

Abstract
Alveolar macrophages (AMs) and epithelial cells (ECs) are the lone resident lung cells positioned to respond to pathogens at early stages of infection. Extracellular vesicles (EVs) are important vectors of paracrine signaling implicated in a range of (patho)physiologic contexts. Here we demonstrate that AMs, but not ECs, constitutively secrete paracrine activity localized to EVs which inhibits influenza infection of ECs in vitro and in vivo. AMs exposed to cigarette smoke extract lost the inhibitory activity of their secreted EVs. Influenza strains varied in their susceptibility to inhibition by AM‐EVs. Only those exhibiting early endosomal escape and high pH of fusion were inhibited via a reduction in endosomal pH. By contrast, strains exhibiting later endosomal escape and lower fusion pH proved resistant to inhibition. These results extend our understanding of how resident AMs participate in host defense and have broader implications in the defense and treatment of pathogens internalized within endosomes.
Keywords: alveolar macrophage, endosome fusion, epithelial cell, extracellular vesicles, influenza
Subject Categories: Membrane & Intracellular Transport; Microbiology, Virology & Host Pathogen Interaction
Extracellular vesicles secreted from alveolar macrophages can promote acidification of endosomes in influenza virus‐infected epithelial cells to inhibit viral replication.

Introduction
Influenza respiratory infection is a global health problem affecting people of all ages. Patients who smoke or have chronic lung disease have enhanced susceptibility and more severe infection (Arcavi & Benowitz, 2004). Influenza is transmitted person‐to‐person to the proximal airways where it infects and replicates within epithelial cells (ECs). As the replication cycle generates infectious virions, these travel distally to infect ECs in the delicate alveolar spaces responsible for gas exchange. The subsequent occurrence of diffuse alveolar damage is a major determinant of the morbidity and mortality associated with influenza (Korteweg & Gu, 2008; Taubenberger & Morens, 2008). Current pharmacologic treatments and vaccination strategies for influenza have limited applications and efficacy (Dobson et al, 2015; CDC, 2020), and improved approaches are needed. Naïve individuals require up to 5 days for the development of specific T‐ and B‐cell responses to influenza (Hermans et al, 2018). Therefore, the resident immune cells of the lung—alveolar macrophages (AMs) and the alveolar ECs (AECs) that comprise the alveolar surface—have an indispensable role in host defense at the early stages of infection. An improved understanding of these early innate immune protective mechanisms and how they become dysregulated is required to inform future therapeutic approaches.
The AM is uniquely adapted to cope with the distinct and numerous challenges of this microenvironment. While resident AMs are now considered to be protective against influenza (Kim et al, 2008, 2013; Purnama et al, 2014; Schneider et al, 2014; Cardani et al, 2017), the mechanisms responsible remain to be elucidated. Electron microscopic morphometric analysis of human (Fehrenbach et al, 1994) and mammalian (Hyde et al, 2004) lungs as well as recent live microscopic studies of the mouse lung (Westphalen et al, 2014) demonstrate that there are fewer than one AM per each pulmonary alveolus. Moreover, and contrary to traditional notions, live microscopy has suggested that AMs may remain relatively stationary in vivo (Westphalen et al, 2014). Taken together, these observations strongly favor an important role for paracrine communication in the ability of AMs to protect AECs from influenza infection.
Extracellular vesicles (EVs) represent one mode of paracrine intercellular communication whose importance is increasingly appreciated over the last decade. EVs are small (< 1 μm) membrane‐delimited structures emanating from endosomal or plasma membranes of a wide range of cell types and organisms. Surface proteins on EVs can neutralize extracellular antigens or engage surface receptors on target cells (Lima et al, 2009; Atay et al, 2011). Additionally, biologically active vesicular cargo (lipids, nucleic acids, proteins) can exert intracellular actions in recipient cells upon internalization of EVs (Pitt et al, 2016; Robbins et al, 2016). The packaging of cargo molecules within EVs serves to protect them from degradation in the extracellular space (Zhang et al, 2015). Recent work from our laboratory has revealed that cues from neighboring AECs and external stimuli [e.g., soluble mediators, cigarette smoke (CS), and lipopolysaccharide] can rapidly modify the number of secreted AM‐derived EVs, their cargo, and their uptake, with resulting modulation of inflammatory responses within target AECs (Bourdonnay et al, 2015; Speth et al, 2016; Schneider et al, 2017). These features position EVs as nimble vectors for signaling within the dynamic lung environment.
Despite the public health impact of influenza, little is known about cell communication via EVs in the host response to this infection, and EVs secreted from AMs in influenza have not been considered. Using in vitro and in vivo models of alveolar communication, we investigated the ability of EVs secreted from AMs to inhibit the replication within AECs of a panel of patient‐derived influenza viruses. We report here that AM‐derived EVs are capable of inhibiting only a subset of these influenza strains. Only those strains susceptible to inhibition by EVs exhibited defects in endosomal fusion and egress. This differential strain susceptibility facilitated the determination that AM‐EVs inhibit influenza replication in AECs by enhancing endosomal acidification. These studies provide new insights into the mechanisms by which AMs promote anti‐viral defense within the lung and reveal important characteristics of influenza virus that facilitate evasion of the early innate immune response. In addition, these studies reveal an important consequence of EV uptake within endosomes which has potential relevance to numerous endocytosis‐dependent pathophysiologic processes beyond influenza.
Results
Paracrine inhibitory activity of AMs against laboratory strains of influenza infection localizes to their secreted EVs
Investigation of the paracrine anti‐viral activity of AMs requires a sensitive assay of influenza replication. A previously constructed luciferase reporter was inserted downstream of the PA segment in the genome of the commonly studied laboratory strain of influenza A/WSN/33 (Tran et al, 2013). The resulting Luc‐WSN/33 generates luminescence with high specific activity from infected cells which correlates with virus replication. Luc‐WSN/33 infectivity is similar to that of its unmodified wild‐type (WT) counterpart. This permits a sensitive, time‐integrated, high‐throughput assay of viral replication.
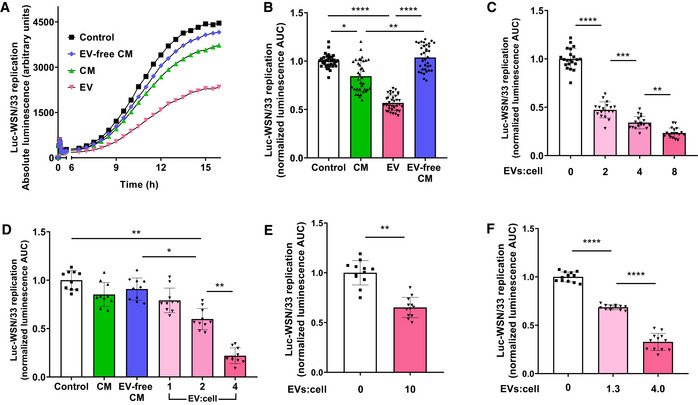
We set out to determine the capacity of substances constitutively secreted by AMs to protect ECs against influenza infection. We therefore collected conditioned medium (CM) from the mouse MH‐S AM cell line which we have previously demonstrated readily sheds EVs with functional and structural characteristics similar to those of primary AMs (Bourdonnay et al, 2015; Schneider et al, 2017). Luc‐WSN/33 infection was initially assayed in MDCK‐SIAT1 cells derived from the standard (Green, 1962) MDCK cells, and which were engineered to overexpress α2,6‐sialic acid for improved infectivity of recent circulating influenza strains (Oh et al, 2008). Indeed, CM from naïve AMs inhibited Luc‐WSN/33 replication in these ECs (Fig 1A and B). This inhibitory effect of CM was localized to ultracentrifugation‐purified EVs contained within CM (Fig 1A and B) and was not shared by the remaining EV‐free CM. This inhibition of EC influenza replication increased with the dose of AM‐EVs provided (Fig 1C) and was not the result of decreased EC viability (Appendix Fig S1). This inhibition of influenza replication in ECs by MH‐S cell EVs was confirmed by using EVs isolated from primary mouse AMs (Fig 1D) or peritoneal macrophages (Fig EV1) both obtained by lavage from naïve C57BL/6 mice and then subjected to retroviral immortalization. Finally, EVs isolated from primary rat AMs isolated by lung lavage (Fig 1E) and human macrophages differentiated from the THP‐1 monocyte cell line (Fig 1F) also demonstrated inhibition of influenza replication. These data demonstrate that the inhibitory activity against influenza constitutively secreted by AMs localizes to EVs.
Figure 1. Paracrine inhibitory activity of AMs against laboratory strains of influenza infection localizes to their secreted EVs.
-
A, BMDCK‐SIAT1 cells co‐incubated with Luc‐WSN/33 and MH‐S CM fractions. Replication was quantified by luminescence. (A) Replication curves display average luminescence reads from 5 wells per condition (measured at 7.5 min intervals—displayed here at 30 min intervals) from 1 experiment representative of 7 independent experiments. (B) Mean calculated area under the luminescence curves (AUC) for individual wells normalized to the corresponding mean of the control condition from these 7 experiments.
-
CLuc‐WSN/33 replication in MDCK‐SIAT1 cells incubated with flow cytometry‐quantified MH‐S AM‐EVs at specified EV:cell ratios.
-
DLuc‐WSN/33 replication in MDCK‐SIAT1 cells incubated with CM fractions of J2 retrovirus‐immortalized primary mouse AMs.
-
ELuc‐WSN/33 replication in MDCK‐SIAT1 cells incubated with primary rat AM‐derived EVs.
-
FLuc‐WSN/33 replication in MDCK‐SIAT1 cells incubated with human THP‐1 macrophage‐derived EVs.
Figure EV1. EVs isolated from immortalized mouse primary peritoneal macrophages inhibit influenza replication.
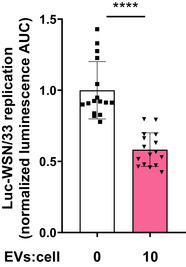
MDCK‐Siat1 cells were co‐incubated with Luc‐WSN/33 and EVs isolated from immortalized primary mouse peritoneal macrophages. Luc‐WSN/33 replication in each condition represented by mean luminescence AUC for individual wells normalized to the mean of the control condition from 4 independent experiments.Data information: Error bars = SD. Unpaired Student's t‐test (****P value of < 0.0001).Source data are available online for this figure.
AM‐EVs inhibit influenza infection of AECs in vitro and in vivo
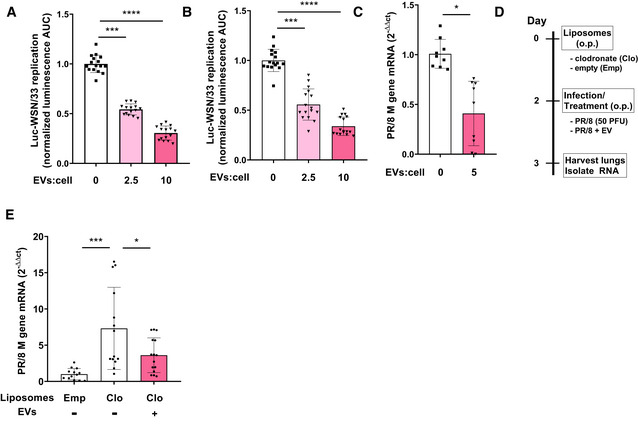
To confirm that inhibition of influenza replication observed in MDCK‐SIAT1 ECs (Fig 1) applies to AECs, we tested the effects of AM‐EVs on the human A549 carcinoma (Fig 2A) and mouse MLE‐12 (Fig 2B) AEC lines infected with Luc‐WSN/33. A dose‐dependent effect of AM‐EVs against influenza replication was also observed in these AEC lines. In separate experiments, we validated the inhibitory effect of AM‐EVs on infection of MLE‐12 AECs with an additional WT laboratory strain PR/8 by more conventionally assessing replication using RT–qPCR analysis of the influenza M1 gene (Fig 2C).
Figure 2. AM‐EVs inhibit influenza replication in AECs in vitro and in vivo .
-
A, BLuc‐WSN/33 replication co‐incubated with MH‐S EVs was assessed in (A) human A549 and (B) mouse MLE‐12 AECs. Data represent mean luminescence AUC from individual wells normalized to the mean of the corresponding control condition from 3 independent experiments.
-
CMLE‐12 AECs incubated with MH‐S EVs infected with PR/8 influenza. Replication was quantified by RT–qPCR at 12 h post‐infection. Data represent mean M gene transcripts normalized to β‐actin from 3 independent experiments.
-
DSchematic outline for AM depletion in lungs of mice with o.p. Clo followed by intrapulmonary delivery of AM‐EVs PR/8 infection.
