

Abstract
Relevance
Lung cancer is the most common malignant tumor with high morbidity (11.6% of the total diagnosed cancer cases) and mortality (18.4% of the total cancer deaths), and its 5-year survival rate is very low (20%). Clarification of any molecular events and the discovery of effective biomarkers will offer increasing promise for lung canner management. N6-methyladenosine (m6A) modification is one of the important RNA modifications that are closely associated with lung cancer, and are tightly regulated by m6A regulators. Elucidation of pathology-specific m6A regulators will directly contribute to lung cancer medical services in the context of predictive, preventive, and personalized medicine (PPPM).
Purpose
To investigate pathology-specific regulators of m6A RNA modifications in lung cancer and further inspect the m6A regulator gene signature as useful tools for PPPM in lung cancers.
Methods
The gene expression data of 19 m6A regulators (m6A-methyltransferases—ZC3H13, KIAA1429, RBM15/15B, WTAP, and METTL3/14; demethylases—FTO and ALKBH5; and m6A-binding proteins—HNRNPC, YTHDF1/2/3, YTHDC1/2, IGF2BP1/2/3, and HNRNPA2B1) and clinical data of 1013 lung cancer patients [511 lung adenocarcinoma (LUAD) and 502 lung squamous carcinoma (LUSC)] and 109 controls (Con) were obtained from the TCGA database. Quantitative real-time PCR (qRT-PCR) was used to verify m6A regulators in lung cancer cell lines. Protein–protein interaction (PPI), gene co-expression, survival analysis, and heatmap were used to analyze these m6A regulators in this set of lung cancer clinical data. Lasso regression was used to optimize the pathology-specific m6A regulator gene signature. Gene set enrichment analysis (GSEA) was used to reveal the functional characteristics of m6A regulators.
Results
Those 19 m6A regulator profiling was significantly differentially expressed in lung cancer tissues relative to control tissues, which was also verified in lung cancer cell lines. Those m6A regulators interacted mutually, and those regulator-based sample clusters were correlated with clinical traits, including survival status, gender, tobacco smoking history, primary disease, and pathologic stage. Further, lasso regression based on the 19 m6A regulators optimized and identified a three-m6A-regulator signature (KIAA1429, METTL3, and IGF2BP1) as independent prognostic factor, which classified 1013 lung cancer patients into high-risk and low-risk groups according to median value (0.84) of the lasso regression risk scores. This three-m6A-regulator signature profiling was significantly related to lung cancer overall survival, cancer status, and the above-described clinical traits. Further, GSEA revealed that KIAA1429, METTL3, and IGF2BP1 were significantly related to multiple biological behaviors, including proliferation, apoptosis, metastasis, energy metabolism, drug resistance, and recurrence, and that KIAA1429 and IGF2BP1 had potential target genes, including E2F3, WTAP, CCND1, CDK4, EGR2, YBX1, and TLX, which were associated with cancers.
Conclusion
This study provided the first view of the pathology-specific regulators of m6A RNA modification in lung cancers and identified the three-m6A-regulator signature (KIAA1429, METTL3, and IGF2BP1) as an independent prognostic model to classify lung cancers into high- and low-risk groups for patient stratification, prognostic assessment, and personalized treatment toward PPPM in lung cancers.
Electronic supplementary material
The online version of this article (10.1007/s13167-020-00220-3) contains supplementary material, which is available to authorized users.
Keywords: Lung cancer, m6A regulators, Gene signature, Prognostic model, KIAA1429, METTL3, IGF2BP1, Biomarker, Clinical traits, Predictive preventive personalized medicine (PPPM), Molecular patterns, Patient stratification
Introduction
Unsatisfactory statistics for current lung cancer management
Lung cancer is a common malignant tumor with high morbidity and mortality, which has aroused a wide concern in the world. Currently, lung cancer is the most commonly diagnosed cancer (11.6% of the total cases) and the leading cause of cancer death (18.4% of the total cancer deaths) [1]. Despite progressions in surgery, radiotherapy, and chemotherapy that have been achieved in lung cancer, its survival improvement is still a big challenge, with a low 5-year survival rate (20%) [2]. The main reasons that result in its low 5-year survival rate are as follows: (i) The early symptoms of lung cancer are commonly concealed, which causes most patients diagnosed in middle or advanced stages; thus, only a partial percentage (30%) of patients has a chance to undergo radical surgery in an early stage [3]. (ii) Lymphatic metastasis and distant metastasis are very common after surgery, and about 50% of the patients have recurrence and metastasis within 5 years [4]. (iii) The number of targeted therapy and immunotherapy is very limited although targeted therapy and immunotherapy are very effective and have achieved some progresses, with the discovery of many molecular targets, especially for the treatment of advanced stage of lung cancers, for example, EGFR-TKI drugs (erlotinib, gefitinib, and afatinib) that inhibit tyrosine kinase at the epidermal growth factor receptor and immunotherapy drugs (pembrolizumab, nivolumab, and durvalumab) that target immune checkpoint (PD-1/PD-L1) for the late stage nonsmall cell lung cancer [5]. There is an urgent need to develop more novel molecular targets for lung cancer.
Role of m6A RNA modification and m6A regulators in pathology-specific molecular signature
The epigenetics about the modifications occurring at DNAs and RNAs have been widely studied in tumor pathogenesis [6, 7]. Of them, m6A modification was one of the emerging research frontiers, which modifies different types of RNAs, including transfer RNAs (tRNAs), messenger RNAs (mRNAs), ribosomal RNAs (rRNAs), small nuclear RNA (snRNA), and long noncoding RNAs (lncRNAs), and others, and which is the most abundant posttranscriptional modification in eukaryotes [8]. A study has found that m6A modification is present in more than 7600 mRNAs and in more than 300 noncoding RNAs (ncRNAs) [9]. m6A modification can change RNA processing such as splicing, export, intracellular distribution stability, and translation [10]. The cellular m6A status is tightly regulated by a series of regulators, including m6A methyltransferases (named as “writers”), demethylases (named as “erasers”), and m6A-binding proteins (named as “readers”) [11]. The methyltransferase complex contains ZC3H13, KIAA1429, RBM15, RBM15B, WTAP, METTL3, and METTL14, which catalyzes m6A methylation [12]. Demethylases include FTO and ALKBH5, which catalyze oxidative demethylation of m6A [13]. The m6A-binding proteins include HNRNPC, YTHDF1, YTHDF2, YTHDF3, YTHDC1, YTHDC2, IGF2BP1, IGF2BP2, IGF2BP3, and HNRNPA2B1 [14]. The current technical methods used to detect m6A modification included high-throughput sequencing [15], colorimetric method, and liquid chromatography–mass spectrometry (LC-MS) [16]. Common methods included MeRIP-seq, miCLIP-seq, LC-MS/MS, and colorimetric method [17]. The multiple functions of m6A modification are associated with the occurrence and worsening of numerous human diseases, including cancer [18]. For example, the m6A eraser FTO promotes the growth of lung cancer cells by regulating the m6A level of ubiquitin-specific protease 7 (USP7) mRNAs, and a study has found that the overexpressed USP7 mRNA level is positively correlated with FTO mRNA level in human lung cancer tissues, and that FTO decreases the m6A level and increases mRNA stability of USP7, which was dependent on the demethylase activity of FTO [19]. The m6A reader IGF2BP1 promotes SRF-dependent transcription in cancer by N6-m6A-dependent manner in cancer, which results in the increased SRF-dependent transcriptional activity and enhances tumor cell proliferation and metastasis [20]. The m6A writer METTL3 promotes tumor proliferation by accelerating pri-miR221/222 maturation in the m6A-dependent manner [21]. Further, a study found that m6A regulators take part in m6A modification of many ncRNAs, such as microRNAs, lncRNAs, and circular RNAs. A review reports that m6A is closely associated with numerous cancers, including lung cancer, prostate cancer, stomach cancer, breast cancer, pancreatic cancer, kidney cancer, colorectal carcinoma, mesothelioma, sarcoma, and leukemia [22]. A study has found that these m6A regulators are the potential biomarkers for prognostic prediction and new therapeutic targets in cancer [23]; for example, the m6A regulator METTL3 is found to associate with mitogen-activated protein kinase cascades, ubiquitin-dependent process, RNA splicing, and regulation of cellular process, which suggests that METTL3 is a potential target for enhancing therapeutic efficacy in pancreatic cancer [24]. Indeed, the findings that m6A regulators are dysregulated and play important roles in various types of cancer suggest that m6A RNA modification could be involved in therapy resistance development [25]. Inhibitors of m6A regulators hold the potential to be combined with chemotherapy or radiotherapy to erase cancer stem cells and achieve complete remission [26]. Such data suggest that m6A RNA modification signatures may serve as predictive markers for personalized cancer treatment and shed light on how to overcome therapy resistance in cancer. The m6A writer KIAA1429 is revealed as an oncogenic factor in breast cancer and hepatocellular carcinoma, but has not been reported in lung cancer [27, 28]. The m6A writer METTL3 shows low expression in lung adenocarcinomas (LUAD) and squamous cell carcinoma (LUSC), when compared to normal lung epithelia, in the TCGA and GTEx data (p < 0.0001) [29]. The m6A reader IGF2BP1 is significantly increased in LUAD tissues (n = 188) compared to adjacent noncancerous tissues (n = 188), and a high expression of IGF2BP1 is correlated with a poor prognosis for lung cancer patients [30]. While m6A plays important roles in different types of cancers, the clinical trait–related m6A regulator study in lung cancer is insufficient. It emphasizes the important scientific merits of m6A regulators in cancer pathogenesis and the scientific necessity of in-depth insights into the relationship of m6A regulators and lung cancers.
Working hypotheses
We hypothesize that the m6A regulator profiling is significantly associated with pathological characteristics of lung cancer, and the optimized m6A regulator signature has benefits for patient stratification, prognostic prediction, and personalized management of lung cancer.
