

Abstract
This review is the counterpart of a 2018 Chemical Reviews article that examined the mechanisms of chemical glycosylation in the absence of stereodirecting participation. Attention is now turned to a critical review of the evidence in support of stereodirecting participation in glycosylation reactions by esters from either the vicinal or more remote positions. As participation by esters is often accompanied by ester migration, the mechanism(s) of migration are also reviewed. Esters are central to the entire review, which accordingly opens with an overview of their structure and their influence on the conformations of six-membered rings. Next the structure and relative energetics of dioxacarbeniun ions are covered with emphasis on the influence of ring size. The existing kinetic evidence for participation is then presented followed an overview of the various intermediates either isolated or characterized spectroscopically. The evidence supporting participation from remote or distal positions is critically examined and alternative hypotheses for the stereodirecting effect of such esters is presented. The mechanisms of ester migration are first examined from the perspective of glycosylation reactions, and then more broadly in the context of partially acylated polyols.
Graphical Abstract

1. Introduction
In a recent issue of this journal we reviewed the mechanisms of glycosylation reactions with emphasis on the experimental evidence for reactions taking place at the SN1-SN2 interface.1 To complement that review we now present and critically assess the evidence in support of glycosylation reactions taking place with participation by a neighboring (vicinal, proximal) or a more remote (distal) ester on the framework of the glycosyl donor, which we term remote or distal participation. As ester migration is frequently a side reaction in glycosylations conducted with the aid of neighboring group participation and more generally in partially acylated carbohydrates, we also review the mechanisms of migration and the evidence supporting them.
The concepts of neighboring group participation and anchimeric assistance are frequently used interchangeably in the literature. In this review we espouse a narrower usage of Winstein’s term,2 as defined in the IUPAC Gold Book and other compendia and reviews of physical organic chemistry,3–5 according to which anchimeric assistance denotes an increase in rate due to neighboring group participation.
The notion of stereodirecting neighboring group participation in glycosylation reactions is long-known and was discussed by Frush and Isbell,6 before the broad general concept was introduced to organic chemistry as a whole by the Winstein laboratory,7 and has been reviewed with varying degrees of rigor multiple times.8–21 It has been practiced as a tool in glycosylation reactions since the discovery of the Koenigs-Knorr reaction22 and as such the number of examples of its use, even in a single year,23 are far too numerous to list here. Stereodirecting participation by more remote groups has also been very widely invoked in glycosylation reactions, since its introduction in the 1970’s,24,25 and has been reviewed in recent years,16,26,27 such that, again, no attempt is made to comprehensively treat all implied examples of it. Rather we present a critical survey of the literature covering the experimental evidence for or against participation, neighboring or distal, and the mechanisms of the associated migrations.28,29 We also discuss alternative mechanisms for the stereodirecting effects of remote esters that do not involve participation in the classical sense. Ester migration between hydroxy groups of partially acylated carbohydrates is an important corollary to migration during glycosylation: it has been briefly reviewed recently28 but is covered here in detail with emphasis on mechanism.
Beyond the concept of ester migration during participation, it is commonly known that acyl groups in organic molecules are prone to intramolecular migration between the hydroxy groups, as first demonstrated for carbohydrates by Fischer in 1920.30 Several studies providing closer insights into the actual migration process have been performed since,28 mostly in monosaccharide derivatives, although also larger carbohydrate molecules have been addressed, albeit less frequently.31–34 The often spontaneous migration, particularly under basic conditions, significantly hampers the use of partially acylated carbohydrate derivatives in synthesis and must always be considered in all isolation, characterization and identification processes of both synthetic compounds and natural products.28,35–39
As esters are central to the entire review we begin with an analysis of the configuration and conformation of the ester group and of the influence of esters on the conformations of pyranoside rings. We continue with an overview of the structure and relative energetics of cyclic dioxocarbenium ions in general before addressing participation by esters in glycosylation reactions from all aspects. Consideration of the evidence presented for and against participation then leads to a critical evaluation of the likelihood of participation by remote esters and the presentation of alternative hypotheses in explanation of the experimental results. We then continue with a discussion of ester migration in the course of glycosylation reactions before addressing the broader issue of ester migration in partially acylated carbohydrates from a mechanistic viewpoint. Finally, a brief conclusion is offered.
2. Conformations of Esters and the Influence of Esters on the Conformational Dynamics of Sugars
It is well established from the examination of crystallographic databases that carboxylate esters overwhelmingly adopt the Z-conformation about the C-O bond (Figure 1).40–42 Albeit not strictly correct, esters in the Z-conformation are commonly called trans-esters. Spectroscopic and computational studies agree with the crystallographic data and suggest Z,E free energy differences of ~5 kcal.mol−1 for methyl formate and ~8 kcal.mol−1 for methyl acetate in the gas phase or in low polarity solvents.43–50 The predominance of the Z-conformation is such that in a 2005 survey of X-ray structures of secondary acetates, only 13 of almost 7000 acetates adopted the E-conformation.42 The difference between the two conformations is less pronounced in polar media owing to the higher polarity of the E-conformation.
Figure 1.
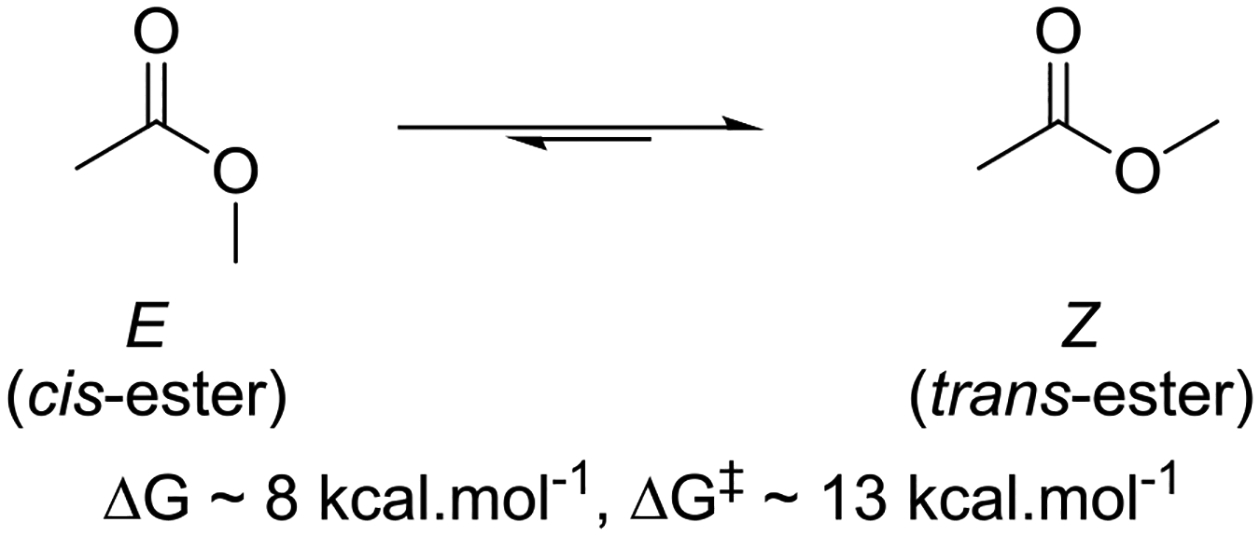
E and Z-conformations of methyl acetate.
Gas phase computations indicate a barrier of ~13 kcal.mol−1 for the interconversion of the Z- and E- conformers of methyl acetate,51 consistent with the experimental barrier of 13.8 kcal.mol−1 separating the Z- and E- conformers of formic acid.52
Following on from a limited 1965 survey by Mathieson,40 Schweitzer and Dunitz surveyed X-ray structures of secondary alkyl esters in the Cambridge Crystallographic Database in 1982.41 They found that in 102 of the approximately 170 structures considered, the α C-H bond of the ester was within 30° of eclipsing the carbonyl oxygen, and was within 60° of it in the remaining structures (Figure 2). This preference for an eclipsed conformation in secondary esters is distinct from that in both primary and tertiary esters, both of which prefer the staggered conformation.41
Figure 2.
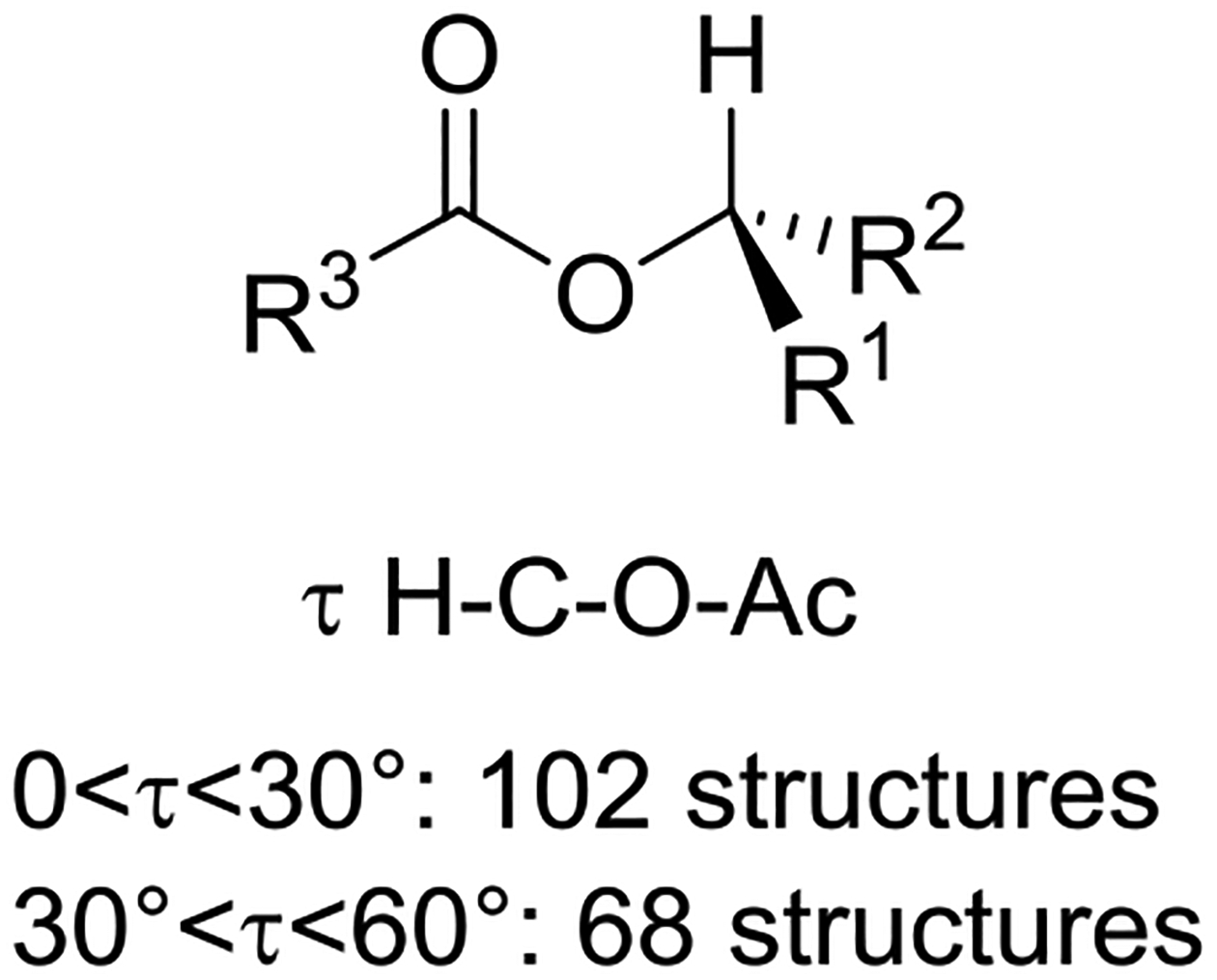
Preferred eclipsed conformation of secondary esters.
A subsequent 2005 survey by Anderson and coworkers covered some 1546 structures of cyclic secondary alkyl acetates resulting in the conclusion that the H-C-O-Ac torsion angle τ is a function of the degree of substitution of the two vicinal positions flanking the ester function.42 In the absence of such substitution, the distribution of τ values displayed in Figure 3 was found indicating the typical H-C-O-C=O torsion angle to be between 30 and 40° in such cases. When one flanking equatorial substituent was present the mean torsion angle τ was 18.8° for the 192 cases when the CO group was rotated toward the substituent, and 27.8° for the 330 cases when it was rotated away from the substituent.
Figure 3.

Distribution of H-C-O-C(=O) torsion angles τ in secondary cyclic acetates lacking flanking substituents.
The presence of a flanking equatorial substituent on either side of the ester function, as is commonly the case in carbohydrate esters, afforded a much narrower distribution of τ that approached the eclipsed conformation. Thus, for the 493 equatorial acetates the mean τ was 11.9° while for the 63 cases of axial acetates it was 17.8° (Figure 4).
Figure 4.
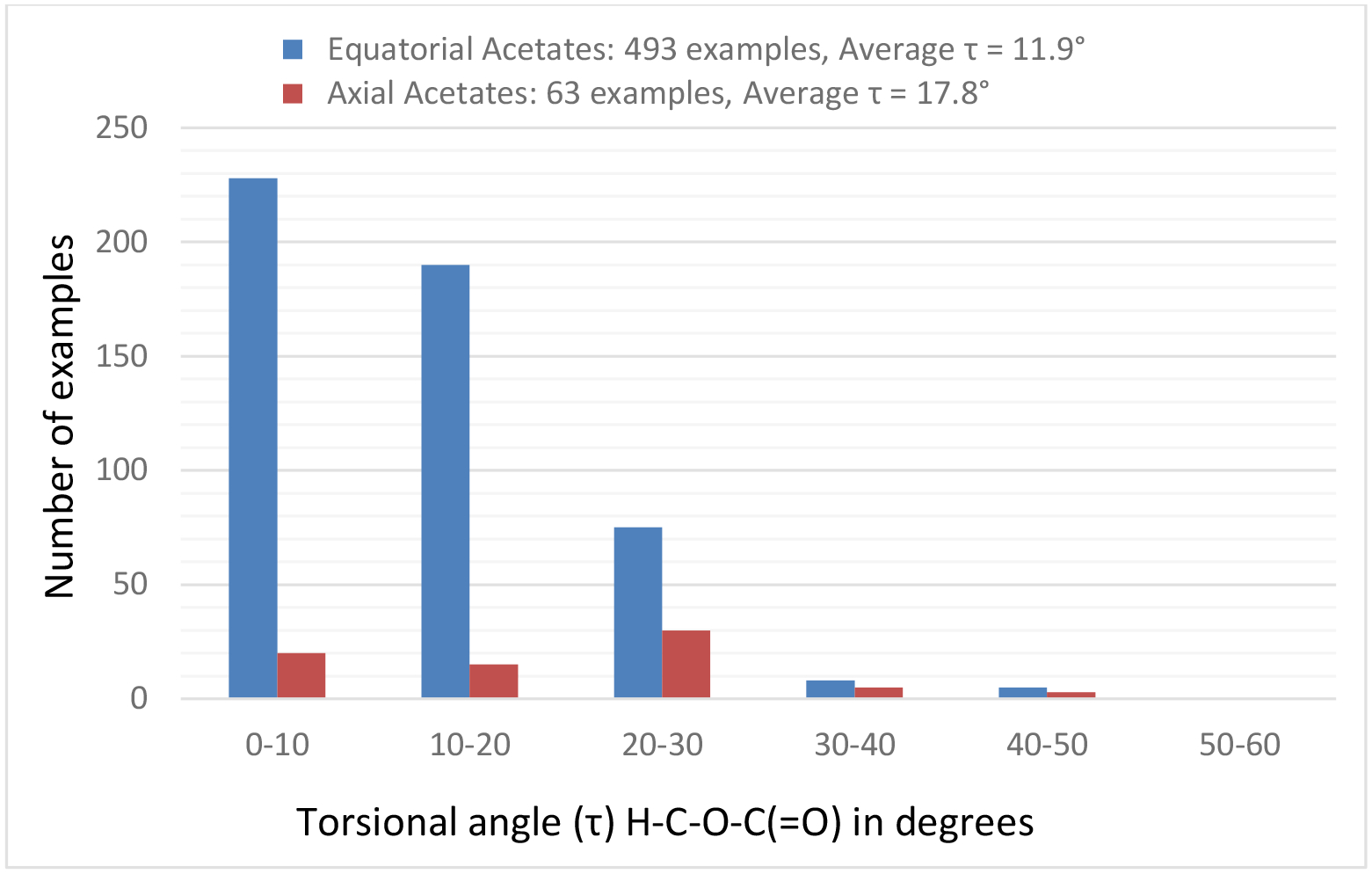
Distribution of H-C-O-C(=O) torsion angles τ in secondary cyclic acetates with two flanking substituents.
Calculated energy barriers for rotation through a perfectly eclipsed conformation were in all cases studied ≤1 kcal.mol−1, whereas those for rotation to the much less common anti-conformation of the ester bond in which τ = 180° varied between 4 and 12 kcal.mol−1 depending on the degree of substitution.42
Finally, with the aid of computed conformations for a limited range of cyclohexyl acetates with differing substitution patterns at the vicinal positions, a Karplus relationship was derived for the 3J heteronuclear coupling constant between the α-proton in the ester moiety and the carbonyl carbon 3J(1H-C-O-13C=O).42 However, as the range of coupling constants for 0<τ<60° was small (2.5–4.6 Hz) caution should be exercised in the extension of these values to more complex systems, particularly to systems carrying electronegative substituents in view of the well-known influence of the latter on the magnitude of coupling constants.53,54
It is well established that cyclohexane rings carrying acyloxy substituents exhibit a greater propensity for the axial conformation than do the corresponding alkoxy substituted systems. For simple ester derivatives of cyclohexanol, Kleinpeter and coworkers established by a combination of low temperature NMR studies and ab initio computations that the proportion of the axial conformer increases with the electronegativity of the ester, such that the cyclohexyl benzoates contain approximately 20% of the axial conformer.55,56 It was reasoned that the axial conformation is stabilized by hyperconjugative interactions with the axial vicinal protons (σC-H → σ*C-O hyperconjugation), which are greater than the corresponding hyperconjugative interactions with the corresponding C-C bonds in the equatorial conformer (σC-C → σ*C-O hyperconjugation) (Figure 5). Further, it was found in a series of aliphatic esters that increasing the steric bulk of the ester results in an increased population of the axial conformer.55 While the exact nature of the steric interactions destabilizing the equatorial ester are not clear, it is evident that they are greater than any increased steric destabilization of the axial conformer, consistent with the minimally strained nature of cyclohexanes carrying axial C-O bonds found in a survey of X-ray crystal structures.57
Figure 5.

σC-H → σ*C-O and σC-C → σ*C-O Hyperconjugative stabilization of axial and equatorial esters.
These electronic trends were accentuated in the esters of trans-1,4-cyclohexanediol, which was attributed by the authors to increased polarity,58 and by Alabugin to unfavorable double hyperconjugation in the equatorial conformer where two σ*C-O orbitals compete for hyperconjugation with the same σC-C orbital. In contrast, esterification of trans-1,2-cyclohexanediol leads to an enhanced preference for the diequatorial conformer, with respect to the corresponding dimethyl ether, because of the reinforcement of the gauche effect by the increased electronegativity.59 To our knowledge, there is no published work on the cyclohexane-1,3-diols. However, by extrapolation of the trends seen in the corresponding dihalides, where computational work suggests that 1,3-cis-difluoride exhibits a significantly greater diaxial population than the corresponding dichloride and dibromide,60 it can be concluded that esterification in the cis-series will lead to an increased population of the diaxial conformer.
The presence of a sp2-hybridized atom in the cyclohexyl moiety as in 4-exo-methylenecyclohexyl benzoate also reduces the energy difference between the axial and equatorial conformers, with respect to the saturated system. In the 4-acyloxycyclohexanones the axial conformer is even further stabilized: it has been found that 4-(4-chlorobenzoyloxy)cyclohexanone is favored by 0.71 kcal.mol−1 over the equatorial conformer in deuteriochloroform solution, and crystallizes as the axial conformer.58 Clearly, the flattening of the ring by the insertion of the sp2 center has a stabilizing effect on the axial conformer, and this is accentuated by the electron-withdrawing effect of the ketone, leading the authors to suggest a polar effect as the main factor at play. Importantly, in the crystal structure the ester moiety of 4-(4-chlorobenzoyl)cyclohexanone adopts the standard conformation with the carbonyl group eclipsing the C-H bond (Figure 6). Any electrostatic stabilization of this axial conformation therefore involves the electron density on the ester oxygen and not that on the carbonyl oxygen. Pertinently, the presence of the sp2 center also serves to lower the barrier to ring inversion in both the 4-exo-methylene and 4-keto series of cyclohexyl esters.58
Figure 6.
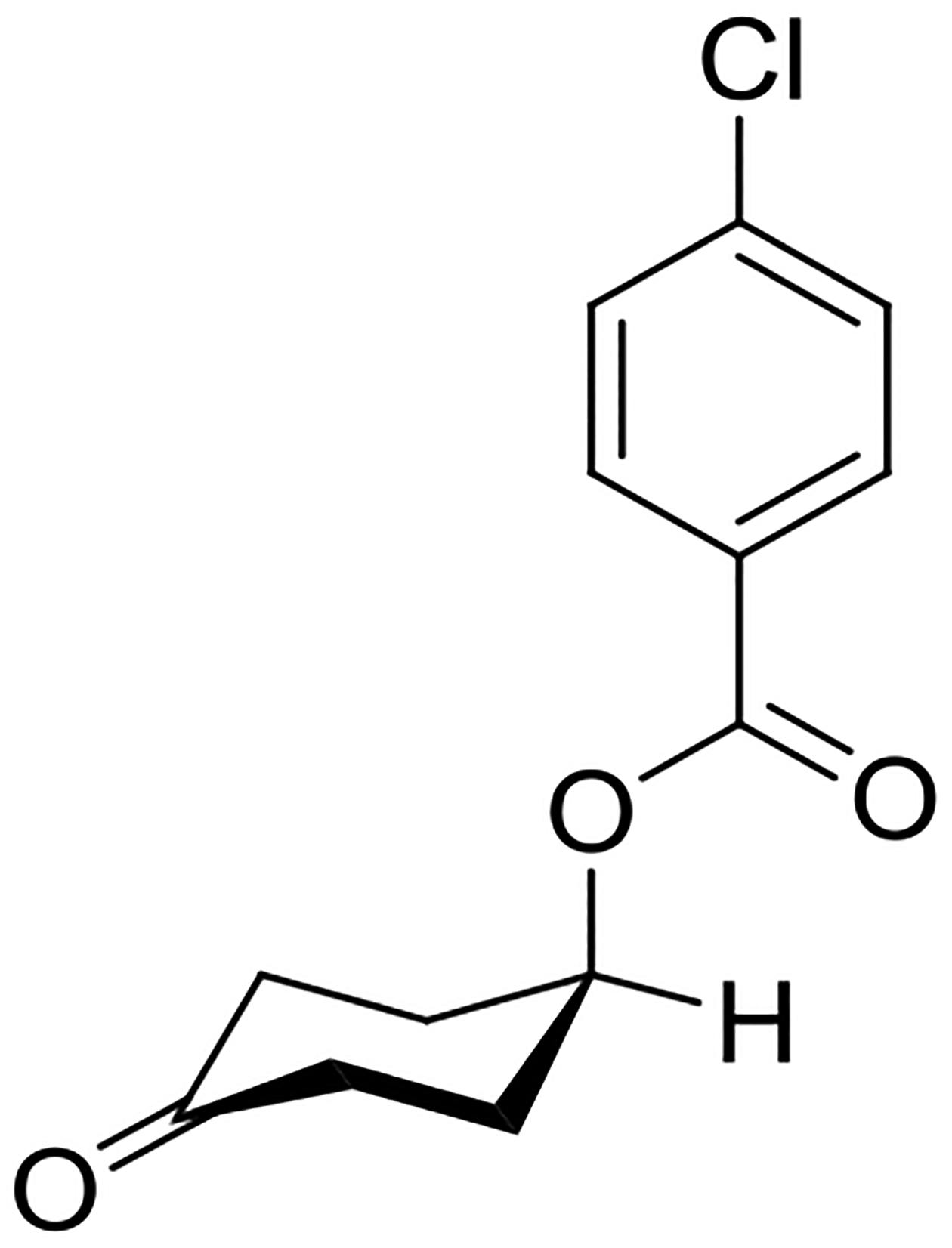
Conformation of 4-(4-chlorobenzoyloxy)cyclohexanone adopted in the crystal structure.
The increased propensity of saturated six-membered rings for conformations carrying axial ester groups relative to comparable ethers manifests itself most clearly in peresterified pentopyranosyl and related structures.61–63 Thus, methyl 2,4-di-O-methyl-3-deoxy-β-L-erythropentopyranoside preferentially adopts the all equatorial conformation, whereas the corresponding diacetate takes up a predominant conformation that is close to the all axial one, in all solvents studied (Figure 7).64
Figure 7.
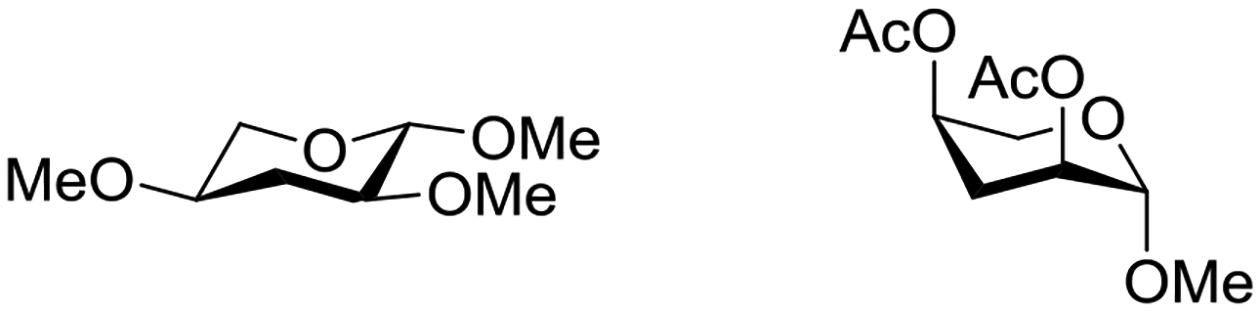
Approximate conformations of the dimethyl ether and diacetyl ester of methyl 3-deoxy-β-L-erythropentopyranoside.
The effect is most widely studied in the peresterified β-D-xylopyranosyl halides, for which it has long been reported that the 1C4 conformation with all substituents axial is preferred in solution.61,62 This early and widespread assignment of the 1C4 conformation for many xylopyranosides was critically re-examined by Lichtenthaler and Lindner who found substantial deviation from the ideal chair conformation in most cases.65 Thus, for example, per-O-benzoyl β-D-xylopyranosyl chloride and the corresponding methyl glycoside adopt the 2So twist boat conformation in the crystal, while the fluoride, bromide and benzoate approach the ideal 1C4 chair but are nevertheless distorted toward the 5Ho half-chair as manifested by substantial deviations (≤42°) from the ideal 180° 1,2-trans-diaxial torsion angles (Figure 8). In solution, on the basis of the averaged vicinal coupling constants, the same authors considered the various per-O-benzoyl β-D-xylopyranosyl derivatives to exist as interconverting mixtures of 4C1 and 1C4 conformers whose relative proportions depended on the anomeric substituent and the solvent.65 In a more recent treatise, Grindley reported on the percentage of the 4C1 conformer present for each member of a series of pentopyranosyl derivatives (α- and β-anomers, with and without esterification) without specifying the other conformations that make up the balance (Table 1).63 Whatever the actual nature of the conformations adopted in the various β-D-pentopyranosyl systems, it is clear that the anomeric effect is the main force away from the 4C1 and toward the 1C4 conformation. This conclusion is reinforced by the fact that per-O-benzoyl-1,5-anhydroxylitol, which lacks an anomeric substituent, takes up the 4C1 conformation with all substituents equatorial in the crystal and only populates the inverted 1C4 conformation to the extent of 19% in d6-acetone solution.66
Figure 8.
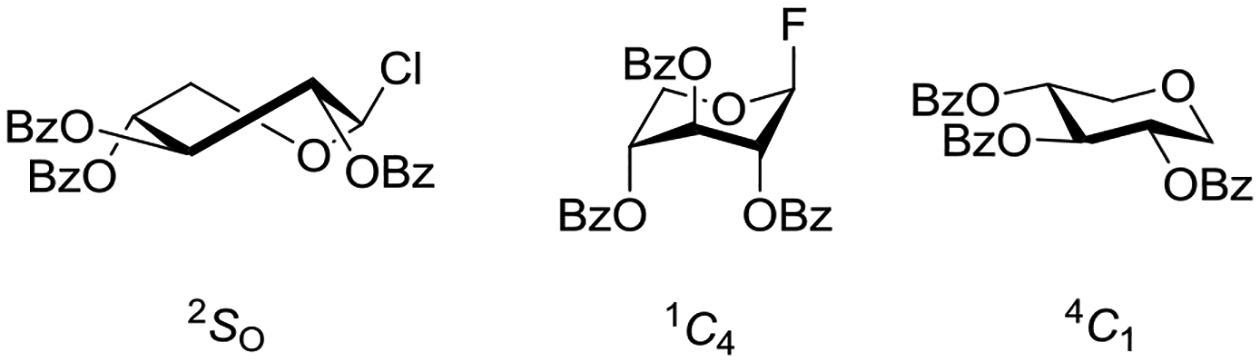
X-ray crystallographically-derived conformations of per-O-benzoyl xylopyranosyl derivatives highlighting the importance of the anomeric substituent.
Table 1.
| Configuration | Methyl 2,3,4,-tri-O-acetyl | 1,2,3,4-Tetra-O-acetyl | 2,3,4-Tri-O-acetyl chloride |
|---|---|---|---|
| β-arabino | 3 | 4 | 2 |
| β-ribo | 39 | 43 | 6 |
| β-xylo | 81 | 72 | 21 |
Inverted conformations placing the anomeric substituent in a pseudoequatorial position and the esters in pseudoaxial ones have also been widely reported for peresterified pyranosyl pyridinium and related imidazolinium ions, and are the origin of the now abandoned concept67–71 of the reverse anomeric effect.61–63,72 Thus for example, tri-O-acetyl-α-D-xylopyranosyl pyridinium bromide is reported to adopt the 1C4 conformation in the crystal to accommodate the bulky pyridinium group in an equatorial position, thereby placing all three esters axial (Figure 9).73 Closer inspection, however, reveals distortion from the ideal chair conformation such that the O3-C3-C4-O4 torsion angle is ~170°. Likewise, neutral tri-O-acetyl-α-D-xylopyranosyl imidazole adopts a distorted 1C4–like conformation in the crystal, so as to place the bulky imidazole ring (unprotonated) in an equatorial position; the distortion from the ideal chair of this structure is apparent from the O3-C3-C4-O4 torsion angle of 161°.74 It is noteworthy that all of the ester groups in the pyridinium salt adopt the standard conformation in which the carbonyl group is more or less eclipsed with the C-H bond. The corresponding unprotected xylopyranosyl pyridinium salts populate the inverted conformations to a lesser extent.75
Figure 9.
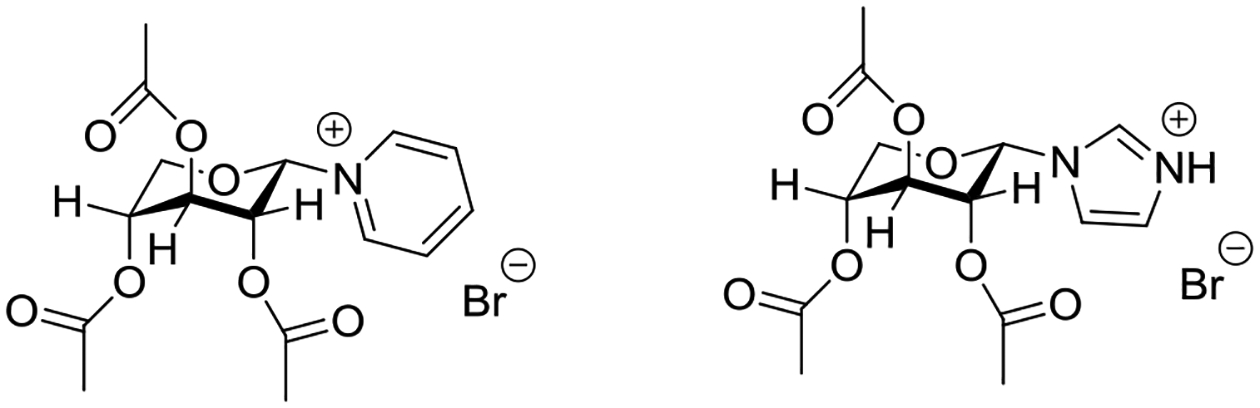
X-ray crystallographic conformations of tri-O-acetyl-α-D-xylopyranosyl pyridinium bromide and tri-O-acetyl-α-D-xylopyranosyl imidazole
Careful study of the solution conformations of the glycosyl imidazolinium ions by Perrin and coworkers led to the conclusion that the populations of 1C4 conformations in such molecules assigned by previous investigators are not reliable.68 Inaccurate limiting coupling constants for the different conformations, required to compute the relative populations, are one likely reason for the apparent errors in the earlier work as noted earlier by Finch and Nagpurkar.75 Other contributing factors include distortions from the ideal 1C4 chair, either by twisting or flattening as apparent in the various X-ray structures, and the differing basicities of the pseudo-axial and equatorial anomeric heterocycles.68
It is appropriate to recall that the preferred diequatorial conformation of trans-vicinal esters in the cyclohexanes is reinforced by the gauche effect,59 and that the inverted diaxial conformer only benefits from a single σC-H → σ*C-O hyperconjugation that stabilizes a single axial ester group.55 Taken to the extreme in the all axial conformation of a 1,2,3-tri-acyloxy substituted system, such as the 1C4 conformation of the xylopyranosyl derivatives, the central ester does not benefit at all from σC-H → σ*C-O hyperconjugation. As such, although the xylopyranoside configuration is the most widely studied class of inverted chair conformations, perhaps because it is the most striking with a full set of pseudo-axial esters, it should not be the configuration in which the effect is most pronounced. The corresponding inverted arabino and ribopyranosyl derivatives retain both the stabilizing gauche effect and at least one σC-H → σ*C-O hyperconjugation to an axial C-O bond in the 1C4 conformation, as is apparent from the much reduced population of the 4C1 conformers manifested by the β-arabino and β-ribo-pyranosides as compared to their xylo-isomers (Table 1).61,63 This same tendency toward the increased population of an “inverted” conformation is seen in the protonated α-mannopyranosyl imidazolides as compared to the corresponding α-glucopyranosyl imidazolides.75
Structural studies of trichloroacetimidates are rare, but an X-ray crystal structure of an iduronic acid donor reveals a standard ester-like conformation in which the C=NH double bond eclipses the anomeric C-H bond (Figure 10). As expected, all esters in this molecule are in the standard trans-conformation with the carbonyl C=O eclipsing the equatorial C-H bond on the ring.
Figure 10.
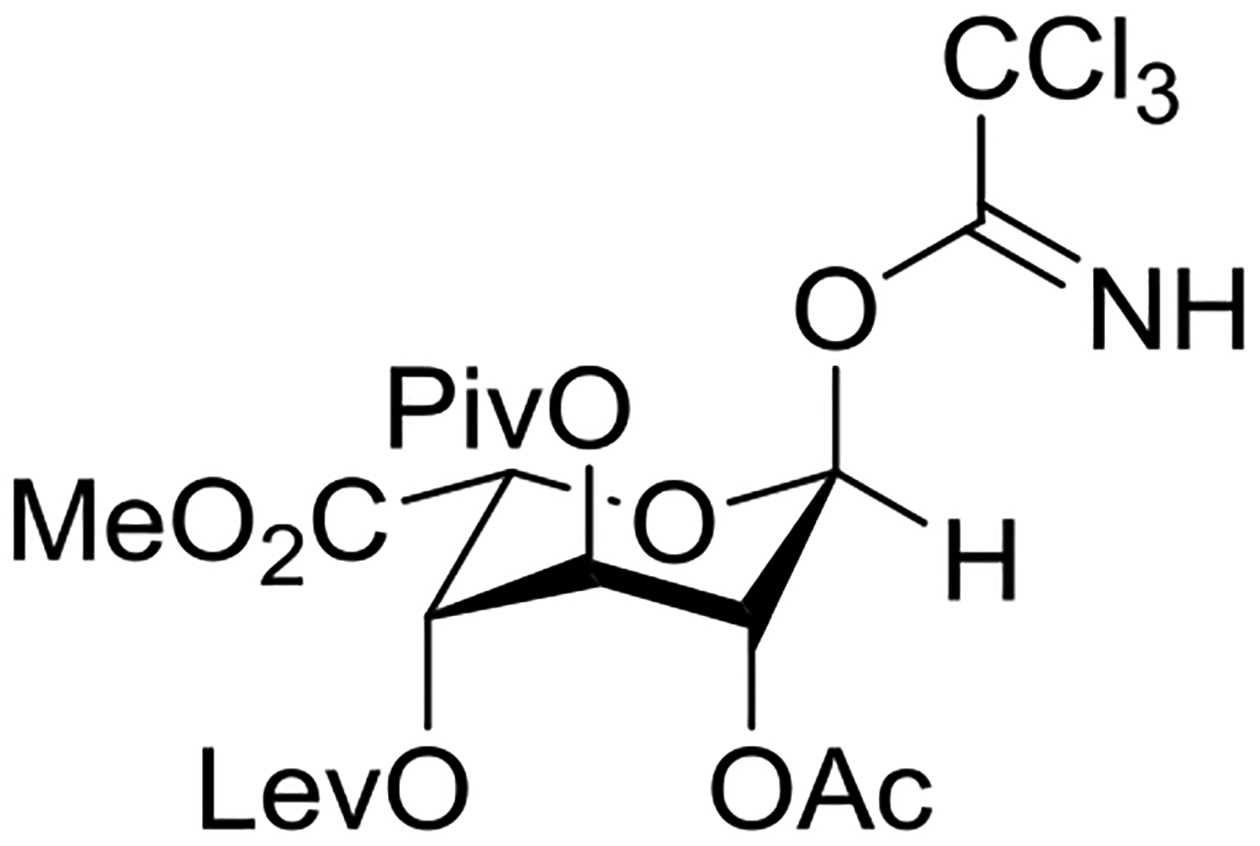
X-ray crystallographically-derived structure of an iduronyl trichloroacetimidate showing the conformation of the trichloroacetimdate moiety.
3. Structures of Dioxocarbenium Ions
The X-ray crystal structure of the 2-methyl-1,3-dioxolan-2-ylium ion, the parent structure common to vicinal participation by acetate esters, was determined by Paulsen and Dammeyer as its perchlorate salt.76 Obtained by treatment of acetoxy-2-bromoethane with silver perchlorate in toluene, this salt was crystallized from acetonitrile by trituration with ether and tetrachloromethane and had a melting point of 176.5–178°C. In the solid state, the dioxolanylium ring is planar and carries two identical sets of eclipsed vicinal C-H bonds. In deuterioacetonitrile solution C2 resonates at δ 191.8 in the 13C NMR spectrum (Figure 11). The 13C NMR spectra of multiple other mono-, di-, tri- and tetra-substituted 2-methyl-1,3-dioxolan-2-ylium ions prepared in a similar manner were also reported by Paulsen and Dammeyer,76 and more detailed analyses were presented subsequently by Paulsen and Schüttpelz.77 cis-Fusion of the parent structure to cyclopentane affords a structure, whose tetrafluoroborate salt revealed little distortion from planarity in the heterocyclic ring (O1-C5-C4-O3 = 5°) and an envelope conformation for the all carbon ring with an endo-pucker of the flap in its X-ray crystal structure (Figure 11).78 cis-Fusion to a cyclohexane ring on the other hand results in substantial distortion of both rings: the heterocyclic ring of the perchlorate was found to adopt a twist conformation with a O1-C5-C4-O3 torsion angle of 25.9° while the cyclohexane ring was distorted from a chair toward a half-chair.79 It has been reported that 1,3-dioxolan-2-ylium rings trans-fused to six-membered rings have been obtained by dehydration of the corresponding trans-2-acetoxy alcohols with trifluoromethanesulfonic acid or directly from the corresponding trans-1,2-diacetoxy systems also with trifluoromethanesulfonic acid.80 However, the formation of these substances was not confirmed directly, but rather inferred from their hydrolysis products. The 1H and 13C NMR parameters of 2-methyl-1,3-dioxan-2-ylium cations obtained from acetoxy-3-bromoalkanes were also investigated by Paulsen and Schüttpelz and interpreted with the aid of semi-empirical calculations (Figure 11).77 The parent system was considered to adopt a rapidly inverting envelope conformation in which the flap was raised some 10° out of the plane of the other five atoms. Substitution in the propylene chain of the 1,3-dioxan-2-ylium ring was considered to lead to a preference for conformations with pseudoequatorial groups, but this preference was computed to be considerably lower than in the corresponding cyclohexanes.
Figure 11.

Preferred conformations of monocyclic and cis-fused bicyclic dioxalan-2-ylium ions and of a monocyclic dioxan-2-ylium ion and their key 13C NMR resonances.
In an impressive series of studies summarized in a 1976 review article, Paulsen and coworkers studied the rearrangements of peresterified polyols and the related intermediate acyloxyalkyl dioxolanylium and dioxanylium ions.81 Such experiments enabled them to determine, inter alia, the relative stabilities of various 2-substituted-1,3-dioxolan-2-yl-ium ions as a function of the substituent at the 2-position, and also to tease out the influence of ring size on stability. Pertinently, the equilibrium between the five and six-membered cyclic ions was found to favor the smaller to the extent that only one form was observable, leading to the conclusion that dioxolanylium rings are more stable than dioxanylium rings (Figure 12).81,82 A similar conclusion regarding the relative stabilities of dioxolanylium and dioxanylium ions was reached by Larsen and Ewing who measured the heats of formation of the cyclic ions on dissolution of the corresponding open chain acyloxyalkenes.83
Figure 12.
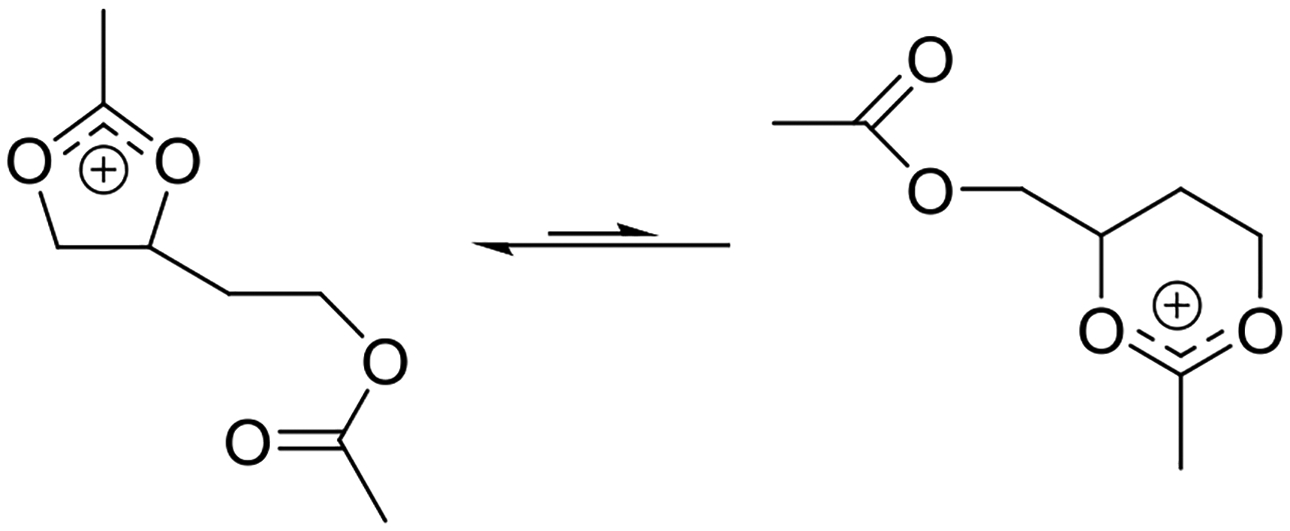
Equilibration reveals the thermodynamic stability of a dioxolanylium ion over a dioxanylium ion.
Ramsey and Taft recorded the NMR spectra of a series of mono-, di-, and trialkoxyalkyl carbenium ions in sulfuric acid, sulfur trioxide in sulfuric acid and in trifluoroacetic acid and measured a barrier to rotation of 11±4 kcal.mol−1 in the 1,1-dimethoxyethyl carbenium ion.84 Further NMR studies of tetrafluoroborate salts were conducted by Borch in deuteriochloroform solution, and corroborated by Dusseau and coworkers who generated the dialkoxyalkyl cations in the form of bromide salts from the reaction of bromine with the corresponding orthoesters in liquid sulfur dioxide.85 The NMR spectra of the 1,1-diethoxyethyl cation at −30°C showed a single isomer with non-equivalent ethyl groups consistent with the E,Z-isomer (Figure 13). The NMR spectra of the corresponding dialkoxymethyl cations displayed a ~ 90:10 mixture of the predominant E,Z and a minor isomer, presumably the E,E-isomer.86 According to Deslongchamps the Z,Z-isomer benefits from two stereoelectronic interactions arising from the antiperiplanar nature of the remaining oxygen lone pairs and the geminal C-O bonds, but suffers from unfavorable steric interactions. The E,E-isomer on the other hand suffers from a repulsive interaction between the two lone pairs; consequently the E,Z-isomer in which both steric and dipolar interactions are minimized, and one stereoelectronic interaction retained, is the most stable configuration.87
Figure 13.
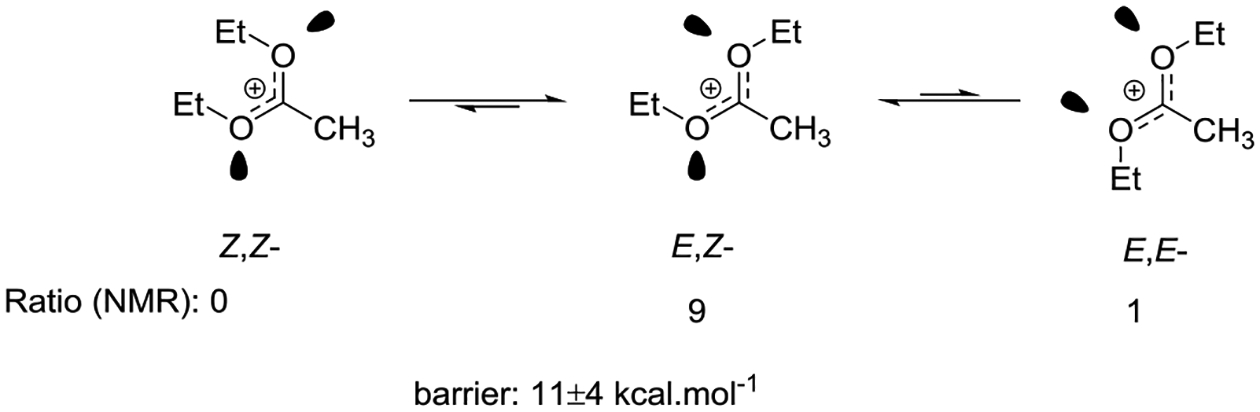
Relative energies and barrier to inversion in the 1,1-diethylethyl carbenium ion.
Woerpel and co-workers prepared and obtained the X-ray crystallographic structures of two dialkoxycarbenium ions; both adopted the E,Z-configuration and not the alternative E,E-isomer (Figure 14).88 DFT-computational work by Whitfield, Boons and their coworkers with a series of related six-membered cyclic 1-alkoxy oxocarbenium ions are consistent with this observation, and indicate that the E,Z-isomer is preferred over the E,E-configuration by >2 kcal.mol−1 in all cases studied.89 Of further note in the X-ray (and NMR solution) structure of the benzyloxy-substituted ion is the pseudoaxial orientation adopted by the remote benzyl ether. As no evidence for actual bridging was observed, this is considered evidence for the through-space electrostatic stabilization of the positive charge by the C-O bond88 to which we return later.
Figure 14.