-
ETotal lung RNA was isolated from mice on day 1 post‐infection and virus was quantified by RT–qPCR. Data represent mean M gene transcripts from individual mice normalized to β‐actin from 3 independent experiments (4–5 mice per group).
Our findings that AM‐EVs protect AECs against influenza offers a possible mechanistic explanation for previous in vivo studies in multiple species in which depletion of AMs leads to exacerbation of influenza‐related pathology (Kim et al, 2008, 2013; Purnama et al, 2014). We therefore assessed whether intrapulmonary delivery of MH‐S cell EVs could rescue the impaired host defense against influenza exhibited by mice depleted of AMs following intrapulmonary instillation of clodronate‐loaded liposomes (Clo) (Fig 2D). As we have reported previously (Zaslona et al, 2014), Clo resulted in 80% depletion of AMs at day 2 compared to empty liposomes (Emp) (Appendix Fig S2). At this time point, AM‐depleted mice were infected oropharyngeally (o.p.) with the PR/8 influenza strain with and without co‐administration of MH‐S AM‐EVs. Consistent with previous reports (Tate et al, 2010), Clo depletion of AMs resulted in higher viral transcript levels in the lungs of influenza‐infected mice compared to mice treated with Emp (Fig 2E). Indeed, o.p. treatment with AM‐EVs significantly lowered influenza transcript levels in lungs from AM‐depleted influenza‐infected mice, confirming that AM‐EVs inhibit influenza replication in vivo.
Cell‐type specificity, modulation, and characteristics of the AM‐EV activity against influenza
That AECs were protected against influenza infection by AM‐EVs suggests that these EVs contain bioactive constituents not elaborated by AECs themselves. To test this hypothesis, EVs were isolated in parallel from MH‐S AMs and A549 cells, quantified by flow cytometry, and incubated at equal concentrations with Luc‐WSN/33‐infected MDCK‐SIAT1 cells (Fig 3A). In contrast to the effects of MH‐S‐EVs (Fig 3A, middle bar), the same number of A549‐EVs had no significant inhibitory action in MDCK‐SIAT1 cells (Fig 3A, right bar), demonstrating that the inhibitory capacity of AM‐EVs against influenza is not a property shared by all alveolar cell‐derived EVs.
Figure 3. Cell‐type specificity, modulation, and characteristics of the AM‐EV activity against influenza.
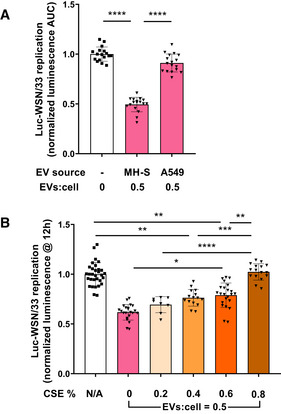
- EVs isolated from MH‐S AMs and A549 AECs were co‐incubated in parallel with Luc‐WSN/33 (MOI = 0.5) in MDCK‐SIAT1 cells. Data represent mean luminescence AUC from individual wells normalized to the mean of the corresponding control condition from 3 independent experiments.
- EVs isolated from MH‐S cells treated with increasing concentrations of CSE were co‐incubated with Luc‐WSN/33 in MLE‐12 cells. Data represent mean luminescence at 12 h post‐infection from individual wells normalized to the mean of untreated AM‐EVs from 3 independent experiments per condition.
Both experimental and epidemiologic studies have concluded that cigarette smoking (CS) and exposure both increase susceptibility to various infections, including influenza (Arcavi & Benowitz, 2004). This vulnerability occurs despite intact humoral immunity (Robbins et al, 2006), suggesting that CS dysregulates important innate immune responses to influenza. Therefore, we tested the effect of CS exposure of AMs on the anti‐viral function of their secreted EVs using an established in vitro model of CS exposure (Phipps et al, 2010), aqueous CS extract (CSE). EVs were isolated from MH‐S cells incubated with increasing concentrations of CSE, quantified by flow cytometry, and delivered at equal concentrations to Luc‐WSN/33‐infected MLE‐12 cells (Fig 3B). CSE treatment of AMs dose‐dependently attenuated the inhibitory actions of their EVs on Luc‐WSN/33 replication in AECs. With exposure of MH‐S cells to 0.8% CSE, their secreted EVs lost all activity against Luc‐WSN/33 replication.
The role of coding and non‐coding RNA species in mediating the biological actions of EVs has gained substantial interest (Mateescu et al, 2017). We therefore developed a means to selectively deplete RNA within AM‐EVs to examine the impact of this macromolecule class on the anti‐viral actions of EVs. Subjecting plasma membranes to multiple freeze‐thaw cycles is known to result in their permeabilization (Mardones & Gonzalez, 2003) and has been shown to provide access to constituents contained within EVs (Genschmer et al, 2019). This permeabilization method was used to facilitate incorporation of RNase A (Fig EV2) to permit the selective degradation of RNA within EVs. This protocol yielded the same number of EVs by flow cytometry, depleted ~ 90% of total RNA within EVs (Fig EV2, right column vs. middle column), and yet failed to abrogate the inhibitory effect of AM‐EVs on Luc‐WSN/33 replication in ECs. These data thus implicate non‐RNA cargo within EVs as the predominant source of activity against influenza. Of note, the vast majority of characterized endogenous inhibitors of influenza infection are proteins (Iwasaki & Pillai, 2014). Moreover, we previously demonstrated that CS disrupts the packaging of selected proteins into AM‐EVs (Bourdonnay et al, 2015). We therefore speculated that protein cargo may mediate the “anti‐viral” actions of constitutively released AM‐EVs and that CSE might result in a reduction in the incorporation of this protein cargo within AM‐EVs.
Figure EV2. AM‐EV inhibitory activity against influenza is not sensitive to RNase A.
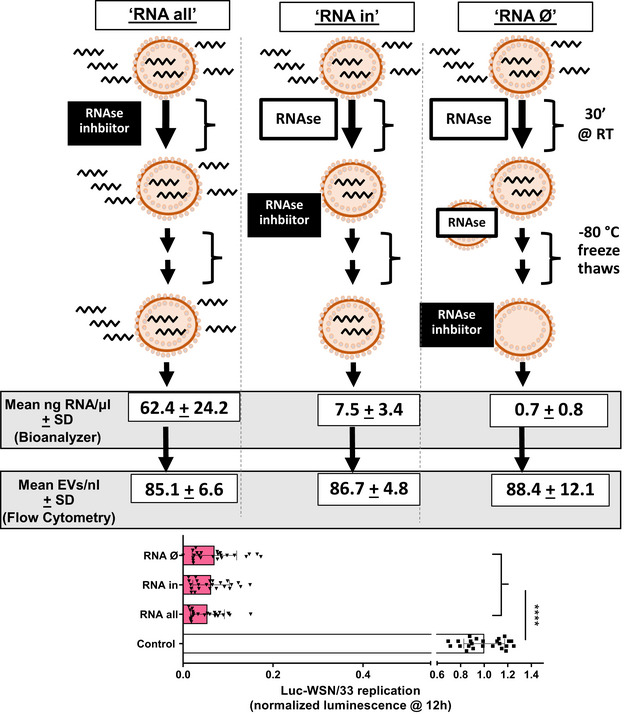
Schematic outline of the strategy to deplete RNA within EVs. Three groups of EVs were isolated from equal volumes of MH‐S CM, subsequently treated with either RNase A, and ultimately subjected to gentle permeabilization by repeated freeze‐thaw cycles at −80°C to permit RNase A entry into EVs. This permeabilization occurred before (right column) or after (left and middle columns) addition of an RNAse inhibitor. Analysis of EVs in each condition included RNA quantification with Agilent Bioanalyzer 2100 and TapeStation Analysis Software 3.1.1 to quantify the RNA present within EVs (n = 2) or quantified with flow cytometry (n = 2). These EVs were co‐incubated with Luc‐WSN/33 in MDCK‐SIAT1 cells, and replication was assessed using luminescence. Data represent mean luminescence at 12 h post‐infection from individual wells normalized to control from 4 independent experiments.Data information: Error bars = SD. One‐way ANOVA (****P value of < 0.0001).Source data are available online for this figure.
To identify candidate proteins within AM‐EVs that might mediate the inhibitory activity against influenza replication within AECs and whose quantity was reduced by CSE, we assessed the differential abundance of proteins from control‐treated and 0.8% CSE‐treated AM‐EVs using tandem mass tag mass spectrometry (TMT‐MS) (McAlister et al, 2014). A schematic outline of the TMT workflow is shown in Fig EV3. Of the ~ 4,500 EV proteins identified with high confidence, only eight proteins exhibiting statistically significant differential abundance (−log10 q value ≥ 2, Fig EV3B, y‐axis) and > 25% absolute difference in abundance ratio (Fig EV3B, x‐axis), and all were upregulated with CSE. Therefore, this method failed to identify any EV protein candidates that could account for the activity of naïve AM‐derived EVs against influenza and whose abundance is reduced by CSE.
Figure EV3. Differential abundance of proteins within AM‐EVs in response to CSE .
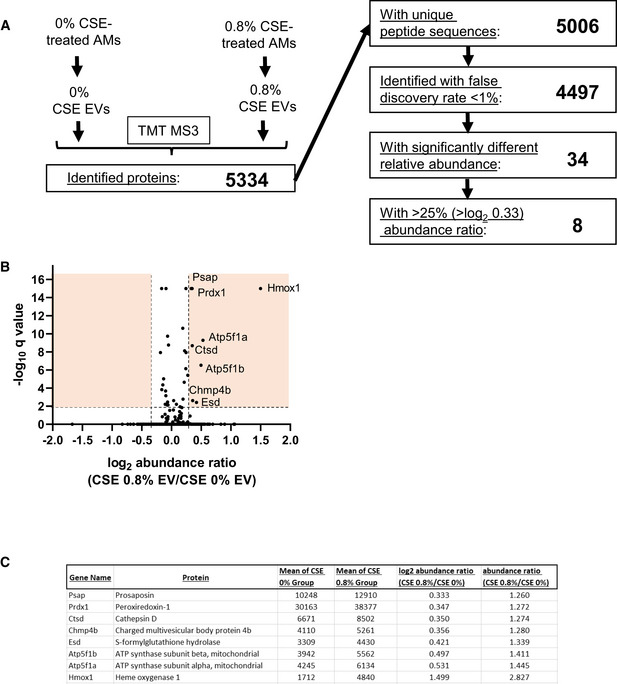
- Flowchart demonstrating the number proteins within each EV condition compared using TMT‐MS and each of the listed criteria.
- Volcano plot of all 4,497 proteins identified in both EV conditions. Orange boxes collectively denote the proteins with significantly different relative abundance (a −log10 q value ≥ 2) and with > 25% difference in abundance ratio.
- List of the 8 proteins identified that exhibited > log2 0.33 in abundance in EVs isolated from 0.8% CSE‐treated AMs compared to 0% CSE‐treated AM‐EVs.
Patient‐derived influenza isolates exhibit strain‐dependent sensitivity to the inhibitory effects of AM‐EVs
The WSN/33 and PR/8 influenza strains utilized thus far have been subjected to decades of adaptation to mice and laboratory cell lines through serial passaging. Consequently, these strains no longer cause disease in wild mice or humans (Staeheli et al, 1988; Thangavel & Bouvier, 2014), and their virulence mechanisms vary significantly from those of seasonal and pandemic strains isolated from infected humans (Gitelman et al, 1984; Kaverin et al, 1989; Brown et al, 2001; Xu et al, 2010, 2011). Thus, the clinical relevance of results obtained with these laboratory strains requires validation from recent patient‐derived isolates (listed in Key Resources Table). MDCK‐SIAT1 cells were infected with these strains in the absence and presence of AM‐EVs (Fig 4A), and replication in each condition was quantified by RT–qPCR. Replication of the laboratory‐adapted strain WSN/33, like PR/8 (Fig 2C), was sensitive to inhibition by AM‐EVs using this readout. The same was true for three of the patient‐derived influenza strains (WI/05, WY/03, and CA/09). In contrast, two of the remaining strains (Sing/16 and HK/14) were completely resistant to AM‐EVs.
Figure 4. Patient‐derived influenza isolates exhibit strain‐dependent sensitivity to the inhibitory effects of AM‐EVs.