Study design
The mRNA expressions of 19 m6A regulators described above and the complete clinical data were obtained from 1013 human lung cancer tissues (511 LUADs and 502 LUSCs; these tissues were obtained from thoracic surgery of white American or African-American patients with lung cancer) and 109 normal control lung tissues (these tissues were postmortem tissues from donors who were healthy white American or African-American before death) from the TCGA database, and 19 m6A regulators were verified with qRT-PCR in 2 LUAD cell lines and 2 LUSC cell lines relative to the normal human bronchial epithelial cell line (HBE) cells. Protein–protein interaction (PPI), gene co-expression, survival analysis, and headmap were used to analyze the relationship of these m6A regulators and lung cancer clinical data. Lasso regression was used to optimize pathology-specific m6A regulator gene signature. Gene set enrichment analysis (GSEA) was used to reveal the functional characteristics of m6A regulators.
Expected impacts in the context of predictive, preventive, and personalized medicine
We expect the lasso regression-optimized pathology-specific m6A regulator gene signature will be helpful for predictive diagnosis or disease prognosis, stratifying patients, and guiding clinical therapy for personalized treatment of lung cancers in the context of predictive, preventive, and personalized medicine (PPPM) [31].
Materials and methods
TCGA data of lung cancer patients and preprocessing
TCGA (http://cancergenome.nih.gov/) was a data sharing platform for large-scale cancer patient information, including RNA expression, protein expression, mutation, RNA splicing, and corresponding clinical data. The mRNA expression data of those 19 m6A regulators and clinical data were obtained from 1013 lung cancer patients (LUAD: n = 511; and LUSC: n = 502) and control patients (n = 109) from lung cancer level 3 RNA-seq V2 transcriptomics data in the TCGA database. Those 19 m6A regulators included 7 writers (ZC3H13, KIAA1429, RBM15, RBM15B, WTAP, METTL3, and METTL14), 2 erasers (FTO and ALKBH5), and 10 m6A-binding proteins (HNRNPC, YTHDF1, YTHDF2, YTHDF3, YTHDC1, YTHDC2, IGF2BP1, IGF2BP2, IGF2BP3, and HNRNPA2B1). Reversible m6A modification on mRNA was shown (Fig. 1). The clinical data included primary disease (LUAD and LUSC), age (from 33 to 90 years), gender (male and female), tobacco smoking history (current reformed smoker with ≤ 15 years’ smoking, current reformed smoker with > 15 years’ smoking, current reformed smoker, and lifelong nonsmoker), pathologic T (T represented tumor size, including T1, T2, T3, T4, and TX), pathologic M (M represented tumor metastasis, including M0, M1, and MX), pathologic N (N represented tumor lymph node metastasis, including N0, N1, N2, and NX), pathologic stage (stages I, II, III, and IV), cancer status (tumor and tumor-free), tumor residual (R0, R1, R2, and RX), and status (alive and dead). PPI was performed with the String network (http://string-db.org/cgi/input.pl). The co-expressions among 19 m6A regulators were analyzed by Spearman correlation coefficient with R package.
Fig. 1.

Reversible m6A modification on RNAs. Methyltransferase complex (writers) contained ZC3H13, KIAA1429, RBM15, RBM15B, WTAP, METTL3, and METTL14. Demethylases (erasers) included FTO and ALKBH5. The m6A-binding proteins (readers) included HNRNPC, YTHDF1, YTHDF2, YTHDF3, YTHDC1, YTHDC2, IGF2BP1, IGF2BP2, IGF2BP3, and HNRNPA2B1. ZC3H13, zinc finger CCCH-type containing 13; KIAA1429, Vir-like m6A methyltransferase associated; RBM15, RNA-binding motif protein 15; RBM15B, RNA-binding motif protein 15B; WTAP, WT1-associated protein; METTL3, mRNA mA methyltransferase; METTL14, methyltransferase-like 14; FTO, FTO alpha-ketoglutarate-dependent dioxygenase; ALKBH5, AlkB homolog 5, RNA demethylase; HNRNPC, heterogeneous nuclear ribonucleoprotein C; YTHDF1, YTH N6-methyladenosine RNA-binding protein 1; YTHDF2, YTH N6-methyladenosine RNA-binding protein 2; YTHDF3, YTH N6-methyladenosine RNA-binding protein 3; YTHDC1, YTH domain containing 1; YTHDC2, YTH domain containing 2; IGF2BP1, insulin-like growth factor 2 mRNA-binding protein 1; IGF2BP2, insulin-like growth factor 2 mRNA-binding protein 2; IGF2BP3, insulin-like growth factor 2 mRNA-binding protein 3; HNRNPA2B1, heterogeneous nuclear ribonucleoprotein A2/B1
Cell lines and cell culture
Two LUAD cell lines (A549 and LTEP-a-2), two LUSC cell lines (H520 and SK-MES-1), and one normal HBE were purchased from the Cell Bank of Chinese Academy of Science (Shanghai, China). A549 was cultured in F-12K medium with 10% fetal bovine serum. LTEP-a-2 and H520 were cultured in RPMI-1640 medium with 10% fetal bovine serum. SK-MES-1 was cultured in Eagle’s minimum essential medium with 10% fetal bovine serum. HBE was cultured in DMEM medium (Corning, NY, USA) supplemented with 10% fetal bovine serum (FBS, GIBCO, South America, NY, USA). All these cells were maintained in 5% CO2 atmosphere at 37 °C.
RNA extraction and quantitative real-time PCR verification
Total RNAs of each cell line (A549, LTEP-a-2, H520, SK-MES-1, and HBE) were extracted with TRIzol reagent (Invitrogen) according to the manufacturer’s instructions. The cells were seeded in 6-well plates at 30–50% density. After the cell density was up to 90%, RNAs were isolated as follows: (i) medium was discarded; (ii) cells were washed with PBS (2×); (iii) a volume (1 ml) of TRIzol reagent and 200 μl chloroform were added in each well for 10 min; (iv) TRIzol reagent was collected on a tube followed by standing on ice for 15 min; (v) samples were centrifuged (15 min, 12,000g, 4 °C); (vi) supernatant was mixed with equal volume of isopropanol followed by standing on ice for 15 min; (vii) samples were centrifuged (15 min, 12,000g, 4 °C); (viii) supernatant was discarded followed by adding a volume (1 ml) of 75% ethyl alcohol; (ix) samples were centrifuged (5 min, 8000g, 4 °C); and (x) supernatant was discarded followed by adding 20 μl RNA enzyme-free water. Total RNAs were reversely transcribed into cDNAs, and then cDNAs were used to perform quantitative real-time PCR (qRT-PCR) with SYBR Premix ExTaq (TaKaRa). GAPDH was used as an internal control for gene quantification. Primer sequences were synthesized by Sangon Biotech (Shanghai, China) for 19 m6A regulators’ mRNAs (RBM15, KIAA1429, ZC3H13, HNRNPC, YTHDC2, METTL14, YTHDC1, WTAP, METTL3, FTO, ALKBH5, YTHDF1, YTHDF2, RBM15B, YTHDF3, HNRNPA2B1, IGF2BP1, IGF2BP2, and IGF2BP3) (Table 1) and were used for qTR-PCR analysis of those 19 m6A regulators in these 5 cell lines.
Table 1.
Primer sequences for qRT-PCR analysis of 19 m6A regulators in lung cancer cell lines (note: F = forward, R = reverse). GAPDH was used as control
| Primer name | Primer sequence (from 5′ to 3′) |
|---|---|
| ALKBH5-F | GCAAGGTGAAGAGCGGCATCC |
| ALKBH5-R | GTCCACCGTGTGCTCGTTGTAC |
| FTO-F | GTTCACAACCTCGGTTTAGTTC |
| FTO-R | CATCATCATTGTCCACATCGTC |
| HNRNPA2B1-F | GCTTAAGCTTTGAAACCACAGA |
| HNRNPA2B1-F | GCTTAAGCTTTGAAACCACAGA |
| IGF2BP1-R | GGGGTGGAATATTTCGGATTTG |
| IGF2BP1-F | GATGAAGGCCATCGAAACTTTC |
| IGF2BP2-F | GATGAACAAGCTTTACATCGGG |
| IGF2BP2-R | GATTTTCCCATGCAATTCCACT |
| IGF2BP3-F | GAGGCGCTTTCAGGTAAAATAG |
| IGF2BP3-R | AATGAGGCGGGATATTTCGTAT |
| KIAA1429-F | GCAACTTCAGGCATTAAGTTCA |
| KIAA1429-R | GTATTGCCTTGTCGAATCTGTC |
| METTL14-F | CAGGCTGGCTCACAGTTGGAC |
| METTL14-R | TTCCACCTCTTCCTCCACCTCTG |
| METTL3-F | CTTCAGCAGTTCCTGAATTAGC |
| METTL3-R | ATGTTAAGGCCAGATCAGAGAG |
| RBM15-F | GGCTGCCTGAGGAGAGTGGAG |
| RBM15-R | CGGCTACTGCTCAATTCTGGACTG |
| RBM15B-F | ATCTTTCAGAGTACGCTCAGAC |
| RBM15B-R | CTAGGATATGCATAGACGTGGG |
| WTAP-F | CTGACAAACGGACCAAGTAATG |
| WTAP-R | AAAGTCATCTTCGGTTGTGTTG |
| YTHDC1-F | AGTGACTCTGGTTCTGAATCTG |
| YTHDC1-R | CTGGTTTGATCTTTTCGGACAG |
| YTHDC2-F | GAGAATTGGGCTGTCGTTAAAG |
| YTHDC2-R | TGAAGCAGGATGAAATCGTACT |
| YTHDF2-F | ACTTCTCAGCATGGGGAAATAA |
| YTHDF2-R | TATTCATGCCAGGAGCCTTATT |
| YTHDF3-F | TCAACCACCACAACCACAGCAG |
| YTHDF3-R | TGAAGCACTGACAGGTACAACACC |
| ZC3H13-F | GATCAGTTAAAGCGTGGAGAAC |
| ZC3H13-R | CTCTCTGTCGTGTTCATATCGA |
| YTHDF1-F | ATGACAATGACTTTGAGCCCTA |
| YTHDF1-R | AGGGAGTAAGGAAATCCAATGG |
| HNRNPC-F | ACAGATCCTCGCTCCATGAACTCC |
| HNRNPC-R | TTCTGCCATCCTCTCCTGCTACAG |
| GAPDH-F | CTGCACCACCAACTGCTT |
| GAPDH-R | TTCTGGGTGGCAGTGATG |
Consensus clustering of lung cancer tissue samples
Lung cancer tissues with mRNA expression information of those 19 m6A regulators and corresponding clinical data were clustered with hierarchical agglomerative consensus clustering based on the method of Ward’s linkage and Euclidean distance. Unsupervised clustering methods used the proportion of ambiguous clustering (PAC) as a simple and powerful method to infer optimal K (K-means) for identifying and classifying patients for further analysis [32]. Cluster analysis was performed using the ConsensuClusterPlus R package with cycle computation for 1000 times to ensure the stability and reliability of classification [33]. The Kaplan–Meier method was used for the overall survival analysis in different clusters. Clinic correlation was performed with pheatmap R package.
