

Abstract

Indoleamine-2,3-dioxygenase 1 (IDO1) inhibition and its combination with immune checkpoint inhibitors like pembrolizumab have drawn considerable attention from both academia and the pharmaceutical industry. Here, we describe the discovery of a novel class of highly potent IDO1 heme-displacing inhibitors featuring a unique bicyclo[1.1.1]pentane motif. Compound 1, evolving from an ALIS (automated ligand identification system) hit, exhibited excellent potency but lacked the desired pharmacokinetic profile due to extensive amide hydrolysis of the benzamide moiety. Replacing the central phenyl ring in 1 with a bicyclo[1.1.1]pentane bioisostere effectively circumvented the amide hydrolysis issue, resulting in the discovery of compound 2 with a favorable overall profile such as excellent potency, selectivity, pharmacokinetics, and a low predicted human dose.
Keywords: indoleamine-2,3-dioxygenase 1; IDO1; bicyclo[1.1.1]pentane; cancer immunotherapy
Research and development of immune checkpoint inhibitors as therapeutics for cancer treatment have evolved significantly in the past few years and become one of the frontiers in the field of cancer immunotherapy.1,2 For example, multiple monoclonal antibodies targeting either programmed cell death 1 (PD-1) receptor or its ligand PD-L1, including pembrolizumab, nivolumab, atezolizumab, avelumab, and durvalumab, have been recently approved by the FDA and have become increasingly powerful tools for clinical oncologists.3 This type of agent functions by recruiting the body’s immune system to fight against the growth of cancer cells, and these therapies have exhibited meaningful and durable clinical efficacy across multiple solid tumors.1,2 However, one of the biggest challenges facing immune checkpoint inhibition has been the low rate of response and often only a fraction of patients with a given type of cancer can benefit from these therapies.4 Local immunosuppression in the tumor microenvironment is believed to be one of the major perpetrators contributing to such resistance against immune checkpoint blockade.5
Indoleamine-2,3-dioxygenase 1 (IDO1) is an intracellular enzyme that catalyzes the initial and rate-limiting step of tryptophan catabolism, resulting in the formation of kynurenine and several other downstream metabolites.6 IDO1 has been shown to be upregulated in many different tumors. Overexpression and activation of IDO1 in the tumor microenvironment can lead to local immunosuppression due to accumulation of immunosuppressive kynurenine and its metabolites.7,8 In addition, IDO1 also promotes immune evasion through down-regulating the activity of natural killer cells.9,10 Thus, IDO1 inhibition and its combination with checkpoint inhibitors have attracted enormous attention across both academia and the pharmaceutical industry.11,12
A number of IDO1 inhibitors have been reported and several of them have entered into clinical trials including epacadostat and BMS-986205 (Figure 1).13,14 These two inhibitors represent two different modes of IDO1 inhibition: heme-binders and heme-displacers. While epacadostat binds directly and reversibly to the heme moiety present in the IDO1 active site,15 BMS-986205 competes with the heme moiety and binds to the apo form of the IDO1 protein.16 In a cellular assay, BMS-986205 was shown to be significantly more potent than epacadostat (IC50: 2.8 nM vs 12.5 nM in our Hela assay), suggesting that this mode of inhibition has the potential to achieve much higher level of IDO1 inhibition in the tumor. The greater potency of BMS-986205 is likely due to its larger interaction interface with IDO1 due to the absence of the bulky heme moiety.
Figure 1.

Examples of known IDO1 heme-binding and heme-displacing inhibitors.
The combination of epacadostat and pembrolizumab showed promising clinical benefit in early clinical developments. However, the early clinical results were not substantiated in a PhIII outcome trial in melanoma patients (ECHO-301/KEYNOTE-252). Multiple factors could have contributed to the PhIII failure, and insufficient IDO1 inhibition in the tumor microenvironment and potential induction of the aromatic hydrocarbon receptor (AhR) by epacadostat are potential leading causes.17,18 Thus, interest in this target persists, and several clinical trials are ongoing, particularly involving BMS-986205 and nivolumab in muscle invasive bladder cancer.19 It is thus highly desirable to discover and develop IDO1 inhibitors that can achieve unprecedented level of target engagement while maintaining a minimum potential for confounding off-target activities. We report herein the discovery and preclinical characterization of such potent heme-displacing inhibitors.
