

Abstract
The incidence of nonmelanoma skin cancer (NMSC) has been increasing worldwide. Most studies have highlighted the importance of cancer-associated fibroblasts (CAFs) in NMSC progression. However much less is known about the communication between normal fibroblasts and epithelia; disruption of this communication affects tumor initiation and the latency period in the emergence of tumors. Delineating the mechanism that mediates this epithelial–mesenchymal communication in NMSC could identify more effective targeted therapies. The nuclear receptor PPARβ/δ in fibroblasts has been shown to modulate adjacent epithelial cell behavior, however, its role in skin tumorigenesis remains unknown. Using chemically induced skin carcinogenesis, we showed that FSPCre-Pparb/dex4 mice, whose Pparb/d gene was selectively deleted in fibroblasts, had delayed emergence and reduced tumor burden compared with control mice (Pparb/dfl/fl). However, FSPCre-Pparb/dex4-derived tumors showed increased proliferation, with no difference in differentiation, suggesting delayed tumor initiation. Network analysis revealed a link between dermal Pparb/d and TGF-β1 with epidermal NRF2 and Nox4. In vitro investigations showed that PPARβ/δ deficiency in fibroblasts increased epidermal Nox4-derived H2O2 production, which triggered an NRF2-mediated antioxidant response. We further showed that H2O2 upregulated NRF2 mRNA via the B-Raf-MEK1/2 pathway. The enhanced NRF2 response altered the activities of PTEN, Src, and AKT. In vivo, we detected the differential phosphorylation profiles of B-Raf, MEK1/2, PTEN, Src, and AKT in the vehicle-treated and chemically treated epidermis of FSPCre-Pparb/dex4 mice compared with that in Pparb/dfl/fl mice, prior to the first appearance of tumors in Pparb/dfl/fl. Our study revealed a role for fibroblast PPARβ/δ in the epithelial–mesenchymal communication involved in cellular redox homeostasis.
Subject terms: Cancer microenvironment, Oncogenes
Introduction
Skin cancer is one of the most common malignancies affecting humans worldwide. Furthermore, the incidence of nonmelanoma skin cancer (NMSC), comprising of basal cell of carcinoma and squamous cell carcinoma (SCC), has been increasing worldwide [1]. The development of NMSC is not a cell-autonomous process. Tumor-associated stromal cells in the reactive tumor microenvironment (TME) are now recognized as an important modulator and even a driver of tumorigenicity [2, 3]. Cancer-associated fibroblasts (CAFs), which differ from normal fibroblasts, are the dominant stromal cells within the TME of many tumors [4]. These CAFs reinforce the pro-tumorigenic communication with adjacent tumor cells, exacerbating the hallmarks of cancer and accelerating tumor malignancy [2, 3]. Recent transcriptional profiling of CAFs from clinical cutaneous SCCs revealed a unique signature profile of nuclear hormone receptors (NRs) [3, 5]. The study showed that targeting specific NRs in CAFs with cognate ligands as concurrent therapy can inhibit the emergence of chemoresistant tumors [3]. To date, most studies have focused on CAFs in the TME of preexisting tumors. However much less is known about the communication between normal fibroblasts and epithelia, whose disruption affects tumor initiation and the latency period in the emergence of tumors. Delineating the specific mechanism that mediates this epithelial–mesenchymal communication that contributes to NMSC could identify more effective targeted therapies to inhibit and/or prevent this disease.
Peroxisome proliferator-activated receptors (PPARs) are NRs that have attracted attention due to their crucial role in lipid homeostasis and tissue repair. PPARs are highly amenable for therapeutic intervention because their activities can be directly modulated by agonists and antagonists [6]. The PPARβ/δ isotype has been implicated in NMSC development, but its functions remain controversial as both oncogenic and tumor-suppressive roles have been reported [7–10]. Using chemically induced skin carcinogenesis, Kim et al. showed that Pparb/dex8 (deleted exon 8) knockout mice developed an increasing number of skin tumor [10]. PPARβ/δ was also reported to promote Hras-induced senescence and tumor suppression by repressing AKT activation [11]. In contrast, Pparb/dex4 (deleted exon 4) knockout mice developed fewer and smaller tumors when challenged with UV [9]. The study showed that UV induced PPARβ/δ activity, which increased Src kinase and EGFR/Erk1/2 activities, resulting in increased expression of epithelial-to-mesenchymal transition markers. This discrepancy has been attributed to several differences, including tumor initiation procedures, genetic disruption strategies of the Pparb/d gene, and the genetic background of the mouse [7, 8]. Importantly, these studies used whole-body Pparb/d knockout mice, thus the contribution of fibroblast PPARβ/δ in NMSC was not investigated.
Evidence for a role for fibroblast PPARβ/δ in skin physiology is emerging. Fibroblast PPARβ/δ has been shown to curb excessive epidermal proliferation during wound healing via the production of a secreted IL-1 receptor antagonist [12]. Wang et al. showed that ligand-activated fibroblast PPARβ/δ reduced oxidative stress in the diabetic wound microenvironment via the upregulation of Gpx1 and catalase, thus facilitating wound healing [13]. Recently, Sng et al. showed that the skin of FSPCre-Pparb/dex4 mice, a fibroblast-selective deletion of the Pparb/d gene, recapitulated various aspects of the fibroproliferative II subtype of human scleroderma, which involves the upregulation of leucine-rich alpha-2-glycoprotein-1 (Lrg1) by fibroblast PPARβ/δ that in turn acts in an autocrine and paracrine manner to modulate tissue-specific TGFβ1 responses [14]. However, the role of fibroblast PPARβ/δ in skin carcinogenesis remains unknown. Given that stromal fibroblasts have a significant influence on skin cancer initiation and development, we investigated the role of fibroblast PPARβ/δ in chemically induced skin carcinogenesis in FSPCre-Pparb/dex4 and wild-type Pparb/dfl/fl mice. Our findings revealed that a reduced activation of epidermal NRF2 by fibroblast PPARβ/δ resulted in increased skin tumor load.
Materials and methods
Cell culture and siRNA knockdown
Primary keratinocytes and fibroblasts were isolated and cultured as previously described [3, 15]. Co-cultures of keratinocyte and fibroblast were conducted as described [12]. Fibroblasts were transfected with SMARTpool small interfering RNAs (siRNAs) targeting either Pparb/d, Nox4, or LRG1 using DharmaFECT1 according to manufacturer’s recommendation (Dharmacon, USA) [4]. Co-transfection of Pparb/d siRNA and Pparb/d expression plasmids (Addgene, USA) was performed using Lipofectamine 2000 (Invitrogen, USA) according to manufacturer’s protocol.
ARE-luciferase reporter assay
The activity of NRF2 in the cells was determined using antioxidant response element (ARE) luciferase reporter vector (BPSBiosciences, USA). This reporter contains a firefly luciferase gene under the control of multimerized ARE responsive elements located upstream of a minimal promoter. Cells were transfected using FugeneHD according to manufacturer’s recommendation (Promega, USA). Reporter activity was measured using Dual-Glo Luciferase Assay System (Promega, USA).