The m6A regulator gene signature in lung cancer identified with lasso regression
Lasso regression based on 19 m6A regulators was used to identify the m6A regulator gene signature that was associated with high lung cancer risk, which was performed with the glmnet R package that fits a GLM with lasso or elasticnet regularization and a generalized linear model via penalized maximum likelihood. Lasso regression was a regression analysis method that performed both variable selection and regularization in order to enhance the prediction accuracy and interpretability of the statistical model it produced. The best subset selection and the connections between lasso coefficient estimates can be identified to construct the prognostic model [34]. The lasso regression-identified m6A regulator gene signature model generated a risk score for each lung cancer tissue sample to associate with pathology-related clinical characteristics. Thus, 1013 lung cancer patients were divided into high- vs. low-risk groups based on the median value = 0.84 of all risk scores. Further, clinical characteristics associated with overall survival were analyzed in lung cancer patients using Cox regression in univariate and multivariate models, and the Kaplan–Meier method was used to evaluate the availability of the prognostic model.
GSEA of KIAA1429, METTL3, and IGF2BP1 in lung cancer tissues
GSEA was widely used to analyze the genomic or proteomic data, which linked disease phenotypes with many different functional gene sets. According to median RNA expression value of each m6A regulator (KIAA1429, METTL3, and IGF2BP1), 1013 lung cancer tissue samples were divided into the corresponding high vs. low expression groups based on the expression median value of KIAA1429 (median value = 10.36), METTL3 (median value = 9.18), or IGF2BP1 (median value = 3.46), respectively, followed by GSEA based on these two high vs. low expression groups of each m6A regulator. GSEA was also conducted based on those two high- vs. low-risk groups that were derived from the lasso regression model (median value = 0.84 of risk scores), which was actually the combination effects of the three-m6A-regulator gene signature (KIAA1429, METTL3, and IGF2BP1).
Statistical analysis
Spearman correlation coefficient was calculated for the molecule pair between m6A regulator genes. Student’s t test in SPSS 13.0 (SPSS Inc., Chicago, USA) was used to assess the expression differences of each m6A regulator in LUAD and LUSC tissues compared to normal tissues and in different lung cancer cells compared to the control HBE cell. Each experiment was repeated at least three times. The relationship between clusters and different risk score groups was studied with chi-square test. In all cases, p < 0.05 was considered as statistically significant. Benjamini–Hochberg for multiple testing and false discovery rate (FDR) were calculated to correct the p value for GSEA.
Results
The mRNA expression profile of m6A regulators in lung cancer tissues and cell lines
The mRNA expressions of 19 m6A regulators (Supplementary Table 1), namely 7 methyltransferases—writers (ZC3H13, KIAA1429, RBM15, RBM15B, WTAP, METTL3, and METTL14), 2 demethylases—erasers (FTO and ALKBH5), and 10 m6A-binding proteins—readers (HNRNPC, YTHDF1, YTHDF2, YTHDF3, YTHDC1, YTHDC2, IGF2BP1, IGF2BP2, IGF2BP3, and HNRNPA2B1), were used to construct the expression heatmap between 1013 lung cancer and 109 control tissues (Fig. 2a). KIAA1429, RBM15, HNRNPC, YTHDF1, HNRNPA2B1, IGF2BP1, IGF2BP2, and IGF2BP3 were significantly increased in lung cancer compared to control tissues (Fig. 2b). ZC3H13, METTL3, METTL14, and FTO were significantly decreased in lung cancer compared to control tissues (Fig. 2b). Moreover, qRT-PCR analysis also found that the mRNA expression profile of 19 m6A regulators was changed in human LUAD cells (A549 and LTEP-a-2) and LUSC cells (H520 and SK-MES-1) compared to the control HBE cell (Fig. 3). These results clearly demonstrated the dysregulated gene expression profile of m6A regulators between human lung cancer and control tissues and between human lung cancer and control cells.
Fig. 2.

The mRNA expression profile of 19 m6A regulators in 1013 lung cancer tissues (T) and 109 controls (N). a The heatmap of mRNA expressions of 19 m6A regulators (ZC3H13, KIAA1429, RBM15, RBM15B, WTAP, METTL3, METTL14, FTO, ALKBH5, HNRNPC, YTHDF1, YTHDF2, YTHDF3, YTHDC1, YTHDC2, IGF2BP1, IGF2BP2, IGF2BP3, and HNRNPA2B1) in lung cancer tissues based on the TCGA database. T = lung cancer (red). N = normal controls (green). The color from green to red bar on the right means the level of gene expression, gradually changed green bar means downregulation gene, and gradually changed red bar means upregulation of gene. b The mRNA expression levels of each m6A regulator among normal (n = 109), lung squamous cell carcinoma (n = 502), and lung adenocarcinoma (n = 511) tissues. *p < 0.05; **p < 0.01; ***p < 0.001
Fig. 3.

qRT-PCR analysis of 19 m6A regulators in human lung adenocarcinoma cells (A549 and LTEP-a-2) and lung squamous cell carcinoma cells (H520 and SK-MES-1) compared to control HBE cells. NS = no significance. *p < 0.05; **p < 0.01; ***p < 0.001
Further, PPI and correlation analysis revealed the relationships among those 19 m6A regulators (Fig. 4). Some highly interacting m6A regulator protein pairs (combined score ≥ 0.99) were identified, including IGF2BP1 and IGF2BP3, WTAP and KIAA1429, HNRNPC and HNRNPA2B1, WTAP and ZC3H13, METTL14 and METTL3, KIAA1429 and ZC3H13, METTL14 and WTAP, WTAP and METTL3, METTL14 and KIAA1429, and METTL3 and KIAA1429 (Supplementary Table 2; Fig. 4a). Some highly correlated m6A regulator gene pairs (|correlation coefficient| ≥ 0.4, p < 0.05) were identified, including ZC3H13 and YTHDC1, METTL14 and ZC3H13, KIAA1429 and YTHDF3, HNRNPA2B1 and YTHDC1, IGF2BP1 and IGF2BP2, IGF2BP1 and IGF2BP3, and IGF2BP2 and IGF2BP3 (Fig. 4b).
Fig. 4.

