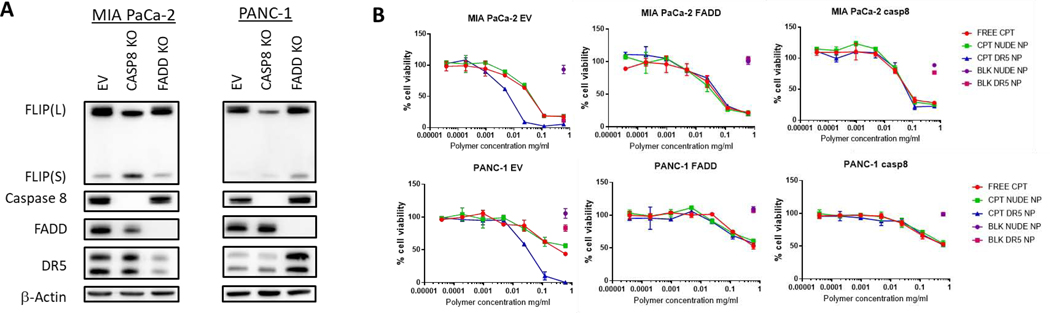
(A) Western blots validating k nock out of FADD and procaspase 8 in MIA PaCa-2 and PANC-1 cell lines (B) Cell viability of MIA PaCa-2 and PANC-1 CRISPR FADD, Caspase 8 k nock out clones and empty vector in response to CPT DR5 NP and controls measured using CellTiter-Glo® at a 48-hr time point. Representative of n = 3
In vivo assessment of CPT-loaded αDΡ5-NPs.
To determine whether the in vitro potency of CPT-loaded αDΡ5-NPs was retained in vivo, we generated PANC-1 and MIA PaCa-2 xenografts and compared the effects of intravenous delivery of CPT-loaded nude NPs with CPT-loaded αDΡ5-NPs. BxPC-3 was not pursued in vivo due to its KRas wild type status which is unlike the vast majority of pancreatic cancers. In the PANC-1 cell line, both CPT-loaded NP formulations retarded tumor growth, with a significant additional inhibitory effect for the αDΡ5 coated NPs (Figure 7A). The effects were more dramatic in the MIA PaCa-2 cell line where delivery of CPT-loaded αDΡ5-NPs caused significant tumor regressions, whereas the CPT-loaded nude NPs only caused a growth retardation (Figure 7B). Moreover, the CPT-loaded nude NPs were well-tolerated as indicated by monitoring mouse behavior and weight (Figures 7C/D). We have previously found that free CPT is toxic in mouse models and therefore it was considered unethical to use this control. Collectively, these in vitro and in vivo studies demonstrate the potential of CPT-loaded αDΡ5-NPs for the treatment of pancreatic cancer.
Figure 7. Tumor volume change over time of cell line xenografts in SCID mice during and after 6 treatments of CPT DR5 NP and appropriate controls.
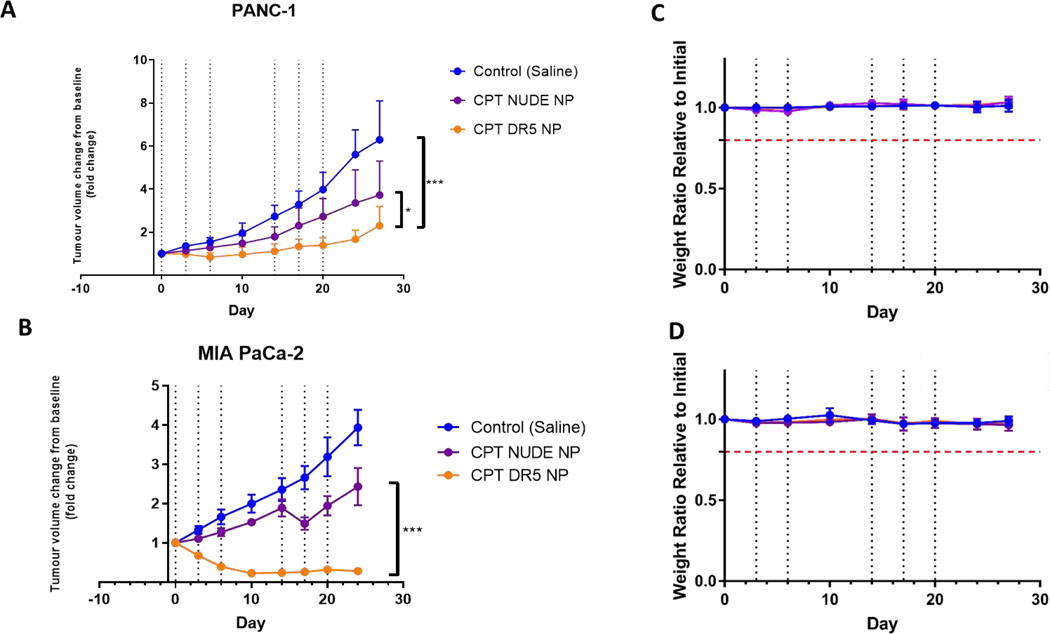
Cells were implanted subcutaneously into SCID mice and allowed to reach 300 mm3 before being randomized into separate treatment groups (n=6 per group). Tumor volumes were monitored during and after treatment. Treatment was 2 mg (100 mg/k g) of nanoparticle or appropriate control delivered intravenously via tail vein. The lower panels show animal weight ratios and Kaplan-Meier survival curves. (brok en red line represents 80% weight cut off). Dashed lines are treatment days
Discussion
Pancreatic cancer (PDAC) is the 4th leading cause of cancer deaths. Most patients have advanced disease at diagnosis, and 2-year survival is a dismal 6%. Multiple factors hinder treatment. One is tumor genetic heterogeneity and the presence of tumor cell populations that are prone to metastasis and intrinsically resistant to chemotherapy. A second is poor tumor drug penetration because of stromal amplification, low micro density, and poor vascular perfusion/permeability. These characteristic features constitute a formidable drug delivery barrier and create the difficult clinical reality that even drugs active against the cancer cells may fail clinically because of poor tumor deposition. Thus, radical pharmacological approaches are needed for the treatment of this disease.