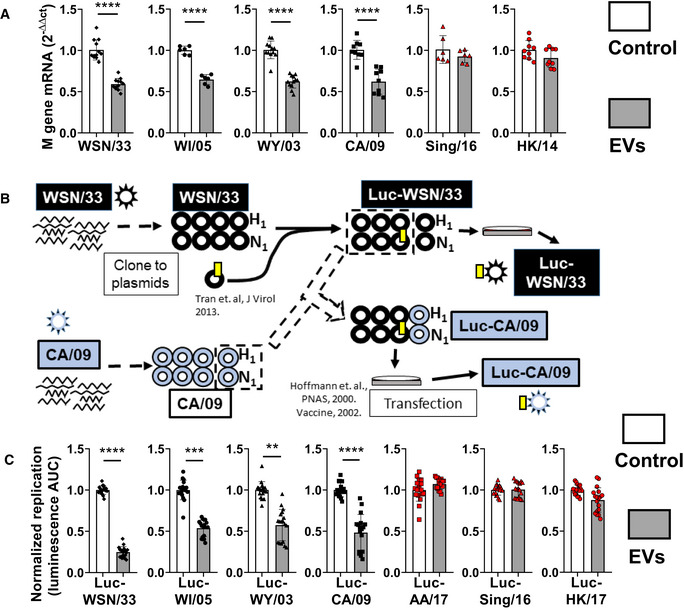
- MLE‐12 AECs co‐incubated with the listed WT strains (MOI = 0.4) plus AM‐EVs (gray bars, EV:cell = 1). Data represent mean M gene transcripts at 12 h post‐infection relative to the mean of the corresponding untreated strain (white bars), each normalized to β‐actin from 2 to 4 independent experiments per strain.
- Schematic flow diagram of the generation of luciferase‐expressing patient‐derived influenza viruses. The generation of the novel Luc‐CA/09 strain is shown as a representative example.
- MH‐S EVs were co‐incubated with the listed Luc‐expressing strains plus AM‐EVs (gray bars, EV:cell = 2) in MDCK‐SIAT1 cells. Data represent mean luminescence AUC from individual wells normalized to the means of the corresponding untreated strain (white bars) from 3 independent experiments per strain.
To explore these differences in sensitivity to AM‐EVs in a higher‐throughput manner, we generated novel Luc‐expressing patient‐derived strains. As schematized in Fig 4B, this was accomplished by combining the hemagglutinin (HA) and neuraminidase (NA) from respective patient‐derived strains with the remaining six genome segments of the Luc‐WSN/33 strain. These resulting strains were incubated in the absence and presence of AM‐EVs in MDCK‐SIAT1 cells and replication was quantified by luminescence (Fig 4C). As with the corresponding WT strains, replication of Luc‐expressing WSN/33, WI/05, WY/03, and CA/09 strains was sensitive, whereas that of Luc‐expressing Sing/16, HK/14, and AA/17 was resistant to inhibition by AM‐EVs. Because the Luc‐expressing strains differ only in their HA and NA proteins (Fig 4B), these results indicate that AM‐EVs inhibit influenza infection in AECs at a stage that is HA‐ or NA‐dependent.
AM‐EVs prevent the fusion of influenza virus with endosomal membranes in susceptible strains
The influenza virus replication cycle involves nine distinct stages defined in ECs (Matsuoka et al, 2013). Those stages which depend on HA and NA include plasma membrane binding (HA‐ and NA‐dependent), endosomal fusion (HA‐dependent), virion assembly (HA‐ and NA‐dependent), and release from the plasma membrane (NA‐dependent). Non‐AM‐EVs have previously been reported to bind influenza virus extracellularly and thereby prevent infection of ECs (Kesimer et al, 2009; Suptawiwat et al, 2017). By contrast, we have previously reported that effects of these AM‐EVs on AEC signaling require their internalization (Schneider et al, 2017). To directly address whether an extracellular interaction between influenza and EVs was required for inhibition in our model, MDCK‐SIAT1 cells were preincubated with AM‐EVs for 8 h, after which the remaining free EVs were washed away and cells were then infected with Luc‐WSN/33 (Fig 5A). Inhibition of Luc‐WSN/33 infection by AM‐EVs was nonetheless evident even when delivered prior to virus, thus excluding any direct extracellular interaction. To directly assess whether EVs inhibited binding of influenza to the plasma membrane, MLE‐12 AECs were co‐incubated with PR/8 virus and EVs at 4°C (allowing virion binding but not internalization), and virus bound to AECs was quantified by RT–qPCR (Fig 5B) or visualized by immunofluorescence microscopy (Fig 5C); no difference in bound influenza virus was detected by either means, indicating that EVs did not inhibit binding of virions to the plasma membrane. These results are consistent with close inspection of our Luc‐WSN/33 replication curves generated with or without EVs (example shown in Fig EV4). These consistently reveal a significant divergence in replication that occurs exclusively after 1 h post‐infection. Given that the length of the influenza replication cycle is 6–8 h (Baccam et al, 2006; Frensing et al, 2016), these data collectively suggest that EVs inhibit an early, intracellular, HA‐/NA‐dependent stage of the influenza replication cycle.
Figure 5. AM‐EVs prevent the fusion of influenza virus with endosomal membranes in susceptible strains.
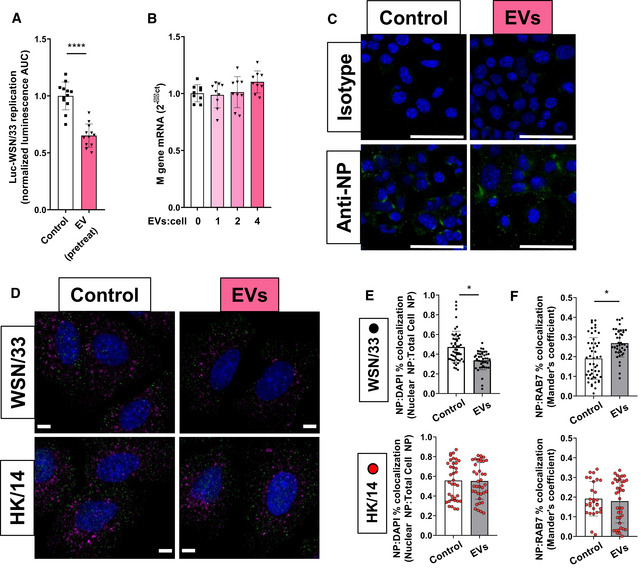
-
AMDCK‐SIAT1 cells were pretreated with MH‐S EVs (EV:cell = 2, “EV pretreat”) or control for 8 h, washed, and subsequently incubated with Luc‐WSN/33 (MOI = 1). Data represent mean luminescence AUC from individual wells normalized to the means of the control condition from 3 independent experiments.
-
B, CMLE‐12 cells were incubated with WT PR/8 ± AM‐EVs at 4°C (to permit surface binding but not entry) for 1 h, washed, and processed for (B) RNA isolation or (C) immunofluorescence microscopy. (B) Data represent mean M gene transcripts relative to the means of the corresponding untreated condition, each normalized to β‐actin from 3 independent experiments. (C) Cells in both conditions were fixed and stained with anti‐NP or isotype control Ab. Images are representative of two independent experiments. Scale bars = 50 μm.
-
DA549 cells were incubated with WSN/33 or HK/14 ± AM‐EVs for 40 min, washed, fixed, and permeabilized. Cells were subsequently stained with anti‐NP (green), anti‐Rab‐7 (purple) Abs, and DAPI (blue). Images shown are representative of 8 independent experiments. Scale bars = 5 μm.
-
E, FCo‐localization analysis of (E) NP:DAPI and (F) NP:Rab7 for each condition was performed on Z‐stack confocal images of WSN/33 (black) or HK/14 (red) infected A549s. Data represent the mean NP:DAPI co‐localization ratios and the mean NP:Rab7 Mander's co‐localization coefficient for individual cells from these 8 experiments.
Figure EV4. AM‐EVs inhibit Luc‐WSN/33 replication within 3 h.
Representative example of measured luminescence from Luc‐WSN/33‐infected MDCK‐SIAT1 cells with (triangles) and without (squares) AM‐EVs (EV:cell = 4). Curves shown from this representative experiment display average luminescence reads of 6 wells per condition.Data information: Mean AUC between curves between 0–1 h were not statistically different, whereas mean AUC between curves 1–3 h were significantly different (P < 0.0001, unpaired Student's t‐test).Source data are available online for this figure.
Of the stages previously mentioned, HA‐dependent endosomal fusion would fulfill all of these criteria. Consistent with observations in other recipient cell types (Mulcahy et al, 2014), we have reported that AM‐EVs are internalized by AECs into endosomes (Schneider et al, 2017), thus providing EVs the opportunity to inhibit the HA‐dependent endosomal fusion of susceptible strains. To evaluate this, A549 cells were co‐incubated with WSN/33 (susceptible to inhibition by EVs) and AM‐EVs for 40 min. This time point was chosen to permit determination of both endosomal and nuclear co‐localization prior to the synthesis and assembly of new viral genes (Bayer et al, 1998; Qin et al, 2019). The nuclear localization of WSN/33 virus (demonstrating successful endosomal fusion) was imaged using immunofluorescence confocal microscopy, quantified with FIJI software, and compared to nuclear localization of HK/14‐infected (shown to be resistant to inhibition by EVs in Fig 4) cells treated with AM‐EVs. AM‐EVs reduced the nuclear localization of influenza NP protein in AECs infected with WSN/33 (Fig 5D and E). In contrast, AM‐EVs failed to alter the nuclear localization of virus in HK/14‐infected cells (Fig 5D and F). Consistent with these changes in nuclear entry, AM‐EVs increased the number of WSN/33 virions within late endosomes as measured by NP co‐localization with the late endosomal marker Rab7 (Fig 5D and E). By contrast, there was no difference in NP/Rab7 co‐localization in HK/14‐infected cells treated with EVs (Fig 5D and F). These data suggest that AM‐EVs inhibit the HA‐dependent endosomal fusion of susceptible strains of influenza in AECs.
Influenza strain susceptibility to AM‐EVs is associated with its timing of endosomal escape and sensitivity to endosomal acidification inhibitors
In order for HA to bind and fuse to internal host endosomal membranes, it requires priming by host transmembrane serine proteases. Generally, these proteases cleave multiple HA subtypes (Bottcher et al, 2006; Chaipan et al, 2009), but host protease specificity to certain HA subtypes has been reported (Galloway et al, 2013). However, in the majority of cases, this proteolytic activation occurs extracellularly (Skehel & Wiley, 2000) and is artificially bypassed in standard in vitro influenza models (including our model) by the addition of TPCK‐treated trypsin in the viral growth medium. Therefore, the HA strain‐dependent intracellular inhibition of endosomal fusion by AM‐EVs observed in our model is unlikely to be explained by the modulation of proteolytic activation of HA by host protein(s). Following proteolytic activation, virions are internalized into endosomes which undergo progressive acidification as they transition from early to late endosomes (Wiley & Skehel, 1987). This results in a pH‐dependent conformational change in HA which facilitates its fusion with endosomal membranes, resulting in endosomal escape and subsequent trafficking of the viral genome into the nucleus. Each unique HA has a defined pH range of fusion (Galloway et al, 2013; Costello et al, 2015; Russier et al, 2016; Singanayagam et al, 2019).
To investigate whether the divergent susceptibility of these influenza strains to the inhibitory effects of AM‐EVs is associated with differences in their pH of fusion, the dynamics of their escape from endosomes was assayed with timed addition of a high dose of the endosomal acidification inhibitor bafilomycin A1 (Baf‐A1). Figure 6A and B show representative examples of endosomal escape assays performed on strains susceptible to AM‐EVs (gray bars) in direct comparison to strains which resist inhibition by AM‐EVs (white bars). When treated with high dose Baf‐A1 at the time of infection (time 0), all strains show > 80% inhibition demonstrating that each strain requires some degree of endosomal acidification to permit its fusion. Calculated as the slope of change in inhibition across multiple experiments (Fig 6C), the EV‐susceptible Luc‐WSN/33 and Luc‐WI/05 strains demonstrated significantly more escape from endosomes within 20 min when compared to the EV‐resistant strains Luc‐AA/17 and Luc‐HK/14. In contrast, Luc‐AA/17 and Luc‐HK/14 still exhibited endosomal escape between 60 and 80 min, yet Luc‐WSN/33 and Luc‐WI/05 had no detectable escape at this time point (Fig 6D). These data demonstrate that the strains susceptible to AM‐EV inhibition fuse with endosomes early (within 20 min) whereas resistant strains are still fusing with endosomes between 60 and 80 min. Because it is known that viruses with a relatively higher fusion pH generally fuse earlier in endosomes (White & Whittaker, 2016), these data suggest that strains susceptible to inhibition by AM‐EVs fusing earlier in endosomes are doing so at a relatively higher pH.
