

Abstract
The bisubstrate analog strategy is a promising approach to develop potent and selective inhibitors for protein methyltransferases. Herein, the interactions of a series of bisubstrate analogs with protein N-terminal methyltransferase 1 (NTMT1) were examined to probe the molecular properties of the NTMT1 active site. Our results indicate that a 2-C to 4-C atom linker enables its respective bisubstrate analog to occupy both substrate and cofactor binding sites of NTMT1, but the bisubstrate analog with a 5-C atom linker only interacts with the substrate binding site and functions as a substrate. Furthermore, the 4-C atom linker is the optimal and produces the most potent inhibitor (Ki, app = 130 ± 40 pM) for NTMT1 to date, displaying over 3,000-fold selectivity over other methyltransferases and even to its homolog NTMT2. This study reveals the molecular basis for the plasticity of the NTMT1 active site. Additionally, our study outlines a general guidance on the development of bisubstrate inhibitors for any methyltransferases.
Graphical Abstract
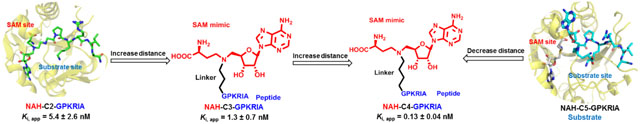
Introduction
Protein methylation, a widespread epigenetic modification that is catalyzed by protein methyltransferases (PMTs), plays critical roles in many cellular processes such as gene expression, transcriptional regulation, DNA replication and damage repair. Dysregulation of PMT expression has been implicated in a variety of diseases including cancer, cardiovascular, neurodegenerative, and other diseases.1–5 Therefore, there have been emerging interests in development of selective inhibitors for PMTs as potential therapeutic agents. Recently, several inhibitors of PMTs have been developed and entered human clinical trials, such as DOT1-like protein inhibitor EPZ-5676 (Phase I/II), protein arginine methyltransferase 5 inhibitor GSK3326595 (Phase I/II), enhancer of zeste homolog 2 inhibitors EPZ-6438 (Phase I/II) and CPI-1205 (Phase I/II).2
PMTs utilize the cofactor S-adenosyl-L-methionine (SAM) as a methyl donor to transfer the methyl group from SAM onto the substrate with concomitant generation of S-adenosine-L-homocysteine (SAH) and methylated substrate.2 The PMT-catalyzed methylation reaction typically undergoes a Bi-Bi mechanism and involves the formation of a ternary complex.6 Bisubstrate analogs that occupy both substrate and cofactor binding sites have the potential to achieve higher binding affinities than a substrate analog.7–10 Furthermore, bisubstrate analogs may offer superior selectivity for their targeted enzyme.9,11,12 On account of a shared SAM cofactor binding pocket across PMT families, it is rational to hypothesize that bisubstrate analogs can be a general approach to offer potent and specific inhibitors for each individual PMT. Recent progresses have been made in the development of bisubstrate analogs for a few PMTs, such as protein N-terminal methyltransferase 1 (NTMT1)11,13,14, coactivator-associated arginine methyltransferase 1 (CARM1)15, protein arginine methyltransferase 1 (PRMT1)16, and PRMT617. These bisubstrate analogs were created by connecting a SAM analog to a substrate or a functional group of the corresponding substrate. However, the potency of those bisubstrate analogs for their respective targets varied from nanomolar to micromolar. This variable potency imposed the need of explicit instruction on design of bisubstrate analogs for PMTs.
The aforementioned need has prompted us to investigate how the linker length would affect the interaction of bisubstrate analog to its target PMT. Here, we used NTMT1 as a model system to examine the optimal length of the linker, with the intention to provide a general guide to facilitate the design of optimal bisubstrate analogs for other PMTs. NTMT1 recognizes a unique X-P-K/R motif at the protein alpha-N-terminus.18–20 It methylates centromere-specific histone H3 variants centromere protein A/B, regulator of chromatin condensation 1, damaged DNA-binding protein 2 and poly(ADP-ribose) polymerase 3.21–26 Hence, NTMT1 has been implicated in regulating chromosome segregation, mitosis, and DNA damage repair.22,24,27 We have previously developed bisubstrate analogs for NTMT1. Among them, compound NAH-C3-PPKRIA showed an IC50 value of 35 ± 2 nM in a MALDI-MS based assay, and displayed more than 100-fold selectivity against other methyltransferases (Figure 1A).11 In addition, compound NAH-C3-GPRRRS showed an IC50 value of 0.94 ± 0.16 μM for NTMT1.14 Both compounds have a propylene linker, which raises the question if a propylene linker is optimal. To address this question, we design and synthesize a series of NTMT1 bisubstrate analogs with variable linker lengths to probe the active site of NTMT1.
Figure 1.
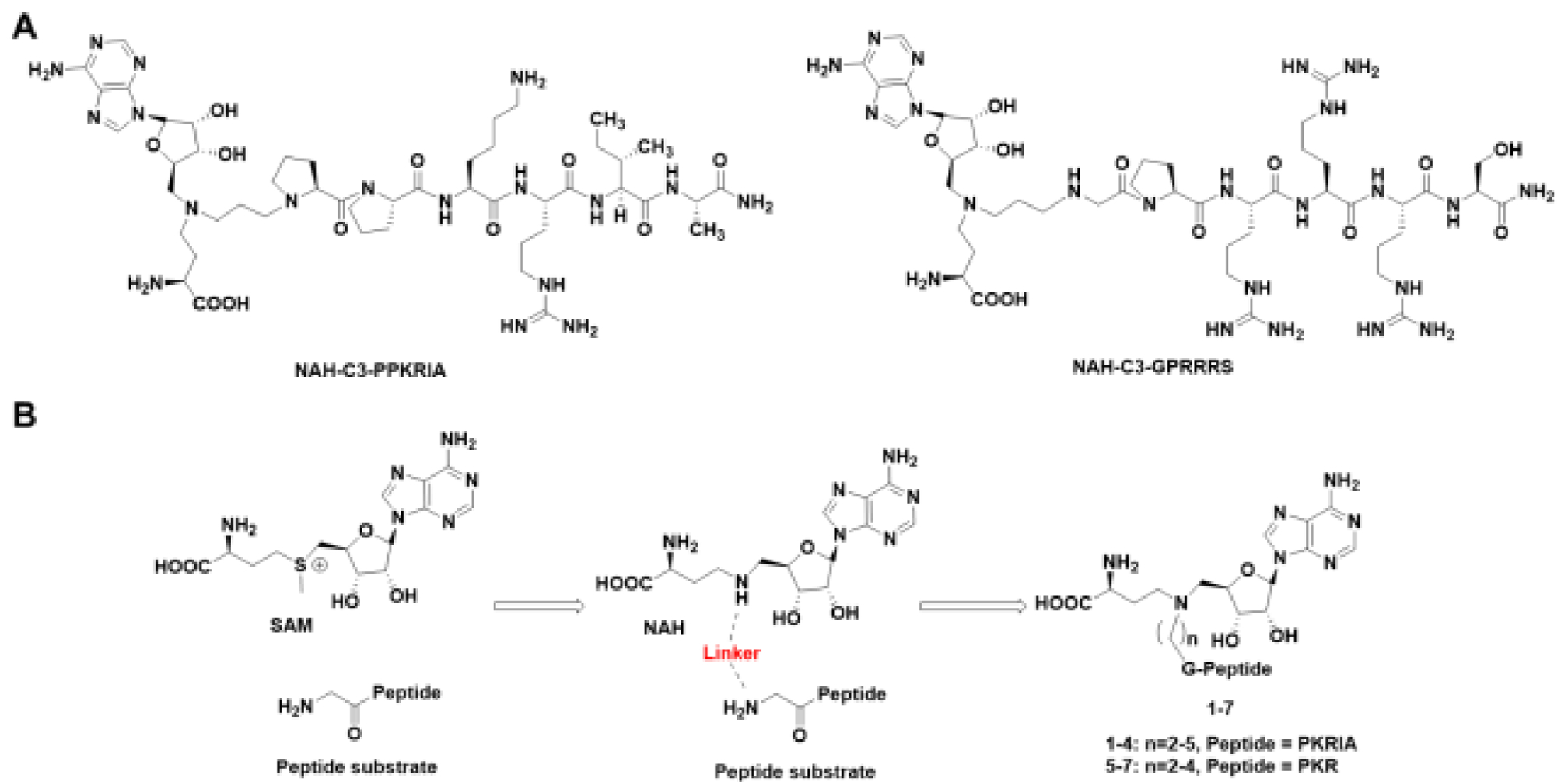
(A) Chemical structures of NTMT1 inhibitors. (B) Design of the bisubstrate analogs for NTMT1.
RESULTS AND DISCUSSION
Design and Synthesis.
The linear distance between the sulfur atom of SAH and the nitrogen atom of substrate ranges 4.6 Å to 5.5 Å in the ternary structures of NTMT1.18,19 A propyl linker was able to offer high potent inhibitor for NTMT1.11 Herein, we proposed to vary the linker from 2 to 5 methylene groups to explore the optimal length to build bisubstrate analogs 1-7 (Figure 1B, Scheme 1). In addition, the peptide sequence was also altered to ensure that the effect of different linkers was not sequence-specific. We chose the peptide starting with Gly due to its synthetic versatility. Meanwhile, compounds 8-12 were designed to investigate the contribution of each part of the bisubstrate analog to the inhibitory activity. The synthesis of compounds 1-7, and 10–12 were accomplished in a convergent manner by reacting the primary amines (16–19, and 24–26) with α-bromo peptide on resin and followed by acid treatment (Schemes 1–2).14 Compounds 8 and 9 were obtained by acidic deprotection of 14 and 18, respectively (Scheme 1). The primary amines 18 and 19 were prepared through reductive amination of a common starting material 14 with respective aldehydes followed by removal of the protection group (Scheme 1).28,29 Commercially available amine 20 was subjected to reductive amination with the aldehyde 21 to provide compound 22. The primary amine 24 was obtained after removal of Fmoc group of compound 23 (Scheme 2).
Scheme 1. Synthetic route for compounds 1–9.
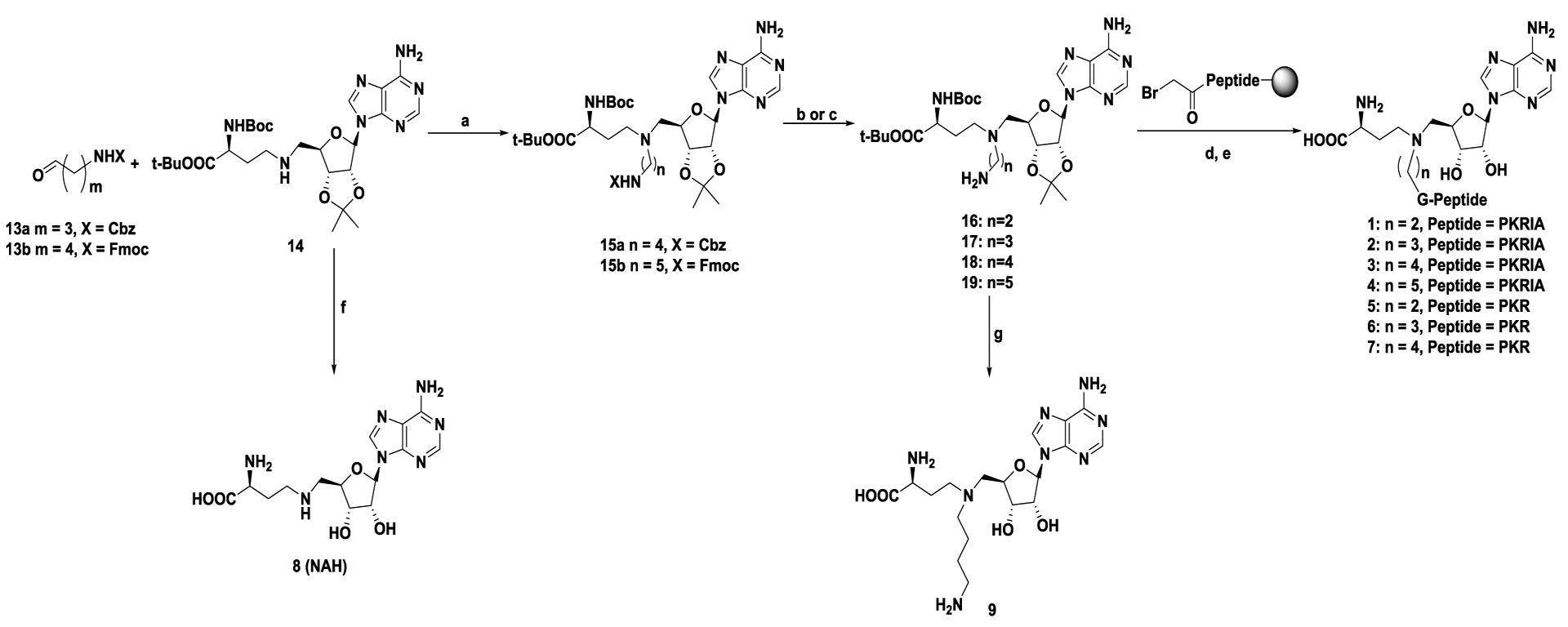
Reagents and conditions: a) NaBH3CN, CH3OH, r.t., 38–68%; b) 10% Pd/C, H2, MeOH, 56%; c) DBU, CH3CN, 0 °C to r.t., 71%; d) α-bromo peptides, DIPEA, DMF, r.t.; e) TFA: DODT: H2O: TIPS = 94: 2.5: 2.5: 1; f) TFA, CH2Cl2, 0 ℃ to r.t., 42%; g) 4N HCl in dioxane, H2O, 0 °C to r.t., 45%.
