

Abstract
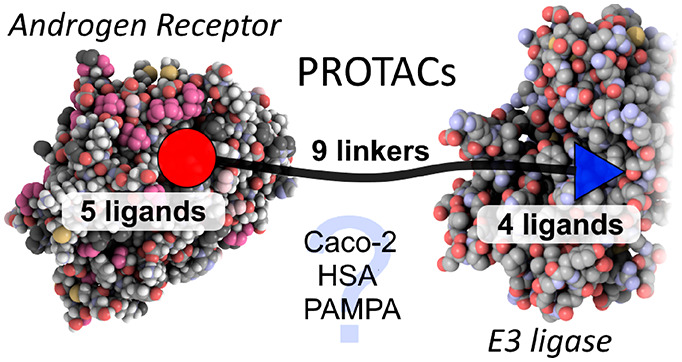
Bifunctional molecules known as PROTACs simultaneously bind an E3 ligase and a protein of interest to direct ubiquitination and clearance of that protein, and they have emerged in the past decade as an exciting new paradigm in drug discovery. In order to investigate the permeability and properties of these large molecules, we synthesized two panels of PROTAC molecules, constructed from a range of protein-target ligands, linkers, and E3 ligase ligands. The androgen receptor, which is a well-studied protein in the PROTAC field was used as a model system. The physicochemical properties and permeability of PROTACs are discussed.
Keywords: PROTACs, drug discovery, androgen receptor, medicinal chemistry
Chemically induced, targeted protein degradation is an emerging pharmacological strategy for manipulation of intracellular protein levels.1 One way this has been achieved is through the design of bifunctional molecules that simultaneously bind to a protein of interest and an E3 ubiquitin ligase. By bringing these two proteins into proximity, PROteolysis TArgeting Chimeras (PROTACs) facilitate the ubiquitinylation and proteasomal degradation of the protein of interest. Early PROTACs relied on short peptide sequences to bind the E3 ligase protein, and their low permeability necessitated microinjection into cells;2 however, recent identification of small molecule ligands for E3 ligases has opened the possibility of cell-permeable nonpeptidic PROTACs. The ability to degrade the entire protein in a cell, rather than simply inhibit the catalytic domain, distinguishes PROTACs from traditional small-molecule inhibitors. Further, the removal of additional roles of the protein, such as complex formation and scaffolding with other proteins, means that PROTACs can access unique pharmacology. PROTACs are also potentially catalytic; each molecule can direct degradation of multiple target proteins. PROTACs have been developed for numerous protein classes, including nuclear receptors,3 epigenetic factors,4 and kinases.5 These compounds have been used to study the effect of enforced degradation of the target protein in a number of cell lines and also in in vivo studies.6,7 While this work has the potential to offer a new pharmacological paradigm, clinical utility is yet to be demonstrated and a number of challenges still remain.8 A recent in vivo study of a potent PROTAC (DC50 0.3 nM) indicated that despite an impressive systemic knock-down of the desired FKBP12 protein, the brain was the only tissue analyzed where protein levels were unaffected, indicating that the blood–brain barrier may intrinsically present a greater challenge to PROTACs than other biological membranes.7
PROTAC molecules are rule-breaking in respect to traditional medicinal chemistry guidelines;8 they possess high molecular weight and high TPSA.9 Permeability is key for activity against intracellular targets, and in general, permeability drops off drastically with increasing molecular weight. Yet despite this, some impressive cell potencies for PROTACs have been reported.6 Other classes of small-molecules, also beyond the rule of 5, have been reported to have high cell permeability and oral bioavailability.10 Notably, cyclic peptides and natural products such as rifamycins are thought to adopt conformations that allow formation of intramolecular hydrogen bonds, reducing the effective polarity of the molecules and allowing passage through membranes.11,12 We hypothesized that in a similar fashion, it might be possible for PROTAC molecules to possess rule-breaking permeability, either by linker-mediated conformational collapse in water or by the two functional ends of the PROTAC migrating through a membrane in a pseudoindependent fashion by virtue of the length and flexibility of the linker so disguising their high molecular volume. PROTAC molecules are typically composed of two rigid protein-binding small molecules, connected by a linker. As we focus on flexible linkers in this work, further linker types with more rigidity remain to be explored. Recently Foley et al. have published observations on permeability using a chloroalkane penetration assay, focusing on the permeability of a chloroalkane modified BRD4 PROTAC.13 A wider study to evaluate the impact of linker, protein–ligand, and E3 ligase ligand on the permeability of PROTACs is warranted.
