

Abstract
Small molecule ligand binding to the ATAD2 bromodomain is investigated here through the synergistic combination of molecular dynamics and protein crystallography. A previously unexplored conformation of the binding pocket upon rearrangement of the gatekeeper residue Ile1074 has been found. Further, our investigations reveal how minor structural differences in the ligands result in binding with different plasticity of the ZA loop for this difficult-to-drug bromodomain.
Keywords: ATAD2 bromodomain, conformational selection, fuzzy binding, protein X-ray crystallography, molecular dynamics
Contemporary drug discovery often relies on the availability of structural data to guide the design of ligands and hit optimization.1 This task becomes challenging when the protein target is flexible and its conformational changes need to be considered.2,3 Proper understanding of protein flexibility and conformational selection in small molecule binding events can be leveraged to the benefit of lead discovery campaigns, in particular with the help of atomistic simulation approaches.4−14 Reader domains involved in epigenetic regulation (regulation of gene expression without alterations to the DNA sequence), are highly flexible and adaptive receptor targets.15−17 Given their potential implication in the control of genetic information transfer within the cell, the quest for potent and selective ligand binders that interact with these proteins has been the focus of intense efforts both in academia and industry over the past decade.
A particularly difficult-to-drug target in this series is the ATAD2 (ATPase family AAA domain-containing 2) bromodomain, a highly flexible and challenging protein–protein interaction domain implicated in multiple cancers.18−23 Despite its high therapeutic potential, only two probes have been recently reported.24−27 ATAD2 is considered poorly druggable mainly due to a shallower, more polar, and flexible KAc binding pocket as compared to other bromodomains. Here, we report a detailed structural investigation on the binding interactions to the acetyl-lysine (KAc)’s reader ATAD2 through a combination of molecular dynamics (MD) simulations and protein crystallography for several de novo designed ligands. Our study sheds light on the multiple factors underlying the plasticity of this bromodomain, including the local disorder of the hydrophobic side chains involved in binding as well as the remarkable flexibility and adaptability of the ZA loop.
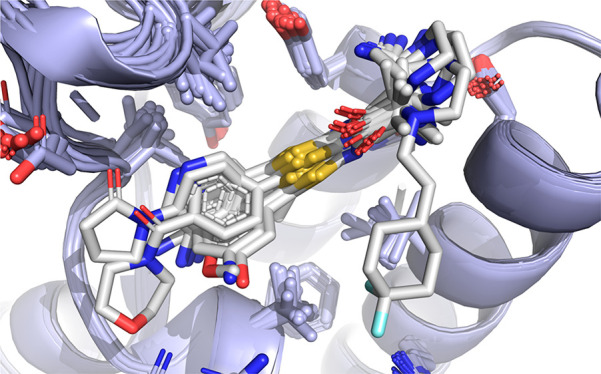
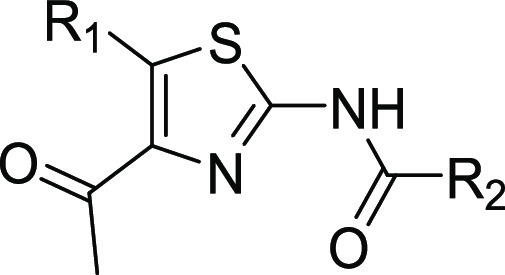
Our investigation commenced with acetylthiazole hit 1 (Table 1), which was identified by protein structure-based pharmacophore search in the available commercial chemical space.28 While several subclasses of 2-amino thiazole (2-AT) are considered Pan Assay INterference CompoundS (PAINS), additional experimental and computational evidence summarized herein below made us confident in our choice of this fragment for further optimization.29,30 Our key discovery hypothesis was the interaction between the acetylthiazole and the conserved asparagine residue (Asn1064), as well as between the primary ammonium group of the alanine moiety and Asp1071 (Figure 1). The stability of these interactions was confirmed by MD simulations (for a movie of the MD simulation, see the SI), and the binding of compound 1 was validated both experimentally via TR-FRET (336 μM, see the SI) and crystallographically (Figure 1A). As predicted, 1 occupies the KAc binding pocket of ATAD2 surrounded by the hydrophobic residues Val1008, Val1013, Val1018, Tyr1021, Tyr1063, and Ile1074. The amino group of the alanine moiety of 1 indeed reaches out to Asp1071, making a relatively weak salt bridge (3.6 Å).
Table 1. Binders of ATAD2 Bromodomain Described in This Study.

