

Abstract
Full differentiation potential along with self‐renewal capacity is a major property of pluripotent stem cells (PSCs). However, the differentiation capacity frequently decreases during expansion of PSCs in vitro. We show here that transient exposure to a single microRNA, expressed at early stages during normal development, improves the differentiation capacity of already‐established murine and human PSCs. Short exposure to miR‐203 in PSCs (miPSCs) induces a transient expression of 2C markers that later results in expanded differentiation potency to multiple lineages, as well as improved efficiency in tetraploid complementation and human–mouse interspecies chimerism assays. Mechanistically, these effects are at least partially mediated by direct repression of de novo DNA methyltransferases Dnmt3a and Dnmt3b, leading to transient and reversible erasure of DNA methylation. These data support the use of transient exposure to miR‐203 as a versatile method to reset the epigenetic memory in PSCs, and improve their effectiveness in regenerative medicine.
Keywords: differentiation, epigenetics, microRNAs, pluripotency, stem cells
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; RNA Biology; Regenerative Medicine
Developmental potency of pluripotent stem cells is enhanced by short‐term exposure to a single microRNA.

Introduction
Pluripotent stem cells (PSCs) provide an important promise for regenerative medicine due to their self‐renewal potential and ability to differentiate into multiple cell lineages. During the last years, multiple efforts have been put into improving the potential of these cells either by expanding their differentiation capacity into a wider variety of cell lineages or by improving maturation properties into specific functional cell types (Li & Izpisua Belmonte, 2016; Takahashi & Yamanaka, 2016). PSCs are commonly cultured in the presence of Mek1/2 and Gsk3 inhibitors together with the cytokine Lif (2i/L conditions Ying et al, 2008). Although these conditions may improve the maintenance of pluripotency in vitro, recent evidences suggest that prolonged inhibition of Mek1/2 may limit the developmental and differentiation capacity of PSCs in vivo, in part by inducing irreversible demethylation of imprinting control regions (ICRs) (Choi et al, 2017a; Yagi et al, 2017). Recent data suggest that genetic ablation of miR‐34 in PSCs results in improved potential to form embryonic and extraembryonic tissues in part by promoting Gata2 expression (Choi et al, 2017b), although the conditions for applying this information to favor the differentiation potential of PSCs remain to be established. Finally, a recent alternative proposal suggests the use of a chemical cocktail of inhibitors that also enhances the developmental potential of PSCs (Yang et al, 2017); however, the applicability of this method is still limited due to the lack of mechanistic details.
Here, we identified miR‐203 as a microRNA preferentially expressed in the 2C‐morula stages during pre‐implantation development in the mouse embryo. By using a variety of in vitro and in vivo approaches, we report that transient exposure of already‐established induced pluripotent stem cells (iPSCs) or ESCs to miR‐203 enhances the ability of these PSCs to differentiate into multiple cell lineages and to reach further maturation properties. Transient expression of miR‐203 in PSCs (miPSCs) leads to greater developmental potential in tetraploid complementation assays, and significantly improves the efficiency of human iPSCs in contributing to interspecies human–mouse conceptuses. Mechanistically, these effects are at least partially mediated through the miR‐203‐dependent control of de novo DNA methyltransferases Dnmt3a and Dnmt3b, thereby regulating the DNA methylation landscape of these miPSCs. These observations suggest that the developmental and differentiation potential of already‐established PSCs can be readily enhanced by transient exposure to a single microRNA.
Results
miR‐203 improves the differentiation potency of PSCs
miR‐203 was originally proposed to limit the stemness potential of skin progenitors (Yi et al, 2008) and to display tumor‐suppressive functions in multiple cancers (Bueno et al, 2008; Michel & Malumbres, 2013), suggesting a role in the balance between stemness and differentiation. However, its expression during early development remained undefined. A first analysis of miR‐203 levels during normal murine and bovine development suggested a modest but specific wave of expression during early development (blastocyst stage in murine and more particularly hatched blastocysts in bovine embryos), whereas its expression was lost in cultured embryonic stem cells (Yang et al, 2008; Goossens et al, 2013). Interestingly, our quantitative PCR analysis in mouse embryos isolated at different early developmental stages showed that miR‐203 expression was low in oocytes, slightly induced at the 2‐cell stage, and displayed a gradual reduction in morulas and blastocysts (Fig 1A).
Figure 1. Effects of transient induction of miR‐203 in iPSC and ESC pluripotency and differentiation potential.
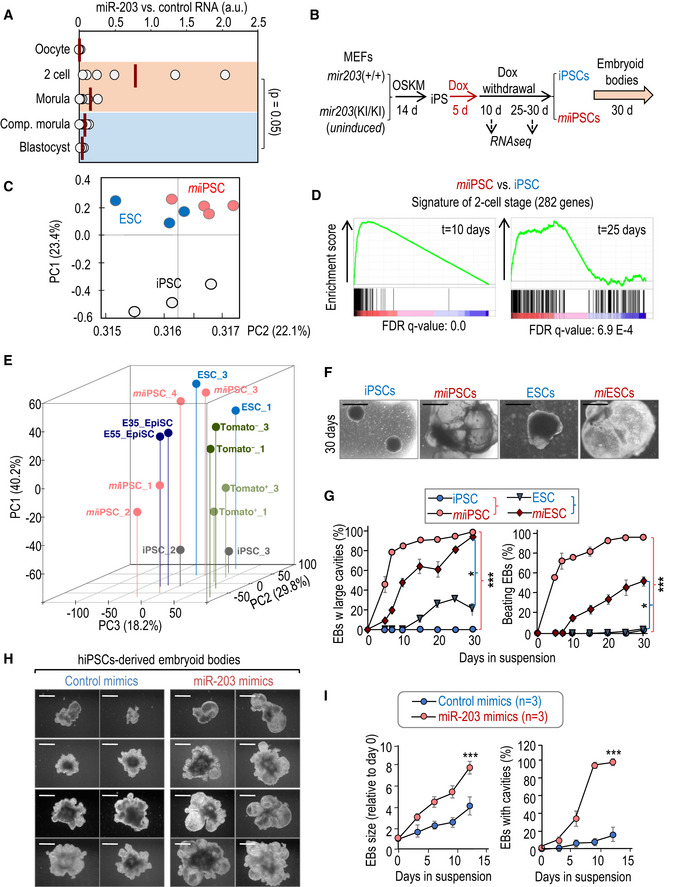
- miR‐203 expression, as determined by qPCR, in five temporal different stages of normal early development: oocyte, 2‐cell embryo, morula, compacted morula, and blastocyst. RNA was extracted from 30 different embryos and pooled in two independent groups for analysis by qPCR. RNA expression is normalized by a housekeeping miRNA (miR‐16) that maintained invariable during early embryogenesis. Data represent the mean of 6 different qPCR measures (red bars). P = 0.05 (Student's t‐test) comparing 2C/morula versus compacted morula/blastocyst.
- Protocol for reprogramming of miR‐203 mutant MEFs into pluripotent iPSCs and subsequent differentiation into embryoid bodies. MEFs were transduced with lentiviruses expressing Oct4, Sox2, Klf4, and cMyc (OSKM) in a constitutive manner. The resulting iPSCs were then treated with doxycycline (Dox) 1 μg/ml during 5 days to induce miR‐203 expression. “miiPSCs” refers to iPSCs in which miR‐203 was transiently expressed during the indicated 5 days. Dox was removed for 15–30 days before starting the embryoid body generation protocol. Samples for RNA sequencing were taken 30 days after Dox withdrawal.
- Principal component analysis of RNAseq data from wild‐type iPSCs (n = 3 clones), miiPSCs (n = 4), and wild‐type ESCs (n = 3).
- Enrichment plots of the 282‐gene 2‐cell signature (Biase et al, 2014) in miiPSCs 10 and 25 days after Dox withdrawal.
- PCA of RNAseq and microarray data from miiPSCs and known already‐published PSC types. Log2 expression values were normalized to mESCs in each study. Data from miiPSCs (this study), mES cells (each study), 2C‐like cells (MacFarlan et al, 2012), and epiblast stem cells (Najm et al, 2011 ) were analyzed, and a total of 17,243 genes were selected. For miiPSCs, clones 1 and 2 were analyzed 10 days after miR‐203 exposure, while clones 3 and 4 were analyzed 25 days after miR‐203 exposure (see schematic shown in Fig 1B). For Tomato PSCs, Tomato fluorescence is associated with 2C‐like retrotransposon expression, and thus, Tomato‐positive cells are identified as 2C‐like cells (Macfarlan et al, 2012).
- Representative images of embryoid bodies (EBs) derived from wild‐type iPSCs or ESCs, or from miiPSC and miESCs at day 30 of differentiation. Scale bars, 500 μm.
- Quantification of the percentage of EBs from panel (F) presenting internal large cavities and EBs beating during the indicated time course. Data are represented as mean ± SEM (n = 3 independent experiments). *P < 0.05; ***P < 0.001 (Student's t‐test).
- Representative images of EBs derived from human iPSCs transiently transfected with either control (left) or miR‐203 mimics (right), at different time points during the differentiation process. Scale bars, 500 μm.
- Left panel shows the quantification of EBs size derived from human iPSCs transiently transfected with either control or miR‐203 mimics as in panel (H) at different time points during the differentiation process. The percentage of EBs presenting internal large cavities during the indicated time course of differentiation is shown in the right panel. Data are mean ± SEM (n = 3 independent experiments). ***P < 0.001 (Student's t‐test).
Source data are available online for this figure.
To easily manipulating miR‐203 levels in vitro and in vivo, we generated a tetracycline‐inducible knock‐in model in which the miR‐203‐encoding sequence was inserted downstream of the type I collagen gene and expressed under the control of a tetracycline‐responsive element [ColA1(miR‐203) allele] in the presence of tetracycline reverse transactivator expressed from the Rosa26 locus [Rosa26(rtTA) allele] (Fig EV1A). Treatment of ColA1(miR‐203/miR‐203); Rosa26(rtTA/rtTA) mouse embryonic fibroblasts (MEFs) or ESCs with doxycycline (Dox) led to a significant induction of miR‐203 levels (Fig EV1B). We also generated iPSCs from wild‐type or un‐induced ColA1(miR‐203/miR‐203); Rosa26(rtTA/rtTA) MEFs by using lentiviral vectors expressing Oct4, Sox2, Klf4, and cMyc (Takahashi & Yamanaka, 2006). Similar to MEFs or ESCs, these mutant iPSCs showed a significant induction of miR‐203 after treatment with doxycycline (Fig EV1B). Although previous reports have suggested that this system suffers of relative leakiness (Stadtfeld et al, 2010b), miR‐203 expression significantly reduced and was undetectable a few days after Dox withdrawal (Fig EV1C).
Figure EV1. Mouse alleles generated and used in this work, transcriptomic analysis of miiPSCs, and validation of miR‐203 mimics and vectors on EBs differentiation.
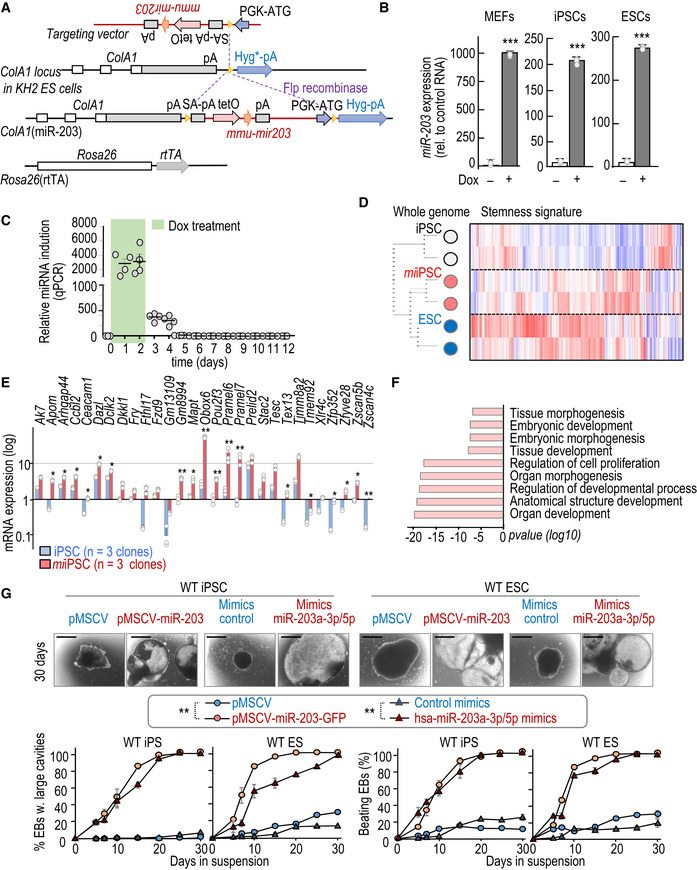
- Schematic representation of the miR‐203‐inducible knock‐in model generated in this work. In the ColA1(miR‐203/miR‐203); Rosa26(rtTA/rtTA) model, the reverse tetracycline transactivator is expressed from the Rosa26 locus, whereas miR‐203 is driven by the tetracycline operator downstream of the ColA1 locus.
- miR‐203 expression, as determined by quantitative PCR, in ColA1(miR‐203/miR‐203); Rosa26(rtTA/rtTA) MEFs, iPSCs, and ESCs treated or not with doxycycline (Dox). miR‐203 expression is normalized by a control miRNA (miR‐142). Data are mean ± SD (n = 3 independent experiments). ***P < 0.001 (Student's t‐test).
- Time course of miR‐203 expression, as determined by qPCR. RNA expression is normalized by a housekeeping miRNA (miR‐16) that maintained invariable. Data are mean ± SD (n = 4 independent replicates). P < 0.001 (Student's t‐test) comparing Dox treatment (green box) versus Dox withdrawal.
- Unbiased clustering of genome‐wide RNAseq data (left) and heat map plot showing the comparative expression of 450 genes associated with pluripotency (Chung et al, 2012); 2 clones per sample.
- Early expression (RPKM) of the indicated transcripts included in the 2C signature. Data are mean ± SEM (n = 3 independent experiments). *P < 0.05; **P < 0.01 (Student's t‐test).
- Top categories in the Gene Ontology analysis of the genes significantly upregulated in miiPSCs versus un‐induced iPSCs (4 independent clones were analyzed; see also Dataset EV1). P‐values were calculated by Fisher's exact test.
- (Upper panels) Representative images of EBs derived from either wild‐type iPSCs (left) or ESCs (right) transduced with empty pMCSV vector, pMCSV‐miR‐203, or transfected with control mimics or miR‐203 mimics, at day 30 of the differentiation process. Scale bars, 500 μm. (Lower panels) Quantification of EBs with large cavities and beating EBs during the differentiation process. Data are mean ± SEM (n = 3 independent experiments). **P < 0.01 (in both iPS and ES cells; Student's t‐test).
Source data are available online for this figure.
We next studied the long‐term effects of miR‐203 by treating ColA1(miR‐203/miR‐203); Rosa26(rtTA/rtTA) iPSCs (generated in the absence of Dox) transiently with Dox for 5 days (microRNA transiently induced iPSCs or miiPSCs; Fig 1B). RNA sequencing of these iPSC clones as well as wild‐type ESCs was analyzed 10 days or 1 month after miR‐203 induction (Fig 1B) and surprisingly revealed that miiPSCs were transcriptionally closer to ESCs than Dox‐treated wild‐type iPSCs both at the genome‐wide level (Fig 1C) and when considering a pluripotency‐associated signature defined previously (Chung et al, 2012) (Fig EV1D). In addition, almost every transcript included in a 282‐gene signature of 2C blastomeres (Biase et al, 2014) was induced by miR‐203 and sustained during 10 days after Dox withdrawal (Figs 1D and EV1E), including genes such as Zscan4c previously related to the zygotic transcriptional program (Eckersley‐Maslin et al, 2019). Importantly, we also assessed the transcriptomes of different clones of miiPSCs, mESCs, 2‐cell‐like mESC subpopulations (MacFarlan et al, 2012; GSE 33923), and epiblast stem cells (Najm et al, 2011; GSE 26814) (Fig 1B and E). Principal component analysis revealed a gene expression pattern of miiPSC clones 1 and 2 (analyzed 10 days after miR‐203 exposure) similar to that observed for Tomato‐positive cells, characterized as 2C‐like ESCs by MacFarlan and collaborators, while miiPSC clones 3 and 4 (analyzed 25 days after miR‐203 exposure) exhibited a profile more similar to ESC clones, corroborating the observations showed in Fig 1D. An additional comparison of the expression profiles in miiPSCs versus un‐induced iPSCs at day 30 after doxycycline withdrawal suggested higher expression of genes related to tissue morphogenesis and embryonic development in those clones in which miR‐203 had been transiently induced (Figs 1B and EV1F and Dataset EV1).