In our study, the androgen receptor (AR) was selected as a model system. A panel of selective AR modulators (SARMs) have previously been developed, and many PROTACs for the AR protein have also been reported, employing a variety of E3 ligase ligands.2,3,14,15 As such it is a derisked, well-studied system. The AR is a DNA-binding transcription factor nuclear hormone receptor that, through binding to the endogenous ligand testosterone, mediates growth factor and cell-signaling dependent gene expression. The ligand binding domain (LBD) provides a defined binding pocket for endogenous steroidal ligands including testosterone. A number of SARMs are in clinical trials for a variety of indications.14,16 Furthermore, polyglutamine expansion of the AR causes the neuromuscular disease spinal and bulbar muscular atrophy (SBMA or Kennedy’s disease).17 A PROTAC designed to catalyze the degradation of this neurotoxic form of AR could offer a strategy for intervention in this disease.
In designing a set of PROTACs, the three components of the PROTAC molecule were systematically varied; the AR ligand, the E3 ubiquitin ligase ligand, and the linker region. To select suitable ligands for the AR, SARMs were identified that were predicted to have good CNS permeability using MPO scoring.18 SARMs 1–5 all bind in the LBD of the AR (Figure 1A). An analysis of the binding modes of available crystal structures reveals SARMs present a similar vector to exit the AR binding site, in a position that has been successfully exploited by previously reported AR PROTACs.3 The structures of AR ligands 1–5 are shown in Figure 1B. SARM 1 is a close analogue of the clinical compounds andarine and ostarine. The hydantoin SARM 2 possesses high affinity for AR (EC50 1.6 nM).19 The thiohydantoin derivative 3 has been developed into the clinical compound enzalutamide and incorporated into PROTACs.20 Pyrrolidine-based SARM 4 represents a very ligand-efficient class of modulators, of which Ligandrol is a compound in clinical development.21 The cyclobutane SARM 5 has also been previously reported.22
Figure 1.

(A) AR ligand 2 in complex with AR ligand binding domain (pdb id: 3v49). The linker attachment point is circled in red. (B) Structures of compounds 1–5, derived from AR ligands, for incorporation into PROTACs. The linker attachment point is indicated in red. (C) Proteolysis directing groups 6–9; the attachment point when incorporated into PROTACs is indicated in blue. (D) Lenalidomide bound to Cereblon E3 ubiquitin ligase (pdb id: 4ci2). The linker attachment point is circled in blue. (E) General AR PROTAC structure of PROTAC Sets 1 and 2.
X-ray crystal structures of SARMs bound to AR reveal the ligand completely enclosed by helix 12 (Figure 2A). In order for AR PROTACs to bind and the linker to exit the AR protein, some protein conformational change must occur. Gryder et al. have previously reported a set of AR-binding bifunctional molecules that also bind histone deacetylases (HDACs) and suggest that helix 12 (H12) forms a “lid” over SARMs, that can move to accommodate extended AR ligands.23 Indeed, the related estrogen receptor (ER) demonstrates large changes in the conformation of H12 when bound to an antagonist;24 hence, we created a homology model of the AR with an H12-open conformation, based upon an H12-open ER protein complex (Figure 2B and Supporting Information). Docking PROTACs into this model successfully reproduced the binding mode of the SARM and allowed the linker to extend past H12 into the solvent, permitting a rational design of linker length and linker–SARM attachment points.
Figure 2.
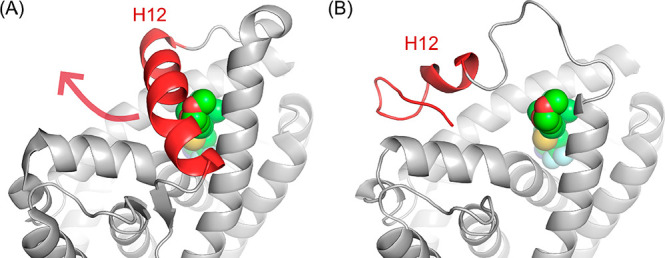
(A) Helix 12 (H12, red) of AR protein (pdb id: 2pnu) occludes the entrance to the ligand-binding domain. The docked small molecule 3b (Figure S1) is indicated by green spheres. (B) A homology model of AR with helix-12 in an open conformation (red helix) was generated. The remodeled helix 12 allows access from the ligand-binding domain to the bulk solvent and accommodation of PROTAC linkers.