RNA extraction, reverse transcription, and quantitative real-time PCR (qPCR)
Total RNA was extracted using TRIzol® Reagent followed by the PureLink™ Micro-to-Midi Total RNA Purification System according to the manufacturer’s protocol (Invitrogen, USA). Total RNA was quantified based on the A260/A280 absorbance using the Nanodrop ND1000 (Thermo scientific, USA). Total RNA was reversed transcribed using iScript Reverse Transcription Supermix for RT-PCR (Bio-Rad, USA). qPCR was performed as previously described [16] using the respective primers (Tables S1–S3).
Immunoblotting
Cells were lysed using ice-cold M-PER Mammalian Protein Extraction Reagent (Thermo Scientific, USA). Total protein lysates were resolved using 10% SDS-PAGE and electrotransferred onto Immobilon-FL PVDF membrane (Merck Millipore, USA). Membranes were blocked with 1× Odyssey Blocking buffer (LI-COR Biotechnology, USA) for 1 h at room temperature. The membrane was then incubated overnight at 4 °C with the indicated primary antibodies in 1× Odyssey Blocking buffer containing 0.05% Tween-20: NOX4 (sc-21860), PPARβ/δ (sc-1987), Erk1/2 (sc-94), phospho-Erk1/2 (sc-16982), and β-tubulin (sc-9104) from Santa Cruz Biotechnology Inc. (USA), NRF2 (D1Z9C), PTEN (9552S), Src (2108S), B-Raf (D9T6S), MEK1/2 (L38C12), phospho-PTEN (9551S), phospho-Src (6943S), phospho-B-Raf (Ser445), and phospho-MEK1/2 (Ser217/221) (41G9) from Cell Signaling Technology (USA), and LRG1 (ab178698) from Abcam (UK). Membranes were washed thrice with TBST (50 mM Tris–HCl, pH 7.6, 150 mM NaCl, 0.05% Tween-20), and incubated with appropriate IRDye®680- or 800-conjugated anti-IgG secondary antibodies in 1× Odyssey Blocking buffer containing 0.05% Tween-20 and 0.01% SDS for 1 h at room temperature. Protein bands were revealed using the Odyssey®CLx Infrared Imaging System, and signals were quantified using ImageStudio Software (LI-COR Biotechnology, USA).
Immunostaining
Five micrometre thick paraffinized sections were deparaffinized in xylene for 20 min and hydrated through a graded alcohol series. The hydrated sections were washed with 1× PBS buffer. Antigen retrieval was conducted in the GTC Buffer (0.01 mM guanidine hydrochloride, 0.01 mM ammonium thiocyanate, 25% glycerol, pH 6.0) using the Aptum Biologics 2100 Antigen Retriever. The sections were blocked with 3% normal goat serum for an hour. Anti-cytokeratin 1 (CK1, Abcam, UK) antibody was added to the sections and incubated 4 °C overnight. The following day, the sections were washed with PBS and incubated cognate secondary antibodies (Thermo Scientific, USA). Next, the sections were washed with PBS buffer, and the procedure was repeated with the anti-cytokeratin 8 antibody (CK8, Abcam, UK). Finally, the sections were washed, counterstained with Hoechst dye for nuclei, and imaged using JuLI™ Stage Real-Time Cell History Recorder. Immunofluorescent staining for Ki67 was performed as above with some modifications. Antigen retrieval was done in sodium citrate buffer (10 mM sodium citrate, 0.05% Tween, pH 6.0) and the sections were blocked using 1% bovine serum albumin. All immunostaining performed without primary antibodies served as negative controls. For hematoxylin and eosin (H&E) staining, the sections were washed in PBS buffer and stained with H&E as recommended by the manufacturer (Sigma-Aldrich, USA). The sections were dehydrated and mounted using the Eukitt® Quick-hardening Mounting Medium. Images of the sections were captured with the Zeiss Axio Scan.Z1 and analyzed with the Zen-Lite software.
PEG-switch assay
PEG-switch assay was performed as previously described [4]. Cells were lysed using M-PER mammalian protein extraction reagent (Thermo Fisher Scientific, Waltham, MA, USA). Oxidized thiols were reduced using DTT and alkylated using 2 mM PEG-maleimide with 0.5% SDS for 2 h at room temperature. The alkylation reaction was quenched using 100 mM Tris–HCl buffer pH 6.8, 4% SDS, 20% glycerol, and 0.01% bromophenol blue and 100 mM maleimide and the samples were resolved by SDS-PAGE and subjected to immunoblot. Oxidatively modified PTEN and Src were analyzed as previously described [17, 18]. The separated proteins were subjected to immunoblot analysis using antibodies against PTEN and Src.
Extracellular H2O2 and intracellular ROS measurement
Extracellular H2O2 and intracellular ROS were measured as previously described [13, 19]. The specificity of the assay for H2O2 was verified with catalase, and the degradation of H2O2 or inhibition of the assay system by the sample was analyzed by determining the recovery of exogenously added H2O2.
Chemical-induced skin carcinogenesis
Male 6-week old FSPCre-Pparb/dex4 and Pparb/dfl/fl mice of C57BL/6 background were used for this study [14, 20]. Chemical-induced skin carcinogenesis was performed in a double-blinded manner as previously described [21]. Briefly, mice were shaved with surgical clippers 2 days before topical initiation of the back with 200 µL of acetone containing 100 nmol of 7,12-dimethylbenz[a]anthracene (DMBA). One week after initiation, mice were promoted thrice weekly with 30 mg benzoyl peroxide (BP). The average number of dorsal skin tumors was recorded at 1-week intervals from 10th to 19th-week post treatment. Skin tumors were harvested at 19 weeks. The tumor size was calculated using the modified ellipsoid formula: ½ (length × width2). The greatest longitudinal diameter (length) and the greatest transverse diameter (width) of each tumor was determined using a digital caliper. The size for each tumor was classified under three categories: <5 mm3 (small), 5–10 mm3 (medium), and >10 mm3 (large). Each tumor from FSPCre-Pparb/dex4 and Pparb/dfl/fl mice measured and categorized accordingly in a double-blinded manner. Outliers were identified using interquartile range method and excluded from analysis. Animal studies were approved by Institutional Animal Care and Use Committee of Nanyang Technological University Singapore (ARF-SBS/NIE-A0112AZ, A0216AZ, A0324, and A0321).
Statistical analysis
Unless mentioned otherwise, animal sample size was determined by power analysis, statistical differences were evaluated with two-tailed Mann–Whitney U-test or one-way analysis of variance test with SPSS software where appropriate. P values < 0.05 indicate statistical significance.