X-ray crystallographically-derived conformations of two tetrahydropyran-based dioxocarbenium ions.
4. Kinetic Studies on Participation
The importance of kinetics in the determination of reaction mechanisms cannot be over-emphasized, with glycosylation reactions being no exception to the rule.1 With regards to the effects of participation in glycosylation reactions, in recent years efforts have focused mainly on the determination of ratios of recovered starting material or of products in competition reactions. Earlier investigators, however, did conduct extensive kinetic studies on solvolysis reactions of esterified donors as summarized in various books and reviews.8,13,18 Unfortunately, these early studies were conducted under solvolytic conditions that are not relevant to modern oligosaccharide synthesis. Pertinent examples are nevertheless discussed here as they illustrate how mechanisms can change significantly with changes in reagents and conditions.
Schroeder and coworkers determined initial rate constants for the solvolysis of 2,3,4,6-tetra-O-acetyl-α-D-glucopyranosyl bromide in a series of primary and secondary alcohols as solvent, and investigated the influence of temperature and of added salts on the kinetic parameters.90 It was found that added lithium bromide (0.1 M) increased the rate of consumption of the substrate approximately twice as much as the identical concentration of added lithium chloride, while at the same time increasing the proportion of the α-glycosides in the reaction mixture. It was suggested that the added bromide might act by promoting orthoester formation, but a more reasonable explanation involves catalysis by formation of the more reactive β-bromide. In the absence of strong electrophilic catalysts, this β-bromide would be susceptible to anchimeric assistance from the 2-O-acetate resulting in a significant overall rate increase. Direct solvolysis of the β-bromide by the alcohol explains the increased formation of the α-glycoside (Table 2).
Table 2.
Effect of Added Lithium Bromide and Lithium Perchlorate on the Solvolysis of 2,3,4,6-Tetra-O-acetyl-α-D-glucopyranosyl Bromide
 | ||
|---|---|---|
| Additive (0.1 M) | ksubs consumption (106k s−1) | Initial β:α ratio |
| - | 24.2 | 18–43:5 |
| LiClO4 | 30.2 | 22–39:5 |
| LiBr | 55 | 23:19 |
In a further study of the influence of acyl protecting groups on the solvolysis of α-D-glucopyranosyl bromides, Eby and Schuerch investigated the behavior of 6-O-acetyl-2,3,4-O-(N-phenylcarbamoyl)-α-D-glucopyranosyl bromide and its per-N-methylated derivative in an acetone/methanol mixture at 23°C.91 Pseudo first order rate constants for the two derivatives showed only a minor acceleration of the N-phenyl derivative over the N-methyl-N-phenyl analog, but the product ratio was dramatically different with the former affording only the β-glycoside and the latter a 7:3 β:α mixture (Table 3). The authors considered that the N-phenyl derivative controlled the stereochemical outcome of the reaction by neighboring group participation following oxocarbenium ion generation, whereas the N-methyl-N-phenyl derivative was unable to participate, resulting in the observed stereoisomeric mixture of products. The differences in rate, which cannot be attributed to anchimeric assistance in this 1,2-cis series, suggest stabilization of an oxocarbenium ion-like transition state for solvolysis by the participating N-phenylcarbamoyl group.
Table 3.
Rate Constants and Product Ratios in the Solvolysis of N-Phenyl and N-Methyl-N-phenyl carbamoyl α-D-glucopyranosyl Bromides
 | ||
|---|---|---|
| R | k (s−1) | β:α ratio |
| NHPh | 3.24 × 10−5 | 1:0 |
| NMePh | 2.60 × 10−5 | 7:3 |
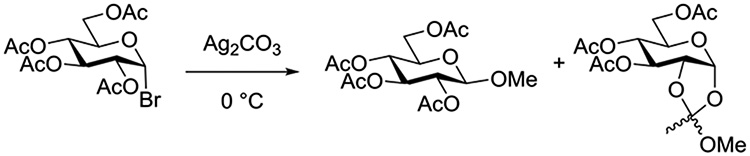
Wulff and Röhle studied the activation of acetobromoglucose by insoluble silver salts in a range of solvents. The time profile of consumption of acetobromoglucose was studied at 0°C enabling half-lives to be determined in the presence and absence of a single molar equivalent of methanol (Table 4). The differences in half-lives in the presence and absence of methanol in acetone, ethyl acetate and especially diethyl ether revealed the involvement of methanol in the rate determining step of the reaction, leading the authors to propose a bimolecular mechanism taking place on the surface of the insoluble silver salt (Figure 15).92 Reactions conducted in dichloromethane, the most widely applied solvent in modern oligosaccharide synthesis, also exhibited methanol dependent half-lives indicative of a bimolecular mechanism. The rates of reactions conducted in tetrahydrofuran were not sensitive to the addition of methanol. In further work on reactions conducted in THF with silver salicylate as promoter, Wulff and Schmidt isolated 4-bromobutyl 2,3,4,6-tetra-O-acetyl-β-D-glucopyranoside leading them to propose the participation of THF via an O-glycosyl oxonium ion (Figure 16).93 Indeed, this and a related paper by Helferich and Zirner, who isolated the same THF-derived glycoside when conducting mercuric bromide-activated glycosylations with acetobromoglucose in THF,94 provide some of the most direct and convincing evidence for the oft-proposed participation of ethers in glycosylation reactions. In contrast, the very significant change in half-lives recorded in diethyl ether (Table 4) argues strongly against participation by ether, at least, under this set of reaction conditions.
Table 4.
Half-lives and Product Distributions for the Consumption of Acetobromoglucose by Silver Carbonate With and Without Equimolar Amounts of Methanol
 | ||||
|---|---|---|---|---|
| Solvent | Product with methanol | t1/2 with MeOH (min) | t1/2 without MeOH (min) | |
| Glucoside (%) | Orthoester (%) | |||
| Et2O | 40 | 1 | 15 | >1000 |
| THF | 0 | 50 | 70 | 70 |
| EtOAc | 10 | 10 | 10 | 25 |
| CH2Cl2 | 30 | 10 | 15 | 50 |
| acetone | 5 | 30 | 10 | 15 |
Figure 15.
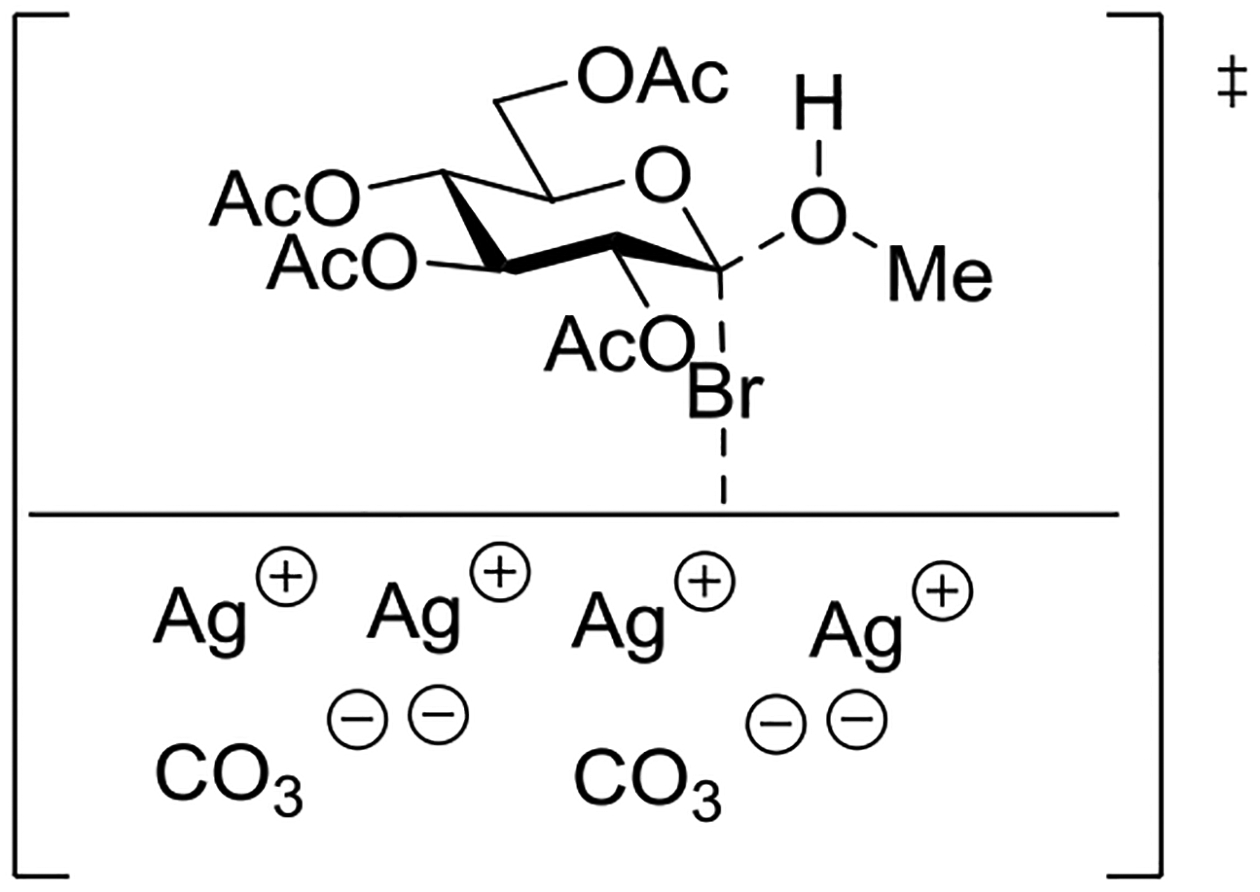
Proposed transition state for the reaction of methanol with acetobromoglucose promoted by insoluble silver salts.
Figure 16.
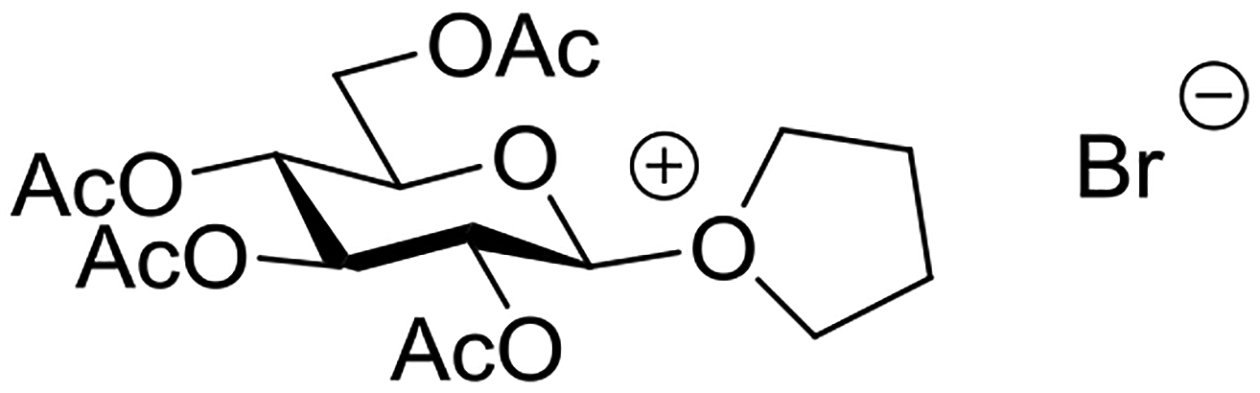
Oxonium ion intermediate proposed for participation by THF.
Wallace and Schroeder made a careful study of the reaction of mercuric cyanide promoted coupling of cyclohexanol with 2-O-acetyl-3,4,6-tri-O-methyl-α-D-glucopyranosyl bromide in 1:1 nitromethane:benzene.95 The reaction was determined to be first order in both the glycosyl bromide and mercuric cyanide but zero order in cyclohexanol, leading the authors to postulate Hg(II) assisted dissociation of the bromide ion to afford an oxocarbenium ion/bromide ion pair as the rate determining step. Analysis of the product distribution as a function of the alcohol concentration, however, revealed the importance of acceptor concentration in the product forming step: higher concentrations of cyclohexanol afforded greater proportions of the β-glycoside at the expense of those of the orthoester and of the α-glycoside which decreased. On this basis it was proposed that stereoselective trapping of an initial contact ion pair by the acceptor affording the β-glycoside competes with ring closure to the dioxalenium ion, affording the orthoester, and equilibration of the ion pair with solvent, leading to formation of the α-glycoside (Scheme 1). A time course study of the reaction conducted with 6 × 10−3 M substrate and promoter but 9 × 10−2 M cyclohexanol revealed a build-up of the orthoester in the early stages of the reaction, followed by its gradual conversion to the β-glycoside after complete consumption of the substrate (Scheme 1).95
Scheme 1.
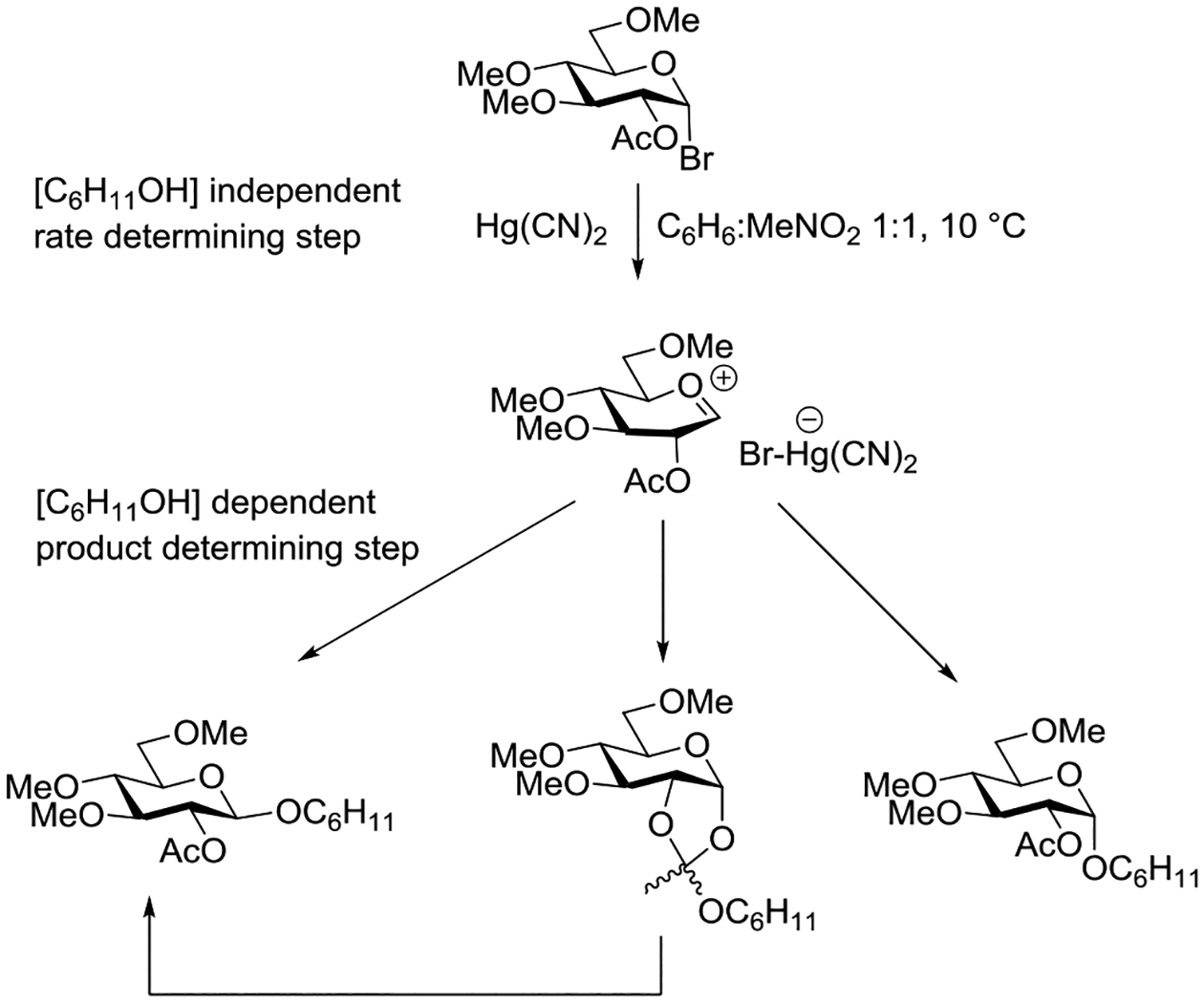
Mercuric Cyanide Promotion of the Reaction of Cyclohexanol with 2-O-Acetyl-3,4,6-tri-O-methyl-α-D-glucopyranosyl Bromide in 1:1 Nitromethane:Benzene
The contrast between the mechanism for β-glycoside formed in Table 4 and Scheme 1 from the identical substrate is striking and illustrates the critical role played by solvent and promoter in glycosylation reactions. What is perhaps even more striking is the fact that the concentration of the acceptor plays a critical role in both reactions, in the one case intervening directly in the rate determining step and so influencing the reaction kinetics and its stereochemical outcome, and in the other case only in the product forming step where it nevertheless impinges directly on stereoselectivity. Thus, whether the reaction proceeds via an SN1 or SN2-like mechanism the concentration of the acceptor correlates with stereoselectivity.
An early study of the influence of remote esters on glycosylation rates was conducted by Glaudemans and Fletcher who studied the rates of methanolysis of 5-O-(4-nitrobenzoyl)-2,3-di-O-benzyl-α-D-arabinofuranosyl chloride and of 3,5-di-O-(4-nitrobenzoyl)-2-O-benzyl-α-D-arabinofuranosyl chloride in comparison to that of 2,3,5-tri-O-benzyl-α-D-arabinofuranosyl chloride in dichloromethane under pseudo-first order conditions.96 This study is prescient in that it raises the possibility of remote stereodirecting participation by the esters at the 5- and especially 3-position of the donor, but also notes the reduced likelihood of participation by the electron-deficient p-nitrobenzoates employed. We are not aware of any further kinetic measurements on the influence of remote esters on the rates of glycosylation reactions.
5. Orthoesters as Intermediates in Glycosylation Reactions Directed by Vicinal Esters
The trapping of dioxalenium ions arising from the participation of carboxylate esters at the 2-position of glycosyl donors by a wide variety of nucleophiles is one of the classical reactions of carbohydrate chemistry, with many hundreds of examples reported in the literature, and constitutes some of the strongest and most long-standing evidence for neighboring group participation.9,13–15,97–99 Orthoesters can be isolated in high yield from a variety of systems, provided activation is carried out in the presence of a non-nucleophilic base.100–103 This process, which is a prerequisite to the use of orthoesters as donors in glycosylation reactions, also provides a practical means of regioselective protection of pyranoses,104 but is perhaps most widely known as a problematic side reaction in neighboring group assisted glycosylation reactions under basic conditions and with unhindered primary alcohols.105 Other than to note that participation by 2-O-carboxylate esters leading to the formation of intermediate dioxalenium ions and orthoesters applies even to sterically bulky esters, such as pivalates,98 and to electron-deficient esters including the chloro- and bromoacetates106 albeit not necessarily to the very electron-deficient trichloro and trifluoroacetates,107 no attempt is made here to catalogue the many descriptions of orthoester isolation, nor of their rearrangement to glycosides.
It is generally considered that such orthoesters are the kinetic products of glycosylation under conditions of neighboring group participation, with the nucleophilic alcohol most rapidly attacking the dioxalenium ion at the site of maximum positive charge density.95,101–103 This hypothesis is supported by computational work by Whitfield and coworkers.108 In the absence of a buffering base, rearrangement then takes place to give the glycoside. The stability of glycosyl orthoesters and the rate at which they rearrange to the corresponding glycosides is a function of the type of ester (acetate, benzoate, etc), with some authors indicating that benzoates are less problematic than acetates.102,109,110
Several groups have studied the influence of steric and electronic effects in the participating ester on conversion of the intermediate dioxalenium ion to the glycoside with a view to suppressing orthoester formation and other deleterious side reactions including erosion of stereoselectivity, hydrolysis, and acyl transfer.106,110–119 In this regard, a recent report by Sun and coworkers according to which the 2-O-[2,2-dimethyl-2-o -nitrophenyl)acetyl group provides complete stereoselectivity in formation of glycosides, without competing orthoester formation, is noteworthy.120 On the basis of the proximity of the nitro group to the carbonyl carbon in X-ray crystal structures, the experimental requirement for an o-, as opposed to a p-nitro group, and of quantum mechanical calculations, it was suggested that the nitro group intercepts and stabilizes the bridging dioxacarbenium ion (Scheme 2). Unfortunately, no direct comparison was made of the relative reactivity of anomerically pure α- and β-trichloroacetimidates, such that it is not possible to comment on the presence or absence of anchimeric assistance in this system. Interestingly, computational analysis of participation by the same ester group from the 6-position revealed the lowest energy structure to involve direct participation by the nitro group at the anomeric center (Figure 17).120 It must be noted, however, that these computations (Scheme 2 and Figure 17) were conducted in the absence of the obligatory triflate counterion, and therefore it is not possible to estimate the stability of the various bridged intermediates relative to the alternative glycosyl triflate.
Scheme 2.
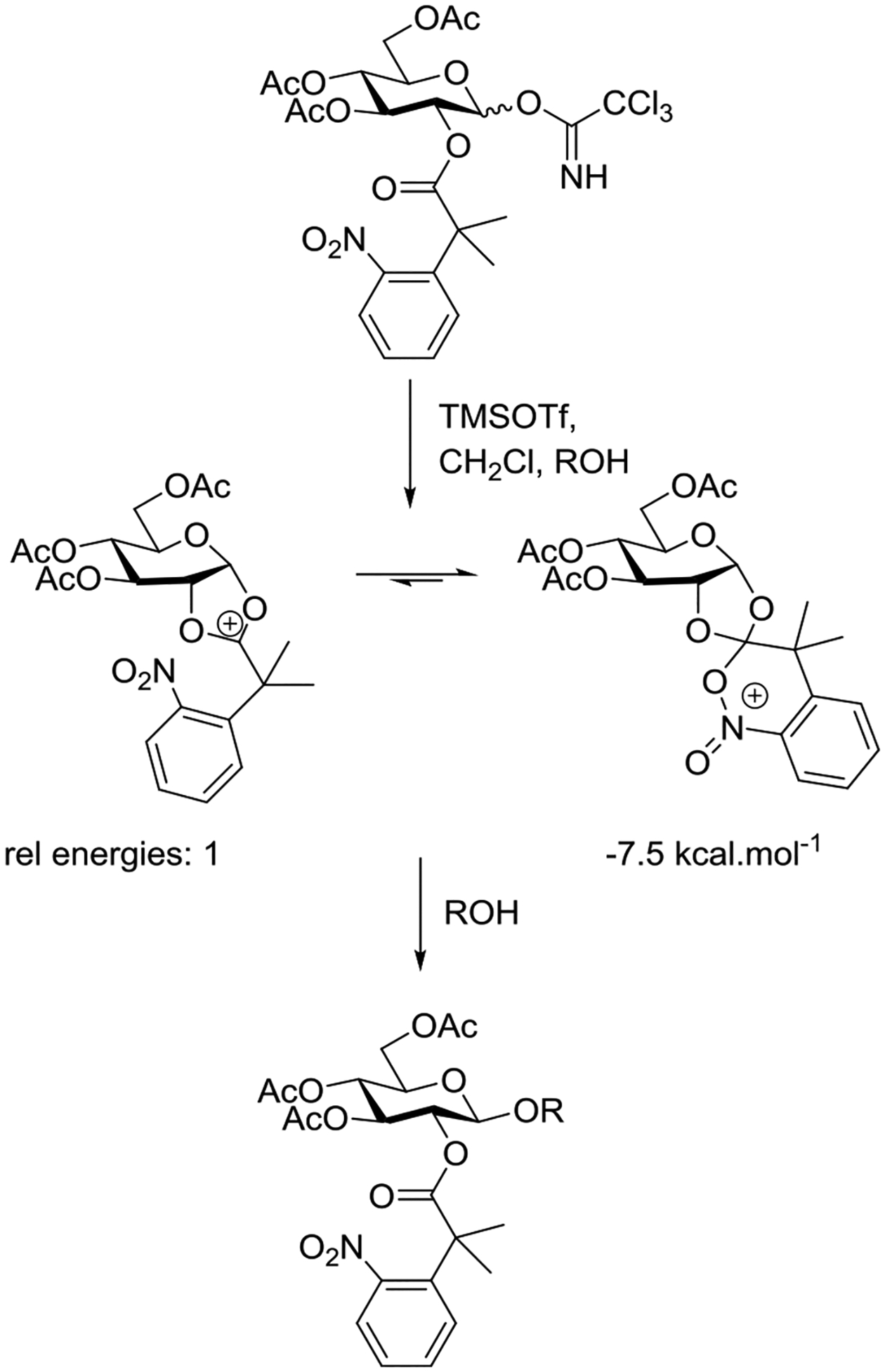
Intramolecular Orthoester Formation by a 2-O-[2-(o-Nitrophenyl)isobutyroyl]glucopyranosyl Donor
Figure 17.

DFT computed relative energies of alternative participation modes by a 6-O-[2-(o-nitrophenyl)isobutyroyl] ester.
Kinetic measurements of the tetramethylurea-buffered, triflic acid-catalyzed rearrangement of a methyl L-rhamnopyranosyl orthoester in deuteriochloroform to the corresponding 1,2-trans-glycoside revealed first order kinetics with respect to the orthoester and a large negative entropy of activation. On this basis it was suggested that the rearrangement follows an intramolecular path via a contact ion pair in which the migrating alcohol remains in close contact with the dioxalenium ion before ultimate recombination with ring opening to give the glycoside (Scheme 3).121 DFT computational work by Whitfield and coworkers on the rearrangement of a methyl orthoester in the 6-O-acetyl-3,4-O-isopropylidene-D-galactopyranose series using continuum dielectric model for solvation arrived at an analogous conclusion, albeit no counterion was included in the computation.108
Scheme 3.

Kinetically-Derived Intramolecular Mechanism for Orthoester to Glycoside Rearrangement
The alternative mechanism is one involving complete dissociation of the alcohol, or its complex with the activating Lewis acid, from the dioxalenium ion prior to recombination with ring opening and glycosidic bond formation. This latter mechanism was demonstrated by the Yu laboratory by means of an elegant cross-over experiment (Scheme 4) for rearrangement mediated by TMSOTf.122 The higher proportions of the two galactopyranosyl products were attributed to the inherently greater reactivity of galactosyl donors over their glucopyranosyl counterparts.
Scheme 4.
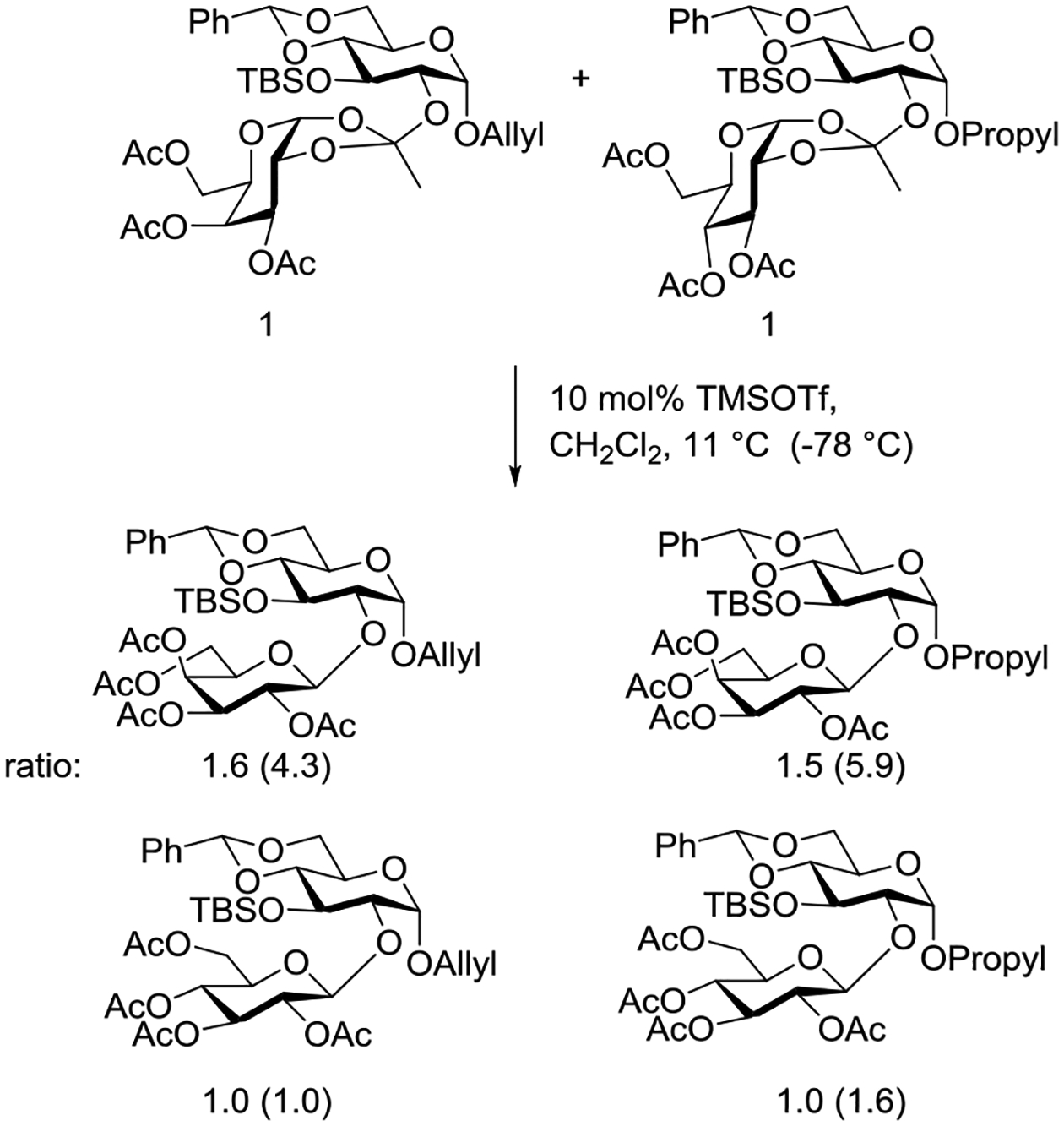
Cross-over Experiment Demonstrating Intermolecular Rearrangement of Glycosyl Orthoester to the Corresponding Glycoside at 11°C (and at −78°C)
We are not aware of any examples of orthoesters arising from the trapping of dioxenium or larger bridging dioxacarbenium ions by external nucleophiles in the course of participation from distal ester groups.
6. Anchimeric Assistance
It has long been recognized on the basis of kinetic and isotopic exchange reactions that 1,2-trans-per-O-acetyl glycopyranoses and 1,2-trans-per-O-acetyl glycopyranosyl halides undergo anomerization more rapidly than the corresponding cis-isomers.12,123,124 As discussed by Lemieux, this phenomenon is best interpreted by anchimeric assistance from the trans-ester in the displacement of the anomeric leaving group resulting in the formation of the cis-fused dioxalenium ion as intermediate. As also discussed by Lemieux, reaction of the cis-isomer is retarded by the presence of the electron-withdrawing β-acetoxy group.12 In a similar vein, Lemieux and Morgan deduced that orthoesters are formed in the presence of pyridine and related bases from the per-O-acyl α-glucosyl halides by initial halide-catalyzed epimerization to the corresponding per-O-acyl β-glucosyl halides followed by ring closure to afford the dioxalenium ions (Scheme 5).98,125,126
Scheme 5.

Halide Ion Catalyzed Orthoester Formation from α-Acetobromoglucose
Paulsen and Herold studied the formation of the fused acetoxonium ion from both anomers of acetochloroglucose and of peracetyl glucopyranose and of related benzoates with SbCl5 in a variety of solvents.127 In tetrachloromethane solution the β-chloride was rapidly converted to the acetoxonium ion, which precipitated in 80–90% yield. With the corresponding α-chloride the yield of precipitated acetoxonium ion was reported as 25–40% under the same conditions (Scheme 6).
Scheme 6.
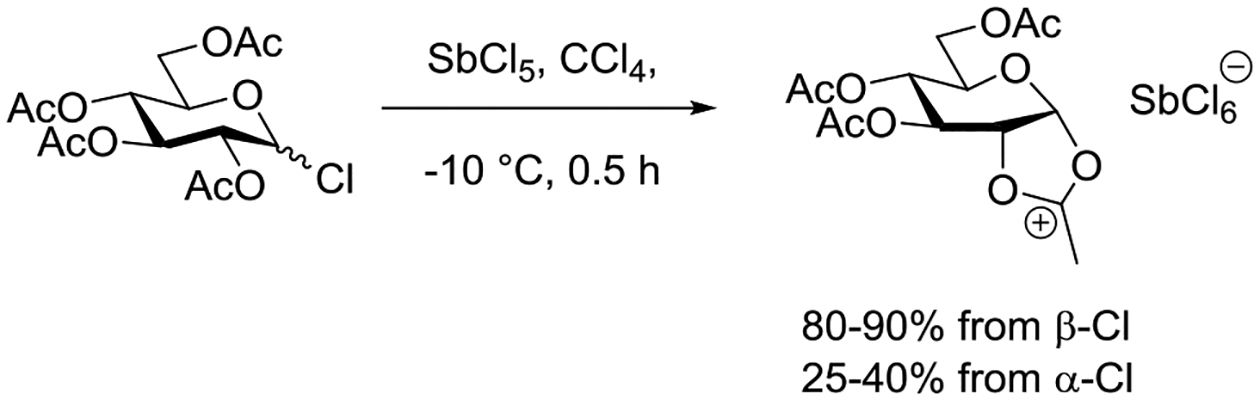
Anomer-Dependent Formation of an Acetoxonium Ion from Acetochloroglucose in Tetrachloromethane
When the β-chloride was activated with SbCl5 in dichloromethane solution on the other hand, the D-ido-configured 4,6-acetoxonium salt crystallized from solution in 94–98% yield (Scheme 7). This observation was rationalized in terms of initial formation of the 1,2-gluco acetoxonium ion followed by a series of rearrangement reactions involving displacement with inversion by the proximal acetate, with the equilibrium driven by crystallization of the less soluble ido-isomer. The α-chloride and the β-acetate were reported to behave similarly. In contrast, the α-acetate did not provide the product under these reaction conditions.127 Clearly, anchimeric assistance is required to activate the anomeric acetate under these conditions, just as it facilitates activation of the β-chloride. The fact that both anomers of the chloride are activated by SbCl5, albeit at apparently different rates, while only the β-acetate is activated under the same conditions, suggests that anchimeric assistance plays a greater role in more weakly activated systems. When the reaction was conducted in nitromethane at 20°C, and when all components were soluble, the equilibrium ratio of the ions was determined to be gluco:manno:altro:ido = 53:18:7:17, clearly indicating the greater stability of the 1,2-dioxalenium ion over the dioxapenium ion.
Scheme 7.

Leaving Group and Configuration Dependent Formation of Acetoxonium Ions from Per-O-acetyl-D-glucopyranosyl Derivatives
Konradsson reported the relative rates of activation by N-bromosuccinimide of pairs of anomers in a series of pentenyl glycosides. It was observed that donors carrying esters at the 2-position showed a greater difference in relative rates of activation than systems protected with benzyl ethers (Table 5, comp of entries 1 and 2, and of 3 and 4), which was attributed to neighboring group participation (anchimeric assistance) by the ester in the case of the 1,2-trans-series.128
Table 5.
Relative Rates of Activation of Anomeric Pairs of Pentenyl Glycosides by NBS
| Entry | Pentenyl Glycosides | Rel Rates (β/α) of Reaction |
|---|---|---|
| 1 |  |
1.70 |
| 2 |  |
5.19 |
| 3 |  |
1.45 |
| 4 | 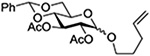 |
3.71 |
Boons and coworkers reported that glycosylation of methyl 2,3,4-tri-O-benzyl-α-D-glucopyranoside by a 1:1 anomeric mixture of dicyclohexylmethyl per-O-benzoyl-D-thioglucopyranoside with N-iodosuccinimide and triflic acid, under conditions in which anomerization of the thioglycosides was not observed, afforded 45% of the expected disaccharide as a single equatorial anomer and 52% of the recovered donor in the form of a 3:1 α:β mixture, indicating the β-donor to be the more reactive of the two (Scheme 8).129 As in the corresponding tetra-O-benzyl thioglucosides, where the α-anomeric donor was the more reactive, these results were interpreted in terms of anchimeric assistance facilitating the activation of the benzoylated β-donor.
Scheme 8.