When bound by its ligand TRAIL (expressed by immune effector cells) or by agonistic therapeutic antibodies, the DR5 death receptor forms a complex known as the DISC (death-inducing signaling complex) at which the adaptor protein FADD recruits procaspase-8. Homo-dimerization of procaspase-8 at the DISC leads to its processing into its active form, which then cleaves substrates such as procaspase-3 and BID to initiate apoptosis. However, the caspase-8 paralog FLIP can also be recruited into the DISC disrupting formation of procaspase-8 homodimers, thereby inhibiting apoptosis induction. In the presence of FLIP, alternative non-apoptotic signaling pathways can be activated, such as the NFκB and MAP kinase pathways (36). These tumor-promoting effects have been confirmed in vivo, with therapeutic administration of TRAIL promoting metastasis to the liver of pancreatic cancer xenografts (37). Moreover, murine death receptor has been shown to promote growth and metastasis of autochthonous mutant KRAS-driven models of non-small cell lung cancer (NSCLC) and pancreatic cancer (38). These findings are consistent with our finding (Figure 1) that high expression of DR5 and the rest of the core DISC components correlated with poor prognosis of pancreatic cancer patients. This is also consistent with the findings of others that high DR5 expression correlates with lymphatic invasion of pancreatic cancers.
We hypothesized that we could take advantage of the maintenance of high expression of the DR5 pathway in pancreatic cancer by devising a NP-based therapeutic that would activate the receptor to promote apoptosis rather than metastasis and growth of PDAC cells.
This approach required a NP that would induce: (1) sufficient clustering of the receptors; and (2) downregulation of FLIP (39). Lack of DR5 super-clustering (a prerequisite for efficient apoptosis induction) and failure to overcome FLIP-mediated inhibition (FLIP being frequently overexpressed in multiple poor prognosis tumor types) were two of the main reasons for the failure of the 1st generation of DR5 agonists, including AMG 655.
To address the first issue (receptor super-clustering), we formulated a nanoparticle conjugated to AMG 655. The conjugation of AMG 655 to the surface of the nanoparticle massively enhanced its ability to induce apoptosis of PDAC cells when compared to its free form. These results are significant in light of the phase 2 trial that used unconjugated AMG 655 in pancreatic cancer patients. The results from this trial were ultimately disappointing with only small improvements observed in 6 months survival (4). Second generation multivalent DR5 binding agents are now emerging that induce receptor super-clustering. These highly potent 2nd generation DR5 agonists mirror the effects we have achieved by conjugating AMG 655 to the surface of NPs, thereby presenting a high density of anti-DR5 paratopes that drive cell surface clustering of DR5 and initiate apoptosis. Moreover, with scalable manufacturing in mind, we have devised a simplified one-step conjugation method for attaching antibodies to the surface of PLGA NPs using an NHS-modified PLGA in the polymer blend.
To address the second issue (high FLIP expression), we assessed the impact of a number of chemotherapeutics and targeted therapeutics on expression of FLIP in pancreatic cancer cells. Although previously reported to downregulate FLIP, none of the MAP kinase and PI3-kinase inhibitors we examined, nor PPARγ ligands had any effect on FLIP. We found here that CPT could downregulate both FLIP isoforms across all cell lines examined and, as hydrophobic molecules, were efficiently entrapped in PLGA NPs. A more potent topo-I inhibitor than its derivative irinotecan, which is approved for the treatment of pancreatic cancer as a component of the FOLFIRINOX regimen, CPT has poor bioavailability and considerable systemic toxicity. For these reasons, novel delivery approaches are needed to capitailize on the potent antitumor properties of CPT. Notably, we found that the entrapment of CPT improved the potency of the αDΡ5-NPs in vitro and in vivo, with tumor regression observed in the MIA PaCa-2 model. The mechanism-of-action was confirmed to involve FLIP down-regulation and enhanced FADD- and caspase-8-mediated apoptosis.