Only a limited number of E3 ligases have been targeted by PROTAC molecules, though recent studies have focused on expanding the repertoire.25 We employed four different proteolysis targeting groups 6–9, with distinct physicochemical properties (Figure 1C). The immunomodulatory drugs thalidomide, and analogues pomalidamide and lenalidomide, have been identified as ligands of the E3 ligase Cereblon (Figure 1D), and structural analogues 6 and 7 have been successfully incorporated into PROTACs.26 Small-molecule ligands of the von Hippel Lindau (VHL) E3 ligase, such as 9, have also been developed and incorporated into PROTACs.4−6 Another approach to chemically target protein degradation has been to covalently27 or noncovalently28 associate a hydrophobic tag with a protein and thus promote its clearance. It has been suggested in the context of AR clearance that the presentation of the hydrophobic adamantane moiety 8 on the AR surface is recognized as unfolded protein by the Hsp70/CHIP system, leading to AR clearance.28
For the first half of this work, a focused set of PROTACs (PROTAC Set 1, Figures 1E and S2) was synthesized by linking AR ligands and smaller E3 ligands with a PEG linker (10–19, Schemes S3 and S4), predicted by calculations to have a neutral effect on the logD (Figure S4). In the second half of the study, a further set of compounds (PROTAC Set 2, Figures 1E and S3) focused on the VHL E3 ligase was synthesized (20a–i, Scheme S6). In this set, the linker composition was varied to evaluate the impact of linker on permeability and functional activity. Fluorine was also incorporated into the linker, which has not been reported in PROTAC linkers previously. The effects of addition of fluorine may be wide-ranging, potentially altering properties such as the electrostatics of the linker, interaction with proteins, linker conformation,29 permeability and microsomal stability. All PROTAC structures are shown in full in the Supporting Information (Figures S2 and S3).
With both sets of PROTACs in hand, the PEG-linked PROTACs (Set 1: 10–19, Figure 3) and the linker-varied VHL set (Set 2: 20a–i, Table 2), a panel of ADMET parameters was selected for their potential to impact cell activity; passive permeability (PAMPA), human serum albumin (HSA) binding, and chromLogD7.4. PROTACs 10–19 (Set 1, Figure 3) represent a model set of PROTACs with physicochemical properties that minimize molecular weight and TPSA and are not atypical to other PROTACs in terms of these parameters. The chosen AR ligands are small and potent, the proteolysis directing groups have low molecular weight, and the PEG-linker represents a typical reasonable linker length and distribution of polar atoms.3,22 The PAMPA Papp for the AR ligands gives a wide range of permeability: low for ligand 1 (1.4 × 10–6 cm s–1) and high for ligand 4 (13.3 × 10–6 cm s–1) (Figure 3A). Given the reported activity of PROTACs in the literature, reasonable permeability for this PROTAC set might be expected, especially when combining the most permeable AR ligand and E3 ligase ligand. However, almost without exception, the permeability of the final PROTAC molecules in the PAMPA assay was very low and below the limit of quantification (Figure 3A and Table 2). It is apparent that the high molecular weight and high TPSA, which is intrinsic to PROTAC molecules, is driving poor permeability for this class of molecules in this assay. Recovery in the PAMPA assay was moderate, i.e. 40–80% in most cases (Table S2). Of note is PROTAC 19, which combines ligandrol derivative 4 and adamantyl degron 8 and has a PAMPA permeability of 2.3 × 10–6 cm s–1, the highest PAMPA permeability measured for our bifunctional molecules (Figure 3A). The high logD for this PROTAC, a feature of PROTACs with the lipophilic adamantyl headgroup, presumably contributes to the moderate permeability (Figure 3C).
Figure 3.

Structure, biophysical properties and biological activities of PEG-linked PROTAC Set 1. Coloring is added to aid visualization. (A) The general structure of a set of PROTACs (10–19, Set 1) is shown in the top left, comprised of AR ligands 1–4, proteolysis directing groups 6–8, and a PEG-linker. The PAMPA permeability for compounds 1–4 and 6–8 is shown in the outer edge of the table. The main body of the table shows the PROTAC compound number in bold (10–19), composed from AR ligand 1–4 and proteolysis directing group 6–8, followed by PAMPA permeability. The same table format is used in (B–C). BLQ = Below limit of quantification. (B) HSA % free determined by HPLC and as described in methods section. (C) chromLogD7.4 determined by HPLC as described in the methods section.
Table 2. Structure, Biophysical Properties, and Permeability of VHL-Targeting PROTACs (PROTAC Set 2).
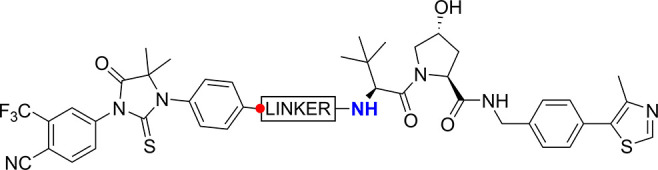

HSA % free, determined by HPLC method.
chromLogD7.4, determined by HPLC method.