The differentiation potential of miiPSCs was directly tested in the embryoid body (EB) formation assay in vitro. Transient induction of miR‐203 for 5 days followed by 2–4 weeks of Dox withdrawal in either iPSCs (miiPSCs) or ESCs (miESCs) resulted in a significant increase in EB size accompanied by the formation of large internal cavities (Fig 1F and G, Appendix Fig S1A–C). Furthermore, miiPSC‐ and miESC‐derived EBs showed beating with higher efficiency and at earlier time points than their untreated counterparts (Fig 1G, Appendix Fig S1C). miiPSC‐derived EBs displayed a complex organization with high expression of primitive endoderm (Gata4), mesoderm (Cd34), and ectoderm (Pax6) or neuroectoderm (Nestin) markers (Appendix Fig S1D and E and Movie EV1).
To further validate these observations beyond the genetic model, we tested the effect of transient expression of ectopic miR‐203 using a CMV‐driven retroviral vector or RNA mimics (Fig EV1G and Appendix Fig S1F and G). These two strategies also resulted in improved EB formation (large cavities, beating) in miiPSCs or miESCs (Fig EV1G), indicating that these effects were not unique to the inducible genetic model.
Since the combination of the MEK inhibitor PD0325901 and the GSK3 inhibitor CHIR99021 with LIF (2i/L conditions) has been previously shown to render iPSCs closer to ESCs (Ying et al, 2008), we also tested the effect of miR‐203 under 2i/L conditions. miiPSCs grown in 2i/L also displayed an enrichment in the transcription of stemness factors and developmental pathways when compared to wild‐type iPSCs grown in the same conditions (Appendix Fig S2A). In addition, pre‐treatment with Dox for 5 days, 2–4 weeks prior to EB assays, of miiPSCs cultured in 2i/L conditions promoted a significant increase in EB size, formation of large internal cavities, and beating (Appendix Fig S2B and C), suggesting an additional effect of miR‐203 over the 2i/L conditions.
The expression of miR‐203 at the 2‐cell stage inspired us to further analyze the early expression of 2C‐related endogenous retroviruses in cultured miPSCs. Transient expression of miR‐203‐GFP in wild‐type ES cells significantly increased the number of 2C‐like cells as determined by the expression of the murine endogenous retrovirus with leucine tRNA primer (MuERV‐L; 2C::Td‐Tomato reporter; MacFarlan et al, 2012) analyzed 24 h after miR‐203 addition (Appendix Fig S3A). In line with these observations, exposure to miR‐203 induced the primary expression of genes harboring a proximal upstream or an intronic MERVL element (Appendix Fig S3B).
Of interest, transient exposure to miR‐203 mimics also rendered human pluripotent cells to a ground naive state (Appendix Fig S3C and D), as measured by the expression of the family HERVH of human endogenous retroviruses (HERVs) involved in the maintenance of human naive pluripotency (Wang & Gao, 2016). miR‐203 mimics induced the expression of the HERVH‐GFP reporter in a significant number of colonies, in many cases not only in the periphery but also in almost the totality of the cells of the colony, and such expression was sustained for 5 days in culture. When the differentiation potential of these cultures was tested (several weeks after miR‐203 transient expression), human miiPSCs generated significantly bigger EBs with larger internal cavities than the control counterparts (Fig 1H and I), suggesting altogether that the effect of miR‐203 can be achieved using delivery systems with independence of the genetic knock‐in system and it is also functional in human cells.
miR‐203 expands developmental potential and plasticity of PSCs
We then tested the potential of miiPSCs (in which the expression of miR‐203 had only been induced for 5 days in vitro) to form teratomas either after subcutaneous or intraperitoneal injection. miiPSCs formed significantly bigger tumors in these assays (Fig 2A). Intriguingly, these teratomas were not only bigger but also they contained tissues that are not typically found in control iPSC‐induced teratomas, such as bone marrow, pancreas, or cartilage, as well as trophoblast giant cells as confirmed by the expression of PL‐1 (Figs 2B and EV2). Transcriptomic studies in these complex structures suggested upregulation of genes involved in embryonic development and organ morphogenesis when derived from miiPSCs (Appendix Fig S4A and Appendix Table S1). Immunohistochemistry studies showed elevated expression of multiple differentiation markers representing ectoderm, mesoderm, and endoderm in miiPSC‐derived teratomas (Appendix Fig S4B and C). Interestingly, these teratomas also showed elevated levels of proliferation (as scored by Ki67; Appendix Fig S4D) or pluripotency markers such as Nanog, Oct4, or Sox2 in vivo (Appendix Fig S4B), suggesting that these structures contained a complex mixture of undifferentiated cells, expressing stemness markers, together with differentiated cells.
Figure 2. Transient exposure to miR‐203 in vitro improves the in vivo developmental potential of iPSCs and ESCs.
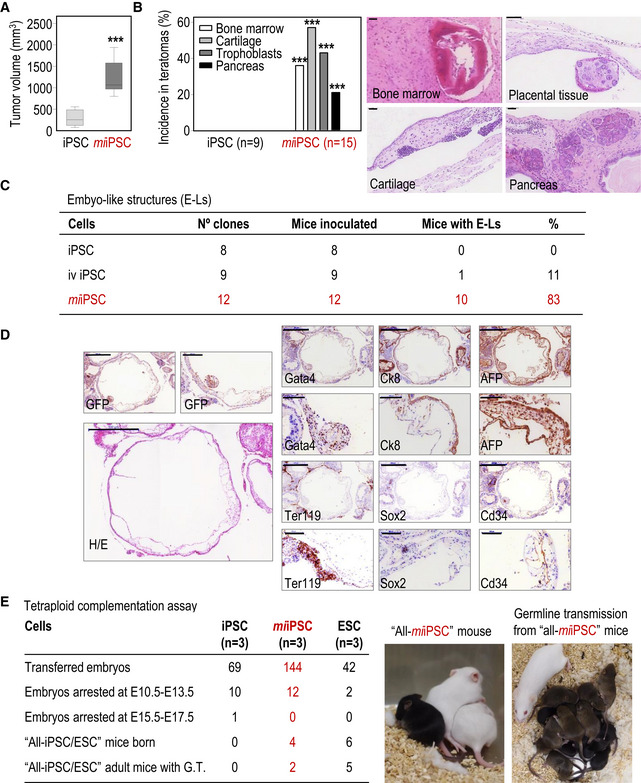
- Teratoma volume (mm3) 20–25 days after subcutaneous injection of wild‐type iPSCs or miiPSCs expressing GFP. Data are represented as mean ± SEM (n = 8 tumors per genotype; 292.8 ± 66.67 versus 1,216 ± 140.1; difference between means: 923.5 ± 155.2; 95% confidence interval: 1,256 to −590.7); ***P < 0.001 (Student's t‐test). Representative images are shown in Fig EV2.
- Incidence and representative images of specific highly differentiated tissues in teratomas. The number of tumors included in the analysis is indicated in the panel. ***P < 0.001 (Student's t‐test). Scale bars, 50 μm.
- Table showing the frequency of nude mice with embryo‐like structures (E‐Ls) in their abdominal cavity 20–30 days after intraperitoneal (i.p.) injection of 400,000–500,000 in vivo (iv)‐generated wild‐type iPSCs, un‐induced iPSCs, or miiPSCs. The number of independent clones tested per condition is indicated in the panel (each animal was inoculated with a different clone).
- Representative example of E‐Ls generated after i.p. injection of GFP‐expressing miiPSCs. H/E, Hematoxylin and eosin. The following antigens were detected by immunohistochemistry: Sox2 (ectoderm), Cd34 (mesoderm), Gata4 (endoderm), AFP, and CK8 (visceral endoderm of the yolk sac) and Ter119 (nucleated erythroid cells). Scale bars, 500 and 100 μm for higher magnifications.
- Embryo tetraploid complementation assays with un‐induced iPSCs, miiPSCs, or wild‐type ESCs (n = 3 clones per condition). Pictures on the right show a representative example of a viable “all‐miiPSC” mouse (black) generated from miiPSCs and litters obtained from “all‐miiPSC” adult mice, which efficiently contributed to germline transmission.
Source data are available online for this figure.
Figure EV2. Histopathological analysis of teratomas generated from miiPSCs.
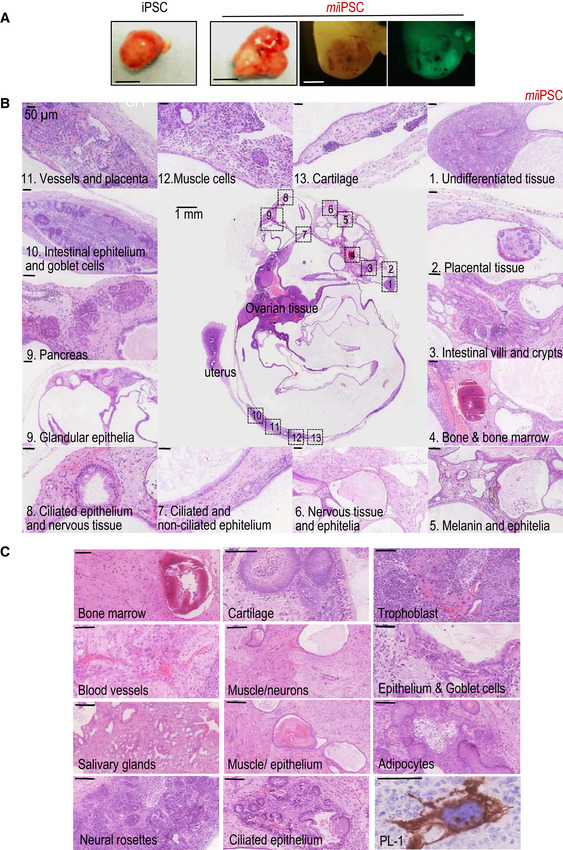
- Representative images of teratomas 20–25 days after subcutaneous injection of wild‐type iPSCs or miiPSCs expressing GFP, as indicated in Fig 2A. Images on the right show an example of a miiPSC‐derived embryo‐like structure, analyzed 20 days after intraperitoneal (i.p.) injection of miiPSCs expressing GFP. Scale bar, 5 mm (two left panels) and 1 mm (two right panels).
- Representative example of a highly differentiated teratoma generated from miiPSCs after i.p. injection in nude mice. Most of these complex teratomas were detected in the proximity of the uterus or as ovarian cysts in the host mice. The panel shows higher magnifications (H&E staining) of several differentiated tissues and cells observed in the teratoma. Scale bars, 1 mm (central image) or 50 μm (insets).
- Histopathological examples (H&E staining) of specific tissues found in miiPSC‐derived teratomas. Scale bars, 100 μm. A magnification of a trophoblast giant cell stained against placental lactogen‐1 (PL‐1) is also shown (scale bar, 50 μm).
It has been reported that iPSCs generated in vivo are able to form small embryo‐like structures, containing tissues derived from the three germinal layers in the presence of extraembryonic membranes, when inoculated intraperitoneally (Abad et al, 2013). In our hands, wild‐type iPSCs generated in vivo were able to form embryo‐like structures in 11% of injected mice, with an efficiency similar to the one reported previously (Abad et al, 2013). However, miiPSCs generated in vitro were much more efficient, with 83% of injected mice showing embryo‐like structures (Fig 2C). As in the case of in vivo‐formed iPSCs, miiPSC embryo‐like structures were positive for specific markers of the three embryonic layers (Fig 2D). Brief exposure to miR‐203 in vitro also improved the formation of chimeras from both miiPSCs and miESCs, achieving 100% of chimerism efficiency in miESCs (19 chimeras were obtained out of 19 pups born; Appendix Fig S5). The number of chimeras that reached adulthood and contributed to germline transmission was also notably increased (5–10 times, respectively) in miiPSC‐ and miESC‐derived mice compared with their respective untreated controls.
The performance of miiPSCs was also tested in the tetraploid complementation assay, one of the most stringent tests for developmental potency. Few iPSCs are efficient in this assay in which any born animal develops uniquely at the expense of exogenous diploid iPSCs (or ESCs) incorporated into a tetraploid embryo. Whereas we did not obtain all‐iPSC mice in control iPSCs, the very same clones were able to form all‐miiPSC mice after transient in vitro exposure to miR‐203 (Fig 2E). Importantly, half (2/4) of these “all‐miiPSC” mice reached adulthood healthy and were proficient for germline transmission (Fig 2E). Although specific clones of iPSCs have been previously found to contribute to all‐iPSC mice (Li et al, 2011), it is important to consider that the effect of miR‐203 is additive on already‐established PSCs providing an easy protocol for enhancing available PSCs.
miR‐203 increases human–mouse interspecies chimera competency
The high efficiency of miPSCs in contributing to mouse chimeras led us to test their effect in the formation of interspecies human–mouse conceptuses. Td‐Tomato fluorescent‐labeled human iPSCs were transiently transfected with pMCSV‐miR‐203‐GFP (human miiPSCs), and cells were maintained at least 1 week in culture to ensure that miR‐203 expression was transient and absent before cell injection. To compare the effect of miR‐203 in the context of the most efficient protocol reported so far, we cultured these human cells in the presence of human LIF, CHIR99021, (S)‐(+)‐dimethindene maleate, and minocycline hydrochloride (LCDM), a cocktail recently proposed to enhance the potential of PSCs (Yang et al, 2017). We initially injected 15 human iPSCs into 8C‐stage mouse embryos and examined its chimeric contribution after 48–60 h of culture in vitro. As shown in Appendix Fig S6, pre‐exposure of human miiPSCs to miR‐203 resulted in 100% of the mouse blastocysts presenting Td‐Tomato‐positive human cells (also identified with anti‐human OCT4 antibodies), whereas this efficiency was only 60% when using control human iPSCs. We next examined the contribution of human iPSCs to interspecies chimeras in similar assays in which injected 8C‐stage embryos were allowed to implant (Appendix Fig S7). The presence of human cells in mouse E10.5 conceptuses was identified by immunostaining with anti‐human nuclei (HuNu) antibody or by direct detection of fluorescent Td‐Tomato. Both HuNu and Td‐Tomato‐positive areas were significantly increased in embryos injected with miiPSCs when compared to control iPSCs (Appendix Fig S7), suggesting a more efficient contribution of human iPSCs to mouse embryos after brief exposure to miR‐203.