Scheme 2. Synthetic route for compound 10–12a.
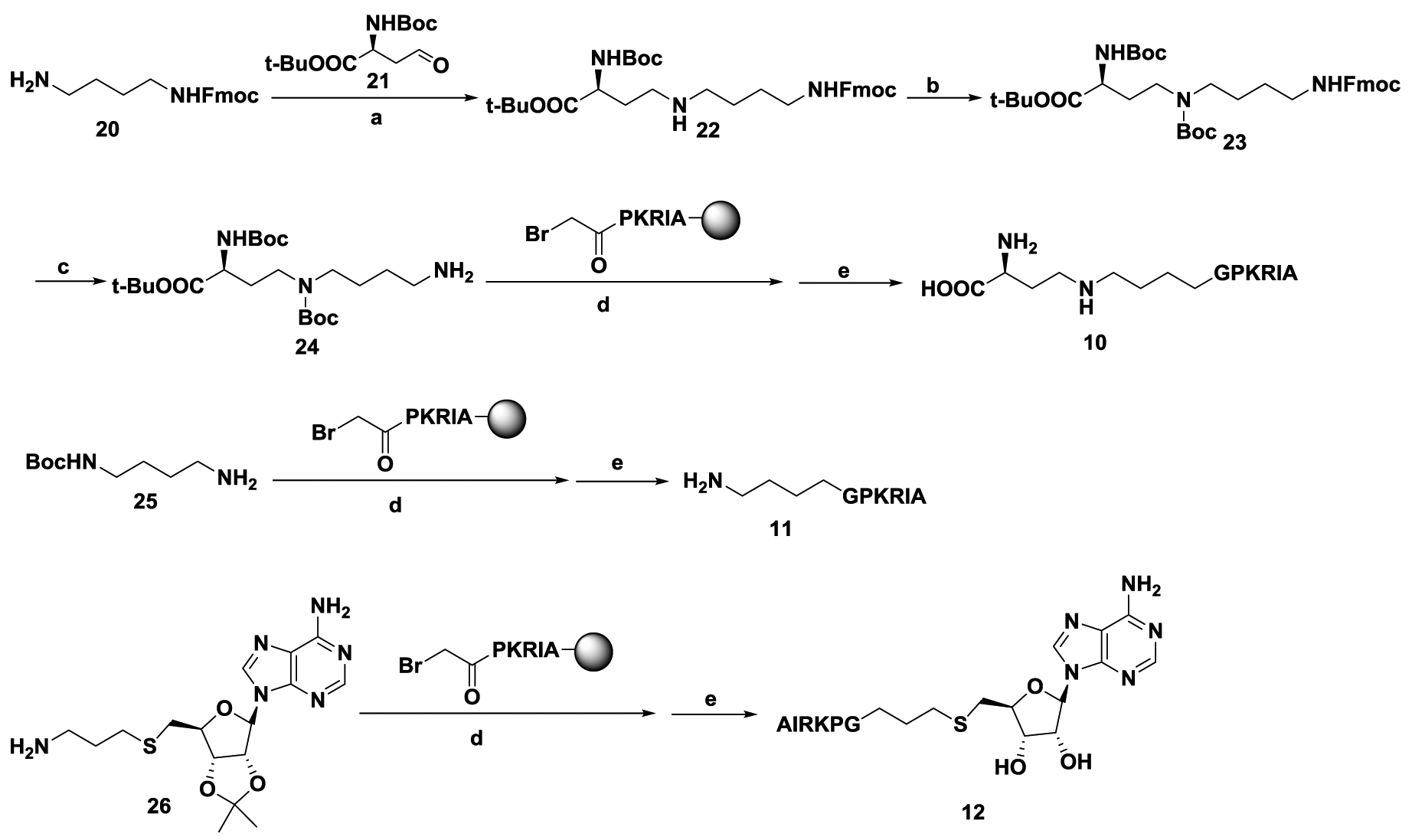
Reagents and conditions: a) NaBH3CN, CH3OH, r.t., 71%; b) (Boc)2O, NaHCO3, THF, H2O, 85%; c) DBU, CH3CN, 0 °C to r.t., 50%; d) α-bromo peptides, DIPEA, DMF, r.t.; e) TFA: DODT: H2O: TIPS = 94: 2.5: 2.5: 1.
MALDI-MS methylation assay.
Compounds 1-4 were first subject to methylation assay catalyzed by NTMT1 in the presence of SAM to first check if they may act as possible substrates. Surprisingly, compound 4 containing the 5-C atom linker was able to be methylated by NTMT1 (Figure 2), indicating 4 as a substrate for NTMT1.
Figure 2.
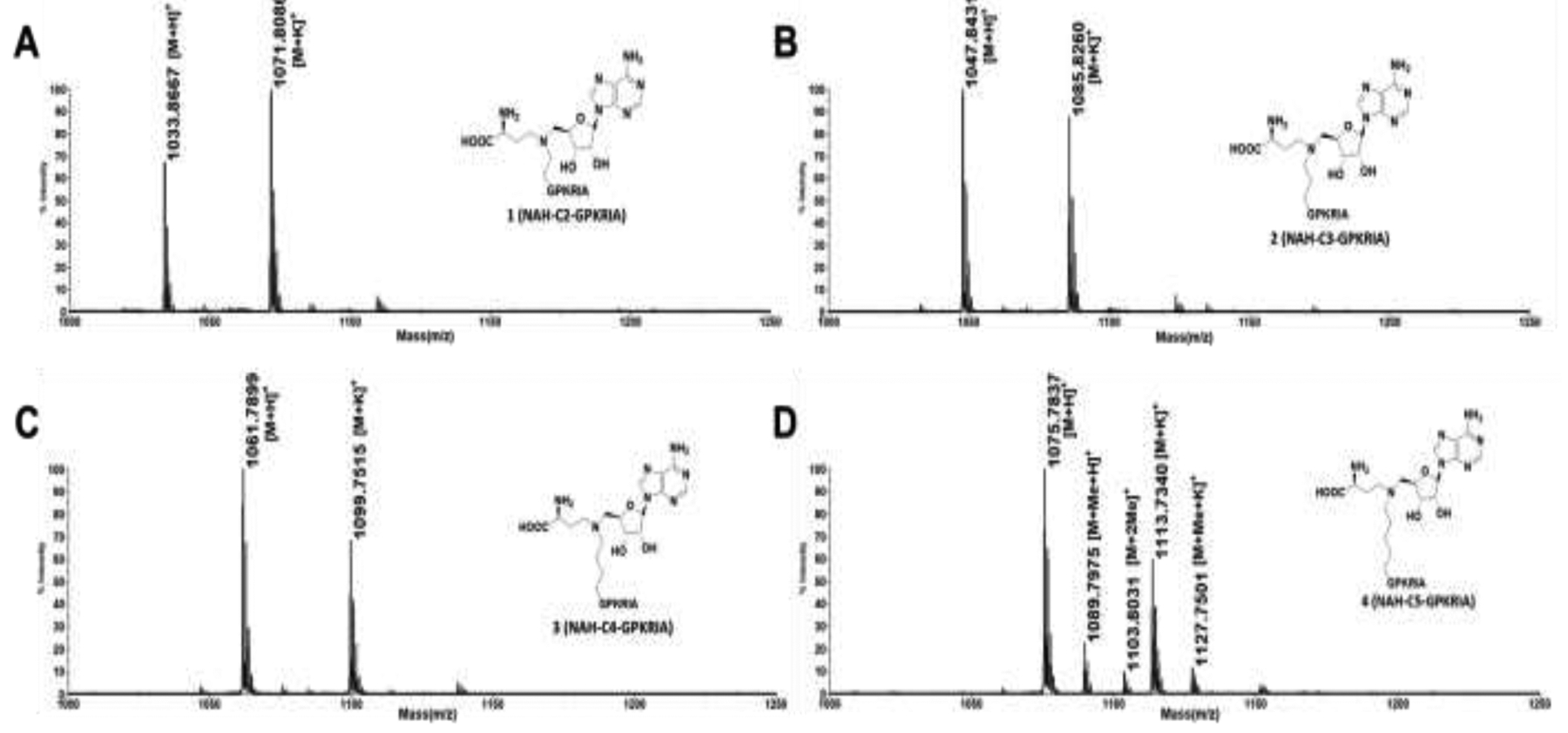
MALDI-MS methylation assay for the bisubstrate analogs 1-4.
Inhibition Evaluation.
The inhibitory activities of compounds 1-3 and 5–7 were evaluated in the SAH hydrolase (SAHH)-coupled fluorescence assay with 10 minutes of preincubation time.30 Both SAM and the peptide substrate GPKRIA were at their respective Km values. In order to compare the inhibitory activity under the same condition, we resynthesized and purified 1, 2 and 6, respectively. Their IC50 values were lower than previous values14, which may be caused by an improved S/N ratio with a different GPKRIA peptide substrate and fluorescent dye ThioGlo4. Among the NAH-GPKRIA series, compounds 3 (IC50 = 82 ± 17 nM) containing a 4-C atom length displayed the best inhibition for NTMT1 (Table 1). When the linker length decreased from a 4-C atom to a 3-C and 2-C, compound 2 (IC50 = 130 ± 40 nM) and 1 (IC50 = 260 ± 70 nM) exhibited a 1.5-fold and 3-fold decrease compared to 3, respectively. Therefore, a 4-C atom length is optimal for the NAM-GPKRIA series. To further validate the optimal linker length, we synthesized three additional bisubstrate analogs that contained a tetrapeptide instead of hexapeptide in the substrate portion. For NAH-GPKR bisubstrate analogs, compounds 5 and 6 showed less than a 2-fold activity decrease compared to their respective NAH-GPKRIA compounds 1 and 2, while compound 7 displayed comparable inhibition with compound 3 (Table 1). Furthermore, the 4-C atom linker is also the best inhibitor as 7 (IC50 = 84 ± 12 nM) showed 2-fold and 4-fold better activity than 6 and 5, respectively. Therefore, the 4-C atom linker is the most favorable length for NTMT1 bisubstrate analogs in both the NAH-GPKRIA and NAH-GPKR series.
Table 1.
IC50 values of bisubstrate compounds
| Compound ID | IC50 (nM)a |
|---|---|
| 1 (NAH-C2-GPKRIA) | 260±70 |
| 2 (NAH-C3-GPKRIA) | 130±40 |
| 3 (NAH-C4-GPKRIA) | 82±17 |
| 4 (NAH-C5-GPKRIA) | N.A. |
| 5 (NAH-C2-GPKR) | 320±80 |
| 6 (NAH-C3-GPKR) | 200±30 |
| 7 (NAH-C4-GPKR) | 84±12 |
triplicates (n = 3). N.A.: not applied.
The bisubstrate analogs contained three parts: a SAM analog 8 (NAH), linker, and peptide substrate. To understand the contribution of each component of the bisubstrate analog to the inhibitory activity for NTMT1, a deconstruction strategy was applied to design and synthesize only partial structures of the bisubstrate analog (Figure 3): 1) compound 8 contained only the NAH part; 2) compound 9 with only the NAH and linker; 3) compound 10 without the adenine portion; 4) compound 11 without the NAH part; 5) compound 12 without the aspartic acid moiety of the SAH. Compounds 10-12 were determined to be substrates for NTMT1 (Figure S1). Compound 9 displayed an IC50 value higher than 10 μM in the SAHH-coupling assay. Since 8 is a potent inhibitor (IC50 = 23 ± 2 nM) for SAHH, but did not exhibit significant inhibition against NTMT1 (IC50 > 10 μM) through an orthogonal MALDI-MS inhibition assay (Figure S2). These results suggested that each part of the bisubstrate inhibitor was critical for its inhibitory actvity for NTMT1.
Figure 3.