PAMPA permeability/10–6 cm s–1.
Permeability/10–6 cm s–1.
Efflux Ratio. ND = Not determined.
Given the low passive permeabilities measured in the PAMPA assay, we also tested a selection of PROTAC molecules for bidirectional permeability in Caco-2 cells (Table 1 and Table 2). Briefly, this more biologically relevant assay uses a polarized monolayer of Caco-2 cells and hence allows assessment of permeability in two opposing directions (“A2B” and “B2A”), also permitting an assessment of active transport from the ratio of directional permeabilities.30 Typical thresholds for permeability classification are low, <1.0 × 10–6 cm s–1, medium, (1–5) × 10–6 cm s–1, and high, >5 × 10–6 cm s–1.31 While Caco-2 permeability is a useful measurement, in particular to predict oral bioavailability, inference of efflux ratios in other cell types may be complicated by different transporter profiles in different cell types.32
Table 1. Summary of PAMPA and Caco-2 Permeability of Selected PROTACs with Varying Proteolysis Directing Group.


Permeability/10–6 cm s–1.
Efflux ratio.
From the Caco-2 data it is possible to make comparisons to discern the effect of the proteolysis directing group in the context of AR ligand 3 and a PEG-linker (Table 1). Interestingly, cereblon ligand-containing PROTAC 14 has the best A2B permeability measured at 1.7 × 10–6 cm s–1. The B2A rate is high for this compound, 14.1 × 10–6 cm s–1, clearly indicating that transporter efflux is an issue (efflux ratio = 8.4). Replacing the cereblon ligand with the small, hydrophobic adamantane degron in PROTAC 18 leads to a 10-fold decrease in A2B and a 64-fold decrease in B2A (0.15 and 0.22 × 10–6 cm s–1, respectively) and no evidence of transporter efflux (ER = 1.5). Substituting with the larger, more polar VHL ligand in PROTAC 20d gives an A2B permeability below the limit of quantification for this compound, but as the B2A rate is 9.6 × 10–6 cm s–1, it is evident that an efflux issue is present (ER > 12).
The effect of the linker on Caco-2 permeability was explored with a set of PROTACs from Set 2 (VHL ligand containing PROTACs) (Table 2). For the PROTACs studied, A2B either could be quantified as low (<1.0 × 10–6 cm s–1) or could not be measured at all. Interestingly, a range of B2A rates were observed. For PROTAC 20b, A2B and B2A were measured as 0.35 and 0.24 × 10–6 cm s–1, respectively, indicating no significant transporter efflux. It is noteworthy that the linkers of PROTACs 20b, 20c, and 20g bearing different numbers of fluorine atoms lead to PROTACs that are all rather similar in terms of permeability; A2B and B2A rates are low. Contrastingly, PROTAC 20d demonstrates a high sensitivity to the change to a PEG-linker, B2A is high (8.6 × 10–6 cm s–1) and the efflux ratio is high (>12). In summary, low passive permeability in PAMPA assays is typically observed for PROTACs, but a marked structural influence of the linker and E3 ligase ligand on A2B, B2A, and efflux ratio in Caco-2 cells can be observed. The recovery of compounds in the Caco-2 assay was reasonable, 60–100% (Table S3). Of note, all PROTACs were relatively unbound to HSA protein, between 6 and 15% (Figure 3B and Table 2). More work is required to investigate this beyond the compound set studied here.
The biological efficacy of the PROTACs to degrade endogenous levels of the AR receptor was assessed using a Western blotting technique in LNCaP cells. LNCaP (androgen-dependent human prostate adenocarcinoma) cells are a well-characterized model for AR expression and are commonly used in AR PROTAC studies. For effective clearance, a stable tertiary complex must form and surface lysines on the target protein must be presented in the correct manner for efficient ubiquitin transfer.25 Briefly, PROTAC activities can generally be described by two numbers: DCMAX is the percentage of protein clearance observed, and the DC50 is the concentration required to clear half of the DCMAX. Previously reported22 PROTAC 20a was chosen as a positive control for the clearance assay (Figure 4A). In general our PROTACs displayed mostly weak (DCMAX < 30%) or no clearance effects in the cell assay (not shown). To disperse the concern that the generally poor activity observed in cells might be due to the chosen attachment points in the PROTACs preventing AR binding, representative PROTACs 20a and 20i were tested for AR binding using a radioligand displacement assay (Figure S5). Both PROTACs retained low nM affinity for the androgen receptor, despite the different AR ligands, the different linker, and the attachment of the VHL ligand. This is supportive of our docking model (Figure 2B) and indicates that the phenolic attachment point of the AR ligands in our PROTACs is compatible with AR binding in vitro. The amide attachment point of the VHL ligand has been well validated in other PROTAC studies.4−6
Figure 4.