Anchimeric Assistance in NIS/TfOH-Mediated Glycosylation by Dicyclohexylmethyl Per-O-benzoyl-D-thioglucopyranoside
Crich and coworkers studied the glycosylation of cyclohexanol with both anomers of phenyl 2,3,4-tri-O-benzoyl-D-thioxylopyranoside activating with benzenesulfenyl triflate at low temperature in the presence of the hindered base DTBMP.103 All reactions smoothly afforded an orthoester product in high yield (Scheme 9). When the corresponding anomeric bromides were activated with silver triflate in dichloromethane at −40°C the β-xylopyranosides were formed cleanly from both anomers with no significant configuration-dependent differences in reactivity (Scheme 9). Clearly, in these more reactive pentopyranosyl systems (lacking the extra electron-withdrawing C-O bond of the corresponding hexopyranosides), with activation by the powerful sulfenyl triflate,130 triflic anhydride131 or silver triflate mediators, anchimeric assistance does not play a significant role. The difference in the product spectrum between the two lines of Scheme 9 was demonstrated to be due to the presence or absence of DTBMP in the reaction mixture: the initial orthoester is stabilized in the presence of this mild base, but rapidly rearranges to the glycoside in its absence.103
Scheme 9.
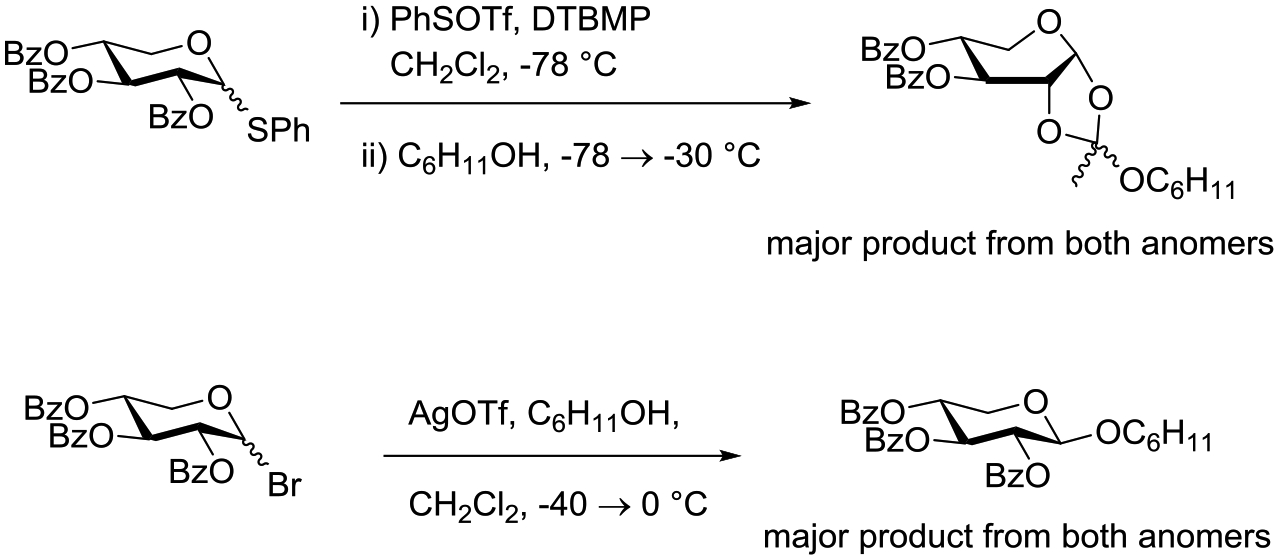
Configuration Independent Activation of Anomeric Per-O-benzoyl Xylopyranosyl Thioglycosides and Bromides with Benzenesulfenyl Triflate and Silver Triflate
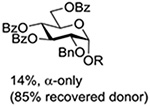
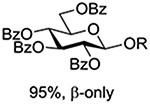
Following up on work by Demchenko and coworkers,132 Crich and Li studied the influence of anomeric configuration in the coupling of benzoxazolyl thioglucosides to a standard glycosyl acceptor with activation by copper triflate over a standard 14 h time period (Table 6).133 Comparison of entries 1 and 2 of Table 6 reveals the lack of influence of anomeric configuration on the rate of reaction in the 2-O-benzyl series, with both anomers affording comparably low yields of the product. Replacement of the 2-O-benzyl ether by a benzoate ester in the 1,2-trans-configured donor results in a dramatic increase in conversion as well as the complete reversal of anomeric configuration in the product (Table 5, cf entries 1 and 3). In this weakly activated system the 2-O-benzoate ester is therefore “arming”134 in the 1,2-trans-configuration. In contrast, the corresponding 1,2-cis-donor carrying a 2-O-benzoate is completely unreactive (Table 6, entry 4). These results clearly reveal the importance of anchimeric assistance in weakly activated systems and warn against the uncritical application of the Fraser-Reid armed-disarmed concept.
Table 6.
Coupling of Benzoxazolyl Thioglycosides to 1,2;3,4-Di-O-isopropylidenegalactopyranose with Activation by Cu(OTf)2 in CH2Cl2 at Room Temperaturea
| Entry | Donor | Product, % yield, β:α ratio |
|---|---|---|
| 1 | 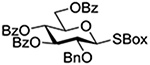 |
 |
| 2 | 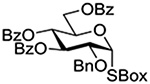 |
 |
| 3 | 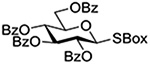 |
 |
| 4 | 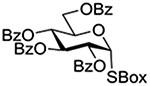 |
No reactio (95% recovered donor) |
ROH = 1,2;3,4-di-O-ispropylidene-α-D-galactopyranose
Extrapolating from the results presented in Table 5, the selective activation of a disaccharyl S-benzothiazolyl type donor and its coupling to a monosaccharyl acceptor bearing an S-benzoxazolyl group (Scheme 10) is clearly the result of anchimeric assistance in a weakly activated system,133 and not of differential stabilization of an intermediate oxocarbenium by the ring oxygen as proposed originally.132 It was subsequently demonstrated that the preferential activation of 1,2-trans 2-O-benzoyl-3,4,6-tri-O-benzyl over per-O-benzyl benzoxalzoyl thioglucosides extends to the galactopyranosyl series when using the mild dimethyl(methylthio)sulfonium triflate (DMTST) activating system; a consistent but much smaller difference in reaction was observed in the 1,2-trans-configured mannopyranose series.135,136
Scheme 10.
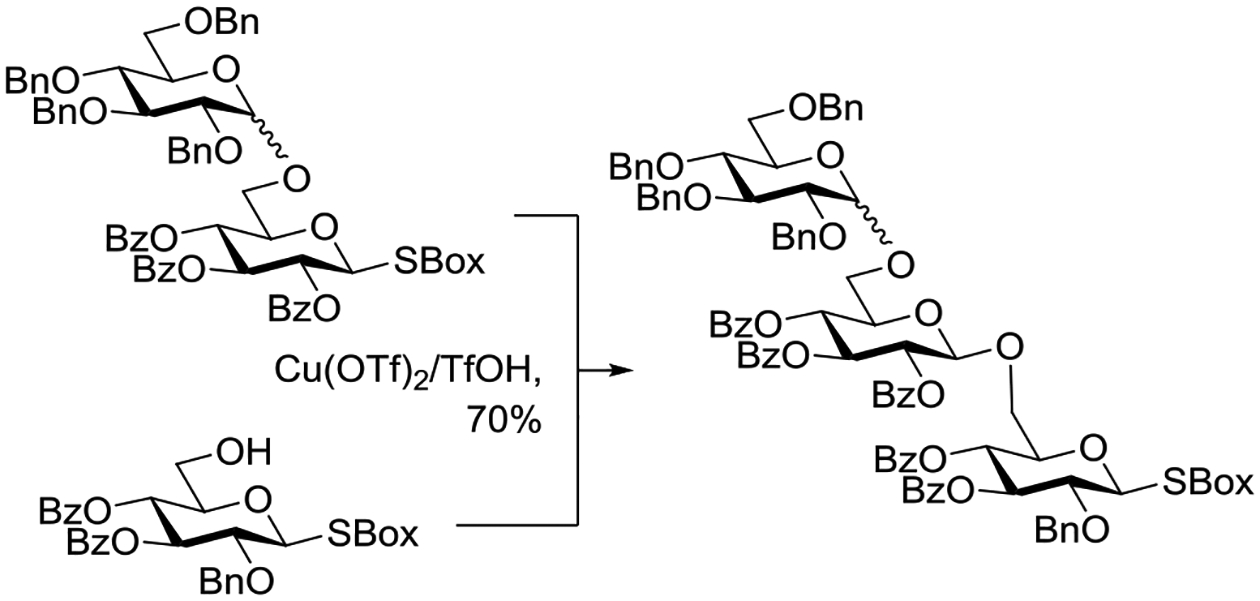
Selective Activation of a 2-O-Benzoyl S-Box Donor in the Presence of a 2-O-Benzyl Donor Due to Anchimeric Assistance.
It was subsequently shown by Demchenko and coworkers that the preferential activation of 1,2-trans-3,4,6-tri-O-benzyl-2-O-benzoyl-D-glucopyranosyl donors relative to the corresponding per-O-benzyl series benefits from anchimeric assistance in some but not all classes of donor/activator pairs (Table 7).137
Table 7.
Donor/Activator Pairs Benefitting from Anchimeric Assistance
 | |
|---|---|
| XR | Activator |
| O-(Benz-1,3-oxazol-2-yl) | Cu(OTf)2 and DMTST |
| O-5-pentenyl | IDCP, but not NIS, NBS, or NIS/TfOH |
| S-Et | MeOTf, DMTST, I2, but not IDCP |
| S-Ph and S-tolyl | I2, no significant difference |
| S-(1,3-thiazolin-2-yl) | I2, no significant difference |
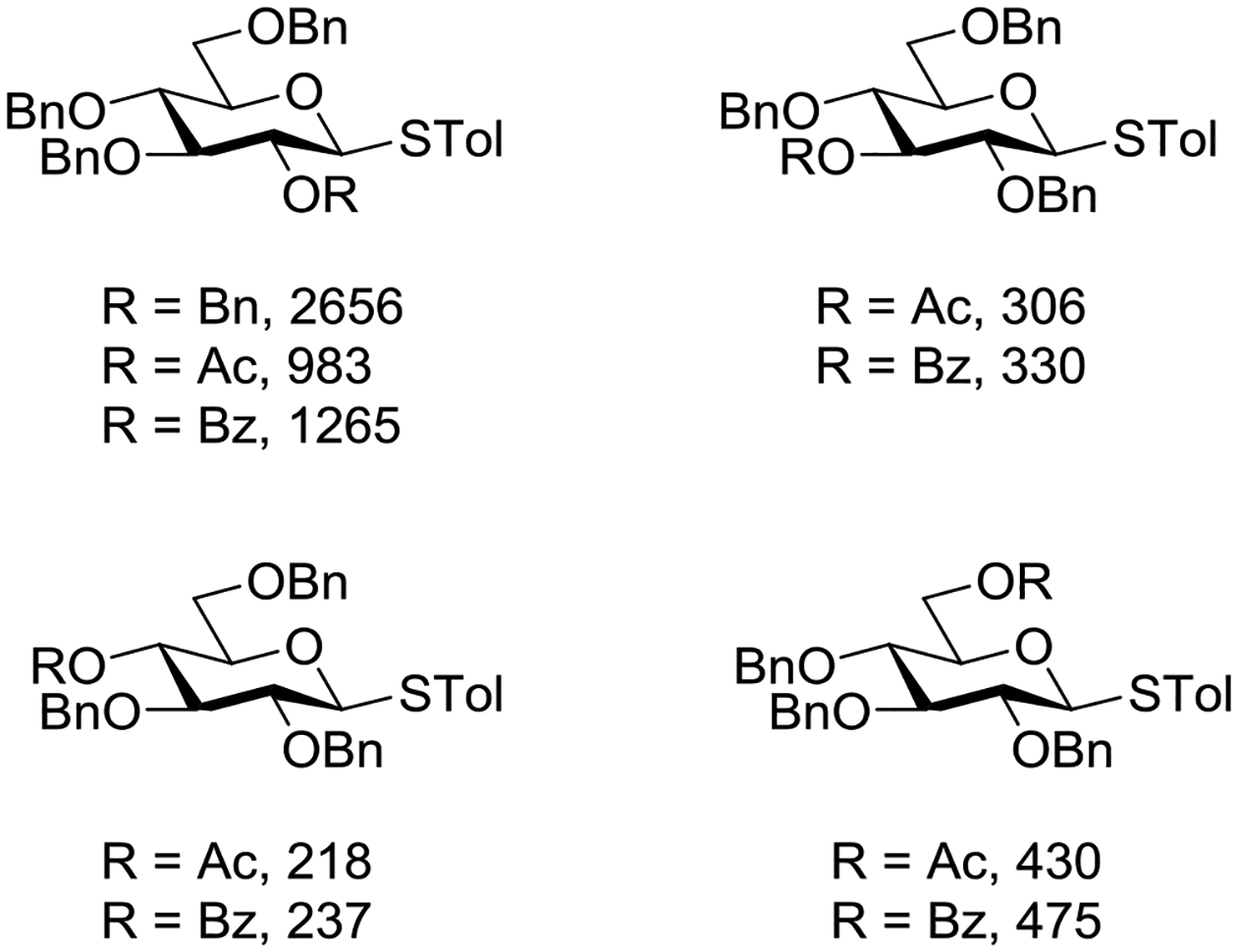
Wong and coworkers determined the relative reactivity values (RRVs)138,139 of tolyl tri-O-benzyl-β-D-thioglucopyanosides carrying a single acetyl or benzoyl group at different positions (Figure 18). Although none of the systems studied had an RRV comparable to the per-O-benzylated donor, the most reactive monoesters had the ester at the 2-position. As the strongly electron-withdrawing effect of the ester will be maximized when the ester is located at the 2-position, these results strongly support anchimeric assistance by the 2-O-acetyl and 2-O-benzoyl groups in these 1,2-trans-configured donors under the NIS/TfOH conditions used for RRV determination.140 Comparing their results to those of Demchenko and coworkers with the analogous benzoxazolyl thioglucosides (Table 6), the authors commented that the arming tendency of the 2-O-acyl group is strongly modulated by the leaving group and probably by the solvent and activator as well.
Figure 18.
Relative reactivity values (RRVs) for the activation of assorted tolyl mono-O-acyl-tri-O-benzyl-β-D-thioglucopyranosides by NIS and TfOH in dichloromethane at 0°C (all values are relative to tolyl tetra-O-acetyl-α-D-thiomannopyranoside).
Zhu and coworkers investigated the relative reactivity of a series of ethyl D-thioglucopyranosyl donors carrying various combinations of benzyl ethers, acetate and benzoate esters, with activation by NIS and trimethylsilyl triflate in dichloromethane between −78 and −20°C and established a reactivity sequence (Figure 19).141 Consistent with the work of Crich and Liu and the intervention of anchimeric assistance,133 the 1,2-trans-configured donors are more reactive than their 1,2-cis-counterparts when carrying an ester at the 2-position. On the other hand, for donors with a 2-O-benzyl ether it is the 1,2-cis thioglycosides that are the more reactive.
Figure 19.
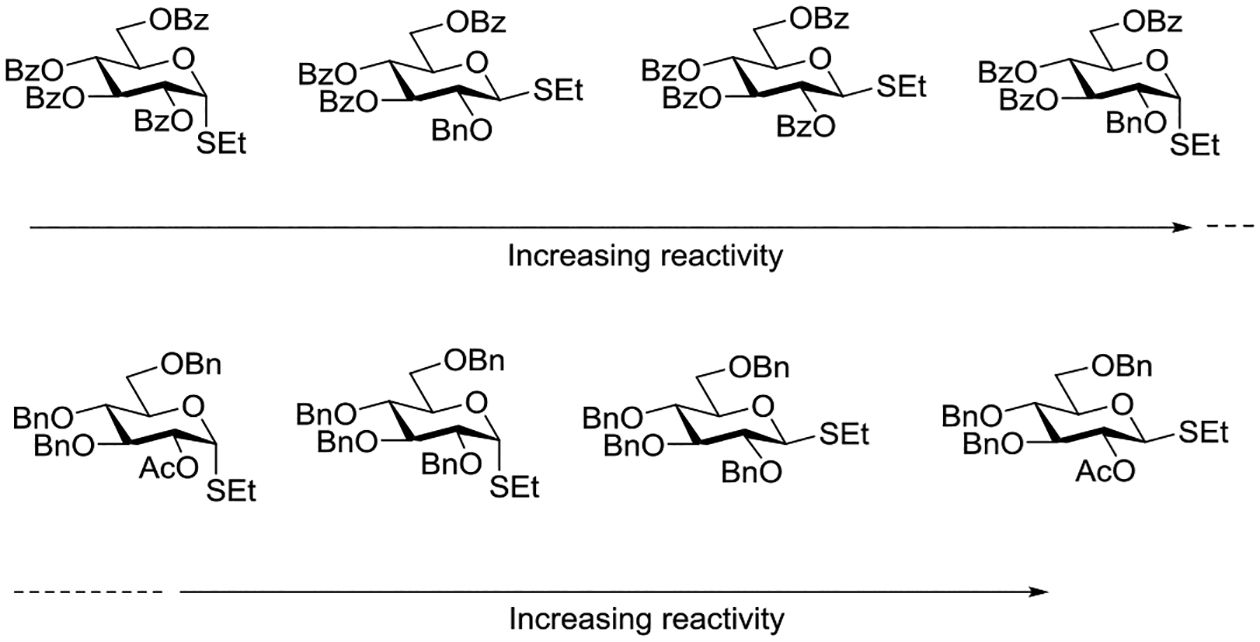
Reactivity sequence in a series of ethylthio glucopyranosides revealing the role of anchimeric assistance.
Following up on the work of Demchencko and coworkers (Table 7),137 Jensen and coworkers examined the relative reactivity of pairs of 1,2-trans-3,4,6-tri-O-benzyl-thio-D-glucopyranosides, carrying either benzyl ethers or benzoate esters at the 2-position by different activating systems. They found that in the S-phenyl and S-ethylthio series the use of NIS/TfOH as promotor results in preferential activation of the 2-O-benzyl ethers over the 2-O-benzoate esters (Table 8).142 In contrast, in the S-(benzoxazolyl) series the 2-O-benzoate was activated preferentially over the corresponding 2-O-benzyl ether by NIS/TfOH, DMTST, TfOH, and MeOTf (Table 8). Whether studying the S-phenyl or S-Box series, the authors found that the relative rates of activation between the 2-O-benzoyl esters and the 2-O-benzyl ethers on treatment with the mild DMTST reagent varied significantly depending on the presence or absence of the mild base TTBP. Strikingly, in the presence of TTBP neither of the S-Box donors were activated at all, leading the authors to suggest that S-Box activation is dependent on the presence of triflic acid.142 Whatever the mechanism of activation, it is clear that in the S-Box series anchimeric assistance from the 2-O-benzoate ester accelerates activation to the extent that the 2-O-ester is more reactive than the 2-O-ether. For the more reactive S-phenyl and S-ethyl series any increase in rate provided by the presence of a 1,2-trans-ester is not sufficient to outweigh the generally more electron-withdrawing effect of the ester. In contrast to Demchenko and coworkers, and indeed to Crich and Li (Table 6), the Jensen lab found that Cu(OTf)2 did not result in more rapid activation of the 2-O-benzoyl S-Box donor over the corresponding 2-O-benzyl, rather the inverse was observed. In view of the importance of triflic acid revealed by the Jensen lab for the activation of S-Box donors, it seems likely that the lack of reproducibility from lab to lab is a function of the type, quality and quantity of molecular sieves added to the reactions.1,143,144
Table 8.
Comparison of Thioglycoside Reactivities by the Jensen Lab
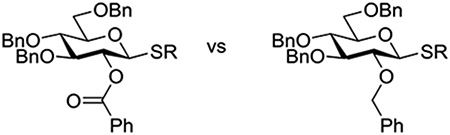 | ||
|---|---|---|
| R | Activator (temp) | Rel Reactivity |
| Ph | NIS/TfOH (−78 → 0°C) | Bn>Bz |
| Ph | DMTST (0°C) | Bn<Bz |
| Ph | DMTST + TTBP (0°C) | Bn>Bz |
| Et | NIS/TfOH (−78 → 0°C) | Bn>Bz |
| Box | NIS/TfOH (0°C → rt) | Bn<Bz |
| Box | DMTST (0°C) | Bn<Bz |
| Box | DMTST + TTBP (0°C) | No reaction |
| Box | TfOH (0°C → rt) | Bn<Bz |
| Box | MeOTf (0°C → rt) | Bn<Bz |
| Box | Cu(OTf)2, rt | Bn>Bz |
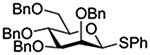
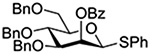
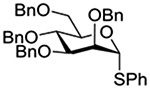
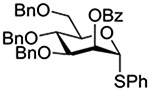
The Jensen lab then conducted a more complete set of competition experiments with both anomers of the ethyl and phenyl 3,4,6-tri-O-benzyl-D-thioglucopyranosides using the NIS/TfOH activating system (Table 9).145 Reaffirming their initial results, it was found that any anchimeric assistance afforded in the 1,2-trans-series is not sufficient to overcome the electron-withdrawing effect of the benzoate ester (Table 9, entry 1). The apparent rate acceleration due to anchimeric assistance on switching from the 1,2-cis to the 1,2-trans-series of 2-O-benzoates is greater for the ethyl thioglucoside donor than for the corresponding phenyl thioglucoside (Table 9, entry 2). This difference however is due to multiple factors and not simply to anchimeric assistance. Thus, while it was found that for the 2-O-benzyl ethers the axial anomer is more reactive than the equatorial one, the difference in rate was demonstrated to be less pronounced for the ethyl thioglucoside than for the phenyl thioglucoside (Table 9, entry 3). Furthermore, the extra electron-withdrawing effect of the benzoate ester over the benzyl ether is more important in the 1,2-cis-series than in the 1,2-trans-series, and is significantly larger for the ethyl thioglycosides than for the phenyl thioglycosides (Table 9, entry 4).
Table 9.
Ratio of Recovered Donors in Competition for Activation by Substoichiometric NIS and Triflic Anhydride in Dichloromethane between −78 and 0°C
| Donor 1 | Donor 2 | Ratio recovered 1:2 | |
|---|---|---|---|
| R = Ph | R = Et | ||
 |
 |
1:2 = 1:2 | 1:2 = 1:2 |
 |
 |
1:2 = 1:2 | 1:2 = 1:20 |
 |
 |
1:2 = 1:4 | 1:2 = 1:1.1 |
 |
 |
1:2 = 1:12 | 1:2 = 1:60 |
 |
 |
1:2 = 1:24 | - |
 |
 |
1:2 = 1:13 | - |
 |
 |
1:2 = 1:1.2 | - |
 |
 |
1:2 = 1:2.5 | - |
An analogous series of competition experiments was then conducted in the mannopyranose series.145 As in the glucopyranosyl series, the simple switch of a 2-O-benzyl ether for a 2-O-benzoate ester results in a significant drop in relative reaction rate, for both the axial or equatorial phenyl thiomannosides (Table 9, entries 5 and 6). The difference in reaction rates between the axial and equatorial thiomannosides in the 2-O-benzoyl series is small (Table 9, entry 7), but interpretation is complicated by the fact that, in contrast to the glucopyranoside series, the equatorial thiomannoside is more reactive than its axial anomer in the presence of a 2-O-benzyl ether (Table 9, entry 8).
Overall, while these experiments from the Jensen laboratory clearly demonstrate the existence of anchimeric assistance, they underline the difficulty of deconvoluting its magnitude from that of the inherent electron-withdrawing effect of the ester.145 They also nicely bring out the importance of not extrapolating from one class of donor to another, with even ethyl and phenyl thioglycosides showing marked differences in otherwise identical systems.
In a further study, Demchenko and coworkers conducted a competition experiment in which an equimolar mixture of the two anomers of phenyl 6-O-benzyl-2-O-benzoyl-3,4-di-O-tert-butyldimethylsilyl-D-thioglucopyranoside, both of which were judged to exist in a twist boat conformation, was activated with a single molar equivalent of N-iodosuccinimide and catalytic triflic anhydride in the presence of excess cyclohexanol as acceptor, with quantification of the unreacted recovered thioglycosides. Only 20% of the 1,2-trans-configured donor was recovered whereas 90% of the 1,2-cis-donor did not react, clearly indicating the 1,2-trans-system to be the more reactive of the two (Scheme 11). However, as a further competition between the 2-O-benzoyl trans-configured donor and the corresponding 2-O-benzyl ether revealed the latter to be the more reactive, it was concluded that the difference in reactivity between the two epimeric 2-O-acyl thioglycosides could not be due to anchimeric assistance. This conclusion rests on the assumption that anchimeric assistance necessarily increases the rate of activation to a greater extent than replacement of the participating ester by a less electron-withdrawing benzyl ether, for which there is no basis. The conclusion was further supported by the unlikely argument that backside attack by the ester on the activated thioglycoside was not possible owing to the twist boat conformation, despite the observation of excellent 1,2-trans-selectivity in actual coupling reactions.146 It appears more likely that the difference in rates of activation of the two anomeric 2-O-acyl thioglycosides is due to the action of anchimeric assistance in the 1,2-trans-system, albeit the acceleration due to this anchimeric assistance in this highly activated, indeed super-armed to use the author’s terminology, is only modest.
Scheme 11.
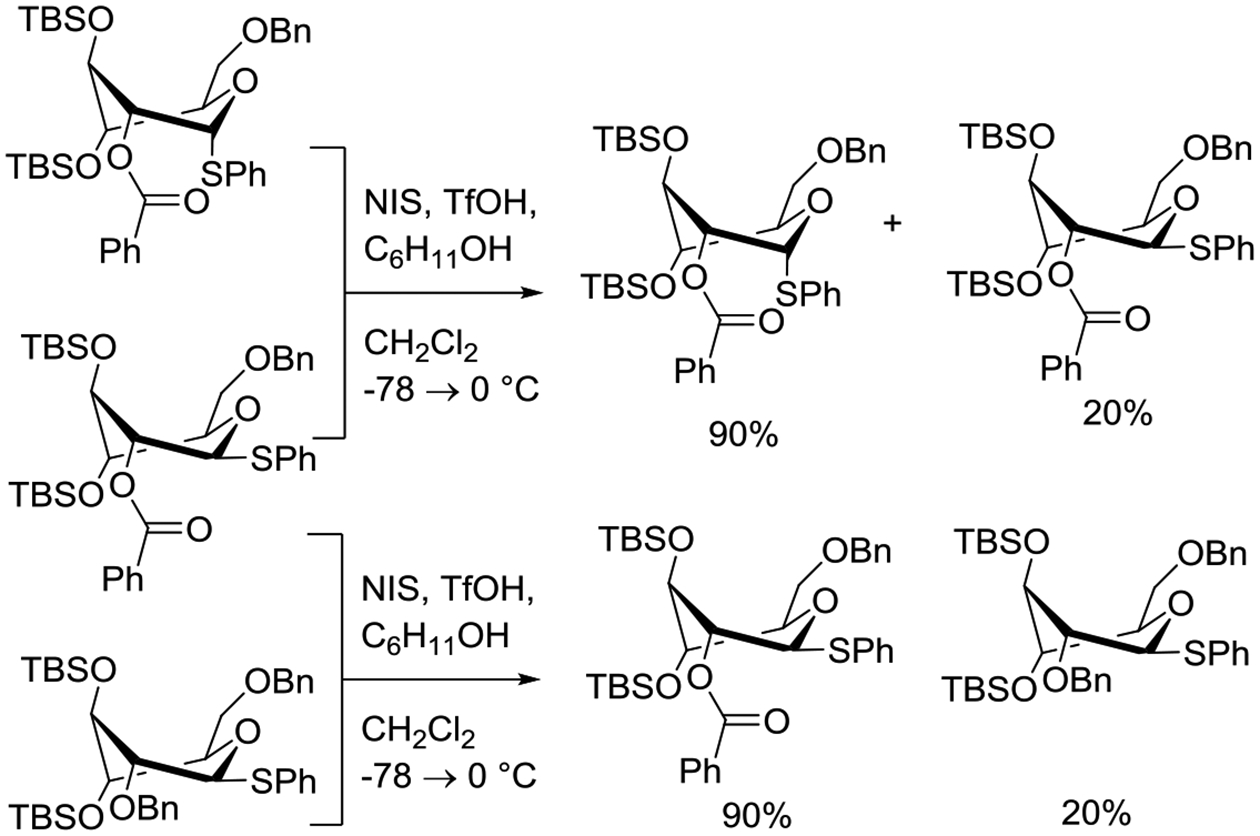
Influence of Anomeric Configuration and Protecting Group at the 2-Position on the Relative Rate of Consumption of Super-armed Thioglucopyranosides
To probe the intervention of anchimeric assistance in the activation of phenyl 3,4,6-tri-O-benzyl-2-O-benzoyl-β-D-glucopyranosides, shown above to be less reactive than the corresponding tetra-O-benzyl donors on activation with NIS and triflic anhydride at low temperature, Jensen and coworkers studied the relative rates of activation of a series of p-substituted 2-O-benzoates by competition methods.147 In the series of compounds studied, only the 2-O-(4-pyrrolidinobenzoyl) system was more reactive than the corresponding per-O-benzyl-protected donor. Nevertheless, the negative log10 of the relative rate constant ratios displayed a linear correlation with the Hammett σp constant indicative of the build-up of positive charge on the ester moiety during the rate determining step, and so strongly supportive of anchimeric assistance (Figure 20). The possibility that the observed trend arose from the difference in electron-withdrawing abilities of the different esters as opposed to anchimeric assistance was discarded on the grounds that the observed trends were much greater in the 1,2-trans-series than in the corresponding 1,2-cis-series where anchimeric assistance is not possible.147
Figure 20.
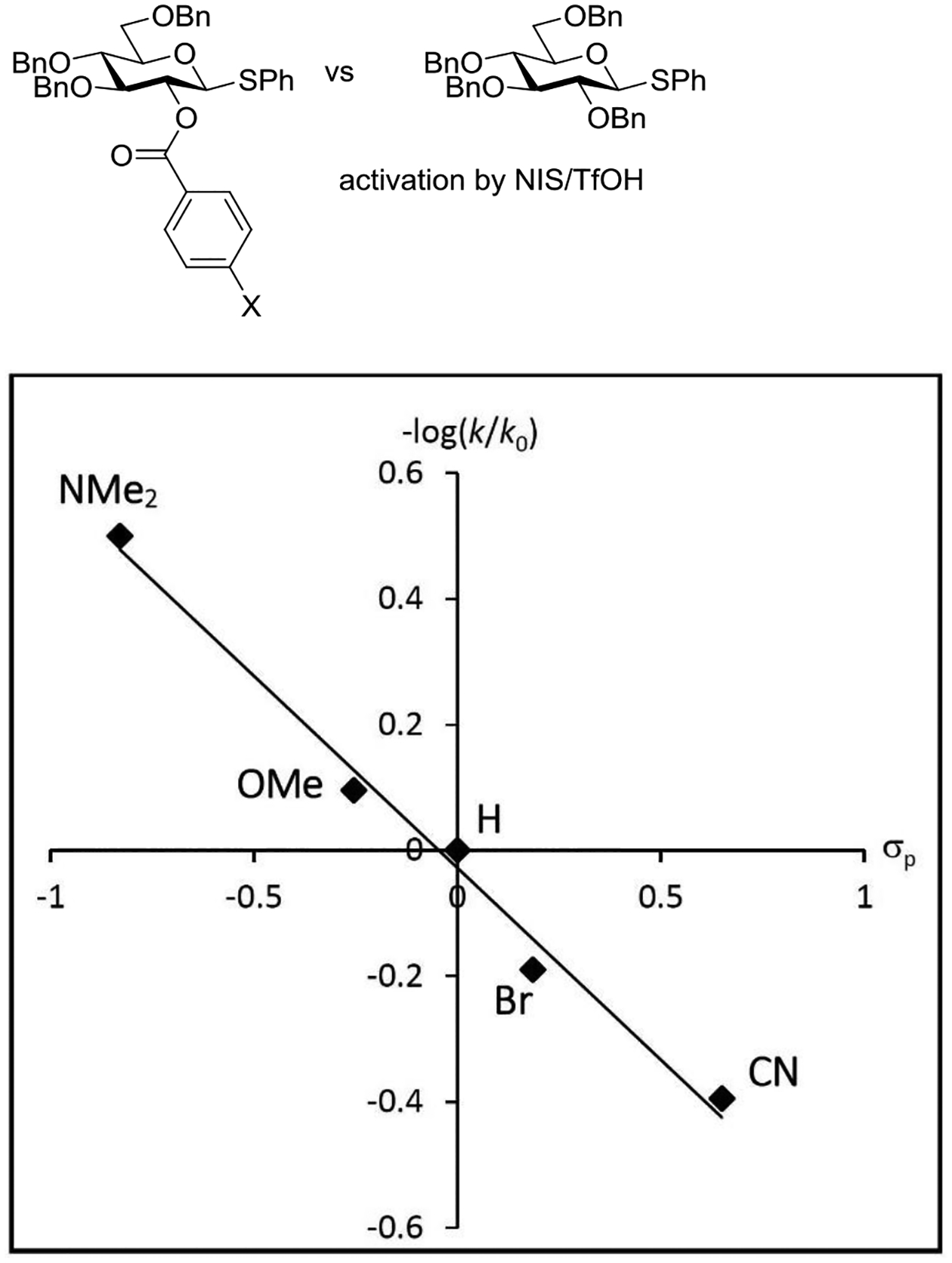
Hammett correlation of the logarithmic rate constant ratio with σp supportive anchimeric assistance in the activation of the phenyl 3,4,6-tri-O-benzyl-2-O-(4-substituted-benzoyl)-β-D-thioglucopyranosides.
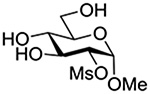
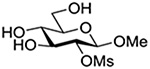
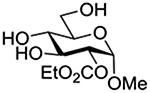
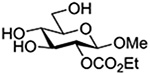
Finally Bols and coworkers, noting that anchimeric assistance is potentially applicable in the hydrolysis of glycosidic bonds as well as in their formation, studied the acid catalyzed hydrolysis of both anomers of a series of methyl glucopyranosides carrying assorted functionality at the 2-position (Table 9).148 For every pair of anomers, the equatorial glycoside was hydrolyzed more quickly than the axial glycoside consistent with the anomeric effect. Hydrolysis was strongly retarded by the presence of a 2-O-mesylate consistent with the strongly inductive electron-withdrawing effect of this group and the absence of participation. More generally, the absence of any correlation of the hydrolysis rates with Hammett parameters or field effects (F) pointed to the intervention of another factor beyond simple inductive effects. To separate the retarding inductive electron-withdrawing effect of the substituents from any accelerating effect due to anchimeric assistance, the authors estimated the rates of hydrolysis based solely on the inductive effect of the different groups. Comparison of these theoretical numbers with the experimental rates then afforded a measure of the rate acceleration due to participation. The accelerations determined in this manner (Table 10) ranged from 2–45 and were mostly compensated for by the inductive electron-withdrawing effect of the various groups. As there was only a minor difference in the estimated rate accelerations between any given pair of anomers, the authors concluded that these effects were due to stabilization of the developing positive charge rather than to any push from the participating group.
Table 10.
Relative Rates of DCl-Catalyzed Hydrolysis in D2O at 60°C, and Estimated Accelerations from Participation
| Substrate | Rel Rate | Field Effect Parameter | Acceleration due to Participation |
|---|---|---|---|
 |
1 | 0 | - |
 |
1.5 | 0 | - |
 |
0.5 | 0.01 | - |
 |
1.3 | 0.01 | - |
 |
0.02 | 0.59 | - |
 |
0.04 | 0.59 | - |
 |
0.3 | 0.34 | 3 |
 |
0.4 | 0.34 | 2 |
 |
1.3 | 0.26 | 7 |
 |
4.1 | 0.26 | 13 |
 |
2.5 | 0.35 | 25 |
 |
7.8 | 0.35 | 45 |
Overall, as discussed previously by ourselves and others,140,149 anchimeric assistance appears to play a greater role in more weakly activated systems, whether due to a weak leaving group, weak coordination between the promoter and leaving group, or inadequate transition state stabilization by the solvent or counter ion. In such cases the extra push from the vicinal ester helps to overcome the barrier for loss of the leaving group. The corollary of this observation is that it is in such weakly activated systems that vicinal esters capable of anchimeric assistance are least likely to be disarming.
7. Anchimeric Assistance by Distal Esters
Albeit anchimeric assistance from a 4-O-benzoyl ester in the course of glycosylation by a fucopyranosyl donor has recently been suggested,150 we are not aware of any published examples of kinetic evidence demonstrating anchimeric assistance from a distal ester in any glycosylation reaction. Rather, the comparable yields and levels of conversion in entries 1 and 2 of Table 6 indicate the absence of anchimeric assistance from the 3-O-benzoate in the 1,3-trans-system (Entry 2) and from the 4-O-benzoate in the 1,4-trans-system (Entry 1). This is noteworthy as it is in such a weakly activated system that anchimeric assistance would be most expected. Similarly, Wong’s RRV values of donors carrying esters at the 3-position of otherwise perbenzylated glucopyranosyl donors are significantly smaller than that of the fully benzylated donor (Figure 18), and so argue against anchimeric assistance from that position.
8. Isolation of Cyclic Intermediates and Products
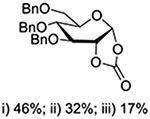
Crich and coworkers introduced the tert-butyloxycarbonate group as a probe for participation by remote esters. In this system cyclization affords an initial cyclic trioxocarbenium species, which then loses the tert-butyl cation to afford the observed cyclic carbonates.151 The viability of this probe was established by isolation of a 1,2-O-cyclic carbonate in 75% yield on activation of a 2-O-Boc protected glucopyranosyl thioglycoside (Table 11, entry 1) under typical glycosylation conditions. More recently Mikula and coworkers, based on precedent from the Descotes and other laboratories,152,153 observed analogous cyclizations from 2-O-benzyloxycarbonyl protected glucosyl donors, be they thioglycosides (Table 11, entry 2) or a trichloroacetimidate (Table 11, entry 3) in the course of a series of exclusively β-selective glycosylation reactions directed by the Cbz group.154 In the glucopyranosyl series, Crich and coworkers, building on precedent work by Lemieux and Hindsgaul,155 similarly employed the 2-O-(o-carboxybenzoate) as a further probe of participation and isolated the anticipated spirocyclic ortho ester in high yield (Table 11, entry 4).151 The 2-O-Boc system also proved its effectiveness in the mannopyranosyl series of donors (Table 11, entry 5).151 Turning to the possibility of participation by esters at the 3-position, particularly in the 4,6-O-benzylidene-protected mannopyranosyl donors where they have such a striking effect on anomeric selectivity,156 α-selective glycosylation was not interpreted by cyclic carbonate formation leading the authors to rule out participation (Table 11, entry 6).151 In more conformationally flexible systems, however, it is established that both equatorial amides and trichloroacetimidates at the 3-position are able to capture activated donors with the formation of bridged bicyclic oxazines under typical glycosylation conditions (Table 11, entries 7 and 8).157,158
Table 11.
Use of Carbonates, Carbamates, Esters, and Imidates as Probes for Stereodirecting Participation
| Entry | Donor | Promoter | Conditions | Product (% yield) | Lit. |
|---|---|---|---|---|---|
| 1 |  |
NIS, AgOTf | CH2Cl2, −10°C | 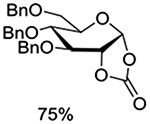 |
151 |
| 2 |  |
i. I2 ii. AgOTf iii. NIS, TfOH |
R = Et, CH2Cl2, 20°C R = 2-pyrimidinyl, CH2Cl2, 0°C R = Et, CH2Cl2, −10°C |
 |
154 |
| 3 | 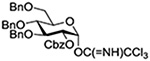 |
TMSOTf | CH2Cl2, −10°C | 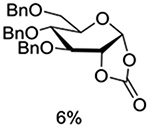 |
154 |
| 4 | 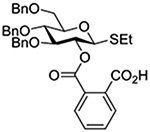 |
NIS, TMSOTf | CH2Cl2, −30°C | 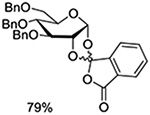 |
151 |
| 5 | 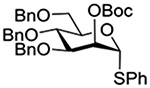 |
BSP, Tf2O | CH2Cl2, −60°C |  |
151 |
| 6 | 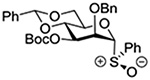 |
Tf2O, TTBP, cC6H11OH | CH2Cl2, −60°C |  |
151 |
| 7 |  |
NBS | CH2Cl2, rt | 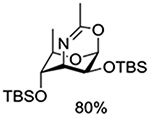 |
157 |
| 8 | 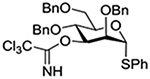 |
BSP, Tf2O | CH2Cl2, −60→0°C |  |
158 |
| 9 | 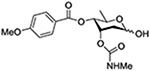 |
AcOH or TsOH | Δ or 1:1 C6H6:CH2Cl2, rt |  |
159 |
| 10 | 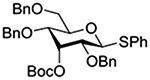 |
BSP, Tf2O, cC6H11OH | CH2Cl2, −60°C | 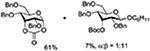 |
151 |
| 11 | 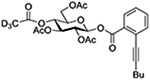 |
Tf2NAuPPh3 | CH2Cl2, rt |  |
160 |
| 12 | 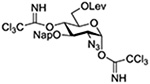 |
TMSOTf | CH2Cl2, 0°C |  |
161 |
| 13 | 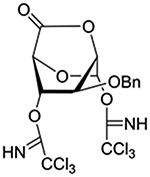 |
TMSOTf | CH2Cl2, 0°C | 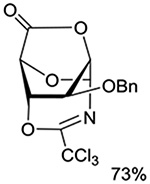 |
162 |
| 14 | 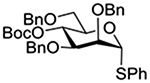 |
BSP, Tf2O, TTBP, iPrOH | CH2Cl2, −40°C | 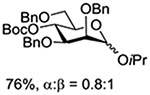 |
151 |
| 15 | 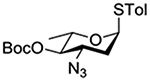 |
BSP, Tf2O, TTBP | i) CH2Cl2, −60°C ii) Ac2O |
 |
151 |
| 16 | 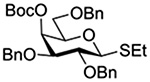 |
BSP, Tf2O, TTBP | i) Et2O, −60°C ii) Ac2O |
 |
151 |
| 17 |  |
BSP, Tf2O, TTBP, cC6H11OH | CH2Cl2, −60°C | 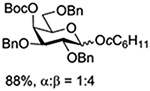 |
151 |
| 18 | 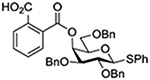 |
NIS, TMSOTf | CH2Cl2, −30°C |  |
151 |
| 19 |  |
NIS, TfOH, iPrOH | CH2Cl2, 0°C |  |
151 |
| 20 | 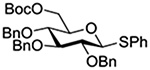 |
NIS, TfOH | CH2Cl2, 0°C |  |
151 |
| 21 | 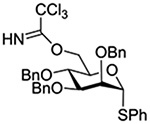 |
BSP, Tf2O | CH2Cl2, −60→0°C | 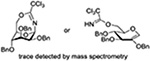 |
158,163 |
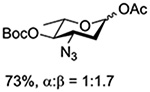
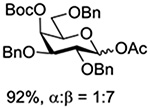
Turning to participation from axial derivatives at the 3-position, carbamates and carbonates proved to be suitable probes and established the stereoelectronic feasibility of participation in such systems (Table 11, 9 and 10).151,159 By means of an isotopically labelled probe, Yu and coworkers demonstrated that the formation of a tricyclic orthoester isolated in the course of glycosylation reactions with a per-O-acetyl glucopyranosyl donor is initiated by participation of an acetate at the 4-position (Table 11, entry 11).160 Subsequently, it was determined that a 4-O-trichloroacetimidate can capture an activated glucopyranosyl donor resulting in the formation of a bridged bicyclic imidate in high yield (Table 11, entry 12). However, the authors of this publication were careful to point out that this cyclic intermediate did not function as a glycosyl donor, thus ruling out its role as an actual glycosylation intermediate under the conditions employed.161 A bridged bicyclic imidate was also isolated in good yield on activation of a 2,6-mannuronolactone-based donor carrying an trichloroacetimidate group at the 4-position (Table 11, entry 13).162 It was noted, however, that the analogous system carrying a Boc group at the 4-position did not undergo cyclization to a bridged bicyclic carbonate thereby establishing the superior nucleophilicity of the imidate group and raising concerns about its use as a probe of remote participation. This latter observation is consistent with the results of Crich and coworkers who found that an equatorial 4-O-Boc group did not intervene in glycosylation reactions (Table 11, entries 14 and 15).151
Turning to 4-substituted galactopyranosyl donors, it was found by Crich and coworkers that a Boc group is not able to capture an activated donor in the absence (Table 11, entry 16) or presence (Table 11, entry 17) of an acceptor.151 Similarly, the o-carboxybenzoate probe failed to provide evidence for the formation of a bridged tricyclic orthoester derived by participation of the benzoate functionality. Rather, a bridged bicyclic system arising from participation by the o-carboxy moiety pendant to the benzoate was observed (Table 11, entry 18).151 Finally, probing for participation from the 6-position, Crich and coworkers employed a 6-O-Boc protected glucopyranosyl donor. In the presence of an acceptor alcohol only the glycoside was observed (Table 11, entry 19), while in the absence of an acceptor a macrocyclic dicarbonate was isolated indicating that intermolecular participation by the Boc group was favored over the intramolecular variant (Table 11, entry 20).151 Kim and coworkers attempted to take advantage of the superior nucleophilicity of the trichloroacetimidate group over the Boc moiety and employed a 6-O-imidoyl probe and reported the isolation of trace amounts of a cyclic product (Table 11, entry 21). However, as the only evidence in support of the bridged bicyclic product was the observation of the molecular ion in the mass spectrum, this result must be viewed with caution as simple proton loss from the oxocarbenium ion to afford a glycal is equally consistent with the reported data.158,163
In a further attempt to probe the interaction of axial esters of pyranosides with the anomeric center, Crich and coworkers attempted trapping of the putative intermediate bridging dioxacarbenium ion with 18O-labelled water (Scheme 12).151 Trapping of the bridging dioxacarbenium ion by the labelled water was anticipated to provide a labelled hemiorthoester that would undergo decomposition to two regioisomeric esters, one at the 4-position and one at the anomeric position, both with the label in the carbonyl oxygen. In the event, and arguing strongly against the likelihood of productive interaction of the ester with the anomeric center, the labelled oxygen atom was only found in the hemiacetal oxygen of the hydrolyzed product, which necessarily arose from direct capture of an intermediate glycosyl triflate or a closely related ion pair.
Scheme 12.
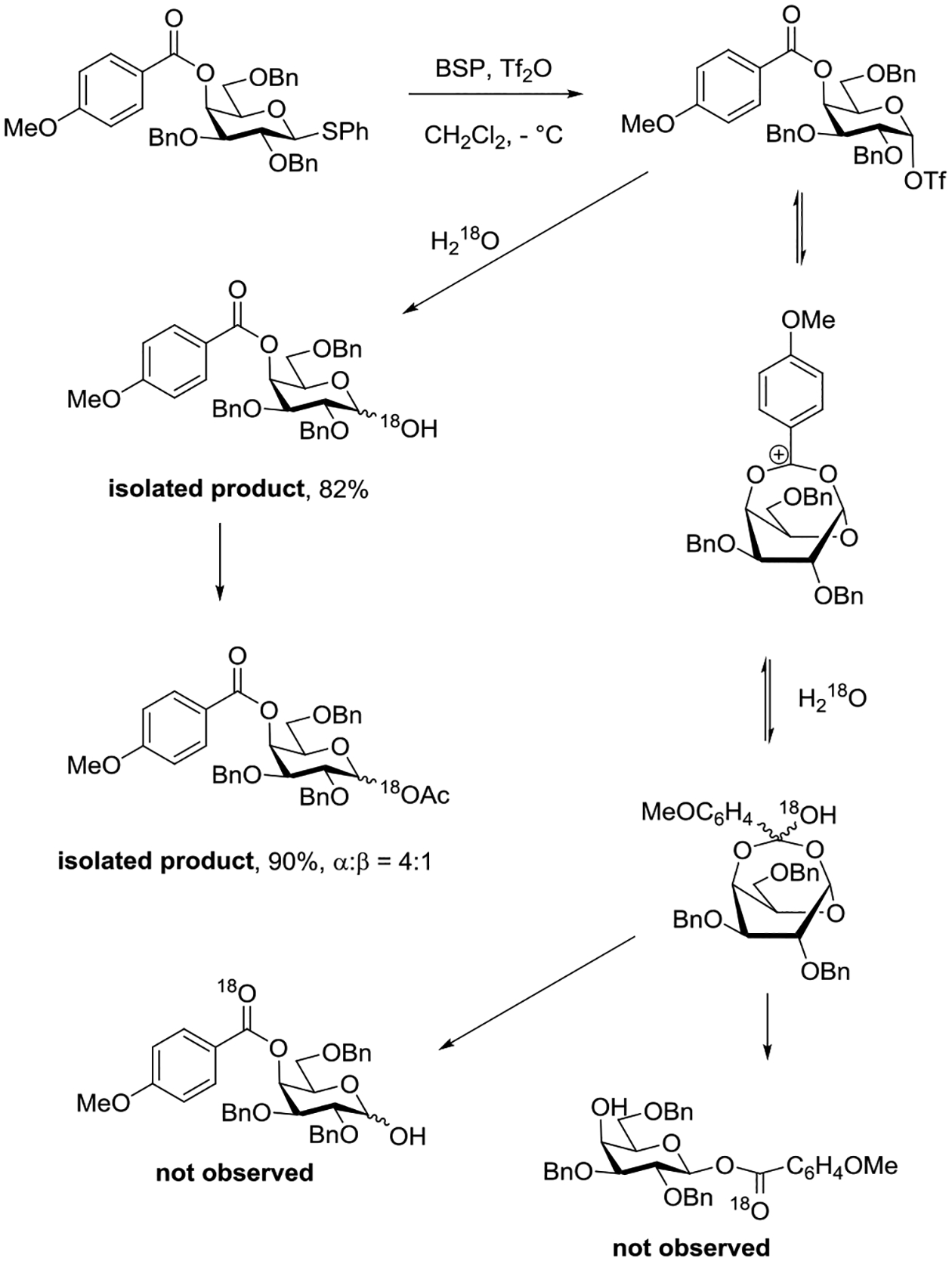
18O-Labelling Experiment Discounting Remote Participation form the 4-Position in the Galactopyranosides
9. Participation by 2-O-Alkoxyacetyl Ethers and Phenacyl Ethers
Boons and coworkers conducted a series of glycosylation reactions with glucopyranosyl donors, bearing R- or S-configured ethoxycarbonylbenzyl ethers at the 2-position capable in principle of participating through six-membered cyclic dioxenium ions.89,164 It was envisaged that the R-isomer would preferentially form a cis-fused decalinoid bridging structure leading ultimately to the stereoselective formation of an equatorial glycoside, while the S-isomer would participate via a trans-decalinoid system eventually providing the axial glycoside. In both cases the preferred ring fusion, cis- or trans-, of the anticipated intermediate bridging ion derives from the equatorial location of the phenyl group in the chiral auxiliary (Scheme 13). These predictions of selectivity were largely borne out in a series of glycosylations conducted by activation of the glycosyl trichloroacetimidates at −78°C in dichloromethane by the addition of TMSOTf followed by warming to room temperature (Scheme 13). Thus, for donors bearing acetyl, benzoyl, or allyloxycarbonyl groups at the 3-position, excellent selectivity for formation of the axial glycosides was observed in the presence of the S-configured auxiliary, whereas their R-configured diastereomers gave the equatorial glycosides albeit typically with somewhat reduced selectivity. With the corresponding donors protected with an allyl ether at the 3-position, the same overall trends were observed but selectivities did not exceed 5:1 for either the R- or S-configured system, and in the case of a less reactive glucosyl 4-OH acceptor were even inverted in the S-series. Although only limited examples were studied and selectivities were modest, two related R- and S-configured galactosyl donors carrying 3-O-acetates gave analogous results to those seen in the glucosyl series.89 DFT calculations were conducted to complement the experimental work and were generally supportive of it, albeit the triflate counter ion was not included.
Scheme 13.
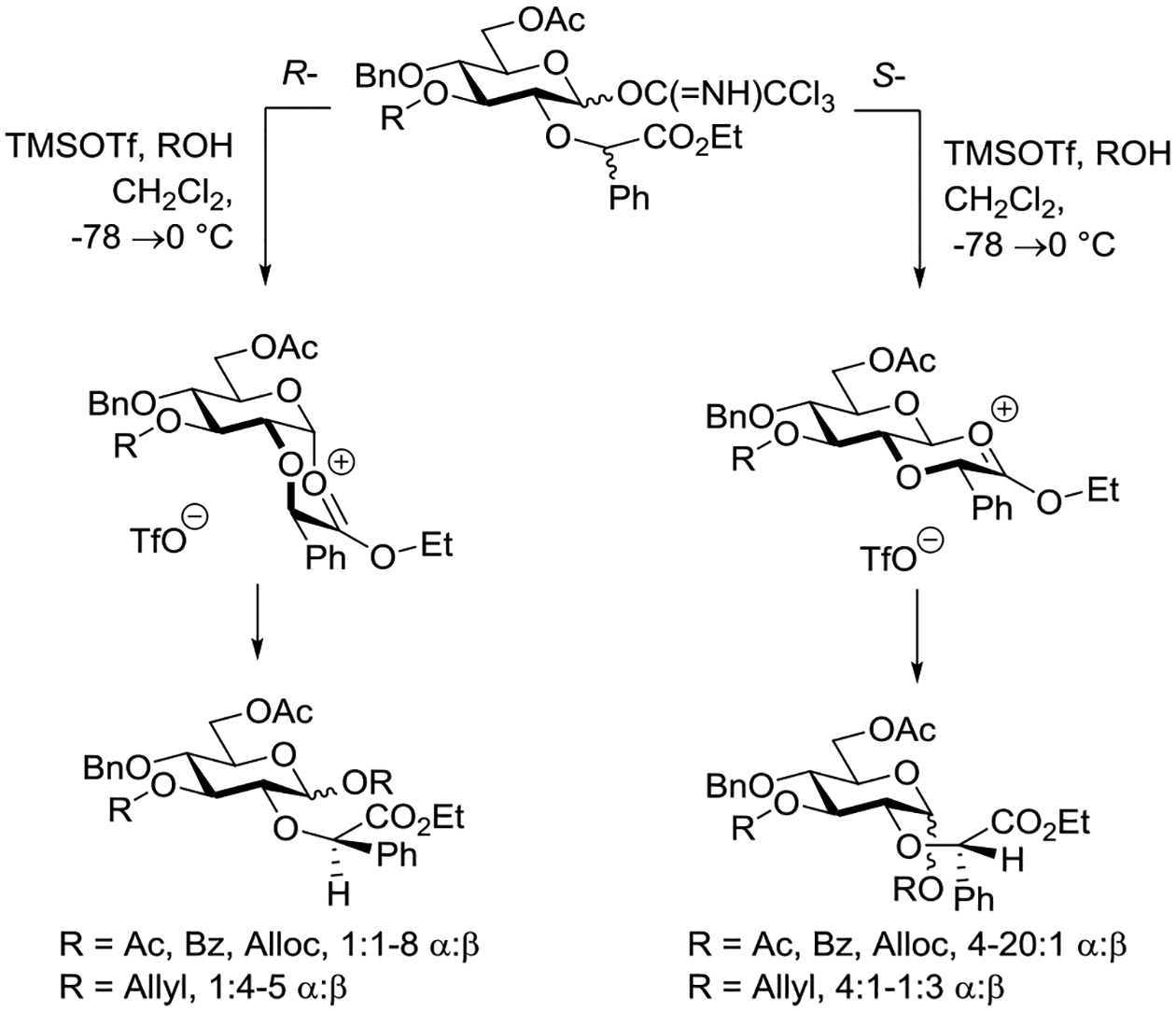
Extent of Stereodirecting Participation from the 2-Position via Six-Membered Cyclic Dioxenium Ions is Dependent on the Functionality at the 3-Position
Building on the work of the Boons laboratory, Wen and Crich investigated a series of mannosyl donors carrying various 2-(oxoalkyl)ethers in which the intended nucleophilic carbonyl group is poised to intercept the activated glycosyl donor without need for extensive conformational distortion of the pyranoside ring.165 In these experiments stereodirecting participation was envisioned to afford the axial glycosides, while its absence was expected to afford the equatorial glycosides by the well-established glycosyl triflate mechanism. In the event, activation of the 2-O-phenacyl-mannosyl thioglycoside with triflic anhydride and diphenyl sulfoxide at −78°C in dichloromethane followed by the addition of 1-adamantanol resulted in the isolation of a cyclic product in 33% yield, thereby establishing that the stereoelectronic requirements for participation were met by this class of probe (Scheme 14). With the more electron-deficient 2,4-trifluoromethylphenacyl analog activation was achieved with 1-benzenesulfinyl piperidine and triflic anhydride and the glycoside was observed as the major reaction product. Comparable results were observed on activation of the corresponding glycosyl sulfoxides with triflic anhydride at low temperature. The excellent equatorial selectivity observed in these reactions ruled out participation as a major stereodirecting pathway.
Scheme 14.
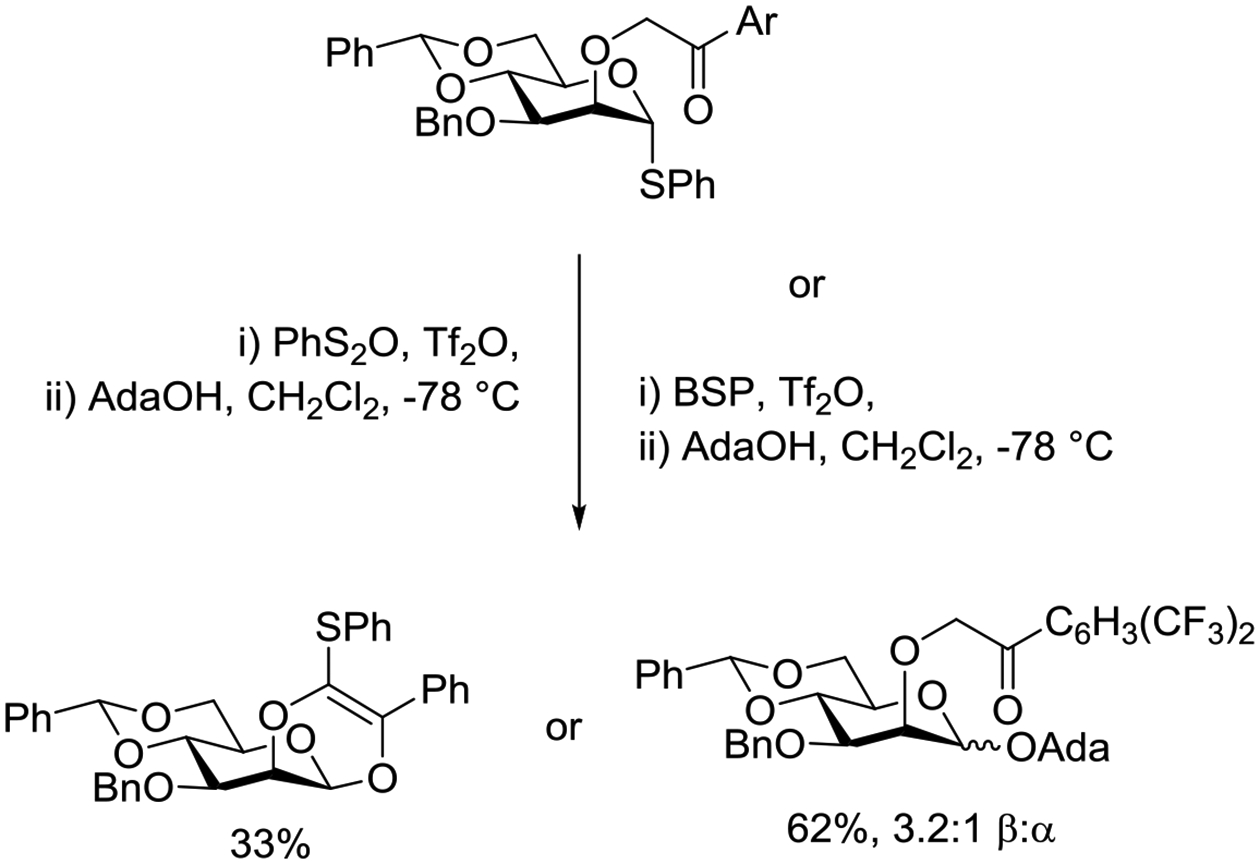
Stereodirecting Participation by 2-O-Phenacyl Groups is Dependent on the Electron Density of the Aryl Group
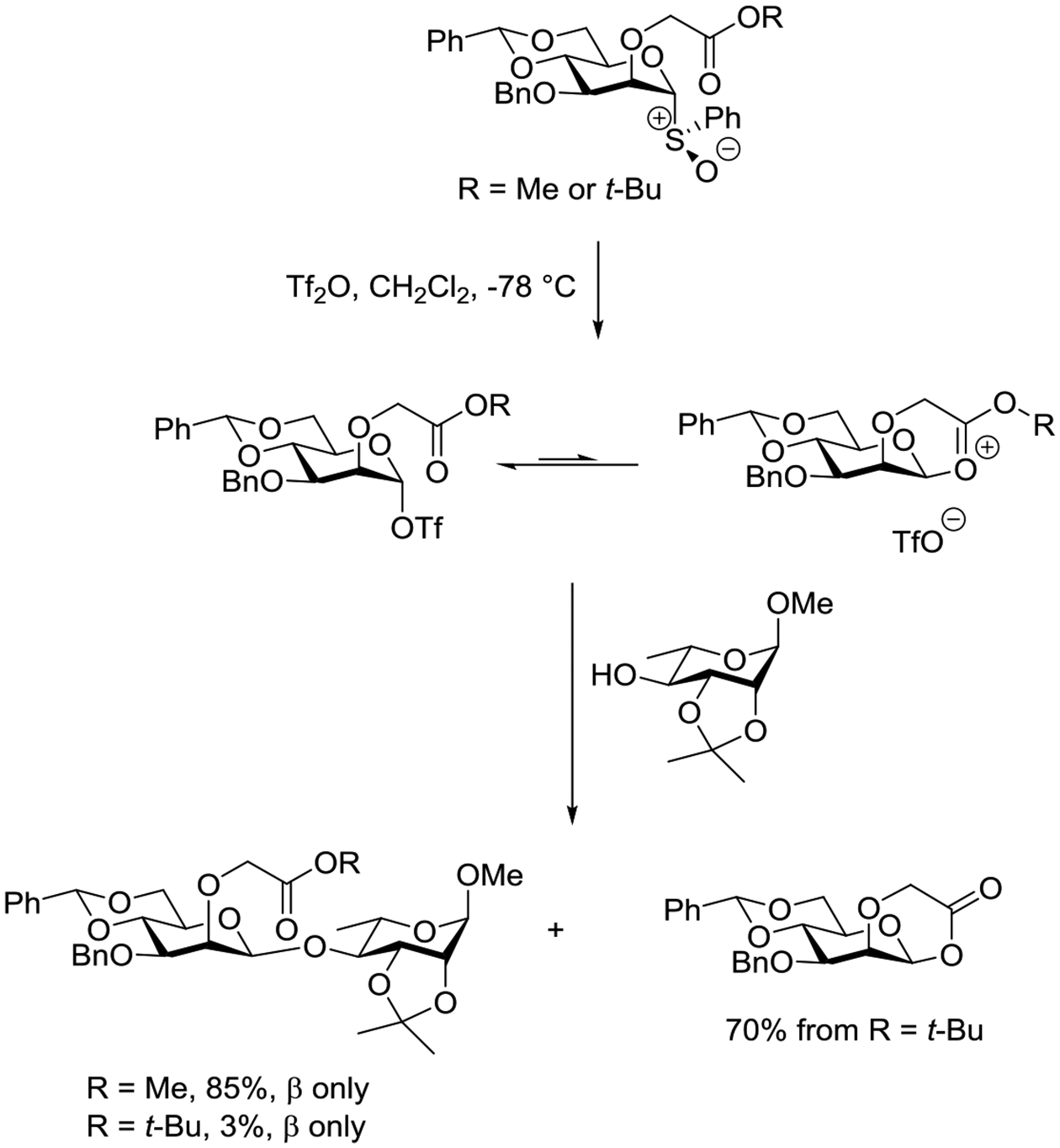
Attention was then turned to the related methoxy and tert-butoxyacetyl ethers, for which initial activation of the glycosyl sulfoxides with triflic anhydride was followed by the addition of a glycosyl acceptor. In the case of the methyl ester, the equatorial glycosides were formed exclusively and in high yield (Scheme 15). Comparable results were observed on activation of the corresponding thioglycoside with benzenesulfinyl piperidine and triflic anhydride. With the tert-butyl ester, the major product, that of ring closure followed by loss of the tert-butyl cation, was accompanied by a low yield of the equatorial glycoside. These results suggest that while participation by the ester is possible, as revealed by the formation of the lactone from the tert-butyl ester, the incipient bond from the carbonyl group to the anomeric position is weak and the main glycosylation pathway flows through the typical glycosyl triflate (Scheme 15).165 This conclusion cannot be extrapolated directly to the analogous Boons system (Scheme 13)89 because of the presence of the additional phenyl substituent in the latter, which potentially accelerates cyclization through a Thorpe Ingold-gem-dimethyl type conformational effect.
Scheme 15.
Trapping of a Cyclic Intermediate by Loss of a tert-Butyl Cation Does Not Guarantee Stereodirecting Participation
10. Spectroscopic and Computational Identification of Intermediates
In addition to the early studies on the characterization of simple 1,3-dioxalenium ions by NMR spectroscopy, such ions were observed in the course of neighboring group participation by the Crich laboratory.103 In this study, a 13C-labelled tri-O-benzoyl xylopyranosyl thioglycoside was activated with benzenesulfenyl triflate in deuteriodichloromethane in both the presence and absence of the mild, sterically hindered base DTBMP at −78 °C. Direct observation of the reaction mixture by 13C NMR spectroscopy at −78 °C revealed the formation of a single predominant species characterized by a sharp resonance at δ 180.3 assigned to the dioxalenium ion, consistent with literature chemical shift values for simple 2-phenyl-1,3-dioxalenium ions.166 When methanol was added to this ion formed in the presence of DTBMP, the signal at δ 180.3 was replaced by one at δ 121.1, taken as indicative of formation of an orthoester. On the other hand, when methanol was added to a solution of the dioxalenium ion prepared in the absence of the base, no orthoester was observed suggesting that the β-glycoside was formed directly (Scheme 16).
Scheme 16.