Measured in a time-resolved (TR)-FRET assay.
Measured by ITC.
Figure 1.

(A–D) Crystal structures of the ATAD2 bromodomain in the complex with compounds 1–4. 2Fo–Fc electron density maps are shown in blue. In all figures, conserved water molecules and water molecules involved in bridging polar interactions (dashed lines) between protein and ligand are shown (red spheres).
Based on the structure of compound 1, a series of derivatives was designed and subsequently prepared to explore the binding landscape of this highly flexible and thus challenging target. The compounds synthesized within this campaign have been summarized in Table 1 (See experimental section. 2D structures, PDB codes for protein–ligand complexes, as well as IC50 or Kd values measured by TR-FRET or ITC can also be found in Table 1 and in the SI). Within this compound series, we first set out to rigidify the structure and improve the salt bridge observed for compound 1 by presenting the secondary amino group to the Asp1071. To this end a compound bearing a piperazine was synthesized (2). The X-ray structure of this ligand in complex with ATAD2 showed the desired interaction of the piperazine group with Asp1071, with its distal nitrogen atom forming a higher quality interaction than the original fragment (2.6 Å; Figure 1B). In contrast, the proximal nitrogen assumed a close position to the amino group of 1 and did not engage with the aspartate residue. A guanidinium derivative of 1 was also prepared (3), which formed a network of hydrogen bonds between Tyr1063 and Asp1071 (Figure 1C). This led to rearrangement of the side chains of the gatekeeper residue Ile1074 as well as that of Val1008, translating into slight remodeling of the binding pocket and tilting of the thiazole ring to maintain hydrophobic interactions with these residues. Similar rearrangement of the binding site was seen in the case of a close derivative 4 with an additional methyl group introduced to improve hydrophobic interactions to Tyr1063 (Figure 1D). While some interactions seemed to have improved with this approach, no significant change in binding affinity could be observed for these compounds.
To better understand the conformational rearrangements within the pocket and their potential impact on binding, we reanalyzed large data sets of all-atom simulations that we previously collected for ATAD2 bromodomain, both of its apo form as well as of the one bound to KAc-containing peptide.31 The rearranged conformations of residues Val1008 and Ile1074 (Figure 2A) were populated in the runs with both apo and holo ATAD2 and appeared independent of the movements of the ZA loop, hinting at a conformational selection mechanism in ligand binding. Moreover, while both observed conformations of Val1008 turned out to appear with similar frequency, there was a clear preference for the conformation of the gatekeeper Ile1074 that opens a remodelled cavity (Figure 2B). While this preference was limited in terms of estimated energy difference (∼1–2 kcal/mol), we envisaged the possibility of attaining better ligands by binding to the more populated conformation of the gatekeeper Ile1074.
Figure 2.

(A) Superposition of structures of compounds 1 and 3 (shown in black). (B) Populations of different conformers of the residues Val1008 and Ile1074 according to the MD simulations. Individual data points represent the conformational states in crystal structures deposited in the PDB as of 01-03-2019. (C) Co-crystal structure of compound 5 bound to ATAD2 bromodomain.
We decided to cyclize the original fragment 1 and “grow” it deeper into the pocket to gain stabilization of the acetyl group, whose high mobility has been observed in the MD simulations (see the Supporting Information for details). Furthermore, most bromodomains, like ATAD2, contain up to four water molecules in their KAc binding pocket. As shown by others32 different acetyl mimics can perturb this water network and induce selectivity over other bromodomains. Therefore, we hypothesized that improved selectivity could be attained for a reshaped pocket by the cyclic acetyl mimic, as suggested by docking. Compound 5 was synthesized confirming this hypothesis according to the X-ray structure shown in Figure 2C. Still, no appreciable affinity improvement could be quantified for this compound, and thus alternative modifications of the acetylthiazole core were subsequently explored.
Based on the initial exploration of the ligand blueprints and interactions within the binding site, the molecules were grown in the direction of the ZA loop. Recently, our groups have reported a novel molecular diversification software named AutoCouple.33 AutoCouple generates a combinatorial virtual library of binder candidates that are novel and synthetically accessible in one formal step by coupling a validated fragment with commercially available building blocks. The library, fairly exhaustive in terms of easily accessible chemical derivative space, is then evaluated for possible binding to the target by the means of tethered docking, and candidates most likely to bind are shortlisted. Guided by these predictions, we prepared a series of derivatives of compound 1 via Pd-catalyzed Suzuki cross-coupling reactions (compounds 6–10) to pick up additional interactions with the ZA loop while maintaining the key interaction cascade to Asp1071 and Asn1067.
This focused library provided insight into conformational changes upon ligand binding. The same rearrangement of the side chains of the gatekeeper residue Ile1074 and Val1008, as well as the increased size of the binding pocket and slight tilting of the thiazole ring were observed for example for compound 6. In addition, the phenol moiety engages a superficial solvent-exposed hydrogen bond to Arg1007 (Figure 3A). Interestingly, compound 7, a close derivative of 6, lacks the stabilizing interaction with Arg1007 due to the methylation of the hydroxyl group, and its aromatic moiety is disordered in the corresponding cocrystal structure (Figure S1A).
Figure 3.