The relationships of 19 m6A regulators in lung cancers. a Protein–protein interaction (PPI) network of 19 m6A regulator proteins. b Co-expressions among 19 m6A regulator genes. X = no statistical significance
Consensus clustering of lung cancer tissue samples based on the mRNA expressions of 19 m6A regulators
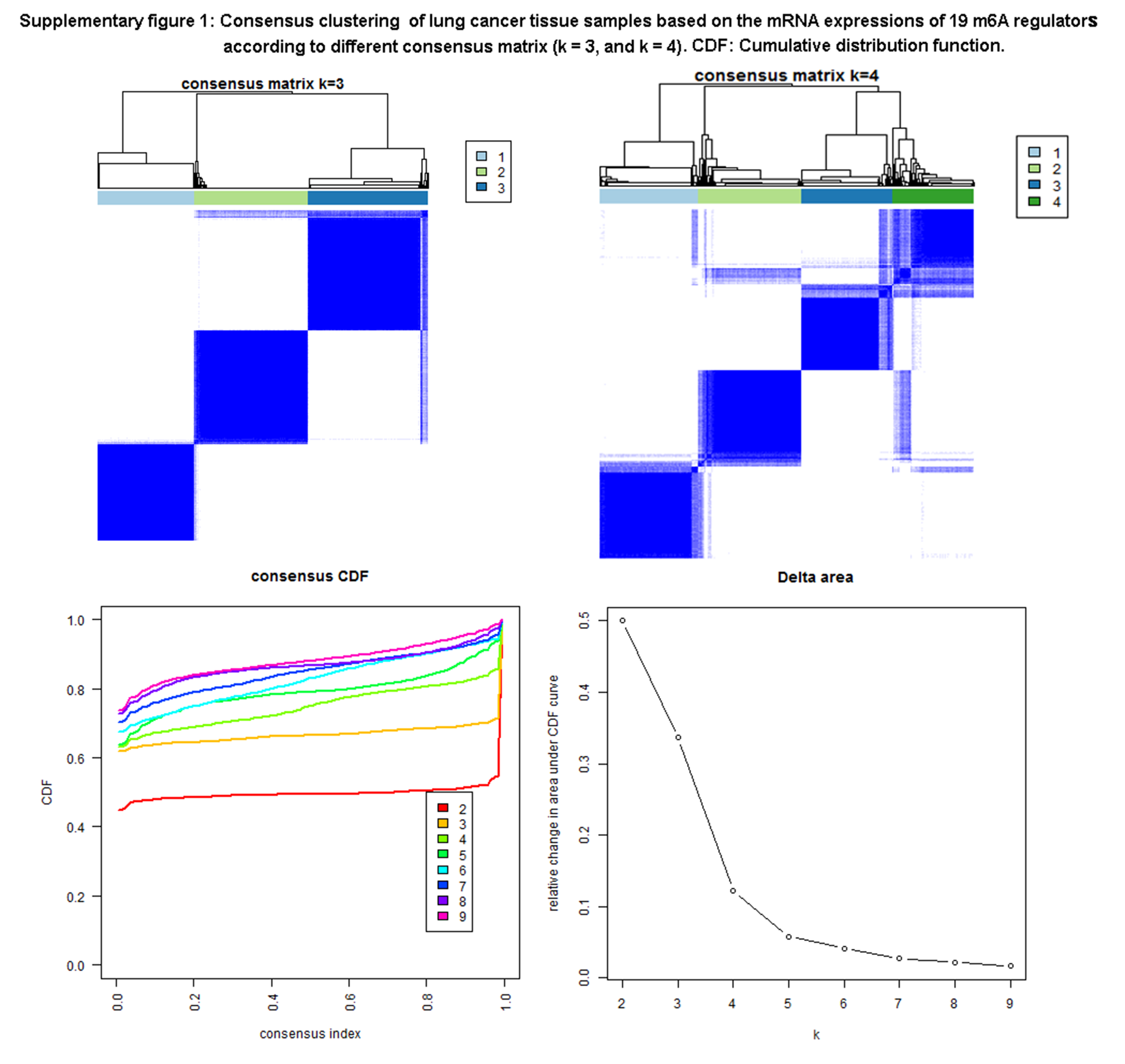
To determine the optimal cluster number, clustering stability was assessed with the ConsensusClusterPlus package (Supplementary Fig. 1; Fig. 5a), which supported the existence of two robust lung cancer sample clusters (Fig. 5a). When the consensus matrix k value was equal to 2, there was no crossover between lung cancer samples; thus, 1013 lung cancer tissue samples were grouped into two clusters (Supplementary Table 3; Fig. 5a). Correlations between clinical feature and m6A regulators were tested with chi-square test based on those two robust lung cancer sample clusters (Supplementary Table 3; Fig. 5a). The sample clusters were significantly related to those clinical characteristics, including primary disease (LUAD and LUSC) (p < 0.001), gender (male and female) (p < 0.001), tobacco smoking history (current reformed smoker with ≤ 15 years’ smoking, current reformed smoker with > 15 years’ smoking, current reformed smoker, and lifelong nonsmoker) (p < 0.001), pathologic stage (stages I, II, III, and IV) (p < 0.001), and status (alive and dead) (p < 0.05) (Fig. 5b). However, the overall survival analysis showed no statistical significance between clusters 1 and 2 (Fig. 5c). Therefore, it was necessary to select other optimization methods.
Fig. 5.

Consensus clustering of lung cancer tissue samples based on the mRNA expressions of 19 m6A regulators according to the optimized consensus matrix k = 2. a The optimal cluster number was two using the ConsensusClusterPlus package. b The heatmap of clusters and lung cancer-related clinical characteristics, including primary disease, age, gender, tobacco smoking history, pathologic T (T represented tumor size, including T1, T2, T3, T4, and TX), pathologic M (M represented tumor metastasis, including M0, M1, and MX), pathologic N (N represented tumor lymph node metastasis, including N0, N1, N2, and NX), pathologic stage, cancer status, tumor residual, and status. c Overall survival analysis between cluster 1 and cluster 2. *p < 0.05; **p < 0.01; ***p < 0.001
Lasso regression identified the prognostic model of the three-m6A-regulator gene signature
To determine the optimal prognostic model, lasso regression was performed with the glmnet R package. Lasso regression was a generalized linear model, and the adjustment degree of lasso regression complexity was controlled by lambda. The optimal prognostic model of the three-m6A-regulator gene signature (KIAA1429, METTL3, and IGF2BP1) was identified when log(lambda) was between − 3 and − 4 (Fig. 6 a and b). The lasso regression coefficient of the three-m6A-regulator gene signature was 0.170 for KIAA1429, − 0.160 for METTL3, and 0.036 for IGF2BP1. The univariate Cox regression analysis also revealed that KIAA1429 (hazard ratio [HR] 1.33; 95% confidence interval [CI] 1.05–1.68; p = 0.018), METTL3 (HR 0.77; 95% CI 0.64–0.92; p = 0.005), and IGF2BP1 (HR 1.34; 95% CI 1.07–1.67; p = 0.011) were correlated significantly with a poor overall survival (Table 2). It clearly demonstrated that the results of lasso regression were consistent with the results of the univariate Cox regression. The univariate Cox regression analysis confirmed the three-m6A-regulator gene signature model. The relationship between the three-m6A-regulator gene signature (KIAA1429, METTL3, and IGF2BP1) and lung cancer risk was calculated; thus, each lung cancer sample obtained a risk score, and finally, lung cancer samples were grouped into high-risk vs. low-risk groups according to the median value = 0.84 of all risk scores (Supplementary Table 4). The overall survival analysis showed statistical significance between high-risk and low-risk groups (p = 1.723E-04) (Fig. 6c). Further, the sample risk groups were significantly related to those clinical characteristics, including primary disease (LUAD and LUSC) (p < 0.001), gender (male and female) (p < 0.001), tobacco smoking history (current reformed smoker with ≤ 15 years’ smoking, current reformed smoker with > 15 years’ smoking, current reformed smoker, and lifelong nonsmoker) (p < 0.001), pathologic stage (stages I, II, III, and IV) (p < 0.001), cancer status (tumor and tumor free) (p < 0.01), and status (alive and dead) (p < 0.05) (Fig. 7).
Fig. 6.

Lasso regression analysis of 19 m6A regulators to identify the prognostic model of three-m6A-regulator gene signature (2 = KIAA1429, 9 = METTL3, and 17 = IGF2BP1) between log(lambda) value from − 4 to − 3. a, b Lasso regression complexity was controlled by lambda using the glmnet R package (1 = RBM15; 2 = KIAA1429; 3 = ZC3H13; 4 = HNRNPC; 5 = YTHDC2; 6 = METTL14; 7 = YTHDC1; 8 = WTAP; 9 = METTL3; 10 = FTO; 11 = ALKBH5; 12 = YTHDF1; 13 = YTHDF2; 14 = RBM15B; 15 = YTHDF3; 16 = HNRNPA2B1; 17 = IGF2BP1; 18 = IGF2BP2; 19 = IGF2BP3). c Overall survival analysis between the high-risk score and low-risk score group derived from lasso regression
Table 2.
Association between each m6A regulator and overall survival in TCGA lung cancer patients analyzed with Cox regression
| m6A regulator | HR | 95% confidence intervals (low) | 95% confidence intervals (high) | p value |
|---|---|---|---|---|
| RBM15 | 1.10 | 0.87 | 1.39 | 0.434 |
| KIAA1429 | 1.33 | 1.05 | 1.68 | 0.018* |
| ZC3H13 | 1.12 | 0.91 | 1.36 | 0.283 |
| HNRNPC | 1.08 | 0.84 | 1.41 | 0.542 |
| YTHDC2 | 0.90 | 0.73 | 1.10 | 0.308 |
| METTL14 | 1.08 | 0.84 | 1.38 | 0.565 |
| YTHDC1 | 0.86 | 0.64 | 1.17 | 0.337 |
| WTAP | 1.09 | 0.87 | 1.35 | 0.456 |
| METTL3 | 0.77 | 0.64 | 0.92 | 0.005** |
| FTO | 1.07 | 0.87 | 1.31 | 0.523 |
| ALKBH5 | 1.01 | 0.79 | 1.29 | 0.943 |
| YTHDF1 | 0.92 | 0.71 | 1.19 | 0.519 |
| YTHDF2 | 0.90 | 0.67 | 1.20 | 0.463 |
| RBM15B | 1.04 | 0.80 | 1.35 | 0.758 |
| YTHDF3 | 1.13 | 0.91 | 1.39 | 0.265 |
| HNRNPA2B1 | 1.04 | 0.79 | 1.37 | 0.769 |
| IGF2BP1 | 1.34 | 1.07 | 1.67 | 0.011* |
| IGF2BP2 | 1.03 | 0.97 | 1.09 | 0.330 |
| IGF2BP3 | 1.03 | 0.98 | 1.08 | 0.199 |
HR = hazard ratio
*p < 0.05; **p < 0.01
Fig. 7.

The heatmap analysis of relationships between clinical characteristics and the model of the three-m6A-regulator gene signature in 1013 lung cancer tissue samples. The risk score of each lung cancer sample was derived from lasso regression risk scores of the three-m6A-regulator gene signature (KIAA1429, METTL3, and 17 = IGF2BP1). These clinical characteristics included primary disease, age, gender, tobacco smoking history, pathologic T (T represented tumor size, including T1, T2, T3, T4, and TX), pathologic M (M represented tumor metastasis, including M0, M1, and MX), pathologic N (N represented tumor lymph node metastasis, including N0, N1, N2, and NX), pathologic stage, cancer status, tumor residual, and status. *p < 0.05; **p < 0.01; ***p < 0.001
Moreover, the lasso regression risk score of each lung cancer sample based on the three-m6A-regulator gene signature (KIAA1429, METTL3, and IGF2BP1) (Supplementary Table 4) was used as a risk factor in combination with clinical features to perform univariate and multivariate Cox regression analyses. The univariate Cox regression analysis revealed that pathologic N (HR 1.350; 95% CI 1.204–1.514; p = 3.00E-07), pathologic T (HR 1.432; 95% CI 1.249–1.643; p = 2.00E-07), pathologic stage (HR 1.469; 95% CI 1.309–1.649; p = 5.00E-11), cancer status (HR 4.096; 95% CI 3.152–5.321; p = 4.72E-26), residual tumor (HR 1.276; 95% CI 1.088–1.497; p = 2.70E-04), and risk score (HR 6.374; 95% CI 2.671–15.211; p = 3.00E-05) were correlated significantly with a poor OS (Fig. 8a). The multivariate Cox regression analysis revealed that pathologic T pathologic stage (HR 1.262; 95% CI 1.015–1.569; p = 3.60E-02), cancer status (HR 3.884; 95% CI 2.875–5.247; p = 9.53E-19), and risk score (HR 5.082; 95% CI 1.460–17.685; p = 1.10E-02) were significantly correlated with a poor overall survival (Fig. 8b). It clearly demonstrated that the risk score factor based on the optimized prognostic model of the three-m6A-regulator gene signature was significant with both univariate and multivariate Cox regression analyses.