Observation of a Xylopyranosyl Dioxalenium Ion and its Trapping Products by 13C NMR spectroscopy
Huang and coworkers studied the activation of a set of three thioglycosides by p-tolylsulfenyl triflate by 1H and 13C NMR spectroscopy in deuteriochloroform at −60 °C.167 With a tetra-O-benzoyl galactosyl system they observed the formation of the α-galactosyl triflate, characterized by its anomeric 13C chemical shift of δ 104.4 and the presence of four resolved carbonyl resonances at δ 166.4, 165.9, 165.8, and 165 ppm. In contrast, with the corresponding 3,4,6-tri-O-benzyl-2-O-benzoyl system the main species observed was the dioxalenium ion arising from participation of the benzoate ester. Finally, with a tetra-O-acetyl glucopyranosyl thioglycoside a temperature-dependent equilibrium of a mixture of an α-glucosyl triflate and a dioxalenium ion was observed, with higher temperatures favoring the covalent species (Scheme 17). Comparable experimental observations (δ values and VT effects) were also recorded by Crich and Sun on activation of the corresponding tetra-O-acetylglycopyranosyl phenylsulfoxide with triflic anhydride in deuteriochloroform.168 Clearly, there is a delicate protecting group-dependent balance between dioxalenium triflates and glycosyl triflates at low temperatures in relatively non-polar solvents, which Huang and coworkers attributed to the destabilization of the charged dioxalenium ions by the non-participating esters, with benzoates being more electron-withdrawing than acetates.167 The contrast between the per-O-benzoyl xylopyranosyl system, which favors the dioxalenium ion, and the corresponding galactopyranosyl system, which favors the covalent triflate, was attributed to the extra electron-withdrawing C-O in the latter and its destabilizing influence on the charged dioxalenium ion.
Scheme 17.
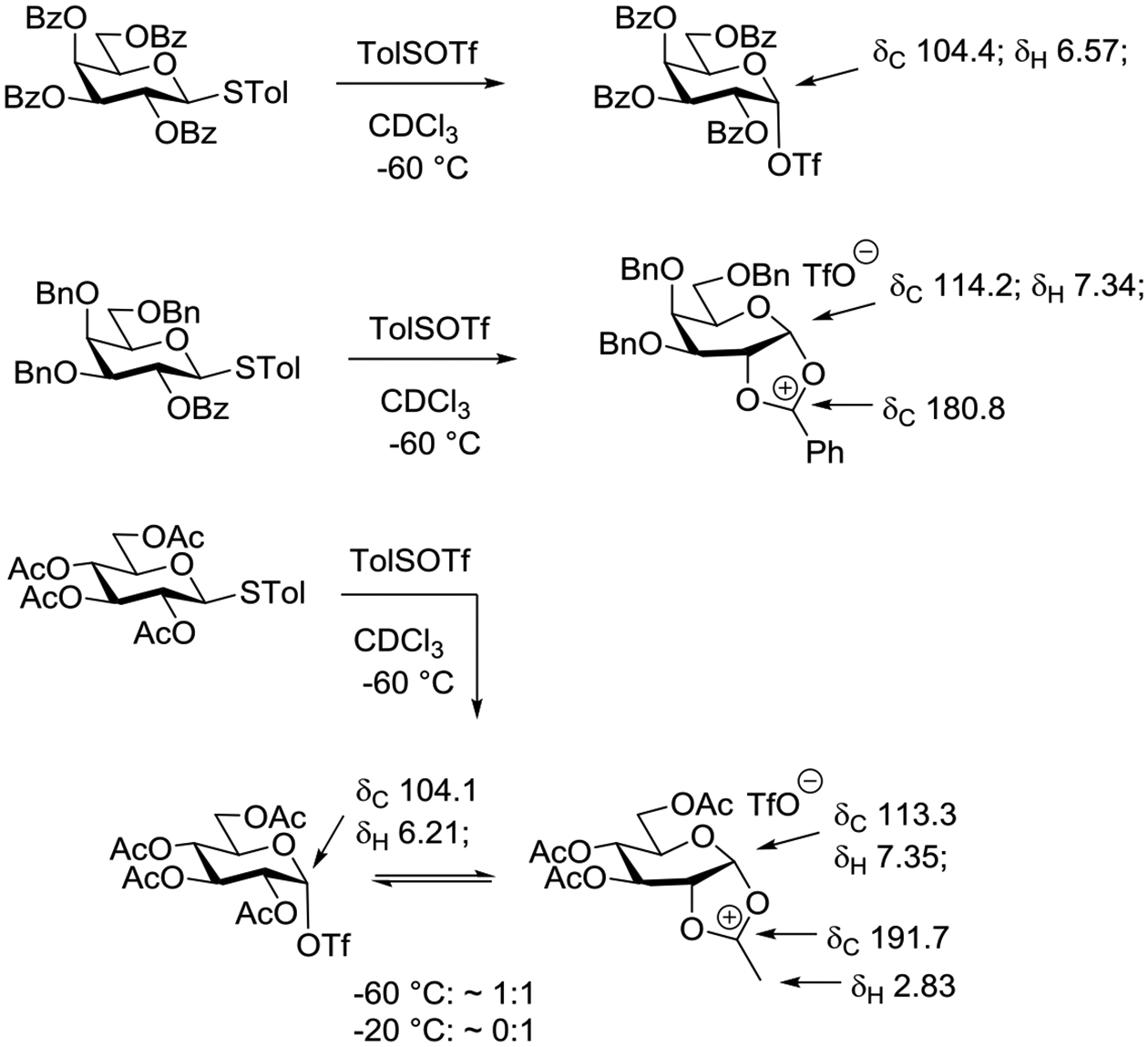
Influence of Protecting groups on the Dioxalenium Ion Glycosyl Triflate Equilibrium
Williams and coworkers studied the activation of tolyl 2,3,4,6-tetra-O-mesitoyl-β-D-thiogalactopyranoside by NIS/TfOH in deuteriochloroform in comparison to that of 3,4,6-tri-O-acetyl-2-O-mesitoyl-β-D-thiogalactopyranoside.114 Activation was conducted at −60°C and was followed by warming to 0°C before observation of the 13C NMR spectra. In the case of the 3,4,6-tri-O-acetyl-2-O-mesitoyl system resonances indicative of both the glycosyl triflate and the bridging dioxalenium ion were observed, whereas with the 2,3,4,6-tetra-O-mesitoyl protected donor only the dioxalenium ion could be detected (Scheme 18). Clearly, there is a subtle balance between the stabilities of the bridging dioxalenium ion and the corresponding glycosyl triflates following activation of the peresterified donors and it cannot be assumed that the dioxalenium ion is the most stable intermediate in each case.
Scheme 18.
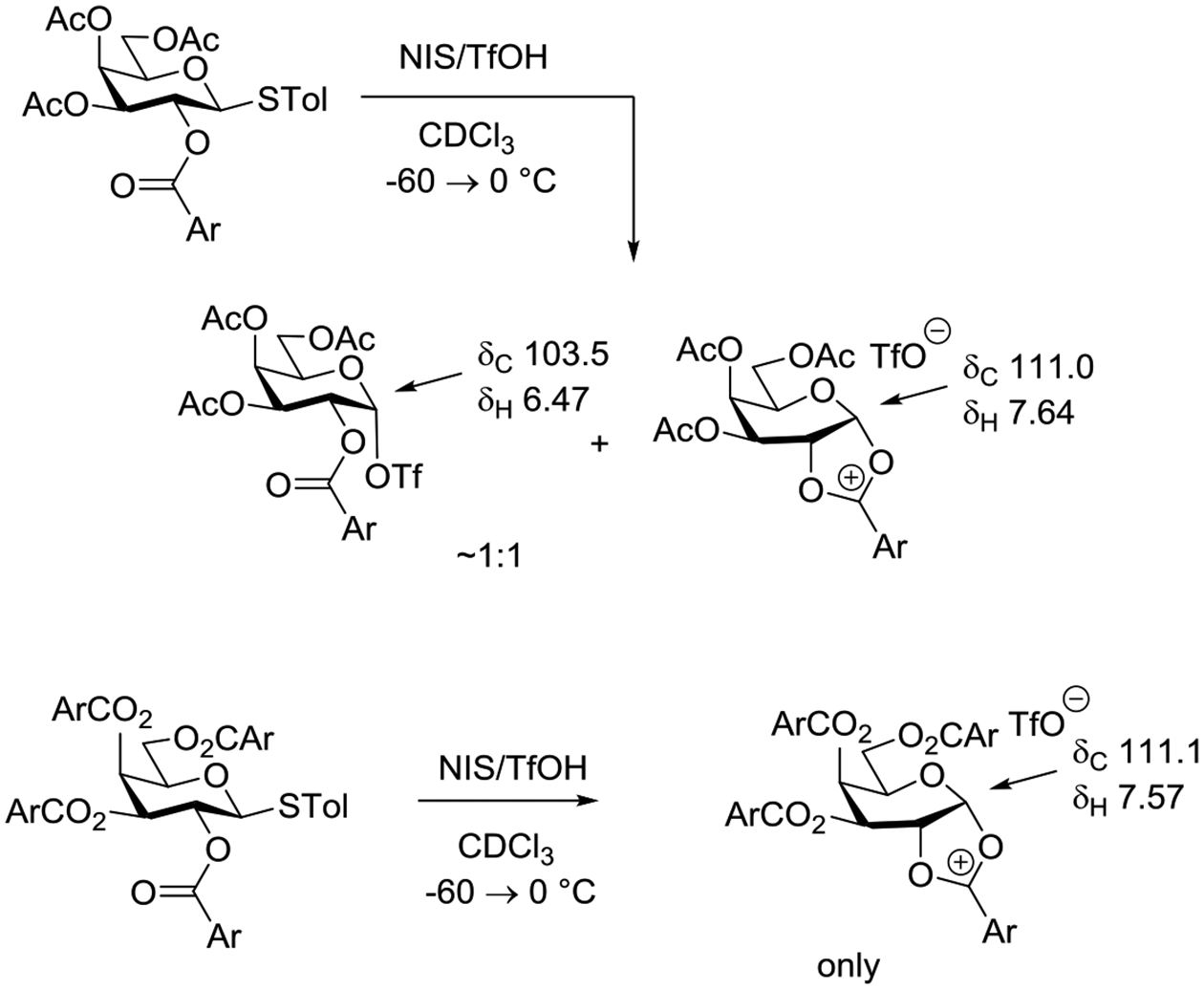
Influence of Distal Esters on the Dioxalenium Ion Glycosyl Triflate Equilibrium in the Galactopyranoside Series
In related work, looking to probe the influence of electron-withdrawing groups at O3 and O4 in a series of otherwise perbenzylated gluco and galactopyranosyl donors, Jeon, Seeberger, Kim and their coworkers activated thioglycosides with 1-benzenesulfinyl piperidine and triflic anhydride in deuteriodichloromethane at −60°C with observation by 1H and 13C-NMR spectroscopy. In all cases the spectra reported were consistent with the predominance of covalent glycosyl triflates: no evidence was provided for the observation of bridging esters from either the 3- or the 4-positions in the glucose series and from the 4-position in the galactose series.169
Nifantiev and coworkers found the α-selectivity of a series of 2-O-benzyl fucopyranosyl donors to be increased by the presence of a benzoate ester at either the 3- or the 4-positions, and especially by the presence of benzoate esters at both the 3- and 4-positions, with respect to the corresponding benzyl ethers (Scheme 19).170,171 They hypothesized that the ester-dependent selectivity arose from participation by the remote esters and provided supporting molecular mechanics and limited DFT calculations, which revealed the greater stability of the bridging ions than of the simple glycosyl oxocarbenium ions (Figure 21). In particular, the correlation between α-selectivity and computed stability of the bridging ion in a small series of p-substituted benzoate esters at the 4-position was advanced in favor of the remote participation mechanism.
Scheme 19.
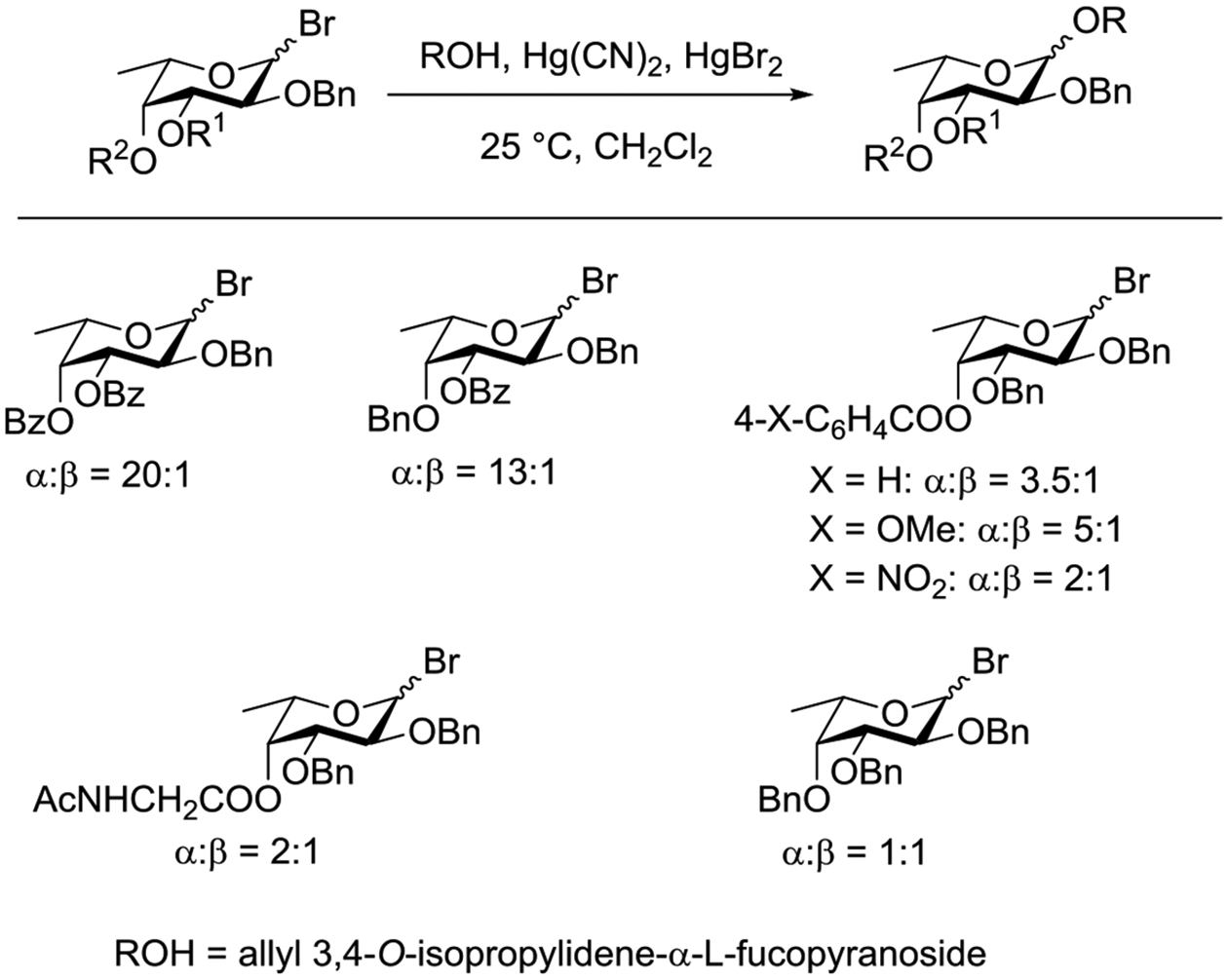
Dependence of Anomeric Selectivity on the Protecting Groups at O3 and O4 in Fucopyranosylation
Figure 21.
MM+ (and DFT) Energies of representative bridging ions relative to the corresponding 3H4 oxocarbenium Ion in the fucopyranosyl series (kcal.mol−1).
In a closely related study, Kalikanda and Li described the influence of distal esters and of temperature on the selectivity of 2-azido-2-deoxy galactosylation reactions.172,173 A series of axial trichloroacetimidate donors were prepared and their glycosylation reactions studied with a standard primary acceptor featuring activation in dichloromethane with TMSOTf at temperatures from −78°C to room temperature (Scheme 20). The authors found the peracetylated donor to be more α-selective than the corresponding perbenzylated donor and to exhibit a greater positive dependence on temperature, with α-selectivity increasing with increased temperature (Scheme 20). Control experiments ruled out equilibration and confirmed the observed selectivities to be kinetic. In common with the work of Nifantiev in the fucose series (Scheme 19), the greatest α-selectivity however was found with the donor carrying only two acetyl groups at the 3- and 4-positions. The least α-selectivity was observed with the donor carrying a single acetyl group at the 6-position. The authors concluded that esters at the 3- and 4-positions promoted α-selectivity by stabilization of the glycosyl oxocarbenium ion by the formation of bridging intermediates, whereas esters at the 6-position were detrimental to the stability of the oxocarbenium ion. A pair of esters at the 3- and 4-positions were considered to act synergistically and improve the overall α-selectivity, albeit no mechanism for this synergy was advanced.
Scheme 20.

Dependence of Anomeric Selectivity on the Protecting Groups at O3 and O4 in Galactopyranosylation
Like Nifantiev and coworkers, in support of their arguments the authors computationally derived relative energies of the anticipated bridging ions relative to the oxocarbenium ion, which in each case was found to prefer the 4H3 conformation (Figure 22).
Figure 22.
DFT Energies of representative bridging ions relative to the corresponding 4H3 oxocarbenium Ion in the galactopyranosyl series (kcal.mol−1).
While the calculations presented in Figures 21 and 22 obviously support the greater stability of dioxocarbenium ions relative to oxocarbenium ions, they do not account for the stabilization of glycosyl oxocarbenium ions in the form of glycosyl triflates, and the evident fine balance observed between bridging ions and covalent glycosyl triflates noted experimentally by the Huang and Williams groups (Schemes 17 and 18). When the stability of the glycosyl triflates and the temperature dependence of the reactions is taken into account a more complex picture necessarily emerges, and the attractiveness of mechanisms involving remote participation are downgraded relative to those based on the displacement of covalent intermediates. In fairness it must be noted that the ability to compute ion pairs of oxocarbenium ions with triflate anions was not developed until several years later.174–176 Unfortunately, neither the Nifantiev group nor Kalikanda and Li made any attempt to study their hypothetical bridged intermediates experimentally. The synergy between a pair of vicinal cis-esters at the 3- and 4-positions in promoting selectivity noted by Kalikanda and Li (Scheme 20 and Figure 22) is presumably related to the stabilization of inverted and non-chair conformers by the presence of multiple esters as discussed earlier.
Blériot and coworkers reported that per-O-acetyl-α-D-glucopyranose and per-O-acetyl-2-acetamido-2-deoxy-α-D-glucopyranose exist as the fully protonated starting materials in superacidic HF/SbF5 solution at −40 °C as determined by NMR spectroscopy. With the corresponding β-anomers, however, the dioxalenium and oxazolinium ions were observed (Scheme 21),177 clearly demonstrating both the importance of solvent polarity and potentially anchimeric assistance in the formation of such cyclic intermediates. These studies were subsequently extended to the β-D-galactopyranosyl and β-D-mannopyranosyl series.178 With respect to the galactosyl system, participation took place from the 2-position and the dioxalenium ion was the only species observed: no evidence was found for long range participation by the axial ester at the 4-position. In the mannosyl series, a mixture of the fully protonated substrate and the dioxalenium ion resulting from participation by the 2-O-acetate was observed (Scheme 21).
Scheme 21.
NMR Spectroscopic Observation of Fused Dioxalenium Ions in a Superacid Medium
IR characterization of participating esters has been realized by the use of hybrid MS/IR techniques. Thus, Boltje and coworkers, following electrospray ionization, used collision-induced dissociation to fragment the ammoniated parent ion from a per-O-acetyl-α-D-mannopyranosyl thioglycoside followed by isolation of the fragment ion in a quadrupole trap and its interrogation by IR spectroscopy. The experimental data fit closely with the computed spectrum for participation by the 2-O-acetyl group, as opposed to participation by the 3-, 4- and 6-O-acetyl groups, resulting in assignment of the dioxalenium ion (Scheme 22).179 Two mannuronate ester thioglycosides were similarly analyzed: in the first of these, which contained only methyl ether protecting groups, the observed IR spectrum was consistent with participation from the ester group through a six-membered methoxypyranium ion. In the second example, which carried an acetate ester at the 4-position in addition to methyl ethers at the 2- and 3-positions, a more complex IR spectrum was obtained. This spectrum was best interpreted as a composite of two species arising from participation by the side chain ester and by the 4-O-acetyl group. The IR spectra of these ions did not contain the stretching absorption anticipated for a simple mannosyl oxocarbenium ion, whose existence was demonstrated experimentally with the per-O-methyl ether (Scheme 22), confirming the stability of the doubly stabilized carbenium ions with respect to the simple oxacarbenium ion.
Scheme 22.
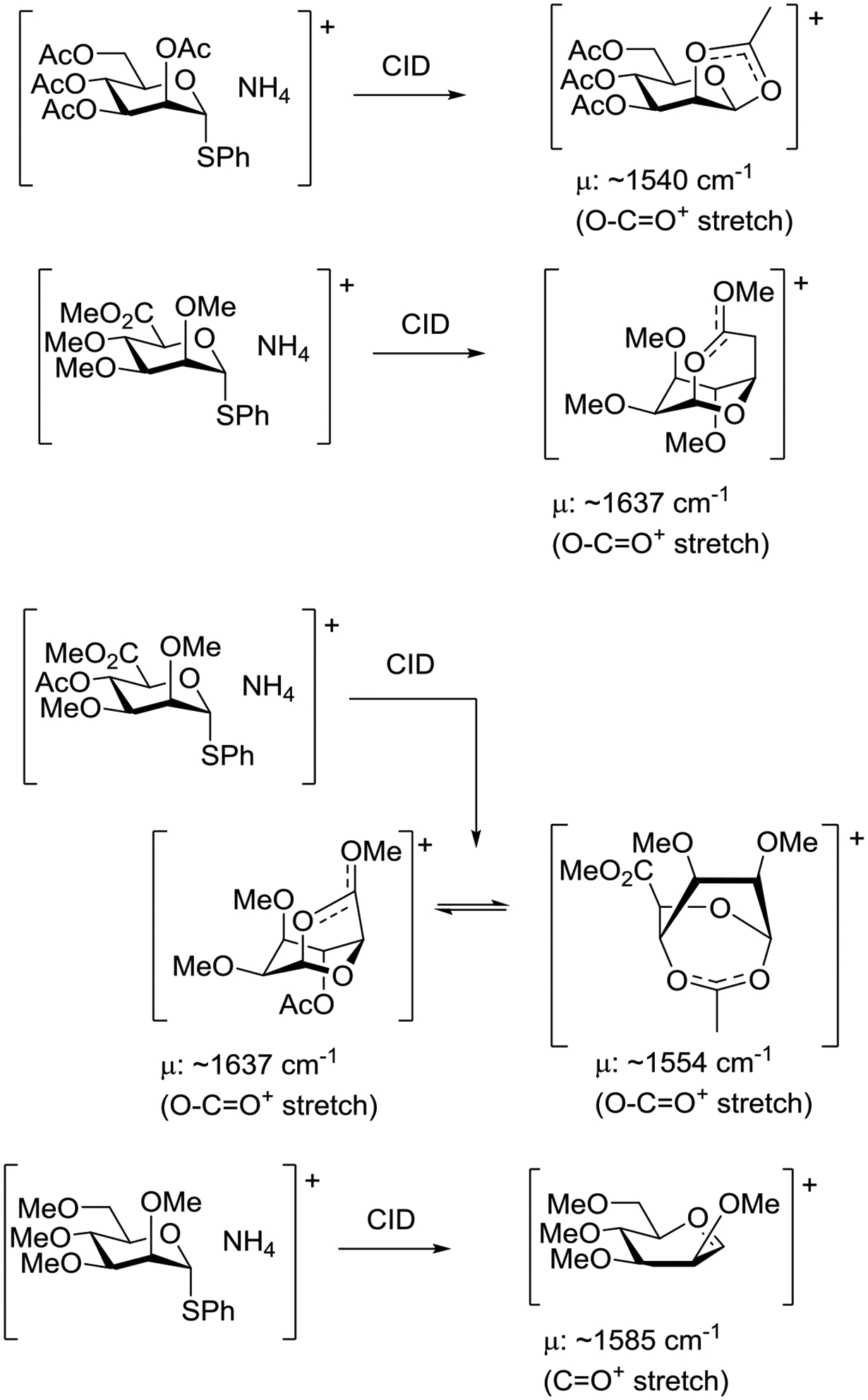
Fused and Bridging Dioxacarbenium Ions Observed by IR Spectroscopy in the Gas Phase
Using an alternate and cryogenic hybrid MS/IR technique Seeberger and coworkers collected fragment ions from the dissociation of the parent thioglycoside-derived molecular ions in a hexapole trap, from which they were then flowed in cryogenic liquid helium droplets at 0.4 K for IR examination.180 In this method IR excitation by multiple photons causes evaporation of the helium droplets and results in the spectrum of the naked gas phase ions at cryogenic temperatures. The 4- and 6-O-acetyl galactopyranosyl thioglycosides carrying benzyl ethers at the non-acylated positions were examined in this manner, and in each case the IR spectra generated supported the formation of bridging ions from participation by the remote acetyl groups as the major species. The spectra were generally of higher resolution than those obtained by Boltje and coworkers, owing to the much lower temperatures employed, but were otherwise in excellent agreement with them when the comparable species were studied.
A purely computational study by Kononov and coworkers employing DFT methods, and taking note of the influence of protecting groups on stereoselectivity in sialidation reactions,181,182 determined the relative energies of two chair conformers of the putative sialyl oxocarbenium ion and the bridging ions derived from participation by a 5-N-trifluoroacetyl group and by a 7-O-acetyl group (Scheme 23).183
Scheme 23.
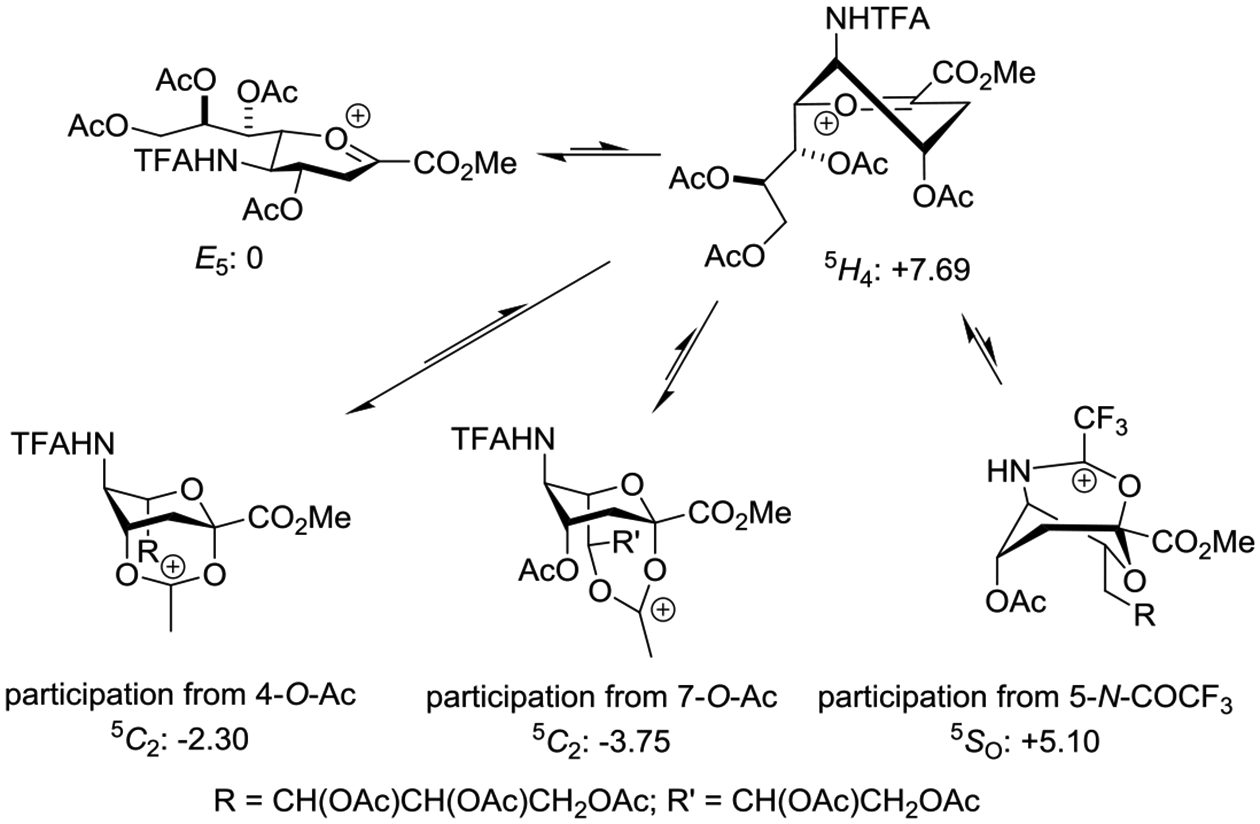
DFT Computed Relative Energies (kcal.mol−1) of Oxocarbenium Ions and of Bridging Ions in the Sialic Acid Series
The lowest energy conformation of the oxocarbenium ion was found to be the E5 conformer, which closely approximates the 2C5 conformation of sialic acid glycosides with all substituents at sp3-hybridized C in the pseudoequatorial configuration. The 5H4 half-chair conformation of the oxocarbenium ion, considered to experience through-space electrostatic stabilization by the pseudoaxial acetoxy and trifluoroacetamido groups,184,185 was found to be some 7.7 kcal.mol−1 higher in energy, presumably because of the pseudoaxial orientation of the bulky side chain. Participation by acetate groups at the 4- and 7-positions, however, was found to provide bridging dioxenium and dioxapenium ions some 2.3 and 3.8 kcal.mol−1 lower in energy than the most stable oxocarbenium ion. Unfortunately, these computations were conducted in the absence of counter ion, and we consider it unlikely that the relative energetics favoring the formation of bridged intermediates would be maintained in the presence of a typical counter ion such as the triflate anion that will differentially stabilize the oxocarbenium ion. Participation by the trifluoroacetamido group was found to be energetically unfavorable even in the absence of a counter ion rendering participation by this group highly unlikely (Scheme 23).
11. Role of the C2-O2 Rotor in Dioxalenium Ion Formation
Following up on their earlier computational study of dioxalenium ion formation and cleavage in the galactopyranosyl series,108 Whitfield and coworkers studied the role of ring conformation and of the C2-O2 rotor in formation of dioxalenium ions from glycosyl oxocarbenium ions. In the 3,4-O-isopropylidene galactopyranose series it was found, by means of dynamic DFT calculations, that the oxocarbenium ion purportedly formed on activation of the donor and undergoes a series of conformational changes leading to a OS2 skew boat in which the C2-O2 bond is pseudoaxial, and, in which the carbonyl group continues to eclipse the C2-H2 bond as in the ground state ester conformation. In this boat conformation, only a minimal rotation of the C2-O2 bond lines the carbonyl group for attack on the oxocarbenium ion and leads to ring closure (Figure 23).186
Figure 23.
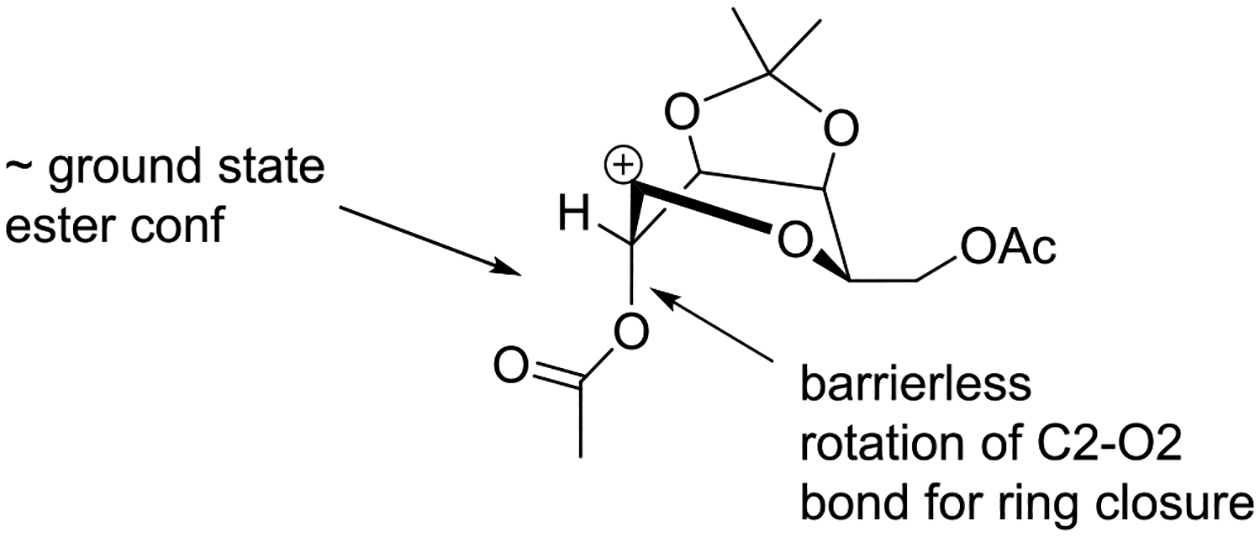
DFT-transition state for dioxalenium ion formation in the 3,4-O-isopropylidene galactopyranosyl system.
In the 3,4,6-tri-O-methyl-glucopyranosyl series on the other hand, the lowest energy conformation of the oxocarbenium ion was found to be a 5S1 conformer in which the carbonyl carbon also eclipses the C2-H2 bond. In order for ring closure to take place, it was determined that a pseudorotation to the 3S1 conformer and a rotation of the C2-O2 bond of 110° was required, with the latter considered the main component of the activation barrier of 6.6 kcal.mol−1 (Figure 24).187 A similar picture emerged for the corresponding mannopyranosyl oxocarbenium ion with rotation about the C2-O2 forming the main component of the barrier to closure of the dioxalenium ion from a preformed oxocarbenium ion.187 Albeit these studies were conducted in the absence of a counterion, and presume the unlikely existence of free oxocarbenium ions, they reveal the importance of the role played by the conformation of the ester in the formation of dioxalenium ions.
Figure 24.

DFT-transition state for dioxalenium ion formation in the 3,4,6-tri-O-methyl-glucopyranosyl system.
12. Stereodirecting Participation and Anchimeric Assistance from the Exocyclic Position of Ketoses
Very few studies have been conducted on the possibility of stereodirecting participation by esters at the exocyclic position of ketoses. Vasella and coworkers, building on early observations by Pacsu,188 observed the formation of two diastereomeric spirocyclic orthoesters as byproducts in the silver-mediated glycosylation of methanol by a per(4-nitrobenzoylated) fructopyranosyl chloride (Scheme 24).189 The authors, however, focused on the rigorous identification of the spirocyclic products and did not address the possibility of stereodirecting participation.
Scheme 24.
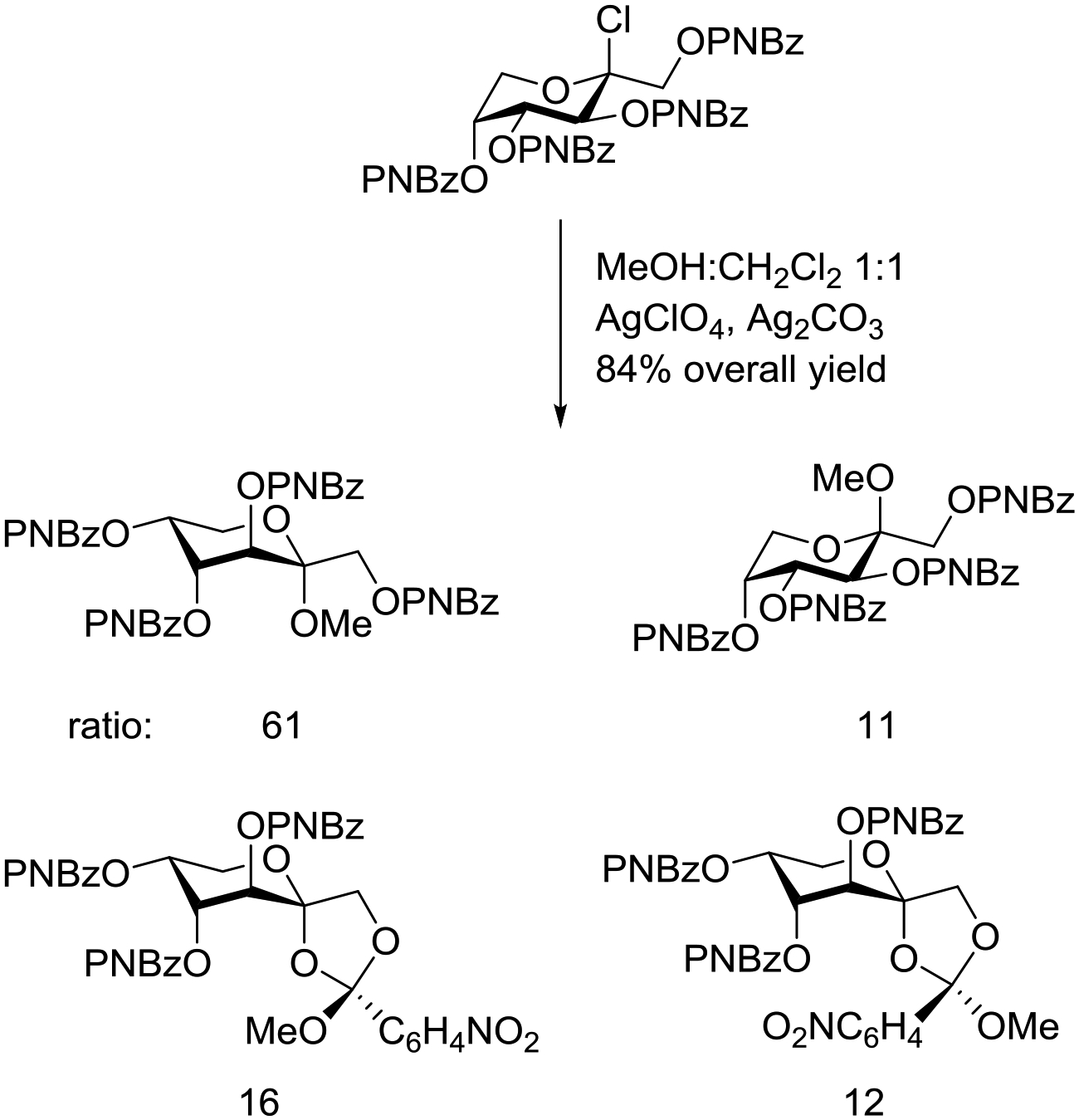
Formation of Spirocyclic Orthoesters Demonstrating the Possibility of Participation from the Exocyclic Position in Fructopyranosyl Systems
Subsequently, Lin and coworkers, operating in the fructofuranosyl series, isolated a pair of orthoesters on the attempted glycosylation of a N-phenyl trifluoroacetimidate donor with allyl alcohol on activation with TMSOTf in dichloromethane between −20°C and room temperature (Scheme 25).190 The possibility of stereodirecting participation by the exocyclic ester was raised by the authors, but as it was not possible to fully elucidate the structure of the orthoester (spirocyclic incorporating the 1-O-benzoate or fused encompassing the 3-O-benzoate), no firm conclusions could be drawn.
Scheme 25.
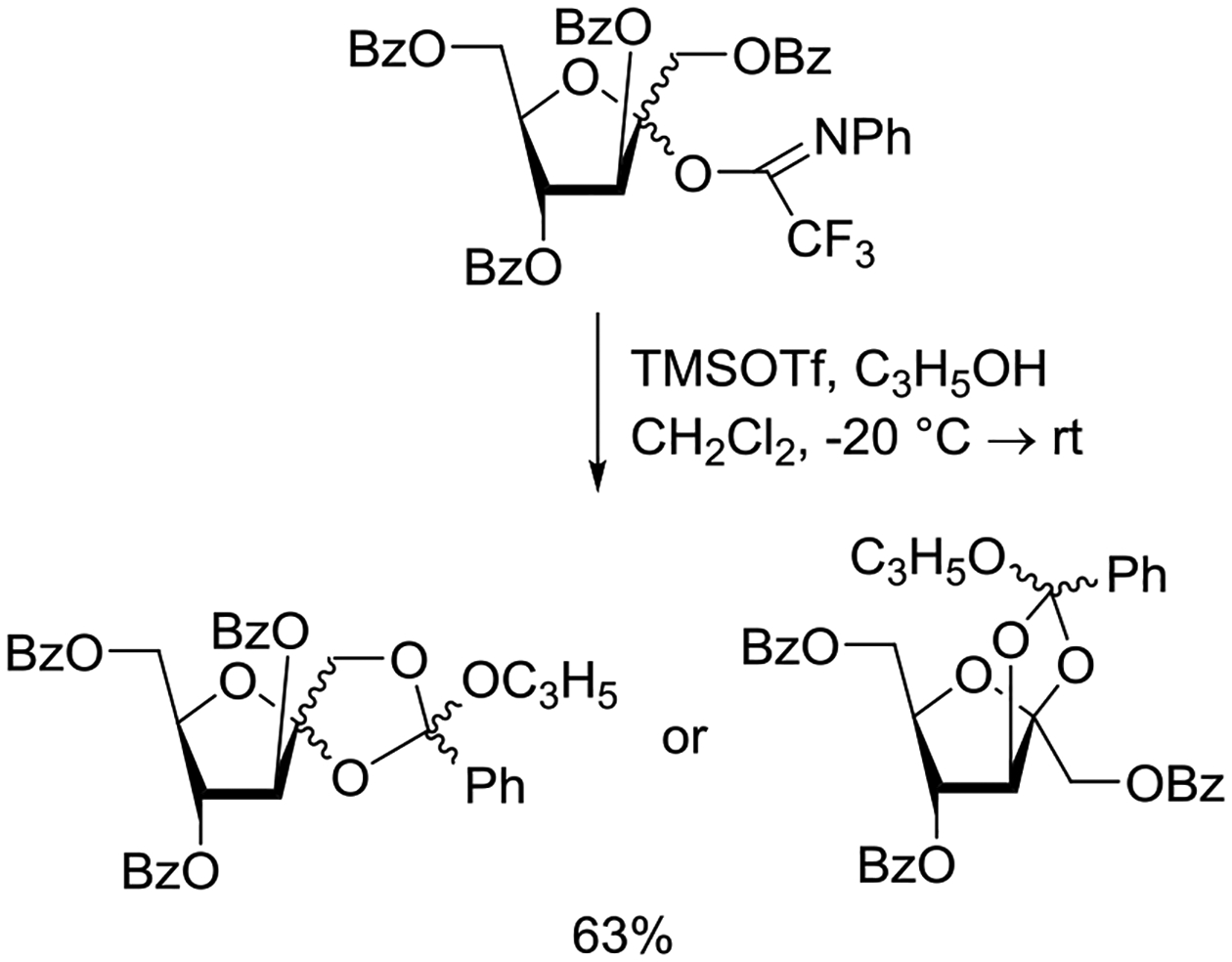
Alternative Structures of an Orthoester Isolated in the Fructofuranosyl System
Working in the N-acetylneuraminic acid series, Haberman and Gin designed an amide-based auxiliary tethered via the acid moiety and intended to provide stereodirecting participation via a six-membered spirocyclic imidonium ion (Scheme 26).191 While moderate to good equatorial selectivities exceeding those seen with the standard methyl esters were obtained with simple and primary carbohydrate acceptors with several activating systems, no physical evidence was provided for the suggested mechanism. Moreover, as each of the reactions was conducted at sub-zero temperatures, the suggested mechanism is unlikely as it requires the C1 carboxylate to adopt the unstable cis-ester configuration. Rather, the enhanced selectivity of the system over that seen with the simple methyl esters likely arises from the inductively electron-withdrawing effect of the appended amide functionality, which destabilizes the oxocarbenium ion and promotes a concerted mechanism either with direct displacement of the activated leaving group or of an intermediate glycosyl triflate. This hypothesis is supported by recent work from Schmidt, Sun, and coworkers who, as a result of extensive experimental and computational investigations, determined that the enhanced stereoselectivities observed in sialidations with picolinyl as compared to methyl esters of 4,7,8,9-tetra-O-acetyl-5-azido-5-deoxyneuraminic acid thioglycosides is likely due to the stabilization of an anomeric triflate rather than to participation by the picolinyl group (Figure 25).192 Indeed, there is increasing experimental evidence from several groups that, at least under some conditions, the phenomenon of hydrogen bond mediated aglycone delivery discovered by the Demchenko laboratory193 is due to protonation of the picolinyl group, or its complexation with a Lewis acid, which renders it more powerfully electron-withdrawing.194–196 This has the effect of destabilizing oxocarbenium ion-like transition states for glycosylation and consequently of promoting bimolecular SN2-like mechanisms.
Scheme 26.
Mechanism for α-Sialidation via Spirocyclic Participation Requiring Adoption of a Disfavored cis-Ester Conformation for Cyclization.
Figure 25.
Sialyl triflate stabilized by a strongly electron-withdrawing protonated picolinyl ester, and the alternative high energy intermediate arising from intramolecular participation.
13. Evidence for participation through 6- and 7-membered rings from the non-carbohydrate literature
Dolby and Schwarz studied the acetolysis of cis- and trans-2-acetoxymethylcyclohexyl brosylate with an 18O-label in the carbonyl group. After saponification of the reaction mixture, 76% of the label was retained implying that a major portion of the product was derived via formation of a six-membered intermediate dioxalenium ion (Scheme 27).197 It was also reported that solvolysis of the corresponding tosylate in ethanol afforded 20% of the ethyl orthoester (Scheme 28), thereby providing further support for the formation of the dioxalenium ion.198 The same ortho ester was also reported, albeit in 70% yield, on solvolysis of the regioisomeric cis-acetoxy tosylate (Scheme 28),199 indicating that the degree of participation is sensitive to substrate structure even when a common ion is formed. Treatment of the latter acetoxy tosylate with silver tetrafluoroborate in dichloromethane enabled isolation of the crystalline dioxenium ion tetrafluoroborate salt in almost quantitative yield (Scheme 28).200
Scheme 27.
18O-Labelling Experiment Demonstrating Participation by Acetate Through a Dioxenium Ion
Scheme 28.
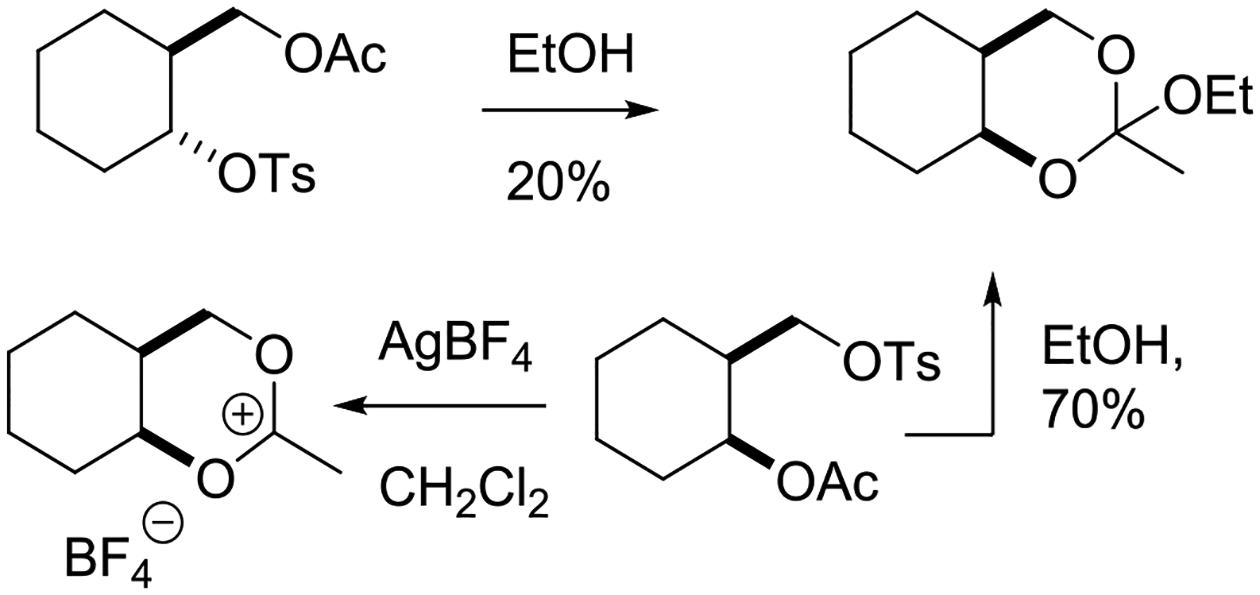
Trapping Experiments and Isolation of a Dioxenium Ion
An 18O-labelling experiment was also employed by Wilen and coworkers to demonstrate the participation of acetate through 5-, 6-, and 7-membered rings. In this experiment a series of diols was acetylated with 18O-labelled acetyl chloride and then exposed to aluminum chloride resulting in the formation of the corresponding acetoxyalkyl chlorides. The extent of participation was judged by the level of incorporation of the label following hydrolysis (Scheme 29). For the ethylene and propylene-based systems, it was estimated that >95% of the label was found in the product, indicative of very high levels of participation via 5- and 6-membered cyclic ions, respectively. For the butylene-based system on the other hand, 18O-incorporation into the diol was estimated at between 20 and 56% suggesting that while participation via a 7-membered dioxapenium ion is one pathway to the product, simple displacement competes effectively. In support of this argument it was noted that the butylene-based substrate was consumed more slowly than the propylene-based substrate, which in turn reacted more slowly than the ethylene glycol derivative.201 Overall this series of experiments, while establishing that participation through a 7-membered dioxapenium ion is possible, clearly demonstrates it to be much slower than participation through the lower homologs to the extent that intermolecular competition readily intervenes
Scheme 29.

Labelling Experiment Demonstrating the Relative Rates of Participation through Dioxalenium, Dioxenium, and Dioxapenium Ions
A more recent example of high yield participation of a carboxylate ester through a 6-membered dioxenium ion was reported in the synthesis of a complex natural product. Thus, acid-mediated dehydration of an allylic alcohol resulted in the formation of a dioxenium ion that was trapped intramolecularly by a vicinal alcohol to yield a cyclic orthoester in 90% isolated yield (Scheme 30).202 The ease of this transformation evidently arises from the pseudoaxial nature of the benzoate group which predisposes it to participation, but presumably also from the tertiary nature of the ester, which eliminates the usual conformation preference of secondary esters.
Scheme 30.
Participation of a Tertiary Benzoate through a Dioxenium Ion
14. Participation by Other Functional Groups
The advancement of participation by a proximal or distal functionality as a rationale for stereocontrol is by no means limited to esters. Indeed, stereodirecting participation by halides, ethers, thioethers, nitrogen heterocycles, and an array of o-functionalized benzyl ethers, through cyclic ions of varying size, has been invoked on many occasions in the literature of glycosidic bond formation, with important work by the Boons,203,204 Fairbanks,205–208 Demchenko,209,210 and Turnbull211–214 laboratories, among many others.215–219 As the focus of this review is on the mechanisms of stereodirecting participation by esters, and as a review has recently appeared on participation by ether type groups,220 this extensive area is not covered here other than to note the parallels with participation by esters. Thus, important issues involve the possible involvement of fused and bridging ions versus that of glycosyl oxocarbenium ions and/or reactive intermediates such as glycosyl triflates in the transition state for glycosidic bond formation, with evidence presented both for, and against, dependent on the system and conditions under study.177,178,221–229 This delicate balance between stereodirecting participation and alternative mechanisms is a prominent feature of the involvement of esters in glycosylation and apparent from the comparison of a recent study on 1,2-trans-glycosylation directed by the presence of 2-O-alkoxymethyl methyl groups,230 and one of the many instances of intramolecular aglycone delivery,231 a common strategy for 1,2-cis glycoside formation (Scheme 31).232–234
Scheme 31.

Neighboring Group Participation versus Intramolecular Aglycone Delivery
15. Analysis of Participation from Distal Positions.
Stereodirecting participation by remote esters has been invoked frequently in glycosylation reactions. The subject has been reviewed recently,26,27 and examples continue to be advanced in the context of a wide variety of glycosyl donors.150,161,162,235–242 Unfortunately, in most cases the reports are limited to observations of changes in selectivity, often relatively minor, when switching between a remote ether type protecting group and an ester. We have suggested elsewhere,151 and maintain here, that such participation by distal esters via bridged intermediates is improbable despite its attractiveness on the basis of extrapolation from participation by proximal esters.
Two extreme mechanisms can be envisaged for participation by remote esters, the first being an SN1 type process with initial formation of a glycosyl oxocarbenium ion followed by cyclization onto the distal ester and formation of the bridging dioxacarbenium ion. The second mechanism is one of direct displacement of the anomeric leaving group by the participating ester leading in a single reaction step to the bridging dioxacarbenium ion. These mechanisms are illustrated for the most commonly invoked case of α-D-galactopyranosides with assistance from the axial ester at O4 in Schemes 32 and 33. The SN1 mechanism must ultimately involve generation of the glycosyl oxocarbenium ion in a suitable conformation, coupled with the population of a high energy conformation of the participating ester, for ring closure to take place. The barrier for ring closure must therefore be a composite of three energetically unfavorable steps, each with its unfavorable equilibrium constant: ionization (K1); conformational reorganization of the oxocarbenium ion (K2); and conformational reorganization of the ester (K3); before the final, ultimately favorable ring closure (K4). The apparent rate constant (k) for ring closure from the glycosyl donor will therefore be a composite of four equilibria, only one of which is favorable (Scheme 32). The exact sequence of the steps in Scheme 32 will depend on the relative energetics of the individual steps, but the overall analysis holds whatever the sequence of events leading up to the final ring closure. It is impossible to escape the conclusion that the formation of bridging ring-closed dioxacarbenium ions from a glycosyl donor is kinetically slow and that consequently, alternative mechanisms will dominate. This analysis does not contradict the spectroscopic observation of bridging ring-closed ions in the gas phase (or perhaps eventually in the condensed phase in superacidic media), but simply emphasizes the fact that the thermodynamic stability of a dioxacarbenium relative to an oxocarbenium ion is only a part of a significantly more complex overall picture.
Scheme 32.
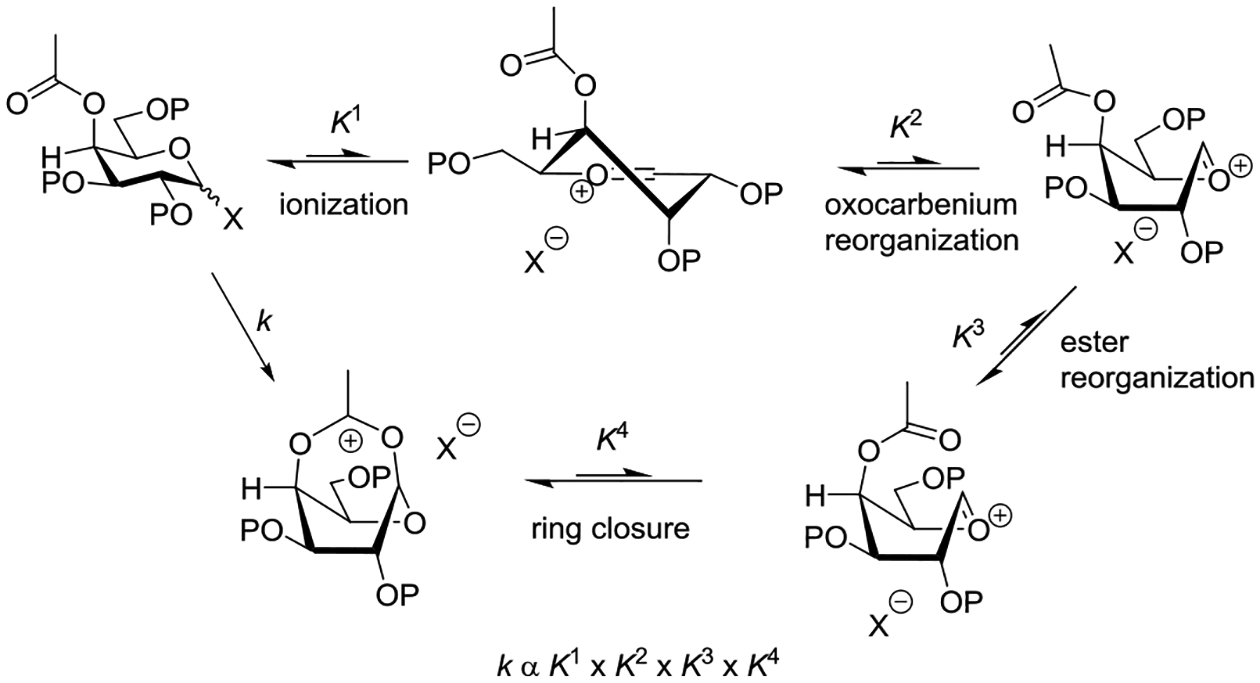
Stepwise Mechanism for Bridging Ion Formation via an Intermediate Oxocarbenium Ion.
Scheme 33.