Previously, we reported a class of inhibitors featuring a unique amino-cyclobutane motif and a heme-binding mode similar to that of epacadostat.20 In addition, we also discovered another class of inhibitors that adopts a heme-displacing binding mode similar to that of BMS-986205.21 Starting from the identification of bisamide hit from ALIS (automated ligand identification system) screening,22,23 SAR exploration through parallel synthesis and structure-based design led to the discovery of compound 1 (compound 3 in ref (21) (Figure 2)). Compound 1 showed excellent potency in both cellular-based and whole blood assays. However, compound 1 suffered from poor metabolic stability in both in vitro and in vivo rat pharmacokinetic studies. Amide bond cleavage of the benzamide of the central aniline moiety was found to be the predominant metabolic pathway as indicated in metID studies. Different strategies were explored to address this issue including amide bioisosteres and saturation or replacement of the central phenyl aniline ring. Most modifications led to significant loss of cell potency or insufficient improvement in metabolic stability. Herein, we disclose our efforts in utilizing a bicyclo[1.1.1]pentane moiety as a bioisotere of the central phenyl ring in 1, which culminated in the discovery of lead compound 2 with excellent potency, selectivity, improved metabolic stability, excellent off-target profile, and a low predicted human dose.
Figure 2.

Strategy to mitigate metabolic stability by employing bicyclo[1.1.1] pentane as a bioisostere of phenyl ring.
Bicyclo[1.1.1]pentanes have been increasingly utilized as a saturated bioisostere of 1,4-disubstituted phenyl rings to successfully overcome different issues including metabolic stability, selectivity, solubility, and physiochemical properties.24−27 Molecular modeling indicated that compounds 3 and 4 could be well tolerated in the binding site of IDO1, therefore providing an opportunity to potentially mitigate amide hydrolysis of 1. It can also avoid the formation of the resulting aniline-moiety from amide cleavage, a known structural alert often associated with anilines such as bioactivation and genotoxicity (Figure 2).
To quickly test our hypothesis, we decided to synthesize compound 4 first based on starting material availability and by following a synthetic route as shown in Scheme 1. Starting from commercially available N-Boc-protected bicyclo[1.1.1] pentane methyl ester 5, the D-pocket28 arylamide moiety 6 was installed readily via two steps. Subsequent ester hydrolysis, Weinreb amide formation, and Grignard addition afforded ketone amide 7. One carbon elongation was achieved through the well-known Van Leusen reaction utilizing p-toluenesulphonylmethyl isocyanide (TOSMIC) as a one-carbon synthon and delivered compound 8.29 Conversion of cyanide 8 into the key carboxylic acid intermediate 9 was obtained in two steps under mild conditions. Finally, amide coupling delivered the desired compound 4, which was separated through chiral separation into its two enantiomers 4-a and 4-b.
Scheme 1. First Generation Synthesis of Bicyclo[1.1.1]pentane Bisamide and Profile of Compound 4-a.

Gratifyingly, compound 4-a showed very good potency in both cellular (HeLa IC50: 3.1 nM) and human whole blood (IC50: 121 nM) assays (Scheme 1). In contrast, its enantiomer 4-b was much less active (HeLa IC50: 230 nM). Encouraged by this promising result, we further investigated the metabolic stability of compound 4-a both in vitro and in vivo. Gratifyingly, compound 4-a exhibited a significantly improved stability in in vitro incubations in hepatocytes, which translated to a good overall pharmacokinetic profile in rat including low clearance, good elimination half-life, and excellent oral bioavailability. Compound 4-a also showed excellent passive permeability and a clean profile in a counter screen for common off-targets such as PXR activity and CYP inhibition. Compound 4-a also exhibited good selectivity for IDO1 over TDO (IC50 > 10 μM).