Results
Fibroblast PPARβ/δ increased skin tumor load
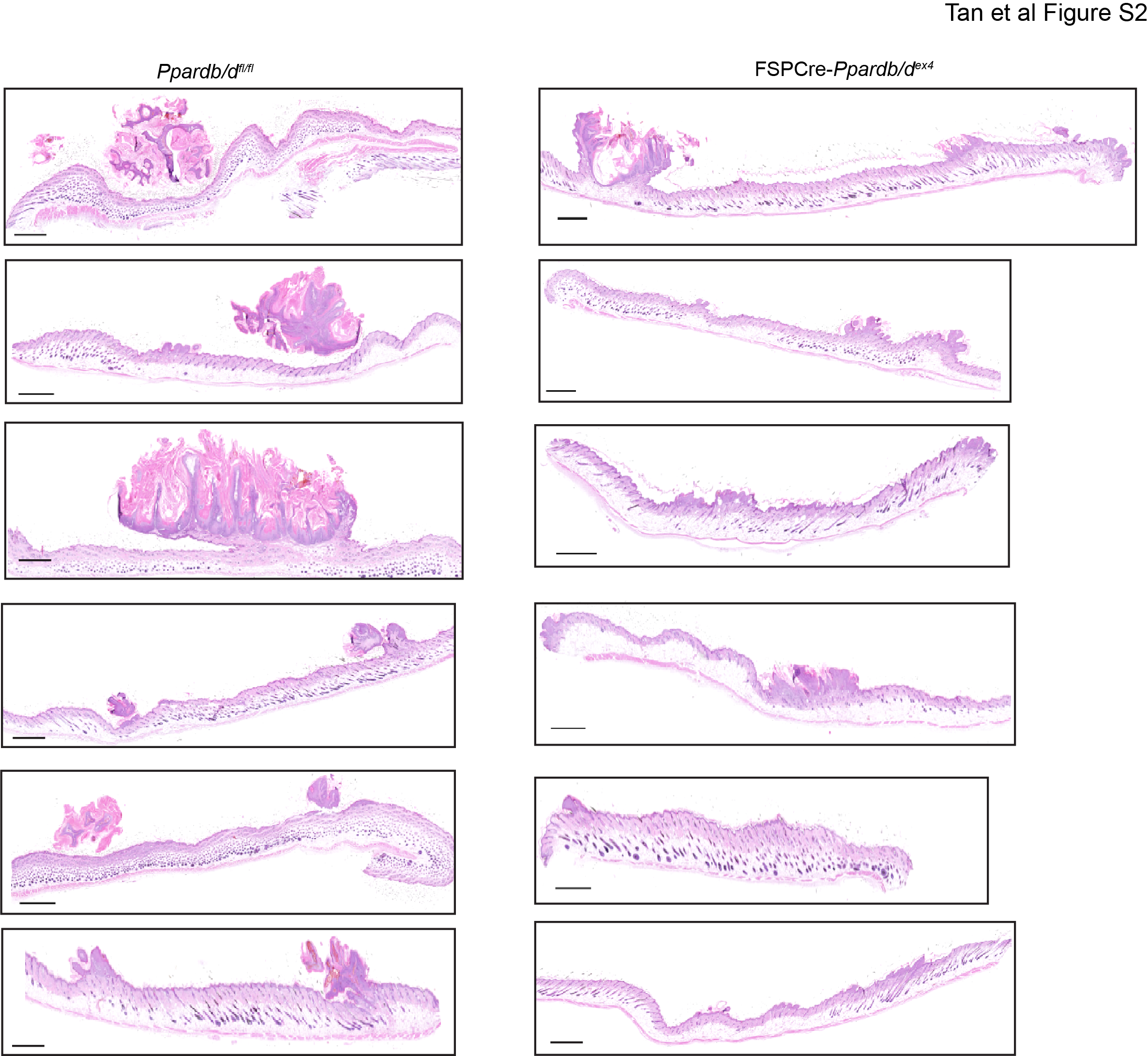
FSPCre-Pparb/dex4 and wild-type Pparb/dfl/fl mice were subjected to the two-stage chemical-induced skin carcinogenesis regimen. The deletion of Pparb/d in the fibroblasts was confirmed by genotyping and western blot analysis (Fig. S1A, B). The number and volume of tumors were monitored weekly throughout the regimen. Palpable tumors were observed after 10–12 weeks of treatment in Pparb/dfl/fl mice compared with 12–15 weeks in FSPCre-Pparb/dex4 mice (Fig. 1a). All Pparb/dfl/fl mice developed tumors by week 10, whereas tumors were observed in all FSPCre-Pparb/dex4 mice at week 13 (Fig. 1b). We segregated the tumor numbers by volume, small (<0.005 cm3), medium (0.005–0.01 cm3), and large (>0.01 cm3) after 19 weeks of treatment. The FSPCre-Pparb/dex4 mice developed fewer and smaller tumors compared with Pparb/dfl/fl mice (Figs. 1c, S1C, and S2). Immunofluorescence staining for Ki67, a proliferation marker, revealed a higher number of Ki67-positive cells in FSPCre-Pparb/dex4-derived tumors than in Pparb/dfl/fl-derived tumor biopsies (Figs. 1d and S1D). We utilized CK1 and CK8 to distinguish the stage of tumor differentiation. CK1 expression is elevated in the hyperplastic epidermis, and the loss of CK1 combined with an aberrant increase in CK8 is characteristic of the progression of mouse skin papilloma to SCCs [22]. As expected, CK1 was expressed in normal and hyperplastic skin from both genotypes. Regardless of tumor size, no difference in CK8 expression between tumors from Pparb/dfl/fl and FSPCre-Pparb/dex4 mice was observed, indicating that PPARβ/δ deficiency in fibroblasts does not affect tumor differentiation (Fig. 1d). Taken together, our observations suggest an impaired tumor initiation, resulting in a longer latency period in the emergence of tumors in FSPCre-Pparb/dex4 mice.
Fig. 1. Reduced skin tumor load in FSPCre-Pparb/dex4 mice.

a Graph showing the mean number of tumors in FSPCre-Pparb/dex4 (open circle) and Pparb/dfl/fl (solid circle) mice starting from week 10 post tumor initiation. b Graph showing the percentage of FSPCre-Pparb/dex4 (open circle) and Pparb/dfl/fl (solid circle) mice that developed palpable tumors after chemical-induced skin carcinogenesis. The duration of tumor emergence in FSPCre-Pparb/dex4 (median = 12.5 weeks) and Pparb/dfl/fl (median = 9 weeks) mice was evaluated with the log-rank test. ***p < 0.001. c Box-and-whisker plot showing the distribution of tumor number and size in FSPCre-Pparb/dex4 (white) and Pparb/dfl/fl (gray) mice. Three categories were used for tumor volume classification: small (<5 mm3), medium (5–10 mm3), and large (>10 mm3). Outliers were identified by using the interquartile range and presented as black dots. The table below shows the weight (mean ± s.d.) of the tumors for each category. Data are represented as mean ± s.d. from n = 15–18 mice. *p < 0.05, ***p < 0.001. d Representative images of normal skin, hyperplastic skin, and tumors from FSPCre-Pparb/dex4 and Pparb/dfl/fl mice. Dual immunofluorescence staining was performed for CK1 (red) and CK8 (green). The nuclei were counterstained with DAPI (blue). The same sections were used for hematoxylin and eosin (H&E) staining. Proliferation marker Ki67 staining was performed with other sections from the same biopsies. Scale bar: 100 μm.
PPARβ/δ deficiency in fibroblasts increased epidermal Nox4 and NRF2 expression
To decipher the underlying mechanism for the delayed emergence of tumors in FSPCre-Pparb/dex4 mice, we interrogated the intrinsic networks that underlie differentially regulated genes using the gene expression microarray data of FSPCre-Pparb/dex4 and Pparb/dfl/fl epidermis and dermis (GSE71419). Using the Ingenuity Pathway Analysis, a gene connectivity network grouped diseases and functions into broad categories, with connective tissue development having the largest coverage, followed by organismal injury and abnormalities and cellular movement (Fig. 2a). A core network linked dermal Pparb/d and TGF-β1 with epidermal NRF2 (nuclear factor erythroid 2-related factor 2) and Nox4 (NADPH oxidase-4) signaling (Fig. 2a, b).