Fig. 8.

The univariate and multivariate Cox regression analyses of risk factors in lung cancer. a The univariate analysis of risk factors in lung cancer. b The multivariate analysis of risk factors in lung cancer
GSEA revealed pathways and potential target genes
According to the GSEA enrichment results of KIAA1429, METTL3, and IGF2BP1 (Supplementary Table 5), gene sets were significantly enriched in drug resistance (antibiotics), proliferation, RNA metabolism, metastasis, energy metabolism (gluconeogenesis), cell cycling, apoptosis, recurrence, DNA replication, inflammatory mediators pathway (IL6 and prostaglandin E2), and epithelial–mesenchymal transition (EMT). It clearly showed that m6A regulator genes affected tumor biology behaviors by several biological mechanisms. Further, the potential target genes in GSEA results of two m6A regulators IGF2BP1 (Fig. 9a) and KIAA1429 (Fig. 9b) included E2F3, WTAP, CCND1, CDK4, EGR2, YBX1, IL6, and TLX, which indicated the potential mechanism of m6A regulators and that m6A widely regulated mRNAs and ncRNAs [20].
Fig. 9.

Gene set enrichment analysis (GSEA) results based on mRNA expressions (high expression vs. low expression) of each m6A regulator (IGF2BP1 and KIAA1429). a The GSEA results of IGF2BP1. b The GSEA results of KIAA1429
GSEA was also carried out in different sample risk score groups derived from the lasso regression prognostic model of the three-m6A-regulator gene signature (KIAA1429, METTL3, and IGF2BP1) according to the median value = 0.84 of all risk scores. The GSEA results showed that the high-risk score group was significantly related to the multiple important biological processes, including ubiquitin-mediated proteolysis, hypoxia via ELK3, adrenocortical tumor markers, liver cancer survival, metastasis, stemness, and TGFB1 targets (Supplementary Table 6 and Fig. 10), which were closely associated with tumorigenesis and development.
Fig. 10.

Gene set enrichment analysis (GSEA) results based on risk score (high-risk score vs. low-risk score) derived from the lasso regression model. a Ubiquitin-mediated proteolysis pathway enriched by GSEA. b Hypoxia via ELK3 pathway enriched by GSEA. c Liver cancer survival enriched by GSEA. d Metastasis pathway enriched by GSEA. e Stemness pathway enriched by GSEA. f TGFB1 targets pathway enriched by GSEA
Discussion
This study analyzed the mRNA expression profile of 19 m6A regulators in 1013 lung cancer tissues relative to 109 normal tissues and in 4 lung cancer cell lines relative to normal control cell line and further analyzed the associations of these m6A regulators and clinical characteristics of lung cancers. An optimized lasso regression prognostic model of the three-m6A-regulator gene signature (IGF2BP1, METTL3, and KIAA1429) was achieved, which was significantly associated with important clinical characteristics of lung cancer and with the poor overall survival of lung cancer. It was a potential biomarker pattern for prognostic prediction, patient stratification, and personalized treatment of lung cancer toward its PPPM practice.
Roles of m6A RNA modification and its regulators in multiple cancers
The m6A modification is the most meaningful and pervasive modification of human RNA molecules. Further studies on the biological functions of m6A RNA modifications have not been paid enough attention until around 2012, when major progress has been made in the transcriptome profiling of m6A RNA modifications through antibody-based immunoprecipitation and high-throughput sequencing [35]. The m6A antibody-based immunoprecipitation has not been reported in lung cancer. The m6A RNA modification is a reversible process through regulation of m6A regulators, including methyltransferases (writers), demethylases (erasers), and m6A-binding proteins (readers) (Fig. 1). The m6A is installed by the methyltransferase complex (ZC3H13, KIAA1429, RBM15, RBM15B, WTAP, METTL3, and METTL14). The identity of demethylases (FTO and ALKBH5) provides reliable evidence that m6A RNA modification is a reversible posttranscriptional modification [36]. m6A RNA modification influences the properties of RNA transcripts, including secondary structure, base-pairing, protein–RNA interactions, and charge, which, in turn, affects gene expression through altering RNA localization, translation, processing, decay, and eventually metabolism. The altered expression levels of m6A regulators substantially affect phenotypes in a cell and organism [37]. Emerging data have demonstrated the important roles of m6A regulators (writers, erasers, and readers) in human cancers [38], including acute myeloid leukemia (METTL3, METTL14, FTO, and YTHDF2), breast cancer (METTL13, METTL14, FTO, and ALKBH5), nonsmall cell lung cancer (METTL3, FTO, ALKBH5, YTHDF1, and YTHDF2), gastric cancer (METTL3, FTO, ALKBH5, and IGF2BP3), bladder cancer (METTL3, METTL14, and ALKBH5), prostate cancer (METTL3 and YTHDF2), osteosarcoma (METTL3), glioblastoma (METTL3, METTL14, FTO, and ALKBH5), renal cell carcinoma (METTL3 and METTL14), hepatocellular carcinoma (METTL3, METTL14, WTAP, VIRMA, YTHDF1, YTHDF2, and IGF2BP1/2/3), pancreatic cancer (METTL3, FTO, ALKBH5, YTHDF2, and IGF2BP2), colorectal cancer (METTL3, METTL14, YTHDF1, YTHDF3, and IGF2BP2), gynecological cancer (FTO, YTHDF2, IGFBP1/2/3, METTL3, METTL14, and ALKBH5), and melanoma (METTL3 and FTO) [25, 39]. It clearly demonstrated the important scientific merits of m6A regulators in different types of cancers. The identification of pathology-specific m6A regulator pattern might directly serve cancer management in the context of predicting prognosis, stratifying patients, and personalizing medical services.
Role of m6A RNA modification and its regulators in lung cancer
Recent studies have found that m6A RNA modification plays important roles in human cancer biology [40]. EMT was the initiation of metastasis in cancer progression. In the EMT process, epithelial cells tend to lose cell–cell adhesion and cell polarity, and gain capacities of migration and invasion to become mesenchymal stem cells. The previous study highlighted the critical roles of m6A RNA modification in regulation of EMT in cancer cells [41]. m6A sequencing in combination with functional analysis confirmed that Snail was modified by m6A in coding the sequence region, which triggered polysome-mediated translation of m6A-regulated EMT by Snail in cancer cells [41]. It is well-known that viruses have close relationships with some kinds of cancers, such as the Epstein–Barr virus (EBV), human papilloma virus (HPV), hepatitis virus B (HBV), and human T cell leukemia/lymphoma virus I (HTLV-1) [42]. Due to the expanded knowledge of m6A, m6A was identified in transcripts encoded by many kinds of viruses, which indicated widespread control over life cycles of viruses [42]. It innovated our comprehension and cognition of the regulatory mechanisms about viral replication [43]. In this study, GSEA showed that the alerted m6A regulators in lung cancers were related to the activations of various biological behaviors, including drug resistance, proliferation, metastasis, energy metabolism, DNA replication, inflammatory mediators pathway, RNA metabolism, cell cycling, apoptosis, recurrence, and EMT. These findings were consistent with many previous studies [20, 34]. With the advancement of tumor immunology and molecular biotechnology, immunotherapy became an effective clinical therapy in the combination therapy of tumor. The relevant roles of m6A RNA methylation in immunoregulation have received great attention. Anti-PD-1/PD-L immune checkpoint therapy was the most popular targets for new anticancer drugs. The m6A demethylase FTO has been proved to promote tumorigenesis and has function of anti-PD-1 resistance. This result indicated that FTO inhibition in combination with anti-PD-1 blockade might be the possible solution to immunotherapy resistance in cancer [44]. Energy metabolism alteration of tumor cells has been proved to be an emerging sign of tumorigenesis since it was closely related to some malignant behaviors of cancer cells. Alternative splicing of energy metabolism-related enzymes may modulate enzyme activity to impact energy reprogramming, for example, the ratio between M1 and M2 isoforms of pyruvate kinase in aerobic glycolysis and splicing programs at different levels of 2-oxoglutarate-dependent oxidases. The effect of m6A on the splicing profile of target RNAs has been identified; for example, the m6A reader YTHDC1 affects splicing isoforms by recruiting splicing factors of the SR family, and the m6A erasers FTO and ALKBH5 were directly involved in RNA splicing [45]. One study also found that m6A regulators (METTL3, FTO, ALKBH5, YTHDF1, and YTHDF2) significantly contributed to the pathogenesis of nonsmall cell lung cancer [25]. These findings clearly demonstrated that m6A regulators play important roles in lung cancer pathogenesis.