Figure 6. Influenza strains susceptible to inhibition by AM‐EVs escape endosomes early and are resistant to inhibitors of endosomal acidification.
-
A–DReplication assessed by luminescence following 10 nM Baf‐A1 addition at the indicated times following synchronized uptake of bound Luc‐expressing virions. (A, B) Shown are examples from 1 experiment representative of a total of 3–11 independent experiments per strain. Data represent mean luminescence AUC normalized to the mean of the corresponding strain incubated in 0.1% DMSO (dashed line). (C, D) The kinetics of endosomal escape by Luc‐WSN/33 and HK/14 strains are depicted by the loss of inhibition by high dose Baf‐A1 between (C) 0–20 min and (D) 60–80 min. Data represent mean slope (change in % inhibition/time interval) from 5 wells per plate from 3 to 11 independent experiments.
-
E, FShown are dose response curves (left) and corresponding IC50 values (right) for effects of the endosomal acidification inhibitors (E) Baf‐A1 and (F) Chl on replication of Luc‐expressing strains. Curves display mean luminescence AUC expressed as a percentage of replication of corresponding strains in control conditions from 3 to 5 independent experiments (mean of 3 wells per condition) for each strain and inhibitor concentration. Bar graphs depict mean IC50 values from these experiments.
In experiments complementary to Fig 6A–D, we further explored the endosomal fusion characteristics of these strains by quantifying their differential sensitivity to inhibition by Baf‐A1 and the additional endosomal acidification inhibitor chloroquine (Chl). We hypothesized that infection by strains fusing at a higher pH would tolerate higher doses of these acidification inhibitors. To test this, MDCK‐SIAT1 cells were pretreated with the indicated doses of Baf‐A1 (Fig 6E) or Chl (Fig 6F) prior to co‐incubation of the inhibitors with the indicated Luc‐influenza strains. Strains susceptible (Fig 6E and F—filled symbols) to inhibition by AM‐EVs (Fig 4) and exhibiting endosomal escape at early time points (Fig 6A–D) all had significantly higher IC50 concentrations for both Baf‐A1 and Chl when compared to strains resistant (open symbols) to AM‐EVs which escape endosomes at later time points. Collectively, these data demonstrate that influenza strains resistant to inhibition by AM‐EVs fuse with endosomes at a lower pH than do susceptible strains. Therefore, we hypothesized that internalized AM‐EVs lower the pH of endosomes outside the optimal fusion pH range of susceptible strains, but within the fusion pH range of resistant strains.
Internalization of AM‐EVs enhances the acidification of endosomes
To assay endosomal pH, we measured the pH‐sensitive change in ratiometric emission spectral intensities of internalized 3 kDa dextran conjugated to FITC (FDx) (Davis & Swanson, 2010; Ma et al, 2017). First, a pH standard curve for FDx internalized into MDCK‐SIAT1 cells was generated. Following overnight incubation with FDx, free FDx was washed away, pH was clamped at values chosen to fit the wide pH range of early to late endosomes which includes the narrower pH range of HA fusion. Emission (525/50) after sequential excitation at the pH‐insensitive 445 and pH‐sensitive 488 nm wavelengths was collected from these cells with confocal live cell microscopy. Distinct intracellular particles were identified, and the ratio of their emission intensities was recorded using FIJI software and subsequently fit to a standard curve (Fig 7A). To detect internalized AM‐EVs as particles distinct from FDx, EVs were labeled with a lipophilic deep red fluorescent marker (DR‐EVs) and incubated with MDCK‐SIAT1 cells. Emission from DR‐EVs internalized into MDCK‐SIAT1 cells was readily detected from a separate 700/75 bandpass filter (Fig 7B) and demonstrated no spectral crossover into the 525/50 filter (used to detect FDx) when excited at 488 nm (Fig EV5). Because sensitive strains demonstrated endosomal escape between 20–60 min (Fig 6A–C), we determined if EVs internalized into endosomes resulted in a measurable endosomal pH change within this time range. MDCK‐SIAT1 cells were pulsed with FDx with and without DR‐EVs for 20 min and subsequently chased for 5 min. Images were captured for the ensuing 35 min.
Figure 7. Internalization of AM‐EVs enhances the acidification of endosomes.
- Data are mean 525em ratio from 488/445 nm excitation intensities of internalized FDx particles vs. pH of clamping buffer fit to a standard curve from two independent experiments.
- Representative images of PBS‐ and DR‐EV‐treated MDCK‐SIAT1 cells. Scale bars for full size images = 50 μm. Scale bars for cropped images = 10 μm. FDx particles are shown green and DR‐EV particles are shown purple. White arrows are FDx endosomes containing DR‐EVs.
- Measured pH of FDx endosomes in PBS‐ (left bar) and DR‐EV‐treated (right bar) MDCK‐SIAT1 cells. The pH of FDx endosomes containing DR‐EVs in DR‐EV‐treated cells (middle bar) (corresponding to white arrows in Fig 7B). Data are the means of individual data points representing the average calculated pH of each endosome population within 1 hpf in each of 5 independent experiments.
Figure EV5. DR‐EVs exhibit no spectral overlap into the emission filter used to detect FDx.
To determine if DR‐EVs exhibited spectral overlap with FDx, the emission intensities of DR‐EVs (488ex/525em, top histogram) were compared to the background emission intensities (488ex/525em, middle histogram) within MDCK‐SIAT1 cells. Using FIJI software, ROIs containing DR‐EV particles internalized into MDCK‐SIAT1 cells were defined using 647ex/700em images. These ROIs corresponding to DR‐EVs were applied to 488ex/525em images to assess their spectral crossover into this emission range, and their corresponding intensities were plotted (top histogram). Particles within the DR‐EV ROIs were then cleared from the 488ex/525em images. To establish the background 488ex/525em intensity, new ROIs with the same dimensions were applied to the remaining area within these cells and their associated intensities were recorded and plotted (middle histogram). These histograms show that DR‐EVs exhibit no 488ex/525em above background (top vs. middle histogram). For comparison, 488ex/525em intensities of FDx particles were recorded and plotted (bottom histogram). Data represent all measured particles from all cells within 4 hpf per condition. Statistics for each histogram are listed in the figure.
In MDCK‐SIAT1 cells incubated with DR‐EVs, endosomes containing EVs exhibited significantly lower pH (Fig 7C, center bar) compared to endosomes from control cells (Fig 7C, left bar) and trended toward lower pH compared to endosomes without EVs from the same cells treated with DR‐EVs (Fig 7C, right bar). These data demonstrate that endosomes containing EVs mature to a lower pH range more rapidly than control endosomes. Overall, this indicates that AM‐EVs may inhibit influenza replication of susceptible strains by lowering endosomal pH.
Discussion
AMs are known to participate in host defense against influenza, but the operative mechanisms remain incompletely defined. Among those reported are their ability to sequester virus yet not support productive replication, ingest apoptotic bodies, clear cellular debris (Duan et al, 2017), and generate pro‐inflammatory cytokines in response to signals from infected AECs (Seo et al, 2004). However, their paracrine elaboration of signals that inhibit viral replication within AECs has received limited attention (Cardani et al, 2017) and a role for EVs in this process remains unexplored. In a series of in vitro and in vivo studies, we demonstrate that EVs secreted constitutively from naïve rodent and human macrophages are taken up within AEC endosomes, where they restrict the fusion of influenza virus, thereby preventing its nuclear entry and resulting replication. While this activity was shared by macrophage populations residing outside the alveolar compartment, it was not shared by AEC‐derived EVs. This activity within EVs was also abolished by exposure of the AM to CSE—providing a possible mechanistic explanation for the increased susceptibility to and severity of influenza infection recognized in smokers. This inhibitory activity was present against clinically relevant, patient‐derived influenza strains—including the pandemic CA/09 virus—which were sensitive to EV‐mediated decreases in endosomal pH. Other strains were resistant to this inhibitory effect providing new and clinically relevant insights into the innate immune repertoire by which AMs defend the lung against infection.
Endocytosis of extracellular components by AECs is a critical mechanism for preservation of gas exchange (Kim & Malik, 2003). We have reported that AECs utilize this pathway to internalize EVs that help to restrain inflammation in the challenging lung microenvironment (Bourdonnay et al, 2015; Schneider et al, 2017). Among other viruses, influenza has hijacked these endosomal entry pathways in order to gain access to the AEC nucleus essential to its replication cycle (Matsuoka et al, 2013). It has been argued that the shuttling of host and viral cargo through a common intercellular pathway represents coevolution rather than coincidence (Nolte‐‘t Hoen et al, 2016). It is therefore not surprising that the host has exploited this same pathway to protect against this viral infection, while certain strains of the virus may have adapted to circumvent this immune defense mechanism.
One theme of the limited investigations to date into EVs in influenza has been their ability to prevent infection by interfering with the virus binding to the surface of ECs (Kesimer et al, 2009; Suptawiwat et al, 2017). In contrast, our results clearly demonstrate an intracellular target (Figs 5 and EV4) for EVs secreted from AMs. To our knowledge, this reduction in endosomal pH (Fig 7) describes a novel functional consequence of EV uptake within a target cell. The other theme of prior research is immune cell activation (Admyre et al, 2006; Testa et al, 2010) or inhibition of infection (Maemura et al, 2018) by EVs secreted from cells artificially engineered to express specific peptides or microRNAs. Our studies reveal that AMs can defend the lung against influenza through secretion of naturally derived EVs. EVs were previously categorized as representing either of two major subsets, namely microvesicles or exosomes. However, it is now understood that there is substantial overlap in the size, biogenesis, and markers typical of EV subsets (Mateescu et al, 2017). Our demonstration that this anti‐viral activity is present within a particular defined population of AM‐EVs does not exclude the possibility that other subsets of AM‐EVs (isolated with methods distinct from those used in this manuscript) may or may not possess this activity. Although our study was focused on influenza, this phenomenon and its resulting impact on endosomal pH have broader implications for host defense and treatment against other pathogens internalized via endocytosis.
The pH of endosomal fusion of influenza strains is a defining characteristic contributing to their transmissibility, virulence, and tropism for certain host species and organs. For example, highly pathogenic avian and pandemic influenza strains (e.g., CA/09) are reported to fuse with endosomes at a higher pH, whereas seasonal strains typically have a lower pH of fusion (Imai et al, 2012; Galloway et al, 2013; Russier et al, 2016). Focusing on the relationship of this property with virulence, strains that escape endosomes relatively early were more capable of evading the effects of the interferon‐responsive IFITM protein‐mediated inhibition of endosomal fusion in ECs (Gerlach et al, 2017). Our study has identified a marked divergence in susceptibilities of different strains to innate immune defense via AM‐EVs that is linked to this fundamental property of the virus. In contrast to their resistance to IFITM‐mediated inhibition, our results demonstrate that viruses that escape the endosome comparatively early (Fig 6A–D) are susceptible to the innate protective effects of AM‐EVs. In this way, previously unrecognized paracrine signals from neighboring AMs which can rapidly endow AECs with resistance to influenza infection may compensate for a critical gap which exists in the better‐studied interferon‐dependent anti‐viral state against high‐fusion pH strains with considerable epidemiologic impact (e.g., highly pathogenic avian, CA/09).
Our results are in line with other studies demonstrating that influenza strains with higher pH of fusion are more susceptible to other early innate protective mechanisms in the lung (Singanayagam et al, 2019). However, strains with a higher pH of fusion may be more capable of infection and/or replication within AMs (Marvin et al, 2017). Whether infection of AMs disrupts their EV‐mediated host defense function is therefore an important subject for future studies. In addition, we observed that while the more recently isolated seasonal H3N2 strains HK/14, Sing/16, AA/17 were resistant to inhibition by AM‐EVs, older seasonal H3N2 isolates WI/05 and WY/03 were not. This indicates that strain susceptibility to AM‐EVs is not merely a function of its HA subtype. Rather, this result implies that a drift in the H3 subtype prior to 2014 changed its pH of fusion with a resulting ability to evade this innate immune defense. Precise differences between these strain H3 subtypes are therefore also worthy of future investigation.
RNA cargo within EVs has been implicated in many of their biological actions (Mateescu et al, 2017). This includes activity against influenza—an RNA virus—targeted by small EV‐derived RNAs in engineered in vitro models (Maemura et al, 2018) or those derived from other body fluids (Khatri et al, 2018). Therefore, it was reasonable to consider that the observed “anti‐viral” activity of AM‐EVs could be explained by RNA cargo. We sought to evaluate this possibility by determining if anti‐viral activity was lost when RNase A was incorporated within the EVs by freeze‐thaw (Fig EV2). To our knowledge, this is a novel experimental approach that may prove useful in other studies investigating the contribution of RNA cargo to EV actions. We found that 90% depletion of RNA within AM‐EVs failed to abrogate their inhibitory effects on influenza replication within ECs. A likely explanation for the lack of involvement of RNA relates to the fact that at the early stage of infection at which AM‐EVs restrict virions within endosomes (40 min, Fig 5), the viral genome is still enveloped and thus unlikely to be available as an inhibitory target for, for example, microRNA.