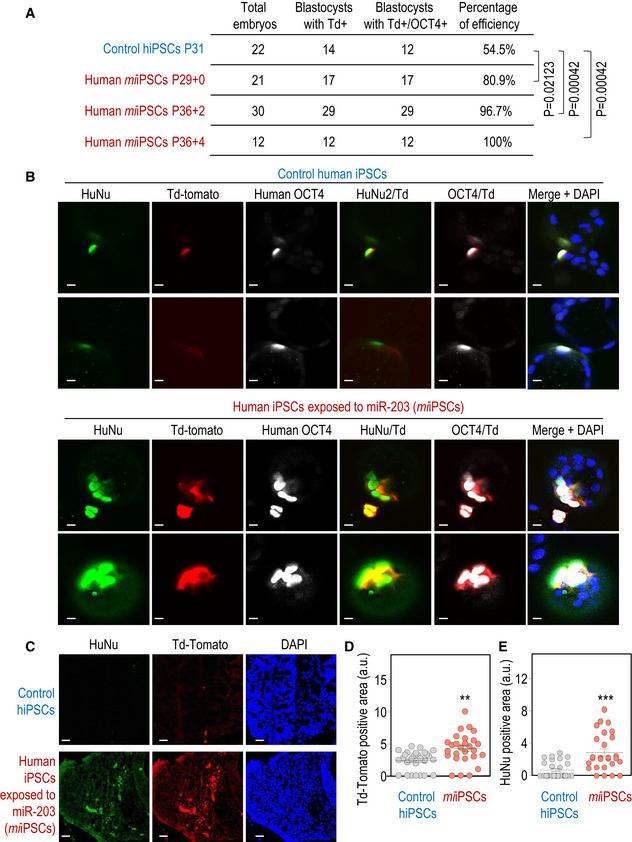
To further challenge the system, we injected one single human iPSCs into 8C‐stage mouse embryos and examined its chimeric contribution after 48–60 h of culture in vitro. As shown in Fig 3A and B pre‐exposure of human miiPSCs to miR‐203 resulted in 80–100% of the mouse blastocysts presenting Td‐Tomato‐positive human cells (also identified with anti‐human OCT4 and HuNu antibodies), whereas this efficiency was only 54% when using control human iPSCs also cultured in the presence of LCDM but not miR‐203. We next examined the contribution of human iPSCs to interspecies chimeras in similar assays in which one human cell‐injected 8C‐stage embryos were allowed to implant. The presence of human cells in mouse E9.5 conceptuses was identified by immunostaining with anti‐human nuclei (HuNu) antibody or by direct detection of fluorescent Td‐Tomato, and their proper integration and differentiation into the mouse embryo were corroborated using embryo markers such as Gata4, Sox2, or hSOX17 (Fig EV3C). Both HuNu‐ and Td‐Tomato‐positive areas were significantly increased in embryos injected with miiPSCs when compared to control iPSCs (Figs 3C–E and EV3A and B). Altogether, these data suggested a more efficient contribution of human iPSCs to mouse embryos after brief exposure to miR‐203.
Figure 3. miR‐203‐exposed human iPSCs efficiently contribute to human–mouse interspecies chimeras.
-
ASummary of chimera assays in which one single Td‐Tomato fluorescent‐labeled human iPSC, either control or transiently transfected with miR‐203‐expressing vectors (miiPSCs), was injected into 8C‐stage mouse embryos and their contribution analyzed 48‐60 h later. P‐values are indicated in the figure (Student's t‐test).
-
BRepresentative images showing the contribution of human control iPSCs or miiPSCs to mouse blastocysts. These structures were co‐immunostained with (human) anti‐OCT4 and anti‐HuNu antibodies. Td‐Tomato, red; Scale bars, 50 μm.
-
CInterspecies chimera assays in which the 8C‐stage mouse embryos indicated in (A, B), injected with one single human iPSCs and cultured in vitro for 48 h to reach the blastocyst stage, were then transfected to 2.5 days post‐coitum pseudo‐pregnant females. The post‐implantation mouse conceptuses were dissected at the E9.5 developmental stage and analyzed by immunofluorescence. Representative images showing the integration of control human iPSCs (top) or miiPSCs (bottom) into mouse E9.5 embryos. Anti‐human HuNu antibody was co‐stained with Td‐Tomato direct fluorescence to detect human cells in E9.5 mouse embryos. Scale bars, 20 μm.
-
D, EQuantification of the Td‐Tomato‐positive (D) or HuNu‐positive (E) area in 40‐50 different cryosections of these embryos randomly and blindly selected for IF analysis. Data are mean ± SEM **P < 0.01; ***P < 0.001 (Student's t‐test).
Source data are available online for this figure.
Figure EV3. miR‐203‐exposed human iPSCs efficiently contribute to human–mouse interspecies chimeras.
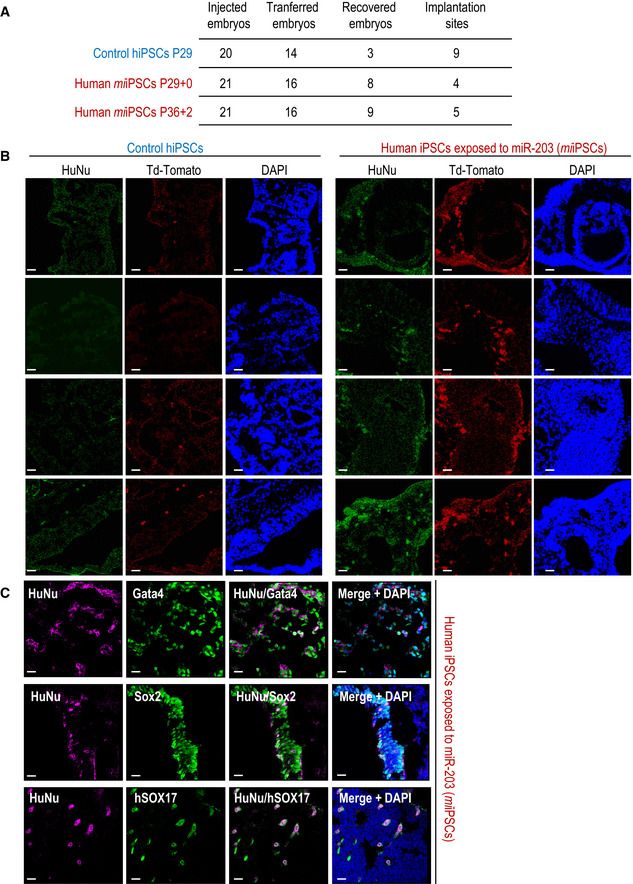
- Summary of interspecies chimera assays in which one single Td‐Tomato fluorescent‐labeled human iPSC, either control or transiently transfected with miR‐203‐expressing vectors (miiPSCs), was injected into 8C‐stage mouse embryos. These embryos were cultured in vitro for 48 h to reach the blastocyst stage and then transfected to 2.5 days post‐coitum pseudo‐pregnant females. The post‐implantation mouse conceptuses were dissected at the E9.5 developmental stage and analyzed by immunofluorescence.
- Representative immunofluorescence images from these E9.5 embryos summarized in panel (A). Anti‐human HuNu antibody was co‐stained with Td‐Tomato direct fluorescence to detect human cells in E9.5 mouse embryos. Scale bars, 20 μm.
- Representative immunofluorescence images from these E9.5 embryos summarized in panel (A), co‐stained with human HuNu antibody and Gata4, Sox2, or hSOX17 antibodies showing differentiation and proper integration of human cells into the mouse embryo. Scale bars, 20 μm.
miR‐203 effects are Dnmt3a/b‐dependent
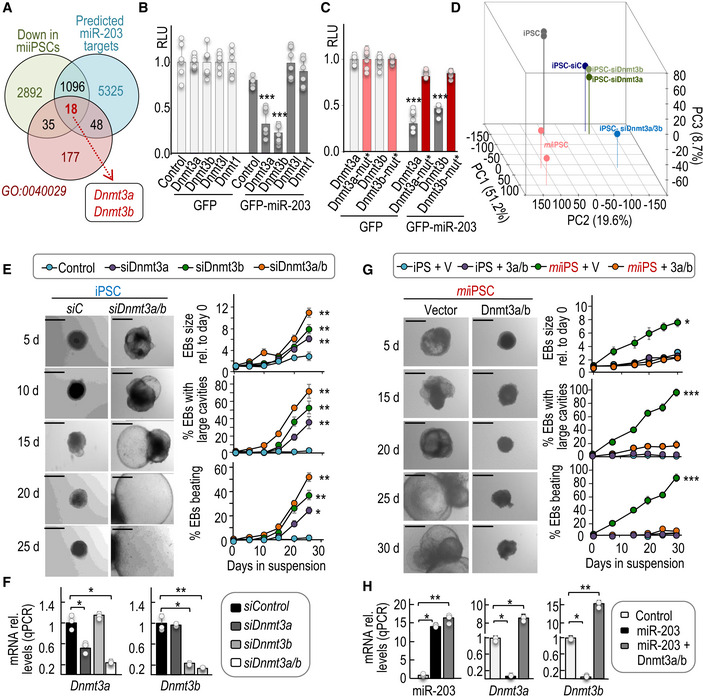
To analyze the mechanism by which miR‐203 enhances the differentiation capacity of PSCs, we searched for predicted miR‐203 targets among the transcripts downregulated in miiPSCs (Fig 4A). As the long‐term effects resulting from transient expression of miR‐203 suggest an epigenetic mechanism, we selected from the previous list those genes involved in epigenetic regulation of gene expression (GO0040029). We found 18 GO0040029 transcripts downregulated in miiPSCs and predicted as miR‐203 targets according to different microRNA target prediction algorithms (Fig 4A and Appendix Table S2). Among these transcripts, the top hits with the highest score as miR‐203 targets corresponded to the de novo DNA methyltransferases Dnmt3a and Dnmt3b. These two transcripts were also significantly downregulated after expression of miR‐203 mimics in wild‐type iPSCs (log2 fold change = −0.24 for Dnmt3a and −0.22 for Dnmt3b transcripts). Human miR‐203 and DNMT3a/b transcripts have been previously shown to display inverse expression profiles in cancer cells (Sandhu et al, 2012; Gasque Schoof et al, 2015), and DNMT3b was recently shown to be a direct miR‐203 target in human colon cancer cells (To et al, 2017). Both Dnmt3a and Dnmt3b transcripts contain conserved miR‐203 sites in their 3′‐UTR (Fig EV4A), and exogenous expression of miR‐203 led to decreased signal of a luciferase construct fused to these sequences, but not to 3′‐UTR sequences from the related genes Dnmt3l or Dnmt1 (Fig 4B and C). The downregulation of these genes was abrogated (Fig 4C) when the putative miR‐203‐binding sites were mutated (Fig EV4B) indicating a direct control of these transcripts by miR‐203. To further confirm the regulation of DNMT3A/B by miR‐203, we also tested their protein levels in 5 different clones of mouse un‐induced iPSCs or miiPSCs as well as one human cell line, transfected with control or miR‐203 mimics, immediately after miR‐203 exposure (Fig EV4E).
Figure 4. DNA methyltransferases 3a and 3b are miR‐203 targets involved in the regulation of PSC potential.
-
AVenn diagrams representing common genes downregulated in miiPSCs, predicted as miR‐203 targets, and also involved in the epigenetic regulation of gene transcription (GO:0040029). The list of the common 18 transcripts (including Dnmt3a and Dnmt3b) is presented as Appendix Table S2.
-
B, CRelative luciferase units (RLU; normalized to Renilla luciferase and relative to DNA amount) in 293T cells transfected with DNA constructs carrying the wild‐type 3′‐UTRs from the indicated transcripts (B) or the mutated versions of Dnmt3a and Dnmt3b 3′‐UTRs, downstream of the luciferase reporter (C). Cells were co‐transfected with Renilla luciferase as a control of transfection, and a plasmid expressing GFP or miR‐203‐GFP. Data are represented as mean ± SD (n = 3 independent experiments).
-
DPrincipal component analysis from RNAseq data including profiles from wild‐type iPSCs, miiPSCs, and wild‐type iPSCs transfected with either control siRNAs (siC), or siRNAs specific against Dnmt3a (siDnmt3a), Dnmt3b (siDnmt3b), or both (siDnmt3a/b).
-
ERepresentative images of embryoid bodies (EBs) derived from wild‐type iPSCs in which the expression of Dnmt3a and Dnmt3b was transiently repressed by siRNAs. Scale bars, 500 μm. Plots show the quantification of the size of EBs and the percentage of EBs with large cavities or beating at different time points during the differentiation process. Data are represented as mean ± SEM (n = 3 independent experiments).
-
FExpression levels of Dnmt3a or Dnmt3b transcripts after transfection of wild‐type iPSCs with specific siRNAs either against Dnmt3a, Dnmt3b, or a combination of both (Dnmt3a/b) (as indicated in E). RNA expression was measured 24 h after the transfection protocols and was normalized by GAPDH mRNA levels. Data are represented as mean ± SEM (n = 3 independent experiments).
-
GRepresentative images of EBs derived from miiPSCs that were transiently and simultaneously transduced with Dnmt3a and Dnmt3b cDNAs or empty vectors, and simultaneously treated with Dox to induce miR‐203 expression. Scale bars, 500 μm. Plots show the quantification of EB size, percentage of EBs with large cavities, and beating EBs at different time points during differentiation. Data are represented as mean ± SEM (n = 3 independent experiments).
-
HExpression levels of miR‐203, Dnmt3a, or Dnmt3b transcripts in iPSCs after induction of miR‐203 (miiPSCs) and transduced with Dnmt3a and Dnmt3b cDNAs or empty vectors (as indicated in G). RNA expression was measured 24 h after the transfection protocols and was normalized by a control miRNA (miR‐142) or GAPDH mRNA, respectively. Data are represented as mean ± SEM (n = 3 independent experiments).
Figure EV4. miR‐203 targets Dnmt3a/b 3′‐UTR sequences.
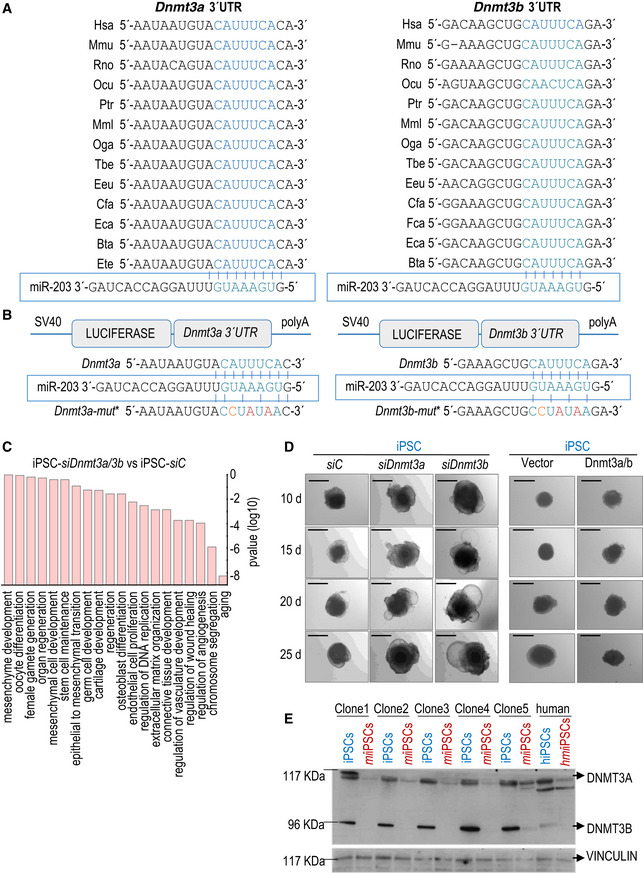
- Dnmt3a and Dnmt3b 3′‐UTR alignment in several representative species. The seed region of the miR‐203 target site contained in these 3′‐UTRs is highlighted in blue and aligned with the corresponding miR‐203 seed sequence.
- Schematic representation of the luciferase reporter, carrying the wild‐type Dnmt3a (left) or Dnmt3b (right) complete 3′‐UTRs or the corresponding mutated versions, downstream of the luciferase gene. The seed region of the miR‐203 target site contained in these 3′‐UTRs is written in blue and aligned with the corresponding miR‐203 seed sequence. The mutated residues are indicated in red.
- Major pathways from the Gene Ontology database upregulated in wild‐type iPSCs treated with specific siRNAs against Dnmt3a and Dnmt3b versus scrambled sequences (siC). P‐values were calculated by Fisher's exact test.
- Representative images of embryoid bodies (EBs) generated from wild‐type iPSCs treated with specific siRNAs either against Dnmt3a or Dnmt3b (left panel) or transfected with a combination of miR‐203‐resistant Dnmt3a/b cDNAs (right panel) at the indicated time points during the differentiation process. Scale bars, 500 μm.