(A–B) Crystal structures of the ATAD2 bromodomain in the complex with compounds 6 and 8, respectively. (C) Different binding modes seen in the all-atom MD simulations of the compound 8–ATAD2 bromodomain complex: left–van der Waals interaction between the side chain of Val1013 and amino-pyridine; right–hydrogen bond between the backbone NH of Asp1014 and the nitrogen atom of the pyridine ring.
Some of these molecules bound to the enlarged conformation of the pocket but without engaging the ZA loop. In these cases, both the loop and aromatic moiety appear to be disordered, as for pyridine-containing molecule 8 (Figure 3B). Significant mobility of the ZA loop is expected, based on previous MD simulations.31 To understand the interaction mode of 8 and the higher mobility of the ZA loop, a series of all-atom simulations of the complex were performed, revealing a larger disorder in the ZA loop, as compared to simulations previously reported by us.31 While the key interactions of 8 in the binding pocket remained stable for more than 150 ns (e.g., salt bridge to Asp1071), multiple interaction modes of the aromatic substituent with the ZA loop emerge from the simulations (Figure 3C). These include the direct hydrogen bond between the nitrogen atom of the pyridine and the main chain NH of Asp1014, and the hydrophobic contact between the aromatic ring and the side chain of Val1013. These interactions engage and disengage over the course of the simulation without a single major defined conformation, which is reminiscent of fuzzy binding. Similarly to compound 8, compound 9 bears a pyridine substituent that cannot be seen in the electron density in the cocrystal structure, along with part of the ZA loop. In contrast, the ZA loop and the R1 substituent are ordered for 10, which bears a 3-aminobenzene substituent and shows a binding mode similar to the phenol derivative 6 (Figure S1C). Thus, the promiscuous fuzzy binding behavior seems to be induced by the pyridine moiety.
It should be noted that fuzzy or disordered binding is usually avoided in ligand design to favor well-defined enthalpy-driven interactions, which provide potency and selectivity. To study the relationship between the compound structure and the fuzzy binding mode further, molecules containing an aminopyridine moiety were extended by merging them with different motifs designed to better engage the Asp1071 residue. As these molecules might be able to select different conformations of the binding pocket, we envisaged it would be worth it to recapitulate how this affects the fuzzy binding behavior. As seen before, compound 4 selects for enlarged conformation of the pocket and would project the aromatic group alongside the same vector as in 8. As expected, the aminopyridine-bearing derivative 11 exhibits very similar fuzzy binding to that seen for 8 (Figure 4). In contrast, both derivatives of 2 and 3 (12 and 13, respectively), show ordered binding of the aminopyridine group as predicted in the original docking efforts. Importantly, 12 binds to the same conformation of the pocket as 2, and introduction of the aromatic moiety does not shift its conformational selectivity, unlike in the case of 1 and 8. It is clear from these results that the fuzzy binding behavior arises as a result of very delicate interplay of multiple factors, so that even small modifications to the ligand may disrupt such binding mode, which further complicates the structure-based design against such a highly mobile target. To slightly modulate interactions with Tyr1063, the alanine moiety was modified with a highly strained cyclobutyl ring (14), which not only led to ordering of the aminopyridine moiety but also shifted the conformational equilibrium of the gatekeeper residue Ile1074 and slightly improved binding affinity for this compound.
Figure 4.