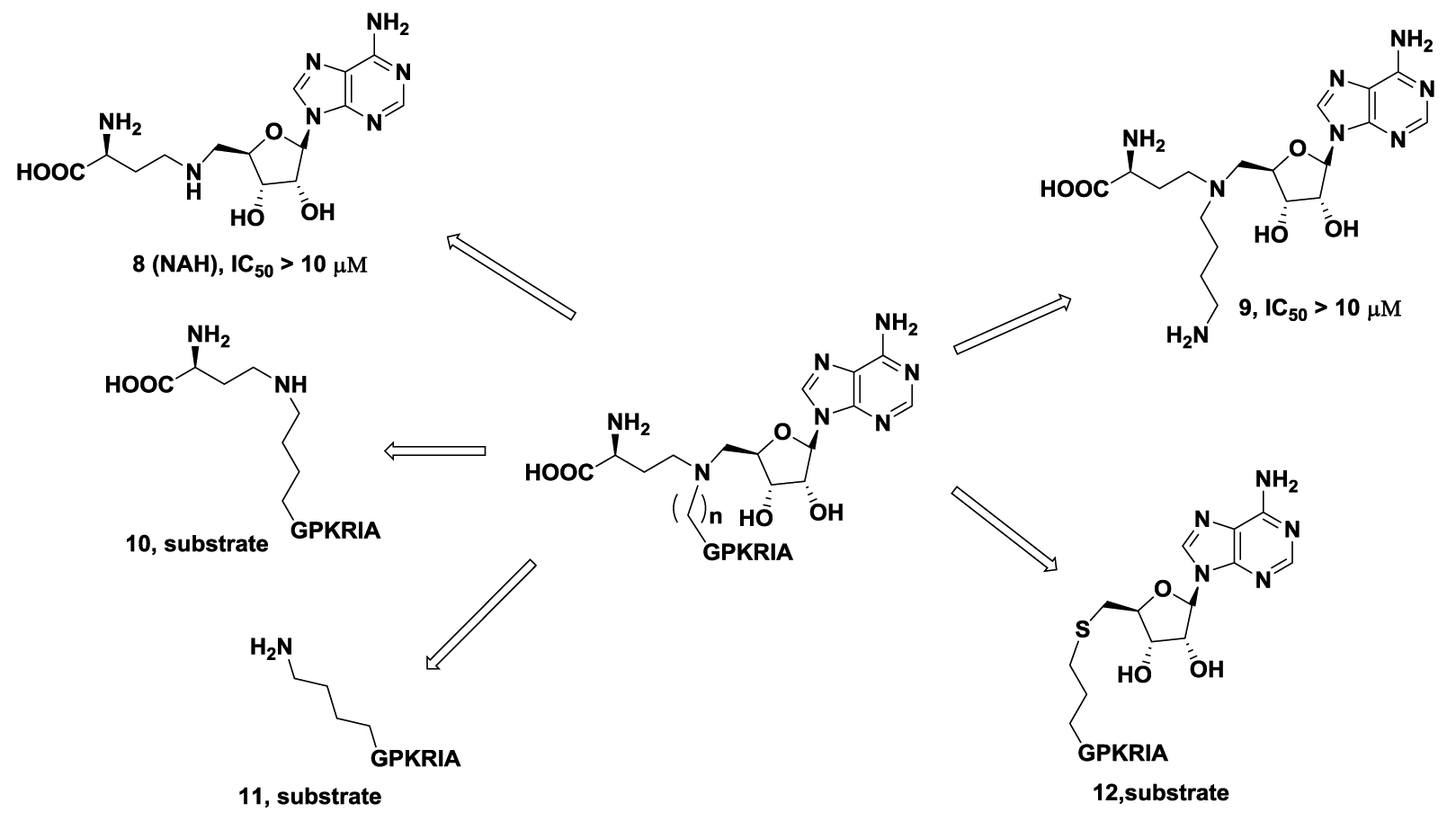
Deconstruction strategy and inhibition evaluation of the compounds. The IC50 value of 8 was determined by MALDI-MS methylation inhibition assay while 9 was determined by the SAHH-coupling assay.
Time-dependent Inhibition Study.
The measured IC50 values for compounds 2, 3 and 7 converged to the enzyme concentration (100 nM) present in the assay (Table 1), suggesting tight-binding inhibition. Therefore, Morrison’s equation is a better method to analyze the concentration-response data for these tight binding inhibitors to define their apparent Ki values.31,32 The tight-binding inhibitors often exhibit slow binding traits, so compounds 1-3 were chosen to investigate the effects of preincubation time on the inhibitory activity of tight-binding inhibitors (Figure 4).31,32 The Ki, app values of the three compounds were inversely proportional to the pre-incubation time (Figure 4B, D and F), indicating their slow binding feature. The results showed the interaction between the compounds and NTMT1 reached equilibrium after 30 min of incubation. Compound 3 containing a 4-C atom linker displayed the lowest Ki, app value (130 ± 40 pM) (Figure 4B) after 120 min, further substantiating its slow and tight binding feature ([E]/Ki, app > 100).31,32 When the linker length decreased from a 4-C atom to a 3-C and 2-C, compound 2 (Ki, app = 1.3 ± 0.6 nM) and 1 (Ki, app = 5.4 ± 2.6 nM) exhibited a 10-fold and 40-fold decrease in comparison with 3, respectively. The above results infer decent plasticity in the transition state of NTMT1 to accommodate a linker length ranging from 2-C to 4-C atom. Among them, a 4-C atom linker is the optimal length to achieve the best inhibition. Notably, a 5-C atom linker would convert a tight-binding bisubstrate analog to a substrate for NTMT1 (Figure 2), indicting an upper limit for the linker length to achieve potent inhibition.
Figure 4.
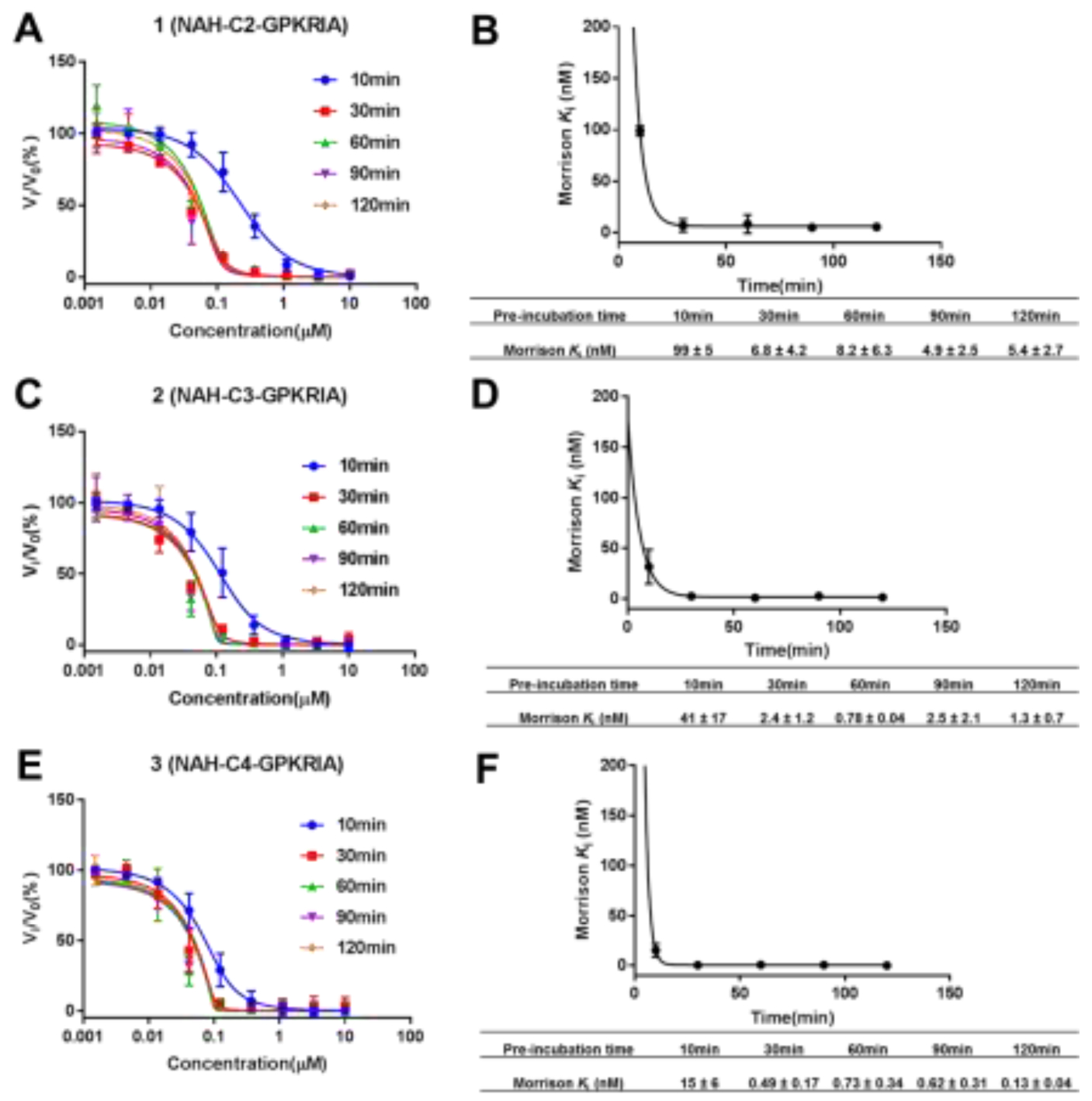
Time-dependent inhibition study of compounds 1–3. All experiments were run with duplicate (n=2), except 10 min of pre-incubation (n=3). (A), (C) and (E) Concentration–response plots for different pre-incubation times fitted to Morrison’s quadratic equation for compounds 1, 2 and 3, respectively ; (B), (D) and (F) Nonlinear regression of Morrison Ki, app values with pre-incubation time for compounds 1, 2 and 3, respectively.
ITC.
NTMT2 is a closely-related homolog of NTMT1, which shares over 50% sequence similarity and the same X-P-K recognition motif.33 To assess the interactions of the bisubstrate anlogs with both enzymes, the binding affinities of 1-4 for both NTMT1 and NTMT2 were determined by isothermal titration calorimetry (ITC) experiment (Figure 5). All four compounds bound to NTMT1, especially compound 3 (Kd1/d2 = 0.31 μM/2.47 nM) showing the tightest binding affinity. Notably, the Kd values of bisubstrate analogs were higher than their Ki, app values except compound 4 (Kd1/d2 = 1.6/0.48 μM). This difference may be caused by endogenous amounts of SAM/SAH binding to recombinant NTMTs and slow binding characteristic of tight-binding inhibitors 1-3.18,33 However, the ITC binding data was still informative, since the bisubstrate analogs at 1 mM showed negligible interactions with NTMT2.33 This delicate selectivity further corroborates the benefits of bisubstrate analogs on account of their high specificity.
Figure 5.