Mechanism for SN2-like Formation of a Bridging Dioxacarbenium Ion
A similar analysis pertains to the SN2-like ring closure, where at the very least, a high energy conformation of the pyranoside suitable for cyclization and a high energy conformation of the ester must be accessed simultaneously (Scheme 33). Bridging ion formation will therefore necessarily be a kinetically slow process and other mechanisms can be expected to predominate.
Similar analyses can be made for bridging ion formation from equatorial esters at the 3-position, equatorial esters at the 4-position and esters at the 6-position of pyranoside rings, leading to the conclusion that each will be kinetically slow, however favorable the relative stabilities of the various ions, and that alternative mechanisms will dominate.
Remote participation by either of the mechanisms illustrated in Schemes 32 and 33 will become increasingly likely as one or more of the equilibria for ionization, conformational reorganization of the carbohydrate framework and of the ester become more favorable. Thus, as furanosyl oxocarbenium ions are more stable than comparable pyranosyl oxocarbenium ions, and as the barriers to pseudorotation of furanosides are lower than those for the inversion of pyranosides, stereodirecting remote participation in the furanoside series is less unlikely. Septanosides,243–245 again because of the lower barriers to conformational equilibration of the ring, can similarly be expected to be moderately more conducive to remote participation. Donors carrying multiple esters, particularly cis-configurated vicinal esters, will have a lower overall barrier to participation because of the greater propensity to the population of non-chair, or inverted chair conformations found in such systems.
Axial esters at the 3-position of pyranosyl donors also present a far more likely case for stereodirecting participation than their equatorial counterparts or esters at the 4- or 6-positions. In the SN1 scenario this is because i) little distortion of the oxacarbenium ion from the usual half-chair or envelope conformations will be required for ring closure, and ii) because a rotation of only ~120°about the C3-O3 bond is required to line the ester carbonyl up for cyclization (Scheme 34). In the SN2 manifold, in addition to the reduced rotation required about the C3-O3 bond (not illustrated), it will be appreciated that the anomeric equilibrium favors the β-anomer of the donor to a greater extent, because of minimization of 1,3-diaxial interactions (Scheme 34). Indeed, a number of likely examples of participation from such esters have been presented.159,246–248 Moreover, it has proven possible to trap such bridging intermediates under conditions of actual glycosylation reactions (Table 10).
Scheme 34.
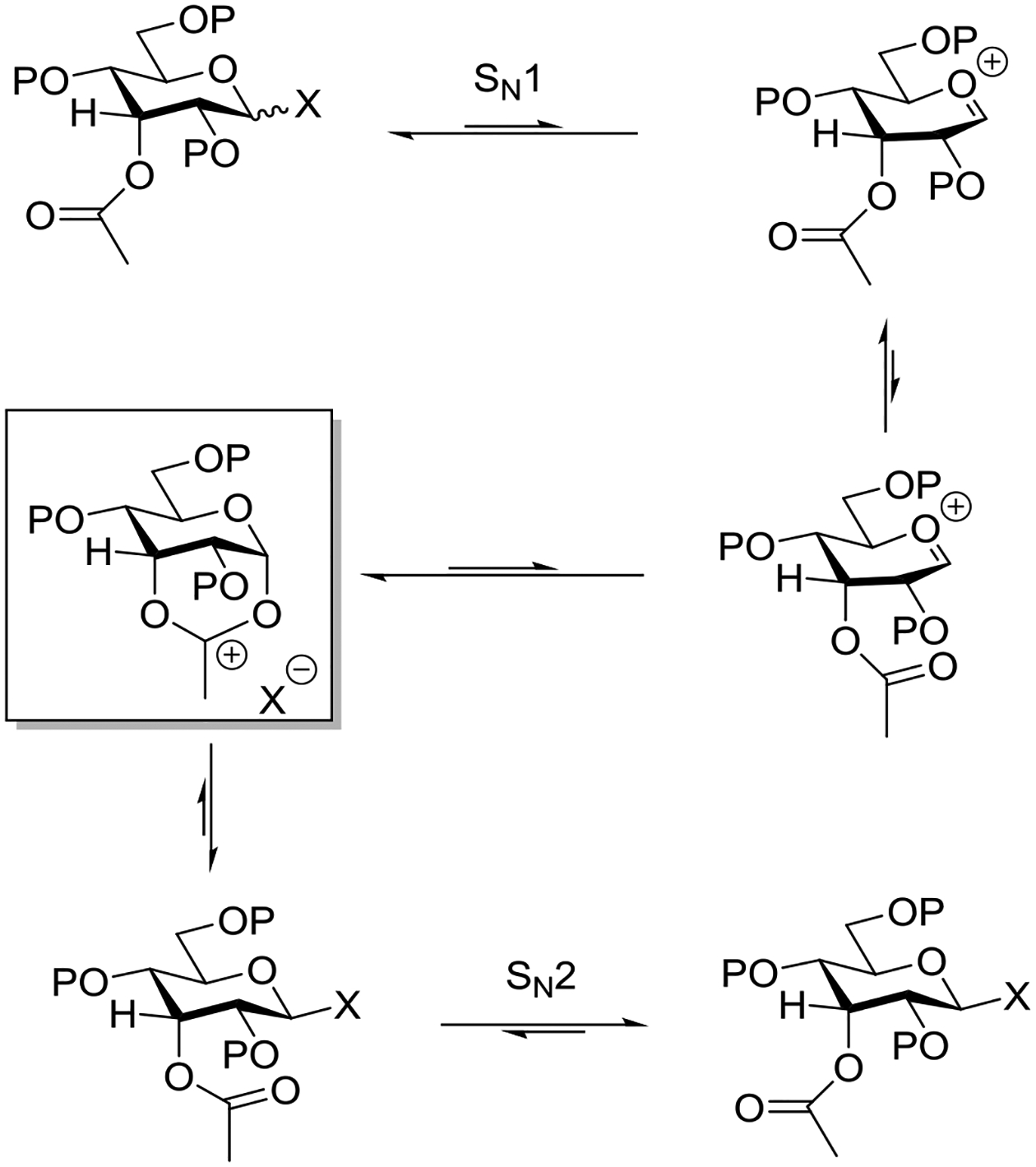
SN1 and SN2 Manifolds for Bridging Ion Formation from an Axial Ester at the 3-Position
16. Comparison of the Relative Ease of Participation by Proximal and Distal Esters.
The most obvious exception to the above analysis of participation by distal esters is that of participation from the 2-position, ie, classical neighboring group participation. Several factors contribute to the relative ease of participation from the proximal position. The first of these is the greater exothermicity of the transformation of a glycosyl oxocarbenium ion to a fused dioxalenium ion. This increase in exothermicity is the result of two factors, the first of which itself can be traced to two factors: first, the evidence that 5-membered cyclic dioxalenium ions are more thermodynamically stable than their higher homologs is incontrovertible both in simple models (Figure 12) and on sugar frameworks (Scheme 7), and is well-supported computationally (Figures 21 and 22, and Schemes 22). The second factor in the greater exothermicity of ring closure of proximal esters onto oxocarbenium ions as opposed to that from distal esters arises from the differential electron-withdrawing, destabilizing influence of proximal and distal esters on glycosyl oxocarbenium ions.: it is very well documented that esters at the 2-position exert a much stronger disarming influence on the oxocarbenium ion than those at more remote positions.96,138,249 Thus, dioxalenium ion formation from a 2-O-acyl oxocarbenium ion begins from a higher energy species and ends with a lower energy species than comparable ring closures involving esters at the 3-, 4-, and 6-positions. The change in entropy on ring closure also plays an important role. Thus fewer degrees of freedom are lost on cyclization of a proximal ester to give a fused bicyclic system, than are lost on the closure of a more remote ester to afford a conformationally more rigid bridged bicyclic system, something that is especially pertinent to participation by esters in the side chain which constrains the previously freely rotating exocyclic bond. Finally, it requires only a minimum rotation about the C2-O2 bond away from the ground state ester conformation in order to achieve suitable overlap with the oxocarbenium ion for ring closure.
17. Alternative Hypotheses for the Influence of Remote Esters
The broadest alternative explanation for the influence of remote esters and other electron-withdrawing protecting groups simply builds on the general mechanism for glycosylation, and the ability of protecting groups to influence the key covalent donor – ion pair equilibria.1 Thus, as first advanced by van Boeckel and coworkers250 building on earlier work by the Paulsen lab,251 the more electron-withdrawing a protecting group is, the more it will destabilize glycosyl oxocarbenium ions and ion pairs incorporating them, and shift glycosylation mechanisms toward the SN2-end of the mechanistic continuum (Scheme 35). The van Boeckel hypothesis is supported in the gluco- and mannopyranoside series by their observation of the correlation of increased β-selectivity with the electron-withdrawing ability of esters at the 4-position. Strong-supporting evidence is also provided by the work of Kim and coworkers.163,252 A comparable analysis holds for the influence of protecting groups at the 6-position.
Scheme 35.
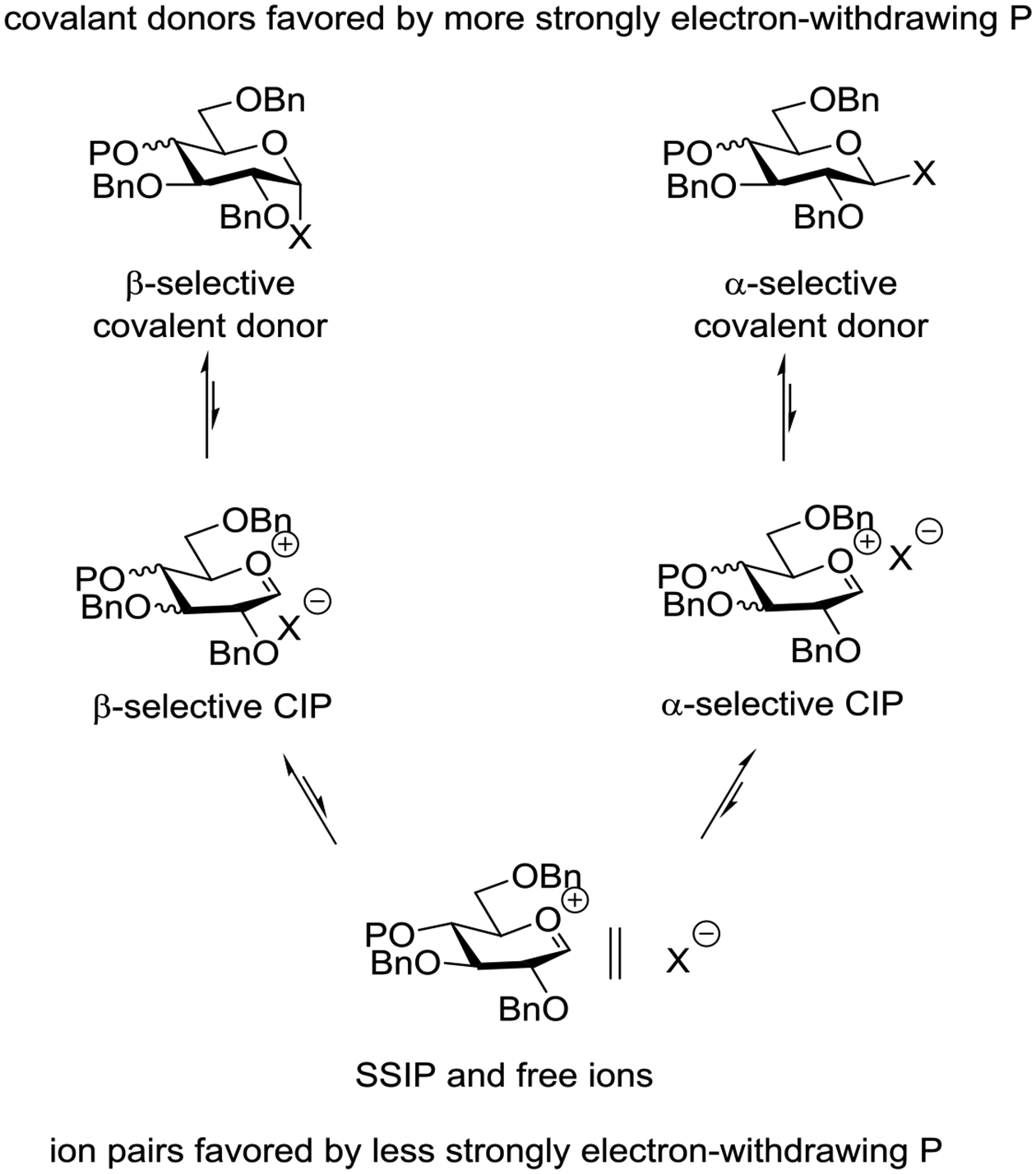
Influence of Remote Protecting Groups on Covalent Donor – Ion Pair Equilibria
In the galacto and related fucosyl and KDO series, esters at the 4-position clearly do enhance α-selectivity. As discussed by van Boom and coworkers, this is still consistent with enhancement of the SN2-like reaction manifold provided it is coupled with a Lemieux-type253 rapid in situ anomerization and preferential displacement of the leaving group from the equatorial position.254 Alternatively, the change in selectivity seen with 4-O-acyl protected donors on going from the gluco and manno series to the galacto series is consistent with a switch in mechanism from SN2-like in the former to SN1-like in the latter.
The most convincing and oft-cited evidence for participation by axial esters at the 4-position comes from the laboratories of Nifantiev and Boons. Nifantiev and coworkers studied the selectivity of series of 4-O-benzoyl-, 4-O-p-methoxybenzoyl-, and 4-O-p-nitrobenzoyl- 2,3-di-O-benzyl-α,β-L-fucopyranosyl bromides toward a common acceptor on activation with a combination of mercuric cyanide and mercuric bromide in dichloromethane.171 They found the p-methoxybenzoate to afford the highest, and the p-nitrobenzoate the lowest levels of α-selectivity, which they argued was consistent with stereodirecting participation by the remote group and for which computational support was advanced (Scheme 19 and Figure 21).
Contemporaneously, Boons and coworkers found that 4-O-benzoyl protected galactopyranosyl donors afforded increased α-selectivity when compared to alkanoyl esters. Furthermore, electron rich benzoates were more α-selective than electron poor ones and, in the alkanoyl series, a pivaloate was more α-selective than an acetate, which in turn was more selective than either a trichloroacetate or a trifluoroacetate (Table 12).255 These observations drove Boons and coworkers to favor an SN1-like mechanism and argue in support of participation by the distal ester as the underlying cause of the observed α-selectivity (Figure 26). However, it can just as easily be argued that the more electron-donating an ester group, the greater will be its ability to stabilize developing positive charge by through space electrostatic interaction (Figure 26). This latter hypothesis, which also applies to the fucopyranosyl and KDO series, builds on the generally accepted explanation1,184,185,256 of the increased reactivity and α-selectivity of galactopyranosyl donors over their glucopyranosyl analogs and importantly does not require the ester to deviate from its low energy conformation. The influence of the electron-donating or withdrawing ability of the p-substituent on the ester is perhaps most easily visualized in terms of the no-bond resonance form involving an acylium ion alkoxide pair. Through space stabilization of electrostatic charge at the anomeric center by the ground state ester is consistent with the enhanced population of the axial conformer of 4-(4-chlorobenzoyloxy)cyclohexanone, relative to the cycloalkane (Figure 6). Finally, electrostatic stabilization of positive charge at the anomeric center by the ground state ester also satisfactorily explains the solvent effects noted by Boons and coworkers, with 1,4-dioxane/toluene mixtures affording better α-selectivity than dichloromethane, and confirmed by numerous groups subsequently.237 Yet others have argued that “electrostatic effects”, rather than remote participation, underlie the marginal increase in α-selectivity observed with a 4-O-acetyl-2-azido-3,6-di-O-benzyl-2-deoxy-β-D-galactopyranosyl fluoride on activation with dicyclopentadienylhafnium dichloride in dichloromethane as compared to the corresponding 4-O-pentafluoropropionyl donor.107,257
Table 12.
Influence of Esters at the Galactopyranoside 4-Position on Anomeric Selectivity
 | ||||||
|---|---|---|---|---|---|---|
| R | % yield | α:β ratio | σp258 | F258 | gg/gt/tg | |
| H | 68 | 1.7/1 | 0 | 0 | 21.9/34.4/43.7 | |
| Me | 90 | 2.9/1 | −0.17 | 0.01 | - | |
| CH2Ph | 91 | 2.2/1 | −0.09 | −0.04 | 13.2/22/6/64.2 | |
| CH2CF3 | 58 | 2.3/1 | 0.09 | 0.15 | 11.7/21.7/66.7 | |
| COMe | 76 | 7.2/1 | 0.50 | 0.33 | 16.4/31.0/52.6 | |
| COCMe3 | 74 | 16/1 | 0.32 | 0.26 | 15.0/28.6/56.4 | |
| COCCl3 | 72 | 4.5/1 | - | - | 6.9/18.8/74.3 | |
| COCF3 | 71 | 3.0/1 | 0.46 | 0.58 | 7.7/19.2/73.0 | |
| COPh | 72 | 17/1 | 0.43 | 0.31 | 15.6/30.5/53.9 | |
| COC6H4-4-Me | 82 | 18/1 | - | - | 15.4/32.0/52.5 | |
| COC6H4-4-OMe | 85 | 33/1 | - | - | 16.2/32.5/51.3 | |
| COC6H4-4-NO2 | 87 | 14/1 | - | - | 17.3/23.5/59.1 | |
Figure 26.
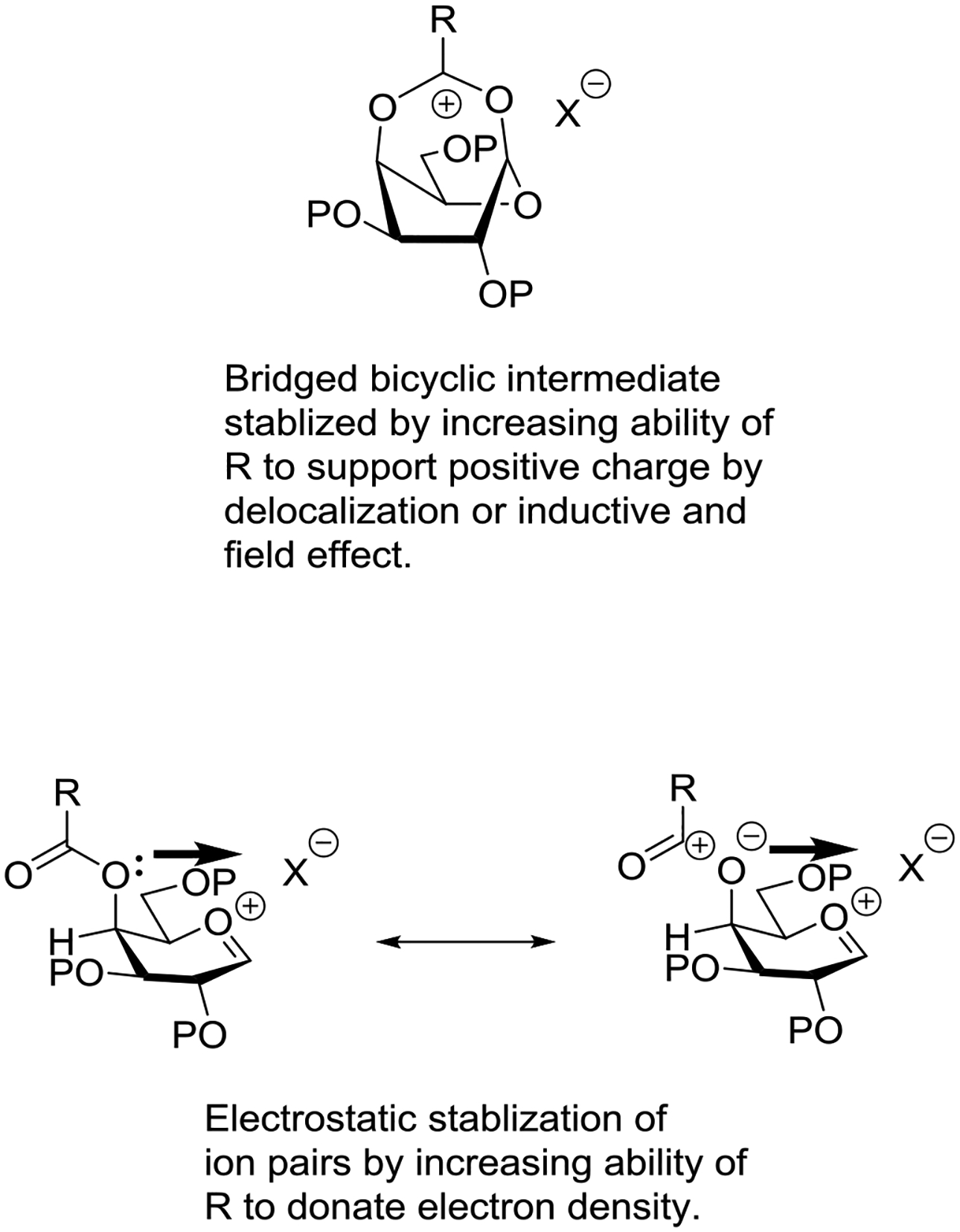
Correlation of α-selectivity with increased electron-donating potential of esters at the galactopyranosyl 4-position is consistent with remote participation and with through space electrostatic stabilization of positive charge at the anomeric position.
A further hypothesis advanced by Crich and coworkers, and based on the well-documented influence of side chain conformation on the reactivity and selectivity of glycosyl donors,259–263 postulates that changes in the protecting group at the 4-position result in changes in the conformation of the side chain and so have an indirect influence on selectivity.264 Indeed, in an NMR spectroscopic study of side chain conformation of the same series of phenyl 2,3,6-tri-O-benzyl-β-D-thiogalactopyranosides employed by Boons and coworkers, distinct changes in the distribution of the side chain conformation among the three staggered gg, gt, and tg conformers on changing the O4 protecting group were observed (Table 12, last column). Of particular note is the high proportion of the strongly disarming tg conformer observed with the highly electron-withdrawing trichloroacetyl and trifluoroacetyl esters; similarly in the series of 4-substituted benzoate esters it is the most electron withdrawing 4-nitrobenzoate that has the highest proportion of the tg conformation. Albeit a different pattern was found, the side chain conformation of phenyl 2,3,6-tri-O-benzyl-β-D-thioglucopyranosides was also found to be modulated by the protecting group at the 4-position. While these effects were out of necessity only observed on covalently bound unactivated donors, it can be anticipated that similar, and likely, more substantial effects will occur in glycosylation transition states as summarized in Figure 27. Clearly, this hypothesis does not extend to the fucopyranosyl series in which the side chain consists of a simple methyl group, nor to the pentopyranosyl series which lacks a side chain altogether.
Figure 27.
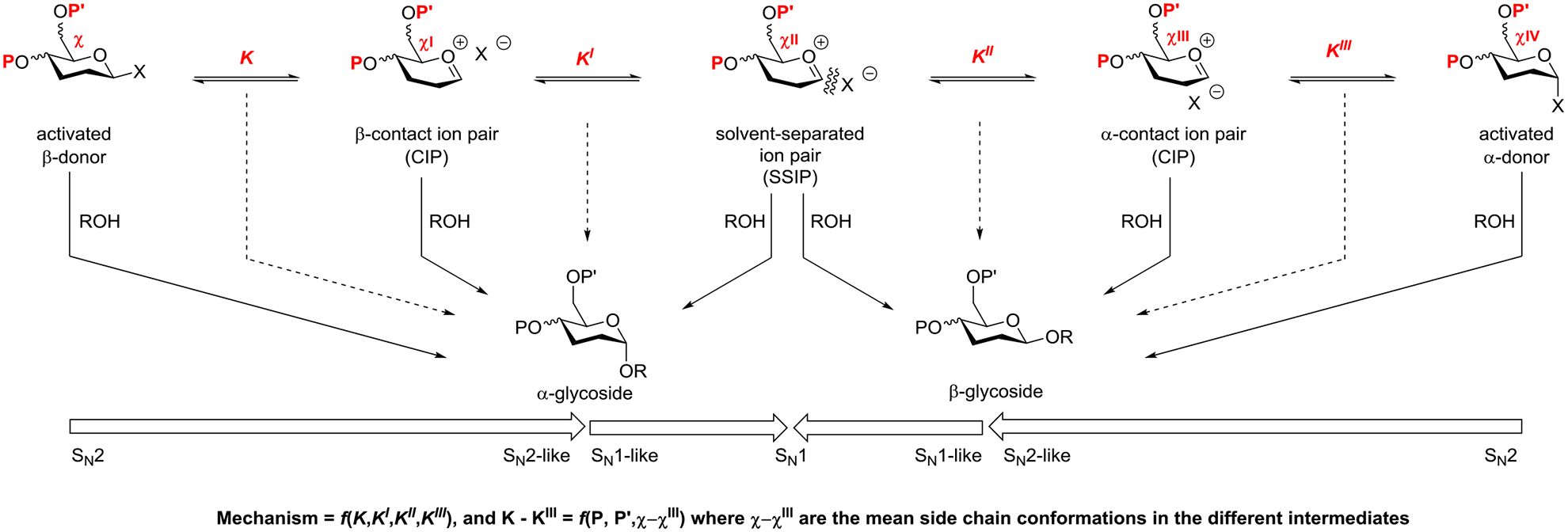
Abbreviated glycosylation mechanism showing the influence of the protecting group at the 4-position on the side chain conformation.
Turning to equatorial esters at the 3-position, significant differences are found in anomeric stereoselectivity with the corresponding donors carrying ethers at the same position. Thus, van Boeckel and coworkers noted in 1984 that the presence of an acetyl or trichloroacetyl protecting group at the 3-position of glucopyranosyl bromides carrying ethers at the 2- and 4-positions gave predominantly the α-glycosides on activation by silver silicate, whereas the corresponding 3-O-benzyl ethers gave the opposite selectivity.250 Numerous related examples in the glucopyranosyl series were later presented by Nifantiev and coworkers, working with thioglycosides and or glycosyl sulfoxides, who also noted that the coupling of protecting group changes at the 3-position with those at the 6-position.265,266 Similar effects have also been noted in the rhamnopyranosyl series, albeit with an unusual solvent dependence.267 However, the most remarkable changes in selectivity due to the change of an ether for an ester protecting group have been noted in the 4,6-O-benzylidene protected mannopyranosylation reaction. Thus, in contrast to the excellent β-selectivity seen with 2,3-di-O-benzyl-4,6-O-benzylidene protected mannopyranosyl donors of all types and attributed to the SN2-like displacement of a covalent α-mannosyl triflate,1,268,269 it was found that the corresponding 3-O-acyl-2-O-benzyl protected donors were highly α-selective.156,270 The extent of this change is most apparent in the course of a synthesis of a repeating unit from a bacterial lipopolysaccharide, when two mannopyranosyl donors differing only in the protecting group at O3 gave completely opposite selectivity (Scheme 36).271
Scheme 36.
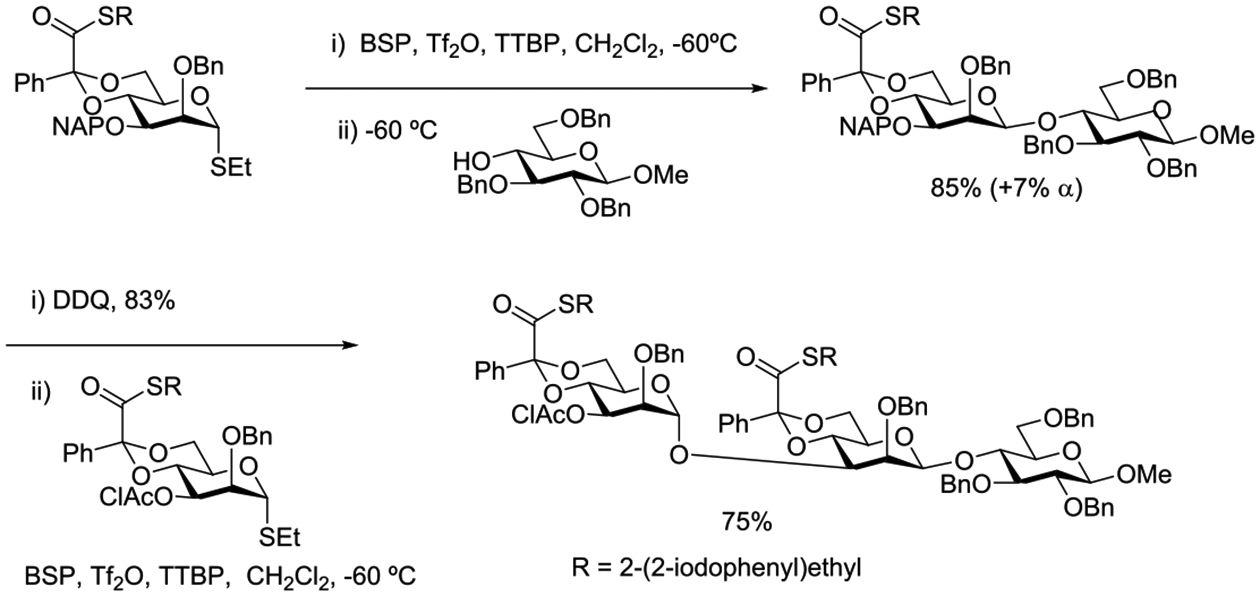
Inversion of Selectivity in Mannopyranosylation on Replacement of a 3-O-Benzyl Ether by a 3-O-Carboxylate Ester
Initially, the α-selectivity imposed by the presence of the carboxylate at the 3-position was considered to be the effect of remote participation by that ester,156 and this explanation is still upheld by some authors in a more general sense.26,27,252 Subsequently, Crich and co-authors suggested that repulsion between the parallel dipoles of the ester at the 3-position and of the anomeric C-O bond would destabilize the usual α-mannosyl triflate and promote alternative mechanisms of a more dissociative nature, or possibly involving the intervention of a transient β-mannosyl triflate (Figure 28).272 Later, recognizing the importance of the ester group at the 3-position in their chiral auxiliary directed α-glucopyranosylation and galactopyranosylation reactions, Boons and coworkers proposed with computational support that the function of the ester is to serve as a hydrogen bond donor to the incoming acceptor alcohol and so direct it to the α-face of the donor (Figure 28).204 Both hypotheses displayed in Figure 28 have the distinct advantage of retaining the ground state conformation of the ester linkage, and a synergy between the two effects seems likely. The role of donor-acceptor hydrogen bonding and proton transfer in glycosidic bond formation has been studied from a computational perspective by Whitfield as summarized in a recent review.273
Figure 28.
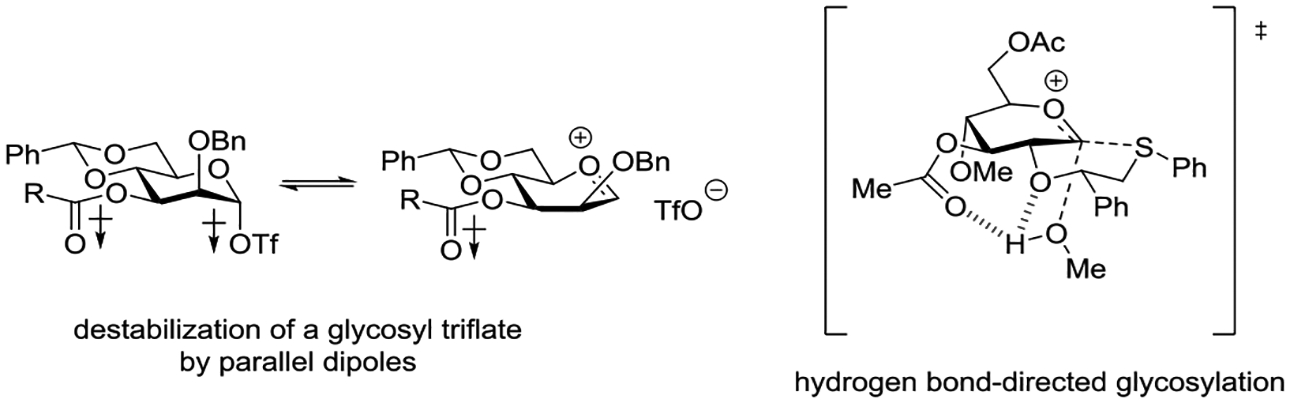
Two hypotheses for the α-directing effect of 3-O-carboxylates.
18. Ester Migration during Participation
Ester migration during the course of glycosylation steered by participation of a vicinal ester is one of the common glycosylation side reactions.105 It can take place intramolecularly to the anomeric position or intermolecularly to the acceptor alcohol; in either case it has a negative impact on yield and complicates product isolation. Many examples have been described in the literature, many of which are listed in the work of Whitfield and coworkers.108,117,274 Efforts to limit acyl group transfer parallel those used to limit orthoester formation, and mainly rely on the use of sterically bulky participating groups.106,110–119 Consistent with this, it has been demonstrated that the relative ease of migration in glycosylations with 2,3,4,6-tetra-O-acyl galactopyranosyl trichloroacetimidates is benzoyl << isobutyryl ~ acetyl.117 In a more extensive series of 2-O-acyl-3,4,6-tri-O-benzyl galactopyranosyl trichloroacetmidates the relative ease of migration of the acyl group was determined to be benzoyl = pivaloyl << levulinoyl < acetyl < formyl.117
Mechanistically, it has been demonstrated through use of regioselective deuterium-labelling that in glycosylations with acetobromogalactose, it is the ester from the 2-position that is transferred (Scheme 37),275 in broad agreement with the idea of acyl transfer as a byproduct of participation by a proximal ester.95,101,102 A related experiment leading to the same conclusion has been described by Kochetkov and coworkers.276
Scheme 37.
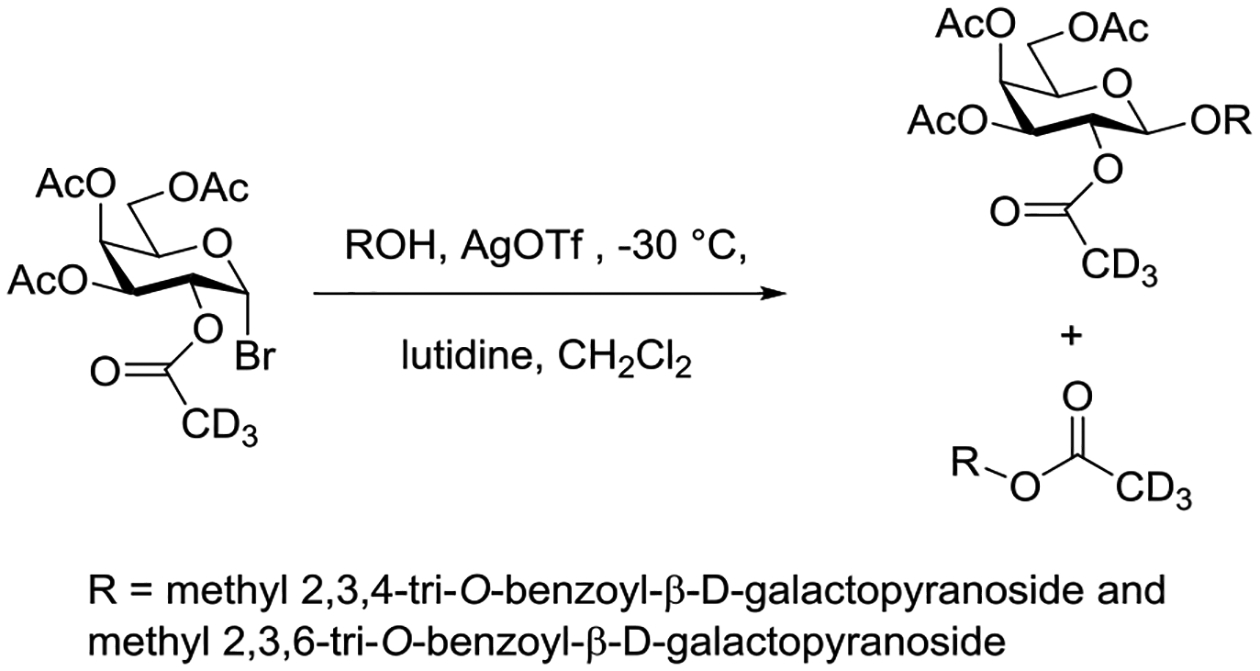
Use of Deuterium Labelling to Demonstrate the Origin of a Migrating Ester Group.
Following on from their work on the acid-catalyzed hydrolysis of 3,4,6-tri-O-methyl-1,2-(ethoxyethylidene)-α-D-glucopyranose,277 a mechanism for acyl transfer was offered by Wallace and Schroeder.95 Thus, according to these authors, acyl transfer is the result of protonation of the orthoester on what was originally the carbonyl oxygen of the migrating ester. Ring opening with assistance from the pyranosyl ring oxygen then affords a glycosyl oxocarbenium ion that is trapped on either face by a further equivalent of the acceptor alcohol resulting overall in the formation of glycosides carrying a hemiorthoester on the 2-position; decomposition of this latter species completes the acyl transfer step (Scheme 38).
Scheme 38.
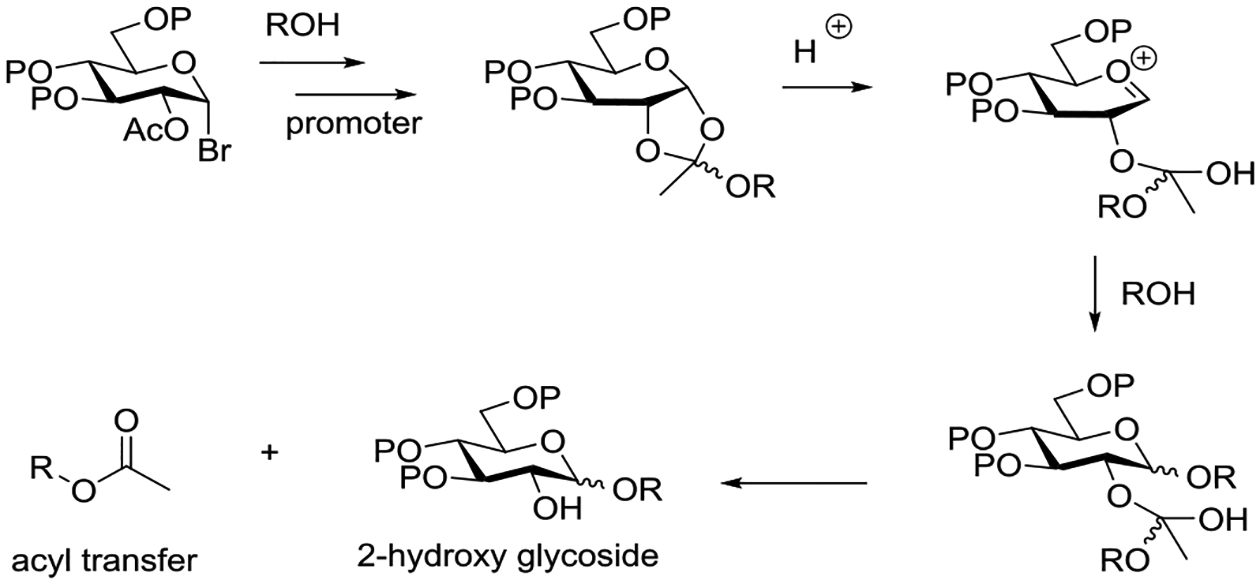
Mechanism Advanced by Wallace and Schroeder for Acyl Transfer
Building on work from Kochetkov and coworkers,278 who noted the greater basicity of O2 (as opposed to O1 in orthoesters) making it the most likely site of protonation, Whitfield and coworkers conducted a DFT computational study of the mechanism of acyl transfer in the 3,4-O-isopropylidene galactose series, which they found to be particularly susceptible to this reaction.117,274 As a result of this computational study it was postulated that the key step in acyl transfer is proton transfer within a three component complex of the dioxalenium ion and two molecules of acceptor (Scheme 39). Important aspects of this relay mechanism were considered to be the initial generation of the acyl transfer product in the higher energy cis-conformation for all cases, except in the transfer of a pivalate group, and the avoidance of an orthoester as a key intermediate. Barriers to the formation of the initial three component hydrogen bonded assembly were found to vary significantly with the type of orthoester and ranged from 7.2 kcal.mol−1 for R = H (orthoformate) to 18.7 kcal.mol−1 (R = Ph), thereby explaining the relative ease of acyl migrations. Conversely, the relative energy of the second transition state ranged from 9.6 – 22.0 kcal.mol−1, with the pivalate having the lowest barrier, consistent with its formation directly in the trans-ester configuration, and the benzoate the highest barrier.
Scheme 39.
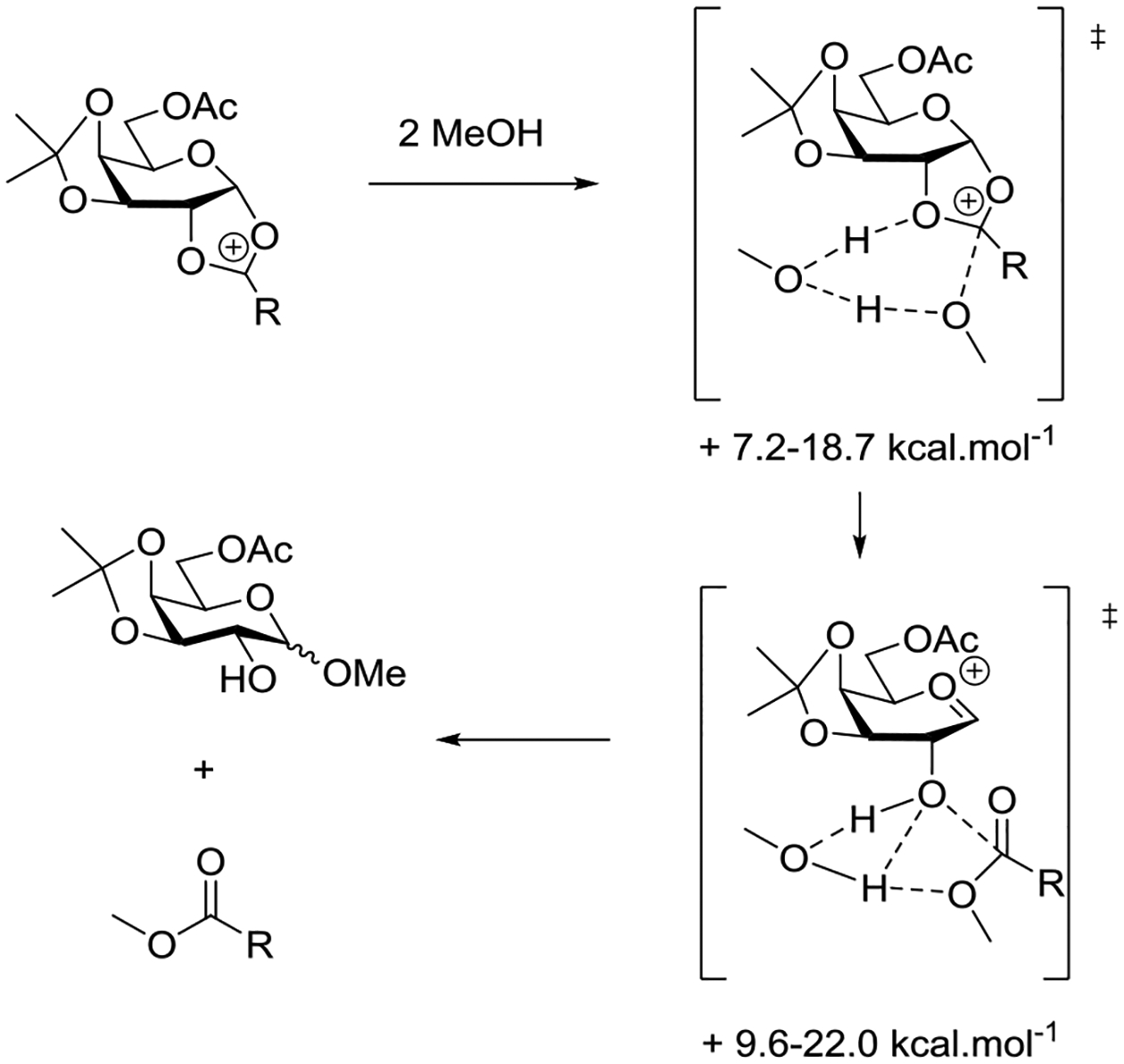
Alternative Mechanism for Acyl Transfer During Glycosylation Eliminating the Need for Prior Orthoester Formation
The mechanism of intramolecular acyl migration to the anomeric position during the course of glycosylation reactions is readily understood277 in terms of attack of water on the intermediate dioxalenium ion and is not further discussed here.
Finally, we are not aware of any examples of acyl migration directly from a remote position to either the aglycone (intermolecular transfer) or the anomeric position (intramolecular transfer) during the course of glycosylation reactions, despite rigorous attempts to identify at least the latter (Scheme 12).151
19. Ester Migration in Partially Acylated Polyols
In addition to migration during the course of neighboring group directed glycosylation reactions, esters also migrate in partially acylated polyols under a variety of acidic, basic, neutral and enzymatic conditions. The actual migration path in monosaccharides has been subject to several investigations; it is often presumed to be intramolecular and clockwise between two adjacent hydroxy groups via cyclic five- or six-membered intermediates. Migrations over one or more monosaccharide hydroxy groups in pyranoses, such as O2→O4 in glucopyranose have been proposed,279,280 but lack direct experimental evidence. Theoretically, when other possible pathways are blocked, non-adjacent migration could take place, provided that a suitably low energy conformation with a sufficient lifetime is accessible. Potential non-adjacent migration was studied, but not observed, in a myo-inositol derivative (Figure 29) in which only transannular migration is possible; in the boat conformation the acetyl group would appear to approach the free hydroxy group across the ring at sufficiently short distance, but consideration of the ground state conformation of the ester group reveals that this is not the case.281 Similarly, in d-mannopyranose the potential O2→O6 migration has been investigated, but not experimentally observed.282
Figure 29.
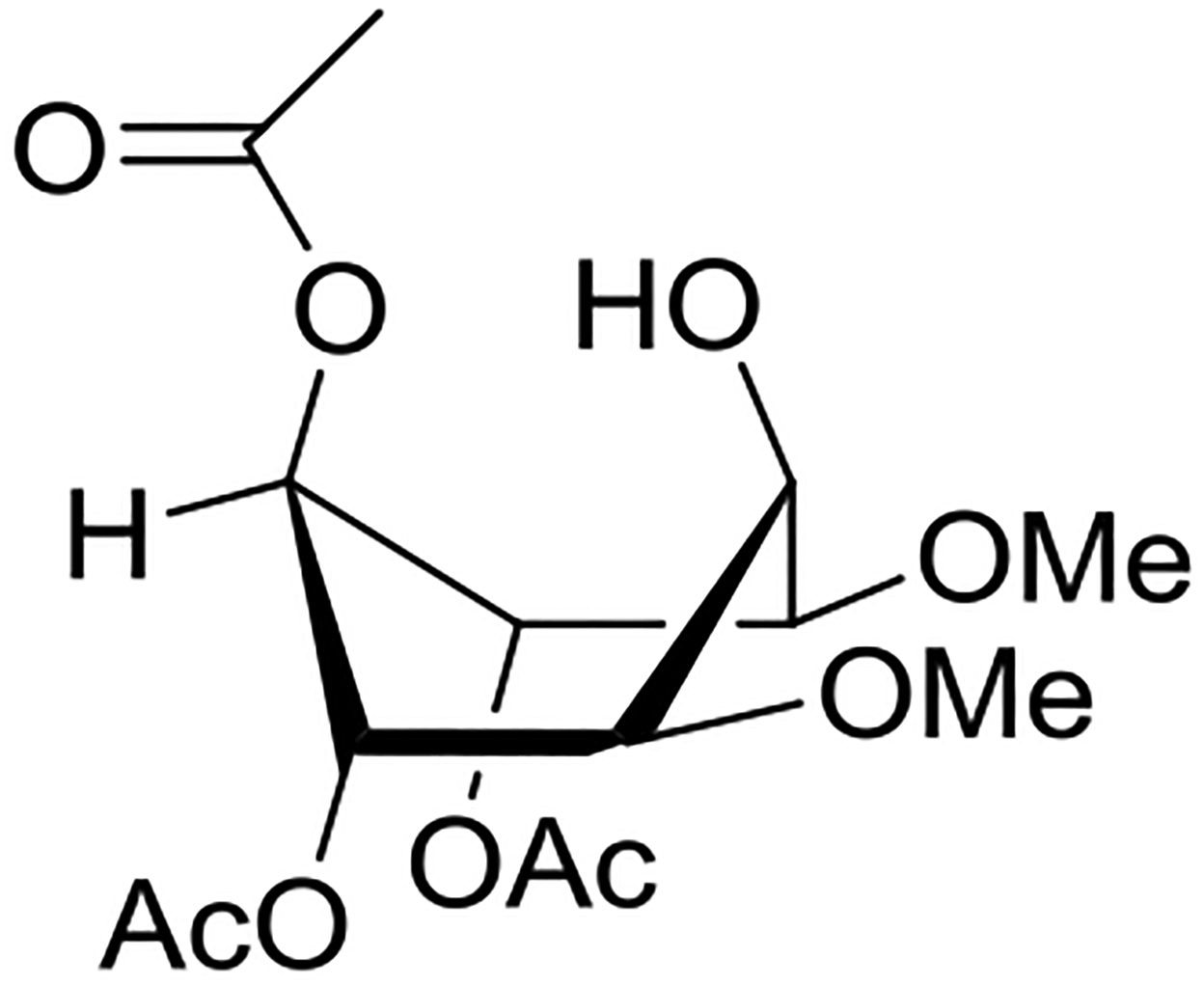
Migration from a distal group is unlikely even in a boat conformation.
In general, the acyl groups in carbohydrates migrate toward a primary hydroxy group, if present, from where hydrolysis may take place. Under strongly acidic conditions, the acyl group may also be cleaved directly without migration. Steric hindrance induced by substitution pattern and the stereochemical axial/equatorial relationships of the secondary hydroxy groups are other contributing factors to the overall migration path. In acetyl α-d-glucopyranose, for example, the ester sequentially migrates to O6 via the O2, O3 and O4 (Scheme 40).283 Similar migrations are observed for O2-acylated d-galactopyranose284 and d-mannopyranose.285
Scheme 40.
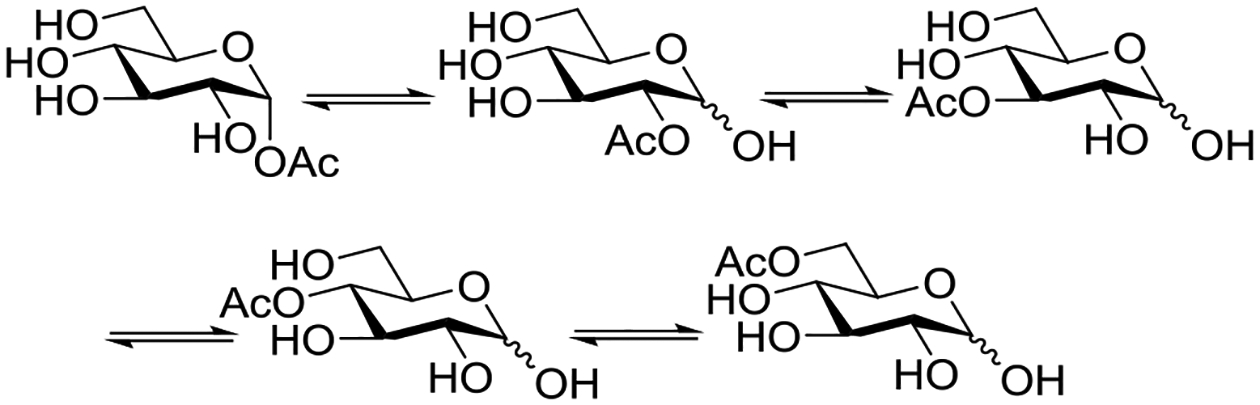
Acetyl Group Migration in d-Glucose in Deuterated buffer at pH 7.2–7.3 and 30°C.
The stability of acyl groups at primary hydroxy groups over secondary ones also becomes evident from studies with neuraminic acid,286,287 1,2-O-isopropylidene-α-d-hexofuranoses,288 and glycerol.289 In the absence of primary hydroxy groups, the positional preference and outcome of migration are less straightforward and depend on steric and electronic factors. Axial verses equatorial influences were investigated early on by Angyal and Melrose who studied the axial verses equatorial preference of acetyl groups in myo-inositol derivatives.281 In a myo-inositol derivative where the only possible migration was between a single axial and an equatorial hydroxy group, only minimal preference for the equatorial position was observed in pyridine:water 50:50 (Scheme 41). In parallel, it was demonstrated that for pentaacetylated myo-inositol the equatorial preference of the acetyl groups increases with increasing amounts of pyridine.
Scheme 41.