The three-m6A-regulator gene signature as biomarker in the context of PPPM in lung cancer
In this study, lasso regression identified the prognostic model of the three-m6A-regulator gene signature (IGF2BP1, METTL3, and KIAA1429) in 1013 human lung cancer tissues. First, many studies showed that IGF2BP1 enhanced tumor progression and metastasis in LUADs and that the increased IGF2BP1 expression was correlated with lower overall survival in lung carcinoma [46]. IGF2BP1 promoted the expression of the transcriptional regulator SRF in a conserved and m6A-dependent manner through competing with the miRNA-dependent manner of SRF mRNA degradation [20]. IGF2BP1 that regulates various target genes in the m6A-dependent manner could be a conserved oncogenic driver network in lung cancer. Second, the METTL3 is one of the key components in the m6A-methyltransferase complex responsible for m6A RNA modification. The endogenous METTL3 change and subsequent alteration of related gene expression profiles might directly influence the growth of human nonsmall cell lung carcinoma cell line (H1299) in vitro and in vivo [47]. Third, the m6A writer KIAA1429 was never reported in lung cancer, but studies in other kinds of cancers were consistent with our findings. KIAA1429 was highly expressed in breast cancer tissues compared to noncancerous breast tissues. The high expression of KIAA1429 was a predicting factor of poor prognosis and was associated with cancer proliferation and metastasis by regulating CDK1 in an m6A-independent manner [27]. The activities and molecular mechanisms of m6A-methyltransferases–writers were an important field for lung cancer research. Here we have to note that it does not make sense to take a single molecule as biomarker for PPPM practice in cancer because cancer is a very complex and multifaceted disease [48, 49]. The significant hub-molecule signature model was constructed to assess cancer survival risks, which benefited for patient stratification, prognostic prediction, and personalized treatment of cancer [50]. In our study, the high-risk vs. low-risk groups based on the lasso regression prognostic model of the three-m6A-regulator gene signature (IGF2BP1, METTL3, and KIAA1429) were significantly related to overall survival and clinicopathologic characteristics, which might be an effective biomarker pattern for predictive diagnostics or prognostic assessment, patient stratification, and personalized treatment of lung cancers. However, the sensitivity, specificity, and accuracy of this optimized prognostic model must be investigated further in the large-scale clinical cohort study toward PPPM practice in lung cancers.
The potentially targeted genes of m6A regulators in lung cancer
The m6A RNA modification might influence multiple cellular signaling pathways and lots of RNA (mRNAs and ncRNAs). In this study, GSEA identified a few tumor-related gene sets of proliferative signaling, metastasis, apoptosis by CDKN1A via TP53, and mitochondrial transcription activity in lung cancer. Meanwhile, many studies showed that m6A-related gene knockdown inhibited total RNA m6A methylation level, as well as capacity of cancer cell proliferation and migration [51]. BNIP3, a pro-apoptosis gene, was identified as a downstream target of eraser FTO-mediated m6A RNA modification by m6A RNA immunoprecipitation sequencing [52]. It clearly showed that m6A RNA modification had functional significance in the apoptosis process. Even though the relationship of m6A and mitochondrial transcription in mammal has not been revealed, comprehensive characterization of m6A modification patterns has been studied in the Arabidopsis mitochondrial transcriptomes, which showed that over 86% of the transcripts were methylated by m6A in the mitochondria [53]. The m6A modification patterns in plant mitochondrial transcription were involved in mitochondrial transcription activity in an m6A-dependent manner. Moreover, in this study, GSEA identified a few tumor-related gene targets, including E2F3, WTAP, CCND1, CDK4, EGR2, YBX1, IL6, and TLX, which have been reported to be associated with lung cancer. For example, CCND1 and CDK4 promoted tumorigenicity of lung cancer [54, 55]. The oncogene YBX1 also proved to affect migration, invasion, and vasculogenic mimicry of lung cancer cells [56]. However, it is not clear whether E2F3, WTAP, CCND1, CDK4, EGR2, YBX1, IL6, and TLX were in the m6A-dependent manner to influence the malignant behavior of lung cancers.
Strength and limitations
m6A RNA methylation that extensively occurs at different types of mRNAs and ncRNAs is a new frontier research hotspot, which is closely associated with cancer pathogenesis and has extensive therapeutic implications in cancer [12–15, 25, 38, 39]. m6A RNA methylation is tightly controlled by the m6A regulator system that includes 7 methyltransferases—writers, 2 demethylases—erasers, and 10 m6A-binding proteins—readers (Fig. 1). The investigation of the relationships between the m6A regulator system and clinically pathological characteristics will directly contribute to clinical research and practice toward PPPM in cancer. The present study was the first link of the m6A regulator system and clinically pathological characteristics in lung cancer and identified the three-m6A-regulator signature (KIAA1429, METTL3, and IGF2BP1) as an independent prognostic model to classify lung cancers into high- and low-risk groups for patient stratification, prognostic assessment, and personalized treatment toward PPPM in lung cancers. Also, the mRNA expressions of 19 m6A regulators were analyzed in a large set of human tissue samples with the corresponding clinical data, including 1013 human lung cancer tissues (511 LUADs and 502 LUSCs) and 109 normal control lung tissues from the TCGA database, and in two LUAD cell lines and two LUSC cell lines relative to the normal HBE cells by qTR-PCR, which assured the reliability of the results in this study. Here, we would like to mention that not every tissue sample had the complete clinical data (Supplementary Table 3), which might be a limitation in this aspect, but from a statistical point of view, it did not play a significant role in this study. Also, these tissue samples in this study were from white and African Americans; thus, whether the findings in this study can be exactly applied in other races, it might need to further study the difference among different races. The status “alive/dead” of lung cancer tissue samples was the follow-up data of patient after thoracic surgery, which might not affect the overall outcome of our findings in this study. Furthermore, one must realize that, in order to deeply clarify the molecular mechanisms and biological roles of the m6A regulator system in lung cancer, (i) the protein expression profile of 19 m6A regulators should be further investigated in lung cancer tissues and cell models, (ii) the targeting RNA molecules and modification sites of m6A regulators should be investigated with m6A RNA sequencing and m6A antibody-based RNA immunoprecipitation in lung cancer, and (iii) the m6A RNA modification data may integrate the large-scale clinical characteristics and other multiomics data to achieve more meaningful findings for lung cancer.
Conclusions and expert recommendations
m6A RNA modification extensively occurs at mRNAs and ncRNAs to regulate transcription and affects the functions of RNAs, which is closely associated with cancer pathogenesis [12–15, 38, 39] and has extensive therapeutic implications in cancer [25]. m6A RNA modification is tightly regulated by m6A regulators. The identification of a pathology-specific m6A regulator pattern might directly serve cancer management in the context of predicting diagnosis, stratifying patients, and personalizing medical services. This study, for the first time, investigated the dysregulated mRNA expression profile of these m6A regulators in lung cancers and analyzed the relationship of the dysregulated mRNA expression profile of m6A regulators and clinical characteristics in human lung cancers. GSEA showed that the alerted m6A regulators in lung cancers were related to the activations of various biological behaviors, including DNA replication, RNA metabolism, EMT, cell cycling, proliferation, apoptosis, energy metabolism, inflammatory mediators pathway, drug resistance, metastasis, and recurrence. Lasso regression optimized and identified the prognostic model of the three-m6A-regulator gene signature (IGF2BP1, METTL3, and KIAA1429), which was significantly associated with clinical characteristics (primary disease, gender, tobacco smoking history, pathologic stage, cancer status, and alive/dead status) and poor overall survival in lung cancer. The lasso regression risk score factor based on this three-m6A-regulator gene signature was a comprehensive independent prognostic factor for prognostic assessment/prediction to stratify lung cancer patients into high- vs. low-risk score groups for personalized management in the context of PPPM practice. We conclude that pathology-specific regulators of m6A RNA modification are effective biomarkers and have important scientific merits in lung cancers.
We strongly recommend to emphasize and popularize the scientific importance of, and strengthen its studies of, the m6A regulator system (writers, erasers, and readers) in combination with clinical characteristics in lung cancer, and transit m6A regulator gene signature into clinical practice in the context of PPPM in lung cancer. We propose, for the further PPPM development and practical application of the altered m6A regulator pattern in lung cancer, the following three aspects:
-
(i)
Potential utility of the m6A regulator gene signature for predictive diagnosis: Many studies have demonstrated that the m6A regulator system plays important roles in cancer pathogenesis [38, 39]. It is a promising approach to integrate the abnormal expression profile of the m6A regulator system (writers, erasers, and readers) and clinical characteristics, for the discovery of a pathology-specific m6A regulator gene signature to optimize lung cancer management in the context of PPPM practice. This study identified a three-m6A-regulator gene signature (IGF2BP1, METTL3, and KIAA1429), which was significantly associated with clinical characteristics and poor overall survival. It is a potential biomarker pattern for predicting prognosis, stratifying patient, and personalizing treatment of lung cancer [57].
-
(ii)
Potential utility of the m6A regulator system for targeted prevention: The m6A regulator system (writers, erasers, and readers) tightly controls the m6A RNA modification to affect the functions and structures of mRNAs and ncRNAs, and is extensively associated with cancer pathogenesis [25]. The abnormal m6A regulator system (writers, erasers, and readers) is the potential therapeutic targets [25] for targeted drug therapy or targeted drug prevention of lung cancer. A study found that abnormal m6A regulators might promote or inhibit cancer [58]. The changed m6A regulators might be the novel therapeutic targets for lung cancer [25]. Thus, it would be an effective therapeutic strategy to restore the abnormal m6A regulators for targeted prevention or personalized therapy of lung cancers.
-
(iii)
Potential utility of the m6A regulator system for personalized treatment algorithms: m6A methylation extensively occurred in mRNAs and ncRNAs to affect the activities of transcripts and closely associate with cancer pathogenesis. The m6A regulator system (writers, erasers, and readers) is the central molecular machine to control the occurrence of m6A methylation at different mRNAs and ncRNAs. The development of different algorithms of the m6A regulator pattern in combination with clinical pathology characteristics will directly contribute to patient stratification for personalized treatment in lung cancer. Furthermore, m6A RNA sequencing and m6A antibody-based RNA immunoprecipitation are the effective methods to discover m6A RNA modification in lung cancer, and the m6A RNA modification data may integrate the large-scale clinical characteristics and other multiomics data of lung cancer [48–50], from a systematic point of view toward PPPM practice.
Electronic supplementary material
{kind=link}
(PNG 13244 kb)
(XLS 472 kb)
(XLS 27 kb)
(XLSX 75 kb)
(XLS 183 kb)
(XLS 26 kb)
(XLSX 11 kb)
Acknowledgments
We would like to thank The Cancer Genome Atlas (TCGA) project organizers as well as all study participants for providing the publicly available TCGA RNA-seq data and clinical data.