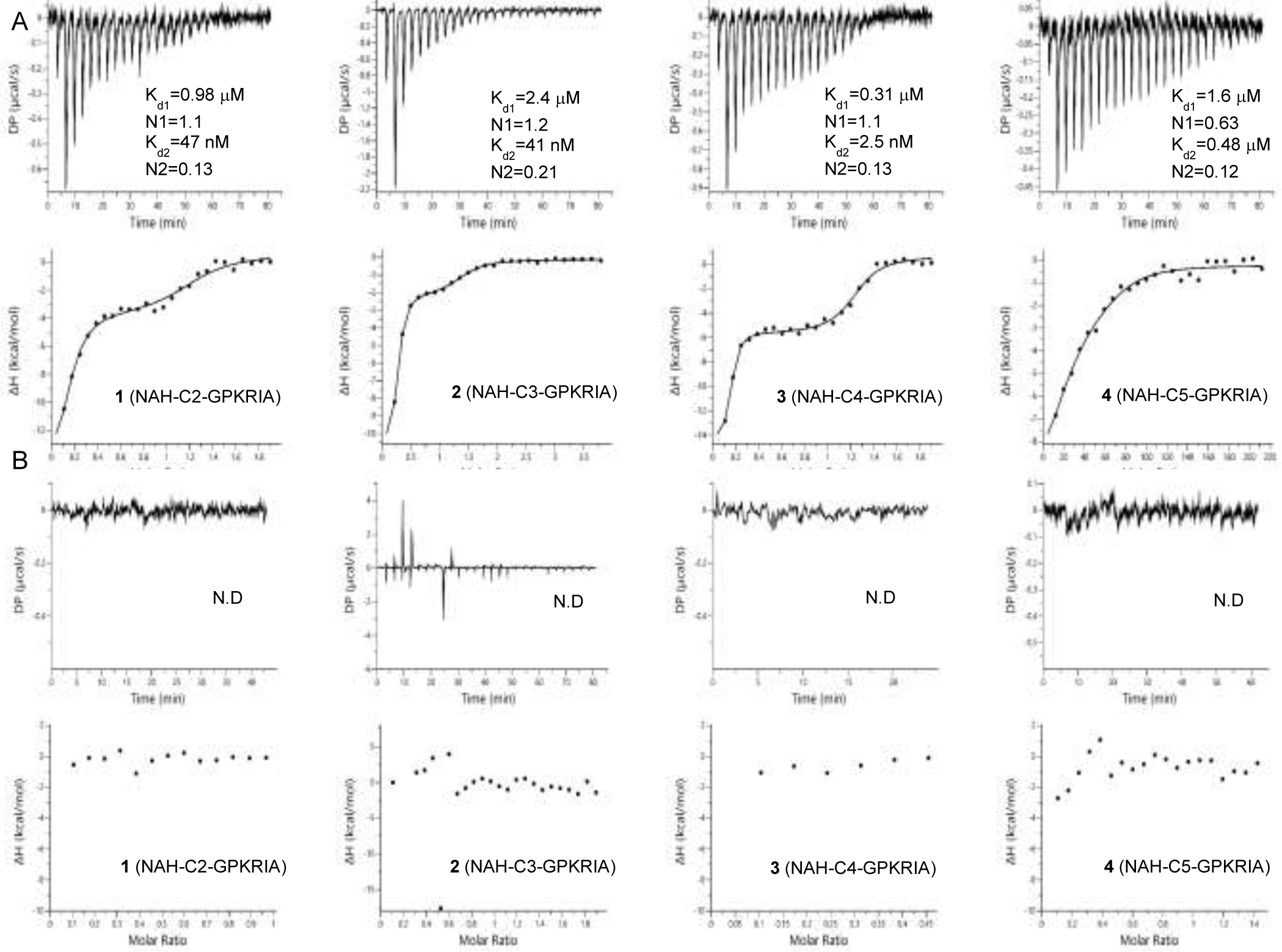
Binding affinity for NTMT1 (A) and NTMT2 (B). N.D.: not detected.
MALDI-MS Methylation Inhibition Assay.
To further confirm the inhibitory activity of bisubstrate inhibitor, the MALDI-MS methylation assay was performed as an orthogonal assay to monitor the production of methylated peptides with both substrate peptide GPKRIA and SAM at their Km values after 10 min of preincubation time (Figure 6A–B). At 50 nM of compound 3, tri-methylation of GPKRIA peptide was substantially reduced. At 200 nM of compound 3, tri-methylation of GPKRIA peptide was barely detected (Figure 6B).
Figure 6.
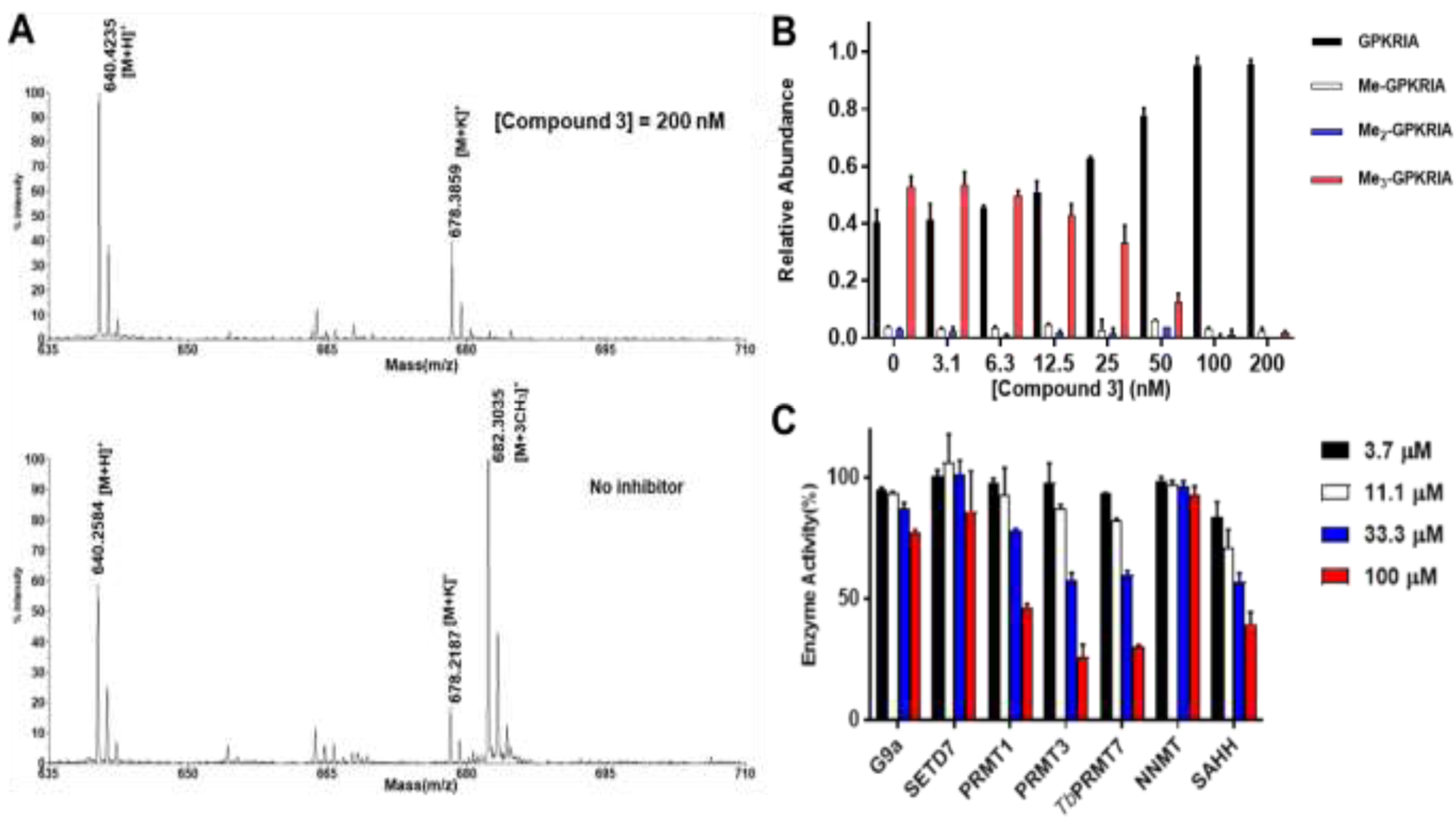
(A) and (B) MALDI-MS methylation inhibition assay compound 3 (n=2). (A) MALDI-MS results of MALDI-MS methylation inhibition assay for compound 3; (B) Quantification of methylation progression of GPKRIA by NTMT1 with 3 at 20 min. (C) Selectivity evaluation of compound 3 (n=2).
Selectivity Assessment.
The selectivity of 3 for NTMT1 was further examined against a panel of methyltransferases that shared a SAM binding pocket, which also included the coupling enzyme SAHH that has a SAH binding site and was used in the fluorescence assay (Figure 6C,table s2). The compound barely showed inhibitory activity over euchromatic histone-lysine N-methyltransferase 2 (G9a), SET domain-containing protein 7 (SETD7) and nicotinamide N-methyltransferase (NNMT) at 100 μM. At 33 μM, 3 demonstrated less than 50% inhibition over protein arginine methyltransferase 1 (PRMT1), protein arginine methyltransferase 3 (PRMT3), Trypanosoma brucei protein arginine methyltransferase 7 (TbPRMT7) and SAHH. Therefore, compound 3 showed more than 1000-fold selectivity over other methyltransferases.
Co-crystal Structures of Compounds 1 and 4 with NTMT1.
To understand the molecular basis of the effect of different linkers, we obtained the co-crystal structures of bisubstrate analogs 1 and 4 with NTMT1 (Figure 7). The binary complex X-ray crystal structure of NTMT1-1 complex (PDB: 6WJ7) illuminates the inhibition mechanism since compound 1 does occupy both cofactor and substrate binding sites of NTMT1. It also overlays well with previously reported bisubstrate inhibitor NAH-C3-PPKRIA in the X-ray crystal structure of NTMT1-NAH-C3-PPKRIA (PDB code: 6DTN) (Figure 7B). The SAM analog moiety of 1 in the binary complex binds almost same as NAH-C3-PPKRIA, which is nearly identical with SAH in the ternary complex of substrate peptide/SAH with NTMT1. When increasing the carbon linker to 5-C atom linker, the peptide part of the compound 4 retained its interaction with the substrate binding pocket and overlays very well with the PPKRIA peptide portion of NAH-C3-PPKRIA (Figure 7D–F). However, the NAH moiety of the compound 4 no longer occupied the SAM binding pocket. Interestingly, the SAM analog part inserted to a new pocket adjacent to the substrate binding pocket, which resulted in exposure of the secondary amine (α-N-terminal amine of the peptide portion) of compound 4 to receive the methyl group from SAM. Nevertheless, it infers the length of 5-C atom linker is too long to allow the bisubstrate analog 4 to simultaneously occupy both SAM and peptide binding sites.
Figure 7.
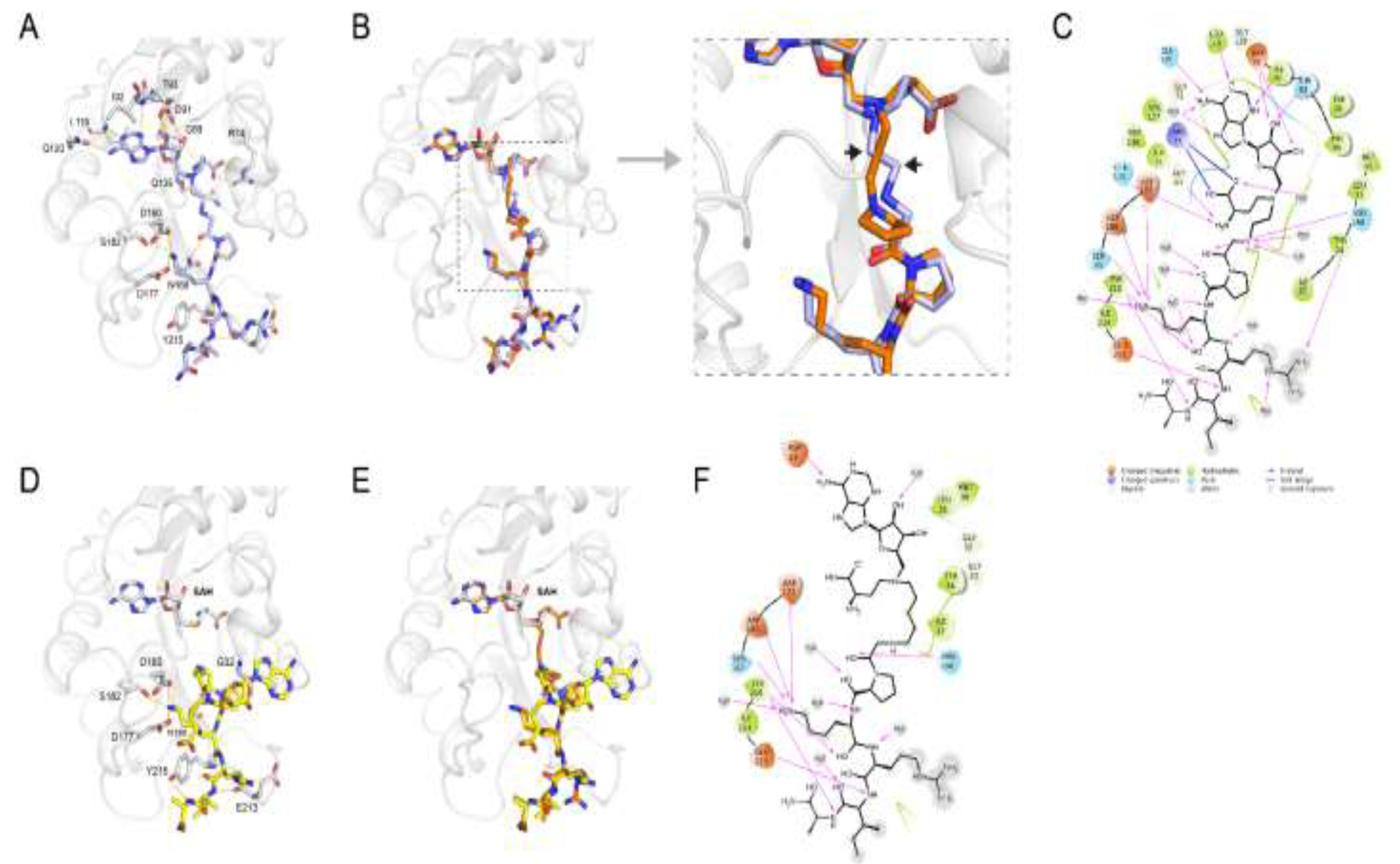
X-ray co-crystal structures. (A) The X-ray crystal structure of compound 1 (purple stick) in complex with NTMT1 (gray ribbon) (PDB code: 6WJ7). H-bond interactions are shown in yellow dotted lines; (B) Overlay of the crystal structure of NTMT1(gray ribbon)-1 (purple stick) complex with the crystal structure of the NTMT1-NAH-C3-PPKRIA (orange stick) binary complex (PDB code: 6DTN); (C) Compound 1 2D interaction diagram (Schrödinger Maestro) with NTMT1; (D) The X-ray crystal structure of NTMT1(gray ribbon)-4 (yellow stick)-SAH (gray stick) (PDB code: 6PVB). H-bond interactions are shown in yellow dotted lines; (E) Overlay of the crystal structure of NTMT1(gray ribbon)-4 (yellow stick)-SAH (gray stick) complex (PDB code: 6PVB) with the crystal structure of NTMT1-NAH-C3-PPKRIA (orange stick) binary complex (PDB code: 6DTN); (F) Compound 4 2D interaction diagram (Schrödinger Maestro) with NTMT1.