Axial Equatorial Equilibrium Ratio in a myo-Inositol Derivative.
Due to its important biological role, several studies have addressed the acyl migration in d-glucuronic acid, a compound lacking primary hydroxy groups. Starting from the β-anomer, migration away from O1 is fast, while the reverse migration back to O1 is typically not observed.290,291 As shown by Blanckaert and coworkers, the O3 position is the most favored followed by O2 and O4.292 Nicholls has demonstrated by means of a series of positional isomers of trifluoromethylbenzoyl esters, that the most favored position in glucuronic acid depends on the nature of the acyl group.293
Mastihubová and Biely investigated acetyl group migration in d-xyloses containing both one and two acetyl groups.294 It was shown that with one acetyl group the order of positional stability was O4 > O3 > O2 in a phosphate buffer at pH 6. A similar pattern was observed for the di-O-acetyl d-xylopyranose derivatives, with O3,O4 being the most favorable positions, followed by O2,O4 and O2,O3. Filice and coworkers have studied tetraacetylated d-glucosamine and d-galactosamine with acetyl groups located at their N, O1, O3 and O4-positions in phosphate buffer at pH = 8.5 at 4°C.295,296 Following the initial migration from O4 to O6, equilibration of the fourth acetyl group between the O3 and O4 positions takes place. For the glucosamine derivative a positional preference for O3 was clearly observed, while in galactosamine the acetyl group partitions between O3 and O4 with only slight preference for the prior (Scheme 42).296
Scheme 42.
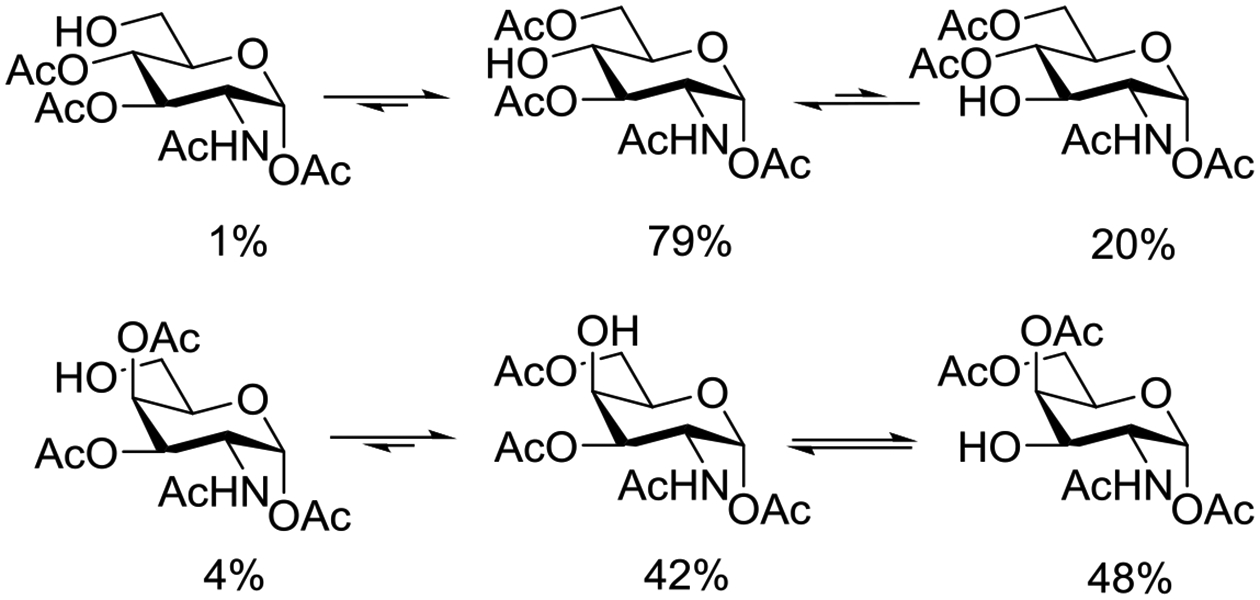
Equilibrium Ratios of Positional Isomers in Partially Acetylated Glucosamine and Galactosamine in Phosphate Buffer at pH 8.5, and 4°C
The rate of acyl migration significantly depends on the relative configuration (cis or trans) of the parent diol. While multiple conformations are available to a primary hydroxy group enabling alignment for possible migration, considerably fewer degrees of freedom are available for secondary functionalities. Illustrative examples are migrations in galactopyranose and mannopyranose. For d-mannopyranose, a study of migration rate constants in D2O buffer at pD = 8 clearly indicated faster migration between the O2 and O3 positions than between O3 and O4, with an average rate constant for migration between the cis-vicinal hydroxy groups 7.5 times that for migration across the trans-diol (Scheme 43).285 For d-galactopyranose on the other hand, the O3–O4 migration is four times faster than the O2–O3 migration under similar conditions.284 These examples clearly demonstrate the importance of favorable distances and close contacts between the adjacent hydroxy groups for the rate of migration.
Scheme 43.
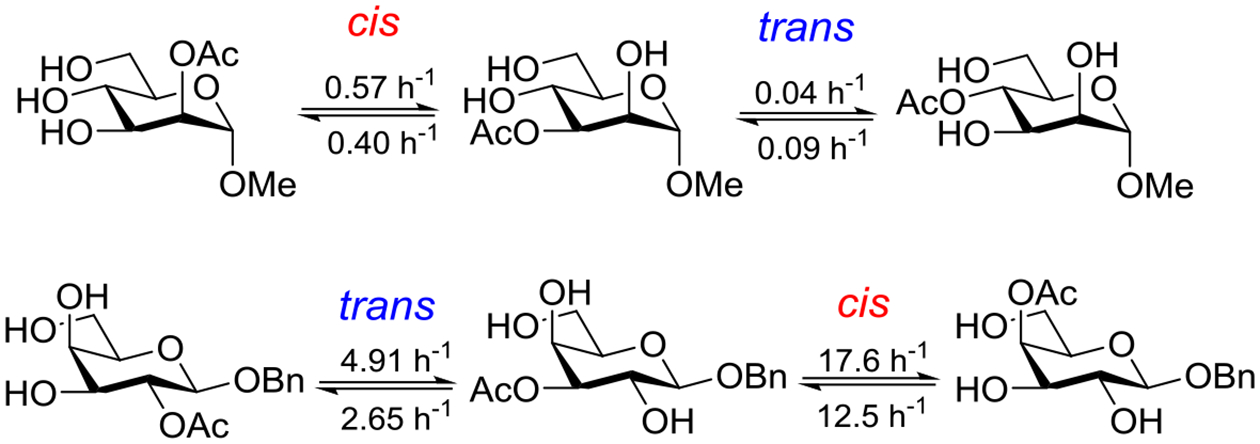
Relative Rate Constants for Migration Between cis- and trans-Vicinal Diols in 10 mM Deuterated Phosphate Buffer at pH 8, and at 25°C.
Migration processes in furanoses have been less investigated. Chevallier and Migaud studied the potential O1→O5, O2→O5 and O3→O5 migration in ribose, the corresponding O2→O5 migration in arabinose, and O3→O5 migration in xylose by blocking other potential migration pathways (Scheme 44).297 By changing the migration conditions, solvent and the base, it was shown that the O1→O5 migration takes place when the O1 and C5 share a cis-relationship, but also that the O3→O5 migration requires a cis-relationship between O3 and C5. This is due to the rigid structure of the isopropylidene constrained furanoses, which prevents the trans migration between trans-diols.
Scheme 44.
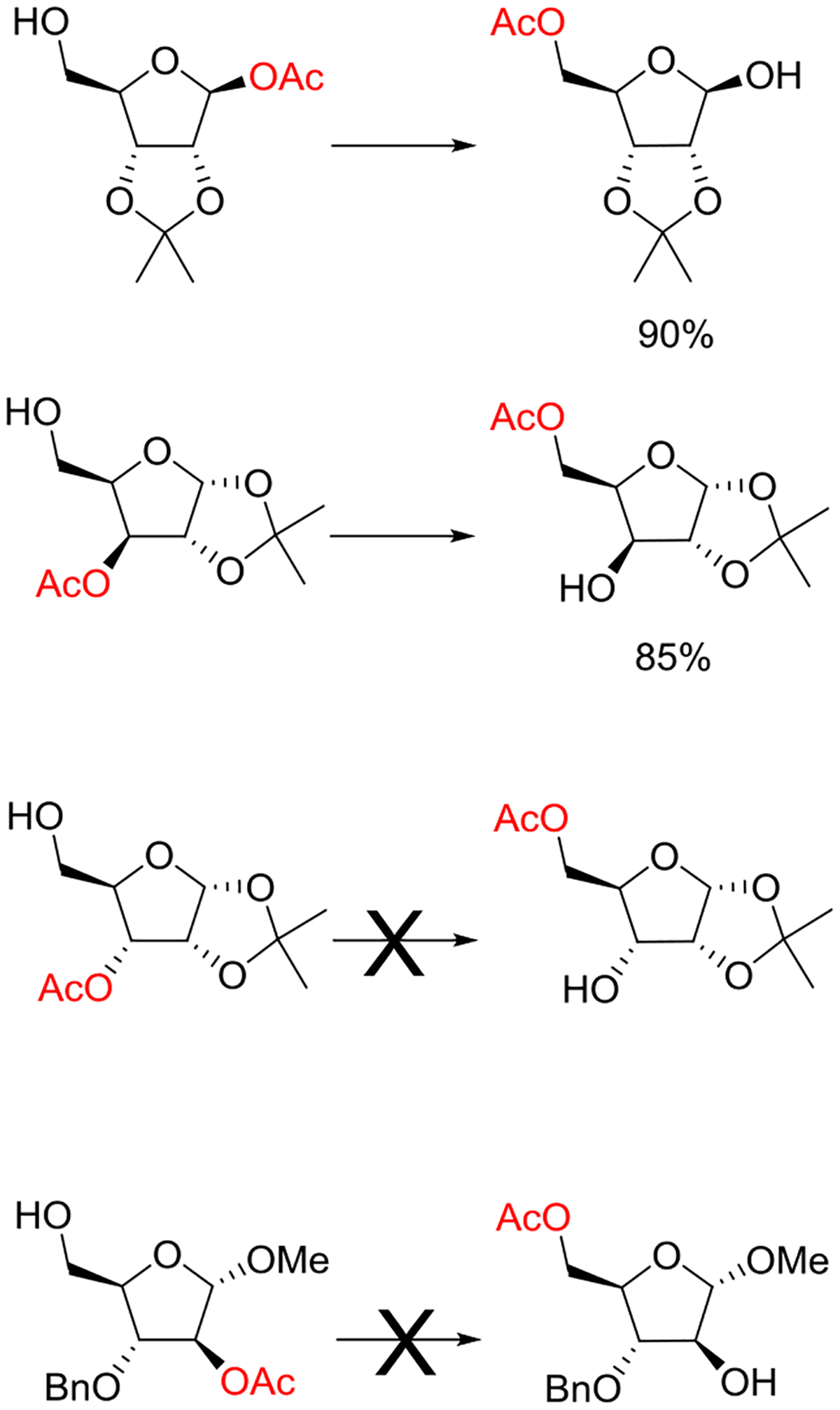
Long Range Migrations in Furanoses in THF in the Presence of Tetrabutylammonium Fluoride.
In a mannodisaccharide, an equilibrium between O2 and O3 is established in 5 hours at 25°C in a phosphate buffer with pD 8.285 In the corresponding trisaccharide, however, slow inter-residue migration to the non-adjacent primary hydroxy group was observed recently, as evidenced by NMR spectroscopy (Scheme 45).
Scheme 45.
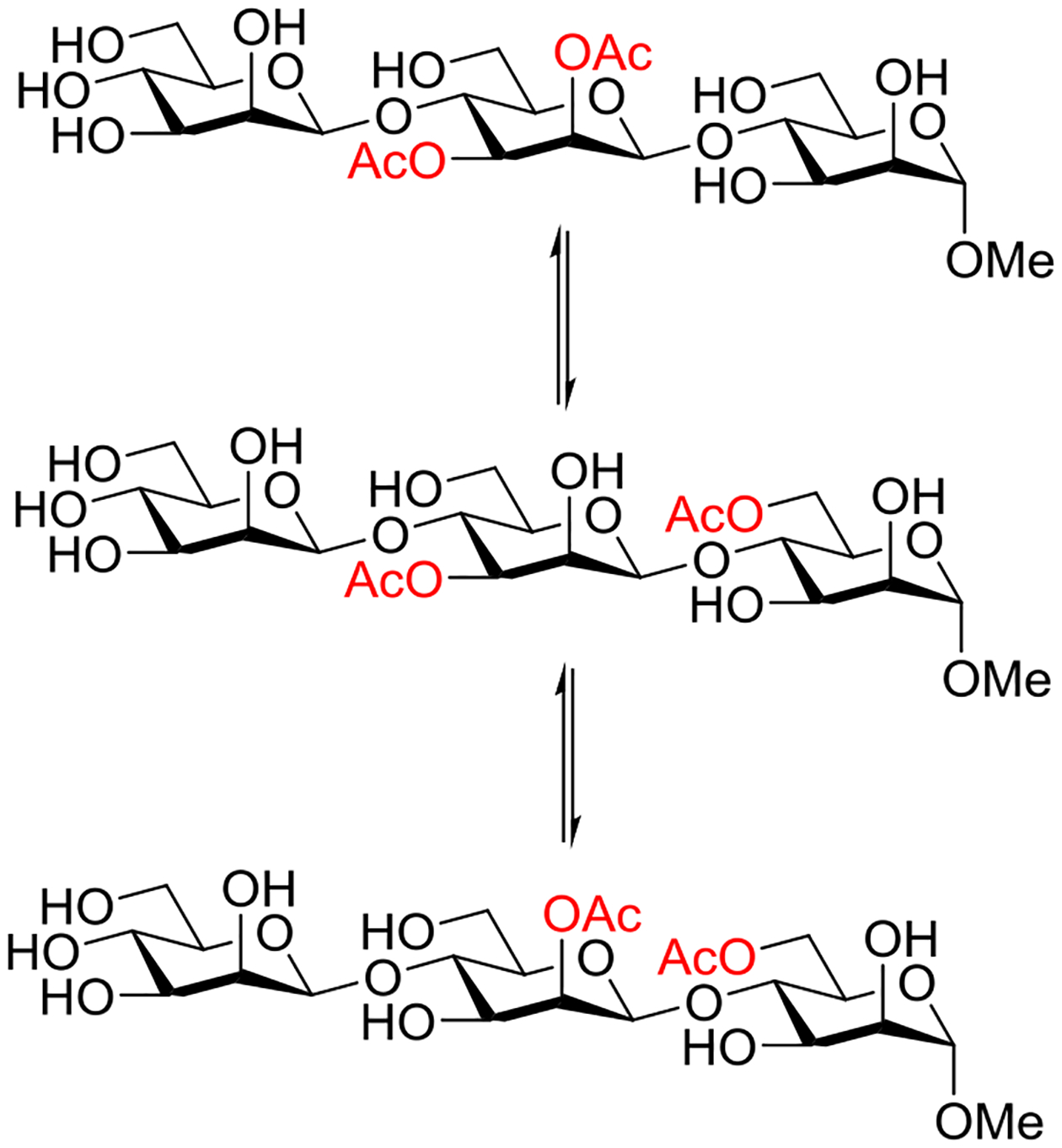
Inter and Intra-Residue Acetyl Migration in a Mannotriose in 10 mM Deuterated Phosphate Buffer at pH 8 and Room Temperature.
20. Mechanism of Ester Migration in Polyols
While several hypotheses have long been presented for the acyl group migration,30 experimental evidence has been lacking. In recent years computational calculations have backed up some of the suggested mechanisms,285,298,299 and experimental evidence has begun to emerge.285,300 Most studies address O→O acyl group migration, but corresponding S→O and S→N migrations have also been observed.301 In buffer, one main mechanism has been proposed under basic conditions,302 while in organic solvents two mechanisms have been suggested: one under acidic conditions,299 and one using silver or cesium salts in combination with iodine.303
Under basic conditions, subtly different mechanisms have been suggested. In 1972, Oesterling and Metzler proposed deprotonation of the acyl-receiving hydroxy group as the first step, followed by cyclization to a hemiorthoester (Scheme 46).302 Notably, a comparable orthoester intermediate was suggested by Fischer in the original publication on acyl migration.301 Further, according to Casinovi et al.,304 the negative charge on the deprotonated hydroxy group would be stabilized by acceptance of a hydrogen bond from an adjacent hydroxy group, or for an O2 alkoxide, by the electron-withdrawing effect of the anomeric carbon.
Scheme 46.
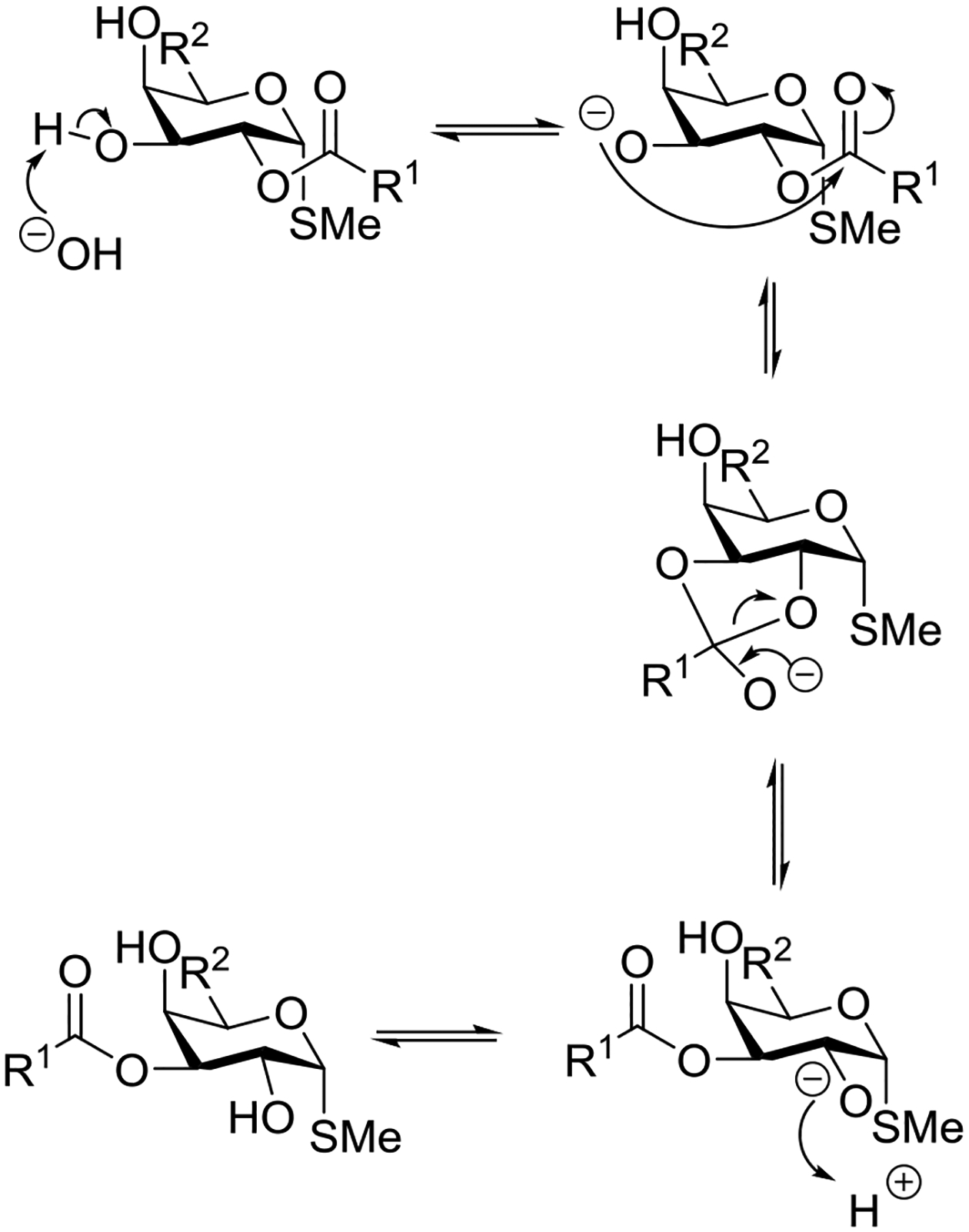
Anionic Mechanism for Acyl Group Migration.
Experimental evidence has been acquired to support the hemi-orthoester and deprotonation components of the mechanism. A hemi-orthoester has been isolated by Petrović, demonstrating that an O2,O3-orthoester in galactopyranose is a stable intermediate in pivaloyl migration in PBS buffer (pH = 7.2 at 37°C).300 This intermediate could then be acetylated and isolated, so confirming the identity of the product and supporting the notion of orthoester formation during the migration process. Evidence for deprotonation as the first mechanistic step in migration was recently acquired from kinetic isotope effect studies.285 By investigating the acyl migration rates of methyl 2-O-acetyl-α-d-mannopyranoside in both D2O and H2O buffers, it was shown that the average rate was 2.5 times faster for the protio than for the deuterio-substrate (Scheme 47).
Scheme 47.
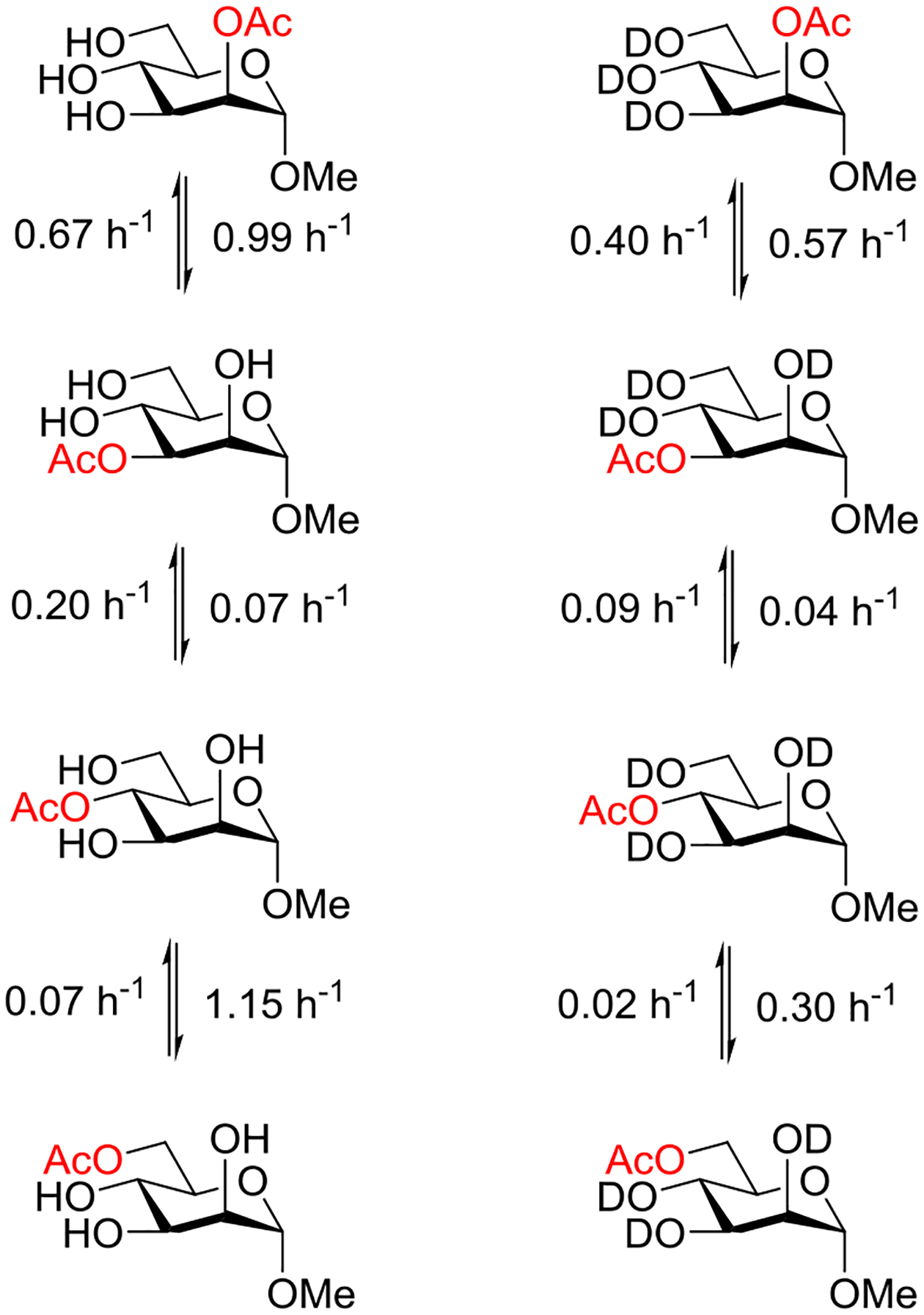
Kinetic Isotope Effects Consistent with Deprotonation as Rate Determining Step in 10 mM Phosphate Buffer (Deuterated or Non-Deuterated) at pH 8, and 25°C.
It has been further demonstrated in buffers, that the migration rates are highly dependent on the pH of the reaction medium.284,305,306 Leino and coworkers investigated acetyl, benzoyl and pivaloyl group migration in benzyl α-d-galactopyranoside in buffers at varying pH.284 They found that for all acyl groups the migration is retarded as the reaction mixture pH approaches neutral and that no migration takes place at pH 1; conversely at pH 10 the migration is very fast. Illing and Wilson have demonstrated that the migration of isoxepac in d-glucuronic acid takes place at pH 6 and accelerates with increasing pH.306 Furthermore, Khan and coworkers have shown that for d-glucuronic acid some migration of ifetroban takes place at pH = 5 but not at pH ≤ 4.307 In a study by Brecker et al., even small differences in pH were shown to influence the migration rate, with a clear difference in migration rates in O-acetyl glucopyranose between pH = 7.2 and 7.3.283 A study by Mortensen and coworkers showed the differences in the migration rates of R- and S-naproxen 1-O-acyl-β-D-glucuronic acid in buffers with pH:s 7.00, 7.40, and 8.00 (Figure 30).305 Here, the rate increases exponentially with increasing pH. Overall, concentration of the OH ions in the buffer is important for the migration rate and, therefore, the deprotonating ability of the solution is of significance.
Figure 30.

pH Profile of migration rate constants for R- and S-naproxen 1-O-acyl-β-d-glucuronic acid in 100 mM phosphate buffer and at 37°C.
Computational studies have further contributed to the understanding of the migration mechanism under basic conditions. Rangelov et al. investigated formyl migration in a model system, cis-tetrahydrofuran-3,4-diol, by DFT methods.298 Three possible mechanisms, concerted, stepwise and monoanionic were considered in addition to the influence of solvent (Scheme 48). The suggested stepwise mechanism proceeds via a hemi-orthoester intermediate, while the concerted mechanism takes place in one step. The stepwise mechanism is preferred over the concerted one by 10.8–11.5 kcal.mol−1, although the activation energy is high for a spontaneous reaction (45 kcal.mol−1) in vacuum. When solvents (chloroform, acetonitrile, methanol and water) were considered in the calculations, all energies decreased. While the monoanionic mechanism has a higher activation energy than the stepwise one in vacuum, it is favored when solvents are taken into consideration due to stabilization of the hemiorthoester-like transition state. By introduction of the solvents, the ortho acid intermediate is 3.6–7.2 kcal.mol−1 more stable than either the initial or final alkoxides. Consequently, these calculations provide support for the monoanionic mechanism being the most likely. The addition of ammonia and trimethylamine to the energy calculations of the mechanism in acetonitrile further lowered the deprotonation energy to 32.8 and 22.4 kcal.mol−1, respectively, from the initial 66.3 kcal.mol−1. This indicates that the ability of solutions to generate alkoxides is essential for migration to take place.
Scheme 48.
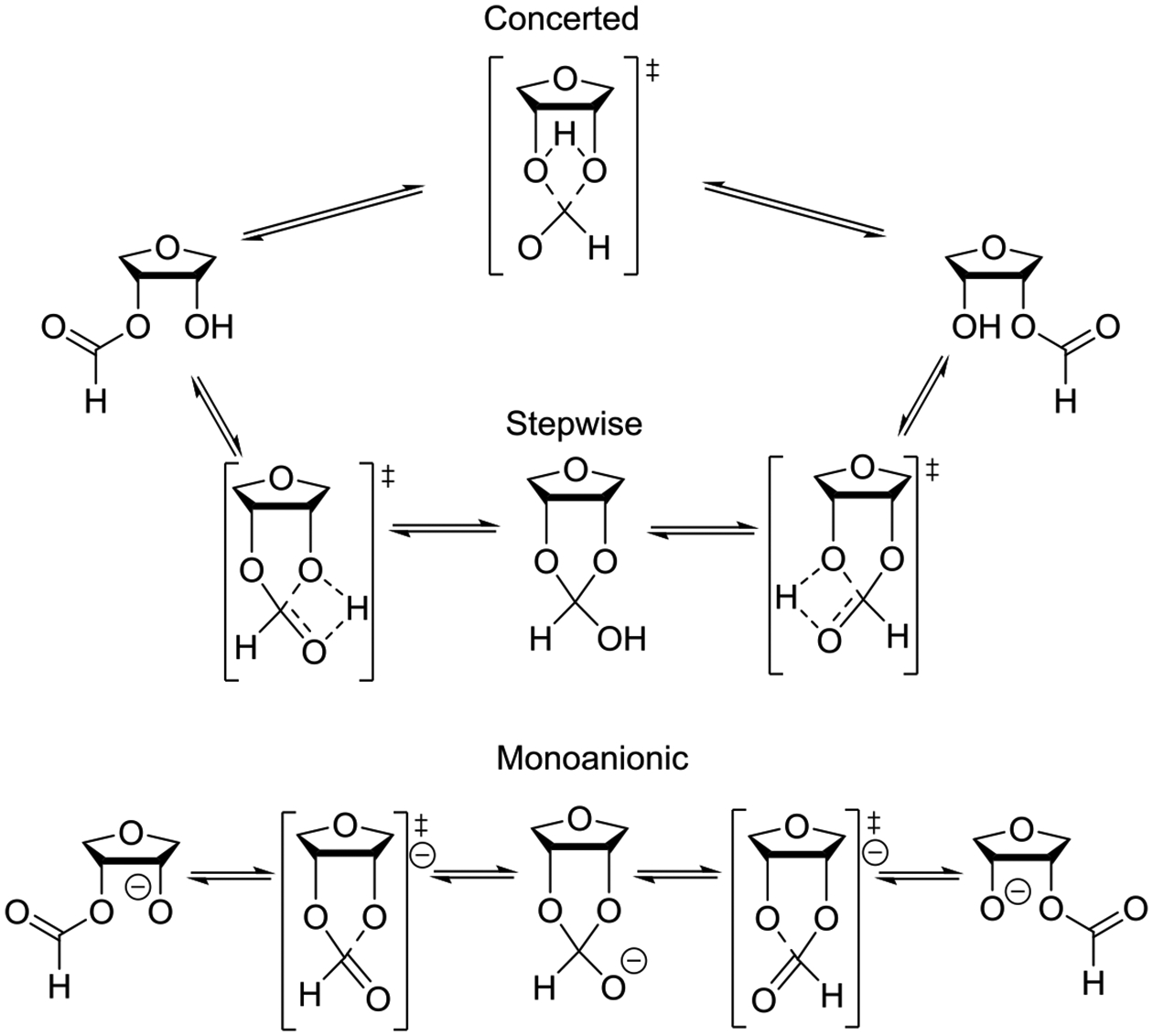
Concerted, Stepwise, and Monoanionic Mechanisms for Migration.
Martin and Hedrick have suggested a similar mechanism for S→O acetyl group migration as observed for O→O migration under basic conditions.301 Furthermore, a mechanism for S→N migration, a critical component in the synthesis of peptides and proteins by native chemical ligation,308 was presented where deprotonation of the amine takes place after the N-C bond has been formed, followed by sequential protonations at O and S. Finally, deprotonation of the hydroxy group takes place followed by cleavage of the S-C bond (Scheme 49).301
Scheme 49.
Proposed Mechanisms for S→O and S→N Acetyl Migration.
Several migration mechanisms have been suggested for acyl migration under acidic conditions. Horrobin and coworkers selectively removed the 6-O-acetyl group of peracetyl glucopyranose and then followed the subsequent acetyl migrations in toluene:AcOH (100:1) to obtain 1,2,3,6-tetra-O-acetyl-d-glucopyranose. Analogous studies were conducted in the galactopyranose, mannopyranose, and methyl 2,3,6-tri-O-acetyl-α-d-glucopyranoside, galactopyranoside and mannopyranoside series.299 It was hypothesized that under acidic conditions an oxonium ion is formed in the rate-limiting step, followed by nucleophilic attack by the acetyl-accepting hydroxy group (Scheme 50). Energy calculations at the MMX level, suggested more favorable O4→O6 migration in glucopyranose compared to O3→O4 by 4.3 kcal.mol−1, due to the highly strained nature of the trans-fused five-membered ring required for the latter.
Scheme 50.
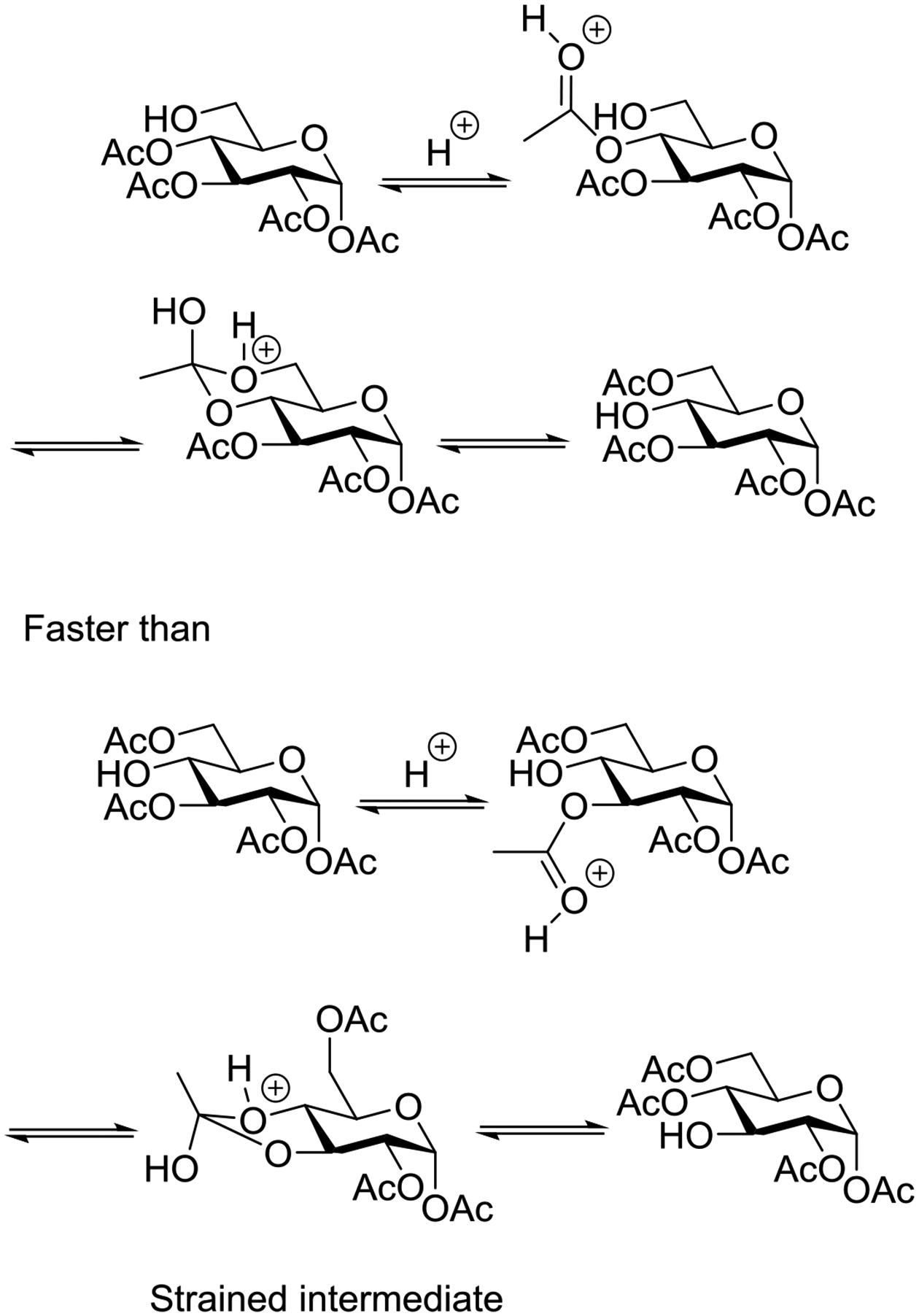
Acid-Catalyzed O4→O6 and O3→O4 Acetyl Migration in the Glucopyranose Series.
In another mechanistic investigation, Deng and coworkers studied promotion by iodide in combination with CsO2CCF3 or Ag2O in DMF.303 The salts act as a Lewis base and acid to allow the migration to take place. The suggested mechanism commences by nucleophilic addition of the hydroxy group to the carbonyl functionality (Scheme 51). A Lewis base then deprotonates the O2, followed by nucleophilic attack of iodide to the Lewis acid-hemiorthoester complex, reforming the carbonyl functionality and breaking the C-O bond.
Scheme 51.