The use of active targeting as a strategy in nanomedicine remains unproven as no actively targeted nano-formulation has yet reached the market, unlike non-targeted formulations, which mainly take advantage of the enhanced permeability and retention (EPR) effect and which have been on the market for over 20 years. Wilhelm et al. conducted a meta-analysis of the current literature and reported that overall, actively targeted nanoparticles accumulated in the tumor 50% more efficiently compared a separate set of passively targeted nanoparticles in in vivo models (i.e. 0.9% vs 0.6% ID) (40). Moreover, antibody-conjugated nanoparticles are an attractive alternative to antibody-drug conjugates as they offer reduced toxicity due to their less potent drug payloads (41,42). There is also potential added benefit when the targeting strategy induces its own therapeutic effect as has been observed in this study where the nanoparticle delivers a chemotherapy that synergizes with the targeting antibody that can induce apoptosis. However, the potential offered by these approaches can only be overcome with improved manufacturing and characterization techniques.
While the in vivo models used here can only be considered proof-of-concept, it will be important to explore biodistribution in a pancreatic model that exhibits more pancreatic tumor microenvironment-like characteristics. However, on the basis that the potential of nanotechnology in pancreatic cancer has already been exemplified by the approval of Nab-paclitaxel (Abraxane®) and liposomal irinotecan (Onivyde®) (43,44), the approach explored here may have potential for the treatment of DR5-positive, poor prognostic pancreatic cancer.
Materials and methods
Nanoparticle formulation – single emulsion method
Polymeric nanoparticles were formulated using the single emulsion evaporation method. Polymer blends (20 mg, 75% PLGA RG502H, 25% PLGA-PEG-NHS (Polyscitech® AI64) or PLGA-PEG-COOH (Polyscitech® AI34)) were dissolved in 1 ml of dichloromethane. Camptothecin (200 μg in vitro, 600 μg in vivo) dissolved in DMSO at 10 mg/ml was added to the organic phase and mixed if required. This solution was then injected into 7 ml of PVA (2.5% w/v polyvinyl alcohol in MES 50 mmol/l buffer at pH5) stirring at 600 rpm to form an emulsion. The emulsion was the sonicated using a sonicator probe controlled by a Fischer automated sonicator for 90 seconds pulsing at 50% amplitude. After sonication the emulsion was left overnight stirring at 600 rpm. The nanoparticles were then isolated by centrifugation at 20,000 x g at 4°C for 20 minutes and resuspended in MES buffer (50 mmol/l, pH5) using brief sonication. This was then repeated another 2 times. If antibody conjugation was required, particles were then resuspended at 1mg/ml in MES buffer. 50 μg of antibody per mg of polymer was then added to this solution and left to conjugate for 2½ hours stirring at 90 rpm. This was then centrifuged as above and resuspended in PBS.
Nanoparticle characterization
Nanoparticle size and polydispersity were analyzed by dynamic light scattering using a NanoBrook Omni (Brookhaven Instruments Corporation, NY, USA). The particles were resuspended in dH2O at a concentration of 0.1 mg/ml and pipetted into a cuvette. Zeta potential was measured by phase analysis light scattering, resuspending the nanoparticles as above. Nanoparticle size was also measured by nanoparticle tracking analysis using the NanoSight NS300 (Malvern Instruments, UK) in PBS at a concentration of 0.1 mg/ml in PBS. The nanoparticles were imaged by scanning electron microscopy (SEM) by washing and resuspending in dH2O (5 mg/ml) this was then added dropwise to double sided copper tape affixed to aluminum stubs and allowed to dry overnight in a laminar flow cabinet. The next morning nanoparticle were sputter coated with gold and imaged using a FEI Quanta 250 FEG - Environmental Scanning Electron Microscope (E-SEM).
Validation and quantification of antibody conjugation
Antibody conjugation was confirmed and quantified using a Micro BCA Protein Assay Kit obtained from ThermoFisher Scientific, UK. Nanoparticles were resuspended at 1 mg/ml and 50 μl of sample was added per well of a clear non-sterile 96well plate and a standard curve was prepared using blank particles and free antibody. 100 μl of Micro BCA reagent was added to each well. This was then left to develop for 1 hour incubated at 37°C. The sample was then read at a wavelength of 562 nm using a BioTek synergy HT plate reader.