(A) AR clearance activity of previously reported22 PROTAC 20a. (B) AR clearance activity of PROTACs 14 and 15. Plots show normalized AR protein levels as a function of PROTAC concentration. PROTAC 14 is inactive in this cell assay. A minor change of the Cereblon ligand leads to higher activity of PROTAC 15: a DCMAX of 33% and a DC50 of 10 nM. N = 5 (or more) independent experiments performed in triplicate (individual data points shown). Statistical significance by ANOVA (Dunnett’s) **** = p < 0.0001.
The activity data for two structurally related PROTACs, 14 and 15, are shown in Figure 4B and serve to exemplify how small changes in PROTACs can lead to profound changes in cell activity. PROTAC 15 is active (DCMAX = 33%, DC50 = 10 nM), but PROTAC 14 is inactive, despite possessing the highest A2B permeability measured, albeit with a high efflux ratio. The only structural difference between the two PROTACs is whether the E3 ligase ligand possesses a carbonyl group in the cereblon-binding moiety. Interestingly, Wang et al. have reported similar large activity differences regarding this carbonyl group in a set of BET PROTACs.33 There are many reasons why PROTAC AR activity data may show idiosyncratic SAR, beyond permeability. In particular, recent papers demonstrate that in the case of cereblon-binding PROTACs, small changes in PROTAC chemical structure can significantly reduce the intended activity of a bona fide PROTAC and induce off-target GSPT1 degradation, thereby altering the PROTAC’s mode of action.34
In summary, even our simplest PROTAC molecules formed from moderately sized linkers connecting relatively small AR ligands and E3 ligase ligands possess low PAMPA and Caco-2 A2B permeability. If this is a general observation, it is clear that PROTAC permeability is not “rule-breaking”. Caco-2 permeability data sheds a little more light on the permeability profiles of some PROTACs, and evidence of structure-dependent engagement with transport efflux proteins is observed. We recommend that Caco-2 permeability therefore, rather than PAMPA permeability, might be a more useful measurement. A recent publication from Cantrill et al. supports these conclusions.35 Broadly, a catalytic mode of action is supported by our permeability data set, that substoichiometric levels of PROTACs in a cell can catalyze the clearance of a target protein population. Some minimal permeability threshold for PROTACs may exist but is likely to be much lower than is usually recognized for Rule of 5-compliant small molecules. The chemical stability of a PROTAC in a cell over time will also be important for the continued degradation of protein. Recently, methodology to measure concentrations of small molecules in cellular compartments has become increasingly utilized and has already been applied to PROTACs.36 The use of this technology has emerged as a potentially useful tool to provide insights into PROTAC uptake into cells, though interpretation might be complicated by compound adhering to the outer cell membrane. To investigate the real time clearance of proteins, Riching et al. have recently reported methods to monitor PROTAC-induced protein degradation.37 Understanding the cellular uptake of a PROTAC, its stability in the cell over time, and the kinetics of protein clearance will be critical in informing design of better PROTACs. By better understanding the connection between PROTAC structure and cellular efficacy, we will be able to rationally design better molecules and translate PROTAC molecules into the clinic more efficiently.
Acknowledgments
The authors wish to thank Simon Edwards for synthesis of compound 2 and David Rubinsztein for helpful discussions.
Glossary
Abbreviations
- AR
androgen receptor
- PROTACs
PROteolysis TArgeting Chimeras
- PAMPA
parallel artificial membrane permeability
- TPSA
total polar surface area
- A2B
Apical to Basolateral
- B2A
Basolateral to Apical
- BLQ
Below Limit of Quantification
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00194.
General experimental information; organic synthesis and compound characterization, AR affinity data, logD and HSA data and protocol, cell activity protocol, and AR binding assay (PDF)
Author Present Address
∥ (T.M.) The Francis Crick Institute, 1 Midland Road, London, NW1 1AT, United Kingdom
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was funded by Alzheimer’s Research UK (grant: ARUK-2015DDI-CAM), with support from the ALBORADA Trust. The ALBORADA Drug Discovery Institute is core funded by Alzheimer’s Research UK (registered charity No. 1077089 and SC042474).
The authors declare no competing financial interest.