Abbreviations
- ALKBH5
AlkB homolog 5, RNA demethylase
- CCND1
cyclin D1 [Homo sapiens]
- CDK4
cyclin-dependent kinase 4
- DNA
deoxyribonucleic acid
- E2F3
E2F transcription factor 3
- EBV
Epstein–Barr virus
- EGFR-TKI
epidermal growth factor receptor tyrosine kinase inhibitors
- EGR2
early growth response 2
- EMT
epithelial–mesenchymal transition
- FDR
false discovery rate
- FTO
FTO alpha-ketoglutarate dependent dioxygenase
- GSEA
gene set enrichment analysis
- HBV
hepatitis virus B
- HNRNPA2B1
heterogeneous nuclear ribonucleoprotein A2/B1
- HNRNPC
heterogeneous nuclear ribonucleoprotein C
- HPV
human papilloma virus
- HR
hazard ratio
- HTLV-1
human T cell leukemia/lymphoma virus I
- IGF2BP1
insulin-like growth factor 2 mRNA-binding protein 1
- IGF2BP2
insulin-like growth factor 2 mRNA-binding protein 2
- IGF2BP3
insulin-like growth factor 2 mRNA-binding protein 3
- IL6
interleukin 6
- KIAA1429
Vir-like m6A methyltransferase associated
- lncRNAs
long noncoding RNAs
- LUAD
lung adenocarcinoma, LUSC lung squamous carcinoma
- m6A
N6-methyladenosine
- METTL14
methyltransferase-like 14
- METTL3
mRNA mA methyltransferase
- mRNAs
messenger RNAs
- ncRNAs
noncoding RNAs
- PAC
proportion of ambiguous clustering
- PD-1
programmed cell death 1
- PD-L1
CD274 molecule
- PPI
protein–protein interaction
- qRT-PCR
quantitative real-time polymerase chain reaction
- RBM15
RNA-binding motif protein 15
- RBM15B
RNA-binding motif protein 15B
- RNA
ribonucleic acid
- rRNAs
ribosomal RNAs
- snRNA
small nuclear RNA
- TCGA
The Cancer Genome Atlas
- TLX
nuclear receptor subfamily 2 group E member 1
- tRNAs
transfer RNAs
- USP7
ubiquitin-specific protease 7
- WTAP
WT1-associated protein
- YBX1
Y-box binding protein 1
- YTHDC1
YTH domain containing 1
- YTHDC2
YTH domain containing 2
- YTHDF1
YTH N6-methyladenosine RNA-binding protein 1
- YTHDF2
YTH N6-methyladenosine RNA-binding protein 2
- YTHDF3
YTH N6-methyladenosine RNA-binding protein 3
- ZC3H13
zinc finger CCCH-type containing 13
- Abbreviations for all particular genes and proteins can be found at the following link:
Authors’ contributions
N.L. performed bioinformatic analysis, carried out the experiments, prepared the figures and tables, and designed and wrote the manuscript. X.Z. conceived the concept, instructed experiments, supervised results, coordinated, critically revised/wrote manuscript, and was responsible for its financial supports and the corresponding works. All authors approved the final manuscript.
Funding information
The authors acknowledge the financial supports from the Shandong First Medical University Talent Introduction Funds (to X.Z.), the Hunan Provincial Hundred Talent Plan (to X.Z.), and the Central South University Graduate Student Exploration Innovative Project 2019 (Grant No. 206021701).
Compliance with ethical standards
Competing interests
The authors declare that they have no competing interests.
Ethical approval
All the patients were informed about the purposes of the study and, consequently, have signed their “consent of the patient” form. All investigations conformed to the principles outlined in the Declaration of Helsinki and were performed with permission by the responsible Medical Ethics Committee of Xiangya Hospital, Central South University, China.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Mao Y, Yang D, He J, Krasna MJ. Epidemiology of lung cancer. Surg Oncol Clin N Am. 2016;25:439–445. doi: 10.1016/j.soc.2016.02.001. [DOI] [PubMed] [Google Scholar]
- 3.Aokage K, Yoshida J, Hishida T, Tsuboi M, Saji H, Okada M, Suzuki K, Watanabe S, Asamura H. Limited resection for early-stage non-small cell lung cancer as function-preserving radical surgery: a review. Jpn J Clin Oncol. 2017;47:7–11. doi: 10.1093/jjco/hyw148. [DOI] [PubMed] [Google Scholar]
- 4.Ma L, Qiu B, Zhang J, Li QW, Wang B, Zhang XH, Qiang MY, Chen ZL, Guo SP, Liu H. Survival and prognostic factors of non-small cell lung cancer patients with postoperative locoregional recurrence treated with radical radiotherapy. Chin J Cancer. 2017;36:93. doi: 10.1186/s40880-017-0261-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takamori S, Toyokawa G, Takada K, Shoji F, Okamoto T, Maehara Y. Combination therapy of radiotherapy and anti-PD-1/PD-L1 treatment in non-small-cell lung cancer: a mini-review. Clin Lung Cancer. 2018;19:12–16. doi: 10.1016/j.cllc.2017.06.015. [DOI] [PubMed] [Google Scholar]
- 6.Guccione E, Richard S. The regulation, functions and clinical relevance of arginine methylation. Nat Rev Mol Cell Biol. 2019;20(10):642–657. doi: 10.1038/s41580-019-0155-x. [DOI] [PubMed] [Google Scholar]
- 7.Zhan X, Huang Y, Qian S. Protein tyrosine nitration in lung cancer: current research status and future perspectives. Curr Med Chem. 2018;25:3435–3454. doi: 10.2174/0929867325666180221140745. [DOI] [PubMed] [Google Scholar]
- 8.Pandey RR, Pillai RS. Counting the cuts: MAZTER-Seq quantifies m(6)A levels using a methylation-sensitive ribonuclease. Cell. 2019;178:515–517. doi: 10.1016/j.cell.2019.07.006. [DOI] [PubMed] [Google Scholar]
- 9.Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012;149:1635–1646. doi: 10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang Y, Hsu PJ, Chen YS, Yang YG. Dynamic transcriptomic m(6)A decoration: writers, erasers, readers and functions in RNA metabolism. Cell Res. 2018;28:616–624. doi: 10.1038/s41422-018-0040-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wei J, He C. Site-specific m(6)A editing. Nat Chem Biol. 2019;15:848–849. doi: 10.1038/s41589-019-0349-8. [DOI] [PubMed] [Google Scholar]
- 12.Wang X, Feng J, Xue Y, Guan Z, Zhang D, Liu Z, Gong Z, Wang Q, Huang J, Tang C, Zou T, Yin P. Corrigendum: structural basis of N(6)-adenosine methylation by the METTL3-METTL14 complex. Nature. 2017;542:260. doi: 10.1038/nature21073. [DOI] [PubMed] [Google Scholar]
- 13.Lee M, Kim B, Kim VN. Emerging roles of RNA modification: m(6)A and U-tail. Cell. 2014;158:980–987. doi: 10.1016/j.cell.2014.08.005. [DOI] [PubMed] [Google Scholar]
- 14.Roundtree IA, Evans ME, Pan T, He C. Dynamic RNA modifications in gene expression regulation. Cell. 2017;169:1187–1200. doi: 10.1016/j.cell.2017.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, Sorek R, Rechavi G. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–206. doi: 10.1038/nature11112. [DOI] [PubMed] [Google Scholar]
- 16.Thüring K, Schmid K, Keller P, Helm M. LC-MS analysis of methylated RNA. Methods Mol Biol. 2017;1562:3–18. doi: 10.1007/978-1-4939-6807-7_1. [DOI] [PubMed] [Google Scholar]
- 17.Linder B, Grozhik AV, Olarerin-George AO, Meydan C, Mason CE, Jaffrey SR. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods. 2015;12:767–772. doi: 10.1038/nmeth.3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tuncel G, Kalkan R. Importance of m N(6)-methyladenosine (m(6)A) RNA modification in cancer. Med Oncol. 2019;36:36. doi: 10.1007/s12032-019-1260-6. [DOI] [PubMed] [Google Scholar]
- 19.Li J, Han Y, Zhang H, Qian Z, Jia W, Gao Y, Zheng H, Li B. The m6A demethylase FTO promotes the growth of lung cancer cells by regulating the m6A level of USP7 mRNA. Biochem Biophys Res Commun. 2019;512:479–485. doi: 10.1016/j.bbrc.2019.03.093. [DOI] [PubMed] [Google Scholar]
- 20.Muller S, Glass M, Singh AK, Haase J, Bley N, Fuchs T, Lederer M, Dahl A, Huang H, Chen J, Posern G, Huttelmaier S. IGF2BP1 promotes SRF-dependent transcription in cancer in a m6A- and miRNA-dependent manner. Nucleic Acids Res. 2019;47:375–390. doi: 10.1093/nar/gky1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Han J, Wang JZ, Yang X, Yu H, Zhou R, Lu HC, Yuan WB, Lu JC, Zhou ZJ, Lu Q, Wei JF, Yang H. METTL3 promote tumor proliferation of bladder cancer by accelerating pri-miR221/222 maturation in m6A-dependent manner. Mol Cancer. 2019;18:110. doi: 10.1186/s12943-019-1036-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu X, Sang L, Gong Y. N6-methyladenine RNA modification and cancers. Am J Cancer Res. 2018;8:1957–1966. [PMC free article] [PubMed] [Google Scholar]
- 23.Li T, Hu PS, Zuo Z, Lin JF, Li X, Wu QN, Chen ZH, Zeng ZL, Wang F, Zheng J, Chen D, Li B, Kang TB, Xie D, Lin D, Ju HQ, Xu RH. METTL3 facilitates tumor progression via an m(6)A-IGF2BP2-dependent mechanism in colorectal carcinoma. Mol Cancer. 2019;18:112. doi: 10.1186/s12943-019-1038-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taketo K, Konno M, Asai A, Koseki J, Toratani M, Satoh T, Doki Y, Mori M, Ishii H, Ogawa K. The epitranscriptome m6A writer METTL3 promotes chemo- and radioresistance in pancreatic cancer cells. Int J Oncol. 2018;52:621–629. doi: 10.3892/ijo.2017.4219. [DOI] [PubMed] [Google Scholar]