Fig. 2. Pathway analysis and gene connectivity network.

a A gene connectivity network links Pparb/d, Nox4, NRF2 (also known as NFE2L2), and TGF-β1 signaling with the indicated diseases and functions. Twelve molecules with significant fold change (>1.5 times) and confidence (p < 0.05) identified from the microarray were connected to these signaling networks. Activating (green), deactivating (red), and inconclusive (gray) effects on the various functions are indicated. Arcs surrounding the gene network group diseases and functions into broad categories that include connective tissue development, cancer and free radical scavenging. b Diseases and functions are tabulated and arranged into categories based on their p value. The molecules responsible for each disease and function are also indicated.
Our analysis revealed a hitherto unknown connection linking Pparb/d, NRF2, and Nox4, thus, we focused our investigation on this new network. We detected higher Nox4 and NRF2 in the epidermis of FSPCre-Pparb/dex4 mice than in that of Pparb/dfl/fl mice (Fig. 3a). We also detected higher extracellular H2O2 in conditioned medium from primary FSPCre-Pparb/dex4 keratinocyte cultures than from Pparb/dfl/fl keratinocyte cultures, and this effect could be eliminated by exogenous catalase (Fig. 3b). These changes were consistent with higher intracellular ROS as indicated by the higher mean CellRox fluorescence readings in keratin-positive cells from the dorsal skins of FSPCre-Pparb/dex4 than in those from Pparb/dfl/fl mice (Fig. 3c). A higher basal NRF2 activity, determined using an ARE-luciferase reporter transactivation assay, was also observed in keratinocytes cocultured with primary fibroblasts from FSPCre-Pparb/dex4 mice than those from Pparb/dfl/fl mice (Fig. 3d). We also detected higher mRNA levels of NRF2, along with NRF2-regulated antioxidant enzymes in the FSPCre-Pparb/dex4 epidermis compared with the Pparb/dfl/fl epidermis (Fig. 3e). To understand how ROS, specifically H2O2, increased the mRNA level of NRF2 mRNA, we subjected primary human keratinocytes to a kinase inhibitor screen and examined NRF2 expression at 4 h post H2O2 stimulation. We observed that kinase inhibitors against the B-Raf and MEK kinases could attenuate H2O2-mediated upregulation of NRF2, suggesting that B-Raf-MEK pathway is involved in the transcription of NRF2 (Fig. 3f). Western blot analyses confirmed the increase in the phosphorylation levels of B-Raf, MEK1/2, and ERK1/2 upon H2O2 exposure (Fig. 3g). The increase in NRF2 mRNA was unlikely due to autoregulation as its increase occurred before NRF2 upregulated its target genes such as NQO1 and GPX1 (Fig. S3A).
Fig. 3. Elevated Nox4 stimulates NRF2-related responses to reduce epidermal ROS.

a Representative immunoblots of Nox4 and NRF2 in epidermis from FSPCre-Pparb/dex4 and Pparb/dfl/fl mice. Densitometric quantification plots are shown (top panel). β-tubulin served as a housekeeping protein from the same samples. b Extracellular H2O2 levels in FSPCre-Pparb/dex4 and Pparb/dfl/fl keratinocytes. Extracellular H2O2 was detected by the Amplex Red Assay and normalized to cell number. c Histogram shows the mean intracellular ROS (CellRox fluorescence readings) in cytokeratin-positive cells from the skin of FSPCre-Pparb/dex4 (red) and Pparb/dfl/fl skin (gray). d Fold change in ARE-dependent luciferase activities in keratinocytes cocultured with primary fibroblasts from FSPCre-Pparb/dex4 or Pparb/dfl/fl mice. For the reporter assay, firefly luciferase activity was normalized to Renilla luciferase activity. e Relative mRNA levels of the indicated antioxidant genes in the epidermis from FSPCre-Pparb/dex4 and Pparb/dfl/fl mice. Ribosomal protein L27 was used as a housekeeping gene. f Relative mRNA level of NRF2 in human primary keratinocytes treated with vehicle and kinase inhibitors against B-Raf and MEK1/2 at 4 h post H2O2 stimulation. Ribosomal protein L27 was used as a housekeeping gene. g Representative immunoblots of total and phosphorylated B-Raf, MEK1/2, and ERK1/2 in human keratinocytes treated with H2O2 (left panel). A densitometric quantification plot is shown (right panel). Total B-Raf, MEK1/2, and ERK1/2 as cognate normalizing proteins were from the same samples. h Representative immunoblots of Nox4 and NRF2 in human keratinocytes (Kers) cocultured with either dermal human fibroblasts knockdown of Pparb/d (FsiPparb/d) or scrambled siRNA (Fscrambled) (bottom panel). β-tubulin served as housekeeping protein and was from the same samples. A densitometric quantification plot is shown (top panel). Data are represented as the mean ± s.d. For a and d–h, n = 3 independent experiments. For b, c, n = 5 independent experiments. *p < 0.05, **p < 0.01 and n.s. denotes non-significant.
Consistent with the above observation, we observed higher Nox4 and NRF2 expression in human keratinocytes (Kers) cocultured with human dermal fibroblasts whose endogenous PPARβ/δ was suppressed by siRNA (FsiPparb/d) than in control fibroblasts (Fscrambled). The overexpression of PPARβ/δ in FsiPparb/d fibroblasts reduced Nox4 and NRF2 expression (Figs. 3h and S3B, C). In summary, PPARβ/δ deficiency in fibroblasts increased Nox4 expression and triggered an enhanced antioxidant NRF2 response in the adjacent epidermis.
TGF-β1 elevated Nox4 expression in keratinocytes and was attenuated by LRG1
The epithelial–mesenchymal communication between dermal Pparb/d and TGF-β1 has been previously reported [14]. PPARβ/δ in fibroblasts upregulates the expression of Lrg1, which attenuates epidermal and dermal responses to TGFβ1. We investigated the potential contribution of TGF-β1 signaling, which was a key player in our IPA analysis, to the increased epidermal expression of Nox4 and NRF2. We showed that the expression of Nox4 (Fig. 4a) and the activity of NRF2 (Fig. 4b) were stimulated by TGF-β1 in Pparb/dfl/fl keratinocytes. The FSPCre-Pparb/dex4 keratinocytes showed a higher basal ARE-dependent luciferase activity that was further increased by exogenous TGF-β1 (Fig. 4b). PPARβ/δ transcriptionally upregulates the expression of LRG1 in fibroblasts [14]. LRG1 attenuates TGF-β1 signaling in the skin and lung to suppress fibrosis [14, 23]. We observed that recombinant LRG1 dose-dependently attenuated the expression of Nox4 and the activity of the reporter luciferase. The suppression of endogenous Nox4 (siNox4) or the exposure to the antioxidant N-acetyl-cysteine (NAC) attenuated ARE-dependent luciferase activity (Fig. 4b).
Fig. 4. Fibroblast PPARβ/δ modulates epidermal Nox4 via LRG1-TGFβ1 signaling.