- Detection of Dnmt3a (117 KDa) and Dnmt3b (96 KDa) protein levels by Western blot, in 5 independent clones of mouse un‐induced iPSCs or miiPSCs and one human cell line, transfected with mimics control (hiPSCs) or miR‐203 mimics (hmiiPSCs). Protein extracts were evaluated immediately after miR‐203 exposure, to detect the modulation of Dnmt3a/b protein levels. Vinculin levels are included as loading control.
To test to what extent downregulation of Dnmt3a/b could mimic the effect of miR‐203, we knocked down these de novo DNA methyltransferases by RNA interference means (siDnmt3a/b). RNA sequencing of these samples (following the protocols explained above) revealed that siDnmt3a/b iPSCs exhibited a transcriptomic profile closer to miiPSCs (Figs 4D and EV4C). In addition, downregulation of Dnmt3a/b induced the growth, formation of long cavities, and increased beating of EBs to a similar extent to that observed in miiPSC‐derived EBs (Fig 4E and F), whereas individual knockdown of these transcripts displayed partial effects (Figs 4E and F, and EV4D). Importantly, the overexpression of miR‐203‐resistant Dnmt3a and Dnmt3b cDNAs, simultaneously to the transient doxycycline treatment, prevented the overgrowth of miiPSC‐derived EBs (Figs 4G and H, and EV4D).
At shorter time points, knockdown of Dnmt3a/b also increased the 2C‐like population in wild‐type ESCs as scored by the expression of the MERVL element (Appendix Fig S8), although to lesser extent than miR‐203. Whether these differences are due to variable efficiencies in the control of gene expression of additional miR‐203 targets is unclear at present. Moreover, the expression of miR‐203‐resistant Dnmt3a/b cDNAs significantly prevented the expression of the 2C‐related marker, as measured 5 days after transfections (Appendix Fig S8). Altogether, these observations demonstrate that Dnmt3a/b de novo methyltransferases are miR‐203 targets, in both mouse and human PSCs, responsible of at least part of the miR‐203‐mediated effects.
miR‐203 expression results in global and reversible DNA hypomethylation in PSCs
Given the critical role of Dnmt3a/b as de novo DNA methyltransferases, we next analyzed the genome‐wide methylation profile of control (un‐induced) iPSCs and miiPSCs (after transient induction with Dox) as well as embryoid bodies derived from them (Fig 5A). Wild‐type and mutant iPSCs displayed similar levels of methylation before Dox, and wild‐type cells were not affected by this treatment. In contrast, transient induction of miR‐203 for 5 days resulted in a genome‐wide hypomethylation in miiPSCs, which was strikingly more prominent 20 days after withdrawal of Dox (t = 25; Fig 5B–E and Datasets EV2 and EV3), a time point in which Dnmt3a/b transcript levels had already recovered after their repression in the presence of Dox (Fig EV5A). Notably, the number of DNA methylation valleys (DMVs; (Xie et al, 2013) and partially methylated domains (PMDs; (Lister et al, 2009) was increasingly higher in miiPSCs with time (Fig 5B and Dataset EV3); for instance, 131 PMDs were found at t = 0, while 548 and 6,555 PMD were found at t = 10 and t = 25, respectively. DNA methylation comparison between groups showed that 128 differentially methylated regions (DMRs) out of 131 total DMRs (97.7%; t = 10 versus t = 0) and 12,542 out of 12,549 (99.9%; t = 25 versus t = 0) DMRs were hypomethylated in miiPSCs as a consequence of previous exposure to miR‐203 (Fig 5E). These gradual changes in methylation spread over a significant proportion of transcription start sites (TSSs) in a genome‐wide manner (Fig 5F), and also affected genes of the 2C signature (Fig 5G and Dataset EV4), in line with the observed upregulation in the expression levels of these 2C markers. Further transcriptional analysis of these samples suggested that deregulated genes with affected DMRs were significantly enriched in chromatin regulators, or genes involved in DNA replication and cell division or embryonic morphogenesis among other pathways (Appendix Fig S9 and Appendix Tables S5 and S6).
Figure 5. Transient expression of miR‐203 induces genome‐wide hypomethylation in iPSCs.
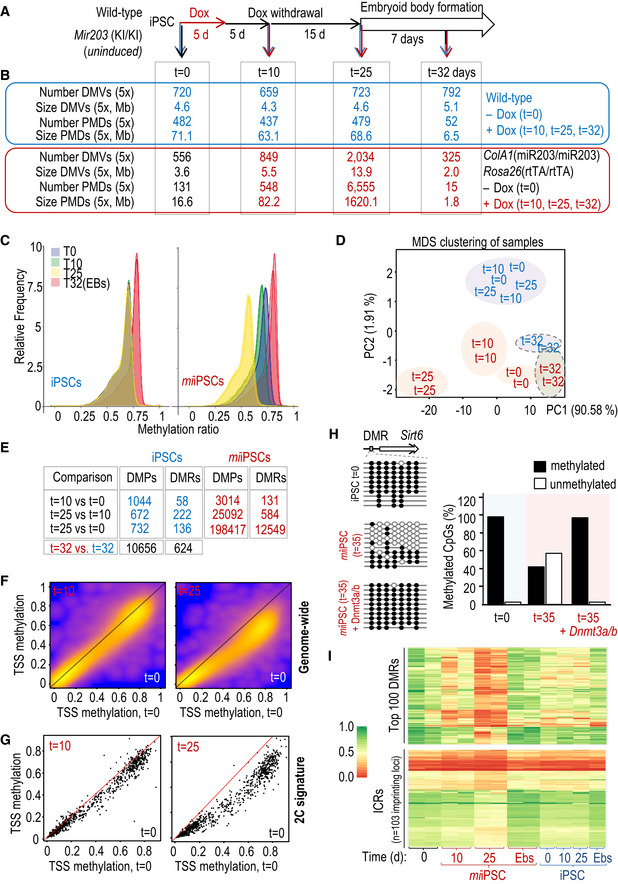
- Experimental design for the genome‐wide DNA methylation analysis of wild‐type and miiPSC as well as embryoid bodies (EBs) derived from them. Cells (two independent miiPSC clones and two iPSC technical replicates) were transiently treated with Dox for 5 days and then subjected to Dox withdrawal for 20 additional days before starting the EB formation protocol. Samples for DNA and RNA analysis were collected at the indicated time points before Dox (t = 0), 5 days after Dox withdrawal (t = 10), 20 days after Dox withdrawal (t = 25), or 7 days after starting the EB generation protocol (t = 32 days).
- Genome‐wide DNA methylation data showing the number and total size of DNA methylation valleys (DMVs) and partially methylated domains (PMDs).
- DNA methylation distribution of the indicated samples, smoothed over 100‐kb blocks.
- Principal component analysis showing the distribution of methylation profiles in the indicated samples (color code as in panel B).
- Number of differentially methylated single CpG sites (DMPs) and differentially methylated regions (DMRs) in the indicated comparisons (color code as in panel B).
- Heat scatterplots representing the mean methylation over all transcripts (transcription start sites, TSSs) in mm10, pairwise for “t = 10 versus t = 0” (left panel) and “t = 25 versus t = 0” (right panel). Note that especially for “t = 25 versus t = 0,” most of the points are below the diagonal, indicating broad hypomethylation across most of the genome.
- Heat scatterplots representing the mean methylation over the TSS of the 282 genes included in the 2‐cell signature (as in Fig 1D). Analysis as in (F).
- The specific differentially methylated regions (DMRs) at the Sirt6 locus were analyzed by PCR amplification and sequencing of bisulfite‐modified DNA. Ten independent clones were sequenced per condition. The quantification of methylated (filled circles) versus unmethylated (empty circles) CpGs is shown in the histogram in the indicated conditions.
- Heat maps representing the methylation levels at the top differentially methylated regions (DMRs; n = 100; upper panel) or the imprinting control regions (ICRs; n = 103 different imprinting loci; lower panel) in the indicated samples (referred to panels A, B). Color code and scale of DNA methylation, from 0.0 (red) to 1.0 (green), are applicable to both heat maps.
Source data are available online for this figure.
Figure EV5. Genome‐wide methylation of iPSCs or embryoid bodies after transient exposure to miR‐203.
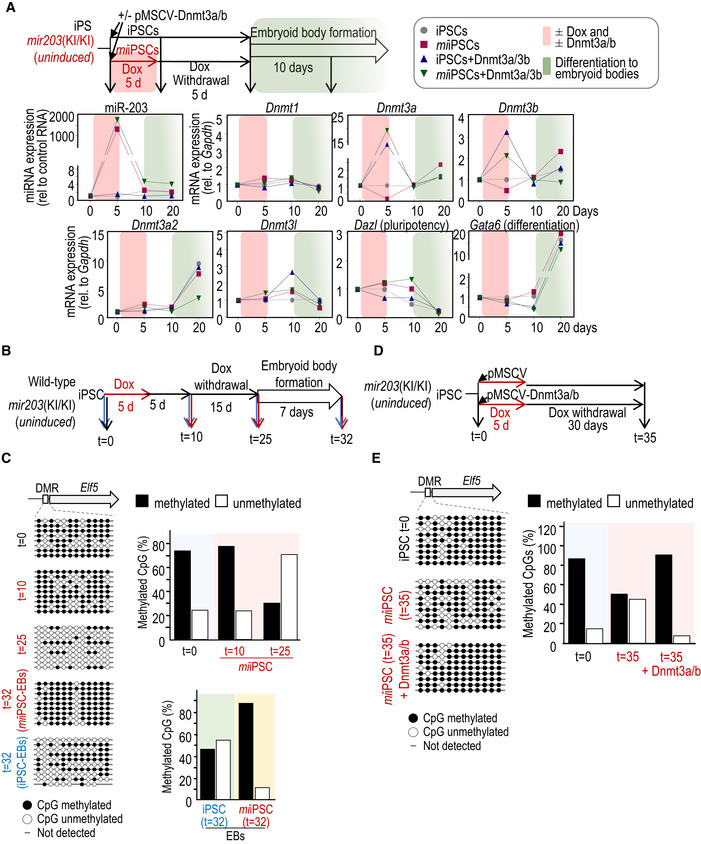
- Expression levels, as determined by quantitative PCR, of miR‐203, and transcripts for the DNA methyltransferases Dnmt1, Dnmt3a, Dnmt3b, Dnmt3a2, and Dnmt3l, or pluripotency (Dazl) and differentiation (Gata6) markers. ColA1(miR‐203/miR‐203); Rosa26(rtTA/rtTA) iPSCs treated or not with doxycycline (Dox) and simultaneously transduced with Dnmt3a/b cDNAs or empty vector were used as shown in the schematic representation of the experimental design. The pink shadow indicates the time lapse in which the cells were treated or not with Dox and transduced or not with Dnmt3a/b cDNAs. The green shadow indicates the differentiation process to embryoid bodies. Data are represented as mean of three technical replicates per experiment (n = 2 independent experiments).
- Experimental design used for the validation of methylation data in the indicated Elf5 differentially methylated region (DMR).
- DNA was isolated as indicated and sequenced after bisulfite modification. Eight to ten independent clones were sequenced per condition. Histograms show the percentage of DNA methylation at the Elf5 DMR in the different conditions.
- Experimental protocol followed to test DNA methylation rescue by miR‐203‐resistant Dnmt3a/b cDNAs.
- The specific differentially methylated regions (DMRs) at the Elf5 locus were analyzed by PCR amplification and sequencing of bisulfite‐modified DNA. The quantification of methylated versus unmethylated CpGs is shown in the histogram.
Source data are available online for this figure.
As validation of the genome‐wide methylation analysis, bisulfite modification followed by clonal sequencing confirmed miR‐203‐dependent hypomethylation of the promoter region of the gene encoding the E74‐like ETS transcription factor 5 gene (Elf5), a protein involved in the differentiation of epithelial cells and trophoblast stem cell renewal and differentiation (Latos et al, 2015) (Fig EV5B–E). Interestingly, the embryoid bodies derived from miiPSCs displayed higher global levels of DNA methylation and lower number of PMDs (Fig 5B–E) in agreement with the upregulation of Dnmt3a and Dnmt3b transcripts observed after induction of differentiation (Fig EV5A). In line with the observations at a genome‐wide scale, the Elf5 promoter was hypermethylated in embryoid bodies generated from miiPSCs during the differentiation process (Fig EV5B and C). Previous data suggest that interfering with DNA methyltransferase expression or activity results in global hypomethylation in the genome (Chen et al, 2003; Jackson et al, 2004; Mikkelsen et al, 2008; Blattler et al, 2014; Liao et al, 2015). Expression of exogenous, miR‐203‐resistant forms of Dnmt3a/b rescued the hypomethylation observed after miR‐203 induction both in the Elf5 DMRs (Fig EV5D and E) as well as in a DMR located at the histone deacetylase Sirt6 gene (Fig 5H), suggesting that these de novo DNA methyltransferases are critical targets of miR‐203 in inducing genome‐wide hypomethylation.
Given the recent findings showing that a widespread loss of methylation in PSC cultures might be deleterious when accompanied by massive erasure of genomic imprints (Choi et al, 2017a; Yagi et al, 2017), we tested the methylation levels at 103 different genes controlled by imprinting control regions (ICRs) in miiPSCs. Whereas miiPSCs displayed a progressive demethylation of genes (red signal at t = 10 and t = 25 in Fig 5I; upper panel), demethylation of ICRs was very limited at t = 10 and moderate at t = 25 in the same samples (lower panel). Importantly, demethylation was in all cases fully recovered upon differentiation (miiPSC‐derived EBs; Fig 5I), suggesting that demethylation of miiPSCs, in both DMRs and ICRs, is manageable and reversible, and does not compromise neither the quality of miiPSCs nor their competence to differentiate.
Transient exposure to miR‐203 improves differentiation and maturation into cardiomyocytes in a Dnmt3a/b‐dependent manner
Since transient expression of miR‐203 improves the function of pluripotent cells in several assays, we next decided to directly testing the effect of expressing this miRNA during differentiation of PSCs into cardiomyocytes. The effect of miR‐203 was first tested in primary cardiomyocytes isolated from neonatal (P1) rats that undergo further expansion and differentiation when cultured in vitro. miR‐203 mimics triggered a transient burst of cell proliferation as measured by incorporation of the nucleotide analogue EdU (Fig EV6A) and mitotic markers such as cyclin B1 (Fig EV6B). Importantly, this increase in proliferation occurred in cardiac troponin T (cTnT)‐positive cells (Fig EV6C) and resulted in cells with increased ratio of Myh6 versus Myh7 myosin heavy chain genes (Fig EV6B), a developmentally regulated switch that correlates with cardiomyocyte maturation and cardiac performance (Miyata et al, 2000).
Figure EV6. Improved cardiomyocyte differentiation and maturation after transient expression of miR‐203.
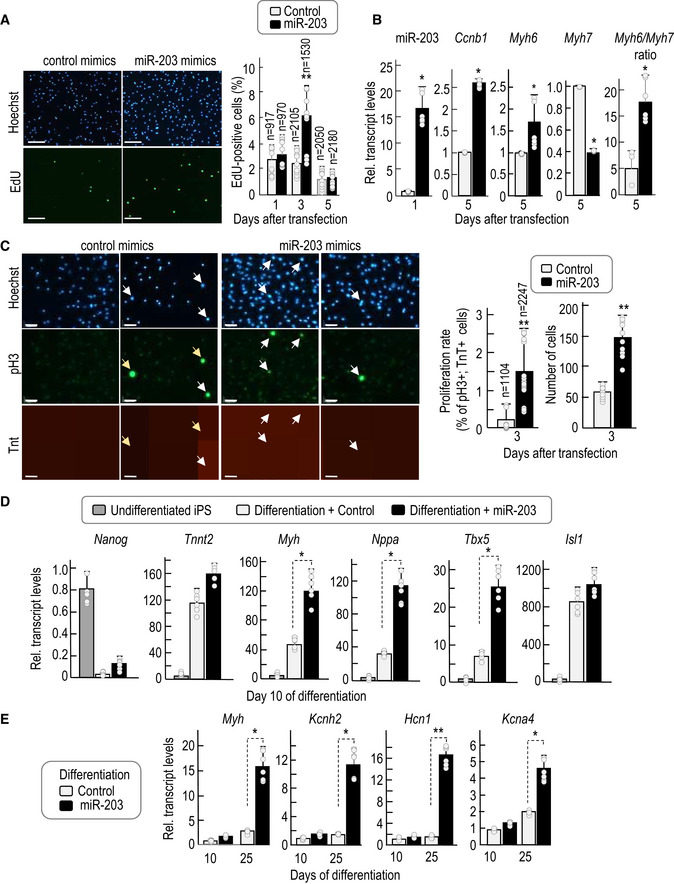
- Representative immunofluorescence showing EdU (green) and nuclei (Hoechst, blue) staining of primary cardiomyocytes extracted at post‐natal day 1 and transiently transfected with control or miR‐203 mimics 24 h after extraction. Pictures were taken 3 days after transfection. Scale bar, 60 μm. The histogram shows the percentage of EdU‐positive cells at different days post‐transfection. Data are mean ± SD (total number of cells counted is indicated in the figure; the dots represented in the bars correspond to n = 2 independent experiments, with 5 and 4 different replicates, respectively.