Tissue culture
PANC-1 and MIA PaCa-2 cells were obtained from ATCC and cultured in DMEM supplemented with 10% fetal bovine serum (Gibco), 50 units/ml penicillin streptomycin and 1 mM sodium pyruvate. BxPC-3 cells, obtained from ATCC and were cultured in RPMI supplemented with L-glutamine, 10% fetal bovine serum, 50 unit/ml penicillin streptomycin, 1 mM sodium pyruvate and 10 mM HEPES. AsPC-1 cells were obtained from ATCC and were cultured in RPMI supplemented with L-glutamine, 10% fetal bovine serum and 50 unit/ml penicillin streptomycin.
Cells were grown in t25, t75 or t175 flasks. Cells were allowed to reach 70–95% confluency depending on the line. They were kept in an environment of constant temperature at 37°C and 5% CO2.
Western blot
Cells were lysed in RIPA buffer and analyzed for protein content using a BCA assay (ThermoFischer Scientific, UK). Gel electrophoresis was performed using tris-glycine gels of varying percentages depending on the size of the protein of interest. Gels were run at 125 V for 90 minutes. Semi-dry transfer was used to transfer protein onto an activated polyvinylidene difluoride membrane. Nonspecific binding sites were blocked in blocking buffer for 1 hour at room temperature. The membrane was then incubated in primary antibody at 4OC overnight under agitation. The blot was then washed for 5 minutes 6 times in TBS tween and incubated for one hour in HRP (horseradish peroxidase) conjugated secondary antibody. The wash steps were repeated and pierce ECL plus kit protocol was followed to image blot using a BioRad XRS+ imager.
Assessment of cell viability by CellTiter-Glo assay
CellTiter-Glo® luminescent cell viability assay uses a luciferase reaction in the presence of ATP and oxygen. Spent media was aspirated to leave 25 μL of media in 96 well white tissue culture treated plates (Corning 734–1665). This was then combined with 25 μL of CellTiter-Glo reagent and mixed on plate shaker for 3 minutes to ensure cell lysis. Luminescence was then read using a BioTek Synergy HT plate reader.
Caspase 3/7 and 8 activity assays
Caspase-Glo® 8 and 3/7 are also luciferase-based assays. Spent media was removed from the wells to be analyzed on 96 well white tissue culture treated plates (Corning 734–1665) leaving 25 μL of media. This was then incubated with 25 μL of Caspase-Glo® reagent for 45 minutes under gentle agitation at room temperature and protected from light then the luminescence was read using a BioTek Synergy HT plate reader.
Flow cytometry – cell surface staining
Cells were seeded in a 6-well plate at 200,000 cells/well and left to adhere overnight in a 5% CO2 incubator at 37°C. The next morning spent media was aspirated and the cells were washed with ice cold PBS three times. After detachment and isolation by centrifugation, cells were resuspended in 400 μl of ice cold 5% FCS (fetal calf serum) in PBS and incubated for 30 minutes on ice with 5 μl (0.25 μg) DR5 PE stained antibody (ebiosciences 12–9908), with 5 μl (0.5 μg) of PE stained IgG1k isotype control antibody (ebiosciences 12–4714) or are left untreated. Post incubation the cells were then centrifuged as above and resuspended in 1 ml of ice cold 5% FCS PBS and this is repeated twice before samples are read by the BD FACS Calibur flow cytometer or a BD Accuri C6 plus flow cytometer.
Flow cytometry – Annexin V/PI
Cells seeded at 400000 cells per well in a 6 well plate were treated with 0.4 mg/ml CPT DR5 NP or equivalent control for 24 hours. Media and cells detached using a scraper were stained with 3 μl Annexin V FITC antibody (BD pharmingen) and 2 μl of propidium iodide. The samples were then analyzed using a BD Accuri C6 plus flow cytometer.
Flow cytometry – TMRE
Cells were seeded at 400000 cells per well in a 6 well plate and treated with 0.4 mg/ml CPT DR5 NP. Cells were stained with 25 nM Tetramethylrhodamine (TMRE) for 15 minutes at 37OC, then gently detached using a cell scraper and washed once in PBS. The samples were then analyzed using a BD Accuri C6 flow plus cytometer.
Cell line xenografts
All animal studies were performed in collaboration with the Straubinger group in Roswell Park Cancer Institute (RPCI) and State University of New York at Buffalo. All experiments were carried out in line with New York state and US federal law. SCID mice were obtained from an in-house colony in RPCI. MIA PaCa-2 and PANC-1 cells were implanted subcutaneously with Matrigel into SCID mice and treatment commenced upon tumors reaching sufficient size. Mice were randomized into groups based on tumor size. Where nanoparticles were used as monotherapy, treatment was administered intravenously on day 0,3,6,14,17 and 20. Tumor volumes were measured on alternate days and plotted over time. Mice were sacrificed when tumors reached a threshold volume.