a Relative mRNA (top panel) and protein (bottom panel) levels of Nox4 in Pparb/dfl/fl keratinocytes treated with either TGFβ1 or LRG1 proteins. Representative immunoblot for Nox4 is shown. β-tubulin served as a housekeeping protein from the same samples. A densitometric quantification plot is shown. b Fold change in ARE-dependent luciferase activities in FSPCre-Pparb/dex4 and Pparb/dfl/fl keratinocytes treated with either TGFβ1 (10 ng/mL) or LRG1 (50–100 ng/mL). Keratinocytes whose endogenous Nox4 expression was suppressed by siRNA were denoted by siNox4. Antioxidant NAC was used at 500 µM. For the reporter assay, firefly luciferase activity was normalized to Renilla luciferase activity. c Representative immunoblots of Nox4 and NRF2 in human keratinocytes (Kers) cocultured with either dermal human fibroblasts knockdown of Pparb/d (FsiPparb/d) or LRG1 (FsiLRG1) (bottom panel). β-tubulin served as housekeeping protein and was from the same samples. A densitometric quantification plot is shown (top panel). d Relative mRNA levels of the indicated antioxidant genes in Kers cocultured with FsiPparb/d or Fscrambled. Ribosomal protein L27 was used as a housekeeping gene. e Mean intracellular ROS level in Kers cocultured with either FsiPparb/d (red) or Fscrambled (gray). The histogram shows mean CellRox fluorescence readings in keratinocytes. f Relative mRNA levels of Nox4 and NRF2 in Kers treated with either TGFβ1, LRG1, or NAC. Ribosomal 18S was used as a housekeeping gene. g Representative immunoblots of Nox4 and NRF2 in primary wild-type mouse keratinocytes transfected with various Nox4 mutants. Densitometric quantification plots are shown (right panel). β-tubulin served as a housekeeping protein from the same samples. h Fold change in ARE-dependent luciferase activities in mouse keratinocytes overexpressing wild-type, Nox4ΔD-loop, and Nox4ΔB-loop Nox4 mutants. Empty vector served as the control. For the reporter assay, firefly luciferase activity was normalized to Renilla luciferase activity. All data are represented as the mean ± s.d. For a and e, n = 5 independent experiments. For b–d and f–h, n = 3 independent experiments. **p < 0.01.
Cocultured of Kers with FsiPparb/d or FsiLRG1 and treatment with exogenous LRG1 reduced the expression of Nox4 and NRF2 in Kers (Figs. 4c and S3D). This observation suggests that LRG1-TGF-β1 signaling is involved in regulating Nox4 and NRF2 expression in keratinocytes. In human Kers cocultured with FsiPparb/d, the mRNA levels of several antioxidant enzymes were also elevated (Fig. 4d), but the intracellular ROS level remained higher than that in Kers cocultured with Fscrambled (Fig. 4e). These results are consistent with our coculture experiment of keratinocytes with primary fibroblasts from either Pparb/dfl/fl or FSPCre-Pparb/dex4 mice (Fig. 3c, e). Human keratinocytes treated with TGF-β1 alone showed a mild increase in Nox4 and NRF2 expression accompanied by an increase in the expression of antioxidant enzymes (Fig. S3E), indicating that NRF2 is a downstream mediator of TGF-β1. The observed upregulation of Nox4 and NRF2 was attenuated by LRG1 (Fig. 4f). Notably, the increase in NRF2, but not Nox4, was abolished by NAC suggesting that NRF2 expression was dependent on Nox4-derived H2O2 (Fig. 4f). Nox4 increases oxidative stress by producing H2O2 [24], whereas an increased NRF2 response elevates the expression of many antioxidant genes [25]. To understand the relationship between Nox4 and NRF2, we examined the effect of wild-type Nox4 and various Nox4 mutants on NRF2 expression and activity. The ectopic expression of wild-type Nox4 and the H2O2-producing Nox4ΔD-loop mutant increased NRF2 expression and activity (Fig. 4g, h). In contrast, the overexpression of the Nox4ΔB-loop mutant, which is defective in H2O2 production, stimulated neither NRF2 expression nor NRF2 activity (Fig. 4g, h). In summary, we showed that PPARβ/δ-deficient fibroblasts increased Nox4 expression in adjacent keratinocytes which involved paracrine LRG1/TGF-β1 signaling. The elevated epidermal Nox4 increased intracellular oxidative stress that triggered an enhanced NRF2 response as indicated by the higher expression of antioxidant/detoxification genes in the epidermis.
Differential phosphorylation kinetics of B-Raf, MEK1/2, PTEN, Src, and AKT prior to tumor formation
The epidermis of FSPCre-Pparb/dex4 has an enhanced NRF2 response. The inhibition of either B-Raf or MEK1/2 kinases attenuated the mRNA expression of NRF2 via H2O2. To gain further insight into the mechanism in vivo, we examined the expression of B-Raf, MEK1/2, and NRF2 in the epidermis of vehicle- or DMBA/BP-treated Pparb/dfl/fl and FSPCre-Pparb/dex4 mice between weeks 4 and 8, prior to the emergence of palpable tumors at week 10 post treatment in Pparb/dfl/fl mice. The expression profiles of phospho-B-Raf and phospho-MEK1/2 were elevated in the epidermis of vehicle-treated FSPCre-Pparb/dex4 mice, concomitant with the higher expression of NRF2, when compared with cognate Pparb/dfl/fl mice (Fig. 5a). The expression of phospho-B-Raf, phospho-MEK1/2, and NRF2 increased more rapidly in DMBA/BP-treated FSPCre-Pparb/dex4 mice than in Pparb/dfl/fl mice (Fig. 5a). Together with the in vitro data, this observation indicates that the upregulation of NRF2 mRNA is mediated via the B-Raf-MEK1/2 pathway.
Fig. 5. Protein expression profiles of NRF2, B-Raf, MEK1/2, PTEN, Src, and AKT in the epidermis of FSPCre-Pparb/dex4 and Pparb/dfl/fl mice.

a Representative immunoblots of NRF2, total and phosphorylated B-Raf, MEK1/2, PTEN, Src, and AKT in the epidermis of vehicle control- and DMBA/BP-treated FSPCre-Pparb/dex4 and Pparb/dfl/fl mice between weeks 4 and 8 (top panel). Densitometric quantification plots are shown (bottom panel). Total B-Raf, MEK1/2, PTEN, Src, and AKT served as cognate normalizing controls from the same samples. Values are represented as the mean ± s.d. from n = 3 independent experiments. *p < 0.05 and **p < 0.01 compared with vehicle-control treatment of the same week. b Schematic illustration of the mechanism. Fibroblast PPARβ/δ increases the expression of LRG1 [14]. Secreted LRG1 attenuates TGF-β1 signaling, which is required for the upregulation of Nox4 in keratinocytes. Less Nox4-derived H2O2 production reduces the NRF2 expression and response, and consequently reduces the expression of antioxidant/detoxification genes and increases intracellular ROS. Higher levels of phosphorylated PTEN, Src, and AKT were present in the epidermis of Pparb/dfl/fl mice compared with FSPCre-Pparb/dex4 mice and contributed to the earlier appearance of tumors.