- RNA expression as determined by quantitative PCR of miR‐203 (24 h after transfection) and Ccnb1, Myh6, and Myh7 transcripts (5 days after transfection). The Myh6/Myh7 ratio is calculated as an indicator of cardiomyocyte maturation. Data are mean ± SD (the dots represented in the bars correspond to n = 3 independent experiments, with 2 replicates each).
- Representative immunofluorescence showing phospho‐histone 3 (Ser‐10; pH3) in green, cardiac Troponin T (cTnT) in red, and Hoechst for nuclei staining in blue, in primary cardiomyocytes extracted at post‐natal day 1 and transiently transfected with control or miR‐203 mimics 24 h after extraction. Images were taken 3 days after transfection. Scale bars, 64 μm. White arrows point to cardiomyocytes positive for pH3. Yellow arrows point to cells positive for pH3 but not properly differentiated to cardiomyocytes, according to cardiac Troponin T staining. Middle histogram shows the proliferation rate measured as the percentage of pH3‐positive cells respect to the total number of cTnT‐positive cells at day 3 post‐transfection. Data are mean ± SD Total number of cells counted is indicated in the figure; the dots represented in the bars correspond to n = 2 independent experiments, with 8 different replicates each. The plot on the right shows the total number of cells at day 3 post‐transfection. Data are mean ± SD. The dots represented in the bars correspond to n = 2 independent experiments with 5 replicates each.
- mRNA levels as determined by quantitative PCR of the indicated transcripts at different time points before and during cardiomyocyte differentiation. iPSCs were transfected either with control mimics or with miR‐203 mimics, maintained during 15 days in culture, and then differentiated in vitro. Data are mean ± SD The dots represented in the bars correspond to n = 2 independent experiments, with 3 replicates each.
- mRNA levels of the indicated transcripts at different time points during cardiomyocyte differentiation. Data are mean ± SD The dots represented in the bars correspond to n = 2 independent experiments, with 3 replicates each.
We also tested a cardiomyocyte differentiation protocol from wild‐type iPSCs (Kattman et al, 2011). Mouse iPSCs were transiently transfected with miR‐203 or control mimics, and 15 days later, they were differentiated into cardiomyocytes using specific media and culture conditions (Fig 6A). Transient exposure to miR‐203 was accompanied by increased expression of cardiomyocyte differentiation transcripts such as myosin heavy chain (Myh), atrial natriuretic peptide (Nppa), cardiac troponin T (encoded by the Tnnt2 gene), and markers for cardiac progenitors such as insulin gene enhancer transcription factors Isl1 and Tbx5 in iPSCs previously treated with miR‐203 mimics (Figs 6B and EV6D). Importantly, transient exposure to miR‐203 resulted in higher expression of not only differentiation but also maturation markers such as the potassium channel components encoded by the Kcnh2, Hcn1, and Kcna4 genes (Otsuji et al, 2010) that were only minimally induced in cells treated with control mimic RNAs (Fig EV6E). In line with these data, the beating frequency in cardiomyocytes derived from miR‐203‐treated iPSCs was significantly higher suggesting enhanced functionality (Fig 6C and Movies EV1 and EV2). Expression of miR‐203‐resistant forms of Dnmt3a and Dnmt3b in parallel to miR‐203 (Fig 6A) prevented the upregulation of these differentiation and maturation markers (Fig 6B and D) as well the increase in beating frequency (Fig 6C), suggesting the relevance of the miR‐203‐Dnmt3a/b axis in functional differentiation and maturation of cardiomyocytes from pluripotent cells.
Figure 6. Transient exposure to miR‐203 enhances differentiation into mature cardiomyocytes and improves cardiac regeneration.
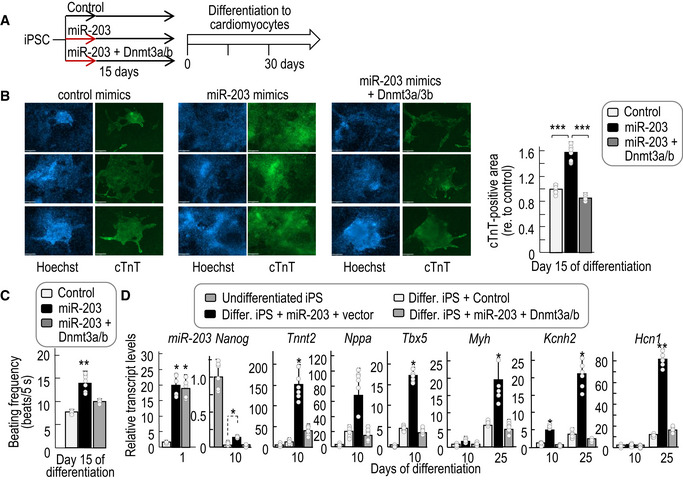
- Experimental protocol followed for the differentiation of cardiomyocytes from iPSCs in the absence or presence of miR‐203 mimics and Dnmt3a/b cDNAs.
- Representative immunofluorescences showing cardiac Troponin T (cTnT, green) and nuclei (Hoechst, blue) staining of in vitro‐generated cardiomyocytes derived from wild‐type iPSCs transiently transfected with either control mimics, miR‐203 mimics, or miR‐203 mimics + Dnmt3a/b cDNAs. Pictures were taken at day 15 of differentiation. Scale bars, 68 μm. The cTnT‐positive area in these cardiomyocytes is shown in the right histogram. Data are represented as mean ± SD (n = 2 independent experiments with 6 replicates each).
- Beating frequency (measured as number of beats per 5 s) of these cardiomyocytes at day 15 of differentiation. Data are represented as mean ± SD (n = 8 independent replicates).
- miRNA or mRNA levels as determined by quantitative PCR of the indicated transcripts at different time points during cardiomyocyte differentiation in the indicated samples. Data are represented as mean ± SD (n = 2 independent experiments with 6 replicates each).
Discussion
Multiple efforts in the last decade have focused on improving the developmental and differentiation potential of PSCs with the ultimate goal of using these cells in regenerative medicine. PSCs, and in particular iPSCs, are inefficient in the most stringent assays such as tetraploid complementation (Kang et al, 2009; Zhao et al, 2009) and human–mouse interspecies assays (Mascetti & Pedersen, 2016; Wu et al, 2016). Improving culture conditions has been a major subject of research offering multiple options (Choi et al, 2017a; Yagi et al, 2017; Yang et al, 2017). The use of 2i/L clearly improves some properties of PSCs in vitro and has been widely used in the last years, although recent data indicate that prolonged use of these conditions results in reduced developmental potential in vivo (Choi et al, 2017a; Yagi et al, 2017). Recent alternatives include the use of Src inhibitors or the LCDM cocktail whose targets require further validation (Choi et al, 2017a; Yagi et al, 2017; Yang et al, 2017). Despite these advances, the long‐term effects of chemicals that perturb major signaling pathways in the cell remain uncertain. In particular, it will be critical to study how these treatments affect the differentiation potential of PSCs toward specific cell lineages and the functional properties of the resulting cells.
In this work, we report that transient expression of a single microRNA can expand functionality and differentiation plasticity of either mouse or human PSCs both in vitro and in vivo. Already‐established PSCs display enhanced expression of a signature of the top ~280 transcripts characteristic of the 2‐cell stage during embryonic development (Biase et al, 2014) after brief exposure to miR‐203. Importantly, transient exposure to this microRNA results in (i) enhanced differentiation into multiple tissues (e.g., including pancreas, bone marrow, or trophoblast) in teratomas generated in vivo; (ii) complex embryo‐like structures observed after the injection of these cells into mice; and (iii) augmented efficiency in stringent tests for assessing developmental potency, such as tetraploid complementation assays and human–mouse interspecies chimeras. Remarkably, half of those all‐iPSC mice produced with miiPSCs by tetraploid complementation reached the adulthood showing efficient germline transmission, which has been rarely found in previous reports (Okita et al, 2007; Kang et al, 2009; Zhao et al, 2009; Stadtfeld et al, 2010a; Leitch et al, 2013; Choi et al, 2017a; Yagi et al, 2017; Yang et al, 2017). Similarly, human iPSCs transiently exposed to miR‐203 contributed to interspecies human–mouse conceptuses with high efficiency (up to 100% contribution in vitro) compared with previous reports (Mascetti & Pedersen, 2016; Theunissen et al, 2016; Wu et al, 2016). Importantly, miR‐203 was able to improve the potential of enhanced pluripotent stem (EPS) cells generated in special culture conditions (Yang et al, 2017), suggesting that exposure to this microRNA may be used in combination with other strategies aimed to optimize PSC culture conditions.
Endogenous miR‐203 levels are transiently induced during early pre‐implantation development, suggesting that this microRNA may contribute to the specific expression profile of early blastomeres. The fact that brief exposure to miR‐203 sequences improves the long‐term developmental potential of miPSCs even many weeks and passages after its expression suggests an epigenetic mechanism. In fact, miPSCs display a hypomethylated state in chromatin that is self‐sustained and accentuated several weeks after exposure to this microRNA implying a replication memory. Among the multiple miR‐203 targets, de novo DNA methyltransferases Dnmt3a and Dnmt3b likely play an important role in this effect. In cancer cells, miR‐203 expression has been previously shown to decrease DNMT3B protein levels leading to de novo methylation of cancer‐related genes and decrease expression of their products including the xenobiotic transporter ABCG2 (Gasque Schoof et al, 2015; To et al, 2017). In ESCs, lack of these DNA methyltransferases is known to induce progressive hypomethylation of chromatin with passages (Chen et al, 2003; Liao et al, 2015) and knockdown of both Dnmt3a and Dnmt3b transcripts mimicked the effect of miR‐203 in several assays. In addition, the expression of miR‐203‐resistant Dnmt3a and Dnmt3b cDNAs significantly rescued the phenotypes induced by miR‐203 including differentiation to mature cardiomyocytes. Of note, whereas the severe hypomethylation observed in Dnmt3a/b knockout cells blocks differentiation (Okano et al, 1999; Jackson et al, 2004), the hypomethylated state induced by miR‐203 is reversible and differentiation is highly improved, when compared to untreated cells, and accompanied of potent de novo DNA methylation. Whereas the basis for these differences is unclear, given the promiscuity of microRNAs, miR‐203 may have additional targets that could also cooperate with Dnmt3a/b repression in the effects of this microRNA in pluripotent cells.
Maintaining methylation of imprinted loci during pre‐implantation development is a process mediated by Dnmt1, rather than Dnmt3a or Dnmt3b (Branco et al, 2008; Hirasawa et al, 2008), in agreement with our data showing a limited effect in the demethylation of ICRs in miPSCs, in which Dnmt1 expression is not modified. Conditions in which ICRs are also demethylated are not appropriate for PSC function as ICR hypomethylation is frequently irreversible (Theunissen et al, 2016). In fact, the use of 2i/L conditions results in a widespread loss of DNA methylation including massive erasure of genomic imprints that do not recover methylation after differentiation protocols (Yagi et al, 2017) (Yagi et al, 2017), leading to defective developmental potential in chimerism studies or tetraploid complementation assays (Choi et al, 2017a; Yagi et al, 2017). On the contrary, transient exposure to miR‐203 induces moderate and reversible ICR hypomethylation that likely contributes to the differentiation and development potential of these PSCs when submitted to differentiation stimuli.
Altogether, our data present miR‐203 sequences as an easy‐to‐use and suitable tool to improve PSCs in vitro, by expanding their differentiation and developmental potential in vivo. In contrast to other protocols aimed to improve reprogramming into pluripotent cells (Mikkelsen et al, 2008; Chen et al, 2013; Bar‐Nur et al, 2014; Li & Izpisua Belmonte, 2016; Takahashi & Yamanaka, 2016), the effects induced by miR‐203 can be easily achieved through brief exposure to specific mimics in already‐established pluripotent cultures. Together, we conclude that transiently modulating the DNA methylation landscape of miPSCs by short exposure to miR‐203 may enhance the performance of these cells in multiple functional assays, including the efficient differentiation and maturation to specific cellular lineages of interest in regenerative medicine.
Materials and Methods
Animal models and procedures
Animal experimentation was performed according to protocols approved by the CNIO‐ISCIII Ethics Committee for Research and Animal Welfare (CEIyBA). The miR‐203‐inducible model was generated by cloning a 482‐bp genomic mmu‐mir203 sequence into the pBS31 vector for recombination into the ColA1 locus in ES cells following the strategy reported previously (Beard et al, 2006). The resulting knock‐in allele [ColA1(miR‐203)] was combined with a Rosa26‐M2rtTA allele [Rosa26(rtTA)] for doxycycline‐dependent induction as described previously (Beard et al, 2006) (Fig EV1A). These animals were maintained in a mixed C57BL6/J × 129 × CD1 genetic background. For subcutaneous teratomas, iPSCs were trypsinized and 2–3 million cells were suspended in 100 µl PBS supplemented with 0.1% glucose and were subcutaneously injected into both flanks of athymic nude mice (provided by Charles River). Teratomas were daily controlled and measured by a caliper and finally isolated when their diameters reached 1.5 cm. Animals were euthanized at that point, and teratomas were weighed and processed for RNA extraction or histopathological analysis. For intraperitoneal injections, wild‐type athymic mice were injected with 4–5 × 105 iPSCs resuspended in 100 µl PBS supplemented with 0.1% glucose. Mice were supervised daily from the day of the injection. Usually, 30 or 40 days after injection mice were euthanized and the visceral teratomas and embryo‐like structures were isolated and processed, either for RNA extraction or for histopathological analysis.
For chimera generation, iPSCs or ESCs (5–7 cells per embryo, 10 passages) were microinjected into C57BL/6J‐Tyrc‐2J/J blastocysts and transferred to Crl:CD1 (ICR) pseudo‐pregnant females. For tetraploid complementation studies, 2‐cell stage Hsd:ICR(CD‐1) embryos were harvested from pregnant females at E1.5 and electrofused in 0.3 M mannitol using a BLS CF‐150/B cell fusion instrument with a 250‐µm electrode chamber. The electric pulse conditions were 30 V amplitude, 31 µs width, and 1.5 AC voltage. One to two hours later, 1‐cell (tetraploid) embryos were selectively picked and cultured in KSOM overnight. Next day, 4‐cell stage embryos were selected and aggregated with ESCs or iPSCs. Aggregated embryos were transferred to pseudo‐pregnant females 24 h later. To study germline contribution, black ES‐iPS mice (from tetraploid complementation assays) were crossed with Albino C57BL/6J‐Tyrc‐2J/J females and hair coat pigmentation was monitored in the progeny.