In addition, we also were able to obtain a cocrystal structure of compound 4-a with IDO1 protein (Figure 3). By comparing this structure with that of our previously reported benzamide analog of compound 1,21 we can see that both compounds bind very similarly, and consistently with modeling prediction. In addition, it can be expected that compound 4-a likely binds to a similar binding pocket as that of BMS-986205 based on the structural overlay we described previously.21 Both compounds 4-a and 1 form hydrogen bond interactions in the A-pocket region, from the amide NH directly to the side chain of Ser-167 and a water-bridged interaction between the amide carbonyl oxygen and the side chain of His-346. The fluoro-aniline moiety sits in the lipophilic A-pocket, which is surrounded by the side chains of Tyr-126, Phe-163, Phe-164, and Leu-234. On the opposite end of the molecule, the Cl-substituted arylamide moiety overlays closely in both compounds and lies in the lipophilic D-pocket,28 bound in part by the side chains of Ala-174, Phe-273, and Leu-342. There are two water-mediated hydrogen bond interactions from the aryl amide to Ser-267 and Arg-343. From this structure, the central phenyl or bicyclo[1.1.1]pentane moiety appears to serve mainly as a linker between the two aryl amide moieties, which explains the minimal change in potency when switching from phenyl for bicyclo[1.1.1]pentane.
Figure 3.
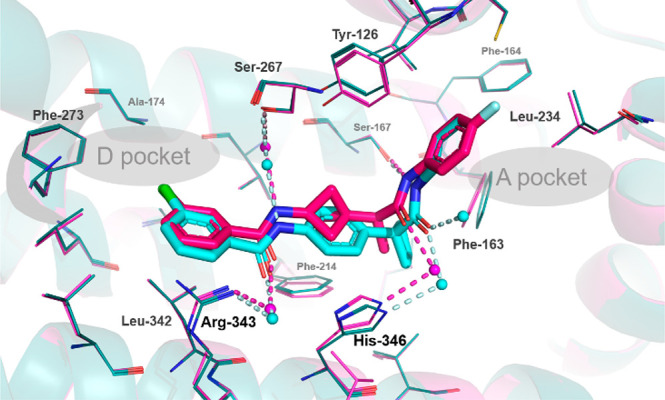
Overlay of IDO1 cocrystal structures of 4-a (PDB code 6WJY) (magenta) and aryl bisamide (PDB code 6V52) (cyan). Residues involved in direct or water-mediated hydrogen bond interactions with the compound are indicated, and their interactions are color-coded to match the structure. The general locations of pockets A and D are labeled.
Encouraged by these results, we conducted some additional exploration of SAR around 4-a to seek opportunities for further improvements (i.e., potency) while maintaining other good properties including metabolic stability, selectivity, etc. Our SAR optimization focused on three areas including alpha-Me replacement, and further modifications of both the A-pocket and D-pocket aryl amides.
First, we focused on exploring additional substitutions alpha to the aniline-based A-pocket amide. To accelerate this exploration, a more flexible and universal synthetic route was developed to allow quick access to several key intermediates (12–1, 12–2, and 12–3) (Scheme 2). Starting from commercially available bicyclo[1.1.1]pentane primary alcohol compound 10, one-carbon elongation was achieved through SN2 displacement of the corresponding mesylate with NaCN. Subsequent alkylation to introduce different substituents at the α position of the nitrile group was achieved through one pot deprotonation and quenching with various alkyl halides. Hydrolysis of the corresponding alkyl cyanides furnished the desired key carboxylic acid intermediates 12–1, 12–2, and 12–3. From these intermediates, the remaining syntheses were straightforward and proceeded smoothly through two amide coupling steps via intermediates (13–1, 13–2, 13–3) to afford the desired analogs 14–1, 14–2, and 14–3.
Scheme 2. Second Generation Synthetic Procedure for Bicyclo[1.1.1]pentane Bisamide Formation.

As shown in Table 1, bis-α-methyl analog 15 and α-cyclobutyl analog 3 did not show obvious advantage relative to compound 4-a. To avoid adding unproductive lipophilicity, we decided to focus our additional explorations only within the α-methyl context. A small set of analogs featuring different A-pocket arylamide moieties was explored. It was found that replacing the 4-fluoro substitution in 4-a with a 4-chloro substitution (16) resulted in a boost in potency in cellular assay (∼3×). However, this led to increased lipophilicity and no improvement in whole blood assay. Attempts to incorporate heteroatoms into the aryl ring to reduce AlogP led to a drop of potency, which is consistent with our previous observations21 and the lipophilic nature of the A-pocket.