The higher intracellular ROS in Pparb/dfl/fl modifies the activities of tumor suppressors or oncogenes. Direct oxidative modification of products of tumor suppressors or oncogenes can have a direct impact on tumor formation. Previous studies showed that oxidation increases the autophosphorylation of PTEN and Src, which attenuates the tumor-suppressive catalytic activity of PTEN [18, 26] and triggers the oncogenic functions of Src [17, 27]. PTEN and Src have been implicated in SCC formation [28, 29]. To this end, we examined the oxidative state of the tumor suppressor PTEN and oncogenic Src in FSPCre-Pparb/dex4 and -Pparb/dfl/fl mice. The PEG-switch assay is a semiquantitative method for measuring target reversible cysteine oxidation in complex protein mixtures derived from tissue or cultured cells [30]. Quantification of pegylated PTEN and Src against the reduced form showed a significant increase in PTEN and Src oxidation, indicating reduced PTEN and enhanced Src activities in Pparb/dfl/fl keratinocytes compared with FSPCre-Pparb/dex4 keratinocytes (Fig. S3F).
We propose that the difference between the phosphorylation levels of PTEN and Src between FSPCre-Pparb/dex4 and Pparb/dfl/fl mice also contributes to the delayed emergence of tumors in the former. There was no significant difference in the phosphorylation levels of PTEN and Src in the epidermis of vehicle-treated FSPCre-Pparb/dex4 and Pparb/dfl/fl mice (Fig. 5a), suggesting that these pathways may not be responsible for the already elevated NRF2 levels in FSPCre-Pparb/de4 mice (Fig. 5a). We observed a progressive increase in phosphorylated PTEN and Src in the epidermis of Pparb/dfl/fl mice upon DMBA/BP treatment, indicating that the altered activities of PTEN and Src preceded the emergence of tumors (Fig. 5a). Phosphorylation of PTEN at Ser380 suppresses its activity by controlling its recruitment into the PTEN-associated complex as well as its stability [31, 32]. With reduced PTEN activity, we observed higher levels of phosphorylated AKT in the epidermis of Pparb/dfl/fl mice than in the epidermis of FSPCre-Pparb/dex4 mice. This observation is consistent with the higher tumor burden in Pparb/dfl/fl than FSPCre-Pparb/dex4 (Fig. 5a). In contrast, the increase in phosphorylated PTEN and Src in the epidermis of FSPCre-Pparb/dex4 was muted and was only significant compared with the cognate vehicle control from week 6 onwards (Fig. 5a). Taken together, the late tumor emergence and reduced tumor load in FSPCre-Pparb/dex4 mice compared with Pparb/dfl/fl mice can be attributed in part to altered NRF2, B-Raf, MEK1/2, PTEN, Src, and AKT activation profiles during tumor initiation.
Discussion
CAFs are the major cell population within the tumor stroma and are involved in cancer progression [2]. Emerging studies have shown that concurrent therapies targeting CAFs increase the efficacy of conventional anticancer chemotherapeutics [3]. NRs have surfaced as ideal drug targets because their activity is precisely modulated by the binding of small lipophilic molecules. In this context, PPARβ/δ is of great interest because it has been implicated in a variety of diseases, such as cardiovascular diseases and diabetes. However, the biological function of PPARβ/δ in NMSC remains controversial, due in part to a poor understanding of the role of fibroblast PPARβ/δ in NMSC [7]. Furthermore, previous studies mainly focused on CAFs in preexisting tumors [5], rather than in the epithelial–mesenchymal communication between normal fibroblasts and epithelia, whose disruption facilitates tumor development.
We observed delayed emergence and reduced tumor burden in FSPCre-Pparb/dex4 mice compared with Pparb/dfl/fl mice using a chemically induced skin carcinogenesis model. However, FSPCre-Pparb/dex4-derived tumors showed increased proliferation, with no difference in differentiation compared with similarly sized Pparb/dfl/fl-derived tumors, suggesting a delayed tumor initiation. The underlying mechanism involves enhanced epidermal NRF2 and Nox4 expression, which is in line with a previous study that reported a protective effect of NRF2 against UV-induced cutaneous SCCs [33]. In contrast, the tumor-promoting activity of NRF2 in virus-induced skin tumorigenesis has been reported [34]. The mixed evidence suggests a complex and context-dependent role of NRF2 in tumor progression. In our study, the activation of NRF2 and Nox4 integrates into the PPARβ/δ and LRG1-TGFβ1 networks (Fig. 5b). Fibroblast PPARβ/δ transcriptionally upregulates the expression of LRG1, which enhances TGFβ1 signaling in adjacent epidermis [14]. We showed that the expression of Nox4 was stimulated by TGF-β1 and that PPARβ/δ-deficient fibroblasts also triggered an enhanced NRF2-mediated response, which was dependent on Nox4-derived H2O2, i.e., oxidative stress. We showed that H2O2 rapidly upregulated NRF2 mRNA via the B-Raf-MEK1/2 pathway. The enhanced NRF2-related response altered the intracellular ROS that modulate the activities of tumor suppressor PTEN, oncogene Src, and AKT. These findings supported a pro-tumorigenic role for fibroblast PPARβ/δ.
Although the genetic background, knockout mouse model, and tumor induction protocol were different, our experimental outcome was consistent with the outcome of the UV-induced skin carcinogenesis protocol performed on full-body hairless SKH1/Pparb/dex4 mice. The induction of skin cancer by DMBA/BP involves molecular mechanisms that are different from those associated with skin cancer that is attributable to chronic UV exposure [35, 36]. These independent studies have underscored a pro-tumorigenic role for PPARβ/δ in skin cancers. Lending support, the long-term treatment of mice with the PPARβ/δ agonist GW501516 caused rapid induction of cancers in several organs [37]. Our findings were at odd with cancer studies using whole-body Pparb/dex8 mice, whose Pparb/d gene was inactivated by insertion of the phosphoribosyltransferase II gene at the last exon. It has been proposed that whole-body Pparb/dex8 displays a hypomorphic phenotype, i.e., a partial loss of function of PPARβ/δ [7, 8, 38]. There are also some limitations to our study; we did not explore other tumor induction protocols such as UV irradiation or use FSPCre-Pparb/dex4 mice from a different genetic background. It is conceivable that different mouse strains may exhibit different phenotypic severities or outcomes owing to differences in vulnerabilities to oxidative stress and carcinogens. FSP1 is a key marker of a specific subset of macrophages in the liver during fibrosis and injury, although no report has described confounding issues in other organs [39]. Based on existing technology, it is not possible to achieve pure fibroblastic gene deletion or any tissue-specific deletion with absolute certainty. In addition, we were unable to generate an epidermis-specific NRF2 and fibroblast-specific PPARβ/δ double conditional knockout based on current Cre-lox technology alone. Nevertheless, to the best of our knowledge, this is the first study to examine the role of fibroblast PPARβ/δ in NMSC.