Inhibition Mechanism Studies.
Despite additional efforts to screen and optimize the conditions, co-crystal structure of NTMT1 in complex with 2 or 3 was not obtained. Hence, Inhibition mechanism studies were performed to confirm the binding modes for 4-C atom linker compound. Since the co-crystal structures of NAH-C3-PPKRIA containing the 3-C linker revealed that the compound could simultaneously occupy SAM and peptide binding sites, it was reasonable to assume that compound 2 with 3-C linker could also interact with both SAM and peptide binding sites. So, inhibition mechanism studies for compound 3 was performed along with compounds 1 and 2 as the control under the condition of 2 h of pre-incubation (Figure 8). Even at both 6 Km and 12 Km concentrations of peptide substrate, there were slight competition for compound 3. Moreover, ~10% of enzymatic activity was rescued in presence of 0.3 μM or higher concentration of compounds 2 and 3. When competing peptide substrate concentration increased to 50 Km, right shift of the IC50 curves was observed for 2 and 3. In addition, ~20% enzymatic activity was rescued, further reflecting their tight and slow-binding character. When increasing the concentration of SAM from 6 Km to 25Km, similar trends were observed. The catalytic activity of NTMT1 could be rescued with higher concentration of SAM or peptide, suggesting the compounds could be competitive with peptide and SAM. Those results indicated that all three bisubstrate inhibitors 1-3 occupied both SAM and peptide substrate binding sites in a reversible manner.
Figure 8.
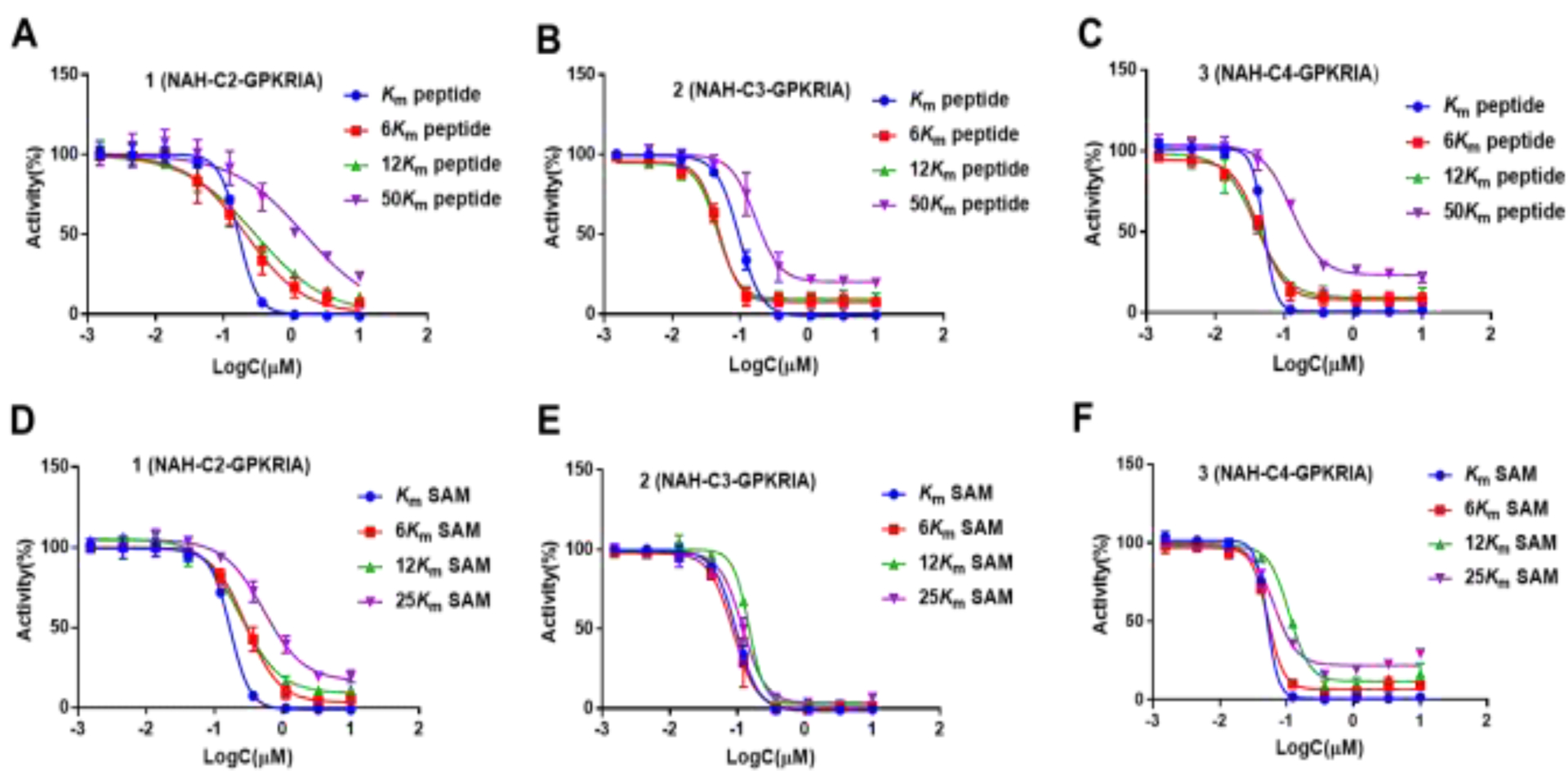
Inhibition Mechanism Studies for compounds 1-3. (A-C) IC50 curves of 1-3 at varying concentrations of peptide with fixed concentration of SAM; (D-F) IC50 curves of 1 - 3 at varying concentrations of SAM with fixed concentration of peptide. The pre-incubation time for the assay was 120 min, and all the experiment were run in duplicates (n = 2).
Conclusion
This series of tight-binding and selective bisubstrate analogs to NTMT1 represent valuable tools to shed light on the plasticity in the transition state of NTMT1 since it can accommodate linker length from 2-C to 4-C atom. Among them, a 4-C atom linker of compound 3 is the optimal length to achieve the best inhibition with a Ki, app value of 130 pM. Notably, a 5-C atom linker of compound 4 would convert a tight-binding bisubstrate analog to a substrate for NTMT1, suggesting the upper limit of the aforementioned plasticity. The co-crystal structure of 4 in complex with NTMT1 (PDB code: 6PVB) implied that extending the linker to a 5-C atom linker may cause tension to prevent the bisubstrate analog from engaging with both binding sites. Given the transient nature of transition state, these bisubstrate analogs provide dynamic snapshots of the reorganization within the NTMT1 active site during the catalysis. Furthmore, top inhibitor 3 (NAH-C4-GPKRIA) containing a 4-C atom linker showed picomolar inhibition (Ki, app = 130 ± 40 pM) for NTMT1, and more than 100,000-fold selectivity over other MTases. In addition, all bisubstrate analogs 1-3 demonstrate no interation with its close homolog NTMT2 despite of the similar catalytic sites of NTMT1/2.33 This delicate selectivity further corroborates the benefits of bisubstrate analogs on account of their high specificity to offer tight and selective inhibitors for individual target. However, increasing the carbon linker length to a 5-C atom resulted in a substrate instead of an inhibitor for NTMT1, which defined the scope or limit of plasticity. To our knowledge, this is the first case that delineates the optimal linker range of MTase bisubstrate analog and provide the molecular basis for the obliterated activity for longer linkers.
The linear distance between the sulfonium ion and the nitrogen atom that accepting methyl group is very similar for most PMTs, which ranges from 3.5 Å to 5.5 Å among the ternary complex of all examined PMTs including NTMT1, SET domain-containing protein 8, and euchromatic histone-lysine N-methyltransferase 1.18,19,34–37 Therefore, we propose that a 3-C or 4-C atom linker would serve as a general and feasible starting point to build bisubstrate analogs for any MTase. The optimal linker length has also been corroborated by successful inhibitors for RNA MTase, as a 3-C atom linker showed better inhibition compared to a 2-C atom linker.38 There have been reported small molecule inhibitors targeting either SAM or substrate binding sites for MTase.2,39 Future directions will focus on applying such linkers to conjugate small molecule inhibitors that target substrate binding site with SAM mimic or SAM competitive small molecule inhibitors to achieve higher selectivity and cellular potency for respective methyltransferases.
Experimental Section
Chemistry General Procedures.
The reagents and solvents were purchased from commercial sources (Fisher) and used directly. The intermediates and products were purified by using an ISCO CombiFlash system and prepacked SiO2 cartridges. Final compounds were purified on preparative high-pressure liquid chromatography (RP-HPLC) was performed on Agilent 1260 Series system. Systems were run with 0–20% methanol/water gradient with 0.1% TFA. NMR spectra were acquired on a Bruker AV500 instrument (500 MHz for 1H-NMR, 126 MHz for 13C-NMR). TLC-MS were acquired using Advion CMS-L MS. Matrix-assisted laser desorption ionization mass spectra (MALDI-MS) data were acquired in positive-ion mode using a Sciex 4800 MALDI TOF/TOF MS. The peptides were synthesized using a CEM Liberty Blue Automated Microwave Peptide Synthesizer with the manufacturers standard coupling cycles at 0.1 mmol scale. The purity of final compounds was confirmed by Waters LC-MS system and/or Agilent 1260 Series HPLC system. Systems were run with 0–40% methanol/water gradient with 0.1% TFA. All the purity of target compounds showed >95% in RP-HPLC.
α-bromo peptides (G*PKRIA).
Active bromo acetic acid (139mg, 1mmol) by DIC (155μL, 1mmol) for 20min. Then added the solution into the peptide PKRIA on resin (0.1 mmol). The mixture was shaken overnight at r.t. The suspension was filtered, and the resin was washed with DMF (3ml × 3). Repeated the procedures again. Do test cleavage (TFA: TIPS: H2O=95:2.5:2.5) and MALDI-MS to check the progress of the reaction.
NAH-C2-GPKRIA (1). To a suspension of G*PKRIA on resin (0.1 mmol) in DMF (3 mL) was added compound 8 (91mg, 0.15 mmol) and DIPEA (26 μL, 0.15 mmol). The mixture was shaken at room temperature overnight. The suspension was filtered, and the resin was subsequently washed with DMF (3 mL × 3), MeOH (3 mL × 3), and CH2Cl2 (3 mL × 3). The peptide conjugates were cleaved from the resin in the presence of a cleavage cocktail (10 mL) containing trifluoroacetic acid (TFA)/2,2’-(Ethylenedioxy)-diethanethiol (DODT) /triisopropylsilane (TIPS) /water (94:2.5:1:2.5 v/v) at room temperature for 4–5 h and resin was rinsed with a small amount of TFA. Volatiles of the filtrate were removed under N2 and the residue was precipitated with 10 vol. cold anhydrous ether, and collected by centrifugation. The supernatant was discarded and the pellet was washed with chilled ether and air-dried. The white precipitate was dissolved in ddH2O and purified by reverse phase HPLC using a waters system with a XBridge (BEH C18, 5μm, 10 X 250 mm) column with 0.1% TFA in water (A) and MeOH (B) as the mobile phase with monitoring at 214 nm and 254 nm. MALDI-MS (positive) m/z: calcd for C44H77N18O11 [M + H]+ m/z 1033.6019, found m/z 1033.7422.
NAH-C3-GPKRIA (2). Compound was prepared according to the procedure for NAH-C2-GPKRIA (1) and purified by reverse phase HPLC. MALDI-MS (positive) m/z: calcd for C45H79N18O11 [M + H]+ m/z 1047.6176, found m/z 1047.8483.