Generation of FLIP (L), FLIP (S) and XIAP knockdowns using siRNA
FLIP (L) and FLIP (S) siRNAs were purchased from Eurofins. FLIP(T) and XIAP was purchased from Dharmacon. Transfections were performed using lipofectamine RNAiMAX (Life Technologies). Initially 900000 cells were seeded in a p90 and left overnight. The next morning 9 μl of RNAiMAX was added to 600 μl of OptiMEM while 1.8 μl (10nM stock) of siRNA was added to 600 μl of OptiMEM. Both solutions were mixed together and left to incubate at room temperature for 15 minutes. After incubation, 2.4 ml of growth medium was added to the mixture and was added to cells once spent media was aspirated.
This transfection media was incubated with the cells for 4 hours then 6.4 ml of normal growth media was added to the cells and left incubating at 37ºC until harvested.
Generation of CRISPR FADD and caspase 8 knockout cell lines
MIA PaCa-2 and PANC-1 FADD (guide seq: GCGGCGCGTCGACGACTTCG) and Caspase 8 (guide seq: AAGTGAGCAGATCAGAATTG) CRISPR knockouts were generated using lentiviral infection using pLentiCRISPRV2. Initially, HEK 293T cells were seeded in P90’s and left overnight. The next day vector, envelope and packaging DNA were transfected using GeneJuice® (MerckMillipore, UK) and left for 6 hours. The transfection medium was then removed, and normal growth medium was added and left to incubate for 48 hours. The resultant media was then collected and stored at −20ºC. This media was then added to the desired cell line and left overnight. The next morning cells were washed with PBS twice and media added with puromycin as a selecting reagent. Once a reference plate has been completely killed by puromycin clones were generated for each gene knockout and selected for favorable protein expression levels in comparison to the empty vector mixed population.
Gene correlation plots and calculation of disease-free survival
Disease free survival was calculated using GEPIA.com which compares RNAseq data from The Cancer Genome Atlas Program to The Genotype-Tissue Expression project (30). Disease free survival was calculated using a median group cutoff. Correlation plots were calculated using the Pearson correlation coefficient.
Data analysis
Statistical tests were carried out with GraphPad Prism software version 7.0. Parametric tests were used where data was normally distributed and of sufficient sample size. To assess significance between two groups the students t-test was employed. Where three or more groups were to be assessed for significant difference one way or two-way analysis of variance (ANOVA) were employed. The null hypothesis was rejected when p < 0.05. Levels of significance were indicated as follows: * p < 0.05, ** p < 0.01, *** p < 0.001. Combination indexes were calculated using CompuSyn software (45).
Supplementary Material
The death receptor 5 pathway is upregulated in pancreatic cancer and correlates with poorer prognosis.
Conjugation of AMG 655 to the nanoparticle surface renders it capable of inducing apoptosis via death receptor 5 in pancreatic cancer cell lines.
FLIP downregulation increases response to TRAIL and nanoparticle conjugated AMG 655.
Camptothecin entrapment causes downregulation of FLIP.
CRISPR targeting shows conjugated AMG 655 efficacy is FADD and caspase 8 dependent
Acknowledgements
Thanks Amgen for the AMG 655 necessary to do this work. Thanks to Serratore A and Muppavarapu B for their help with this work and thanks to Minx C for their technical assistance. Financial Support: This work was funded by the Medical Research Council UK (MRC grant MC_PC_15013, MRC grant 1598124) and through a US-Ireland R&D Partnership grant awarded by HSCNI (STL/5010/14). Support was provided by NIH/NCI grants R01CA198096, and R21CA234775 to RMS, and pilot seed funding from NIH/National Center for Advancing Translational Sciences grant UL1TR001412 to the University at Buffalo, State University of New York. Comprehensive Cancer Center Support grant NIH/NCI P30CA016056 to Roswell Park Comprehensive Cancer Center provided shared resources utilized in the work.