Dual Acid and Base Promotion of Acyl Migration.
21. Acyl Group Scope in Migration Along Polyols
On a general level, the migration rate has been shown to depend on both the steric and electronic properties of the acyl group and the glycone. The influence of the acyl group on the migration rate has been most extensively studied in glucuronides, where the degradation rate (migration rate) is of great significance for evaluating the performance and safety profiles of drugs.293,307,309,310 One of the most detailed studies to date was conducted by Yoshioka and Baba in 2009.311 They studied the degradation kinetics in a large set of structurally diverse carboxylate esters of glucuronic acid. In all cases, the ester was located at the anomeric position in the β-configuration; the acyl groups included four NSAIDs, sixteen substituted benzoates, three arylacetates, six 2-arylpropionates, 3-phenylpropionate and 4-phenylbutyrates. The reaction kinetics were reported to be first-order and the rate constants varied significantly (60-fold) within the studied library. Similar findings were reported for a large number of 1-O-glucuronide drugs in which the half-lives varied between 0.26 and 79 h.312 These studies revealed that electron withdrawing substituents enhance the electrophilicity of the carbonyl group (increase in acidity of the corresponding carboxylic acid) and are accompanied by accelerated migration rates. In addition, increasing the steric bulk close to the ester carbonyl group (e.g., the o-position in benzoates and bulky substituents at the α-position of aliphatic esters) was shown to decrease the migration rate. These findings are consistent, considering that a nucleophilic attack from a neighboring hydroxy group/alkoxide ion would be favored by increased electrophilicity at the carbonyl carbon and disfavored by steric hindrance. A more surprising finding, also reported by other investigators,290,313 is the difference in migration rate between stereoisomers in which the α-carbon of the acyl group is mono-substituted and constitutes a stereogenic center. In general, the (S)-isomers degrade more slowly than the corresponding (R)-isomers. This phenomenon has been observed in 1-O-glucuronide derivatives of ibuprofen, naproxen, fenoprofen, benoxaprofen, flunoxaprofen and beclobric acid.290,305,311,312 The difference in migration rates has been proposed to be due to a difference in energies of the diastereomeric transition states, for which computational support has been provided.313
While most of the studies on 1-O-acyl glucuronides have focused on the degradation rate of the parent molecules, some have also addressed subsequent migrations that take place following the initial shift to give a 2-O-acyl pyranose derivative.293,309 In all cases, the half-lives of the 2-O-acyl, 3-O-acyl and 4-O-acyl derivatives are longer than those of the initial 1-O-glucuronide. The relative stability order of the migration products is, however, not as clear as for the 1-O-acyl derivatives. While the acyl groups have been reported to be most stable at either the O2 or O3 positions, the relative stability is greatly influenced by the structural features of the acyl groups.309
Structural changes in the carbohydrate core have been reported to have a marginal effect on the migration rates. This was confirmed by Stachulski et al. in a comparison of the acyl migration kinetics between glucosides and glucuronides.291 A large number of migration studies have been performed on carbohydrates other than glucuronic acid. Most of the studies have focused on the migration of acetyl, benzoyl or pivaloyl groups which are widely applied temporary protective groups in carbohydrate chemistry.295,296,314–320 Acetylated carbohydrates are found in nature, which provides additional motivation to study their explicit migration behavior.32,285,321–326 Reports on the migration of fatty acid esters,31,303,327 stilbene,328 levulinoyl,329 alkyloxycarbonyl,330 formyl,283 and benzoyl derivatives33 can be found in the literature. However, only a few of these studies have featured a direct comparison between the migration behavior of different acyl groups. Leino et al. compared the migration behavior of acetyl, benzoyl and pivaloyl groups in 2-O-acyl substituted galactopyranose derivatives.284 The pivaloyl group was found to have the slowest migration rates while similar rates were observed for benzoyl and acetyl groups at all stages except in the O4→O6 migration and the subsequent hydrolysis (Table 13). It is puzzling that the O4→O6 migration is considerably slower for the benzoyl group, while the O6 hydrolysis rate is significantly faster. Apparently, the electronic and steric properties of the acyl groups and the surrounding chemical environment affect the stability order at various positions in the carbohydrate backbone. A comparison of the migration rates of formyl and acetyl groups has been reported in glucosides, but the results were compromised by the rapid hydrolysis of the formates.283 Unfortunately, most other reported studies have been conducted under differing experimental conditions such that direct comparisons cannot be made. In order to shed light on the electronic and steric factors that govern the acyl migration, a more coherent series of acyl groups would need to be screened under similar experimental conditions on a wider set of glycones.
Table 13.
Relative Migration Rate Constants for Acetyl, Benzoyl and Pivaloyl Groups in 2-O-Acyl Substituted Galactopyranose Derivatives at pD = 8.0. n.d. = not determined.
| k (h–1) | Acetyl | Benzoyl | Pivaloyl |
|---|---|---|---|
| k2→3 | 4.91 ± 0.032 | 2.92 ± 0.07 | 0.06 ± 0.004 |
| k3→2 | 2.65 ± 0.068 | 4.11 ± 0.32 | 0.10 ± 0.04 |
| k3→4 | 17.6 ± 0.44 | 18.8 ± 2.1 | 0.61 ± 0.13 |
| k4→3 | 12.5 ± 0.46 | 10.3 ± 1.56 | 0.26 ± 0.09 |
| k4→6 | 10.4 ± 0.14 | 1.91 ± 0.06 | n.d. |
| k6→4 | 2.48 ± 0.06 | 0 | n.d. |
| k6→hydrolysis | 0.04 ± 0.005 | 1.54 ± 0.1 | n.d. |
22. Solvents Effects on Ester Migration in Polyols
While the effect of pH on the acyl migration reaction has been well documented (see Scheme 38 and associated discussion),283,284,305,307 there are relatively few systematic studies addressing the effects of solvents.298,331 It can be expected that solvents that stabilize the charged orthoester intermediate should be accompanied by an increase in the migration rate, while solvents that destabilize the ionic species should have an adverse detrimental effect on the migration rate. Despite these theoretical underpinnings, the conducted studies have yielded conflicting results.
In myo-inositol dibenzoates,331 the effect of solvent polarity was studied using pyridine and sodium carbonate as bases, and toluene, water, DMF and THF as solvents. Based on these studies it was concluded that the tetrahedral intermediate is better stabilized in a polar solvent environment, which enhances the migration rate. The effect of water on the reaction rate was proposed to be due to its polarity rather than its protic nature. The amount of pyridine needed has been studied by Angyal and Melrose281 in partially acetylated substrates where water:pyridine mixtures in different proportions ranging from 5:95–95:5 were screened. Increasing the pyridine concentration above 90% slowed down the migration significantly. In the same study, the migration was reported to be significantly slower with silver oxide in chloroform than with pyridine in water, however, equilibrium could be achieved in both cases. This is an indication that solvent polarity does indeed affect the migration rate. Further evidence was supplied from a study of ester migrations on the methyl 6-O-trityl-glucopyranoside framework in benzene solution with 0.2 M sodium hydrogen carbonate as base. Long reaction times were needed for equilibrium to be reached, for which the apolar nature of the solvent was suggested as a possible reason. In addition, acetyl migration has been reported to be slower in DMSO than in water.332 While these experimental studies all support the postulation that solvent polarity may have a significant effect on the migration rate, one of the few computational studies reported on the subject did not provide significant supporting evidence.298 In comparing the influence of water, methanol, acetonitrile and chloroform, the results revealed that the energy barrier for migration decreases with increasing dielectric constant of the solvent (vacuum > chloroform > acetonitrile > methanol > water). This is an indication of the ability of the solvent to stabilize the charge (Scheme 52), however, the relative stabilities of the intermediates were not influenced by the solvent’s presence. In addition, the solvent effect was reported to be negligible when compared to the effect of prior deprotonation of a hydroxy group. Consequently, the migration rates appear to be governed by the acidity or basicity of the reaction mixture rather than the solvent alone.
Scheme 52.
Influence of Solvent Polarity on Migration in Tetra-O-acetyl glucopyranose
23. Effect of Temperature on Ester Migration in Polyols
The literature results suggest a major contribution of temperature to the acyl migration rate with even small changes having a potentially large effect. Blanckaert and coworkers have studied acyl group migration in glucuronic acid, stored in buffer at pH 7.8: the migration rate was reduced significantly on cooling from 37 to 0°C.292 Approximately 10% of the starting 1-O-acyl glucuronic acid remained after 3 h at 37°C, while at 0°C 70% of the starting material was unchanged after 22 h. In another study, for triolein the migration rate for mono- and disubstituted glycerides increased 6–10 fold on increasing the temperature from 30°C to 55°C (Table 14).333
Table 14.
Migration in Mono- and Disubstituted Glycerides in 0.1 mM Phosphate Buffer at pH 6.8.a
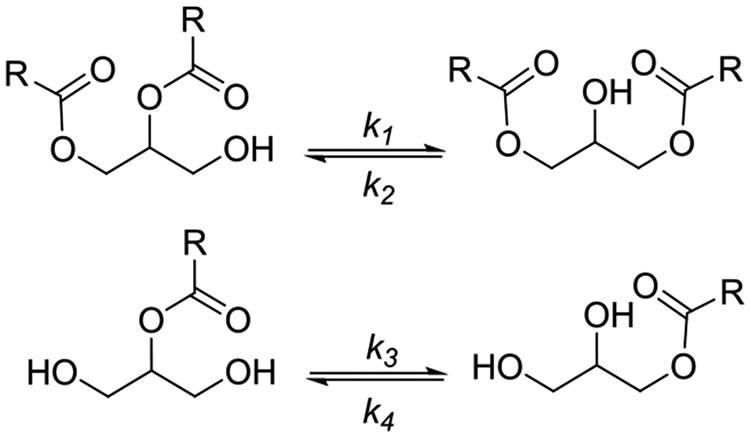 | ||||
|---|---|---|---|---|
| T (°C) | k1 (h−1) | k2 (h−1) | k3 (h−1) | k4 (h−1) |
| 30 | 0.013 | 0.008 | 0.045 | 0.005 |
| 37 | 0.025 | 0.015 | 0.152 | 0.007 |
| 45 | 0.045 | 0.027 | 0.274 | 0.013 |
| 55 | 0.128 | 0.078 | 0.581 | 0.028 |
In the N-acetylneuraminic acid series acetyl group migration from O7 to O9 was investigated by Kamerling and coworkers.286 It was shown that at 0°C almost no migration takes place after 100 min, while at 27°C 50% of the starting compound is consumed and at 37°C only 30% of the initial ester remained. Filice and coworkers have investigated the O6→O4 and O4→O3 migration in tetraacetylated glucopyranosamine in order to obtain the free hydroxy group at O4.295 Two temperatures 4°C and 22°C were studied at pH 9.5 and 8.5. At the higher temperature of 22°C, 98% conversion of the starting material was reached in 10–15 min, while at 4°C 90 min was required for the migration to reach a 93─98% conversion. Under both pH values, the migration progressed further to the C3-OH at 22°C, clearly illustrating the significant role of temperature in facilitating the acyl migration process. Hasegawa et al. have demonstrated, using both (2ʹR)- and (2ʹS)-1-O-(2ʹ-phenylpropanoyl)-β-d-glucuronide in a phosphate buffer with pH 7.4, that an increase in temperature from 27 to 47°C increased the average rate of O1→O2 migration 9–10 times.290 The migration rate from O1 to O2 doubles between 27 and 32°C, and increases further by 1.6 fold between 42°C and 47°C. This indicates an exponential increase in the migration rate with increasing temperature (Figure 31).
Figure 31.
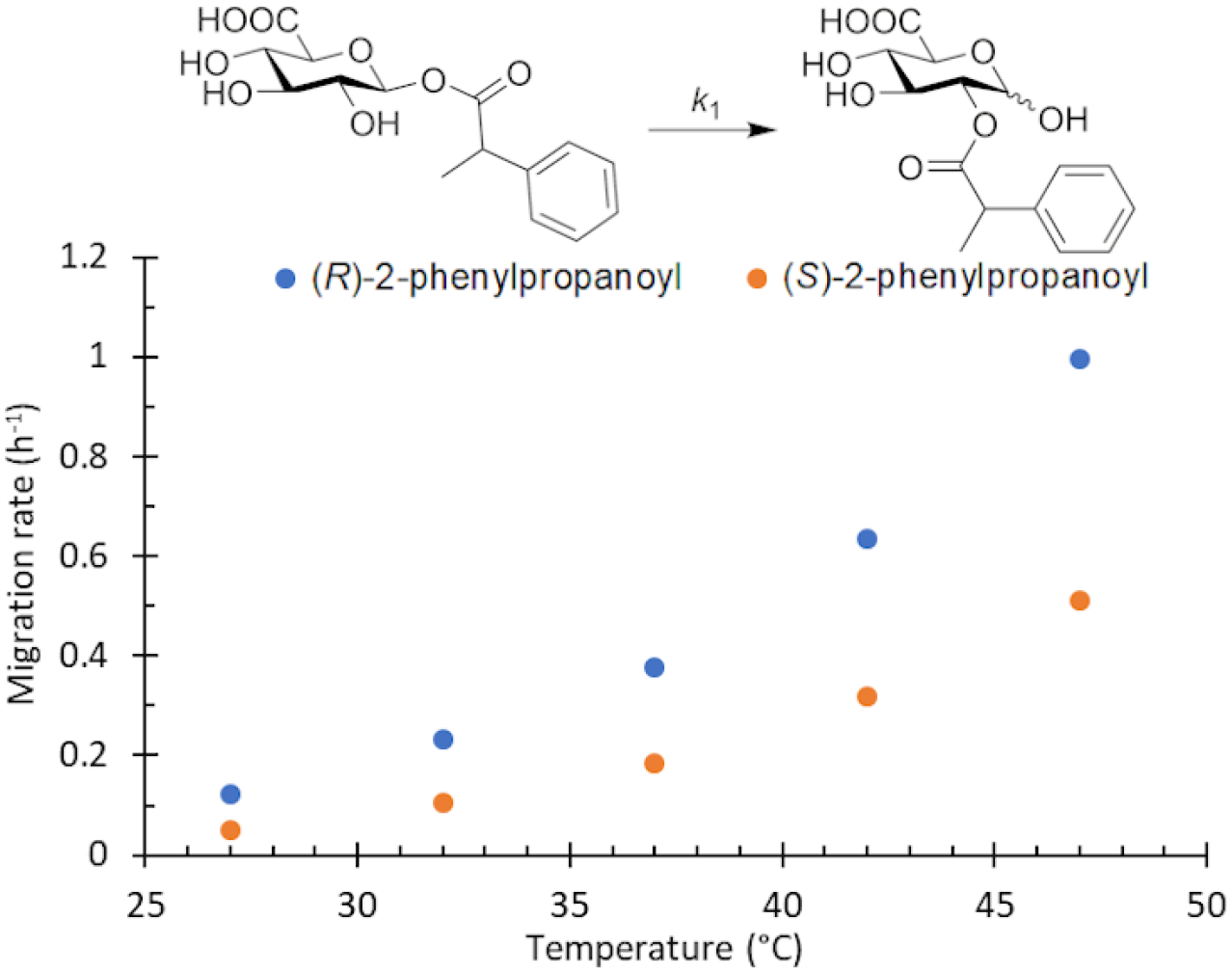
Influence of temperature on the O1→O2 migration of R- and S-2-phenylpropionate in the glucuronide.
For S→O acetyl migrations a similar trend is observed, as investigated by Martin and Hedrick with S-acetylmercaptoethanol and the corresponding acetylthio propanol.301 It was shown that the migration rate for S-acetylmercaptoethanol increases 14 fold when the temperature is increased from 15 to 35°C, and for S-acetylmercaptopropanol 8 fold when the temperature increases from 18.5 to 35°C. Similarly, the S→N acetyl group migration was also investigated in the same study using S-acetylmercaptoethylamine. By varying the pH and the temperature (15, 25 and 35°C), it was shown that at lower pH the rate increase is much higher (13 times) compared to high pH (3 times), when the temperature was increased from 15°C to 35°C.
24. Effect of Additives on Ester Migration in Polyols
Most studies of migration have been carried out in buffers or with a base in organic solvents. Typically, however, such migrations are not sufficiently selective for synthetic purposes, and consequently additives, such as metal salts and boranes, have been used in order to improve the selectivities. The addition of salts may influence the equilibrium position in a migration process. Deng et al. used various additives, including silver, zinc, copper, iron and cesium salts with TBAI in DMF in an investigation of acyl migrations between O2 and O3 in d-glucopyranose and d-galactopyranose derivatives.303,327 It was found that Ag2O and CsO2CCF3 were the best alternatives for directing the O3→O2 acyl group migration in phenyl 4,6-O-benzylidene-1-thio-β-d-galactopyranoside and the O2→O3 acyl group migration in phenyl 4,6-O-benzylidene-1-thio-α-d-glucopyranside (Table 15). Good yields of the 2-O-acyl galactopyranose derivatives and the 3-O-acyl glucopyranose derivatives were obtained with a wide range of acyl groups. In a similar study, Ren et al. studied the use of Ag2O additive with NaBr in acetonitrile for influencing the migration between O2 and O3 in mannopyranose, galactopyranose and glucopyranose derivatives.319 Acetyl and benzoyl groups located at O3 in the starting compounds, and in the case of methyl α-d-glucopyranoside at O2, were investigated. In the absence of either Ag2O or NaBr no migration took place, and for selective reaction the combination was needed. In most cases, fair to good yields were obtained (Table 16). While in Deng’s work 1 equivalent of Ag2O was used,303 in the study by Ren 0.1 equivalents were employed,319 yielding nevertheless similar results, especially for the mannopyranose and glucopyranose derivatives.
Table 15.
The O3→O2 Migration in Protected Acyl Galactopyranosides and the O2→O3 Migration in Protected Acyl Glucopyranosides.a
| Compound | Methoda | Temp. (°C) | Time (h) | Product | Yield (%) |
|---|---|---|---|---|---|
 |
A | 75 | 7 |  |
80 |
| B | 75 | 8 | 83 | ||
 |
A | 40 | 10 |  |
77 |
| B | 40 | 12 | 79 | ||
 |
A | 60 | 7 |  |
76 |
| B | 50 | 12 | 76 | ||
 |
A | r.t. | 48 |  |
68 |
| B | 45 | 24 | 71 | ||
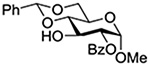 |
A | 75 | 7 | 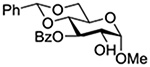 |
74 |
| B | 50 | 17 | 78 | ||
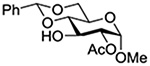 |
A | r.t. | 48 | 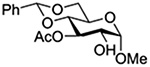 |
60 |
| B | r.t. | 24 | 67 | ||
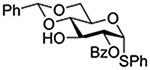 |
A | r.t. | 48 | 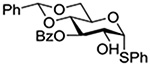 |
70 |
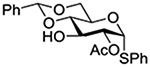 |
A | 50 | 24 | 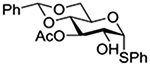 |
71 |
| B | 50 | 12 | 72 |
Method A: Ag2O, TBAI, DMF; Method B: CsO2CCF3, TBAI, DMF.
Table 16.
Acetyl and Benzoyl Migration in Protected Carbohydrates.a
| Compound | Acyl group | Product | Yield (%) |
|---|---|---|---|
 |
Ac | 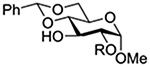 |
63 |
| Bz | 83 | ||
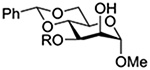 |
Ac | 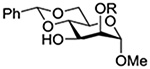 |
69 |
| Bz | 50 | ||
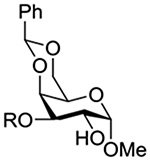 |
Ac |  |
32 |
| Bz | 44 | ||
| Ac |  |
65 | |
| Bz | 84 |
Reaction conditions: Ag2O, NaBr, r.t.−40°C, AcN, 24 h.
Ahn and Chang investigated the influence of added boronic acid derivatives on benzoyl migration in chiro-inositol derivatives.334 Starting from 1,4-di-O-benzoyl-chiro-inositol, by use of large excess of phenylboronic acid, 3,4-di-O-benzoyl-chiro-inositol was isolated in 82% yield after 7 h under basic conditions. The phenylboronic acid additive favored the cis-relationship upon coordination to the hydroxy groups, thereby leaving only two positions free for the benzoyl group migration (Scheme 53).
Scheme 53.
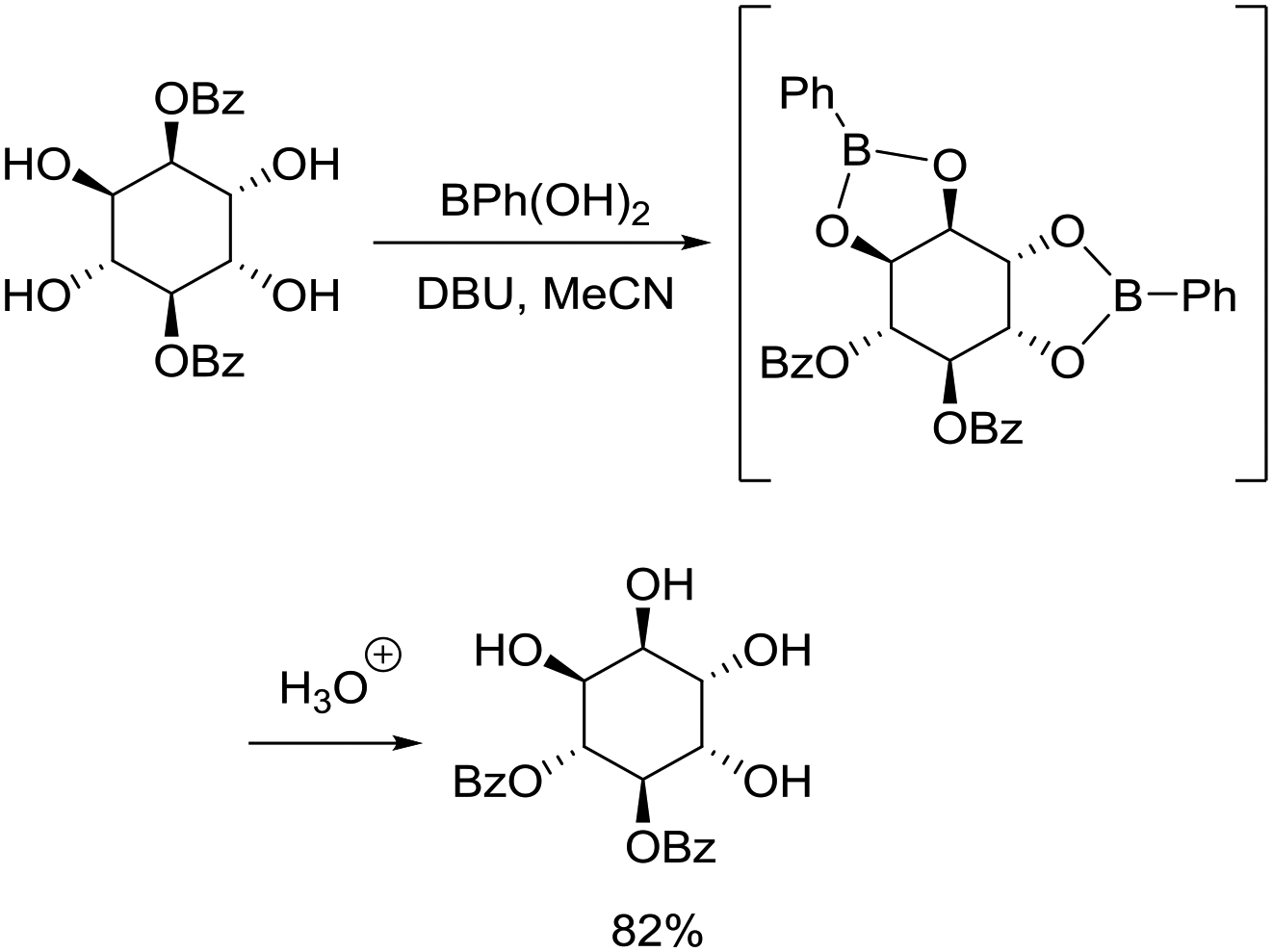
Phenylboronic Acid-Controlled Benzoate Migration in a chiro-Inositol Derivative.
For myo-inositol, the same authors also demonstrated that different boranes could yield different products.335 Starting from 1,4,5-tri-O-benzoyl-myo-inositol, a 94% yield of 2,4,6-tri-O-benzoyl-myo-inositol was obtained by use of boronic acid as additive. With phenylboronic acid, however, 1,4,6-tri-O-benzoyl-myo-inositol was first obtained as the main product in 82% yield, but after 7 h, 2,4,6-tri-O-benzoyl-myo-inositol was the main product in 96% yield (Scheme 54). This observation is consistent with 1,4,6-tri-O-benzoyl-myo-inositol being the kinetically favored product when phenylboronic acid is used as an additive while 2,4,6-tri-O-benzoyl-myo-inositol is favored thermodynamically.
Scheme 54.
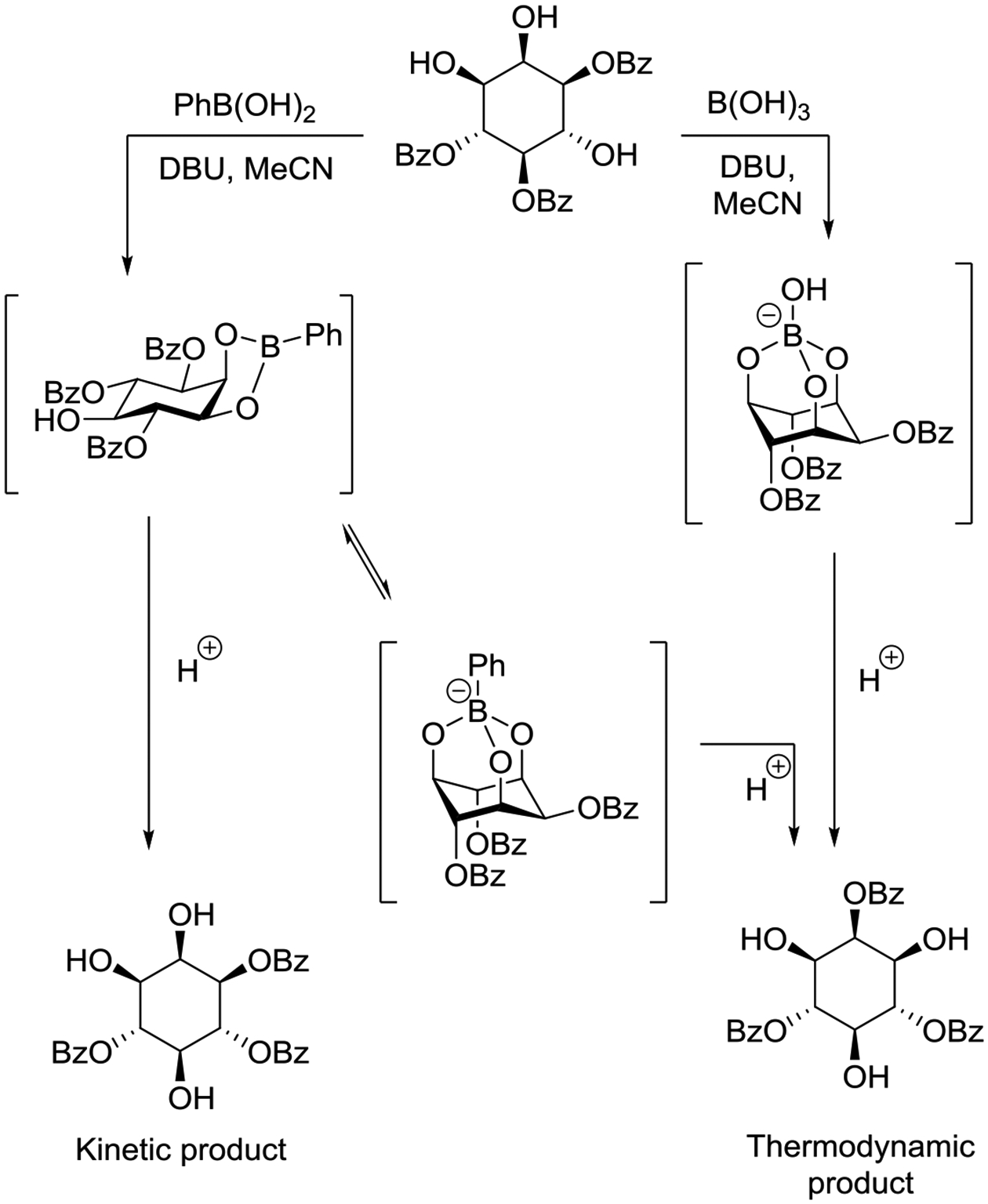
Kinetic and Thermodynamic Pathways in Phenylboronic Acid-Mediated Benzoate Migration in Tri-O-benzoyl-myo-inositiol.
25. Conclusions
An overview of the structures of carboxylate esters and of dioxacarbenium ions provides the background for a critical review and analysis of all aspects of stereodirecting participation by esters, whether proximal or distal, in glycosylation reactions. It is concluded that while long range participation is structurally feasible on the basis of the isolation and characterization of various intermediates, it is necessarily kinetically slow and subject to competition by other mechanisms. A major consideration in the analysis of both participation and migration, and one that is frequently overlooked by workers in the field, is the kinetic barrier provided by the ground state conformation of the ester in which the carbonyl bond brackets a conformation in which it eclipses the RCO2-C(-H)R1R2 C-H bond. Overall, it is clear that modern carbohydrate chemistry has gained much from and will continue to benefit from the critical investigation and analysis of reaction mechanisms and from their juxtaposition with the broader field of organic chemistry in general.
Acknowledgments
R. La. gratefully acknowledges a Ph.D. scholarship from the Magnus Ehrnrooth Foundation. F. S. E. is grateful for financial support from the Jane and Aatos Erkko Foundation, the Cancer Foundation, the Swedish Cultural Foundation, the Ruth and Nils-Erik Stenbäck Foundation and the University of Helsinki research funds. D. C. thanks the NIH (GM62160 and GM125271) for generous support of his programs in glycochemistry, and his long-standing collaborator Dr Luis Bohé at the Insititut de Chimie des Substances Naturelles, Gif-sur-Yvette, France, for the many insightful and stimulating discussions leading up to this review and in general.
Biographies
Asiri Hettikankanamalage obtained his B.Sc. degree from the University of Colombo, Sri Lanka in 2011. He completed his M.Phil. degree, which was based on Natural Products Chemistry with Prof. Dilip E. de Silva, at the University of Colombo, Sri Lanka and Prof. Raymond. J. Andersen at the University of British Columbia, Canada in 2016. He is continuing research toward the Ph.D. degree under the guidance of Prof. David Crich in the Department of Chemistry and in the Department of Pharmaceutical and Biomedical Sciences at the University of Georgia in the fields of carbohydrate chemistry and synthetic methods chemistry, with a focus on hydroxylamines.
Robert Lassfolk received his M.Sc. (2017) in organic chemistry at Åbo Akademi University, where he is currently pursuing his Ph.D. degree with Prof. Reko Leino. His research focuses on elucidating the chemical and biological significance of acyl group migration in complex mono-, oligo- and polysaccharides.
Filip Ekholm studied organic chemistry at Åbo Akademi University, where he obtained his M.Sc. (2008) and Ph.D. (2012) under the supervision of Prof. Reko Leino. During this period, he also worked as a visiting scientist in the laboratories of Prof. Jesús Jiménez-Barbero at the CIB-CSIC in Spain and Prof. Ferenc Fülop at the University of Szeged in Hungary. He worked as a senior scientist at the biotechnology company Glykos Finland Ltd. during 2012–2015 from where he transferred to the University of Helsinki, where he currently holds a University Lecturer position and leads the Biomolecular Chemistry group. The research in the Biomolecular Chemistry group is focused on solving challenges at the interface of chemistry, biology and medicine.
Reko Leino studied chemical engineering at Åbo Akademi University where he joined the research group of Professor Jan H. Näsman in 1993 graduating with M.Sc. in chemical technology and D.Sc. in technology in 1998. Following a postdoctoral year at Stanford University with Professor Robert M. Waymouth he briefly worked in the biopharmaceutical industry at Carbion Ltd. before joining the faculty of Åbo Akademi University in 2003 as a professor of synthetic organic chemistry. His research interests are in organic and organometallic chemistry, catalysis, carbohydrates and glycobiology.
David Crich is Georgia Research Alliance and David Chu Eminent Scholar in Drug Design at the University of Georgia, where he is Professor of Pharmaceutical and Biomedical Sciences, Professor of Chemistry, and Adjunct Member of the Complex Carbohydrate Research Center. A more detailed biography can be found in previous Chemical Reviews, most recently in 2018.
Footnotes
The authors declare no competing financial interest
References
- (1).Adero PO; Amarasekara H; Wen P; Bohé L; Crich D The experimental evidence in support of glycosylation mechanisms at the SN1-SN2 interface. Chem. Rev 2018, 118, 8242–8284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Winstein S; Lindegren CR; Marshall H; Ingraham LL Neighboring carbon and hydrogen. XIV. Participation in solvolysis of some primary benzenesulfonates. J. Am. Chem. Soc 1953, 75, 147–155. [Google Scholar]
- (3).IUPAC compendium of chemical technology; 2nd Edition “The Gold Book” ed.; McNaught AD; Wilkinson A, Eds.; Blackwell Scientific Publications: Oxford, 1997. [Google Scholar]
- (4).Muller P Glossary of terms used in physical organic chemistry. Pure & Appl. Chem 1994, 66, 1077–1184. [Google Scholar]
- (5).Page MI The energetics of neighboring group participation. Chem. Soc. Rev 1973, 2, 295–323. [Google Scholar]
- (6).Frush HL; Isbell HS Sugar acetates, acetylglycosyl halides and orthoacetates in relation to the Walden inversion. J. Research Natl. Bur. Standards 1941, 27, 413–428. [Google Scholar]
- (7).Winstein S; Buckles RE The role of neighboring groups in replacement reactions. 1. Retention of configuration in the reactions of some dihalides and acetoxyhalides with silver acetate. J. Am. Chem. Soc 1942, 64, 2780–2786. [Google Scholar]
- (8).Capon B Mechanism in carbohydrate chemistry. Chem. Rev 1969, 69, 407–498. [Google Scholar]
- (9).Capon B; McManus SP Neighboring group participation; Plenum: New York, 1976. [Google Scholar]
- (10).Guo J; Ye X-S Protecting groups in carbohydrate chemistry: Influence on stereoselectivity of glycosylations. Molecules 2010, 15, 7235–7265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Brabham R; Fascione MA In Selective glyocsylations: Synthetic methods and catalysts; Bennett CS, Ed.; Wiley-VCH: Weinheim, 2017. [Google Scholar]
- (12).Lemieux RU Some implications in carbohydrate chemistry of theories relating to the mechanisms of replacement reactions. Adv. Carbohydr. Chem 1954, 9, 1–57. [DOI] [PubMed] [Google Scholar]
- (13).Bochkov AF; Zaikov GE Chemistry of the O-glycosidic bond; Pergamon: Oxford, 1979. [Google Scholar]
- (14).Kong F Recent studies on reaction pathways and applications of sugar orthoesters in synthesis of oligosaccharides. Carbohydr. Res 2007, 342, 345–373. [DOI] [PubMed] [Google Scholar]
- (15).Fraser-Reid B; López JC In Handbook of chemical glycosylation; Demchenko AV, Ed.; Wiley-VCH: Weinheim, 2008. [Google Scholar]
- (16).Yang B; Yang W-B; Ramadan S; Huang X Pre-activation-based stereoselective glycosylations. Eur. J. Org. Chem 2018, 1075–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Igarashi K The Koenigs-Knorr reaction. Adv. Carbohydr. Chem. Biochem 1977, 34, 243–283. [Google Scholar]
- (18).Haynes LJ; Newth FH The glycosyl halides and their derivatives. Adv. Carbohydr. Chem 1955, 10, 207–256. [DOI] [PubMed] [Google Scholar]
- (19).Garegg PJ Synthesis and reactions of glycosides. Adv. Carbohydr. Chem. Biochem 2004, 59, 69–134. [DOI] [PubMed] [Google Scholar]
- (20).Green LG; Ley SV In Carbohydrates in Chemistry and Biology; Ernst B;Hart GW;Sinaÿ P, Eds.; Wiley-VCH: Weinheim, 2000; Vol. 1. [Google Scholar]
- (21).Ghosh G; Kulkarni SS Adavnces in protecting groups for oliogsaccharide synthesis. Chem. Asian J 2020, 15, 450–462. [DOI] [PubMed] [Google Scholar]
- (22).Koenigs W; Knorr E Derivatives of dextrose. Sitzungsber. Bayr. Akad. Wiss 1900, 103–105. [Google Scholar]
- (23).Barresi F; Hindsgaul O Chemically synthesized oligosaccharides, 1994. A searchable table of glycosidic linkages. J. Carbohydr. Chem 1995, 14, 1043–1087. [Google Scholar]
- (24).Dejter-Juszynski M; Flowers HM Studies on the Koenigs-Knorr reaction. Part III. A stereoselective synthesis of 2-acetamido-2-deoxy-6-O-α-L-fucopyranosyl-D-glucose. Carbohydr. Res 1972, 23, 41–45. [Google Scholar]
- (25).Dejter-Juszynski M; Flowers HM The effect of participating groups on the stereochemistry of disaccharide formation. Carbohydr. Res 1973, 61–74. [Google Scholar]
- (26).Komarova BS; Ustyuzhanina NE; Tsvetkov YE; Nifantiev NE In Modern synthetic methods in carbohydrate chemistry; from monosaccharides to complex glycoconjugates; Werz DB;Vidal S, Eds.; Wiley: Weinheim, 2014. [Google Scholar]
- (27).Komarova BS; Tsvetkov YE; Nifantiev NE Design of α-selective glycopyranosyl donors relying on remote anchimeric assistance. Chemical Record 2016, 16, 488–506. [DOI] [PubMed] [Google Scholar]
- (28).Ekholm FS; Leino R In Protecting groups – strategies and applications in carbohydrate chemistry; Vidal S, Ed.; Wiley-VCH: Weinheim, 2019. [Google Scholar]
- (29).Dimakos V; Taylor MS Site-selective functionalization of hydroxyl groups in carbohydrate derivatives. Chem. Rev 2018, 118, 11457–11517. [DOI] [PubMed] [Google Scholar]
- (30).Fischer E Wanderung von acyl bei den glyceriden. Ber. Dtsch. Chem. Ges 1920, 53, 1621–1633. [Google Scholar]
- (31).Molinier V; Wisniewski K; Bouchu A; Fitremann J; Queneau Y Transesterification of sucrose in organic medium: Study of acyl group migrations. J. Carbohydr. Chem 2003, 22, 657–669. [Google Scholar]
- (32).Kabel MA; De Waard P; Schols HA; Voragen AGJ Location of O-acetyl substituents in xylo-oligosaccharides obtained from hydrothermally treated eucalyptus wood. Carbohydr. Res 2003, 338, 69–77. [DOI] [PubMed] [Google Scholar]
- (33).Wang Z; Lu R Facile direct acylation and acyl migration of β-cyclodextrin on the secondary hydroxyl face. J. Incl. Phenom. Macrocycl. Chem 2009, 63, 373–378. [Google Scholar]
- (34).Wang ZZ; Liu MY; Lu RH; Fu XY; Dai GD Direct regioselective benzoylation of a single C-2 hydroxyl group of β-cyclodextrin and its hydrolysis via benzoyl group migration. Monatshefte fur Chemie 2011, 142, 1283–1287. [Google Scholar]
- (35).Volbeda AG; van der Marel GA; Codée JDC In Protecting groups; Vidal S, Ed.; Wiley: Weinheim, 2019. [Google Scholar]
- (36).Donnier‐Maréchal M; Vidal S; Fiore M In Protecting groups; V. S, Ed.; Wiley: Weinheim, 2019. [Google Scholar]
- (37).Goekjian PG; Vidal S In Protecting groups; Vidal S, Ed.; Wiley: Weinheim, 2019. [Google Scholar]
- (38).Vidal S; Goekjian PG In Protecting groups; Vidal S, Ed.; Wiley: Weinheim, 2019. [Google Scholar]
- (39).Sakonsinsiri C; Turnbull WB In Protecting groups; Vidal S, Ed.; Wiley: Weinheim, 2019. [Google Scholar]
- (40).Mathieson AM The preferred conformation of the ester group in relation to saturated ring systems. Tetrahedron Lett. 1965, 6, 4137–4144. [Google Scholar]
- (41).Schweizer WB; Dunitz JD Structural characteristics of the carboxylic ester group. Helv. Chim. Acta 1982, 65, 1547–1554. [Google Scholar]
- (42).González-Outeiriño J; Nasser R; Anderson JE Conformation of acetate derivatives of sugars and other cyclic alcohols. Crystal structures, NMR studies, and molecular mechanics calculations of acetates. When is the exocyclic C-O bond eclipsed? J. Org. Chem 2005, 70, 2486–2493. [DOI] [PubMed] [Google Scholar]
- (43).Huisgen R; Ott H Die konfiguration der carbonestergruppe und die sondereigenschaften der lactone. Tetrahedron 1959, 6, 253–267. [Google Scholar]
- (44).Oki M; Nakanishi H Conformations of the ester group. Bull. Chem. Soc. Jpn 1970, 43, 2558–2566. [Google Scholar]
- (45).Blom CE; Guenthard HH Rotational isomerism in methyl formate and methyl acetate: A low-temperature matrix infrared study using thermal molecular beams. Chem. Phys. Lett 1981, 84, 267–281. [Google Scholar]
- (46).Arnett EM; Harrelson JA A spectacular example of the importance of rotational barriers: The ionization of Meldrum’s acid. J. Am. Chem. Soc 1987, 109, 809–812. [Google Scholar]
- (47).Wiberg KB; Laidig KE Barriers to rotation adjacent to double bonds. The C-O barrier in formic acid, methyl formate, acetic acid, and methyl acetate. The origin of the ester and amide resonance. J. Am. Chem. Soc 1987, 109, 5935–5943. [Google Scholar]
- (48).Wiberg KB; Laidig KE Acidity of z- and e-methyl acetates: Relationship to Meldrum’s acid. J. Am. Chem. Soc 1988, 110, 1872–1874. [Google Scholar]
- (49).Wang X; Houk KN Theoretical elucidation of the origin of the anomalously high acidity of Meldrum’s acid. J. Am. Chem. Soc 1988, 110, 1870–1872. [Google Scholar]
- (50).Houk KN; Jabbari A; Hall HK; Alemán C Why δ-valerolactone polymerizes and γ-butyrolactone does not. J. Org. Chem 2008, 73, 2674–2678. [DOI] [PubMed] [Google Scholar]
- (51).Spellmeyer DC; Houk KN Force-field model for intramolecular radical additions. J. Org. Chem 1987, 52, 959–974. [Google Scholar]
- (52).Bjarnov E; Hocking WH The structure of the other rotamer of formic acid. cis-HCOOH. Z. Naturforsch 1978, 33A, 610–618. [Google Scholar]
- (53).Van Beuzekom AA; De Leeuw FAAM; Altona C Effect of the orientation of an α-substituent on vicinal carbon-13-proton spin-spin coupling constants. Magn. Reson. Chem 1990, 28, 68–74. [Google Scholar]
- (54).Altona C In Encyclopedia of nmr; Harris RK;Wasylishen RE, Eds.; Wiley: Chichester, 2012; Vol. 9. [Google Scholar]
- (55).Kleinpeter E; Taddei F; Wacker P Electronic and steric substituent influences on the conformational equilibria of cyclohexyl esters: The anomeric effect is not anomalous! Chem. Eur. J 2003, 9, 1360–1368. [DOI] [PubMed] [Google Scholar]
- (56).Kleinpeter E; Bölke U; F., A. Polar substituent effect of the ester group on conformational equilibria of O-mono-substituted cyclohexanes and the para-substituent effect in cyclohexyl benzoates. Tetrahedron 2008, 64, 10014–10017. [Google Scholar]
- (57).Steiner T; Saenger W Relief of steric strain by intramolecular C-H ⋯ O interactions: Structural evidence for the 1,4-disubstituted cyclohexanes. J. Chem. Soc, Perkin Trans 2 1998, 371–377. [Google Scholar]
- (58).Kleinpeter E; Werner P; Linker T Synthesis and NMRs spectroscopic conformational analysis of benzoic acid esters of mono- and 1,4-dihydroxycyclohexane, 4-hydroxycyclohexanone and the -ene analogue - the more polar the molecule the more stable the axial conformer. Tetrahedron 2017, 73, 3801–3808. [Google Scholar]
- (59).Zefirov NS; Samoshin VV; Subbotin OA; Baranenkov VI; Wolfe S The gauche effect. On the nature of the interaction between electronegative substituents in trans-1,2-disubstituted cyclohexanes. Tetrahedron 1978, 34, 2953–2959. [Google Scholar]
- (60).Wiberg KB Conformational studies in the cyclohexane series. 3. The dihalocyclohexanes. J. Org. Chem 1999, 64, 6387–6393. [Google Scholar]
- (61).Durette PL; Horton D Conformational analysis of sugars and their derivatives. Adv. Carbohydr. Chem. Biochem 1971, 26, 49–125. [Google Scholar]
- (62).Kirby AJ The anomeric effect and related stereoelectronic effects at oxygen; Springer-Verlag: Berlin, 1983. [Google Scholar]
- (63).Grindley TB In Glycoscience: Chemistry and chemical biology; Fraser-Reid B;Tatsuta K;Thiem J, Eds.; Springer: Berlin, 2001; Vol. 1. [Google Scholar]
- (64).Lemieux RU; Pavia AA Substitutional and solvation effects on conformational equilibria. Effects on the interaction between opposing axial oxygen atoms. Can. J. Chem 1969, 47, 4441–4446. [Google Scholar]
- (65).Lichtenthaler FW; Lindner HJ On the extent of chair distortion in the “tetra-axial” derivatives of 2,3,4-tri-O-benzoyl-β-D-xylopyranose. Carbohydr. Res 1990, 200, 91–99. [Google Scholar]
- (66).Luger P; Kothe G; Vangehr K; Paulsen H; Heiker FR Konformationen von 1,2,3,4-tetra-O-benzoyl-β-D-xylopyranose, 1,5-anhydro-2,3,4-tri-O-benzoylxylitol und 1,5-anhydro-2,3,4-tri-O-benzoylribitol. Carbohydr. Res 1979, 68, 207–223. [Google Scholar]
- (67).Perrin CL Reverse anomeric effect: Fact or fiction? Tetrahedron 1995, 11901–11935. [Google Scholar]
- (68).Perrin CL; Fabian MA; Brunckova J; Ohta BK Absence of reverse anomeric effect in glycosylimidazoles. J. Am. Chem. Soc 1999, 121, 6911–6918. [Google Scholar]
- (69).Chan SSC; Szarek WA; Thatcher GRJ The reverse anomeric effect in N-pyranosylimidazolides: A molecular orbital study. J. Chem. Soc., Perkin Trans 2 1995, 45–60. [Google Scholar]
- (70).Jones PG; Kirby AJ; Komarov IV; Wothers PD A test for the reverse anomeric effect. Chem. Commun 1998, 1695–1696. [Google Scholar]
- (71).Randell KD; Johnston BD; Green DF; Pinto BM Is there a generalized reverse anomeric effect? Substituent and solvent effects on the configurational equilibria of neutral and protonated N-arylglucopyranosylamines and N-aryl-5-thioglucopyranosylamines. J. Org. Chem 2000, 65, 220–226. [DOI] [PubMed] [Google Scholar]
- (72).Lemieux RU; Morgan AR The abnormal conformations of pyridinium α-glycopyranosides. Can. J. Chem 1965, 43, 2205–2213. [Google Scholar]
- (73).Konitz A; Dmochowska B; Nowacki A; Wojnowski W; Wisniewski A Crystal structure of N-(tri-O-acetyl-α-D-xylopyranosyl)pyridinium bromide. Carbohydr. Res 2001, 333, 257–261. [DOI] [PubMed] [Google Scholar]
- (74).Luger P; Kothe G; Paulsen H Triaxial conformation of 1-(tri-O-acetyl-α-D-xylopyranosyl)imidazole. Chem. Ber 1974, 107, 2626–2634. [Google Scholar]
- (75).Finch P; Nagpurkar AG The reverse anomeric effect: Further observations on N-glycosylimidazoles. Carbohydr. Res 1976, 49, 275–287. [Google Scholar]
- (76).Paulsen H; Dammeyer R Carboxonium compounds in carbohydrate chemistry, xxix. Investigation of charge distribution in 1,3-dioxolan-2-ylium ions Chem. Ber 1976, 109, 1837–1849. [Google Scholar]
- (77).Paulsen H; Schüttpelz E Carboxonium compounds in carbohydrate chemistry. 33. Proton and carbon-13 nmr studies of charge distribution in substituted 1,3-dioxan-2-ylium salts. Org. Magn. Res 1979, 12, 616–623. [Google Scholar]
- (78).Paulsen H; Schüttpelz E Carboxonium compounds in carbohydrate chemistry. XXXV. Endo-envelope conformation in 2-methyl-cis-4,5-trimethylene-1,3-dioxolan-2-ylium tetrafluoroborate. Chem. Ber 1979, 112, 3214–3220. [Google Scholar]
- (79).Paulsen H; Dammeyer R Carboxonium compounds in carbohydrate chemistry, XXVIII. Molecular structure of 2-methyl-4,5-tetramethylene-1,3-dioxolan-2-ylium perchlorate Chem. Ber 1976, 109, 605–610. [Google Scholar]
- (80).Paulsen H; Hoehne H; Durette PL Carboxonium compounds in carbohydrate chemistry, xxvii. Can trans-linked 1,3-dioxolan-2-ylium ions be made? Chem. Ber 1976, 109, 597–604. [Google Scholar]
- (81).Paulsen H Transformations of carboxonium compounds in carbohydrate and polyol chemistry Pure & Appl. Chem 1975, 41, 69–92. [Google Scholar]
- (82).Paulsen H; Meyborg H; Behre H Cyclorearrangement of di-O-pivaloylpentaerythitol-O-pivaloxonium hexachloroantimonate. Angw. Chem., Int. Ed. Engl 1969, 8, 888–888. [Google Scholar]
- (83).Larsen JW; Ewing S Relative heats of formation of cyclic oxonoim ions in sulfuric acid. J. Am. Chem. Soc 1971, 93, 5107–5111. [Google Scholar]
- (84).Ramsey BG; Taft RW The spectral properties of alkoxymethyl cations. J. Am. Chem. Soc 1966, 88, 3058–3063. [Google Scholar]
- (85).Dusseau CHV; Schaafsma SE; Steinberg H; de Boer TJ Alkoxycarbenium ions from acetals and ortho esters with bromine. Tetrahedron Lett. 1969, 10, 467–470. [Google Scholar]
- (86).Borch RF Alkoxycarbenium ions. The structure of O-alkylated esters. J. Am. Chem. Soc 1968, 90, 5303–5305. [Google Scholar]
- (87).Deslongchamps P Stereoelectronic effects in organic chemistry; Pergamon: Oxford, 1983. [Google Scholar]
- (88).Chamberland S; Ziller JW; Woerpel KA Structural evidence that alkoxy substituents adopt electronically preferred pseudoaxial orientations in six-membered ring dioxocarbenium ions. J. Am. Chem. Soc 2005, 127, 5322–5323. [DOI] [PubMed] [Google Scholar]
- (89).Kim J-H; Yang H; Khot V; Whitfield D; Boons G-J Stereoselective glycosylations using (R) or (S)-(ethoxycarbonyl)benzyl chiral auxiliaries at C2 of glycopyranosyl donors. Eur. J. Org. Chem 2006, 5007–5028. [Google Scholar]
- (90).Schroeder LR; Green JW; Johnson DC Alcoholyses of 2,3,4,6-tetra-O-acetyl-α-D-glucopyranosyl bromide; kinetics and products at 25–40 °C. J. Chem. Soc. (B) 1966, 447–453. [Google Scholar]
- (91).Eby R; Schuerch C Solid phase synthesis of oligosaccharides. Methanolysis of N-phenylcarbamoyl and N-methyl-N-phenylcarbamoyl derivatives of α-D-glucopyranosyl bromide. Carbohydr. Res 1973, 27, 63–72. [Google Scholar]
- (92).Wulff G; Roehle G Kinetic investigations on the mechanisms of the Koenigs-Knorr reaction. Chem. Ber 1972, 105, 1122–1132. [DOI] [PubMed] [Google Scholar]
- (93).Wulff G; Schmidt W Uber die orthoesterbildung als konkurrenzreaktion zur glykosylierung. Carbohydr. Res 1977, 53, 33–46. [Google Scholar]
- (94).Helferich B; Zirner J 4-Bromobutyl tetra-O-acetyl-β-D-glucopyranoside. Chem. Ber 1963, 96, 374-. [Google Scholar]
- (95).Wallace JE; Schroeder LR Mechanistic study of mercury (ii) cyanide promoted reactions of 2-O-acetyl-3,4,6-tri-O-methyl-α-D-glucopyranosyl bromide with cyclohexanol in benzene-nitromethane. J. Chem. Soc., Perkin Trans 2 1977, 795–802. [Google Scholar]
- (96).Glaudemans CPJ; Fletcher HG Long range effects of acyl groups in the solvolysis of glycofuranosyl halides. The synthesis of 2,3-di-O-benzyl-5-O-p-nitrobenzoyl-α-D-arabinofuranosyl chloride and of 2-O-benzyl-3,5-di-O-p-nitrobenzoyl-α-D-arabinofuranosyl chloride. J. Am. Chem. Soc 1965, 87, 4636–4641. [Google Scholar]
- (97).Kochetkov NK; Khorlin AJ; Bochkov AF A new method of glycosylation. Tetrahedron 1967, 23, 693–707. [Google Scholar]
- (98).Lemieux RU; Morgan AR The preparation and configurations of tri-O-acetyl-α-D-glucopyranose 1,2-orthoesters. Can. J. Chem 1965, 43, 2199–2204. [Google Scholar]
- (99).Pacsu E Carbohydrate orthoesters. Adv. Carbohydr. Chem 1945, 1, 77–95. [DOI] [PubMed] [Google Scholar]
- (100).Hanessian S; Banoub J Chemistry of the glycosidic linkage. A rapid and efficient synthesis of carbohydrate 1,2-orthoesters. Carbohydr. Res 1975, 44, c14–c17. [Google Scholar]
- (101).Banoub J; Bundle DR 1,2-orthoacetate intermediates in silver trifluoromethanesulfonate promoted Koenigs-Knorr synthesis of disaccharide glycosides. Can. J. Chem 1979, 57, 2091–2097. [Google Scholar]
- (102).Garegg PJ; Konradsson P; Kvarnstrom I; Norberg T; Svensson SCT; Wigilius B Studies on Koenigs-Knorr glycosidations. Acta Chem. Scand 1985, B39, 569–577. [Google Scholar]
- (103).Crich D; Dai Z; Gastaldi S On the role of neighboring group participation and ortho esters in β-xylosylation: 13C-NMR observation of a bridging 2-phenyl-1,3-dioxalenium ion. J. Org. Chem 1999, 64, 5224–5229. [DOI] [PubMed] [Google Scholar]
- (104).Wattanasiri C; Ruchirawat S; Platon M; Boonyarattanakalin S In Carbohydrate chemistry: Proven synthetic methods; van der Marel G;Codée JDC, Eds.; CRC Press: Boca Raton, 2014; Vol. 2. [Google Scholar]
- (105).Christensen HM; Oscarson S; Jensen HH Common side reactions of the glycosyl donor in chemical glycosylation. Carbohydr. Res 2015, 408, 51–95. [DOI] [PubMed] [Google Scholar]
- (106).Joosten A; Boultadakis-Arapinis M; Gandon V; Micouin L; Lecourt T Substitution of the participating group of glycosyl donors by a halogen atom: Influence on the rearrangement of transient orthoesters formed during glycosylation reactions. J. Org. Chem 2017, 82, 3291–3297. [DOI] [PubMed] [Google Scholar]
- (107).Takatani M; Matsuo I; Ito Y Pentafluoropropionyl and trifluoroacetyl groups for temporary hydroxyl group protection in oligomannoside synthesis. Carbohydr. Res 2003, 338, 1073–1081. [DOI] [PubMed] [Google Scholar]
- (108).Nukada T; Berces A; Zgierski MZ; Whitfield DM Exploring the mechanism of neighboring group assisted glycosylation reactions. J. Am. Chem. Soc 1998, 120, 13291–13295. [Google Scholar]
- (109).Seeberger PH; Eckhardt M; Gutteridge CE; Danishefsky SJ Coupling of glycal derived thioethyl glycosyl donors with glycal acceptors. An advance in the scope of the glycal assembly. J. Am. Chem. Soc 1997, 119, 10064–10072. [Google Scholar]
- (110).Deng S; Yu B; Xie J; Hui Y Highly efficient glycosylation of sapogenins. J. Org. Chem 1999, 64, 7265–7266. [Google Scholar]
- (111).Harreus A; Kunz H Stereoselective glycosylierung von steroidalalkoholen mit 2,3,4,6-tetra-O-pivaloyl-α-D-glucopyranosylbromid und 2,3,4,6-tetra-O-(o-toluoyl)-α-D-glucopyranosylbromid. Liebigs Ann. Chem 1986, 717–730. [Google Scholar]
- (112).Kunz H; Harreus A Glycosidsynthese mit 2,3,4,6-tetra-O-pivaloyl-α-D-glucopyranosylbromid. Liebigs Ann. Chem 1982, 41–48. [Google Scholar]
- (113).Brocke C; Kunz H Synthetic glycopeptides of the tandem repeat sequence of the epithelial mucin MUC4 with tumor-associated carbohydrate antigens. Synlett 2003, 2052–2056. [Google Scholar]
- (114).Williams RJ; McGill NW; White JM; Williams SJ Neighboring group participation in glycosylation reactions by 2,6-disubstituted 2-O-benzoyl groups: A mechanistic investigation. J. Carbohydr. Chem 2010, 29, 236–263. [Google Scholar]
- (115).Thompson C; Ge M; Kahne D Synthesis of vancomycin from the aglycone. J. Am. Chem. Soc 1999, 121, 1237–1244. [Google Scholar]
- (116).Ge M; Thompson C; Kahne D Reconstruction of vancomycin by chemical glycosylation of the pseudoaglycon. J. Am. Chem. Soc 1998, 120 (42), 11014–11015. [Google Scholar]
- (117).Bérces A; Whitfield DM; Nukada T; do Santos I; Obuchowska A; Krepinsky JJ Is acyl migration to the aglycon avoidable in 2-acyl assisted glycosylation reactions? Can. J. Chem 2004, 82, 1157–1171. [Google Scholar]
- (118).Brown RT; Carter NE; Mayalarp SP; Scheinmann FP A simple synthesis of morphine-3,6-β-D-glucuronide. Tetrahedron 2000, 56, 7591–7594. [Google Scholar]
- (119).Ito Y; Numata M; Sugimoto M; Ogawa T Highly stereoselctive synthesis of ganglioside GD3. J. Am. Chem. Soc 1989, 111, 8508–8510. [Google Scholar]
- (120).Liu H; Hansen T; Zhou S-Y; Wen G-E; Liu X-X; Zhang Q-J; Codée JDC; Schmidt RR; Sun J-S Dual-participation protecting group solves the anomeric stereocontrol problems in glycosylation reactions Org. Lett 2019, 21, 8713–8717. [DOI] [PubMed] [Google Scholar]
- (121).Banoub JH; Michon F; Rice J; Rateb L Kinetic studies on the rearrangement of 3,4-di-O-benzyl-1,2-(1-methoxyethylidene)-β−L-rhamnopyranose with a catalytic amount of 1,1,3,3-tetramethylurea-trifluoromethanesulfonic acid at different temperatures. Carbohydr. Res 1983, 123, 109–116. [Google Scholar]
- (122).Yang Z; Lin W; Yu B Rearrangement of sugar 1,2-orthoesters to glycosidic products: A mechanistic implication. Carbohydr. Res 2000, 329, 879–884. [DOI] [PubMed] [Google Scholar]
- (123).Lemieux RU; Hayami J-I The mechanism of the anomerization of the tetra-O-acetyl-D-glucopyranosyl chlorides. Can. J. Chem 1965, 43, 2162–2173. [Google Scholar]
- (124).Bonner WA Relative rates of inversion and Cl acetoxy exchange during anomerizations of acetylated aldopyranoses. J. Am. Chem. Soc 1959, 81, 5171–5176. [Google Scholar]
- (125).Lemieux RU; Morgan AR The mechanism for the formation of 1,2-cis-pyridine nucleosides from 1,2-cis-acetohalogenosugars. A novel rearrangement. J. Am. Chem. Soc 1963, 85, 1889–1890. [Google Scholar]
- (126).Lemieux RU; Morgan AR Mechanisms for the reaction of the anomeric tetra-O-acetyl-D-glucopyranosyl bromides with pyridine and 2-pyridone. Can. J. Chem 1965, 43, 2214–2221. [Google Scholar]
- (127).Paulsen H; Herold C-P Untersuchungen zur acetoxonium-umlagerung von D-glucose zu D-idose. Chem. Ber 1970, 103, 2450–2462. [Google Scholar]
- (128).Konradsson P In Glycoscience: Chemistry and chemical biology; Fraser-Reid B;Tatsuta K;Thiem J, Eds.; Springer: Berlin, 2001; Vol. 1. [Google Scholar]
- (129).Geurtsen R; Holmes DS; Boons G-J Chemoselective glycosylations. 2. Differences in size of anomeric leaving groups can be exploited in chemoselective glycosylations. J. Org. Chem 1997, 62, 8145–8154. [DOI] [PubMed] [Google Scholar]
- (130).Crich D; Sun S Direct formation of β-mannopyranosides and other hindered glycosides from thioglycosides. J. Am. Chem. Soc 1998, 120, 435–436. [Google Scholar]
- (131).Kahne D; Walker S; Cheng Y; Engen DV Glycosylation of unreactive substrates. J. Am. Chem. Soc 1989, 111, 6881–6882. [Google Scholar]
- (132).Kamat MN; Demchenko AV Revisiting the armed-disarmed concept rationale: S-benzoxazolyl glycosides in chemoselectivve oligosaccharide synthesis. Org. Lett 2005, 7, 3215–3218. [DOI] [PubMed] [Google Scholar]