NAH-C4-GPKRIA (3). Compound was prepared according to the procedure for NAH-C2-GPKRIA (1) and purified by reverse phase HPLC. MALDI-MS (positive) m/z: calcd for C46H81N18O11 [M + H]+ m/z 1061.6332, found m/z 1062.0251.
NAH-C5-GPKRIA (4). Compound was prepared according to the procedure for NAH-C2-GPKRIA (1) and purified by reverse phase HPLC. MALDI-MS (positive) m/z: calcd for C46H81N18O11 [M + H]+ m/z 1075.6489, found m/z 1075.6666.
NAH-C2-GPKR (5). Compound was prepared according to the procedure for NAH-C2-GPKRIA (1) and purified by reverse phase HPLC. MALDI-MS (positive) m/z: calcd for C35H61N16O9 [M + H]+ m/z 849.4807, found m/z 849.7723.
NAH-C3-GPKR (6). Compound was prepared according to the procedure for NAH-C2-GPKRIA (1) and purified by reverse phase HPLC. MALDI-MS (positive) m/z: calcd for C36H63N16O9 [M + H]+ m/z 863.4964, found m/z 863.6786.
NAH-C4-GPKR (7). Compound was prepared according to the procedure for NAH-C2-GPKRIA (1) and purified by reverse phase HPLC. MALDI-MS (positive) m/z: calcd for C37H65N16O9 [M + H]+ m/z 877.5120, found m/z 877.7390.
(S)-2-amino-4-((((2R,3S,4R,5S)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)amino)butanoic acid (8). To a solution of compound 14 (10mg, 0.018 mmol) in CH2Cl2 was added TFA under ice bath. Continue to stir for 1h under ice bath. Removed the TFA in vacuum, and the residue was purified by cation-iron exchange chromatography (0.1M NH4HCO3 aq. solution). After lyophilization, obtained 2.7 mg (41%) white solid. 1H NMR (500 MHz, Deuterium Oxide) δ 8.13 (dt, J = 12.4, 4.0 Hz, 2H), 5.95 (d, J = 5.0 Hz, 1H), 4.80 – 4.73 (m, 1H), 4.27 (dt, J = 19.5, 4.5 Hz, 2H), 3.62 (t, J = 6.5 Hz, 1H), 3.27 (dd, J = 43.7, 9.1 Hz, 2H), 3.02 (t, J = 7.3 Hz, 2H), 2.00 (ddd, J = 29.3, 14.8, 7.7 Hz, 2H). 13C NMR (126 MHz, Deuterium Oxide) δ 174.19, 155.63, 152.75, 148.71, 140.75, 119.20, 88.61, 80.84, 72.94, 71.43, 53.29, 49.51, 45.30, 28.15. MALDI-MS (positive) m/z: calcd for C14H22N7O5 [M + H]+ m/z 368.1682, found m/z 368.2080.
(S)-2-amino-4-((((2R,3S,4R,5S)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)(4-aminobutyl)amino)butanoic acid (9). To a suspend of compound 18 (20mg, 0.032mmol)in ddH2O was added 4N HCl in dioxane under ice bath. Then stirred for 2h at r.t.. Removed the solvent in vacuum, and the residue was purified by cation-iron exchange chromatography (0.1M NH4HCO3 aq. solution). After lyophilization, obtained 6 mg (45%) white solid. 1H NMR (500 MHz, Deuterium Oxide) δ 8.23 – 8.10 (m, 2H), 5.98 – 5.92 (m, 1H), 4.27 – 4.09 (m, 2H), 3.50 – 3.38 (m, 1H), 2.85 – 2.68 (m, 4H), 2.66 – 2.55 (m, 2H), 2.52 – 2.39 (m, 2H), 1.91 – 1.64 (m, 2H), 1.51 – 1.27 (m, 4H). MALDI-MS (positive) m/z: calcd for C18H31N8O5 [M + H]+ m/z 439.2417, found m/z 439.3303.
(S)-2-amino-4-((4-((2-((S)-2-(((S)-6-amino-1-(((S)-1-(((2S,3S)-1-(((S)-1-amino-1-oxopropan-2-yl)amino)-3-methyl-1-oxopentan-2-yl)amino)-5-guanidino-1-oxopentan-2-yl)amino)-1-oxohexan-2-yl)carbamoyl)pyrrolidin-1-yl)-2-oxoethyl)amino)butyl)amino)butanoic acid (10). Compound was prepared according to the procedure for NAH-C2-GPKRIA (1) and purified by reverse phase HPLC. MALDI-MS (positive) m/z: calcd for C36H70N13O8 [M + H]+ m/z 812.5470, found m/z 812.7323.
(S)-N-((S)-6-amino-1-(((S)-1-(((2S,3S)-1-(((S)-1-amino-1-oxopropan-2-yl)amino)-3-methyl-1-oxopentan-2-yl)amino)-5-guanidino-1-oxopentan-2-yl)amino)-1-oxohexan-2-yl)-1-((4-aminobutyl)glycyl)pyrrolidine-2-carboxamide (11). Compound was prepared according to the procedure for NAH-C2-GPKRIA (1) and purified by reverse phase HPLC. MALDI-MS (positive) m/z: calcd for C32H63N12O6 [M + H]+ m/z 711.4994, found m/z 711.5203.
(S)-N-((S)-6-amino-1-(((S)-1-(((2S,3S)-1-(((S)-1-amino-1-oxopropan-2-yl)amino)-3-methyl-1-oxopentan-2-yl)amino)-5-guanidino-1-oxopentan-2-yl)amino)-1-oxohexan-2-yl)-1-((3-((((2S,3S,4R,5S)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)thio)propyl)glycyl)pyrrolidine-2-carboxamide (12). Compound was prepared according to the procedure for NAH-C2-GPKRIA (1) and purified by reverse phase HPLC. MALDI-MS (positive) m/z: calcd for C41H71N16O9S [M + H]+ m/z 963.5311, found m/z 963.6245.
Tert-butyl (S)-4-((((3aR,4R,6S,6aR)-6-(6-amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)(4-(((benzyloxy)carbonyl)amino)butyl)amino)-2-((tert-butoxycarbonyl)amino)butanoate (15a). To a solution of tert-butyl (S)-4-((((3aR,4R,6S,6aR)-6-(6-amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)amino)-2-((tert-butoxycarbonyl)amino)butanoate (14, 0.5g, 0.89 mmol) in methanol, benzyl (4-oxobutyl)carbamate (13a, 0.26g, 1.15 mmol) was add into the reaction. The mixture stirred for 20 min at r.t.. Then, NaBH3CN (82mg, 1.34 mmol) was added in protions and the reaction continued to stir for 6h at r.t.. After removing the solvent in vacuum, the residue was purified by column chromatography (CH2Cl2: MeOH = 30:1) to obtain 0.46g (68%) white solid. 1H NMR (500 MHz, Methanol-d4) δ 8.25 (s, 1H), 8.22 (s, 1H), 7.39 – 7.24 (m, 5H), 6.16 (d, J = 2.4 Hz, 1H), 5.53 – 5.49 (m, 1H), 5.05 (s, 2H), 5.04 – 5.01 (m, 1H), 4.37 – 4.28 (m, 1H), 4.09 – 4.02 (m, 1H), 3.14 – 3.00 (m, 2H), 2.89 – 2.75 (m, 1H), 2.68 – 2.54 (m, 2H), 2.53 – 2.31 (m, 3H), 1.95 – 1.84 (m, 1H), 1.72 – 1.61 (m, 1H), 1.58 (s, 3H), 1.45 – 1.44 (m, 2H), 1.43 (s, 9H), 1.42 (s, 9H), 1.38 (s, 3H).
Tert-butyl (S)-4-((5-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)pentyl)(((3aR,4R,6S,6aR)-6-(6-amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)amino)-2-((tert-butoxycarbonyl)amino)butanoate (15b). To a solution of tert-butyl (S)-4-((((3aR,4R,6S,6aR)-6-(6-amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)amino)-2-((tert-butoxycarbonyl)amino)butanoate (14, 0.17g, 0.30 mmol) in methanol, (9H-fluoren-9-yl)methyl (5-oxopentyl)carbamate (13b, 0.13g, 0.40 mmol) was add into the reaction. The mixture stirred for 20 min at r.t.. Then, NaBH3CN (26mg, 0.42 mmol) was added in portion and the reaction continued to stir overnight at r.t.. After removing the solvent in vacuum, the residue was purified by column chromatography (CH2Cl2: MeOH = 50:1) to obtain 0.1g (38%) white solid. 1H NMR (500 MHz, Chloroform-d) δ 8.34 (s, 1H), 7.93 (s, 1H), 7.76 (d, J = 7.5 Hz, 2H), 7.60 (d, J = 6.9 Hz, 2H), 7.39 (t, J = 7.4 Hz, 2H), 7.30 (t, J = 7.3 Hz, 2H), 6.09 – 6.01 (m, 1H), 5.67 – 5.43 (m, 3H), 5.03 – 4.91 (m, 1H), 4.49 – 4.32 (m, 3H), 4.31 – 4.08 (m, 2H), 3.31 – 3.01 (m, 2H), 2.83 – 2.21 (m, 6H), 1.99 – 1.84 (m, 1H), 1.70 (ws, 3H), 1.61 (s, 3H), 1.44 (s, 18H), 1.39 (s, 3H), 1.34 – 1.10 (m, 4H).
Tert-butyl (S)-4-((((3aR,4R,6S,6aR)-6-(6-amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)(4-aminobutyl)amino)-2-((tert-butoxycarbonyl)amino)butanoate (18). To a solution of 15a (0.46g, 0.6mmol) in MeOH, 10 % Pd/C was added slowly into the reaction. The mixture was degassed under vacuum and then under H2 atmosphere and stirred overnight. After filtration and concentration, the residue was purified by column chromatography (CH2Cl2: MeOH = 10:1) to get 0.21g (56%) white solid. 1H NMR (500 MHz, Methanol-d4) δ 8.26 (s, 1H), 8.22 (s, 1H), 6.17 (d, J = 2.1 Hz, 1H), 5.54 – 5.51 (m, 1H), 5.08 – 5.01 (m, 1H), 4.37 – 4.26 (m, 1H), 4.13 – 4.00 (m, 1H), 2.84 – 2.74 (m, 1H), 2.66 – 2.52 (m, 4H), 2.50 – 2.39 (m, 2H), 2.38 – 2.28 (m, 1H), 2.00 – 1.86 (m, 1H), 1.69 – 1.60 (m, 1H), 1.59 (s, 3H), 1.46 – 1.41 (m, 20H), 1.38 (s, 3H).
Tert-butyl (S)-4-((((3aR,4R,6S,6aR)-6-(6-amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)(5-aminopentyl)amino)-2-((tert-butoxycarbonyl)amino)butanoate (19). To a solution of 15b (0.1g, 0.11mmol) in CH3CN cooled in ice-bath, DBU (34 μL, 0.22 mmol) was added in dropwise. The reaction stirred for 1h at r.t.. The solvent was removed in vacuum, and the residue was purified by column chromatography (CH2Cl2 : MeOH = 10:1) to get 50 mg (71%) product. 1H NMR (500 MHz, Methanol-d4) δ 8.26 (s, 1H), 8.22 (s, 1H), 6.17 (d, J = 2.2 Hz, 1H), 5.57 – 5.50 (m, 1H), 5.08 – 5.02 (m, 1H), 4.36 – 4.28 (m, 1H), 4.11 – 4.01 (m, 1H), 2.84 – 2.74 (m, 1H), 2.66 – 2.54 (m, 4H), 2.48 – 2.39 (m, 2H), 2.38 – 2.29 (m, 1H), 1.99 – 1.88 (m, 1H), 1.68 – 1.61 (m, 1H), 1.59 (s, 3H), 1.44 (s, 9H), 1.44 (s, 9H), 1.42 – 1.40 (m, 2H), 1.39 (s, 3H), 1.37 – 1.30 (m, 2H), 1.30 – 1.17 (m, 2H).