Table 1. SAR Exploration of Alpha-Me and the A-Pocket Amide Replacement.

IC50 values are the mean of at least two independent assays, see Supporting Information for IC50 curves and standard deviation.
AlogP was calculated according to the method described in ref (30).
Rat in vitro unbound clearance.
At this point, we shifted focus to exploration of the D-pocket. First, a library of aryl amides with various small substituents on the aryl ring was investigated (Table 2). Incorporation of an additional fluoro-substituent was tolerated in general (18, 19, and 20) in terms of potency, but without additional benefit to metabolic stability. However, bis-fluoro substituted analog 2 showed some further improvement potency, in particular, in whole assay, while maintaining good metabolic stability. Incorporation of a heteroatom into the aryl ring (21) was disfavored in either potency or metabolic stability.
Table 2. SAR Exploration of Aryl Amides in D-Pocket.

IC50 values are the mean of at least two independent assays, see Supporting Information for IC50 curves and standard deviation.
AlogP was calculated according to the method described in ref (30).
Rat in vitro unbound clearance.
In addition, we also explored whether it was feasible to replace the amide with other functional groups such as carbamate or N-aryl moieties (Table 3). These compounds were synthesized readily through carbamate formation or N-arylation by utilizing the same amine intermediate used previously as shown in Scheme 2. Small cyclopropyl carbamate 22 showed much weaker activity in cellular assay. Increasing the steric bulkiness to i-Pr or cyclopentyl could improve potency but was accompanied by decreased metabolic stability. Several N-arylated analogs exhibited single digit nanomolar potency in cellular assay (25 and 26). However, both compounds suffered from inferior whole blood potency and poor metabolic stability.
Table 3. Different Functional Group Replacement in D-Pocket.

IC50 values are the mean of at least two independent assays, see Supporting Information for IC50 curves and standard deviation.
AlogP was calculated according to the method described in ref (30).
Rat in vitro unbound clearance.
By virtue of its overall favorable attributes, compound 2 was selected for further profiling. As summarized in Figure 4, compound 2 showed good IDO1 potency and was highly selective against TDO. Compound 2 also demonstrated superior metabolic stability in both in vitro and in vivo studies across species. In particular, 2 exhibited excellent pharmacokinetic profile in rat and dog including low clearance, long elimination half-life, and high oral bioavailability. Compound 2 also showed to be highly permeable and not a substrate of the P-gp efflux transporter. Furthermore, 2 possessed a clean off-target profile against a Eurofin screen panel of 108 off-targets (see Supporting Information). Human PK and dose prediction using allometric scaling suggested that 2 would have very low clearance (0.2 mL/min/kg) and long t1/2 (66 h), and the potential to achieve high target engagement with QD oral dosing at a dose <10 mg.
Figure 4.

Overall profile of lead compound 2.
In conclusion, guided by structure-based drug design (SBDD), a class of novel IDO1 heme-displacing inhibitors featuring a bicyclo[1.1.1]pentane core was discovered. The presence of the bicyclo[1.1.1]pentane motif as a bioisostere of phenyl ring drastically improved metabolic stability through mitigation of amide hydrolysis potential of the D-pocket aryl amide moiety, with only minimal loss in potency relative to the corresponding phenyl containing compound. Subsequent explorations of the A- and D-pocket amides were accelerated by parallel synthesis as well as the development of a second generation more efficient synthetic route that allowed quick access to some key intermediates. These efforts led to the discovery of compound 2, which exhibited a very favorable overall profile including excellent potency, selectivity, pharmacokinetics, and a much lower predicted human dose compared to epacadostat. This high quality molecule would be useful in further testing the clinical efficacy of IDO1. The binding model for this class of molecules was also established unambiguously by an X-ray cocrystal structure of a representative compound 4-a bound to the IDO1 protein.
Acknowledgments
We thank Drs. David Candito and Xiaoshen Ma for proofreading the manuscript and providing constructive suggestions, Drs Leo Joyce, Steve Castro, and Ed Sherer for providing the VCD report for the absolute configuration assignment, and Dr. Kelly Bleasby and the IVT group for providing data and the procedure for the transporter assay. We also thank Dr. Nengxue Wang and colleagues at Wuxi Apptec for preparation of several compounds described here.