- 25.Huang H, Weng H, Chen J. m(6)A modification in coding and non-coding RNAs: roles and therapeutic implications in cancer. Cancer Cell. 2020;37:270–288. doi: 10.1016/j.ccell.2020.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Visvanathan A, Patil V, Arora A, Hegde AS, Arivazhagan A, Santosh V, Somasundaram K. Essential role of METTL3-mediated m6A modification in glioma stem-like cells maintenance and radioresistance. Oncogene. 2017;37:522–533. doi: 10.1038/onc.2017.351. [DOI] [PubMed] [Google Scholar]
- 27.Qian JY, Gao J, Sun X, Cao MD, Shi L, Xia TS, Zhou WB, Wang S, Ding Q, Wei JF. KIAA1429 acts as an oncogenic factor in breast cancer by regulating CDK1 in an N6-methyladenosine-independent manner. Oncogene. 2019;38:6123–6141. doi: 10.1038/s41388-019-0861-z. [DOI] [PubMed] [Google Scholar]
- 28.Cheng X, Li M, Rao X, Zhang W, Li X, Wang L, Huang G. KIAA1429 regulates the migration and invasion of hepatocellular carcinoma by altering m6A modification of ID2 mRNA. Onco Targets Ther. 2019;12:3421–3428. doi: 10.2147/ott.s180954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cayir A, Barrow TM, Guo L, Byun HM. Exposure to environmental toxicants reduces global N6-methyladenosine RNA methylation and alters expression of RNA methylation modulator genes. Environ Res. 2019;175:228–234. doi: 10.1016/j.envres.2019.05.011. [DOI] [PubMed] [Google Scholar]
- 30.Huang H, Wang D, Guo W, Zhuang X, He Y. Correlated low IGF2BP1 and FOXM1 expression predicts a good prognosis in lung adenocarcinoma. Pathol Res Pract. 2019;215:152433. doi: 10.1016/j.prp.2019.152433. [DOI] [PubMed] [Google Scholar]
- 31.Zhan X, Long Y, Lu M. Exploration of variations in proteome and metabolome for predictive diagnostics and personalized treatment algorithms: innovative approach and examples for potential clinical application. J Proteome. 2018;188:30–40. doi: 10.1016/j.jprot.2017.08.020. [DOI] [PubMed] [Google Scholar]
- 32.Lock EF, Dunson DB. Bayesian consensus clustering. Bioinformatics. 2013;29:2610–2616. doi: 10.1093/bioinformatics/btt425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilkerson MD, Hayes DN. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics. 2010;26:1572–1573. doi: 10.1093/bioinformatics/btq170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alhamzawi R, Ali HTM. The Bayesian adaptive lasso regression. Math Biosci. 2018;303:75–82. doi: 10.1016/j.mbs.2018.06.004. [DOI] [PubMed] [Google Scholar]
- 35.Deng X, Su R, Weng H, Huang H, Li Z, Chen J. RNA N(6)-methyladenosine modification in cancers: current status and perspectives. Cell Res. 2018;28:507–517. doi: 10.1038/s41422-018-0034-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mongan NP, Emes RD. Detection and analysis of RNA methylation. F1000Res. 2019;8:559. 10.12688/f1000research.17956.1. [DOI] [PMC free article] [PubMed]
- 37.Meyer KD. m(6)A-mediated translation regulation. Biochim Biophys Acta Gene Regul Mech. 2019;1862:301–309. doi: 10.1016/j.bbagrm.2018.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barbieri I, Kouzarides T. Role of RNA modifications in cancer. Nat Rev Cancer. 2020;20:303–322. doi: 10.1038/s41568-020-0253-2. [DOI] [PubMed] [Google Scholar]
- 39.Zhang B, Wu Q, Li B, Wang D, Wang L, Zhou YL. m6A regulator-mediated methylation modification patterns and tumor microenvironment infiltration characterization in gastric cancer. Mol Cancer. 2020;19(1):53. doi: 10.1186/s12943-020-01170-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fitzsimmons CM, Batista PJ. It’s complicated... m(6)A-dependent regulation of gene expression in cancer. Biochim Biophys Acta Gene Regul Mech. 1862;2019:382–393. doi: 10.1016/j.bbagrm.2018.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin X, Chai G, Wu Y, Li J, Chen F, Liu J. RNA m(6)A methylation regulates the epithelial mesenchymal transition of cancer cells and translation of Snail. Nat Commun. 2019;10:2065. doi: 10.1038/s41467-019-09865-9. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 42.Qian S, Golubnitschaja O, Zhan X. Chronic inflammation: key player and biomarker-set to predict and prevent cancer development and progression based on individualized patient profiles. EPMA J. 2019;10:365–381. doi: 10.1007/s13167-019-00194-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Manners O, Baquero-Perez B, Whitehouse A. m(6)A: widespread regulatory control in virus replication. Biochim Biophys Acta Gene Regul Mech. 2019;1862:370–381. doi: 10.1016/j.bbagrm.2018.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang S, Wei J, Cui YH, Park G, Shah P. m(6)A mRNA demethylase FTO regulates melanoma tumorigenicity and response to anti-PD-1 blockade. Nat Commun. 2019;10:2782. doi: 10.1038/s41467-019-10669-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Biamonti G, Maita L, Montecucco A. The Kreb’s cycle connection: reciprocal influence between alternative splicing programs and cell metabolism. Front Oncol. 2018;8:408. doi: 10.3389/fonc.2018.00408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rosenfeld YB, Krumbein M, Yeffet A, Schiffmann N, Mishalian I, Pikarsky E, Oberman F, Fridlender Z, Yisraeli JK. VICKZ1 enhances tumor progression and metastasis in lung adenocarcinomas in mice. Oncogene. 2019;38:4169–4181. doi: 10.1038/s41388-019-0715-8. [DOI] [PubMed] [Google Scholar]
- 47.Du Y, Hou G, Zhang H, Dou J, He J, Guo Y, Li L, Chen R, Wang Y, Deng R, Huang J, Jiang B, Xu M, Cheng J, Chen GQ, Zhao X, Yu J. SUMOylation of the m6A-RNA methyltransferase METTL3 modulates its function. Nucleic Acids Res. 2018;46:5195–5208. doi: 10.1093/nar/gky156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheng T, Zhan X. Pattern recognition for predictive, preventive, and personalized medicine in cancer. EPMA J. 2017;8:51–60. doi: 10.1007/s13167-017-0083-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lu M, Zhan X. The crucial role of multiomic approach in cancer research and clinically relevant outcomes. EPMA J. 2018;9:77–102. doi: 10.1007/s13167-018-0128-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li N, Zhan X. Signaling pathway network alterations in human ovarian cancers identified with quantitative mitochondrial proteomics. EPMA J. 2019;10:153–172. doi: 10.1007/s13167-019-00170-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu T, Yang S. Dysregulated N6-methyladenosine methylation writer METTL3 contributes to the proliferation and migration of gastric cancer. J Cell Physiol. 2020;235(1):548–562. doi: 10.1002/jcp.28994. [DOI] [PubMed] [Google Scholar]
- 52.Niu Y, Lin Z, Wan A, Chen H, Liang H, Sun L, Wang Y, Li X, Xiong XF, Wei B, Wu X, Wan G. RNA N6-methyladenosine demethylase FTO promotes breast tumor progression through inhibiting BNIP3. Mol Cancer. 2019;18:46. doi: 10.1186/s12943-019-1004-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang Z, Tang K, Zhang D, Wan Y, Wen Y, Lu Q, Wang L. High-throughput m6A-seq reveals RNA m6A methylation patterns in the chloroplast and mitochondria transcriptomes of Arabidopsis thaliana. PLoS One. 2017;12:e0185612. doi: 10.1371/journal.pone.0185612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liao SY, Kuo IY, Chen YT, Liao PC, Liu YF, Wu HY, Lai WW, Wang YC. AKT-mediated phosphorylation enhances protein stability and transcription activity of ZNF322A to promote lung cancer progression. Oncogene. 2019;38:6723–6736. doi: 10.1038/s41388-019-0928-x. [DOI] [PubMed] [Google Scholar]
- 55.Cao J, Zhu Z, Wang H, Nichols TC, Lui GYL, Deng S, Rejto PA, VanArsdale T, Hardwick JS, Weinrich SL, Wei P. Combining CDK4/6 inhibition with taxanes enhances anti-tumor efficacy by sustained impairment of pRB-E2F pathways in squamous cell lung cancer. Oncogene. 2019;38:4125–4141. doi: 10.1038/s41388-019-0708-7. [DOI] [PubMed] [Google Scholar]
- 56.Peng Z, Wang J, Shan B, Li B, Peng W, Dong Y, Shi W, Zhao W, He D, Duan M, Cheng Y, Zhang C, Duan C. The long noncoding RNA LINC00312 induces lung adenocarcinoma migration and vasculogenic mimicry through directly binding YBX1. Mol Cancer. 2018;17:167. doi: 10.1186/s12943-018-0920-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Seifirad S, Haghpanah V. Inappropriate modeling of chronic and complex disorders: how to reconsider the approach in the context of predictive, preventive and personalized medicine, and translational medicine. EPMA J. 2019;10:195–209. doi: 10.1007/s13167-019-00176-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang S, Chai P, Jia R, Jia R. Novel insights on m(6)A RNA methylation in tumorigenesis: a double-edged sword. Mol Cancer. 2018;17:101. doi: 10.1186/s12943-018-0847-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PNG 13244 kb)
(XLS 472 kb)
(XLS 27 kb)
(XLSX 75 kb)
(XLS 183 kb)
(XLS 26 kb)
(XLSX 11 kb)