As an alternate mechanistic direction, we chose to survey the AM‐EV proteome in an effort to identify candidate proteins whose abundance within EVs was downregulated by CSE treatment of AMs, thus explaining the ability of CSE to abrogate the inhibitory activity against influenza (Fig 3B). We also envisioned that subsequent informatics analysis would refine the list of candidates based on the identified target within ECs—here shown to be the acidification of endosomes. The primary protein involved in the acidification of endosomes is the vacuolar ATPase (V‐ATPase) (Marvin et al, 2017). Despite the identification of ~ 4,500 EV proteins with high confidence, only a very small fraction of these were shown to exhibit differential abundance by TMT‐MS identification (Fig EV3), none were downregulated by CSE, and none of the components of the V‐ATPase were identified in either condition with high confidence. Of the eight differentially abundant proteins, four are described to influence intracellular redox tone which is noteworthy in that redox state determines the pump activity of the V‐ATPase (Cipriano et al, 2008). While this provides a plausible explanation for the loss of “anti‐viral” activity in EVs from CSE‐treated AMs, these upregulated candidates shed no light on the “anti‐viral” activity present constitutively within EVs derived from naïve AMs. One obvious limitation to the approach employed herein is that it will not identify proteins within EVs that are functionally inactivated (e.g., oxidation) by CSE, but whose abundance within EVs is preserved. It also remains possible that the baseline activity of EVs reflects the actions of multiple proteins and/or lipids. These results underscore the complexity of possibilities and the need for further research to define the active cargo.
Newer vaccine formulations incorporate adjuvants specifically based on their ability to activate innate immune pathways, which subsequently enhance humoral responses (Levitz & Golenbock, 2012). Thus, this study on fundamental innate protective mechanisms and how they become dysregulated has the potential to inform both the development of new therapeutic approaches as well as their eventual incorporation into optimal vaccination strategies. If the responsible cargo within EVs is ultimately identified, then a translational application of this study would be the encapsulation of this lone cargo within synthetic liposomes for treatment against applicable pathogens. Alternatively, if the activity of EVs described herein is not attributable to lone cargo, there are numerous applications of cell‐derived EVs which are currently undergoing clinical trials for the treatment of various diseases (Wiklander et al, 2019).
Materials and Methods
Reagents and Tools table
| Reagent/Resource | Reference or source | Identifier or catalog number |
|---|---|---|
| Experimental models | ||
| Mouse: MH‐S cells | ATCC | CRL‐2019 |
| Mouse: MLE‐12 cells | ATCC | CRL‐2110 |
| Human: 293T cells | ATCC | CRL‐3216 |
| Human: A549 cells | ATCC | CCL‐185 |
| Human: THP‐1 cells | ATCC | TIB‐202 |
| Canine: MDCK‐SIAT1 cells | Dr. Arnold S. Monto; University of Michigan School of Public Health | N/A |
| AMJ2‐C11 | ATCC | CRL‐2456 |
| J2‐immortalized primary mouse AMs | This paper | N/A |
| J2‐immortalized primary mouse PMs | This paper | N/A |
| Mouse: C57BL/6 | Jackson Laboratory | N/A |
| Rat: Wistar | Charles River | N/A |
| Antibodies | ||
| Rabbit monoclonal anti‐Rab7 | Cell Signaling Technology | Cat#9367; RRID:AB_1904103 |
| Rabbit IgG, polyclonal | Abcam | Cat#ab171870; RRID:AB_2687657 |
| Mouse monoclonal anti‐influenza NP | Abcam | Cat#ab20343; RRID:AB_445525 |
| Mouse IgG2a, kappa monoclonal [MG2a‐53]—Isotype control antibody | Abcam | Cat#ab18415, RRID:AB_2722535 |
| Goat polyclonal anti‐mouse IgG (H+L) Cross‐Adsorbed Secondary Antibody, Alexa Fluor 488 | Thermo Fisher | Cat#A‐11001, RRID:AB_2534069 |
| Goat polyclonal anti‐rabbit IgG (H+L) Cross‐Adsorbed Secondary Antibody, Alexa Fluor 568 | Thermo Fisher | Cat#A11001, RRID:AB_143157 |
| Chemicals, Enzymes and other reagents | ||
| Dextran, Fluorescein, 3000 MW | Thermo Fisher | Cat #D3305 |
| EnduRen Live Cell Substrate | Promega | Cat #E6481 |
| Bafilomycin‐A1 | Sigma‐Aldrich | Cat #B1793 |
| Chloroquine | Sigma‐Aldrich | Cat #C6628 |
| CellMask Deep Red Plasma membrane Stain | Thermo Fisher | Cat #C10046 |
| Clodronate and control liposomes (PBS) | Liposoma BV | Cat #CP‐005‐005 |
| RNAse A | Sigma‐Aldrich | Cat #10109169001 |
| RNAse inhibitor | Invitrogen | Cat #N8080119 |
| Cell‐Titer Glo Luminescent Cell Viability Assay | Promega | Cat #G7571 |
| Tandem Mass Tag 6‐plex Labeling Kit | Thermo Fisher | Cat #90061 |
| QuantiPro BCA Assay Kit | Sigma‐Aldrich | Cat #QPBCA |
| Total RNA Purification Micro Kit | Norgen BioTek | Cat #35300 |
| Primer: influenza M gene Forward: 5′‐GGACTGCAGCGTAGACGCTT‐3′ | IDT | N/A |
| Primer: influenza M gene Reverse: 5′‐CATCCTGTTGTATATGAGGCCCAT‐3′ | IDT | N/A |
| Probe: influenza M gene: 5′‐/56‐FAM/CTCAGTTAT/ZEN/TCTGCTGGTGCA/3IABkFQ/‐3′ | IDT | N/A |
| Primer: mouse β‐actin Forward: 5′‐CCGTGAAAAGATGACCCAGATC‐3′ | IDT | N/A |
| Primer: mouse β‐actin Reverse: 5′‐CACAGCCTGGATGGCTACGT‐3′ | IDT | N/A |
| Probe: mouse β‐actin: 5′‐/56‐FAM/TTTGAGACC/ZEN/TTCAACACCCCA/3IABkFQ/‐3′ | IDT | N/A |
| pHW2000 plasmid | Robert G. Webster; St. Jude Children's Research Hospital | N/A |
| pBD‐PA‐SWAP‐2A‐NLuc plasmid | Tran et al (2013) | N/A |
| Software | ||
| Fiji | Schindelin et al (2012) | https://fiji.sc/ |
| FlowJo, LLC | Tree Star | https://www.flowjo.com/solutions/flowjo/downloads |
| GraphPad Prism 8.0 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Other | ||
| Influenza A/WSN/33 H1N1 (WSN/33) | ATCC | VR‐1520 |
| Influenza A/PR/8/34 H1N1 (PR/8) | ATCC | VR‐95 |
| Influenza A/Wyoming/3/2003 H3N2 (WY/03) | Dr. Arnold S. Monto; University of Michigan School of Public Health | N/A |
| Influenza A/Ann Arbor/2017 (clade 3C.2a2) H3N2 (AA/17) | Dr. Arnold S. Monto; University of Michigan School of Public Health | N/A |
| Influenza A/Hong Kong/4801/2014 (clade 3C.2a) H3N2 (HK/14) | Centers for Disease Control and Prevention International Reagent Resource | FR‐397 |
| Influenza A/Singapore/Infimh‐16‐0019/2016 (clade 3C.2a1) H3N2 (Sing/16) | Centers for Disease Control and Prevention International Reagent Resource | FR‐1590 |
| Influenza A/Wisconsin/67/2005 H3N2 (WI/05) | Centers for Disease Control and Prevention International Reagent Resource | FR‐397 |
| Influenza A/California/07/2009 H1N1 (CA/09) | Centers for Disease Control and Prevention International Reagent Resource | FR‐201 |
| Influenza A Luc‐WSN/33 H1N1 | This paper | N/A |
| Influenza A Luc‐WY/03 H3N2 | This paper | N/A |
| Influenza A Luc‐AA/17 H3N2 | This paper | N/A |
| Influenza A Luc‐HK/14 H3N2 | This paper | N/A |
| Influenza A Luc‐Sing/16 H3N2 | This paper | N/A |
| Influenza A Luc‐WI/05 H3N2 | This paper | N/A |
| Influenza A Luc‐CA/09 H1N1 | This paper | N/A |
Methods and Protocols
Mice and rats
All animal experiments were performed according to National Institutes of Health guidelines for the use of experimental animals with the approval of the University of Michigan Committee for the Use and Care of Animals. Female C57BL6/J mice and Wistar rats were housed in ventilated, monitored cages. At least four mice per experimental group, 8 weeks of age were used in these experiments. Two rats per experimental group were used in these experiments.
Cell lines
MDCK Siat‐1 (Arnold S. Monto at The University of Michigan School of Public Health) and 293T (ATCC CRL‐3216) cells were cultured in DMEM with 10% FBS and penicillin/streptomycin. A549 AECs (ATCC CCL‐185) and MH‐S AMs (ATCC CRL‐2019) were cultured in RPMI 1640 supplemented with 10% FBS and penicillin/streptomycin. MLE‐12 AECs (ATCC CRL‐2110) were cultured in HITES medium which contained F‐12 K medium with l‐glutamine (Gibco), 2% FBS, 0.5% ITS liquid supplement (insulin, transferrin, sodium selenite (Sigma)), 2.5 mg transferrin (Sigma), 5 mM HEPES (Gibco), 10 nM β‐estradiol (Sigma), 10 nM hydrocortisone (Sigma), and penicillin/streptomycin. Human THP‐1 monocytes (ATCC TIB‐202) were maintained in 10% FBS‐supplemented RPMI. For THP‐1 differentiation into macrophages (Volgers et al, 2017), cells were stimulated for 72 h with 100 nM phorbol 12‐myristate 13‐acetate (Sigma). Cell lines were used between passages 5 and 40. Primary mouse AMs were J2 retrovirally immortalized using previously published methods (Zhou et al, 2008) with some modifications. The source of J2 retrovirus was the CM of AMJ2‐C11 cells (ATCC CRL‐2456) clarified through centrifugation (3,000 × g for 15 min @ 4°C) and filtered through a 0.45 μm membrane (Millipore). Primary AMs and peritoneal macrophages were isolated by lung and peritoneal lavage of 8‐week‐old female C57BL/6J mice and cultured overnight in RPMI with 10% FCS and penicillin/streptomycin. Macrophages were then washed and cultured in the clarified J2 retrovirus‐containing AMJ2‐C11 CM for 24 h. Excess virus was washed away, and cells were cultured in fresh RPMI with 5% FCS, 5 mM HEPES, and penicillin/streptomycin for 1 day. This process was repeated once more, and cells were then cultured back in RPMI with FCS and HEPES until immortalization was observed approximately 2 weeks later. These immortalized primary mouse macrophages were subsequently maintained in RPMI plus 10% FBS and 5 mM HEPES and used for EV isolation within five passages.
Primary rat AM isolation
Resident AMs were obtained by lavage of the lungs of pathogen‐free 125–150 g female Wistar rats (Charles River) (Bourdonnay et al, 2015; Speth et al, 2016). AMs isolated by lung lavage were pelleted by centrifugation (500 × g for 5 min at 4°C) and resuspended in serum‐free RPMI to a final concentration of 1 × 106 cells/ml. AMs were adhered to plastic cell culture plates for 1 h and then washed with PBS to remove non‐adherent cells and products liberated from AMs by their activation following adherence to plastic. AMs were placed in fresh, serum‐free RPMI and incubated for 16 h prior to isolation of their EVs.