Interspecies chimeric assays
Human iPSCs were cultured in serum‐free N2B27‐LCDM medium under 20% O2 and 5% CO2 at 37°C (Yang et al, 2017). A total of 500 ml of N2B27 medium was prepared by including 240 ml DMEM/F12 (Thermo Fisher Scientific, 11330‐032), 240 ml Neurobasal (Thermo Fisher Scientific, 21103‐049), 2.5 ml N2 supplement (Thermo Fisher Scientific, 17502‐048), 5 ml B27 supplement (Thermo Fisher Scientific, 12587‐010), 1% GlutaMAX (Thermo Fisher Scientific, 35050‐061), 1% non‐essential amino acids (Thermo Fisher Scientific, 11140‐050), 0.1 mM β‐mercaptoethanol (Thermo Fisher Scientific, 21985‐023), and penicillin–streptomycin (Thermo Fisher Scientific, 15140‐122). To prepare the N2B27‐LCDM medium, small molecules and cytokines were added in the N2B27 medium as indicated at the following final concentrations: 10 ng/ml recombinant human LIF (L, PeproTech, 300‐05), 1 μM CHIR99021 (C, Tocris, 4423), 2 μM (S)‐(+)‐dimethindene maleate (D, Tocris, 1425), and 2 μM minocycline hydrochloride (M, Santa Cruz Biotechnology, sc‐203339). Y‐27632 (5 μM; Tocris, 1254) was added 24 h before and after single‐cell passage. Human iPSCs were cultured on mitomycin C (Sigma‐Aldrich, M4287)‐inactivated mouse embryonic fibroblast (MEF) feeder cells (3*104 cells/cm2). The N2B27‐LCDM medium was changed every day with fresh LCDM medium. To maintain human iPSCs in an undifferentiated state, we used the following criteria: (i) Avoid plating human cells too sparsely; (ii) use the proper quantity of freshly prepared MEF feeder cells; and (iii) do not allow human cells to overgrow. iPSCs were passaged by Accumax (Innovative Cell Technologies) every 3–5 days (normally at a split ratio ranging from 1:3 to 1:10), and the exact passage day and split ratio should be determined for each cell line specifically.
For transient miR‐203 expression, human iPSCs expressing the Tomato reporter were transfected with pMCSV‐miR‐203‐GFP. The cell pellet was resuspended with the nucleofection/plasmid mixture (P3 Primary Cell 4D‐Nucleofector Kit, Lonza): 82 μl solution + 18 μl supplement 1 + 2 μg plasmid. The cells were transferred into a cuvette for nucleofection using a 4D‐Nucleofector (Program CB150, Lonza) and then seeded on DR4 MEFs. The next day, the cells were gently digested into single cells using Accumax. Suspensions were filtered through a cell strainer (40 μm). Then, the samples were loaded and sorted to harvest GFP+ and Tomato+ cells on a Becton‐Dickinson Fusion sorter. After cell collection, they were plated on DR4 MEFs in N2B27‐LCDM medium plus 10 μM Y‐27632. The medium was changed the next day without Y‐27632.
For interspecies chimeric experiments, human cells were used 1 day before passaging, which showed an optimal undifferentiated morphology and proliferated exponentially. At this time point, the colonies were at sub‐confluent density (approximately 70% density of the day the cells should be passaged). 8‐ to 10‐week ICR female mice were superovulated by the treatment of PMSG and 48 h later intraperitoneal administration of hCG. Female mice were then mated with a male for oocyte fertilization. Appearance of plugs was checked in female mice, and 2‐cell stage embryos were harvested 1 day after that. 2‐cell stage embryos were cultured in KSOM for another 24 h and named E2.5 embryos. At this time, most of the embryos were around 8‐cell stage and ready for human EPS cell injection.
Human iPSCs were first rinsed with PBS, then treated with Accumax for 2 min, and subsequently filtered through a cell strainer (40 μm). Cells were then centrifuged at 1,200–1,500 rpm (250–300 g) at room temperature for 3 min. The supernatant was removed, and the cells were resuspended in the culture medium at a proper density (2–6*105 cells/ml). 10 μM Y‐27632 was added into the suspension. The suspension was placed on ice before injection. One individual human cell (or 15 human cells, in Appendix Figs S6 and S7) was microinjected into each E2.5 ICR diploid mouse embryo. Injected embryos were cultured in N2B27‐LCDM medium for the first 4 h (in the presence of 10 μM Y‐27632), followed by culture for 44–60 h in mixed cell culture medium and KSOM medium at a 1:1 ratio. When the embryo reached the blastocyst stage, approximately 10 injected embryos were transferred to each uterine horn of a 2.5 days post‐coitum pseudo‐pregnant ICR female, or fixed with 4% paraformaldehyde for immunostaining. Post‐implantation embryos were collected at E9.5, rinsed in PBS, followed by embedding, freezing, and slicing (10 μm thick) with a Cryostat.
For immunofluorescence, embryos or slides were fixed in 4% paraformaldehyde (Sigma) at room temperature for 20 min and washed with PBST for 30 min. Samples were then permeabilized with 0.2% Triton X‐100 in PBS. After 3 washings, samples were blocked with PBS (Corning, 21‐040‐CVR) that contained 1% BSA and 0.05% TWEEN‐20 at room temperature for 45 min, and then incubated with primary antibodies at 4°C overnight. After 3 washings, secondary antibodies (Jackson ImmunoResearch) were incubated at room temperature for 1 hr. The nuclei were stained with DAPI (Southern Biotech). The following antibodies were used: anti‐OCT4 (C30A3) (1:200; Cell Signaling), anti‐HuNu (1:200; Novus, NBP2‐34342), anti‐CDX2 (1:200, CDX2‐88; Bio‐Genex), anti‐Gata4 (1:200; Santa Cruz, C‐20), anti‐Sox2 (1:200; R&D, AF2108), and anti‐hSOX17 (1:200; R&D, AF1924) (Appendix Table S3). Immunofluorescence images were obtained using a confocal microscope, and signals were then analyzed and quantified using ImageJ.
All the chimeric experiments were reviewed and approved by the Salk Institutional Animal Care and Use Committee (IACUC) and followed the ethical guidelines of the Salk Institute.
Cell culture and gene expression
Primary mouse embryonic fibroblasts (MEFs) were obtained from embryos at E13.5 and cultured in DMEM supplemented with 10% of FBS and penicillin–streptomycin. Cultures were routinely tested for mycoplasma. Regarding the miR‐203‐inducible model‐derived MEFs, reprogramming was promoted on these MEF cultures by non‐inducible Oct4‐Sox2‐Klf4‐cMyc (OSKM) lentiviral transduction. For lentiviral transduction, we transfected HEK293T cells with FUW‐OSKM (Addgene #20328) and packaging vectors using Lipofectamine 2000 (Invitrogen). Viral supernatants were collected twice a day on two consecutive days starting 24 h after transfection and were used to infect MEFs, previously plated at a density of 250,000 cells per well in 6‐well plates. Previous to infection, polybrene was added to the viral supernatants at a concentration of 2 μg/ml. For all those experiments in which miR‐203 was expressed in WT iPSCs by mimic transfection or retrovirus transduction, WT MEF reprogramming to obtain WT iPSCs was performed using inducible OSKM lentiviral transduction (TetO‐FUW‐OSKM; Addgene #20321). Infected MEFs were then cultured in iPSC medium, containing KO‐DMEM (Gibco), 2‐mercaptoethanol 50 mM (Invitrogen), non‐essential amino acid MEM NEAA (Invitrogen), penicillin and streptomycin (5,000 μg/ml, Invitrogen), Lif (leukemia inhibitor factor, ESGRO, Millipore), and 20% knockout serum replacement (KSR, Invitrogen). In this case, doxycycline was also added to the iPSC medium to promote reprogramming only for the generation of WT iPSCs to be treated with miR‐203 mimics or retroviruses (but never in the case of iPSCs derived from the miR‐203‐inducible animal model, in which miR‐203 expression depends on DOX addition). Medium was changed every 24 h, and plates were stained for alkaline phosphatase activity to assure the efficiency of reprogramming (AP Detection Kit, Sigma‐Aldrich). Once colonies were picked, iPSCs were cultured in iPSC media over mitomycin C (Roche)‐inactivated feeder cells. G4 ESCs were cultured over mitomycin C‐inactivated feeders and in the presence of ESC medium containing KO‐DMEM, 2‐mercaptoethanol, non‐essential amino acids, GlutaMAX, penicillin and streptomycin, Lif, and 10% fetal calf serum (HyClone). When indicated, the culture media for pluripotent cells included 2i factors (MEK inhibitor PD0325901 1 μM, Gsk3 inhibitor CHIR99021 3 μM) and mouse LIF (as above) in N2B27 medium, as described previously (Ying et al, 2008). For inducing transient miR‐203 overexpression, ColA1(miR‐203/miR‐203); Rosa26(rtTA/rtTA) iPSC or ESC cultures were treated with Dox (1 μg/ml; Invitrogen) during 5 days. After that, Dox withdrawal was standardized for the cultures during following several passages (10–30 days) unless other time points are indicated in the text. In this inducible system, we always test that insert expression is uniquely dependent on Dox, and becomes absolutely undetectable after Dox withdrawal. As a control of the treatment itself, Dox was also added and tested in wild‐type iPSCs or ESCs.
For overexpression experiments, miR‐203 and full‐length cDNAs of Dnmt3a and Dnmt3b were subcloned into the retroviral vector pMCSV‐PIG (Abad et al, 2013) by restriction‐directed subcloning, using pCMV‐Sport6‐mDnmt3a (MGC clone: 5662) and attB‐mCh‐mDnmt3b‐Poly (A)‐NeoR (Addgene plasmid 65553) as templates, respectively. For retroviral transduction, we transfected HEK293T cells with the respective pMCSV‐PIG vector expressing a GFP reporter (including either only GFP; miR‐203‐GFP; Dnmt3a‐GFP or Dnmt3b‐GFP) and the packaging vector pCL‐ECO, using Lipofectamine 2000 (Invitrogen). Viral supernatants were collected twice a day on two consecutive days starting 24 h after transfection and were used to infect either ESCs or IPSCs, previously plated over feeders in 6‐well plates. Preceding the infection, polybrene was added to the viral supernatants at a concentration of 2 μg/ml. When transduced with these retroviral vectors, both ESCs and iPSCs were sorted by FACS and GFP‐positive cells were selected for subsequent cultures and analysis. For transfection with mimics, we used miRIDIAN microRNA human hsa‐miR‐203a‐5p (C‐302893‐00)/has‐miR‐203a‐3p (C‐300562‐03) mimics or mimic transfection control (scramble sequences purchased from GE Dharmacon) with Dy547 (CP‐004500‐01), all of them from GE Dharmacon. Transfection was performed using either DharmaFECT transfection reagent (Dharmacon) or Lipofectamine RNAiMAX (Invitrogen) following the manufacturer's instructions. Transfection efficiency was evaluated 24 h post‐transfection by Dy547 fluorescence, and experiments were then performed as indicated in the figures.
Regarding the use of the two mature forms of miR‐203 mimics: As happens with many other microRNAs, two mature microRNAs can originate from opposite arms of the same pre‐miRNA, has‐miR‐203a, which are denoted with a ‐3p or ‐5p suffix. However, the mature microRNA found from one arm of the hairpin is usually much more abundant than that found from the other arm, in which case, an asterisk following the name indicates the mature species found at low levels from the opposite arm of a hairpin. In the case of miR‐203, the most abundant mature form is miR‐203a‐3p, while the low abundant form is called miR‐203a‐5p*.
Human miR‐203 is expressed from chromosome 19. Its murine counterpart, mmu‐miR‐203 (Accession in miRbase MI0000246), is expressed from chromosome 14 of Mus musculus. The main mature sequence of mmu‐miR‐203, mmu‐miR‐203‐3p (Accession in miRbase MIMAT0000236), seems to be identical to that of hsa‐miR‐203a‐3p. There are experimental evidences of a second mature sequence, mmu‐miR‐203‐5p* (Accession in miRbase MIMAT0004547), which is shorter than hsa‐miR‐203a‐5p (22 nucleotides instead of 25) and differs slightly from its human counterpart in the rest of the sequence (in position 11, G is replaced by A in hsa‐miR‐203a‐5p*). Both miR‐203a‐3p/‐5p were used in these assays, aiming to faithfully mimic the endogenous scenario in which both mature forms are expressed. However, the most abundant form (miR‐203a‐3p) is responsible for the observed effects, as depicted for example in the Appendix Fig S10.
For RNA interference assays, ON TARGETplus SMARTpool for non‐targeting control siRNA (D‐001810‐01, 02, 03, 04), Dnmt3a siRNAs (J‐065433‐09, 10, 11, 12), and Dnmt3b siRNAs (J‐044164‐05, 06, 07, 08) from Dharmacon were used. Transfection was performed using DharmaFECT transfection reagent (Dharmacon) following the manufacturer's instructions. Transfection efficiency was evaluated 24 h post‐transfection by qPCR, using the primers indicated in Appendix Table S4. A few weeks after transfection, the cells were assessed for differentiation to embryoid bodies.
Luciferase‐reported assays were performed in HEK293T cells. Briefly, 200.000 cells per well were seeded on 6‐well plates, and the day after, cells were transfected using Lipofectamine 2000 (Invitrogen), following the manufacturer's instructions. The 3′‐UTR regions from the murine genes Dnmt3a, Dnmt3b, Dnmt3l, and Dnmt1 were amplified by PCR with specific primers (Dnmt3a_EcoRI‐Fw: 5′‐GAATTCAGGGACATGGGGGCAAACTGAA‐3′ (SEQ ID NO: 55); Dnmt3a_NdeI‐Rv: 5′‐CATATGCTGAGGCAGTCATTTTAGATTCAT‐3′ (SEQ ID NO: 56); Dnmt3b_EcoRI‐Fw: 5′‐GAATTCTTTTAGCTCACCTGTGTGGGG‐3′ (SEQ ID NO: 57); Dnmt3b_NdeI‐Rv: 5′‐CATATGCCAGAAAGGTAAACTCTGGGCA‐3′ (SEQ ID NO: 58); Dnmt3l_EcoRI‐Fw: 5′‐GAATTCGAAATGAATCACCATAAGATGAAAG‐3′ (SEQ ID NO: 59); Dnmt3l_NdeI‐Rv: 5′‐CATATGAACAATCCTATGATATATTTGAAAAA‐3′ (SEQ ID NO: 60); Dnmt1_EcoRI‐Fw: 5′‐GAATTCGTGCTCTCACCCAGAGCCCCA‐3′ (SEQ ID NO: 61); and Dnmt1_NdeI‐Rv: 5′‐CATATGGCTTGACAGAAGCGCTTTATTTTG‐3′) (SEQ ID NO: 62) (Appendix Table S4), using cDNA clones (pCMV‐Sport6‐mDnmt3a, cDNA clone MGC:5662; pBluescript‐mDnmt3l, RIKEN clone: 2410006021; pYX‐Asc‐mDnmt1, MGC clone: 62302) or mouse cDNA (in the case of Dnmt3b). PCR products were verified by sequencing and ligated into the pGL3‐Control vector (Promega), downstream of the luciferase reporter gene. Mutations in the miR‐203‐binding sites were generated by site‐directed mutagenesis and subsequently verified by sequencing. Transfections were performed with either pMCSV‐GFP or pMCSV‐miR‐203‐GFP vectors, in combination with the pGL3‐derived vectors, and Renilla as a control. Luciferase measurement was achieved 48 h post‐transfection using a luminescence microplate reader (Bio‐Tek).
Murine ESCs expressing the 2C::Td‐Tomato reporter to detect endogenous expression of the 2C‐stage retrotransposon MuERV‐L were cultured in 2i media over feeders as reported previously (MacFarlan et al, 2012). Human iPSCs expressing the long terminal repeat (LTR7) of HERVH endogenous retroviruses fused with GFP reporter (Wang & Gao, 2016) were cultured in mTeSRTM1 media (Stem Cell Technologies) over a Matrigel basement (Corning). Experimentation with human cells was performed according to protocols approved by the ISCIII Ethics Committee for Research (CEI; number PI 61_2017) and the Salk Institute Ethics Committee for Research.