Despite a delay in tumor initiation, FSPCre-Pparb/dex4 mice had higher tumor cell proliferation than FSPCre-Pparb/dfl/fl mice. The long-term effect of such enhanced tumor cell proliferation on cancer progression suggests a possible dual role in initiation and progression. TGFβ and NRF2 signaling have been reported to play dual roles in tumorigenesis, differing in tumor initiation and progression [40, 41]. Given that fibroblast PPARβ/δ activates epidermal NRF2 via TGFβ signaling, it is conceivable that fibroblast PPARβ/δ may similarly exhibit dichotomous roles. On the one hand, PPARβ/δ deficiency in fibroblasts led to reduced oxidative stress in the epidermis, delayed tumor initiation, and reduced tumor burden in FSPCre-Pparb/dex4 mice. On the other hand, tumors from FSPCre-Pparb/dex4 mice has a higher number of proliferating Ki67-positive cells. Similarly, the proliferation of human skin cancer cells was reduced when cocultured with fibroblasts overexpressing PPARβ/δ [3]. The mechanism underlying this dichotomy remains unclear, but it tempting to speculate that it could be related to the transformation from normal fibroblasts to CAFs. It was shown that the acquisition of an oxidative, CAF-like state by normal fibroblasts could be achieved by chronic exposure to oxidative stress and attenuated TGF-β signaling [4]. Our study showed that a deficiency in PPARβ/δ in fibroblasts disrupted normal epithelial–mesenchymal communication and enhanced the antioxidant/detoxification response to delay NMSC development.
Supplementary information
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank all individuals who take part in this research. MWYT is a recipient of Interdisciplinary Graduate Scholarship from Nanyang Technological University Singapore. ZSL is a recipient of Research Scholarship from Lee Kong Chian School of Medicine, Nanyang Technological University Singapore.
Funding
This research/project is supported by Start-Up Grant (M4082040) and Ministry of Education, Singapore, under Academic Research Fund Tier 1 (2017-T1-002-103) to NST, (2015-T1-001-034) to WW and Start-Up Grant from the Lee Kong Chian School of Medicine, Nanyang Technological University Singapore, Singapore to WW and XW; the Région Midi-Pyrénées through the Chaire d’Excellence Pierre de Fermat and the Bonizzi-Theler-Stiftung to WW; SERB-DST, Govt. of India funded Ramanujan Fellowship Grant (SB/S2/RJN-087/2014) to MP.
Data availability
The datasets for microarray analysis during the current study are available through the Gene Expression Omnibus Series accession number GSE71419. The data in Fig. S4 are in whole based upon data generated by the TCGA Research Network (http://cancergenome.nih.gov/).
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Consent for publication
All authors have read and approved the final manuscript, and consent to publish.
Ethics approval and consent to participate
Animal experiments were carried out in accordance with the guidelines of the institutional animal care and use committee (ARF-SBS/NIE-A0324, A0321, A0112AZ, and A0216AZ) of Nanyang Technological University Singapore, Singapore.
Footnotes
Edited by E. Candi
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Mark Wei Yi Tan, Ming Keat Sng
Supplementary information
The online version of this article (10.1038/s41418-020-0535-y) contains supplementary material, which is available to authorized users.
References
- 1.Leiter U, Eigentler T, Garbe C. Epidemiology of skin cancer. Adv Exp Med Biol. 2014;810:120–40. doi: 10.1007/978-1-4939-0437-2_7. [DOI] [PubMed] [Google Scholar]
- 2.Liao Z, Tan ZW, Zhu P, Tan NS. Cancer-associated fibroblasts in tumor microenvironment - Accomplices in tumor malignancy. Cell Immunol. 2018;343:103729. doi: 10.1016/j.cellimm.2017.12.003. [DOI] [PubMed] [Google Scholar]
- 3.Chan JSK, Sng MK, Teo ZQ, Chong HC, Twang JS, Tan NS. Targeting nuclear receptors in cancer-associated fibroblasts as concurrent therapy to inhibit development of chemoresistant tumors. Oncogene. 2018;37:160–73. doi: 10.1038/onc.2017.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chan JS, Tan MJ, Sng MK, Teo Z, Phua T, Choo CC, et al. Cancer-associated fibroblasts enact field cancerization by promoting extratumoral oxidative stress. Cell Death Dis. 2017;8:e2562. doi: 10.1038/cddis.2016.492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng HS, Lee JXT, Wahli W, Tan NS. Exploiting vulnerabilities of cancer by targeting nuclear receptors of stromal cells in tumor microenvironment. Mol Cancer. 2019;18:51. doi: 10.1186/s12943-019-0971-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grygiel-Gorniak B. Peroxisome proliferator-activated receptors and their ligands: nutritional and clinical implications-a review. Nutr J. 2014;13:17. doi: 10.1186/1475-2891-13-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tan NS, Vazquez-Carrera M, Montagner A, Sng MK, Guillou H, Wahli W. Transcriptional control of physiological and pathological processes by the nuclear receptor PPARbeta/delta. Prog Lipid Res. 2016;64:98–122. doi: 10.1016/j.plipres.2016.09.001. [DOI] [PubMed] [Google Scholar]
- 8.Peters JM, Gonzalez FJ, Muller R. Establishing the role of PPARbeta/delta in carcinogenesis. Trends Endocrinol Metab. 2015;26:595–607. doi: 10.1016/j.tem.2015.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Montagner A, Delgado MB, Tallichet-Blanc C, Chan JS, Sng MK, Mottaz H, et al. Src is activated by the nuclear receptor peroxisome proliferator-activated receptor beta/delta in ultraviolet radiation-induced skin cancer. EMBO Mol Med. 2014;6:80–98. doi: 10.1002/emmm.201302666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim DJ, Prabhu KS, Gonzalez FJ, Peters JM. Inhibition of chemically induced skin carcinogenesis by sulindac is independent of peroxisome proliferator-activated receptor-beta/delta (PPARbeta/delta) Carcinogenesis. 2006;27:1105–12. doi: 10.1093/carcin/bgi346. [DOI] [PubMed] [Google Scholar]
- 11.Zhu B, Ferry CH, Blazanin N, Bility MT, Khozoie C, Kang BH, et al. PPARbeta/delta promotes HRAS-induced senescence and tumor suppression by potentiating p-ERK and repressing p-AKT signaling. Oncogene. 2014;33:5348–59. doi: 10.1038/onc.2013.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chong HC, Tan MJ, Philippe V, Tan SH, Tan CK, Ku CW, et al. Regulation of epithelial-mesenchymal IL-1 signaling by PPARbeta/delta is essential for skin homeostasis and wound healing. J Cell Biol. 2009;184:817–31. doi: 10.1083/jcb.200809028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang X, Sng MK, Foo S, Chong HC, Lee WL, Tang MB, et al. Early controlled release of peroxisome proliferator-activated receptor beta/delta agonist GW501516 improves diabetic wound healing through redox modulation of wound microenvironment. J Control Release. 2015;197:138–47. doi: 10.1016/j.jconrel.2014.11.001. [DOI] [PubMed] [Google Scholar]