Generation of viruses
To generate the Luc‐WSN/33 strain, a nanoluciferase was previously cloned downstream to the influenza WSN/33 PA genome segment (pBD‐PA‐SWAP‐2A‐NLuc) yielding the Luc‐WSN/33 strain with high specific activity, competent replication, and equal virulence to its WT counterpart (Tran et al, 2013). Molecular clones of Luc‐WSN/33 and all patient‐derived WT viruses listed in Key Resources Table were generated by RT–PCR amplification of insertion of all eight genomic segments into individual pHW2000 plasmids (Robert G. Webster; St. Jude Children's Research Hospital) (Hoffmann et al, 2000, 2001). Using standard reverse genetics methods for the generation of reassortant influenza viruses (Hoffmann et al, 2002), novel Luc‐viruses were recovered after transfecting co‐cultures of 293T and MDCK‐SIAT1 cells (2:1 ratio, 12‐well plates) with six plasmids containing individual gene segments from Luc‐WSN/33 (including pBD‐PA‐SWAP‐2A‐NLuc) and two plasmids containing the remaining HA and NA segments corresponding to individual influenza strains (see Key Resources Table and Fig 4C). Transfections were performed with 0.5 μg of each plasmid using Fugene HD (Promega) according to the manufacturer's instructions. This P0 virus was subsequently recovered, and infections were performed in MDCK‐SIAT1 cells at MOI 0.01 for the generation of working P1 viral stocks whose titers were determined with the TCID/50 method. Corresponding patient‐derived WT strains used for data in Fig 4A were recovered, transfected, expanded, and quantified using the same methods in parallel with Luc‐expressing strains. As indicated in Key Resources Table, the WT PR/8 and WSN/33 laboratory strains were obtained from ATCC.
Reagents
The two lots of CSE used in these studies were generated by bubbling the smoke from five 2R4F cigarettes, secured to the inlet port of a glass impinger (Ace Glass, Vineland, NJ), into a reservoir containing 50 ml of RPMI. Aliquots of this CSE stock were frozen at −80°C and subjected to exactly one freeze‐thaw cycle. CSE prepared with these methods and used at concentrations and incubation times in these studies do not affect the viability of AMs (Phipps et al, 2010). Baf‐A1 and Chl were purchased from Sigma. Baf‐A1 was dissolved in 100% DMSO. Working dilutions of Baf‐A1 and corresponding untreated controls were diluted to a final concentration of 0.1% DMSO with cell culture medium. pH clamping buffers contained 130 mM KCl, 15 mM HEPES, 15 mM MES, 2 mM MgCl2, 10 μM valinomycin (Sigma), 10 μM nigericin (Sigma), and 0.02% NaN3 (Davis & Swanson, 2010).
EV isolation and quantification
EVs were isolated and quantified from the CM of MH‐S AMs, primary rat AMs, immortalized primary mouse AMs, or THP‐1 monocyte‐differentiated macrophages using methods we have previously published (Bourdonnay et al, 2015; Schneider et al, 2017). AMs grown in serum‐containing RPMI were passaged into serum‐free RPMI onto polystyrene flasks overnight. Primary rat AMs were maintained in serum‐free RPMI as above. For THP‐1 macrophages, following the 72 h differentiation step above, adherent cells were washed and placed in serum‐free RPMI for an additional 16 h to collect EVs. To assess changes in EV function in response to CSE, MH‐S cells were adhered to culture dishes in serum‐containing RPMI overnight. Cells were subsequently washed and treated with increasing concentrations of CSE diluted in serum‐free medium for 20 h. In all cases, EVs were subsequently isolated from the resulting CM by serial centrifugation (500 g × 5 min then 2,500 g × 12 min at 4°C) followed by pelleting with ultracentrifugation (17,000 g × 30 min at 4°C). EV pellets were washed twice with a maximum volume of PBS between additional ultracentrifugation spins and ultimately resuspended in PBS. Our laboratory has published that AM‐EVs isolated with this method are shed from the plasma membrane and express phosphatidylserine on their outer surface (Bourdonnay et al, 2015) consistent with larger EVs previously termed “microvesicles” (Mateescu et al, 2017). EVs were subsequently quantified using a BD FACS Fortessa with low set window extension and light scatter channels set to logarithmic gain. Prior to EV quantification, the flow cytometer cell was cleared with 10% bleach for 10 min followed by separate aliquots of 0.22 μm‐filtered PBS until events in the EV gate were detected at less than 2 per second. The quantity of these events detected in filtered PBS was subtracted from each EV measurement. Events counted as EVs exhibited a characteristic size range in reference to fluorescent calibration beads (1.0 μm, Invitrogen) and were distinct from machine noise (identified as detected events common to 0.22 μm‐filtered PBS with lower side scatter values than EVs (Schneider et al, 2017)). All EVs for in vitro and in vivo studies were utilized after exactly one freeze‐thaw cycle following storage at −80°C for less than 3 months. Labeling of EVs for endosomal pH experiments was performed with CellMask Deep Red Plasma membrane Stain (Thermo Fisher) according to the manufacturer's instructions with some modifications as we have published previously for labeling AM‐EV membranes with a fluorescent lipophilic dye (Schneider et al, 2017). Following the incubation of pelleted EVs with the Deep Red stain, excess dye was quenched with an equal volume of 0.22 μm‐filtered FBS, and aggregates were removed by centrifugation (2,500 g × 12 min at 4°C). The resulting supernatant containing labeled EVs was washed twice with PBS between additional ultracentrifugation spins.
RNA depletion within EVs
Three groups of EVs were isolated from equal volumes of MH‐S CM, subsequently treated with RNase A (Roche, 10 μg/ml) for 30 min to deplete RNA outside EVs, and ultimately subjected to gentle permeabilization by repeated freeze‐thaw cycles at −80°C to permit RNase A entry into EVs. This permeabilization occurred before or after addition of an RNAse inhibitor (150 U/ml, Applied Biosystem) in the different conditions. EVs in each condition were subsequently re‐pelleted with ultracentrifugation, washed twice with PBS, resuspended directly in RL Buffer (Total RNA Purification Micro Kit; Norgen Biotek), and EV RNA was isolated using according to the manufacturer's instructions. RNA was quantified in each EV condition with Agilent Bioanalyzer 2100 and TapeStation Analysis Software 3.1.1 (Agilent Technologies).
Differential abundance of proteins within AM‐EVs treated with CSE
EVs were collected from MH‐S AMs cultured in 0% CSE or 0.8% CSE in serum‐free RPMI. EVs from both conditions were resuspended in 8 M Urea + 25 mM HEPES lysis buffer followed by high sensitivity protein estimation (Sigma). 22 μg of protein from each condition in triplicate was labeled with the Tandem Mass Tag 6‐plex labeling kit (Thermo Fisher) according to the manufacturer's instructions. Prior to their identification, labeled proteins were precipitated with trichloroacetic acid/acetone and separated using nano‐ultra high‐pressure liquid chromatography. For identification, MS3 analysis (McAlister et al, 2014) on an Orbitrap Fusion Mass Spectrometer was employed. Candidate proteins were selected based on their differential expression in control EVs vs. EVs isolated from 0.8% CSE‐treated AMs. Data were analyzed with Proteome Discoverer (v2.1 Thermo Fisher), and initial spectra were searched against the Uniprot mouse proteome database (modified 2018 07‐27, 53,857 sequences). Identified proteins and peptides had a false discovery rate of < 1%. Paired t‐test with Benjamini–Hochberg multiple testing correction (Benjamini & Hochberg, 1995) was used on spectra to identify statistically significant (P < 0.05) differences in abundance.
Assessment of influenza replication in Luc and WT strains
Viral infections in MDCK‐SIAT1 and A549 cells were performed in viral growth medium (VGM) which contained DMEM, 2 μg/ml TPCK‐treated trypsin (Sigma), 0.1875% BSA (Sigma), 25 mM HEPES, and penicillin/streptomycin. Infections in MLE‐12 cells were performed in serum‐free HITES medium supplemented with 2 μg/ml TPCK‐trypsin, 0.1875% BSA, and additional HEPES to a final concentration of 25 mM. ECs were seeded into 96‐well plates in serum‐containing medium and grown overnight to a final concentration of ~ 20 k cells/well. Cells were subsequently washed, infected with Luc‐strains in VGM and Enduren luciferase substrate (manufacturer's recommended working concentration) (Promega) with and without AM‐EVs or inhibitors. Luciferase activity indicated viral replication and was quantified from individual wells at 90 s intervals while maintaining cells at 37°C and 5% CO2 with a Synergy H1 Luminescence Reader with CO2 regulator (BioTek) until the first condition reached the end of the exponential growth phase (at least 16 h for all experiments and strains) or until the replication time reached 24 h. AUC measurements of luminescence curves were calculated with Prism 8 software for individual wells. In selected plates, cells were subsequently incubated with Cell Titer‐Glo (Promega) to determine cell viability according to the manufacturer's instructions. The final luminescence value at the end of the replication assay was recorded prior to the addition of Cell Titer‐Glo reagent and subtracted from the cell titer luminescence reading for each individual well. For WT strains, MLE‐12 cells were grown overnight in 12 well plates to 90% confluency. Cells were washed and incubated with WT strains with and without EVs for 12 h. All experiments were performed at a multiplicity of infection (MOI) of 1 and with a EV:cell ratio of 1 unless otherwise specified in figure legends.
Influenza infection of AM‐depleted mice
Clo‐loaded liposomes were generated in the laboratory of Dr. Nico van Rooijen according to a previously established method (van Rooijen & Van Kesteren‐Hendrikx, 2003) (Liposoma BV). On day 0, 50 μl of clodronate liposomes (or control empty liposomes) was administered o.p. to 8‐week‐old anesthetized C57BL/6 wild‐type female mice. Depletion of lavageable macrophages was verified by quantification of modified Wright‐Giemsa‐stained (Diff‐Quik, American Scientific Products) cells subjected to cytospin. On day 2, mice were given 50 plaque‐forming units of PR/8 virus (ATCC) resuspended in 50 μl of sterile PBS with or without AM‐EVs (2e5/μl). This concentration is similar to that of other published reports utilizing intrapulmonary instillation of EVs (Bourdonnay et al, 2015; Genschmer et al, 2019). On day 3, mice were sacrificed, lungs perfused with cold PBS, and snap frozen in liquid nitrogen. Both lungs from each mouse were subsequently thawed in tubes containing Trizol (Invitrogen), minced briefly on ice, and homogenized using a tissue bead beater (MP Bio) and lysing matrix beads (MP Bio).
RT–PCR
Total RNA from mouse lungs or MLE‐12 AECs grown on 12‐well plates was isolated through Trizol extraction and purified in RNA Easy Qiagen columns according to the manufacturer's instructions. Influenza replication was quantified using the Taqman RNA‐to‐Ct 1‐Step kit (Applied Biosystems) on an ABI Prism 7300 Thermocycler (Applied Biosystems). Relative gene expression of influenza M gene was determined using the ΔC t method with β‐actin as the reference gene. Primers and probes used are listed in Key Resources Table.
Immunofluorescence microscopy
Human A549 or MLE‐12 AECs were plated overnight at a density of 100k cells/well on fibronectin (Sigma; 1.25 μg/cm2)‐coated #1.5 coverglass chamber slides (Lab‐Tek, Nunc). Cells were washed, co‐incubated for 40 min with WT virus (WSN/33 or HK/14; MOI = 1) with and without AM‐EVs in VGM. Cells were washed with PBS, fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X‐100 (except in plasma membrane binding experiments shown in 5C), and incubated in blocking solution containing 0.3 M glycine, 2% BSA, and 5% goat serum in PBS. Primary staining was performed as sequential overnight incubations at 4°C first using rabbit anti‐Rab7 for late endosomes (Cell Signaling Technologies, #9367) (1:100) or rabbit IgG isotype control (Abcam, ab171870, 1:100) followed by mouse anti‐influenza NP (Abcam, ab20343) (2.89 μg/ml) or mouse IgG isotype control (Abcam, ab18415) in blocking solution. Secondary staining was performed sequentially with Alexa Fluor (Invitrogen) 488 anti‐mouse IgG (1:1,000) followed by Alexa Fluor 568 anti‐rabbit IgG (1:1,000) for 45 min each at RT. Cells were covered with DAPI Prolong Gold mounting medium (Invitrogen). Images were taken from an SP5X inverted confocal microscope and analyzed with Application Suite Advanced Fluorescence Software (Leica). Each image was captured with laser intensities and amplifier gains set to avoid pixel saturation. Fluorophores were independently excited and detected sequentially. Individual images were captured with a 100× objective with 1.4 numerical aperture, oil immersion, and pinhole set to 1 μm. Z‐stack images were captured at 512 × 512 pixel resolution and were subsequently visualized and created with FIJI software (National Institutes of Health). Virus nuclear localization was quantified as a ratio of NP intensity within the DAPI region divided by total cellular NP intensity. Co‐localization of influenza within late endosomes was quantified as the Manders co‐localization coefficient of NP and Rab‐7 staining using the coloc 2 plugin in FIJI.