Embryoid body generation
Briefly, iPSCs or ESCs were trypsinized and resuspended to a concentration of 200,000 cells/ml in the presence of complete growth medium lacking leukemia inhibitory factor (Lif). Small drops of this suspension (~35 μl) were collected and seeded on the lid of a 10‐mm plate, generating hanging drops of approximately 5,000 cells per drop. After 4 days, the aggregates were already visible at the bottom of the drops and were picked and transferred to a non‐adherent plate, containing DMEM and 10% fetal calf serum. There they were maintained in suspension for the indicated times, and beating, size, and cavity formation were assessed every 5 days as described in the figures. Between 20 and 30 EBs were analyzed per condition, and the percentage of beating EBs or EBs with large cavities was calculated for every time point. EB size was measured using the Image J software.
Immunofluorescence and immunohistochemistry
Cells previously seeded in cover slips were fixed in 4% paraformaldehyde for 15 min, permeabilized using PBS 0.1% Triton X‐100 for 15 min, and blocked in BSA for 1 h at room temperature. Primary antibody incubation was performed overnight at 4°C in all cases, followed by secondary antibody incubation for 1 h at room temperature. Nuclear staining was included in the last PBS wash, using Hoechst or DAPI. Primary antibodies used in this study were against Cd34 (Abcam), Gata4 (Santa Cruz), Pax6 (Abcam), Nestin (Millipore), cTnT (Abcam), and phospho‐histone H3 (Millipore) (Appendix Table S3). Cells were examined under a Leica SP5 microscope equipped with white light laser and hybrid detection.
For immunohistochemistry, tissue samples were fixed in 10% neutral buffered formalin (4% formaldehyde in solution), paraffin‐embedded and cut at 3 μm, mounted in SuperFrost® Plus slides, and dried overnight. Consecutive sections were stained with hematoxylin and eosin (H&E) or subjected to immunohistochemistry using automated immunostaining platforms (Ventana Discovery XT, Roche, or Autostainer Plus Link 48). Antigen retrieval was first performed with high or low pH buffer depending on the primary antibody (CC1m, Roche, or low pH antigen retrieval buffer, Dako), endogenous peroxidase was blocked (peroxide hydrogen at 3%), and slides were incubated with primary antibodies against Nanog (Cell Signaling Biotechnology, 8822) cytokeratin 8 (CK8; CNIO Monoclonal Antibodies Core Unit, AM‐TROMA I), GFP (Roche, 11814460001), Sox2 (Cell Signaling Technology, 3728), alpha‐fetoprotein (AFP; R&D Systems, AF5369), Oct4 (Santa Cruz Biotechnology, sc‐9081), KI‐67 (Master Diagnostica, 0003110QD), Nestin (Millipore, MAB353), Cd31 (Abcam), Cd34 (ABCAM ab8158), Cd73 (Cell Signaling Technology), collagenase type I (Rockland), Gata4 (Santa Cruz Biotechnology, sc‐1237), smooth muscle actin (Thermo Scientific, RB‐9010‐PO), skeletal actin (Dako, M0635), or Ter119 (LY‐76; BD Bioscience, 550565) (Appendix Table S3). Secondary antibodies were conjugated with horseradish peroxidase (OmniRabbit, Ventana, Roche), the immunohistochemical reaction was developed using 3,30‐diaminobenzidine tetrahydrochloride (DAB) as a chromogen (chromoMap DAB, Ventana, Roche, or DAB solution, Dako), and nuclei were counterstained with Carazzi's hematoxylin. Finally, the slides were dehydrated, cleared, and mounted with a permanent mounting medium for microscopic evaluation. The images were acquired with a slide scanner (Axio Scan Z1, Zeiss). Images were captured and quantified using the Zen Software (Zeiss).
Western Blot
For Western blotting, cells were lysed in RIPA or a buffer containing 50 mM Tris–HCl, pH 7.5, 1 mM phenylmethylsulphonyl fluoride, 50 mM NaF, 5 mM sodium pyrophosphate, 1 mM sodium orthovanadate, 0.1% Triton X‐100, 1 μg/ml leupeptin, 1 mM EDTA, 1 mM EGTA, and 10 mM sodium glycerophosphate. Thirty micrograms of total protein was separated by SDS–PAGE and probed with primary antibodies against Dnmt3a (Novus Biological, 64B1446), Dnmt3b (Novus Biological, 52A1018), or vinculin (Sigma, V9131).
Analysis of mRNA and microRNA levels
RNA was extracted from cell or tissue samples with TRIzol (Invitrogen) or by using the mirVana miRNA Isolation Kit (Thermo Fisher), following the manufacturer's recommendations. Retrotranscription into cDNA was performed using M‐MLV Reverse Transcriptase (Promega) following the manufacturer's protocol. Quantitative real‐time PCR was performed using SYBR Green Power PCR Master Mix (Applied Biosystems) in an ABI PRISM 7700 Thermocycler (Applied Biosystems). The housekeeping gene Gapdh was used for normalization. The oligonucleotide primers used in this manuscript are listed in the Appendix Table S4. For reverse transcription of microRNAs, we used the TaqMan small RNA assay (4366596), including the specific oligonucleotides for mmu‐miR‐203‐5p and 3p (002580 and 000507), miR‐16, and the housekeeping RNA sno‐202 or sno‐142. Conditions for miRNA amplification were as follows: 30 min at 16°C; 30 min at 42°C; and a final step of 5 min at 85°C. Quantitative real‐time PCR was then performed using the TaqMan Universal PCR Master Mix (434437) following the manufacturer's instructions in an ABI PRISM 7700 Thermocycler (Applied Biosystems).
For RNAseq, total RNA was extracted using the miRVana miRNA Isolation Kit (Thermo Fisher), following the manufacturer's recommendations. Between 0.8 and 1 μg of total RNA was extracted from iPSCs, ESCs, or teratomas, with RIN (RNA integrity number) numbers in the range of 9 to 10 (Agilent 2100 Bioanalyzer). Poly A+ fractions were purified and randomly fragmented, converted to double‐stranded Cdna, and processed using the Illumina's “TruSeq Stranded mRNA Sample Preparation Part # 15031047 Rev. D” Kit. The adapter‐ligated library was completed by PCR with Illumina PE primers (8–11 cycles), and the resulting directional cDNA libraries were sequenced for 50 bases in a single‐read format (Illumina HiSeq 2000) and analyzed with Nextpresso (Graña et al, 2018). Reads were quality‐checked with FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc) and aligned to the mouse genome (GRCm38/mm10) with TopHat‐2.0.10 (Trapnell et al, 2012), using Bowtie 1.0.0 (Langmead et al, 2009) and SAMtools 0.1.19 (Li & Durbin, 2009), allowing two mismatches and five multihits. Transcripts assembly, estimation of their abundances, and differential expression were calculated with Cufflinks 2.2.1 (Trapnell et al, 2012), using the mouse genome annotation dataset GRCm38/mm10 from the UCSC Genome Browser (Rosenbloom et al, 2015). A false discovery rate (FDR) of 0.05 is used as threshold for significance in differential expression. Heat maps were later built using GENE‐E (http://www.broadinstitute.org/cancer/software/GENE-E/index.html). Published datasets (MacFarlan et al (GSE 33923); Najm et al (GSE 26814) (Yang et al, 2017)) were compared using the “Feature Specific Quantile Normalization” method that allows to perform a cross‐platform normalization (Franks et al, 2018).
Bisulfite conversion, genome‐wide DNA methylation, and validation of DMRs
DNA samples were prepared for whole‐genome bisulfite sequencing using the TrueMethyl® Whole Genome Kit (CEGX®) according to the manufacturer's instructions. Briefly, 200 ng of genomic DNA was sheared to 800 bp using M220 Focused‐ultrasonicator™ (Covaris®). Then, the fragmented DNA was denatured and oxidized by a chemical oxidant to convert 5‐hydroxymethyl cytosine to 5‐formylcytosine (5fC). The purpose of the oxidation was to ensure that we purely captured the information for 5′ methyl cytosine methylation and not an indistinguishable pattern combination of 5′methyl cytosine and 5′hydroxymethyl cytosine. Following oxidation, the DNA was subjected to bisulfite conversion for the deamination of cytosines and 5fC to uracils. Bisulfite‐converted DNA was desulfonated and purified to then proceed to library preparation. In this “post‐bisulfite conversion” library preparation method, the fragmented single‐stranded bisulfite‐converted DNA was adapted with sequencing adaptors at the 3′end followed by an extension step and finally ligation of adaptors at the 5′ end of the molecules. Finally, the libraries were indexed and amplified. The PCR was performed for 10 cycles followed by bead‐based purification. An additional purification and size selection step using Agencourt AMPure XP beads (Beckman Coulter, Cat: A63881) were performed to remove adaptor dimers. The purified library was eluted in a final volume of 14 μl pure water. Quality of the library obtained was checked using the DNA High Sensitivity Chip on the Agilent Bioanalyzer. The library was quantified using Qubit and KAPA Biosystems Library Quantification Kit (#KK4824) according to manufacturer's instructions. A total of 1‐2pMol of multiplexed libraries were sequenced on a 125‐bp paired‐end format (Illumina HiSeq 2500). Adapter sequences were removed using Cutadapt version 1.9.1 (Martin, 2011) in paired‐end mode with parameters “‐m 15 ‐u 6 ‐U 6”. Bwa‐meth (preprint: Pedersen et al, 2014) was then used to align reads to mm10 using default parameters. PCR duplicates were removed using Picard v1.91 (http://broadinstitute.github.io/picard). Count tables of the number of methylated and unmethylated bases sequenced at each CpG site in the genome were constructed using the “tabulate” module of bwa‐meth and BisSNP‐0.82.2 (Liu et al, 2012) with default parameters. All libraries passed basic quality control checks with a minimum of 82.7% of read pairs aligning uniquely. DMRs were called using the WGBS module of DMRcate (Peters et al, 2015) with parameters lambda = 1000 and C = 50; DMPs were called using DSS (Feng et al, 2014); PMDs, LMRs, and UMRs were called using MethylSeekR (Burger et al, 2013); and PMDs were called using the R package “aaRon” (https://github.com/astatham/aaRon).
For the validation of DMRs, clonal bisulfite sequencing was performed at two particular loci: Elf5 (chr2: 103, 423, 778‐103, 424, 180) and Sirt6 (chr10: 81, 624, 595‐81, 625, 547) promoter regions. 100 ng of DNA was bisulfite treated using EZ DNA Methylation‐Lightning™ Kit (Zymo Research) following manufacturer's instructions. Triplicate PCR amplifications were then performed using semi‐nested bisulfite conversion‐specific primers listed in Appendix Table S4, following the PCR conditions previously described for Elf5 (Lee et al, 2011) or using the following protocol: 95°C 4 min; 5 cycles (95°C 45 s, 54°C 1.5 min, 72°C 2 min); 25 cycles (95°C 45 s, 54°C 1.5 min; 72°C 1.5 min); and final extension at 72°C 4 min, for Sirt6. The methylation status of the PCR amplicons was determined by the Sanger clonal sequencing of the pooled PCR products to ensure representative clonal analysis using between 8 and 10 clones per sample. The analysis of the methylation status of the clones was performed using BiQ Analyzer (Bock et al, 2005).
Neonatal cardiomyocyte isolation and differentiation studies
Neonatal mouse and rat cardiomyocytes were prepared as described previously (Huang et al, 2015). Briefly, neonatal cardiomyocytes were isolated by enzymatic disassociation of 1‐day‐old neonatal mouse or rat hearts with the Neonatal Cardiomyocyte Isolation System (Cellutron Life Technology). Cardiomyocytes were pre‐plated for 2 h to remove fibroblasts. Cells were then plated on 1% gelatin‐coated plates in medium containing 10% horse serum and 5% fetal calf serum. Eighteen hours after plating, cells were changed into serum‐free medium and then transfected with 50 nM miR‐203a‐3p and 5p mimics or control mimics (from Dharmacon) by using Lipofectamine RNAiMAX transfection reagent, following the manufacturer's instructions. Six hours later, media with transfection reagent were removed and substituted by media containing 1% serum. Different time points (as indicated) were collected for subsequent RNA extraction and EdU staining/immunofluorescence. For EdU staining, we used the Click‐iT EdU Staining Kit (Invitrogen) following the manufacturer's recommendations. After that, immunofluorescence of cardiomyocyte markers (Troponin T) was performed and Hoechst was finally used for nuclei staining.
For in vitro differentiation from mouse iPSCs to cardiomyocytes, wild‐type mouse iPSCs were transfected with either control or miR‐203 mimics prior to differentiation. In some experiments, additionally pMCSV‐Dnmt3a and 3b or empty pMCSV vector was transiently transduced, 24 h after mimic transfection. iPSCs were then maintained in culture under 2i/Lif media for 15 days, before differentiation started. Then, cells were plated in Matrigel pre‐coated 6‐well plates. Once they become confluent, the media were changed to RPMI 1640/B27 medium (Thermo Fisher Scientific, 61870127 for the RPMI 1640 media and A1895601 for B27) with CHIR99021 5 μM (Stem Cell Technologies, 72054). After 2 days, cells were washed and media without CHIR99021 were added. At day 3 of differentiation, media were supplemented with IWR1 5 μM (Stem Cell Technologies, 72564) and maintained in these conditions for 2 more days. At day 7 of differentiation, the media were changed to RPMI 1640/B27 plus insulin. Then, from day 9 to day 15, cells were cultured in RPMI 1640/B27 and the media were changed every 2 days. From day 16 to day 18 of differentiation, cells were cultured in DMEM without glucose and with lactate 4 mM. From day 18 to day 21, the media were changed to RPMI 1640/B27 and changed every day. Finally, from day 22 to day 28, monolayer cells can be isolated into cell clusters and kept in low attachment for another week.
Statistics
Normal distribution and equal variance were confirmed before using the Student's t‐test (two‐tailed, unpaired) to estimate statistical significance. For contingency tables, we used Fisher's exact test. Statistical analysis was performed using Prism (GraphPad Software, La Jolla, CA).
Author contributions
MS‐R. performed most of the cellular and in vivo assays. MT contributed to the characterization of miPSCs and MA‐F and EZ‐S to the generation of DNA constructs and molecular biology studies. FV‐M, TJP, and SJC performed and analyzed the genome‐wide methylation studies. CZ, YY, and JCI‐B performed the human–mouse interspecies chimeric assays. OG‐C analyzed RNAseq data. MA and MS participated in the evaluation of the data. MJB and MGC. generated the miR‐203 knock‐in mouse model, and JF‐P contributed to its analysis. RS, MAB, and D‐ZW contributed to the experiments of cardiomyocyte differentiation. JM and SO performed and analyzed tetraploid complementation assays, and SO supervised and analyzed embryo work. MS‐R and MM conceived the project, supervised the work, and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Movie EV1
Movie EV2
Movie EV3
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
We thank María Guirola, Sheila Rueda, and the CNIO Histopathology Unit for technical support; Carmen Gómez and Patricia Prieto for help with iPSC/ESC culture; Zsuzsanna Izsvak (Max‐Delbrück‐Center for Molecular Medicine, Berlin, Germany) for reagents; Marta Cañamero (Pathology and Tissue Analytics, Roche Pharma, Basel, Switzerland) and Alba de Martino (Histopathology Unit, CNIO) for pathology advice; William Pu, Zhang‐Peng Huang, and Kai Li (Cardiovascular Research Division, Boston Children's Hospital, USA) for help with the cardiac differentiation experiments; and Dr. Jennifer Franks (Geisel School of Medicine at Dartmouth, NH, USA) for her clarifications regarding the use of the FSQN method. We are indebted to the members of the CNIO Cell Division and Cancer laboratory for their constant support and advice. M.S.R. was supported by the Asociación Española contra el Cáncer (AECC; 2012 AIOA120833SALA and 2018 INVES18005SALA) and a Juan de la Cierva contract from the Ministry of Science, Innovation and Universities (MICIU). M.T. was supported by Fundación La Caixa. M.A.F. was supported by MICIU (SAF2014‐60442‐JIN). J.F.P. was funded by MICIU (RTI2018‐093330‐B/FEDER‐EU), Ramón Areces Foundation (CIVP19S7917), CAM (B2017/BMD‐3778), and AECC (2018, PROYE18054PIRI). F.V.M was supported by the National Breast Cancer Foundation/Cure Cancer Australia Foundation Postdoctoral Training Fellow. Work in S.J.C laboratory was supported by the National Health and Medical Research Council (NHMRC 1063560). M.A.B. laboratory is funded by SAF2013‐45111‐R from MICIU, Fundación Botín and Banco Santander, and Worldwide Cancer Research (WCR16‐1177). Work in S.O. laboratory was funded by MICIU grant (SAF2013‐44866‐R). Work in the M.M. laboratory was supported by grants from the MICIU (RTI2018‐095582‐B‐I00, RED2018‐102723‐T, and SAF2017‐92729‐EXP), and the iLUNG Programme (B2017/BMD‐3884) from the Comunidad de Madrid. The CNIO is a Severo Ochoa Centers of Excellence (MICIU award SEV‐2015‐0510).