Glossary
Abbreviations
- ALIS
automated ligand identification system
- IDO1
indoleamine-2,3-dioxygenase-1
- TDO
tryptophan-2,3-dioxygenase
- P-gp
permeability glycoprotein
- PXR
pregnane X receptor
- CYP
cytochrome P450 enzymes
- PK
pharmacokinetics
- hWB
human whole blood
- KHMDS
potassium bis(trimethylsilyl)amide
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3- triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate
- DIEA
N,N-diisopropylethylamine
- TMSOK
potassium trimethylsilanolate
- DME
1,2-dimethoxyethane
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00195.
Synthetic procedures and analytical data for some key compounds, conditions for biological assays, and data of Eurofins screen panel (PDF)
Accession Codes
The PDB code for 4-a is 6WJY.
Author Present Address
$ H.Z., Cerevel Therapeutics, 131 Dartmouth Street, Suite 502, Boston, Massachusetts 02116, United States.
The authors declare no competing financial interest.
Supplementary Material
References
- Lizée G.; Overwijk W. W.; Radvanyi L.; Gao J.; Sharma P.; Hwu P. Harnessing the power of the immune system to target cancer. Annu. Rev. Med. 2013, 64, 71–90. 10.1146/annurev-med-112311-083918. [DOI] [PubMed] [Google Scholar]
- Yang Y. Cancer Immunotherapy: Harnessing the immune system to battle cancer. J. Clin. Invest. 2015, 125, 3335–3337. 10.1172/JCI83871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J.; Yu J. X.; Hubbard-Lucey V. M.; Neftelinov S. T.; Hodge J. P.; Lin Y. Trial watch: The clinical trial landscape for PD1/PDL1 immune checkpoint inhibitors. Nat. Rev. Drug Discovery 2018, 17, 854–855. 10.1038/nrd.2018.210. [DOI] [PubMed] [Google Scholar]
- Alsaab H. O.; Sau S.; Alzhrani R.; Tatiparti K.; Bhise K.; Kashaw S. K.; Iyer A. K. PD-1 and PD-L1 checkpoint signaling inhibition for cancer immunotherapy: mechanism, combinations, and clinical outcome. Front. Pharmacol. 2017, 8, 561–575. 10.3389/fphar.2017.00561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams J. L.; Smothers J.; Srinivasan R.; Hoos A. Big opportunities for small molecules in immuno-oncology. Nat. Rev. Drug Discovery 2015, 14, 603–622. 10.1038/nrd4596. [DOI] [PubMed] [Google Scholar]
- Holmgaard R. B.; Zamarin D.; Munn D. H.; Wolchok J. D.; Allison J. P. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J. Exp. Med. 2013, 210, 1389–402. 10.1084/jem.20130066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone T. W.; Stoy N.; Darlington L. G. An expanding range of targets for kynurenine metabolites of tryptophan. Trends Pharmacol. Sci. 2013, 34, 136–43. 10.1016/j.tips.2012.09.006. [DOI] [PubMed] [Google Scholar]
- Platten M.; Wick W.; Van den Eynde B. J. Tryptophan catabolism in cancer: beyond IDO and tryptophan depletion. Cancer Res. 2012, 72, 5435–40. 10.1158/0008-5472.CAN-12-0569. [DOI] [PubMed] [Google Scholar]
- Prendergast G. C.; Smith C.; Thomas S.; Mandik-Nayak L.; Laury-Kleintop L.; Metz R.; Muller A. J. Indoleamine 2,3-dioxygenase pathways of pathogenic inflammation and immune escape in cancer. Cancer Immunol. Immunother. 2014, 63, 721–735. 10.1007/s00262-014-1549-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung D. J.; Rossi M.; Romano E.; Ghith J.; Yuan J.; Munn D. H.; Young J. W. Indoleamine 2,3-dioxygenase-expressing mature human monocyte-derived dendritic cells expand potent autologous regulatory T cells. Blood 2009, 114, 555–563. 10.1182/blood-2008-11-191197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricciuti B.; Leonardi G. C.; Puccetti P.; Fallarino F.; Bianconi V.; Sahebkar A.; Baglivo S.; Chiari R.; Pirro M. Targeting indoleamine-2,3-dioxygenase in cancer: Scientific rationale and clinical evidence. Pharmacol. Ther. 2019, 196, 105–116. 10.1016/j.pharmthera.2018.12.004. [DOI] [PubMed] [Google Scholar]