Supplementary Material
References
- Lai A. C.; Crews C. M. Induced protein degradation: an emerging drug discovery paradigm. Nat. Rev. Drug Discovery 2017, 16 (2), 101–114. 10.1038/nrd.2016.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneekloth J. S. Jr.; Fonseca F. N.; Koldobskiy M.; Mandal A.; Deshaies R.; Sakamoto K.; Crews C. M. Chemical genetic control of protein levels: selective in vivo targeted degradation. J. Am. Chem. Soc. 2004, 126 (12), 3748–54. 10.1021/ja039025z. [DOI] [PubMed] [Google Scholar]
- Schneekloth A. R.; Pucheault M.; Tae H. S.; Crews C. M. Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorg. Med. Chem. Lett. 2008, 18 (22), 5904–8. 10.1016/j.bmcl.2008.07.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zengerle M.; Chan K. H.; Ciulli A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol. 2015, 10 (8), 1770–7. 10.1021/acschembio.5b00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan L.; Gray N. S. When Kinases Meet PROTACs. Chin. J. Chem. 2018, 36 (10), 971–977. 10.1002/cjoc.201800293. [DOI] [Google Scholar]
- Bondeson D. P.; Mares A.; Smith I. E.; Ko E.; Campos S.; Miah A. H.; Mulholland K. E.; Routly N.; Buckley D. L.; Gustafson J. L.; Zinn N.; Grandi P.; Shimamura S.; Bergamini G.; Faelth-Savitski M.; Bantscheff M.; Cox C.; Gordon D. A.; Willard R. R.; Flanagan J. J.; Casillas L. N.; Votta B. J.; den Besten W.; Famm K.; Kruidenier L.; Carter P. S.; Harling J. D.; Churcher I.; Crews C. M. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 2015, 11 (8), 611–7. 10.1038/nchembio.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X. Y.; Wang J.; Yao X.; Zheng W.; Mao Y.; Lan T. L.; Wang L. G.; Sun Y. H.; Zhang X. Y.; Zao Q. Y.; Zhao J. G.; Xiao R. P.; Zhang X. Q.; Ji G. J.; Rao Y.. A chemical approach for global protein knockdown from mice to non-human primates. Cell Discovery 2019, 5, 10.1038/s41421-018-0079-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churcher I. Protac-Induced Protein Degradation in Drug Discovery: Breaking the Rules or Just Making New Ones?. J. Med. Chem. 2018, 61 (2), 444–452. 10.1021/acs.jmedchem.7b01272. [DOI] [PubMed] [Google Scholar]
- Edmondson S. D.; Yang B.; Fallan C. Proteolysis targeting chimeras (PROTACs) in ’beyond rule-of-five’ chemical space: Recent progress and future challenges. Bioorg. Med. Chem. Lett. 2019, 29 (13), 1555–1564. 10.1016/j.bmcl.2019.04.030. [DOI] [PubMed] [Google Scholar]
- Doak B. C.; Over B.; Giordanetto F.; Kihlberg J. Oral druggable space beyond the rule of 5: insights from drugs and clinical candidates. Chem. Biol. 2014, 21 (9), 1115–42. 10.1016/j.chembiol.2014.08.013. [DOI] [PubMed] [Google Scholar]
- Agrawal S.; Ashokraj Y.; Bharatam P. V.; Pillai O.; Panchagnula R. Solid-state characterization of rifampicin samples and its biopharmaceutic relevance. Eur. J. Pharm. Sci. 2004, 22 (2–3), 127–44. 10.1016/j.ejps.2004.02.011. [DOI] [PubMed] [Google Scholar]
- Matsson P.; Kihlberg J. How Big Is Too Big for Cell Permeability?. J. Med. Chem. 2017, 60 (5), 1662–1664. 10.1021/acs.jmedchem.7b00237. [DOI] [PubMed] [Google Scholar]
- Foley C. A.; Potjewyd F.; Lamb K. N.; James L. I.; Frye S. V. Assessing the Cell Permeability of Bivalent Chemical Degraders Using the Chloroalkane Penetration Assay. ACS Chem. Biol. 2020, 15 (1), 290–295. 10.1021/acschembio.9b00972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan R.; Mohler M. L.; Bohl C. E.; Miller D. D.; Dalton J. T. Selective androgen receptor modulators in preclinical and clinical development. Nucl. Recept. Signaling 2008, 6, e010 10.1621/nrs.06010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X.; Wang C.; Qin C.; Xiang W.; Fernandez-Salas E.; Yang C. Y.; Wang M.; Zhao L.; Xu T.; Chinnaswamy K.; Delproposto J.; Stuckey J.; Wang S. Discovery of ARD-69 as a Highly Potent Proteolysis Targeting Chimera (PROTAC) Degrader of Androgen Receptor (AR) for the Treatment of Prostate Cancer. J. Med. Chem. 2019, 62 (2), 941–964. 10.1021/acs.jmedchem.8b01631. [DOI] [PubMed] [Google Scholar]