Endosomal escape assay and acidification inhibitor IC50 determination
MDCK‐SIAT1 cells were plated at 90% confluency on flat‐bottom, opaque 96‐well plates and cultured overnight in DMEM plus 10% FBS. For the endosomal escape assay, Luc‐expressing strains were bound in VGM on ice for 45 min. Cells were washed and placed in fresh VGM containing the EnduRen substrate. Synchronized uptake of bound virions was then initiated at time 0 by placing plates at 37°C and 5% CO2. At the specified times indicating the duration of 37°C incubation, wells were supplemented with Baf‐A1 (Sigma) to a final concentration of 10 nM. After the time 80 min Baf‐A1 addition, the cells were placed in the luminometer (set at 37°C and 5% CO2) and replication was determined by luminescence measurements for 4 h and 40 min (six total hour of infection time at 37°C) to limit the assay to one replication cycle (Baccam et al, 2006; Frensing et al, 2016). For IC50 determination, MDCK‐SIAT1 cells were plated overnight, subsequently washed, and preincubated with Baf‐A1 or Chl at the concentrations indicated in figure legends for 30 min. Subsequently, cells were co‐incubated with the same corresponding inhibitors and concentrations in VGM plus EnduRen substrate and Luc‐expressing influenza strains, and replication was assessed by continuous luminescence. Each plate contained (escape assay) two strains with each time point Baf‐A1 addition performed in five replicate wells, or (IC50 determination) 4 strains with each dose of inhibitor performed in triplicate wells.
Endosomal pH standard curve generation and measurement
Endosomal pH was determined with live cell measurement of the pH‐sensitive change in ratiometric excitation spectral intensities of internalized 3 kDa FDx (Thermo Fisher) (Davis & Swanson, 2010). MDCK‐SIAT1 cells were plated overnight on uncoated 35 mm #1.5 coverglass dishes (MatTek Corporation). Cells were subsequently pulsed for 20 min with 3 kDa FITC‐dextran with or without DR‐EVs followed by a 5 min chase with PBS. Cells were imaged using a Nikon A1R inverted confocal microscope with a 63× objective and oil immersion. Ratiometric fluorescence of FDx was acquired with sequential excitation of the pH‐insensitive (445 nm) and pH‐sensitive (488 nm) wavelengths followed by emission collection with a single photomultiplier tube using a 525/50 bandpass filter. DR‐EV fluorescence was acquired with 647 nm excitation and emission was collected with a 700/75 filter. All images were captured within 35 min from the end of the 5 min chase period. Laser intensities and amplifier gains were set to avoid pixel saturation. For generation of the pH calibration curve, cells with internalized FDx alone were maintained in a pH clamping buffer (pH range 5.0–7.0, contents listed above) for 20 min prior to imaging. For standard curve generation and pH measurement, post‐acquisition analysis of the emission intensities of the distinct intracellular particles (DR+ EVs, FDx+ endosomes) and their co‐localization was performed using FIJI software. From confocal images of a single focal plane (213 × 213 μm; 512 × 512 pixels) in the 647ex/700em channel, DR+EVs were selected as regions of interest (ROIs) defined by a size range between 0.2 and 5.0 μm and a constant intensity threshold across all experiments. FDx+ endosomes were defined as separate ROIs in the same manner from the 445ex/525em channel and used to select endosomes within both 525em (445ex and 488ex) images. DR+EV ROIs were then applied to both 525em images and FDx+ emission intensities within (DR+EV and FDx+ colocalized endosomes) and outside (FDx+ endosome) these ROIs were separately measured, recorded as a ratio (488ex:445ex), and applied to the standard curve to return the pH value. Individual data points reported are the average emission ratios of all analyzed endosomes within each condition within 1 hpf. At least five images containing ~ 20 cells per hpf were taken for each condition per experiment.
Quantification and statistical analysis
For transparency and to display variation in control conditions for normalized data, all individual data points across multiple experiments for each condition are displayed in figures unless otherwise specified. However, unless otherwise specified, the statistical comparisons presented are between the means of replicates within each experimental conditions for an individual experiment using one‐way ANOVA with Tukey's multiple comparisons post hoc analysis (or Student's t‐test where appropriate). Half maximal inhibitory concentration (IC50) curves were generated [non‐linear fit, (inhibitor) vs. normalized response—variable slope function] and returned values for mean IC50, standard error, and degrees of freedom for each strain. These input values were used for the one‐way ANOVA multiple comparisons calculation to determine statistical significance between the IC50 values of each strain. All data were analyzed using the Prism 8.0 statistical program from GraphPad software. The following symbols were used to display P value ranges in all main and supplemental figures: *P value of ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
Author contributions
Conceptualization: DJS, ASL, BBM, MP‐G; Methodology: DJS, DML, LRP, MM, JAS, ASL; Formal Analysis: DJS, KAS; Visualization: DJS, KAS; Investigation: DJS, KAS, CEL, CAW, JMS; Resources: DJS, BBM, ASL, MP‐G; Writing—Original Draft: DJS, KAS; Writing—Review & Editing: DJS, JAS, BBM, ASL, MP‐G; Supervision: DJS, JAS, BBM, ASL, MP‐G; Project Administration: DJS, MP‐G; Funding Acquisition: DJS, DML, ASL, MP‐G.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We thank Andrew Mehle for the pBD‐PA‐SWAP‐2A‐NLuc plasmid, Robert Webster for the pHW2000 plasmid, Arnold Monto for the MDCK‐SIAT1 cells and influenza strains, Venkatesha Basrur and Alexey Nesvizhskii for their technical assistance with the mass spectrometry experiments, and Pete Mancuso for providing the CSE. This work was supported by grants from the National Institute of Health 1K08HL149051 (D.J.S.), F31AI140618 (D.M.L.), R01HL125555 and R35HL144979 (M.P.G.) and the American Thoracic Society (D.J.S.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The EMBO Journal (2020) 39: e105057
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository (https://www.ebi.ac.uk/pride/archive/projects/PXD019528) with the dataset identifier PXD019528.
References
- Admyre C, Johansson SM, Paulie S, Gabrielsson S (2006) Direct exosome stimulation of peripheral human T cells detected by ELISPOT. Eur J Immunol 36: 1772–1781 [DOI] [PubMed] [Google Scholar]
- Arcavi L, Benowitz NL (2004) Cigarette smoking and infection. Arch Intern Med 164: 2206–2216 [DOI] [PubMed] [Google Scholar]
- Atay S, Gercel‐Taylor C, Taylor DD (2011) Human trophoblast‐derived exosomal fibronectin induces pro‐inflammatory IL‐1beta production by macrophages. Am J Reprod Immunol 66: 259–269 [DOI] [PubMed] [Google Scholar]
- Baccam P, Beauchemin C, Macken CA, Hayden FG, Perelson AS (2006) Kinetics of influenza A virus infection in humans. J Virol 80: 7590–7599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer N, Schober D, Prchla E, Murphy RF, Blaas D, Fuchs R (1998) Effect of bafilomycin A1 and nocodazole on endocytic transport in HeLa cells: implications for viral uncoating and infection. J Virol 72: 9645–9655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol 57: 289–300 [Google Scholar]
- Bottcher E, Matrosovich T, Beyerle M, Klenk HD, Garten W, Matrosovich M (2006) Proteolytic activation of influenza viruses by serine proteases TMPRSS2 and HAT from human airway epithelium. J Virol 80: 9896–9898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourdonnay E, Zaslona Z, Penke LR, Speth JM, Schneider DJ, Przybranowski S, Swanson JA, Mancuso P, Freeman CM, Curtis JL et al (2015) Transcellular delivery of vesicular SOCS proteins from macrophages to epithelial cells blunts inflammatory signaling. J Exp Med 212: 729–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown EG, Liu H, Kit LC, Baird S, Nesrallah M (2001) Pattern of mutation in the genome of influenza A virus on adaptation to increased virulence in the mouse lung: identification of functional themes. Proc Natl Acad Sci USA 98: 6883–6888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardani A, Boulton A, Kim TS, Braciale TJ (2017) Alveolar macrophages prevent lethal influenza pneumonia by inhibiting infection of type‐1 alveolar epithelial cells. PLoS Pathog 13: e1006140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Center for Disease Control (2020) CDC seasonal flu vaccine effectiveness studies [Online]. Center for Disease Control; Available: https://www.cdc.gov/flu/vaccines-work/effectiveness-studies.htm [Accessed 20 February 2020] [Google Scholar]
- Chaipan C, Kobasa D, Bertram S, Glowacka I, Steffen I, Tsegaye TS, Takeda M, Bugge TH, Kim S, Park Y et al (2009) Proteolytic activation of the 1918 influenza virus hemagglutinin. J Virol 83: 3200–3211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipriano DJ, Wang Y, Bond S, Hinton A, Jefferies KC, Qi J, Forgac M (2008) Structure and regulation of the vacuolar ATPases. Biochim Biophys Acta 1777: 599–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello DA, Whittaker GR, Daniel S (2015) Variations in pH sensitivity, acid stability, and fusogenicity of three influenza Virus H3 subtypes. J Virol 89: 350–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MJ, Swanson JA (2010) Technical advance: Caspase‐1 activation and IL‐1beta release correlate with the degree of lysosome damage, as illustrated by a novel imaging method to quantify phagolysosome damage. J Leukoc Biol 88: 813–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson J, Whitley RJ, Pocock S, Monto AS (2015) Oseltamivir treatment for influenza in adults: a meta‐analysis of randomised controlled trials. Lancet 385: 1729–1737 [DOI] [PubMed] [Google Scholar]
- Duan M, Hibbs ML, Chen W (2017) The contributions of lung macrophage and monocyte heterogeneity to influenza pathogenesis. Immunol Cell Biol 95: 225–235 [DOI] [PubMed] [Google Scholar]
- Fehrenbach H, Riemann D, Wahlers T, Hirt SW, Haverich A, Richter J (1994) Scanning and transmission electron microscopy of human donor lungs: fine structure of the pulmonary parenchyma following preservation and ischemia. Acta Anat (Basel) 151: 220–231 [DOI] [PubMed] [Google Scholar]
- Frensing T, Kupke SY, Bachmann M, Fritzsche S, Gallo‐Ramirez LE, Reichl U (2016) Influenza virus intracellular replication dynamics, release kinetics, and particle morphology during propagation in MDCK cells. Appl Microbiol Biotechnol 100: 7181–7192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galloway SE, Reed ML, Russell CJ, Steinhauer DA (2013) Influenza HA subtypes demonstrate divergent phenotypes for cleavage activation and pH of fusion: implications for host range and adaptation. PLoS Pathog 9: e1003151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genschmer KR, Russell DW, Lal C, Szul T, Bratcher PE, Noerager BD, Abdul Roda M, Xu X, Rezonzew G, Viera L et al (2019) Activated PMN exosomes: pathogenic entities causing matrix destruction and disease in the lung. Cell 176: 113–126.e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlach T, Hensen L, Matrosovich T, Bergmann J, Winkler M, Peteranderl C, Klenk H‐D, Weber F, Herold S, Pöhlmann S et al (2017) pH optimum of hemagglutinin‐mediated membrane fusion determines sensitivity of influenza A viruses to the interferon‐induced antiviral state and IFITMs. J Virol 91: e00246‐17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitelman AK, Kaverin NV, Kharitonenkov IG, Rudneva IA, Zhdanov VM (1984) Changes in the antigenic specificity of influenza hemagglutinin in the course of adaptation to mice. Virology 134: 230–232 [DOI] [PubMed] [Google Scholar]
- Green IJ (1962) Serial propagation of influenza B (Lee) virus in a transmissible line of canine kidney cells. Science 138: 42–43 [DOI] [PubMed] [Google Scholar]
- Hermans D, Webby RJ, Wong SS (2018) Atypical antibody responses to influenza. J Thorac Dis 10: S2238–S2247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann E, Neumann G, Kawaoka Y, Hobom G, Webster RG (2000) A DNA transfection system for generation of influenza A virus from eight plasmids. Proc Natl Acad Sci USA 97: 6108–6113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann E, Stech J, Guan Y, Webster RG, Perez DR (2001) Universal primer set for the full‐length amplification of all influenza A viruses. Arch Virol 146: 2275–2289 [DOI] [PubMed] [Google Scholar]
- Hoffmann E, Krauss S, Perez D, Webby R, Webster RG (2002) Eight‐plasmid system for rapid generation of influenza virus vaccines. Vaccine 20: 3165–3170 [DOI] [PubMed] [Google Scholar]