Tert-butyl (S)-4-((4-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)butyl)amino)-2-((tert-butoxycarbonyl)amino)butanoate (22). To a solution of (9H-fluoren-9-yl)methyl (4-aminobutyl)carbamate (20, 0.2g, 0.58 mmol) in methanol, tert-butyl (S)-2-((tert-butoxycarbonyl)amino)-4-oxobutanoate (21, 0.24g, 0.86 mmol) was added into the reaction. The mixture stirred for 30 min at r.t.. Then, NaBH3CN (51mg, 0.81 mmol) was added in portion and the reaction continued to stir for 6h at r.t.. After removing the solvent in vacuum, the residue was purified by column chromatography (CH2Cl2: MeOH = 20:1) to obtain 0.23g (71%) product. 1H NMR (500 MHz, Chloroform-d) δ 7.75 (d, J = 7.6 Hz, 2H), 7.60 (d, J = 7.4 Hz, 2H), 7.39 (t, J = 7.4 Hz, 2H), 7.34 – 7.28 (m, 2H), 5.63 – 5.55 (m, 1H), 5.27 – 5.20 (m, 1H), 4.43 – 4.29 (m, 2H), 4.21 (t, J = 6.8 Hz, 1H), 4.18 – 4.09 (m, 1H), 3.31 – 3.17 (m, 2H), 3.16 – 2.79 (m, 5H), 2.55 – 2.45 (m, 1H), 1.84 – 1.75 (m, 2H), 1.72 – 1.58 (m, 2H), 1.48 (s, 9H), 1.43 (s, 9H). TLC-MS (ESI+) m/z 568.6 [M + H]+
Tert-butyl (S)-4-((4-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)butyl)(tert-butoxycarbonyl)amino)-2-((tert-butoxycarbonyl)amino)butanoate (23).To a mixture of 22 (0.21g, 0.37mmol) and NaHCO3 (95mg, 1.11mmol) in THF and H2O cooled by ice-bath, (Boc)2O (0.16g, 0.74mmol) were added in portion. Then the reaction stirred at r.t. overnight. The mixture was diluted by CH2Cl2, and the organic layer was separated and dried over anhydrous NaSO4. After filtration, the solvent of organic layer was removed in vacuum. The residue was purified by column chromatography (CH2Cl2: MeOH = 20:1) to get 0.21 g (85%) product. 1H NMR (500 MHz, Chloroform-d) δ 7.74 (d, J = 7.5 Hz, 2H), 7.58 (d, J = 7.3 Hz, 2H), 7.37 (t, J = 7.4 Hz, 2H), 7.28 (t, J = 7.3 Hz, 2H), 5.24 – 5.09 (m, 1H), 4.37 (d, J = 6.2 Hz, 2H), 4.19 (t, J = 6.9 Hz, 1H), 4.17 – 4.10 (m, 1H), 3.35 – 3.07 (m, 6H), 2.03 – 1.96 (m, 1H), 1.90 – 1.75 (m, 1H), 1.47 – 1.42 (m, 31H).
Tert-butyl (S)-4-((4-aminobutyl)(tert-butoxycarbonyl)amino)-2-((tert-butoxycarbonyl)amino)butanoate (24). To a solution of 23 (0.21g, 0.31mmol) in CH3CN cooled by ice-bath, DBU (94 μL, 0.62 mmol) was added in dropwise. The reaction stirred for 1h at r.t.. The solvent was removed in vacuum, and the residue was purified by column chromatography (CH2Cl2 : MeOH = 10:1) to get 69 mg (50%) product. 1H NMR (500 MHz, Chloroform-d) δ 5.27 – 5.13 (m, 1H), 4.18 – 4.03 (m, 1H), 3.38 – 3.02 (m, 4H), 3.02 – 2.85 (m, 2H), 2.06 – 1.92 (m, 1H), 1.85 – 1.73 (m, 1H), 1.68 – 1.59 (m, 2H), 1.60 – 1.52 (m, 2H), 1.52 – 1.33 (m, 27H).
Protein Expression and Purification.
Expression and purification of NTMT1, G9a, SETD7, PRMT1, PRMT3, TbPRMT7 and NNMT was performed as previously described6,18,37,40–43
NTMT1 Biochemical Assays and Enzyme Kinetics Study.
A fluorescence-based SAHH-coupled assay was applied to study the IC50 values of compounds with both SAM and the peptide substrate at their Km values. For 10min pre-incubation, the methylation assay was performed under the following conditions in a final well volume of 40 μL: 25 mM Tris (pH = 7.5), 50 mM KCl, 0.01% Triton X-100, 5 μM SAHH, 0.1 μM NTMT1, 3 μM SAM, and 10 μM ThioGlo4. The inhibitors were added at concentrations ranging from 0.15 nM to 10 μM. After 10 min incubation, reactions were initiated by the addition of 0.5 μM GPKRIA peptide. For 120min pre-incubation, NTMT1 and compounds were pre-incubated for 120min. Then the pre-incubation mixture was added into reaction mixture: 25 mM Tris (pH = 7.5), 50 mM KCl, 0.01% Triton X-100, 5 μM SAHH, 3 μM SAM, and 10 μM ThioGlo4. The reactions were initiated by the addition of 0.5 μM GPKRIA peptide. Fluorescence was monitored on a BMG CLARIOstar microplate reader with excitation 400 nm and emission 465 nm. Data were processed by using GraphPad Prism software 7.0.
For the inhibition mechanism studies, varying concentrations of SAM with 0.5 μM fixed concentration of GPKRIA or varying concentration of GPKRIA with 3 μM fixed concentration of SAM were included in reactions at concentration of inhibitors ranging from 0.15 nM to 10 μM. Fluorescence was monitored on a BMG CLARIOstar microplate reader with excitation 400 nm and emission 465 nm. All experiments were determined in duplicate. Data were processed by using GraphPad Prism software 7.0.
ITC.
Isothermal titration calorimetry (ITC) measurements were performed by VP-ITC MicroCal calorimeter at 25 °C. Titration buffer contains 20 mM Tris-HCl pH 7.5, 150 mM NaCl and 0.5 mM TECP (tris(2-carboxyethyl)phosphine). Purified NTMT1 and NTMT2 were diluted with the ITC buffer to the final concentration of 30–50 μM, and the compounds were dissolved to 0.5–1mM in the same buffer. The compound was titrated into the protein solution with 26 injections of 10 μl each. Injections were spaced 180 sec with a reference power of 15 μcal/sec. The ITC data were processed with MicroCal PEAQ-ITC analysis software using one-site binding or two-site binding model.
Time-dependent Inhibition Study.
The assay was performed under the following conditions in a final well volume of 40 μL: 25 mM Tris (pH = 7.5), 50 mM KCl, 0.01% Triton X-100, 5 μM SAHH, 0.1 μM NTMT1, 3 μM SAM, 0.5 μM GPKRIA and 10 μM ThioGlo4. The inhibitors were added at concentrations ranging from 0.15 nM to 10 μM. NTMT1 and compounds were pre-incubated for 30min, 60min, 90min, and 120min, Then the pre-incubation mixture was added into reaction mixture: 25 mM Tris (pH = 7.5), 50 mM KCl, 0.01% Triton X-100, 5 μM SAHH, 3 μM SAM, and 10 μM ThioGlo4. The reactions were initiated by the addition of 0.5 μM GPKRIA peptide. Fluorescence was monitored on a BMG CLARIOstar microplate reader with excitation 400 nm and emission 465 nm. Data were processed by using GraphPad Prism software 7.0 with Morrison’s quadratic equation.
Selectivity Assays.
The selectivity studies of G9a, SETD7, PRMT1, TbPRMT7 and NNMT were performed as previously described.11,44
For PRMT3, the assay was also performed in a final well volume of 40 μL: 2.5 mM HEPES (pH = 7.0), 25 mM NaCl, 25 μM EDTA, 50 μM TCEP, 0.01% Triton X-100, 5 μM SAHH, 1 μM PRMT3, 25 μM SAM, and 15 μM ThioGlo1. After 10 min incubation with inhibitors, reactions were initiated by the addition of 10 μM H4–21 peptide. All experiments were determined in duplicate. Fluorescence was monitored on a BMG CLARIOstar microplate reader with excitation 380 nm and emission 505 nm.
MALDI-MS Methylation Assay and Methylation Inhibition Assay.
MALDI-MS methylation and inhibition assay were performed and analyzed via a Sciex 4800 MALDI TOF/TOF MS. MALDI-MS methylation assay was performed in a final well volume of 40 μL: 0.1 μM NTMT1, 20 mM Tris (pH = 7.5), 50 mM KCl, 3 μM SAM and 10 μM compounds. After incubation overnight at 37 °C, the samples were quenched in a 1:1 ratio with a quenching solution (20 mM NH4H2PO4, 0.4% (v/v) TFA in 1:1 acetonitrile/water). Samples were analyzed by MALDI-MS with 2,5-dihydroxybenzoic acid matrix solution.
The inhibition assay was performed in a final well volume of 40 μL: 0.1 μM NTMT1, 20 mM Tris (pH = 7.5), 50 mM KCl, 3 μM SAM and various concentrations of compounds at 37 °C for 10 min before the addition of 0.5 μM GPKRIA peptide to initiate the reaction. After incubation for 20 min, the samples were quenched in a 1:1 ratio with a quenching solution (20 mM NH4H2PO4, 0.4% (v/v) TFA in 1:1 acetonitrile/water). Samples were analyzed by MALDI-MS with 2,5-dihydroxybenzoic acid matrix solution. Duplicate was performed for all experiments. Data were processed in Data Explorer.
Co-crystallization and Structure Determination.
For co-crystallization, NTMT1 was mixed with 2 mM compounds incubated for 1 hr at 4°C, diluted 6-fold in 1x TBS, pH 7.6, with 1 mM TCEP, and then re-concentrated to 30 mg/mL. Broad matrix and optimization crystallization screening were performed using a Mosquito-LCP high throughput crystallization robot (TTP LabTech) using hanging-drop vapor diffusion method, incubated at 20°C, and crystals harvested directly from the 96-well crystallization plates. Crystals containing 1 were grown in 32 % (w/v) PEG 4000, 0.8 M Lithium Chloride, 0.1 M Tris·HCl (pH = 8.5), and crystals containing 4 were grown in 30% (w/v) PEG3350, 0.2 M Li2SO4, 0.1 M bis-tris propane (pH = 7.0); all crystals were harvested directly from the drop and flash cooled into liquid nitrogen.
Data were collected on single crystals at 12.0 keV at the GM/CA-CAT ID-D beamline at the Advanced Photon Source, Argonne National Laboratory. The data were processed using the on-site automated pipeline with Fast DP and molecular replacement performed with Phaser-MR (PHENIX)45,46. All model building was performed using COOT and subsequent refinement done using phenix.refine (PHENIX)46,47. Structure factors and model coordinates have been deposited in the Protein Data Bank with PDB IDs: 6WJ7 (1) and 6PVB (4). Structure-related figures were made with PyMOL (Schrödinger) and annotated and finalized with Adobe Photoshop and Illustrator.
Supplementary Material
Acknowledgements
We appreciate Dr. Darrel L. Peterson for purification of SAHH. We thank Krystal Diaz for purifying TbPRMT7. The authors acknowledge the support from NIH grants R01GM117275 (RH), 1R01GM127896 (NN), 1R01AI127793 (NN), and P30 CA023168 (Purdue University Center for Cancer Research). The SGC is a registered charity (number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada through Ontario Genomics Institute [OGI-055], Innovative Medicines Initiative (EU/EFPIA) [ULTRA-DD grant no. 115766], Janssen, Merck KGaA, Darmstadt, Germany, MSD, Novartis Pharma AG, Ontario Ministry of Research, Innovation and Science (MRIS), Pfizer, São Paulo Research Foundation-FAPESP, Takeda, and Wellcome. We also thank supports from the Department of Medicinal Chemistry and Molecular Pharmacology (RH) and Department of Biological Sciences (NN) at Purdue University.
ABBREVIATIONS
- NTMT1
protein N-terminal methyltransferase 1
- PMTs
protein methyltransferases
- SAM
S-5’-adenosyl-L-methionine
- SAH
S-5’-adenosyl-L-homocysteines
- SAHH
SAH hydrolase
- PRMT1
protein arginine methyltransferase 1
- CARM1
coactivator-associated arginine methyltransferase 1
- G9a
euchromatic histone-lysine N-methyltransferase 2
- SETD7
SET domain-containing protein 7
- NNMT
nicotinamide N-methyltransferase
- PRMT3
protein arginine methyltransferase 3
- TbPRMT7
Trypanosoma brucei protein arginine methyltransferase 7
- MTase
methyltransferase
- TCEP
tris(2-carboxyethyl)phosphine; TFA, trifluoroacetic acid
Footnotes
Supporting Information
NMR and MS spectra of compound 15a-b, 18–19 and 22–24; NMR, MALDI-MS and LC-MS/HPLC spectra of compound 1-12; Figure S1. Methylation assay of compounds 10-12 for NTMT1; Figure S2. MALDI-MS methylation inhibition assay compound 8; Table S1. Selectivity evaluation of compound 3; Table S2. Crystallography data and refinement statistics (PDB ID: 6WJ7 and 6PVB).
Molecular formula strings (CSV)
Accession Codes
The coordinates for the structure of human NTMT1 in complex with compound 1 (PDB ID: 6WJ7) and 4 (PDB ID: 6PVB) have been deposited in the Protein Data Bank. Authors will release the atomic coordinates and experimental data upon article publication.