- 14.Sng MK, Chan JSK, Teo Z, Phua T, Tan EHP, Wee JWK, et al. Selective deletion of PPARbeta/delta in fibroblasts causes dermal fibrosis by attenuated LRG1 expression. Cell Discov. 2018;4:15. doi: 10.1038/s41421-018-0014-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Teo Z, Chan JSK, Chong HC, Sng MK, Choo CC, Phua GZM, et al. Angiopoietin-like 4 induces a beta-catenin-mediated upregulation of ID3 in fibroblasts to reduce scar collagen expression. Sci Rep. 2017;7:6303. doi: 10.1038/s41598-017-05869-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lam CR, Tan MJ, Tan SH, Tang MB, Cheung PC, Tan NS. TAK1 regulates SCF expression to modulate PKBalpha activity that protects keratinocytes from ROS-induced apoptosis. Cell Death Differ. 2011;18:1120–9. doi: 10.1038/cdd.2010.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giannoni E, Buricchi F, Raugei G, Ramponi G, Chiarugi P. Intracellular reactive oxygen species activate Src tyrosine kinase during cell adhesion and anchorage-dependent cell growth. Mol Cell Biol. 2005;25:6391–403. doi: 10.1128/MCB.25.15.6391-6403.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee SR, Yang KS, Kwon J, Lee C, Jeong W, Rhee SG. Reversible inactivation of the tumor suppressor PTEN by H2O2. J Biol Chem. 2002;277:20336–42. doi: 10.1074/jbc.M111899200. [DOI] [PubMed] [Google Scholar]
- 19.Zhu P, Tan MJ, Huang RL, Tan CK, Chong HC, Pal M, et al. Angiopoietin-like 4 protein elevates the prosurvival intracellular O2(-):H2O2 ratio and confers anoikis resistance to tumors. Cancer Cell. 2011;19:401–15. doi: 10.1016/j.ccr.2011.01.018. [DOI] [PubMed] [Google Scholar]
- 20.Tan EHP, Sng MK, How ISB, Chan JSK, Chen J, Tan CK, et al. ROS release by PPARbeta/delta-null fibroblasts reduces tumor load through epithelial antioxidant response. Oncogene. 2018;37:2067–78. doi: 10.1038/s41388-017-0109-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reiners JJ, Jr, Nesnow S, Slaga TJ. Murine susceptibility to two-stage skin carcinogenesis is influenced by the agent used for promotion. Carcinogenesis. 1984;5:301–7. doi: 10.1093/carcin/5.3.301. [DOI] [PubMed] [Google Scholar]
- 22.Abel EL, Angel JM, Kiguchi K, DiGiovanni J. Multi-stage chemical carcinogenesis in mouse skin: fundamentals and applications. Nat Protoc. 2009;4:1350–62. doi: 10.1038/nprot.2009.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Honda H, Fujimoto M, Serada S, Urushima H, Mishima T, Lee H, et al. Leucine-rich alpha-2 glycoprotein promotes lung fibrosis by modulating TGF-beta signaling in fibroblasts. Physiol Rep. 2017;5:e13556. doi: 10.14814/phy2.13556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nisimoto Y, Diebold BA, Cosentino-Gomes D, Lambeth JD. Nox4: a hydrogen peroxide-generating oxygen sensor. Biochemistry. 2014;53:5111–20. doi: 10.1021/bi500331y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vomund S, Schafer A, Parnham MJ, Brune B, von Knethen A. Nrf2, the master regulator of anti-oxidative responses. Int J Mol Sci. 2017;18:2772. doi: 10.3390/ijms18122772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kwon J, Lee SR, Yang KS, Ahn Y, Kim YJ, Stadtman ER, et al. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc Natl Acad Sci USA. 2004;101:16419–24. doi: 10.1073/pnas.0407396101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Giannoni E, Chiarugi P. Redox circuitries driving Src regulation. Antioxid Redox Signal. 2014;20:2011–25. doi: 10.1089/ars.2013.5525. [DOI] [PubMed] [Google Scholar]
- 28.Ming M, He YY. PTEN: new insights into its regulation and function in skin cancer. J Invest Dermatol. 2009;129:2109–12. doi: 10.1038/jid.2009.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matsumoto T, Jiang J, Kiguchi K, Ruffino L, Carbajal S, Beltran L, et al. Targeted expression of c-Src in epidermal basal cells leads to enhanced skin tumor promotion, malignant progression, and metastasis. Cancer Res. 2003;63:4819–28. [PubMed] [Google Scholar]
- 30.Burgoyne JR, Oviosu O, Eaton P. The PEG-switch assay: a fast semi-quantitative method to determine protein reversible cysteine oxidation. J Pharm Toxicol Methods. 2013;68:297–301. doi: 10.1016/j.vascn.2013.07.001. [DOI] [PubMed] [Google Scholar]
- 31.Vazquez F, Grossman SR, Takahashi Y, Rokas MV, Nakamura N, Sellers WR. Phosphorylation of the PTEN tail acts as an inhibitory switch by preventing its recruitment into a protein complex. J Biol Chem. 2001;276:48627–30. doi: 10.1074/jbc.C100556200. [DOI] [PubMed] [Google Scholar]
- 32.Vazquez F, Ramaswamy S, Nakamura N, Sellers WR. Phosphorylation of the PTEN tail regulates protein stability and function. Mol Cell Biol. 2000;20:5010–8. doi: 10.1128/mcb.20.14.5010-5018.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Knatko EV, Higgins M, Fahey JW, Dinkova-Kostova AT. Loss of Nrf2 abrogates the protective effect of Keap1 downregulation in a preclinical model of cutaneous squamous cell carcinoma. Sci Rep. 2016;6:25804. doi: 10.1038/srep25804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rolfs F, Huber M, Kuehne A, Kramer S, Haertel E, Muzumdar S, et al. Nrf2 activation promotes keratinocyte survival during early skin carcinogenesis via metabolic alterations. Cancer Res. 2015;75:4817–29. doi: 10.1158/0008-5472.CAN-15-0614. [DOI] [PubMed] [Google Scholar]
- 35.Boukamp P. Non-melanoma skin cancer: what drives tumor development and progression? Carcinogenesis. 2005;26:1657–67. doi: 10.1093/carcin/bgi123. [DOI] [PubMed] [Google Scholar]
- 36.Matsumura Y, Ananthaswamy HN. Short-term and long-term cellular and molecular events following UV irradiation of skin: implications for molecular medicine. Expert Rev Mol Med. 2002;4:1–22. doi: 10.1017/S146239940200532X. [DOI] [PubMed] [Google Scholar]
- 37.Sahebkar A, Chew GT, Watts GF. New peroxisome proliferator-activated receptor agonists: potential treatments for atherogenic dyslipidemia and non-alcoholic fatty liver disease. Expert Opin Pharmacother. 2014;15:493–503. doi: 10.1517/14656566.2014.876992. [DOI] [PubMed] [Google Scholar]
- 38.Bility MT, Devlin-Durante MK, Blazanin N, Glick AB, Ward JM, Kang BH, et al. Ligand activation of peroxisome proliferator-activated receptor beta/delta (PPAR beta/delta) inhibits chemically induced skin tumorigenesis. Carcinogenesis. 2008;29:2406–14. doi: 10.1093/carcin/bgn219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Osterreicher CH, Penz-Osterreicher M, Grivennikov SI, Guma M, Koltsova EK, Datz C, et al. Fibroblast-specific protein 1 identifies an inflammatory subpopulation of macrophages in the liver. Proc Natl Acad Sci USA. 2011;108:308–13. doi: 10.1073/pnas.1017547108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Menegon S, Columbano A, Giordano S. The dual roles of NRF2 in cancer. Trends Mol Med. 2016;22:578–93. doi: 10.1016/j.molmed.2016.05.002. [DOI] [PubMed] [Google Scholar]
- 41.Huang JJ, Blobe GC. Dichotomous roles of TGF-beta in human cancer. Biochem Soc Trans. 2016;44:1441–54. doi: 10.1042/BST20160065. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets for microarray analysis during the current study are available through the Gene Expression Omnibus Series accession number GSE71419. The data in Fig. S4 are in whole based upon data generated by the TCGA Research Network (http://cancergenome.nih.gov/).