Footnotes
Conflicts of interest
Michael C. Johnston declares no potential conflicts of interest. Christopher Scott is a consultant to Fusion Antibodies Ltd. Daniel Longley owns stocks in Fusion Antibodies Ltd.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Micheau O, Shirley S, Dufour F. Death receptors as targets in cancer. Br J Pharmacol. 2013;169:1723–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ashkenazi A, Pai RC, Fong S, Leung S, Lawrence DA, Marsters SA, et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J Clin Invest [Internet]. American Society for Clinical Investigation; 1999. [cited 2019 Jun 4];104:155–62. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10411544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.CRUK. Pancreatic cancer statistics | Cancer Research UK [Internet]. 2017. [cited 2017 Oct 18]. Available from: http://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/pancreatic-cancer
- 4.Kindler HL, Richards DA, Garbo LE, Garon EB, Stephenson JJ, Rocha-Lima CM, et al. A randomized, placebo-controlled phase 2 study of ganitumab (AMG 479) or conatumumab (AMG 655) in combination with gemcitabine in patients with metastatic pancreatic cancer. Ann Oncol [Internet]. 2012. [cited 2017 Nov 2];23:2834–42. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22700995 [DOI] [PubMed] [Google Scholar]
- 5.Kaplan-Lefko PJ, Graves JD, Zoog SJ, Pan Y, Wall J, Branstetter DG, et al. Conatumumab, a fully human agonist antibody to death receptor 5, induces apoptosis via caspase activation in multiple tumor types. Cancer Biol Ther. 2010;9:618–31. [DOI] [PubMed] [Google Scholar]
- 6.Dubuisson A, Micheau O. Antibodies and Derivatives Targeting DR4 and DR5 for Cancer Therapy. Antibodies [Internet]. 2017;6:16 Available from: http://www.mdpi.com/2073-4468/6/4/16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li F, Ravetch J V. Apoptotic and antitumor activity of death receptor antibodies require inhibitory Fcγ receptor engagement. Proc Natl Acad Sci U S A [Internet]. National Academy of Sciences; 2012. [cited 2017 Nov 2];109:10966–71. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22723355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Von Karstedt S, Montinaro A, Walczak H. Exploring the TRAILs less travelled: TRAIL in cancer biology and therapy [Internet]. Nat. Rev. Cancer. Nature Publishing Group; 2017. [cited 2018 May 31]. page 352–66. Available from: http://www.nature.com/doifinder/10.1038/nrc.2017.28 [DOI] [PubMed] [Google Scholar]
- 9.Huet HA, Growney JD, Johnson JA, Li J, Bilic S, Ostrom L, et al. Multivalent nanobodies targeting death receptor 5 elicit superior tumor cell killing through efficient caspase induction. MAbs. 2014;6:1560–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walczak H, Miller RE, Ariail K, Gliniak B, Griffith TS, Kubin M, et al. Tumoricidal activity of tumor necrosis factor–related apoptosis–inducing ligand in vivo. Nat Med [Internet]. Nature Publishing Group; 1999. [cited 2019 Apr 4];5:157–63. Available from: http://www.nature.com/articles/nm0299_157 [DOI] [PubMed] [Google Scholar]
- 11.Ganten TM, Koschny R, Sykora J, Schulze-Bergkamen H, Büchler P, Haas TL, et al. Preclinical Differentiation between Apparently Safe and Potentially Hepatotoxic Applications of TRAIL Either Alone or in Combination with Chemotherapeutic Drugs. Clin Cancer Res [Internet]. American Association for Cancer Research; 2006. [cited 2019 Apr 4];12:2640–6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16638878 [DOI] [PubMed] [Google Scholar]
- 12.Gieffers C, Kluge M, Merz C, Sykora J, Thiemann M, Schaal R, et al. APG350 Induces Superior Clustering of TRAIL Receptors and Shows Therapeutic Antitumor Efficacy Independent of Cross-Linking via Fc Receptors. Mol Cancer Ther. 2013;12. [DOI] [PubMed] [Google Scholar]
- 13.Valldorf B, Fittler H, Deweid L, Ebenig A, Dickgiesser S, Sellmann C, et al. An Apoptosis-Inducing Peptidic Heptad That Efficiently Clusters Death Receptor 5. Angew Chemie Int Ed [Internet]. John Wiley & Sons, Ltd; 2016. [cited 2019 Mar 24];55:5085–9. Available from: http://doi.wiley.com/10.1002/anie.201511894 [DOI] [PubMed] [Google Scholar]
- 14.Schmid D, Fay F, Small DM, Jaworski J, Riley JS, Tegazzini D, et al. Efficient Drug Delivery and Induction of Apoptosis in Colorectal Tumors Using a Death Receptor 5-Targeted Nanomedicine. Mol Ther [Internet]. 2014;22:2083–92. Available from: http://linkinghub.elsevier.com/retrieve/pii/S1525001616302568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fay F, McLaughlin KM, Small DM, Fennell DA, Johnston PG, Longley DB, et al. Conatumumab (AMG 655) coated nanoparticles for targeted pro-apoptotic drug delivery. Biomaterials [Internet]. Elsevier; 2011. [cited 2019 May 29];32:8645–53. Available from: https://www.sciencedirect.com/science/article/pii/S0142961211008623?via%3Dihub [DOI] [PubMed] [Google Scholar]