- Cohen S.; Nathan J. A.; Goldberg A. L. Muscle wasting in disease: molecular mechanisms and promising therapies. Nat. Rev. Drug Discovery 2015, 14 (1), 58–74. 10.1038/nrd4467. [DOI] [PubMed] [Google Scholar]
- La Spada A. R.; Wilson E. M.; Lubahn D. B.; Harding A. E.; Fischbeck K. H. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 1991, 352 (6330), 77–9. 10.1038/352077a0. [DOI] [PubMed] [Google Scholar]
- Wager T. T.; Hou X.; Verhoest P. R.; Villalobos A. Moving beyond rules: the development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem. Neurosci. 2010, 1 (6), 435–49. 10.1021/cn100008c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nique F.; Hebbe S.; Peixoto C.; Annoot D.; Lefrancois J. M.; Duval E.; Michoux L.; Triballeau N.; Lemoullec J. M.; Mollat P.; Thauvin M.; Prange T.; Minet D.; Clement-Lacroix P.; Robin-Jagerschmidt C.; Fleury D.; Guedin D.; Deprez P. Discovery of diarylhydantoins as new selective androgen receptor modulators. J. Med. Chem. 2012, 55 (19), 8225–35. 10.1021/jm300249m. [DOI] [PubMed] [Google Scholar]
- Crews C. M.; Crew A. P.; Dong H.; Wang J.; Qian Y.; Siu K.; Jin M.. Imide-based modulators of proteolysis and associated methods of use. WO/2016/197032, 2016.
- Basaria S.; Collins L.; Dillon E. L.; Orwoll K.; Storer T. W.; Miciek R.; Ulloor J.; Zhang A.; Eder R.; Zientek H.; Gordon G.; Kazmi S.; Sheffield-Moore M.; Bhasin S. The safety, pharmacokinetics, and effects of LGD-4033, a novel nonsteroidal oral, selective androgen receptor modulator, in healthy young men. J. Gerontol., Ser. A 2013, 68 (1), 87–95. 10.1093/gerona/gls078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meizhong J.; Crew A. P.; Dong H.; Wang J.; Siu K.; Ferraro C.; Chen X.; Qian Y.. Compounds and methods for the targeted degradation of the androgen receptor. WO/2016/118666, 2016.
- Gryder B. E.; Akbashev M. J.; Rood M. K.; Raftery E. D.; Meyers W. M.; Dillard P.; Khan S.; Oyelere A. K. Selectively targeting prostate cancer with antiandrogen equipped histone deacetylase inhibitors. ACS Chem. Biol. 2013, 8 (11), 2550–60. 10.1021/cb400542w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiau A. K.; Barstad D.; Loria P. M.; Cheng L.; Kushner P. J.; Agard D. A.; Greene G. L. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell 1998, 95 (7), 927–37. 10.1016/S0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- Ottis P.; Toure M.; Cromm P. M.; Ko E.; Gustafson J. L.; Crews C. M. Assessing Different E3 Ligases for Small Molecule Induced Protein Ubiquitination and Degradation. ACS Chem. Biol. 2017, 12 (10), 2570–2578. 10.1021/acschembio.7b00485. [DOI] [PubMed] [Google Scholar]
- Chessum N. E. A.; Sharp S. Y.; Caldwell J. J.; Pasqua A. E.; Wilding B.; Colombano G.; Collins I.; Ozer B.; Richards M.; Rowlands M.; Stubbs M.; Burke R.; McAndrew P. C.; Clarke P. A.; Workman P.; Cheeseman M. D.; Jones K. Demonstrating In-Cell Target Engagement Using a Pirin Protein Degradation Probe (CCT367766). J. Med. Chem. 2018, 61 (3), 918–933. 10.1021/acs.jmedchem.7b01406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tae H. S.; Sundberg T. B.; Neklesa T. K.; Noblin D. J.; Gustafson J. L.; Roth A. G.; Raina K.; Crews C. M. Identification of hydrophobic tags for the degradation of stabilized proteins. ChemBioChem 2012, 13 (4), 538–41. 