References
- (1).Wang SM; Wang SM; Dowhan DH; Muscat GEO Epigenetic Arginine Methylation in Breast Cancer : Emerging Therapeutic Strategies. J. Mol. Endocrinol 2019, 62, R223–227. [DOI] [PubMed] [Google Scholar]
- (2).Kaniskan HU; Martini ML; Jin J Inhibitors of Protein Methyltransferases and Demethylases. Chem. Rev 2018, 118, 989–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Tsai W; Niessen S; Goebel N; Yates JR; Guccione E; Montminy M PRMT5 Modulates the Metabolic Response to Fasting Signals. Proc. Natl. Acad. Sci 2013, 110, 8870–8875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Liu X; Wang L; Li H; Lu X; Hu Y; Yang X; Huang C; Gu D Coactivator-Associated Arginine Methyltransferase 1 Targeted by MiR-15a Regulates Inflammation in Acute Coronary Syndrome. Atherosclerosis 2014, 233, 349–356. [DOI] [PubMed] [Google Scholar]
- (5).Luo M Chemical and Biochemical Perspectives of Protein Lysine Methylation. Chem. Rev 2018, 118, 6656–6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Richardson SL; Mao Y; Zhang G; Hanjra P; Peterson DL; Huang R Kinetic Mechanism of Protein N-Terminal Methyltransferase 1. J. Biol. Chem 2015, 290, 11601–11610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Khalil EM; De Angelis J; Ishii M; Cole PA Mechanism-based Inhibition of the Melatonin Rhythem Enzyme: Pharmacologic Exploitation of Active Site Functional Plasticity. Proc. Natl. Acad. Sci 1999, 96, 12418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Yu M; Magalha MLB; Cook PF; Blanchard JS Bisubstrate Inhibition: Theory and Application to N-Acetyltransferases. Biochemistry 2006, 45, 14788–14794. [DOI] [PubMed] [Google Scholar]
- (9).Parang K; Cole PA Designing Bisubstrate Analog Inhibitors for Protein Kinases. Pharmacol. Ther 2002, 93, 145–157. [DOI] [PubMed] [Google Scholar]
- (10).Huang R; Martinez-ferrando I; Cole PA Enhanced Interrogation: Emerging Strategies for Cell Signaling Inhibition. Nat. Struct. Mol. Biol 2011, 17, 646–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Chen D; Dong G; Noinaj N; Huang R Discovery of Bisubstrate Inhibitors for Protein N-Terminal Methyltransferase 1. J. Med. Chem 2019, 62, 3773–3779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Brandvold KR; Santos SM; Breen ME; Lachacz EJ; Steffey ME; Soellner MB Exquisitely Specific Bisubstrate Inhibitors of C-Src Kinase. ACS Chemical Biology. 2015, 10, 1387–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Zhang G; Richardson SL; Mao Y; Huang R Design, Synthesis, and Kinetic Analysis of Potent Protein N-Terminal Methyltransferase 1 Inhibitors. Org. Biomol. Chem 2015, 13, 4149–4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Zhang G; Huang R Facile Synthesis of SAM-Peptide Conjugates through Alkyl Linkers Targeting Protein N-Terminal Methyltransferase 1. RSC Adv. 2016, 6, 6768–6771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Haren MJ Van; Marechal, N.; Troffer-charlier, N.; Cianciulli, A.; Sbardella, G. Transition State Mimics Are Valuable Mechanistic Probes for Structural Studies with the Arginine. Proc. Natl. Acad. Sci 2017, 114, 3652–3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Osborne T; Roska RLW; Rajski SR; Thompson PR In Situ Generation of a Bisubstrate Analogue for Protein Arginine Methyltransferase 1. J. Am. Chem. Soc 2008, 130, 4574–4575. [DOI] [PubMed] [Google Scholar]
- (17).Policarpo RL; Decultot L; May E; Kuzmic P; Carlson S; Huang D; Chu V; Wright BA; Dhakshinamoorthy S; Kannt A; Rani S; Dittakavi S; Danarese JD; Gaudet R; Shair MD High-Affinity Alkynyl Bisubstrate Inhibitors of Nicotinamide N‑Methyltransferase (NNMT). J. Med. Chem 2019, 62, 9837–9873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Dong C; Mao Y; Tempel W; Qin S; Li L; Loppnau P; Huang R; Min J Structural Basis for Substrate Recognition by the Human N-Terminal Methyltransferase 1. Genes Dev. 2015, 29, 2343–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Wu R; Yue Y; Zheng X; Li H Molecular Basis for Histone N-Terminal Methylation by NRMT1. Genes Dev. 2015, 29, 2337–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Petkowski JJ; Tooley CES; Anderson LC; Shumilin I a; Balsbaugh, J. L.; Hunt, D. F.; Minor, W.; Macara, I. G. Substrate Specificity of Mammalian N-Terminal α -Amino Methyltransferase NRMT. Biochemistry 2012, 51, 5942–5950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Dorgan KM; Wooderchak WL; Wynn DP; Karschner EL; Alfaro JF; Cui Y; Zhou ZS; Hevel JM An Enzyme-Coupled Continuous Spectrophotometric Assay for S-Adenosylmethionine-Dependent Methyltransferases. Anal. Biochem 2006, 350, 249–255. [DOI] [PubMed] [Google Scholar]
- (22).Bailey AO; Panchenko T; Sathyan KM; Petkowski JJ; Pai P-J; Bai DL; Russell DH; Macara IG; Shabanowitz J; Hunt DF; Black BE; Flotz DR Posttranslational Modification of CENP-A Influences the Conformation of Centromeric Chromatin. Proc. Natl. Acad. Sci 2013, 110, 11827–11832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Dai X; Otake K; You C; Cai Q; Wang Z; Masumoto H; Wang Y Identification of Novel α-N-Methylation of CENP-B That Regulates Its Binding to the Centromeric DNA. J. Proteome Res 2013, 12, 4167–4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Cai Q; Fu L; Wang Z; Gan N; Dai X; Wang Y α-N-Methylation of Damaged DNA-Binding Protein 2 (DDB2) and Its Function in Nucleotide Excision Repair. J. Biol. Chem 2014, 289, 16046–16056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Dai X; Rulten SL; You C; Caldecott KW; Wang Y Identification and Functional Characterizations of N-Terminal α-N-Methylation and Phosphorylation of Serine 461 in Human Poly(ADP-Ribose) Polymerase 3. J. Proteome Res 2015, 14, 2575–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Tooley CES; Petkowski JJ; Muratore-Schroeder TL; Balsbaugh JL; Shabanowitz J; Sabat M; Minor W; Hunt DF; Macara IG NRMT Is an Alpha-N-Methyltransferase That Methylates RCC1 and Retinoblastoma Protein. Nature 2010, 466, 1125–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Bonsignore LA; Tooley JG; Van Hoose PM; Wang E; Cheng A; Cole MP; Schaner Tooley CE NRMT1 Knockout Mice Exhibit Phenotypes Associated with Impaired DNA Repair and Premature Aging. Mech. Ageing Dev 2015, 146–148, 42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Nitrone S; Keirs D; Moffat D; Overton K Enantioselective synthesis of optically pure (R)- and (S)-β-lysine via nitrone cycloaddition. J. Chem. Soc., Chem. Commun 1988, 654–655. [Google Scholar]
- (29).Nasopoulou M; Georgiadis D; Matziari M; Dive V; Cedex GYV A Versatile Annulation Protocol toward Novel Constrained Phosphinic Peptidomimetics. J. Org. Chem 2007, 72, 7222–7228. [DOI] [PubMed] [Google Scholar]
- (30).Allali-hassani A; Wasney GA; Siarheyeva A; Hajian T; Vedadi M; Cheryl H Fluorescence-Based Methods for Screening Writers and Readers of Histone Methyl Marks. J. Biomol. Screen 2012, 17, 71–84. [DOI] [PubMed] [Google Scholar]
- (31).Morrison JF The Slow-Binding and Slow, Tight-Binding Inhibition of Enzyme-Catalysed Reactions. Trends Biochem. Sci 1982, 7, 102–105. [Google Scholar]
- (32).Copeland RA Tight Binding Inhibitors In Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists, 2nd Ed., John Wiley & Sons: New Jersey, 2013; pp245–285. [PubMed] [Google Scholar]
- (33).Dong C; Dong G; Li L; Zhu L; Tempel W; Liu Y; Huang R; Min J An Asparagine/Glycine Switch Governs Product Specificity of Human N-Terminal Methyltransferase NTMT2. Commun. Biol 2018, 1, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Rizzo PA Del; Dirk, L. M. A.; Strunk, B. S.; Roiko, M. S.; Brunzelle, J. S.; Houtz, R. L.; Trievel, R. C. SET7 / 9 Catalytic Mutants Reveal the Role of Active Site Water Molecules in Lysine Multiple Methylation J. Biol. Chem 2010, 285, 31849–31858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Chuikov S; Kurash JK; Wilson JR; Xiao B; Justin N; Ivanov GS; Mckinney K; Tempst P; Prives C; Gamblin SJ; Barlev NA; Reinberg D Regulation of P53 Activity through Lysine Methylation. Nature 2004, 432, 353–360. [DOI] [PubMed] [Google Scholar]
- (36).Couture J; Collazo E; Brunzelle JS; Trievel RC Structural and Functional Analysis of SET8 , a Histone H4 Lys-20 Methyltransferase. 2005, 1455–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Wu H; Min J; Lunin VV; Antoshenko T; Dombrovski L; Zeng H; Allali-Hassani A; Campagna-Slater V; Vedadi M; Arrowsmith CH; Plotnikov AN; Schapira M Structural Biology of Human H3K9 Methyltransferases. PLoS One 2010, 5, e8570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Oerum S; Catala M; Atdjian C; Brachet F; Barraud P; Iannazzo L; Droogmans L; Ethève-quelquejeu M; Tisné C Bisubstrate Analogues as Structural Tools to Investigate m A Methyltransferase Active Sites. RNA Biol. 2019, 16, 798–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Taylor AP; Swewczyk M; Kennedy S; Trush VV; Wu H; Zeng H; Dong A; Freitas R. F. De; Tatlock J; Kumpf RA; Wythes M; Casimiro-Garcia A; Denny RA; Parikh MD; Li F; Barsyte-Lovejoy D; Schapira M; Vedadi M; Schapira M; Brown PJ; Arrowsmith CH; Owen DR Selective, Small-Molecule Co-Factor Binding Site Inhibition of a Su(Var)3 − 9, Enhancer of Zeste, Trithorax Domain Containing Lysine Methyltransferase. J. Med. Chem 2019, 62, 7669–7683. [DOI] [PubMed] [Google Scholar]
- (40).Babault N; Allali-Hassani A; Li F; Fan J; Yue A; Ju K; Liu F; Vedadi M; Liu J; Jin J Discovery of Bisubstrate Inhibitors of Nicotinamide N -Methyltransferase (NNMT). J. Med. Chem 2018, 61, 1541–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Feng Y; Xie N; Jin M; Stahley MR; Stivers JT; Zheng YG A Transient Kinetic Analysis of PRMT1 Catalysis. Biochemistry 2011, 50, 7033–7044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Barsyte-Lovejoy D; Li F; Oudhoff MJ; Tatlock JH; Dong A; Zeng H; Wu H; Freeman SA; Schapira M; Senisterra GA; Kuznetsova E; Marcellus R; Allali-Hassani A; Kennedy S; Lambert P; Couzens AL; Aman A; Gingras Al; Al-Awar R; Fish PV; Gerstenberger BS; Roberts L; Benn CL; Grimley RL; Braam MJ; Rossi FMV; Sudol M; Brown PJ; Bunnage ME; Owen DR; Zaph C; Vedadi M; Arrowsmith CH (R)-PFI-2 Is a Potent and Selective Inhibitor of SETD7 Methyltransferase Activity in Cells. Proc. Natl. Acad. Sci 2014, 111, 12853–12858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Debler EW; Jain K; Warmack RA; Feng Y; Clarke SG; Blobel G A Glutamate/Aspartate Switch Controls Product Specificity in a Protein Arginine Methyltransferase. 2016, 113, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Chen D; Li L; Diaz K; Iyamu ID; Yadav R; Noinaj N; Huang R Novel Propargyl-Linked Bisubstrate Analogues as Tight-Binding Inhibitors for Nicotinamide N-Methyltransferase. J. Med. Chem 2019, 62, 10783–10797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Winter G; Ashton A Fast_dp. Methods Zenodo. 2014, 55, 81–93. [Google Scholar]
- (46).Adams PD; Afonine PV; Bunkóczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung LW; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH PHENIX: A Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr. Sect. D Biol. Crystallogr 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Emsley P; Lohkamp B; Scott WG; Cowtan K Features and Development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.