- 16.De Miguel D, Basáñez G, Sánchez D, Malo PG, Marzo I, Larrad L, et al. Liposomes Decorated with Apo2L/TRAIL Overcome Chemoresistance of Human Hematologic Tumor Cells. Mol Pharm [Internet]. American Chemical Society; 2013. [cited 2019 Apr 5];10:893–904. Available from: http://pubs.acs.org/doi/10.1021/mp300258c [DOI] [PubMed] [Google Scholar]
- 17.De Miguel D, Gallego-Lleyda A, Ayuso JM, Pejenaute-Ochoa D, Jarauta V, Marzo I, et al. High-order TRAIL oligomer formation in TRAIL-coated lipid nanoparticles enhances DR5 cross-linking and increases antitumour effect against colon cancer. Cancer Lett [Internet]. Elsevier; 2016. [cited 2018 Feb 12];383:250–60. Available from: https://www.sciencedirect.com/science/article/pii/S0304383516306127?via%3Dihub [DOI] [PubMed] [Google Scholar]
- 18.Nair PM, Flores H, Gogineni A, Marsters S, Lawrence DA, Kelley RF, et al. Enhancing the antitumor efficacy of a cell-surface death ligand by covalent membrane display. Proc Natl Acad Sci U S A [Internet]. National Academy of Sciences; 2015. [cited 2019 Apr 5];112:5679–84. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25902490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Belkahla H, Haque A, Revzin A, Gharbi T, Alexandru Constantinescu A, Micheau O, et al. Coupling tumor necrosis factor-related apoptosis-inducing ligand to iron oxide nanoparticles increases its apoptotic activity on HCT116 and HepG2 malignant cells: effect of magnetic core size. J Interdiscip Nanomedicine [Internet]. 2019. [cited 2019 May 8];4. Available from: https://onlinelibrary.wiley.com/doi/pdf/10.1002/jin2.55 [Google Scholar]
- 20.Riley JS, Hutchinson R, McArt DG, Crawford N, Holohan C, Paul I, et al. Prognostic and therapeutic relevance of FLIP and procaspase-8 overexpression in non-small cell lung cancer. Cell Death Dis [Internet]. 2013. [cited 2018 Feb 1];4:e951. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24309938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Valente G, Manfroi F, Peracchio C, Nicotra G, Castino R, Nicosia G, et al. cFLIP expression correlates with tumour progression and patient outcome in non-Hodgkin lymphomas of low grade of malignancy. Br J Haematol [Internet]. John Wiley & Sons, Ltd (10.1111); 2006 [cited 2019 Jun 10];132:560–70. Available from: http://doi.wiley.com/10.1111/j.1365-2141.2005.05898.x [DOI] [PubMed] [Google Scholar]
- 22.Ullenhag GJ, Mukherjee A, Watson NFS, Al-Attar AH, Scholefield JH, Durrant LG. Overexpression of FLIPL Is an Independent Marker of Poor Prognosis in Colorectal Cancer Patients. Clin Cancer Res [Internet]. 2007. [cited 2019 Oct 15];13:5070–5. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17785559 [DOI] [PubMed] [Google Scholar]
- 23.Ryu B-K, Lee M-G, Chi S-G, Kim Y-W, Park J-H. Increased expression of cFLIPL in colonic adenocarcinoma. J Pathol [Internet]. 2001. [cited 2019 Oct 15];194:15–9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11329136 [DOI] [PubMed] [Google Scholar]
- 24.Haag C, Stadel D, Zhou S, Bachem MG, Moller P, Debatin K-M, et al. Identification of c-FLIPL and c-FLIPS as critical regulators of death receptor-induced apoptosis in pancreatic cancer cells. Gut [Internet]. 2011. [cited 2018 Feb 1];60:225–37. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20876774 [DOI] [PubMed] [Google Scholar]
- 25.Zhou XD, Yu JP, Liu J, Luo HS, Chen HX, Yu HG. Overexpression of cellular FLICE-inhibitory protein (FLIP) in gastric adenocarcinoma. Clin Sci [Internet]. 2004. [cited 2019 Oct 15];106:397–405. Available from: http://www.ncbi.nlm.nih.gov/pubmed/14636156 [DOI] [PubMed] [Google Scholar]
- 26.Lee SH, Kim HS, Kim SY, Lee YS, Park WS, Kim SH, et al. Increased expression of FLIP, an inhibitor of Fas-mediated apoptosis, in stomach cancer. APMIS [Internet]. 2003. [cited 2019 Oct 15];111:309–14. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12716387 [DOI] [PubMed] [Google Scholar]
- 27.McCourt C, Maxwell P, Mazzucchelli R, Montironi R, Scarpelli M, Salto-Tellez M, et al. Elevation of c-FLIP in Castrate-Resistant Prostate Cancer Antagonizes Therapeutic Response to Androgen Receptor-Targeted Therapy. Clin Cancer Res [Internet]. 2012. [cited 2019 Oct 15];18:3822–33. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22623731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bagnoli M, Ambrogi F, Pilotti S, Alberti P, Ditto A, Barbareschi M, et al. c-FLIPL expression defines two ovarian cancer patient subsets and is a prognostic factor of adverse outcome. Endocr Relat Cancer [Internet]. 2009. [cited 2019 Oct 15];16:443–53. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19321593 [DOI] [PubMed] [Google Scholar]