- Brochez L.; Chevolet I.; Kruse V. The rationale of indoleamine 2,3-dioxygenase inhibition for cancer therapy. Eur. J. Cancer 2017, 76, 167–182. 10.1016/j.ejca.2017.01.011. [DOI] [PubMed] [Google Scholar]
- Röhrig U. F.; Majjigapu S. R.; Vogel P.; Zoete V.; Michielin O. Challenges in the discovery of indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors. J. Med. Chem. 2015, 58, 9421–9437. 10.1021/acs.jmedchem.5b00326. [DOI] [PubMed] [Google Scholar]
- Wang X.; Sun S.; Dong Q.; Wu X.; Tang W.; Xing Y. Recent advances in the discovery of indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors. MedChemComm 2019, 10, 1740–1754. 10.1039/C9MD00208A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis-Ballester A.; Pham K. N.; Batabyal D.; Karkashon S.; Bonanno J. B.; Poulos T. L.; Yeh S. R. Structural insights into substrate and inhibitor binding sites in human indoleamine 2,3-dioxygenase 1. Nat. Commun. 2017, 8, 1693–1700. 10.1038/s41467-017-01725-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelp M. T.; Kates P. A.; Hunt J. T.; Newitt J. A.; Balog A.; Maley D.; Zhu X.; Abell L.; Allentoff A.; Borzilleri R.; Lewis H. A.; Lin Z.; Seitz S. P.; Yan C.; Groves J. T. Immune-modulating enzyme indoleamine 2,3-dioxygenase is effectively inhibited by targeting its apo-form. Proc. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 3249–3254. 10.1073/pnas.1719190115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long G. V.; Dummer R.; Hamid O.; Gajewski T. F.; Caglevic C.; Dalle S.; Arance A.; Carlino M. S.; Grob J. J.; Kim T. M.; Demidov L.; Robert C.; Larkin J.; Anderson J. R.; Maleski J.; Jones M.; Diede S. J.; Mitchell T. C. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): a phase 3, randomised, double-blind study. Lancet Oncol. 2019, 20, 1083–1097. 10.1016/S1470-2045(19)30274-8. [DOI] [PubMed] [Google Scholar]
- Muller A. J.; Manfredi M. G.; Zakharia Y.; Prendergast G. C. Inhibiting IDO pathways to treat cancer: lessons from the ECHO-301 trial and beyond. Semin. Immunopathol. 2019, 41, 41–48. 10.1007/s00281-018-0702-0. [DOI] [PubMed] [Google Scholar]
- Komiya T.; Huang C. H. Updates in the clinical development of Epacadostat and other indoleamine 2,3-dioxygenase 1 inhibitors (IDO1) for human cancers. Front. Oncol. 2018, 8, 423–429. 10.3389/fonc.2018.00423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Liu K.; Pu Q.; Achab A.; Ardolino M. A.; Cheng M.; Deng Y.; Doty A. C.; Ferguson H.; Fradera X.; Knemeyer I.; Kurukulasuriya R.; Lam Y.; Lesburg C. A.; Martinot T. A.; McGowan M. A.; Miller J. R.; Otte K.; Biju P. J.; Sciammetta N.; Solban N.; Yu W.; Zhou H.; Wang X.; Bennett D. J.; Han Y. Discovery of amino-cyclobutarene-derived indoleamine-2,3-dioxygenase 1 (IDO1) inhibitors for cancer immunotherapy. ACS Med. Chem. Lett. 2019, 10, 1530–1536. 