10.1002/cbic.201100793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafson J. L.; Neklesa T. K.; Cox C. S.; Roth A. G.; Buckley D. L.; Tae H. S.; Sundberg T. B.; Stagg D. B.; Hines J.; McDonnell D. P.; Norris J. D.; Crews C. M. Small-Molecule-Mediated Degradation of the Androgen Receptor through Hydrophobic Tagging. Angew. Chem., Int. Ed. 2015, 54 (33), 9659–62. 10.1002/anie.201503720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiehoff C.; Rey Y.; Gilmour R. The Fluorine Gauche Effect: A Brief History. Isr. J. Chem. 2017, 57 (1–2), 92–100. 10.1002/ijch.201600038. [DOI] [Google Scholar]
- van Breemen R. B.; Li Y. Caco-2 cell permeability assays to measure drug absorption. Expert Opin. Drug Metab. Toxicol. 2005, 1 (2), 175–85. 10.1517/17425255.1.2.175. [DOI] [PubMed] [Google Scholar]
- Varma M. V.; Gardner I.; Steyn S. J.; Nkansah P.; Rotter C. J.; Whitney-Pickett C.; Zhang H.; Di L.; Cram M.; Fenner K. S.; El-Kattan A. F. pH-Dependent solubility and permeability criteria for provisional biopharmaceutics classification (BCS and BDDCS) in early drug discovery. Mol. Pharmaceutics 2012, 9 (5), 1199–212. 10.1021/mp2004912. [DOI] [PubMed] [Google Scholar]
- Hilgendorf C.; Ahlin G.; Seithel A.; Artursson P.; Ungell A. L.; Karlsson J. Expression of thirty-six drug transporter genes in human intestine, liver, kidney, and organotypic cell lines. Drug Metab. Dispos. 2007, 35 (8), 1333–40. 10.1124/dmd.107.014902. [DOI] [PubMed] [Google Scholar]
- Qin C.; Hu Y.; Zhou B.; Fernandez-Salas E.; Yang C. Y.; Liu L.; McEachern D.; Przybranowski S.; Wang M.; Stuckey J.; Meagher J.; Bai L.; Chen Z.; Lin M.; Yang J.; Ziazadeh D. N.; Xu F.; Hu J.; Xiang W.; Huang L.; Li S.; Wen B.; Sun D.; Wang S. Discovery of QCA570 as an Exceptionally Potent and Efficacious Proteolysis Targeting Chimera (PROTAC) Degrader of the Bromodomain and Extra-Terminal (BET) Proteins Capable of Inducing Complete and Durable Tumor Regression. J. Med. Chem. 2018, 61 (15), 6685–6704. 10.1021/acs.jmedchem.8b00506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Li Y.; Aguilar A.; Liu Z.; Yang C. Y.; Wang S. Simple Structural Modifications Converting a Bona fide MDM2 PROTAC Degrader into a Molecular Glue Molecule: A Cautionary Tale in the Design of PROTAC Degraders. J. Med. Chem. 2019, 62 (21), 9471–9487. 10.1021/acs.jmedchem.9b00846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantrill C.; Chaturvedi P.; Rynn C.; Petrig Schaffland J.; Walter I.; Wittwer M. B.. Fundamental aspects of DMPK optimization of targeted protein degraders. Drug Discovery Today, published online 13th March 2020; 10.1016/j.drudis.2020.03.012. [DOI] [PubMed] [Google Scholar]
- McCoull W.; Cheung T.; Anderson E.; Barton P.; Burgess J.; Byth K.; Cao Q.; Castaldi M. P.; Chen H.; Chiarparin E.; Carbajo R. J.; Code E.; Cowan S.; Davey P. R.; Ferguson A. D.; Fillery S.; Fuller N. O.; Gao N.; Hargreaves D.; Howard M. R.; Hu J.; Kawatkar A.; Kemmitt P. D.; Leo E.; Molina D. M.; O’Connell N.; Petteruti P.; Rasmusson T.; Raubo P.; Rawlins P. B.; Ricchiuto P.; Robb G. R.; Schenone M.; Waring M. J.; Zinda M.; Fawell S.; Wilson D. M. Development of a Novel B-Cell Lymphoma 6 (BCL6) PROTAC To Provide Insight into Small Molecule Targeting of BCL6. ACS Chem. Biol. 2018, 13 (11), 3131–3141. 10.1021/acschembio.8b00698. [DOI] [PubMed] [Google Scholar]
- Riching K. M.; Mahan S.; Corona C. R.; McDougall M.; Vasta J. D.; Robers M. B.; Urh M.; Daniels D. L. Quantitative Live-Cell Kinetic Degradation and Mechanistic Profiling of PROTAC Mode of Action. ACS Chem. Biol. 2018, 13 (9), 2758–2770. 10.1021/acschembio.8b00692. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.