- 29.Humphreys L, Espona-Fiedler M, Longley DB. FLIP as a therapeutic target in cancer. FEBS J [Internet]. Wiley/Blackwell (10.1111); 2018. [cited 2018 Jun 1]; Available from: http://doi.wiley.com/10.1111/febs.14523 [DOI] [PubMed] [Google Scholar]
- 30.Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res [Internet]. Narnia; 2017. [cited 2019 Aug 5];45:W98–102. Available from: https://academic.oup.com/nar/article-lookup/doi/10.1093/nar/gkx247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ndozangue-Touriguine O, Sebbagh M, Mérino D, Micheau O, Bertoglio J, Bréard J. A mitochondrial block and expression of XIAP lead to resistance to TRAIL-induced apoptosis during progression to metastasis of a colon carcinoma. Oncogene. 2008;27:6012–22. [DOI] [PubMed] [Google Scholar]
- 32.Humphreys LM, Fox JP, Higgins CA, Majkut J, Sessler T, McLaughlin K, et al. A revised model of TRAIL ‐R2 DISC assembly explains how FLIP (L) can inhibit or promote apoptosis. EMBO Rep. 2020;21:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mcdaid WJ, Greene MK, Johnston MC, Pollheimer E, Smyth P, Mclaughlin K, et al. Repurposing of Cetuximab in antibody-directed chemotherapy-loaded nanoparticles in EGFR therapy-resistant pancreatic tumours †. Nanoscale. 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schmid D, Jarvis GE, Fay F, Small DM, Greene MK, Majkut J, et al. Nanoencapsulation of ABT-737 and camptothecin enhances their clinical potential through synergistic antitumor effects and reduction of systemic toxicity. Cell Death Dis [Internet]. Nature Publishing Group; 2014;5:e1454. Available from: http://www.nature.com/doifinder/10.1038/cddis.2014.413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Elmore S. Apoptosis: A Review of Programmed Cell Death. Toxicol Pathol. 2007;35:495–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Siegmund D, Lang I, Wajant H. Cell death-independent activities of the death receptors CD95, TRAILR1, and TRAILR2 [Internet]. FEBS J. 2017. [cited 2019 Nov 22]. page 1131–59. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1111/febs.13968 [DOI] [PubMed] [Google Scholar]
- 37.Trauzold A, Siegmund D, Schniewind B, Sipos B, Egberts J, Zorenkov D, et al. TRAIL promotes metastasis of human pancreatic ductal adenocarcinoma. Oncogene [Internet]. 2006. [cited 2019 Oct 31];25:7434–9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16751802 [DOI] [PubMed] [Google Scholar]
- 38.von Karstedt S, Conti A, Nobis M, Montinaro A, Hartwig T, Lemke J, et al. Cancer cell-autonomous TRAIL-R signaling promotes KRAS-Driven cancer progression, invasion, and metastasis. Cancer Cell. 2015;27:561–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mi P, Cabral H, Kataoka K. Ligand‐Installed Nanocarriers toward Precision Therapy. Adv Mater. 2019;1902604. [DOI] [PubMed] [Google Scholar]
- 40.Wilhelm S, Tavares AJ, Dai Q, Ohta S, Audet J, Dvorak HF, et al. Analysis of nanoparticle delivery to tumours. Nat Rev Mater [Internet]. 2016;1:16014 Available from: http://www.nature.com/articles/natrevmats201614 [Google Scholar]
- 41.Fay F, Scott CJ. Antibody-targeted nanoparticles for cancer therapy. Immunotherapy. 2011;3:381–94. [DOI] [PubMed] [Google Scholar]
- 42.Johnston MC, Scott CJ. Antibody conjugated nanoparticles as a novel form of antibody drug conjugate chemotherapy. Drug Discov Today Technol. 2018;30. [DOI] [PubMed] [Google Scholar]
- 43.Saif MW. U.S. food and drug administration approves paclitaxel protein-bound particles (Abraxane®) in combination with gemcitabine as first-line treatment of patients with metastatic pancreatic cancer [Internet]. J. Pancreas. E.S. Burioni ricerche bibliografiche; 2013. [cited 2018 Jan 29]. page 686–8. Available from: http://www.serena.unina.it/index.php/jop/article/view/2028/2014 [DOI] [PubMed] [Google Scholar]
- 44.CHMP. Onivyde, INN-Irinotecan (pegylated liposomal formulation). 2016. [cited 2018 Feb 12]; Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Summary_of_opinion_-_Initial_authorisation/human/004125/WC500210816.pdf
- 45.Chou TC. Drug combination studies and their synergy quantification using the chou-talalay method [Internet]. Cancer Res. 2010. [cited 2019 Aug 1]. page 440–6. Available from: www.aacrjournals.org [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.