10.1021/acsmedchemlett.9b00344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White C.; McGowan M. A.; Zhou H.; Sciammetta N.; Fradera X.; Lim J.; Joshi E. M.; Andrews C.; Nickbarg E. B.; Cowley P.; Trewick S.; Augustin M.; von Koenig K.; Lesburg C. A.; Otte K.; Knemeyer I.; Woo H.; Yu W.; Cheng M.; Spacciapoli P.; Geda P.; Song X.; Smotrov N.; Curran P.; Heo M. R.; Abeywickrema P.; Miller J. R.; Bennett D. J.; Han Y. Strategic incorporation of polarity in potent and selective heme-displacing inhibitors of indoleamine-2,3-dioxygenase-1 (IDO1). ACS Med. Chem. Lett. 2020, 11, 550–557. 10.1021/acsmedchemlett.0c00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annis D. A.; Nickbarg E.; Yang X.; Ziebell M. R.; Whitehurst C. E. Affinity selection-mass spectrometry screening techniques for small molecule drug discovery. Curr. Opin. Chem. Biol. 2007, 11, 518–526. 10.1016/j.cbpa.2007.07.011. [DOI] [PubMed] [Google Scholar]
- Whitehurst C. E.; Yao Z.; Murphy D.; Zhang M.; Taremi S.; Wojcik L.; Strizki J. M.; Bracken J. D.; Cheng C. C.; Yang X.; Shipps G. W. Jr.; Ziebell M.; Nickbarg E. Application of affinity selection-mass spectrometry assays to purification and affinity-based screening of the chemokine receptor CXCR4 Comb. Chem. Comb. Chem. High Throughput Screening 2012, 15, 473–478. 10.2174/138620712800563945. [DOI] [PubMed] [Google Scholar]
- Stepan A. F.; Subramanyam C.; Efremov I. V.; Dutra J. K.; O’Sullivan T. J.; DiRico K. J.; McDonald W. S.; Won A.; Dorff P. H.; Nolan C. E.; Becker S. L.; Pustilnik L. R.; Riddell D. R.; Kauffman G. W.; Kormos B. L.; Zhang L.; Lu Y.; Capetta S. H.; Green M. E.; Karki K.; Sibley E.; Atchison K. P.; Hallgren A. J.; Oborski C. E.; Robshaw A. E.; Sneed B.; O’Donnell C. J. Application of the bicyclo[1.1.1]pentane motif as a nonclassical phenyl ring bioisostere in the design of a potent and orally active γ-secretase inhibitor. J. Med. Chem. 2012, 55, 3414–3424. 10.1021/jm300094u. [DOI] [PubMed] [Google Scholar]
- Mykhailiuk P. K. Saturated bioisosteres of benzene: where to go next?. Org. Biomol. Chem. 2019, 17, 2839–2849. 10.1039/C8OB02812E. [DOI] [PubMed] [Google Scholar]
- Locke G. M.; Bernhard S.S. R.; Senge M. O. Nonconjugated hydrocarbons as rigid-linear motifs: isosteres for material sciences and bioorganic and medicinal chemistry. Chem. - Eur. J. 2019, 25, 4590–4647. 10.1002/chem.201804225. [DOI] [PubMed] [Google Scholar]
- Kanazawa J.; Uchiyama M. Recent advances in the synthetic chemistry of bicyclo[1.1.1]pentane. Synlett 2019, 30, 1–11. 10.1055/s-0037-1610314. [DOI] [Google Scholar]
- Röhrig U. F.; Reynaud A.; Majjigapu S. R.; Vogel P.; Pojer F.; Zoete V. Inhibition Mechanisms of Indoleamine 2,3-Dioxygenase 1 (IDO1). J. Med. Chem. 2019, 62, 8784–8795. 10.1021/acs.jmedchem.9b00942. [DOI] [PubMed] [Google Scholar]
- Oldenziel O. H.; van Leusen D.; van Leusen A. M. A general one-step synthesis of nitriles from ketones using tosylmethyl isocyanide. Introduction of a One-Carbon Unit. J. Org. Chem. 1977, 42, 3114–3118. 10.1021/jo00439a002. [DOI] [Google Scholar]
- Ghose A. K.; Viswanadhan V. N.; Wendoloski J. J. Prediction of hydrophobic (lipophilic) properties of small organic molecules using fragmental methods: an analysis of AlogP and ClogP methods. J. Phys. Chem. A 1998, 102, 3762–3772. 10.1021/jp980230o. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.