The EMBO Journal (2020) 39: e104324
Contributor Information
María Salazar‐Roa, Email: msalazar@cnio.es.
Marcos Malumbres, Email: malumbres@cnio.es.
Data availability
RNAseq and methylation data have been deposited in the GEO repository (https://www.ncbi.nlm.nih.gov/geo/) under accession number GEO SuperSeries GSE81571 and GSE86653.
References
- Abad M, Mosteiro L, Pantoja C, Canamero M, Rayon T, Ors I, Grana O, Megias D, Dominguez O, Martinez D et al (2013) Reprogramming in vivo produces teratomas and iPS cells with totipotency features. Nature 502: 340–345 [DOI] [PubMed] [Google Scholar]
- Bar‐Nur O, Brumbaugh J, Verheul C, Apostolou E, Pruteanu‐Malinici I, Walsh RM, Ramaswamy S, Hochedlinger K (2014) Small molecules facilitate rapid and synchronous iPSC generation. Nat Methods 11: 1170–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beard C, Hochedlinger K, Plath K, Wutz A, Jaenisch R (2006) Efficient method to generate single‐copy transgenic mice by site‐specific integration in embryonic stem cells. Genesis 44: 23–28 [DOI] [PubMed] [Google Scholar]
- Biase FH, Cao X, Zhong S (2014) Cell fate inclination within 2‐cell and 4‐cell mouse embryos revealed by single‐cell RNA sequencing. Genome Res 24: 1787–1796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blattler A, Yao L, Witt H, Guo Y, Nicolet CM, Berman BP, Farnham PJ (2014) Global loss of DNA methylation uncovers intronic enhancers in genes showing expression changes. Genome Biol 15: 469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock C, Reither S, Mikeska T, Paulsen M, Walter J, Lengauer T (2005) BiQ Analyzer: visualization and quality control for DNA methylation data from bisulfite sequencing. Bioinformatics 21: 4067–4068 [DOI] [PubMed] [Google Scholar]
- Branco MR, Oda M, Reik W (2008) Safeguarding parental identity: Dnmt1 maintains imprints during epigenetic reprogramming in early embryogenesis. Genes Dev 22: 1567–1571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bueno MJ, Perez de Castro I, Gomez de Cedron M, Santos J, Calin GA, Cigudosa JC, Croce CM, Fernandez‐Piqueras J, Malumbres M (2008) Genetic and epigenetic silencing of microRNA‐203 enhances ABL1 and BCR‐ABL1 oncogene expression. Cancer Cell 13: 496–506 [DOI] [PubMed] [Google Scholar]
- Burger L, Gaidatzis D, Schubeler D, Stadler MB (2013) Identification of active regulatory regions from DNA methylation data. Nucleic Acids Res 41: e155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Ueda Y, Dodge JE, Wang Z, Li E (2003) Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Mol Cell Biol 23: 5594–5605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Guo L, Zhang L, Wu H, Yang J, Liu H, Wang X, Hu X, Gu T, Zhou Z et al (2013) Vitamin C modulates TET1 function during somatic cell reprogramming. Nat Genet 45: 1504–1509 [DOI] [PubMed] [Google Scholar]
- Choi J, Huebner AJ, Clement K, Walsh RM, Savol A, Lin K, Gu H, Di Stefano B, Brumbaugh J, Kim SY et al (2017a) Prolonged Mek1/2 suppression impairs the developmental potential of embryonic stem cells. Nature 548: 219–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YJ, Lin CP, Risso D, Chen S, Kim TA, Tan MH, Li JB, Wu Y, Chen C, Xuan Z et al (2017b) Deficiency of microRNA miR‐34a expands cell fate potential in pluripotent stem cells. Science 355, eaag1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung HC, Lin RC, Logan GJ, Alexander IE, Sachdev PS, Sidhu KS (2012) Human induced pluripotent stem cells derived under feeder‐free conditions display unique cell cycle and DNA replication gene profiles. Stem Cells Dev 21: 206–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckersley‐Maslin M, Alda‐Catalinas C, Blotenburg M, Kreibich E, Krueger C, Reik W (2019) Dppa2 and Dppa4 directly regulate the Dux‐driven zygotic transcriptional program. Genes Dev 33: 194–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng H, Conneely KN, Wu H (2014) A Bayesian hierarchical model to detect differentially methylated loci from single nucleotide resolution sequencing data. Nucleic Acids Res 42: e69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franks JM, Cai G, Whitfield ML (2018) Feature specific quantile normalization enables cross‐platform classification of molecular subtypes using gene expression data. Bioinformatics 11: 1868–1874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasque Schoof CR, Izzotti A, Jasiulionis MG, Vasques Ldos R (2015) The roles of miR‐26, miR‐29, and miR‐203 in the silencing of the epigenetic machinery during melanocyte transformation. Biomed Res Int 2015: 634749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goossens K, Mestdagh P, Lefever S, Van Poucke M, Van Zeveren A, Van Soom A, Vandesompele J, Peelman L (2013) Regulatory microRNA network identification in bovine blastocyst development. Stem Cells Dev 22: 1907–1920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graña O, Rubio‐Camarillo M, Fernandez‐Riverola F, Pisano SG, Gonzalez‐Peña D (2018) nextpresso: next generation sequencing expression analysis pipeline. Curr Bioinformatics 13: 583–591 [Google Scholar]
- Hirasawa R, Chiba H, Kaneda M, Tajima S, Li E, Jaenisch R, Sasaki H (2008) Maternal and zygotic Dnmt1 are necessary and sufficient for the maintenance of DNA methylation imprints during preimplantation development. Genes Dev 22: 1607–1616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang ZP, Kataoka M, Chen J, Wu G, Ding J, Nie M, Lin Z, Liu J, Hu X, Ma L et al (2015) Cardiomyocyte‐enriched protein CIP protects against pathophysiological stresses and regulates cardiac homeostasis. J Clin Invest 125: 4122–4134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson M, Krassowska A, Gilbert N, Chevassut T, Forrester L, Ansell J, Ramsahoye B (2004) Severe global DNA hypomethylation blocks differentiation and induces histone hyperacetylation in embryonic stem cells. Mol Cell Biol 24: 8862–8871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang L, Wang J, Zhang Y, Kou Z, Gao S (2009) iPS cells can support full‐term development of tetraploid blastocyst‐complemented embryos. Cell Stem Cell 5: 135–138 [DOI] [PubMed] [Google Scholar]
- Kattman SJ, Witty AD, Gagliardi M, Dubois NC, Niapour M, Hotta A, Ellis J, Keller G (2011) Stage‐specific optimization of activin/nodal and BMP signaling promotes cardiac differentiation of mouse and human pluripotent stem cell lines. Cell Stem Cell 8: 228–240 [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory‐efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latos PA, Sienerth AR, Murray A, Senner CE, Muto M, Ikawa M, Oxley D, Burge S, Cox BJ, Hemberger M (2015) Elf5‐centered transcription factor hub controls trophoblast stem cell self‐renewal and differentiation through stoichiometry‐sensitive shifts in target gene networks. Genes Dev 29: 2435–2448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Hinshelwood RA, Bouras T, Gallego‐Ortega D, Valdes‐Mora F, Blazek K, Visvader JE, Clark SJ, Ormandy CJ (2011) Lineage specific methylation of the Elf5 promoter in mammary epithelial cells. Stem Cells 29: 1611–1619 [DOI] [PubMed] [Google Scholar]
- Leitch HG, McEwen KR, Turp A, Encheva V, Carroll T, Grabole N, Mansfield W, Nashun B, Knezovich JG, Smith A et al (2013) Naive pluripotency is associated with global DNA hypomethylation. Nat Struct Mol Biol 20: 311–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics 25: 1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zhang Q, Yin X, Yang W, Du Y, Hou P, Ge J, Liu C, Zhang W, Zhang X et al (2011) Generation of iPSCs from mouse fibroblasts with a single gene, Oct4, and small molecules. Cell Res 21: 196–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Izpisua Belmonte JC (2016) Looking to the future following 10 years of induced pluripotent stem cell technologies. Nat Protoc 11: 1579–1585 [DOI] [PubMed] [Google Scholar]
- Liao J, Karnik R, Gu H, Ziller MJ, Clement K, Tsankov AM, Akopian V, Gifford CA, Donaghey J, Galonska C et al (2015) Targeted disruption of DNMT1, DNMT3A and DNMT3B in human embryonic stem cells. Nat Genet 47: 469–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti‐Filippini J, Nery JR, Lee L, Ye Z, Ngo QM et al (2009) Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462: 315–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Siegmund KD, Laird PW, Berman BP (2012) Bis‐SNP: combined DNA methylation and SNP calling for Bisulfite‐seq data. Genome Biol 13: R61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacFarlan TS, Gifford W, Driscoll S, Lettieri K, Rowe H, Bonanomi D, Firth A, Singer O, Trono D, Pfaff SL (2012) ES cell potency fluctuates with endogenous retrovirus activity. Nature 487: 57–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011) Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnet J 17: 10–12 [Google Scholar]
- Mascetti VL, Pedersen RA (2016) Human‐mouse chimerism validates human stem cell pluripotency. Cell Stem Cell 18: 67–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel CI, Malumbres M (2013) microRNA‐203: tumor suppression and beyond. MicroRNA 2: 118–126 [DOI] [PubMed] [Google Scholar]
- Mikkelsen TS, Hanna J, Zhang X, Ku M, Wernig M, Schorderet P, Bernstein BE, Jaenisch R, Lander ES, Meissner A (2008) Dissecting direct reprogramming through integrative genomic analysis. Nature 454: 49–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata S, Minobe W, Bristow MR, Leinwand LA (2000) Myosin heavy chain isoform expression in the failing and nonfailing human heart. Circ Res 86: 386–390 [DOI] [PubMed] [Google Scholar]
- Najm FJ, Chenoweth JG, Anderson PD, Nadeau JH, Redline RW, McKay RD, Tesar PJ (2011) Isolation of epiblast stem cells from preimplantation mouse embryos. Cell Stem Cell 8: 318–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano M, Bell DW, Haber DA, Li E (1999) DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99: 247–257 [DOI] [PubMed] [Google Scholar]
- Okita K, Ichisaka T, Yamanaka S (2007) Generation of germline‐competent induced pluripotent stem cells. Nature 448: 313–317 [DOI] [PubMed] [Google Scholar]
- Otsuji TG, Minami I, Kurose Y, Yamauchi K, Tada M, Nakatsuji N (2010) Progressive maturation in contracting cardiomyocytes derived from human embryonic stem cells: qualitative effects on electrophysiological responses to drugs. Stem Cell Res 4: 201–213 [DOI] [PubMed] [Google Scholar]
- Pedersen BS, Eyring K, De S, Yang IV, Schwartz DA (2014) Fast and accurate alignment of long bisulfite‐seq reads. arxiv https://arxiv.org/abs/1401.1129 [PREPRINT] [Google Scholar]
- Peters TJ, Buckley MJ, Statham AL, Pidsley R, Samaras K, Lord RV, Clark SJ, Molloy PL (2015) De novo identification of differentially methylated regions in the human genome. Epigenet Chrom 8: 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbloom KR, Armstrong J, Barber GP, Casper J, Clawson H, Diekhans M, Dreszer TR, Fujita PA, Guruvadoo L, Haeussler M et al (2015) The UCSC genome browser database: 2015 update. Nucleic Acids Res 43: D670–D681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandhu R, Rivenbark AG, Coleman WB (2012) Loss of post‐transcriptional regulation of DNMT3b by microRNAs: a possible molecular mechanism for the hypermethylation defect observed in a subset of breast cancer cell lines. Int J Oncol 41: 721–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126: 663–676 [DOI] [PubMed] [Google Scholar]
- Stadtfeld M, Apostolou E, Akutsu H, Fukuda A, Follett P, Natesan S, Kono T, Shioda T, Hochedlinger K (2010a) Aberrant silencing of imprinted genes on chromosome 12qF1 in mouse induced pluripotent stem cells. Nature 465: 175–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtfeld M, Maherali N, Borkent M, Hochedlinger K (2010b) A reprogrammable mouse strain from gene‐targeted embryonic stem cells. Nat Methods 7: 53–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S (2016) A decade of transcription factor‐mediated reprogramming to pluripotency. Nat Rev Mol Cell Biol 17: 183–193 [DOI] [PubMed] [Google Scholar]
- Theunissen TW, Friedli M, He Y, Planet E, O'Neil RC, Markoulaki S, Pontis J, Wang H, Iouranova A, Imbeault M et al (2016) Molecular criteria for defining the naive human pluripotent state. Cell Stem Cell 19: 502–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- To KK, Leung WW, Ng SS (2017) A novel miR‐203‐DNMT3b‐ABCG2 regulatory pathway predisposing colorectal cancer development. Mol Carcinog 56: 464–477 [DOI] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L (2012) Differential gene and transcript expression analysis of RNA‐seq experiments with TopHat and Cufflinks. Nat Protoc 7: 562–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Gao S (2016) Human naïve embryonic stem cells: how full is the glass? Cell Stem Cell 18: 301–303 [DOI] [PubMed] [Google Scholar]
- Wu J, Greely HT, Jaenisch R, Nakauchi H, Rossant J, Belmonte JC (2016) Stem cells and interspecies chimaeras. Nature 540: 51–59 [DOI] [PubMed] [Google Scholar]
- Xie W, Schultz MD, Lister R, Hou Z, Rajagopal N, Ray P, Whitaker JW, Tian S, Hawkins RD, Leung D et al (2013) Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell 153: 1134–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi M, Kishigami S, Tanaka A, Semi K, Mizutani E, Wakayama S, Wakayama T, Yamamoto T, Yamada Y (2017) Derivation of ground‐state female ES cells maintaining gamete‐derived DNA methylation. Nature 548: 224–227 [DOI] [PubMed] [Google Scholar]
- Yang Y, Bai W, Zhang L, Yin G, Wang X, Wang J, Zhao H, Han Y, Yao YQ (2008) Determination of microRNAs in mouse preimplantation embryos by microarray. Dev Dyn 237: 2315–2327 [DOI] [PubMed] [Google Scholar]
- Yang Y, Liu B, Xu J, Wang J, Wu J, Shi C, Xu Y, Dong J, Wang C, Lai W et al (2017) Derivation of pluripotent stem cells with in vivo embryonic and extraembryonic potency. Cell 169: 243–257 e25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi R, Poy MN, Stoffel M, Fuchs E (2008) A skin microRNA promotes differentiation by repressing “stemness”. Nature 452: 225–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying QL, Wray J, Nichols J, Batlle‐Morera L, Doble B, Woodgett J, Cohen P, Smith A (2008) The ground state of embryonic stem cell self‐renewal. Nature 453: 519–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao XY, Li W, Lv Z, Liu L, Tong M, Hai T, Hao J, Guo CL, Ma QW, Wang L et al (2009) iPS cells produce viable mice through tetraploid complementation. Nature 461: 86–90 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Movie EV1
Movie EV2
Movie EV3
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Data Availability Statement
RNAseq and methylation data have been deposited in the GEO repository (https://www.ncbi.nlm.nih.gov/geo/) under accession number GEO SuperSeries GSE81571 and GSE86653.