- Hyde DM, Tyler NK, Putney LF, Singh P, Gundersen HJ (2004) Total number and mean size of alveoli in mammalian lung estimated using fractionator sampling and unbiased estimates of the Euler characteristic of alveolar openings. Anat Rec A Discov Mol Cell Evol Biol 277: 216–226 [DOI] [PubMed] [Google Scholar]
- Imai M, Watanabe T, Hatta M, Das SC, Ozawa M, Shinya K, Zhong G, Hanson A, Katsura H, Watanabe S et al (2012) Experimental adaptation of an influenza H5 HA confers respiratory droplet transmission to a reassortant H5 HA/H1N1 virus in ferrets. Nature 486: 420–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki A, Pillai PS (2014) Innate immunity to influenza virus infection. Nat Rev Immunol 14: 315–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaverin NV, Finskaya NN, Rudneva IA, Gitelman AK, Kharitonenkov IG, Smirnov YA (1989) Studies on the genetic basis of human influenza A virus adaptation to mice: degrees of virulence of reassortants with defined genetic content. Arch Virol 105: 29–37 [DOI] [PubMed] [Google Scholar]
- Kesimer M, Scull M, Brighton B, Demaria G, Burns K, O'Neal W, Pickles RJ, Sheehan JK (2009) Characterization of exosome‐like vesicles released from human tracheobronchial ciliated epithelium: a possible role in innate defense. FASEB J 23: 1858–1868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatri M, Richardson LA, Meulia T (2018) Mesenchymal stem cell‐derived extracellular vesicles attenuate influenza virus‐induced acute lung injury in a pig model. Stem Cell Res Ther 9: 17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KJ, Malik AB (2003) Protein transport across the lung epithelial barrier. Am J Physiol Lung Cell Mol Physiol 284: L247–L259 [DOI] [PubMed] [Google Scholar]
- Kim HM, Lee YW, Lee KJ, Kim HS, Cho SW, van Rooijen N, Guan Y, Seo SH (2008) Alveolar macrophages are indispensable for controlling influenza viruses in lungs of pigs. J Virol 82: 4265–4274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HM, Kang YM, Ku KB, Park EH, Yum J, Kim JC, Jin SY, Lee JS, Kim HS, Seo SH (2013) The severe pathogenicity of alveolar macrophage‐depleted ferrets infected with 2009 pandemic H1N1 influenza virus. Virology 444: 394–403 [DOI] [PubMed] [Google Scholar]
- Korteweg C, Gu J (2008) Pathology, molecular biology, and pathogenesis of avian influenza A (H5N1) infection in humans. Am J Pathol 172: 1155–1170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitz SM, Golenbock DT (2012) Beyond empiricism: informing vaccine development through innate immunity research. Cell 148: 1284–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima LG, Chammas R, Monteiro RQ, Moreira ME, Barcinski MA (2009) Tumor‐derived microvesicles modulate the establishment of metastatic melanoma in a phosphatidylserine‐dependent manner. Cancer Lett 283: 168–175 [DOI] [PubMed] [Google Scholar]
- Ma L, Ouyang Q, Werthmann GC, Thompson HM, Morrow EM (2017) Live‐cell microscopy and fluorescence‐based measurement of luminal pH in intracellular organelles. Front Cell Dev Biol 5: 71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maemura T, Fukuyama S, Sugita Y, Lopes TJS, Nakao T, Noda T, Kawaoka Y (2018) Lung‐derived exosomal miR‐483‐3p regulates the innate immune response to influenza virus infection. J Infect Dis 217: 1372–1382 [DOI] [PubMed] [Google Scholar]
- Mardones G, Gonzalez A (2003) Selective plasma membrane permeabilization by freeze‐thawing and immunofluorescence epitope access to determine the topology of intracellular membrane proteins. J Immunol Methods 275: 169–177 [DOI] [PubMed] [Google Scholar]
- Marvin SA, Russier M, Huerta CT, Russell CJ, Schultz‐Cherry S (2017) Influenza virus overcomes cellular blocks to productively replicate impacting macrophage function. J Virol 91: e01417‐16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateescu B, Kowal EJK, van Balkom BWM, Bartel S, Bhattacharyya SN, Buzás EI, Buck AH, de Candia P, Chow FWN, Das S et al (2017) Obstacles and opportunities in the functional analysis of extracellular vesicle RNA – an ISEV position paper. J Extracell Vesicles 6: 1286095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka Y, Matsumae H, Katoh M, Eisfeld AJ, Neumann G, Hase T, Ghosh S, Shoemaker JE, Lopes TJS, Watanabe T et al (2013) A comprehensive map of the influenza A virus replication cycle. BMC Syst Biol 7: 97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister GC, Nusinow DP, Jedrychowski MP, Wuhr M, Huttlin EL, Erickson BK, Rad R, Haas W, Gygi SP (2014) MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal Chem 86: 7150–7158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulcahy LA, Pink RC, Carter DRF (2014) Routes and mechanisms of extracellular vesicle uptake. J Extracell Vesicles 3: 24641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolte‐‘t Hoen E, Cremer T, Gallo RC, Margolis LB (2016) Extracellular vesicles and viruses: are they close relatives? Proc Natl Acad Sci USA 113: 9155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh DY, Barr IG, Mosse JA, Laurie KL (2008) MDCK‐SIAT1 cells show improved isolation rates for recent human influenza viruses compared to conventional MDCK cells. J Clin Microbiol 46: 2189–2194 18480230 [Google Scholar]
- Phipps JC, Aronoff DM, Curtis JL, Goel D, O'Brien E, Mancuso P (2010) Cigarette smoke exposure impairs pulmonary bacterial clearance and alveolar macrophage complement‐mediated phagocytosis of Streptococcus pneumoniae. Infect Immun 78: 1214–1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitt JM, Kroemer G, Zitvogel L (2016) Extracellular vesicles: masters of intercellular communication and potential clinical interventions. J Clin Invest 126: 1139–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purnama C, Ng SL, Tetlak P, Setiagani YA, Kandasamy M, Baalasubramanian S, Karjalainen K, Ruedl C (2014) Transient ablation of alveolar macrophages leads to massive pathology of influenza infection without affecting cellular adaptive immunity. Eur J Immunol 44: 2003–2012 [DOI] [PubMed] [Google Scholar]
- Qin C, Li W, Li Q, Yin W, Zhang X, Zhang Z, Zhang XE, Cui Z (2019) Real‐time dissection of dynamic uncoating of individual influenza viruses. Proc Natl Acad Sci USA 116: 2577–2582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins CS, Bauer CM, Vujicic N, Gaschler GJ, Lichty BD, Brown EG, Stampfli MR (2006) Cigarette smoke impacts immune inflammatory responses to influenza in mice. Am J Respir Crit Care Med 174: 1342–1351 [DOI] [PubMed] [Google Scholar]
- Robbins PD, Dorronsoro A, Booker CN (2016) Regulation of chronic inflammatory and immune processes by extracellular vesicles. J Clin Invest 126: 1173–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rooijen N, Van Kesteren‐Hendrikx E (2003) In vivo “depletion of macrophages by liposome‐mediated” suicide. Methods Enzymol 373: 3–16 [DOI] [PubMed] [Google Scholar]
- Russier M, Yang G, Rehg JE, Wong S‐S, Mostafa HH, Fabrizio TP, Barman S, Krauss S, Webster RG, Webby RJ et al (2016) Molecular requirements for a pandemic influenza virus: an acid‐stable hemagglutinin protein. Proc Natl Acad Sci USA 113: 1636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda‐Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B et al (2012) Fiji: an open‐source platform for biological‐image analysis. Nat Meth 9: 676‐682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C, Nobs SP, Heer AK, Kurrer M, Klinke G, van Rooijen N, Vogel J, Kopf M (2014) Alveolar macrophages are essential for protection from respiratory failure and associated morbidity following influenza virus infection. PLoS Pathog 10: e1004053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider DJ, Speth JM, Penke LR, Wettlaufer SH, Swanson JA, Peters‐Golden M (2017) Mechanisms and modulation of microvesicle uptake in a model of alveolar cell communication. J Biol Chem 292: 20897–20910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo SH, Webby R, Webster RG (2004) No apoptotic deaths and different levels of inductions of inflammatory cytokines in alveolar macrophages infected with influenza viruses. Virology 329: 270–279 [DOI] [PubMed] [Google Scholar]
- Singanayagam A, Zambon M, Barclay WS (2019) Influenza virus with increased pH of Hemagglutinin activation has improved replication in cell culture but at the cost of infectivity in human airway epithelium. J Virol 93: e00058‐19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skehel JJ, Wiley DC (2000) Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochem 69: 531–569 [DOI] [PubMed] [Google Scholar]
- Speth JM, Bourdonnay E, Penke LR, Mancuso P, Moore BB, Weinberg JB, Peters‐Golden M (2016) Alveolar epithelial cell‐derived prostaglandin E2 serves as a request signal for macrophage secretion of suppressor of cytokine signaling 3 during innate inflammation. J Immunol 196: 5112–5120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staeheli P, Grob R, Meier E, Sutcliffe JG, Haller O (1988) Influenza virus‐susceptible mice carry Mx genes with a large deletion or a nonsense mutation. Mol Cell Biol 8: 4518–4523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suptawiwat O, Ruangrung K, Boonarkart C, Puthavathana P, Maneechotesuwan K, Charngkaew K, Chomanee N, Auewarakul P (2017) Microparticle and anti‐influenza activity in human respiratory secretion. PLoS ONE 12: e0183717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate MD, Pickett DL, van Rooijen N, Brooks AG, Reading PC (2010) Critical role of airway macrophages in modulating disease severity during influenza virus infection of mice. J Virol 84: 7569–7580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubenberger JK, Morens DM (2008) The pathology of influenza virus infections. Annu Rev Pathol 3: 499–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testa JS, Apcher GS, Comber JD, Eisenlohr LC (2010) Exosome‐driven antigen transfer for MHC class II presentation facilitated by the receptor binding activity of influenza hemagglutinin. J Immunol 185: 6608–6616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thangavel RR, Bouvier NM (2014) Animal models for influenza virus pathogenesis, transmission, and immunology. J Immunol Methods 410: 60–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran V, Moser LA, Poole DS, Mehle A (2013) Highly sensitive real‐time in vivo imaging of an influenza reporter virus reveals dynamics of replication and spread. J Virol 87: 13321–13329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volgers C, Benedikter BJ, Grauls GE, Savelkoul PHM, Stassen FRM (2017) Immunomodulatory role for membrane vesicles released by THP‐1 macrophages and respiratory pathogens during macrophage infection. BMC Microbiol 17: 216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphalen K, Gusarova GA, Islam MN, Subramanian M, Cohen TS, Prince AS, Bhattacharya J (2014) Sessile alveolar macrophages communicate with alveolar epithelium to modulate immunity. Nature 506: 503–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JM, Whittaker GR (2016) Fusion of enveloped viruses in endosomes. Traffic 17: 593–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiklander OPB, Brennan MA, Lotvall J, Breakefield XO, el Andaloussi S (2019) Advances in therapeutic applications of extracellular vesicles. Sci Transl Med: 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley DC, Skehel JJ (1987) The structure and function of the hemagglutinin membrane glycoprotein of influenza virus. Annu Rev Biochem 56: 365–394 [DOI] [PubMed] [Google Scholar]
- Xu L, Bao L, Lv Q, Deng W, Ma Y, Li F, Zhan L, Zhu H, Ma C, Qin C (2010) A single‐amino‐acid substitution in the HA protein changes the replication and pathogenicity of the 2009 pandemic A (H1N1) influenza viruses in vitro and in vivo . Virol J 7: 325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Bao L, Li F, Lv Q, Ma Y, Zhou J, Xu Y, Deng W, Zhan L, Zhu H et al (2011) Adaption of seasonal H1N1 influenza virus in mice. PLoS ONE 6: e28901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaslona Z, Przybranowski S, Wilke C, van Rooijen N, Teitz‐Tennenbaum S, Osterholzer JJ, Wilkinson JE, Moore BB, Peters‐Golden M (2014) Resident alveolar macrophages suppress, whereas recruited monocytes promote, allergic lung inflammation in murine models of asthma. J Immunol 193: 4245–4253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Li S, Li L, Li M, Guo C, Yao J, Mi S (2015) Exosome and exosomal microRNA: trafficking, sorting, and function. Genomics Proteomics Bioinformatics 13: 17–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Imrich A, Kobzik L (2008) Characterization of immortalized MARCO and SR‐AI/II‐deficient murine alveolar macrophage cell lines. Part Fibre Toxicol 5: 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository (https://www.ebi.ac.uk/pride/archive/projects/PXD019528) with the dataset identifier PXD019528.