- (133).Crich D; Li M Revisiting the armed-disarmed concept: The importance of anomeric configuration in the activation of S-benzoxazolyl glycosides. Org. Lett 2007, 9, 4115–4118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (134).Mootoo DR; Konradsson P; Udodong U; Fraser-Reid B “Armed” and “disarmed” n-pentenyl glycosides in saccharide couplings leading to oligosaccharides. J. Am. Chem. Soc 1988, 110, 5583–5584. [Google Scholar]
- (135).Mydock LK; Demchenko AV Superarming the S-benzoxazolyl glycosyl donors by simple 2-O-benzoyl-3,4,6-tri-O-benzyl protection. Org. Lett 2008, 10, 2103–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (136).Mydock LK; Demchenko AV Application of the superarmed glycosyl donor to chemoselective oligosaccharide synthesis. Org. Lett 2008, 10, 2107–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (137).Premathilake HD; Mydock LK; Demchenko AV Super-arming common glycosyl donors by simple 2-O-benzoyl-3,4,6-tri-O-benzyl protection. J. Org. Chem 2010, 75, 1095–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (138).Zhang Z; Ollmann IR; Ye X-S; Wischnat R; Baasov T; Wong C-H Programmable one-pot oligosaccharide synthesis. J. Am. Chem. Soc 1999, 121 (4), 734–753. [Google Scholar]
- (139).Wu C-Y; Wong C-H Programmable one-pot glycosylation. Top. Curr. Chem 2011, 301, 223–252. [DOI] [PubMed] [Google Scholar]
- (140).Hsu Y; Lu X-A; Zulueta MML; Tsai C-M; Lin K-I; Hung S-C; Wong C-H Acyl and silyl group effects in reactivity-based one-pot glycosylation: Synthesis of embryonic stem cell surface carbohydrates Lc4 and IV2Fuc-Lc4 J. Am. Chem. Soc 2012, 134, 4549–4552. [DOI] [PubMed] [Google Scholar]
- (141).Smith RS; Müller-Bunz H; Zhu X Investigation of α‑thioglycoside donors: Reactivity studies toward configuration-controlled orthogonal activation in one-pot systems. Org. Lett 2016, 18, 3578–3581. [DOI] [PubMed] [Google Scholar]
- (142).Heuckendorff M; Poulsen LT; Jensen HH On the generality of the superarmament of glycosyl donors. Org. Biomol. Chem 2018, 16, 2269–2276. [DOI] [PubMed] [Google Scholar]
- (143).Posner GH; Bull DS Molecular sieves promote stereocontrolled α,α-disaccharide formation via direct dimerization of free sugars Tetrahedron Lett. 1996, 37, 6279–6281. [Google Scholar]
- (144).Asakura N; Hirokane T; Hosida H; Yamada H Molecular sieves 5Å as an acidic reagent: The discovery and applications. Tetrahedron Lett. 2011, 52, 534–537. [Google Scholar]
- (145).Heuckendorff M; Poulsen LT; Hedberg C; Jensen HH Dissection of the effects that govern thioglucoside and thiomannoside reactivity. Org. Biomol. Chem 2018, 16, 2277–2288. [DOI] [PubMed] [Google Scholar]
- (146).Heuckendorff M; Premathilake HD; Pornsuriyasak P; Madsen AO; Pedersen CM; Bols M; Demchenko AV Superarming of glycosyl donors by combined neighboring and conformational effects. Org. Lett 2013, 15, 4904–4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (147).Poulsen LT; Heuckendorff M; Jensen HH Effect of 2-O-benzoyl para-substituenets on glycosylation rates. ACS Omega 2018, 3, 7117–7123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).Heuckendorff M; Pedersen CM; Bols M The influence of neighboring group participation on the hydrolysis of 2-O-substituted methyl glucopyranosides. Org. Lett 2011, 13, 5956–5959. [DOI] [PubMed] [Google Scholar]
- (149).Bohé L; Crich D A propos of glycosyl cations and the mechanism of chemical glycosylation. CR Chimie 2011, 14, 3–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (150).Wang L; Fan F; Wu H; Gao L; Zhang P; Sun T; Yang C; Yu G; Cai C Effect of anomeric configuration on stereocontrolled α-glycosylation of L-fucose. Synlett 2018, 29, 2701–2706. [Google Scholar]
- (151).Crich D; Hu T; Cai F Does neighboring group participation by non-vicinal esters play a role in glycosylation reactions? Effective probes for the detection of bridging intermediates. J. Org. Chem 2008, 73, 8942–8953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (152).Gentil E; Potier M; Boulaanger P; Descotes G Utilization de dérivés 2-alkoxycarbonylés dans les réactions de glucosylation. Synthèse stéréosélective d’orthocarbonates. Carbohydr. Res 1990, 197, 75–91. [Google Scholar]
- (153).Morère A; Mouffouk F; Leiris S; Leydet A; Montero J-L Influence of the benzyloxycarbonyl protective group on glycosylation with mannopyranosyl donors. J. Carbohydr. Chem 2006, 25, 451–459. [Google Scholar]
- (154).Weber JT; Svatunek D; Krauter S; Tegl G; Hametner C; Kosma P; Mikula H 2-O-benzyloxycarbonyl protected glycosyl donors: A revival of carbonate-mediated anchimeric assistance for diastereoselective glycosylation. Chem. Commun 2019, 55, 12543–12546. [DOI] [PubMed] [Google Scholar]
- (155).Lemieux RU; Hindsgaul O 1,2-acylspiroorthoesters of 3,4,6-tri-O-acetyl-α-D-glucopyranose. Carbohydr. Res 1980, 82, 195–206. [Google Scholar]
- (156).Crich D; Cai W; Dai Z Highly diastereoselctive α-mannosylation in the absence of participating protecting groups. J. Org. Chem 2000, 65, 1291–1297. [DOI] [PubMed] [Google Scholar]
- (157).Nicolaou KC; Daines RA; Ogawa Y; Chakraborty TK Total synthesis of amphotericin b. The final stages. J. Am. Chem. Soc 1988, 110, 4696–4705. [Google Scholar]
- (158).Baek JY; Lee B-Y; Jo MG; Kim KS β-directing effect of electron-withdrawing groups at O-3, O-4, and O-6 positions and α-directing effect by remote participation of 3-O-acyl and 6-O-acetyl groups of donors in mannopyranosylations. J. Am. Chem. Soc 2009, 131, 17705–17713. [DOI] [PubMed] [Google Scholar]
- (159).Jin H; Tsai R; Wiesner K On the synthesis of cardioactive steroid glycosides. On cardioactive steroids. Can. J. Chem 1983, 61, 2442–2444. [Google Scholar]
- (160).Ma Y; Lian G; Li Y; Yu B Identification of 3,6-di-O-acetyl-1,2,4-O-orthoacetyl-α-D-glucopyranose as a direct evidence for the 4-O-acyl group participation in glycosylation. Chem. Commun 2011, 47, 7515–7517. [DOI] [PubMed] [Google Scholar]
- (161).Yao D; Liu Y; Yan S; Li Y; Hu C; Ding N Evidence of robust participation by an equatorial 4-O group in glycosylation on a 2-azido-2-deoxyglucopyranosyl donor. Chem. Commun 2017, 53, 2986–2989. [DOI] [PubMed] [Google Scholar]
- (162).Xu HH; Chen L; Zhang Q; Feng Y; Zu Y; Chai Y Stereoselective β-mannosylation with 2,6-lactone-bridged thiomannosyl donor by remote acyl group participation. Chem. Asian J 2019, 14, 1424–1428. [DOI] [PubMed] [Google Scholar]
- (163).Baek JY; Lee B-Y; Jo MG; Kim KS β-directing effect of electron-withdrawing groups at O-3, O-4, and O-6 positions and α-directing effect by remote participation of 3-O-acyl and 6-O-acetyl groups of donors in mannopyranosylations. J. Am. Chem. Soc 2010, 132, 7729–7729. [DOI] [PubMed] [Google Scholar]
- (164).Kim J-H; Yang H; Boons G-J Stereoselective glycosylation reactions with chiral auxiliaries. Angew. Chem. Int. Ed 2005, 44, 947–949. [DOI] [PubMed] [Google Scholar]
- (165).Wen P; Crich D Absence of stereodirecting participation by 2-O-alkoxycarbonylmethyl ethers in 4,6-O-benzylidene-directed mannosylation. J. Org. Chem 2015, 80, 12300–12310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (166).Childs RF; Frampton CS; Kang GJ; Wark TA Structures of some dialkoxyphenylmethylium ions; steric inhibition of resonance. J. Am. Chem. Soc 1994, 116, 8499–8505. [Google Scholar]
- (167).Zeng Y; Wang Z; Whitfield D; Huang X Installation of electron-donating protective groups, a strategy for glycosylating unreactive thioglycosyl acceptors using the preactivation-based glycosylation method. J. Org. Chem 2008, 73, 7952–7962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (168).Crich D; Sun S Are glycosyl triflates intermediates in the sulfoxide glycosylation method? A chemical and 1H, 13C, and 19F-NMR spectroscopic investigation. J. Am. Chem. Soc 1997, 119, 11217–11223. [Google Scholar]
- (169).Baek JY; Kwon H-W; Myung SJ; Park JJ; Kim MY; Rathwell DCK; Jeon HB; Seeberger PH; Kim KS Directing effect by remote electron-withdrawing protecting groups at O-3 or O-4 position of donors in glucosylations and galactosylations. Tetrahedron 2015, 71, 5315–5320. [Google Scholar]
- (170).Gerbst AG; Ustuzhanina NE; Grachev AA; Khatuntseva EA; Tsvetkov DE; Whitfield DM; Berces A; Nifantiev NE Synthesis, nmr, and conformational studies of fucoidan fragments. III. Effect of benzoyl group at O-3 on stereoselectivity of glycosylation by 3-O- and 3,4-di-O-benzoylated 2-O-benzylfucosyl bromides. J. Carbohydr. Chem 2001, 20, 821–831. [Google Scholar]
- (171).Gerbst A. g.; Ustuzhanina NE; Grachev AA; Tsvetkov DE; Khatuntseva EA; Nifant’ev NE Effect of the nature of protecting group at O-4 on stereoselectivity of glycosylation by 4-O-substituted 2,3-di-O-benzylfucosyl bromides. Mendeleev Commun. 1999, 9, 114–116. [Google Scholar]
- (172).Li Z Computational study of the influence of cyclic protecting groups in stereoselectivity of glycosylation reactions. Carbohydr. Res 2010, 345, 1952–1957. [DOI] [PubMed] [Google Scholar]
- (173).Kalikanda J; Li Z Study of the stereoselectivity of 2-azido-2-deoxygalactosyl donors: Remote protecting group effects and temperature dependency. J. Org. Chem 2011, 76, 5207–5218. [DOI] [PubMed] [Google Scholar]
- (174).Hosoya T; Kosma P; Rosenau T Contact ion pairs and solvent-separated ion pairs from D-mannopyranosyl and D-glucopyranosyl triflates. Carbohydr. Res 2015, 401, 127–131. [DOI] [PubMed] [Google Scholar]
- (175).Hosoya T; Kosma P; Rosenau T Theoretical study on the effects of a 4,6-O-diacetal protecting group on the stability of ion pairs from D-mannopyranosyl and D-glucopyranosyl triflates. Carbohydr. Res 2015, 411, 64–69. [DOI] [PubMed] [Google Scholar]
- (176).Hosoya T; Takano T; Kosma P; Rosenau T Theoretical foundation for the presence of oxacarbenium ions in chemical glycoside synthesis. J. Org. Chem 2014, 79, 7889–7894. [DOI] [PubMed] [Google Scholar]
- (177).Martin A; Arda A; Désiré J; Martin-Mingot A; Probst N; Sinaÿ P; Jiménez-Barbero J; Thibaudeau S; Blériot Y Catching elusive glycosyl cations in a condensed phase with HF/SbF5 superacid. Nat. Chem, 2016, 8, 186–191. [DOI] [PubMed] [Google Scholar]
- (178).Lebedel L; Ardá A; Martin A; Désiré J; Mingot A; Aufiero M; Font NA; Gilmour R; Jiménez-Barbero J; Blériot Y et al. Structural and computational analysis of 2-halogeno-glycosyl cations in the presence of a superacid: An expanisve platform. Angew. Chem. Int. Ed 2019, 58, 13758–13762. [DOI] [PubMed] [Google Scholar]
- (179).a) Elferink H; Severijnen ME; Martens J; Mensink RA; Berden G; Oomens J; Rutjes FPJT; Rijs AM; Boltje TJ Direct experimental characterization of glycosyl cations by infrared ion spectroscopy. J. Am. Chem. Soc 2018, 140, 6034–6038. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hansen T; Elferink H; van Hengst JMA; Houtuijs KJ; Remmerswaal WA; Kromm A; Berden G; van der Vorm S; Rijs AM; Overkleeft HS; et al. Characterization of glycosyl dioxolenium ions and their role in glycosylation reactions. Nat. Commun 2020, 11, 2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (180).Matrianski M; Mucha E; Greis K; Moon S; Pardo A; Kirschbaum C; Thomas DA; Meijer G; von Helden G; Gilmore k. et al. Remote participation during glycosylation reactions of galactose building blocks: Direct evidence from cryogenic vibrational spectroscopy. Angew. Chem. Int. Ed 2020, 59, 6166–6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (181).De Meo C; Priyadarshani U C-5 modifications in N-acetyl-neuraminic acid: Scope and limitations. Carbohydr. Res 2008, 343, 1540–1552. [DOI] [PubMed] [Google Scholar]
- (182).Navuluri C; Crich D In Glycochemical synthesis: Strategies and applications; Hung S-C;Zulueta MML, Eds.; Wiley: New York, 2016. [Google Scholar]
- (183).Panova MV; Orlova AV; Kononov LO Stabilization of sialyl cation in axial conformation assisted by remote acyl groups. Russ. Chem. Bull 2018, 67, 1573–1579. [Google Scholar]
- (184).Smith DM; Woerpel KA Electrostatic interactions in cations and their importance in biology and chemistry. Org. Biomol. Chem 2006, 4, 1195–1201. [DOI] [PubMed] [Google Scholar]
- (185).Jensen HH; Bols M Stereoelectronic substituent effects. Acc. Chem. Res 2006, 39, 259–265. [DOI] [PubMed] [Google Scholar]
- (186).Berces A; Enright G; Nukada T; Whitfield DM The conformational origin of the barrier to the formation of neighboring group assistance in glycosylation reactions: A dynamical density functional theory study. J. Am. Chem. Soc 2001, 123, 5460–5464. [DOI] [PubMed] [Google Scholar]
- (187).Whitfield DM; Nukada T DFT studies of the role of C-2–O-2 bond rotation in neighboring-group glycosylation reactions. Carbohydr. Res 2007, 342, 1291–1304. [DOI] [PubMed] [Google Scholar]
- (188).Pacsu E Studies in the ketone sugar series. Preparation of new methyl and ethyl fructoside acetates. J. Am. Chem. Soc 1935, 57, 745–747. [Google Scholar]
- (189).Steinlin H; Camarda L; Vasella A Uber die herstellung des methyl-α-D-fructopyranosides und die stucktur der dabei gebildeten orthoester. Helv. Chim. Acta 1979, 62, 378–390. [Google Scholar]
- (190).Lian g.; Gao Q; Lin F Synthesis of fructofuranosides: Efficient glycosylation with N-phenyltrifluoroacetimidate as the leaving group. Carbohydr. Res 2008, 343, 2992–2996. [DOI] [PubMed] [Google Scholar]
- (191).Haberman JM; Gin DY A new C(1)-auxillary for anomeric stereocontrol in the synthesis of α-sialyl glycosides. Org. Lett 2001, 3, 1665–1668. [DOI] [PubMed] [Google Scholar]
- (192).Chen J; Hansen T; Zhang Q-J; Liu D-Y; Sun Y; Yan H; Codée JDC; Schmidt RR; Sun J-S 1-Picolinyl-5-azido thiosialosides: Versatile donors for the stereoselective construction of sialyl linkages. Angew. Chem. Int. Ed 2019, 58, 17000–17008. [DOI] [PubMed] [Google Scholar]
- (193).Yasomanee JP; Demchenko AV Effect of remote picolinyl and picoloyl substituents on the stereoselectivity of chemical glycosylation. J. Am. Chem. Soc 2012, 134, 20097–20102. [DOI] [PubMed] [Google Scholar]
- (194).Mannino MP; Demchenko AV Synthesis of β-glucosides with 3-O-picolyl-protected glycosyl donors in the presence of excess triflic acid: A mechanistic study. Chem. Eur. J 2020, 26. [DOI] [PubMed] [Google Scholar]
- (195).Lu Y-C; Mondal S; Wang CC; Lin C-H; Mong K-KT Diverse synthesis of natural trehalosamines and synthetic 1,1’-disaccharide aminoglycosides ChemBioChem 2019, 20, 287–294. [DOI] [PubMed] [Google Scholar]
- (196).Lu Y-CL; Ghosh B; Mong K-KT Unusually stable picoloyl-protected trimethylsilyl glycosides for nonsymmetrical 1,1’-glycosylation and synthesis of 1,1’-disaccharides with diverse configurations. Chem. Eur. J 2017, 23, 6905–6918. [DOI] [PubMed] [Google Scholar]
- (197).Dolby LJ; Schwarz MJ The mechanism of the Prins reaction. IV. Evidence against acetoxonium ion intermediates. J. Org. Chem 1965, 30, 3581–3586. [Google Scholar]
- (198).Dolby LJ; Lieske CN; Rosencrantz DR; Schwarz MJ The mechanism of the Prins reaction. II. The solvolysis of trans-2-hydroxymethylcyclohexyl brosylate and trans-2-acetoxymethylcyclohexyl brosylate. J. Am. Chem. Soc 1963, 85, 47–52. [Google Scholar]
- (199).Kovács Ö; Schneider G; Láng K Solvolysis of 2-hydroxymethylcyclohexanol derivatives. Proc. Chem. Soc 1963, 374–374. [Google Scholar]
- (200).Schneider G; Kovács OKJ Syntheses of 2-methyl-cis-4,5-tetramethylene-1,3-dioxenium tetrafluoroborate. Chem. Comm 1965, 202–203. [Google Scholar]
- (201).Wilen SH; Delguzzo L; Saferstein R Experimental evidence for AcO-7 neighboring group participation. Tetrahedron 1987, 43, 5089–5094. [Google Scholar]
- (202).Han A; Tao Y; Reisman SEA 16-step synthesis of the isoryanodane diterpene (+)-perseanol Nature 2019, 573, 563–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (203).Kim J-H; Yang H; Park J; Boons G-J A general strategy for stereoselective glycosylations. J. Am. Chem. Soc 2005, 127, 12090–12097. [DOI] [PubMed] [Google Scholar]
- (204).Fang T; Gu Y; Huang W; Boons G-J Mechanism of glycosylation of anomeric sulfonium ions. J. Am. Chem. Soc 2016, 138, 3002–3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (205).Cox DJ; Fairbanks AJ Stereoselective synthesis of α-glucosides by neighbouring group participation via an intermediate thiophenium ion. Tetrahedron: Asymm. 2009, 20, 773–780. [Google Scholar]
- (206).Cox DJ; Singh GP; Watson AJA; Fairbanks AJ Neighbouring group participation during glycosylation: Do 2-substituted ethyl ethers participate? Eur. J. Org. Chem 2014, 4624–4642. [Google Scholar]
- (207).Singh GP; Watson AJA; Fairbanks AJ A chiral 2‑hydroxy protecting group for the stereocontrolled synthesis of 1,2-cis-α-glycosides by six-ring neighboring group participation. Org. Lett 2015, 17, 4376–4379. [DOI] [PubMed] [Google Scholar]
- (208).Watson AJA; Alexander SR; Cox DJ; Fairbanks AJ Protecting group dependence of stereochemical outcome of glycosylation of 2-O-(thiophen-2-yl)methyl ether protected glycosyl donors. Eur. J. Org. Chem 2016, 1520–1532. [Google Scholar]
- (209).Smoot JT; Pornsuriyasak P; Demchenko AV Development of an arming participating group for stereoselective glycosylation and chemoselective oligosaccharide synthesis. Angew. Chem. Int. Ed 2005, 44, 7123–7126. [DOI] [PubMed] [Google Scholar]
- (210).Smoot JT; Demchenko AV How the arming participating moieties can broaden the scope of chemoselective oligosaccharide synthesis by allowing the inverse armed-disarmed approach. J. Org. Chem 2008, 73, 8838–8850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (211).Fascione MA; Adshead SJ; Stalford SA; Kilner CA; Leach AG; Turnbull WB Stereoselective glycosylation using oxathiane glycosyl donors. Chem. Commun 2009, 5841–5843. [DOI] [PubMed] [Google Scholar]
- (212).Stalford SA; Kilner CA; Leach AG; Turnbull WB Neighbouring group participation vs. Addition to oxacarbenium ions: Studies on the synthesis of mycobacterial oligosaccharides. Org. Biomol. Chem 2009, 7, 4842–4852. [DOI] [PubMed] [Google Scholar]
- (213).Fascione MA; Kilner CA; Leach AG; Turnbull WB Do glycosyl sulfonium ions engage in neighbouring-group participation? A study of oxathiane glycosyl donors and the basis for their stereoselectivity. Chem. Eur. J 2012, 18, 321–333. [DOI] [PubMed] [Google Scholar]
- (214).Fascione MA; Webb NJ; Kilner CA; Warriner SL; Turnbull WB Stereoselective glycosylations using oxathiane spiroketal glycosyl donors. Carbohydr. Res 2012, 348, 6–13. [DOI] [PubMed] [Google Scholar]
- (215).Buda S; Gołebiowska P; Mlynarski J Application of the 2-nitrobenzyl group in glycosylation reactions: A valuable example of an arming participating group. Eur. J. Org. Chem 2013, 3988–3991. [Google Scholar]
- (216).Buda S; Nawój M; Gołębiowska P; Dyduch K; Michalak A; Mlynarski J Application of 2‑substituted benzyl groups in stereoselective glycosylation. J. Org. Chem 2015, 80, 770–780. [DOI] [PubMed] [Google Scholar]
- (217).Hoang KLM; Liu X-W The intriguing dual-directing effect of 2-cyanobenzyl ether for a highly stereospecific glycosylation reaction. Nat. Commun 2014, DOI: 10.1038/ncomms6051 10.1038/ncomms6051, 5:5051s. [DOI] [PubMed] [Google Scholar]
- (218).Chao C-S; Lin C-Y; Mulani S; Hung W-C; Mong K-KT Neighboring-group participation by C-2 ether functions in glycosylations directed by nitrile solvents Chem. Eur. J 2011, 17, 12193–11220. [DOI] [PubMed] [Google Scholar]
- (219).Hanashima S; Akai S; Sato K -i. Thioester-assisted α-sialylation reaction. Tetrahedron Lett. 2008, 49, 5111–5114. [Google Scholar]
- (220).Mensink RA; Boltje TJ Advances in stereoselective 1,2-cis glycosylation using C-2 auxiliaries. Chem. Eur. J 2017, 23, 17638–17653. [DOI] [PubMed] [Google Scholar]
- (221).Ayala L; Lucero CG; Romero JAC; Tabacco SA; Woerpel KA Stereochemistry of nucleophilic substitution reactions depending on substituent: Evidence for electrostatic stabilization of pseudoaxial conformers of oxocarbenium ions by heteroatom substituents. J. Am. Chem. Soc 2003, 125, 15521–15528. [DOI] [PubMed] [Google Scholar]
- (222).Beaver MG; Billings SB; Woerpel KA Nucleophilic substitution reactions of 2-phenylthio-substituted carbohydrate acetals and related systems: Episulfonium ions vs. Oxocarbenium ions as reactive intermediates. Eur. J. Org. Chem 2008, 771–781. [Google Scholar]
- (223).Billings SB; Woerpel KA Nucleophilic substitution of sulfur-substituted cyclohexanone acetals: An analysis of the factors controlling stereoselectivity. J. Org. Chem 2006, 71, 5171–5178. [DOI] [PubMed] [Google Scholar]
- (224).Garcia A; Otte DAL; Salamant WA; Sanzone JR; Woerpel KA Acceleration of acetal hydrolysis by remote alkoxy groups: Evidence for electrostatic effects on the formation of oxocarbenium ions. Angew. Chem. Int. Ed 2015, 54, 3061–3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (225).Garcia A; Otte DAL; Salamant WA; Sanzone JR; Woerpel KA Influence of alkoxy groups on rates of acetal hydrolysis and tosylate solvolysis: Electrostatic stabilization of developing oxocarbenium ion intermediates and neighboring-group participation to form oxonium ions. J. Org. Chem 2015, 80, 4470–4480. [DOI] [PubMed] [Google Scholar]
- (226).Garcia A; Sanzone JR; Woerpel KA Participation of alkoxy groups in reactions of acetals: Violation of the reactivity/selectivity principle in a Curtin–Hammett kinetic scenario. Angew. Chem. Int. Ed 2015, 54, 12087–12090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (227).Mensink RA; Elferink H; White PB; Pers N; Rutjes FPJT; Boltje TJ A study on stereoselective glycosylations via sulfonium ion intermediates. Eur. J. Org. Chem 2016, 4656–4667. [Google Scholar]
- (228).Elferink H; Mensink RA; White PB; Boltje TJ Stereoselective β-mannosylation by neighboring-group participation. Angew. Chem. Int. Ed 2016, 55, 11217–11220. [DOI] [PubMed] [Google Scholar]
- (229).Moons SJ; Mensink RA; Bruekers JPJ; Vercammen MLA; Jansen LM; J., B. T. α‑selective glycosylation with β‑glycosyl sulfonium ions prepared via intramolecular alkylation. J. Org. Chem 2019, 84, 4486–4500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (230).Karak M; Joh Y; Suenaga M; Oishi T; Torikai K 1,2-trans Glycosylation via neighboring group participation of 2‑O‑alkoxymethyl groups: Application to one-pot oligosaccharide synthesis. Org. Lett 2019, 21, 1221–1225. [DOI] [PubMed] [Google Scholar]
- (231).Chayajarus K; Chambers DJ; Chughtai MJ; Fairbanks AJ Stereospecific synthesis of 1,2-cis-glycosides by vinyl-mediated IADs. Org. Lett 2004, 6, 3797–3800. [DOI] [PubMed] [Google Scholar]
- (232).Barresi F; Hindsgaul O In Modern methods in carbohydrate synthesis; Khan SH;O’Neill RA, Eds.; Harwood Academic Publishers: Amsterdam, 1996. [Google Scholar]
- (233).Jung KH; Muller M; Schmidt RR Intramolecular O-glycoside bond formation. Chem. Rev 2000, 100, 4423–4442. [DOI] [PubMed] [Google Scholar]
- (234).Ishiwata A; Lee YJ; Ito Y Recent advances in stereoselective glycosylation through intramolecular aglycon delivery. Org. Biomol. Chem 2010, 8, 3596–3608. [DOI] [PubMed] [Google Scholar]
- (235).Guo L; Plietker B β-ketoesters as mono- or bisnucleophiles: A concise enantioselective total synthesis of (−)-englerin A and B. Angew. Chem. Int. Ed 2019, 58, 8346–8350. [DOI] [PubMed] [Google Scholar]
- (236).Eradi P; Ghosh S; Andreana PR Total synthesis of zwitterionic tetrasaccharide repeating unit from bacteroides fragilis ATCC 25285/NCTC 9343 capsular polysaccharide ps a1 with alternating charges on adjacent monosaccharides. Org. Lett 2018, 20, 4526–4530. [DOI] [PubMed] [Google Scholar]
- (237).Elferink H; Pedersen CM L-Rhamnosylation: The solvent is the solution. Eur. J. Org. Chem 2017, 53–59. [Google Scholar]
- (238).Wang D-P; Zhuge W; Guo Z; Gu G Synthesis of a disaccharide repeating unit of the O-antigen from burkholderia ambifaria and its oligomers. Carbohydr. Res 2017, 442, 41–51. [DOI] [PubMed] [Google Scholar]
- (239).Huang W; Zhou Y-Y; Pan X-L; Zhou X-Y; Lei J-C; Liu D-M; Chu Y; Yang JS Stereodirecting effect of C5-carboxylate substituents on the glycosylation stereochemistry of 3‑deoxy‑D-manno-oct-2-ulosonic acid (KDO) thioglycoside donors: Stereoselective synthesis of α- and β‑KDO glycosides. J. Am. Chem. Soc 2018, 140, 3574–3582. [DOI] [PubMed] [Google Scholar]
- (240).Zhang Y; Zhang H; Zhao Y; Guo Z; Gao J Efficient strategy for α‑selective glycosidation of D‑glucosamine and its application to the synthesis of a bacterial capsular polysaccharide repeating unit containing multiple α‑linked GlcNAc residues. Org. Lett 2020, 22, 1520–1524. [DOI] [PubMed] [Google Scholar]
- (241).Hayashi T; Kehr G; Bergander K; Gilmour R Stereospecific α-sialylation by site-selective fluorination. Angew. Chem. Int. Ed 2019, 58, 3814–3818. [DOI] [PubMed] [Google Scholar]
- (242).Danishefsky SJ; Gervay J; Peterson JM; McDonald FE; Koseki K; Griffith DA; Oriyama T; Marsden SP Application of glycals to the synthesis of oligosaccharides: Convergent total syntheses of the Lewis x trisaccharide sialyl Lewis x antigenic determinant and higher congeners J. Am. Chem. Soc 1995, 117, 1940–1953. [Google Scholar]
- (243).Saha J; Peczuh MW Synthesis and properties of septanose carbohydrates Adv. Carbohydr. Chem. Biochem 2011, 66, 121–186. [DOI] [PubMed] [Google Scholar]
- (244).Vannam R; Peczuh MW How to homologate your sugar: Synthetic approaches to septanosyl containing carbohydrates. Eur. J. Org. Chem 2016, 1800–1812. [Google Scholar]
- (245).Dey S; Jayaraman N Analysis of the conformations of septanoside sugars. Pure Appl. Chem 2014, 86, 1401–1419. [Google Scholar]
- (246).Tsai TYR; Jin H; Wiesner K A stereoselective synthesis of digitoxin. Can. J. Chem 1984, 62, 1403–1405. [Google Scholar]
- (247).Wiesner K; Tsai T. y. R.; Jin H On cardioactive steroids. Stereoslective β-glycosylation of digitoxose: The synthesis of digitoxin. Helv. Chim. Acta 1985, 68, 300–314. [Google Scholar]
- (248).Chiba S; Kitamura M; Narasaka K Synthesis of (−)-sordarin. J. Am. Chem. Soc 2006, 128, 6931–6937. [DOI] [PubMed] [Google Scholar]
- (249).Douglas NL; Ley SV; Lucking U; Warriner SL Tuning glycoside reactivity: New tool for efficient oligosaccharide synthesis. J. Chem. Soc., Perkin Trans 1 1998, 51–65. [Google Scholar]
- (250).van Boeckel CAA; Beetz T; van Aelst SF Substituent effects on carbohydrate coupling reactions promoted by insoluble silver salts. Tetrahedron 1984, 40, 4097–4107. [Google Scholar]
- (251).Paulsen H; Richter A; Sinnwell V; Stenzel W Darstellung selektiv blockierter 2-azido-2-desoxy-D-gluco und galactopyranosylhalogenide: Reaktivitat und 13C-nmr-spektren. Carbohydr. Res 1978, 64, 339–362. [Google Scholar]
- (252).Kim KS; Suk D-H Effect of electron-withdrawing protecting groups at remote positions of donors on glycosylation stereochemistry. Top. Curr. Chem 2011, 301, 109–140. [DOI] [PubMed] [Google Scholar]
- (253).Lemieux RU; Hendriks KB; Stick RV; James K Halide ion catalyzed glycosidation reactions. Synthesis of α-linked disaccharides. J. Am. Chem. Soc 1975, 97, 4056–4062. [Google Scholar]
- (254).Smid P; de Ruiter GA; van der Marel G; van Boom JH Iodonium-ion assisted stereospecific glycosylation: Synthesis of oligosaccharides containing α(1–4)-linked L-fucopyranosyl units. J. Carbohydr. Chem 1991, 10, 833–849. [Google Scholar]
- (255).Demchenko AV; Rousson E; Boons G-J Stereoselective 1,2-cis-galactosylation assisted by remote neighboring group participation and solvent effects. Tetrahedron Lett. 1999, 40, 6523–6536. [Google Scholar]
- (256).Miljkovic M; Yeagley D; Deslongchamps P; Dory YL Experimental and theoretical evidence of through-space electrostatic stabilization of the incipient oxocarbenium ion by an axially oriented electronegative substituent during glycopyranoside acetolysis. J. Org. Chem 1997, 62, 7597–7604. [Google Scholar]
- (257).Ishiwata A; Ohta S; Ito Y A stereoselective 1,2-cis-glycosylation toward the synthesis of a novel N-linked glycan from the Gram-negative bacterium, Campylobacter jejuni. Carbohydr. Res 2006, 341, 1557–1573. [DOI] [PubMed] [Google Scholar]
- (258).Hansch C; Leo A; Taft RW A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev 1991, 91, 165–195. [Google Scholar]
- (259).Jensen HH; Nordstrøm LU; Bols M The disarming effect of the 4,6-acetal group on glycoside reactivity: Torsional or electronic. J. Am. Chem. Soc 2004, 126, 9205–9213. [DOI] [PubMed] [Google Scholar]
- (260).Moumé-Pymbock M; Furukawa T; Mondal S; Crich D Probing the influence of a 4,6-O-acetal on the reactivity of galactopyranosyl donors: Verification of the disarming influence of the trans-gauche conformation of C5-C6 bonds. J. Am. Chem. Soc 2013, 135, 14249–14255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (261).Kancharla PK; Crich D Influence of side chain conformation and configuration on glycosyl donor reactivity and selectivity as illustrated by sialic acid donors epimeric at the 7-position. J. Am. Chem. Soc 2013, 135, 18999–19007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (262).Dhakal B; Buda S; Crich D Stereoselective synthesis of 5-epi-α-sialosides related to the pseudaminic acid glycosides. Reassessment of the stereoselectivity of the 5-azido-5-deacetamidosialyl thioglycosides and use of triflate as nucleophile in the Zbiral deamination of sialic acids. J. Org. Chem 2016, 81, 10617–10630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (263).Dhakal B; Crich D Synthesis and stereocontrolled equatorially selective glycosylation reactions of a pseudaminic acid donor: Importance of the side chain conformation, and regioselective reduction of azide protecting groups. J. Am. Chem. Soc 2018, 140, 15008–15015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (264).Dharuman S; Amarasekara H; Crich D Interplay of protecting groups and side chain conformation in glycopyranosides. Modulation of the influence of remote substituents on glycosylation? J. Org. Chem 2018, 83, 10334–10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (265).Ustyuzhanina N; Komarova B; Zlotina N; Krylov V; Gerbst A; Tsvetkov Y; Nifantiev N Stereoselective α-glycosylation with 3-O-acetylated D-gluco donors. Synlett 2006, 921–923. [Google Scholar]
- (266).Komarova BS; Orekhova MV; Tsvetkov YE; Nifantiev NE Is an acyl group at O-3 in glucosyl donors able to control α-selectivity of glycosylation? The role of conformational mobility and the protecting group at O-6. Carbohydr. Res 2014, 384, 70–86. [DOI] [PubMed] [Google Scholar]
- (267).Nielsen MM; Pedersen CM Catalytic glycosylations in oligosaccharide synthesis. Chem. Rev 2018, 118, 8285–8358. [DOI] [PubMed] [Google Scholar]
- (268).Crich D Mechanism of a chemical glycosylation. Acc. Chem. Res 2010, 43, 1144–1153. [DOI] [PubMed] [Google Scholar]
- (269).Aubry S; Sasaki K; Sharma I; Crich D Influence of protecting groups on the reactivity and selectivity of glycosylation: Chemistry of the 4,6-O-benzylidene protected mannopyranosyl donors and related species. Top. Curr. Chem 2011, 301, 141–188. [DOI] [PubMed] [Google Scholar]
- (270).Bousquet E; Khitri M; Lay L; Nicotra F; Panza L; Russo G Capsular polysaccharide of streptcoccus pneumoniae type 19F: Synthesis of the repeating unit. Carbohydr. Res 1998, 311, 171–181. [DOI] [PubMed] [Google Scholar]
- (271).Crich D; Yao Q The benzylidene acetal fragmentation route to 6-deoxy sugars: Direct reductive cleavage in the presence of ether protecting groups permitting the efficient, highly stereocontrolled synthesis of ß-D-rhamnosides from D-mannosyl glycosyl donors. Total synthesis of α-D-Gal-(1–3)-α-D-Rha-(1–3)-ß-D-Rha-(1–4)-ß-D-Glu-OMe, the repeating unit of the antigenic lipopolysaccharide from Escherichia hermannii ATCC 33650 and 33652. J. Am. Chem. Soc 2004, 126, 8232–8236. [DOI] [PubMed] [Google Scholar]
- (272).Bhunia A; Vivekanandan S; Eckert T; Burg-Roderfeld M; Wechselberger R; Romanuka J; Bachle D; Kornilov AV; von der Lieth C-W; Jiménez-Barbero J et al. Why structurally different cyclic peptides can be glycomimetics of the HNK-1 carbohydrate antigen. J. Am. Chem. Soc 2010, 132, 96–105. [DOI] [PubMed] [Google Scholar]
- (273).Whitfield DM; Guo J Proton transfer and hydrogen bonding in glycosylation reactions. J. Carbohydr. Chem 2017, 36, 59–99. [Google Scholar]
- (274).Nukada T; Berces A; Whitfield DM Acyl transfer as a problematic side reaction in polymer-supported oligosaccharide synthesis. J. Org. Chem 1999, 64, 9030–9045. [Google Scholar]
- (275).Ziegler T; Kovác P; Glaudemans CPJ Transesterification during glycosylation promoted by silver trifluoromethanesulfonate. Liebigs Ann. Chem 1990, 613–615. [Google Scholar]
- (276).Bochkov AF; Betaneli VI; Kochetkov NK Sugar orthhtoesters X. Structure of byproducts during glycosylation by orthoesters. Izv. Akad. Nauk SSSR. Ser. Khim 1974, 6, 1299–1306. [Google Scholar]
- (277).Schroeder LR; Hultman DP; Johnson DC Acid-catalyzed hydrolyses of 3,4,6-tri-O-methyl-1,2-O-(1-alkoxyethylidene)-α-D-glucopyranoses. J. Chem. Soc., Perkin Trans 2 1972, 1063–1071. [Google Scholar]
- (278).Bochkov AF; Betaneli VI; Kochetkov NK Sugar orthoesters XV. Dependence on the conditions and mechanims of the proton-catalyzed reactions of substituted α-D-glucopyranose 1,2-(alkyl orthoacetate)s in media of low polarity. Bioorg. Khim 1976, 2, 671. [Google Scholar]
- (279).Yoshimoto K; Tsuda Y General path of O-acyl migration in d-glucose derivatives: Acyl migration of methyl mono-O-myristoyl-α- and β-D-glucopyranosides and mono-O-myristoyl-D-glucopyranoses. Chem. Pharm. Bull 1983, 31, 4324–4334. [Google Scholar]
- (280).Yoshimoto Y; Tsuda K On the possibility of direct O-1-β→−6 acyl migration in 1-O-acyl-β-D-glucose derivatives. Chem. Pharm. Bull 1983, 31, 4335–4340. [Google Scholar]
- (281).Angyal SJ; Melrose GJH Cyclitols. Part XVIII. Acetyl migration: Equilibrium between axial and equatorial acetates. J. Chem. Soc 1965, 6494–6500. [Google Scholar]
- (282).Ekholm FS; Ardá A; Eklund P; André S; Gabius HJ; Jiménez-Barbero J; Leino R Studies related to Norway spruce galactoglucomannans: Chemical synthesis, conformation analysis, nmr spectroscopic characterization, and molecular recognition of model compounds. Chem. Eur. J 2012, 18, 14392–14405. [DOI] [PubMed] [Google Scholar]
- (283).Brecker L; Manut M; Schwarz A; Nidetzky B In situ proton nmr study of acetyl and formyl group migration in mono-O-acyl D-glucose. Magn. Reson. Chem 2009, 47, 328–332. [DOI] [PubMed] [Google Scholar]
- (284).Roslund MU; Aitio O; Wärnå J; Maaheimo H; Murzin DY; Leino R Acyl group migration and cleavage in selectively protected β-D-galactopyranosides as studied by nmr spectroscopy and kinetic calculations. J. Am. Chem. Soc 2008, 130, 8769–8772. [DOI] [PubMed] [Google Scholar]
- (285).Lassfolk R; Rahkila J; Johansson MP; Ekholm FS; Wärnå J; Leino R Acetyl group migration across the saccharide units in oligomannoside model compound. J. Am. Chem. Soc 2019, 141, 1646–1654. [DOI] [PubMed] [Google Scholar]
- (286).Kamerling JP; Schauer R; Shukla AK; Stoll S; Van Halbeek H; Vliegenthart JFG Migration of O-acetyl groups in N,O-acetylneuraminic acids. Eur. J. Biochem 1987, 162, 601–607. [DOI] [PubMed] [Google Scholar]
- (287).Reinhard B; Faillard H Regioselective acetylations of sialic acid α-ketosides. Liebigs Ann. Chem 1994, 193–203. [Google Scholar]
- (288).Kefurt K; Kefurtová Z; Jarý J; Horáková I; Marek M Enzymic deacetylation of derivatives of 1,2-O-isoproylidene-α--hexofuranoses. Carbohydr. Res 1992, 223, 137–145. [Google Scholar]
- (289).Doerschuk AP Acyl migrations in partially acylated, polyhydroxylic systems. J. Am. Chem. Soc 1952, 74, 4202–4203. [Google Scholar]
- (290).Hasegawa H; Akira K; Shinohara Y; Kasuya Y; Hashimoto T Kinetics of intramolecular acyl migration of 1β-O-acyl glucuronides of (R)- and (S)-2-phenylpropionic acids. Biol. Pharm. Bull 2001, 24, 852–855. [DOI] [PubMed] [Google Scholar]
- (291).Iddon L; Richards SE; Johnson CH; Harding JR; Wilson ID; Nicholson JK; Lindon JC; Stachulski AV Synthesis of a series of phenylacetic acid 1-β-O-acyl glucosides and comparison of their acyl migration and hydrolysis kinetics with the corresponding acyl glucuronides. Org. Biomol. Chem 2011, 9, 926–934. [DOI] [PubMed] [Google Scholar]
- (292).Blanckaert N; Compernolle F; Leroy P; Van Houtte R; Fevery J; Heirwegh KP The fate of bilirubin-IXα glucuronide in cholestasis and during storage in vitro: Intramolecular rearrangement to positional isomers of glucuronic acid. Biochem. J 1978, 171, 203–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (293).Nicholls AW; Akira K; Lindon JC; Duncan Farrant R; Wilson ID; Harding J; Killick DA; Nicholson JK Nmr spectroscopic and theoretical chemistry studies on the internal acyl migration reactions of the 1-O-acyl-β-D-glucopyranuronate conjugates of 2-, 3-, and 4-(trifluoromethyl)benzoic acids. Chem. Res. Toxicol 1996, 9, 1414–1424. [DOI] [PubMed] [Google Scholar]
- (294).Mastihubová M; Biely P Lipase-catalysed preparation of acetates of 4-nitrophenyl β-D-xylopyranoside and their use in kinetic studies of acetyl migration. Carbohydr. Res 2004, 339, 1353–1360. [DOI] [PubMed] [Google Scholar]
- (295).Filice M; Ubiali D; Fernandez-Lafuente R; Fernandez-Lorente G; Guisan JM; Palomo JM; Terreni M A chemo-biocatalytic approach in the synthesis of β-O-naphtylmethyl-N-peracetylated lactosamine. J. Mol. Catal. B Enzym 2008, 52–53, 106–112. [Google Scholar]
- (296).Filice M; Bavaro T; Fernandez-Lafuente R; Pregnolato M; Guisan JM; Palomo JM; Terreni M Chemo-biocatalytic regioselective one-pot synthesis of different deprotected monosaccharides. Catal. Today 2009, 140, 11–18. [Google Scholar]
- (297).Chevallier OP; Migaud ME Investigation of acetyl migrations in furanosides. Beilstein J. Org. Chem 2006, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (298).Rangelov MA; Vayssilov GN; Petkov DD Quantum chemical model study of the acyl migration in 2’(3’)-formylnucleosides. Int. J. Quantum Chem 2006, 103, 1346–1356. [Google Scholar]
- (299).Horrobin T; Tran CH; Crout D Regioselective 6-deacylation of hexopyranose per-acetates, acid-catalysed rearrangement to the 4-deprotected products and conversions of these into hexose 4- and 6-sulfates. J.Chem. Soc., Perkin Trans 1 1998, 1069–1080. [Google Scholar]
- (300).Petrović V; Tomić S; Matanović M Synthesis and intramolecular transesterifications of pivaloylated methyl α-D-galactopyranosides. Carbohydr. Res 2002, 337, 863–867. [DOI] [PubMed] [Google Scholar]
- (301).Martin RB; Hedrick RI Intramolecular S-O and S-N acetyl transfer reactions. J. Am. Chem. Soc 1962, 84, 106–110. [Google Scholar]
- (302).Oesterling TO; Metzler CM Acidic and alkaline isomerization and hydrolysis of lincomycin monoesters. Carbohydr. Res 1970, 15, 285–290. [DOI] [PubMed] [Google Scholar]
- (303).Deng S; Chang CWT Cesium trifluoroacetate or silver oxide mediated acyl migration for the construction of disaccharide building blocks. Synlett 2006, 756–760. [Google Scholar]
- (304).Casinovi CG; Framondino M; Randazzo G; Slani F Acetyl migration in the monoacetates of methyl 6-O-trityl-α-D-glucopyranoside. Carbohydr. Res 1974, 36, 67–73. [Google Scholar]
- (305).Mortensen RW; Sidelmann UG; Tjørnelund J; Hansen SH Stereospecific pH-dependent degradation kinetics of R- and S-naproxen-β−1-O-acyl-glucuronide. Chirality 2002, 14, 305–312. [DOI] [PubMed] [Google Scholar]
- (306).Illing HPA; Wilson ID Ph dependent formation of β-glucuronidase resistant conjugantes from the biosynthetic ester glucuronide of isoxepac. Biochem. Pharmacol 1981, 30, 3381–3384. [DOI] [PubMed] [Google Scholar]
- (307).Khan S; Teitz DS; Jemal M Kinetic analysis by hplc - electrospray mass spectrometry of the pH-dependent acyl migration and solvolysis as the decomposition pathways of ifetroban 1-O-acyl glucuronide. Anal. Chem 1998, 70, 1622–1628. [Google Scholar]
- (308).Dawson PE; Muir TW; Clark-Lewis I; Kent SBH Synthesis of proteins by native chemical ligation. Science 1994, 266, 776–779. [DOI] [PubMed] [Google Scholar]
- (309).Bradow G; Kan LS; Fenselau C Studies of intramolecular rearrangements of acyl-linked glucuronides using salicylic acid, flufenamic acid, and (S)-and (R)-benoxaprofen and confirmation of isomerization in acyl-linked Δ9–11-carboxytetrahydrocannabinol glucuronide. Chem. Res. Toxicol 1989, 2, 316–324. [DOI] [PubMed] [Google Scholar]
- (310).Dickinson RG; Baker PV; King AR Studies on the reactivity of acyl glucuronides—phenolic glucuronidation of isomeres of difunsial acyl glucuronide in the rat. Biochem. Pharmacol 1991, 42, 2289–2299. [DOI] [PubMed] [Google Scholar]
- (311).Yoshioka T; Baba A Structure-activity relationships for degradation reaction of 1-β-O-acyl glucuronides: Kinetic description and prediction of intrinsic electrophilic reactivity under physiological conditions. Chem. Res. Toxicol 2009, 22, 158–172. [DOI] [PubMed] [Google Scholar]
- (312).Stachulski AV; Harding JR; Lindon JC; Maggs JL; Park BK; Wilson ID Acyl glucuronides: Biological activity, chemical reactivity, and chemical synthesis. J. Med. Chem 2006, 49, 6931–6945. [DOI] [PubMed] [Google Scholar]
- (313).Stachulski AV; Berry NG; Iddon L; Iqbal M; Meng X; Jayapal P; Johnson CH; Nicholson JK; Lindon JC; Harding JR et al. Synthesis, transacylation kinetics and computational chemistry of a set of arylacetic acid 1β-O-acyl glucuronides. Org. Biomol. Chem 2009, 7, 2525–2533. [DOI] [PubMed] [Google Scholar]
- (314).Bonner WA C1-C2 acetyl migration on methylation of the anomeric 1,3,4,6-tetra-O-acetyl-D-glucopyranoses. J. Org. Chem 1959, 24, 1388–1390. [Google Scholar]
- (315).Tejima S; Fletcher HG Syntheses with partially benzylated sugars. II. The anomeric 1-O-benzoyl-L-arabinopyranoses and 1-O-benzoyl-L-arabinofuranoses and their tendencies to undergo acyl migration. J. Org. Chem 1963, 28, 2999–3004. [Google Scholar]
- (316).Johnston GAR Acetyl migration in derivatives of 3β-D-xylosyluracil. Tetrahedron Lett. 1967, 28, 2679–2683. [Google Scholar]
- (317).Tomić S; Ljevaković D; Tomašić J Esterase catalyzed regioselective hydrolyses of acetylated monosaccharides. Tetrahedron 1993, 49, 10977–10986. [Google Scholar]
- (318).Ljevaković D; Tomić S; Tomašić J Syntheses of partially pivaloylated D-glucopyranoses: New substrates for the esterase from rabbit serum. Carbohydr. Res 1992, 230, 107–115. [Google Scholar]
- (319).Ren B; Wang M; Liu J; Ge J; Dong H Enhanced basicity of Ag2O by coordination to soft anions. ChemCatChem 2015, 7, 761–765. [Google Scholar]
- (320).Albert R; Dax K; Stütz AE; Weidmann H Acetyl migration in partially acetylated D-glucopyranosides and acylamidohexopyranosides. J. Carbohydr. Chem 1983, 2, 279–292. [Google Scholar]
- (321).Hoffman M; Jia Z; Peña MJ; Cash M; Harper A; Blackburn AR; Darvill A; York WS Structural analysis of xyloglucans in the primary cell walls of plants in the subclass asteridae. Carbohydr. Res 2005, 340, 1826–1840. [DOI] [PubMed] [Google Scholar]
- (322).Teleman A; Lundqvist J; Tjerneld F; Stålbrand H; Dahlman O Characterization of acetylated 4-O-methylglucuronoxylan isolated from aspen employing 1H and 13C NMR spectroscopy. Carbohydr. Res 2000, 329, 807–815. [DOI] [PubMed] [Google Scholar]
- (323).Jia Z; Cash M; Darvill AG; York WS NMR characterization of endogenously O-acetylated oligosaccharides isolated from tomato (Lycopersicon esculentum) xyloglucan. Carbohydr. Res 2005, 340, 1818–1825. [DOI] [PubMed] [Google Scholar]
- (324).Xing X; Cui SW; Nie S; Phillips GO; Goff HD; Wang Q Study on dendrobium officinale O-acetyl-glucomannan (dendronan®): Part i. Extraction, purification, and partial structural characterization. Bioact. Carbohydrates Diet. Fibre 2014, 4, 74–83. [Google Scholar]
- (325).Willför S; Sjöholm R; Laine C; Roslund M; Hemming J; Holmbom B Characterisation of water - soluble galactoglucomannans from Norway spruce wood and thermomechanical pulp. Carbohydr. Polym 2003, 52, 175–187. [Google Scholar]
- (326).Nunes FM; Domingues MR; Coimbra MA Arabinosyl and glucosyl residues as structural features of acetylated galactomannans from green and roasted coffee infusions. Carbohydr. Res 2005, 340, 1689–1698. [DOI] [PubMed] [Google Scholar]
- (327).Deng S; Gangadharmath U; Chang C-WT Sonochemistry: A powerful way of enhancing the efficiency of carbohydrate synthesis. J. Org. Chem 2006, 71, 5179–5185. [DOI] [PubMed] [Google Scholar]
- (328).Kanaya A; Takashima Y; Harada A Double-threaded dimer and supramolecular oligomer formed by stilbene modified cyclodextrin: Effect of acyl migration and photostimuli. J. Org. Chem 2011, 76, 492–499. [DOI] [PubMed] [Google Scholar]
- (329).Rej RN; Glushka JN; Chew W; Perlin AS Chromic acid oxidation in the synthesis of uronic acids. Use of the O-levulinoyl group to minimize acyl migration. Carbohydr. Res 1989, 189, 135–148. [Google Scholar]
- (330).Dvorakova M; Pribylova M; Pohl R; Migaud ME; Vanek T Alkyloxycarbonyl group migration in furanosides. Tetrahedron 2012, 68, 6701–6711. [Google Scholar]
- (331).Chung S-K; Chang Y-T Base-catalysed acyl migrations in myo-lnositol dibenzoates. Chem. Commun 1995, 13–14. [Google Scholar]
- (332).Reicher F; Corrêa JBC; Gorin PAJ Location of O-acetyl groups in the acidic D-xylan of Mimosa scabrella (bracatinga). A study of O-acetyl group migration. Carbohydr. Res 1984, 135, 129–140. [Google Scholar]
- (333).Li W; Du W; Li Q; Sun T; Liu D Study on acyl migration kinetics of partial glycerides: Dependence on temperature and water activity. J. Mol. Catal. B Enzym 2010, 63, 17–22. [Google Scholar]
- (334).Ahn Y-H; Chang Y-T Molecular evolution on chiro-inositol dibenzoate using intramolecular acyl migration and selection by phenyl boronic acid. J. Comb. Chem 2004, 6, 293–296. [DOI] [PubMed] [Google Scholar]
- (335).Ahn Y-H; Chang Y-T Molecular evolution using intramolecular acyl migration on myo-inositol benzoates with thermodynamic and kinetic selectors. Chem. Eur. J 2004, 10, 3543–3547. [DOI] [PubMed] [Google Scholar]