(A–D) Crystal structures of the ATAD2 bromodomain in complex with compounds 11–14.
A second AutoCouple campaign was launched to design derivatives that would engage the ZA loop (see Supporting Information Figure 2 for cocrystal structures of ATAD2 bromodomain with compounds 15 and 16). Unfortunately, no significant gain in affinity was observed, which showcased the intrinsic difficulty associated with ordering this highly flexible loop (i.e., overcoming a high entropic penalty).
Next, we concentrated on further extending these molecules so that they could pick up interactions with Arg1077, in line with improved affinities observed for compounds previously reported by others (see Supporting Information for IC50 in our TR-FRET assay).24 The proximal nitrogen atom in compound 2, which does not engage Asp1071, was alkylated with an ethyl-linked cyclohexyl ring scaffold yielding ligand 17. This derivative forms a favorable charge–dipole interaction with Arg1077 through its CF2 group (Figure 5). The rigidity of the ethyl difluorocyclohexyl group also allowed us to maintain the Ile1074 conformation with a smaller opening of the pocket. This molecule is also the most potent derivative in this series, with IC50 of 15 μM in our TR-FRET assay and Kd of 25 μM in ITC. In contrast to observations made for the more potent previously reported chemical probe,24 further modifications tackling the interactions with the ZA loop by introduction of an aminopyridine moiety (compound 18) substantially lowered the potency, thus highlighting how not only the extensions, but the molecule’s core need to synergize to achieve substantial gain in affinity for the ATAD2 bromodomain.
Figure 5.

Crystal structure of the ATAD2 bromodomain in the complex with compound 17 (A) and the GSK8814 chemical probe (B). (C) Superposition of compound 17 and the GSK8814 chemical probe (shown in black) in the binding site. (D) 2D structure of GSK8814 and binding affinity determined in house via TR-FRET assay.
In conclusion, we have extensively explored conformational selection of the ATAD2 bromodomain. The MD trajectories and crystal structures allow us to formulate the following conclusions. First, the key hydrophobic residues in the KAc binding site, i.e., the gatekeeper Ile1074, Val1008, and Val1013, can assume multiple orientations for different ligands and even for the same ligand. The local disorder of the hydrophobic side chains involved in binding is observed in the simulations (carried out with compounds 5 and 8) and the crystal structures with ligands 7, 8, 9, and 11. Such disorder is congruent with suboptimal interactions of high micromolar affinity binders. Second, the intrinsic plasticity of the ZA loop results in fuzzy binding or well-defined packing onto the ligand depending on fine details of the latter. As an example, compounds 8 and 10 differ by a single atomic element and show fuzzy binding and single well-defined pose, respectively. Note that this variability is not a consequence of differences in packing between protein molecules in the crystal as all structures belong to the same space group. While the multiple orientations of the hydrophobic side chains have not been reported previously, the formation of a fuzzy complex and adaptable interface of binding, which is mainly due to the flexibility of the ZA loop, has been observed in atomistic simulations of the histone H4 peptide/ATAD2 bromodomain complex.31 The coupling of the ZA-loop rigidification and the rearrangement of the hydrophobic side chains involved in binding can be explored next as a potential strategy to attain potent and selective ligands for this highly mobile target.
Acknowledgments
The use of beamlines at the Swiss Light Source (Paul Scherrer Institut, Villigen, Switzerland) is gratefully acknowledged. We thank Cassiano Langini for help with analysis of the MD data and Maurus Mathis and Johanne Ling for their help with the synthesis and characterization of the compounds on the outset of the project. We are grateful to ChemAxon for providing academic licence for Marvin version 16.2.15.0 (2016), which was used for drawing, displaying, and characterizing chemical structures and protonation states, as well as generating conformers (www.chemaxon.com).
Glossary
Abbreviations
- ATAD2
ATPase family AAA domain-containing 2
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00080.
Author Present Address
∥ J.D.: Astex Therapeutics, Cambridge, UK.
Author Contributions
§ A.D. and L.B. contributed equally.
This work was funded by the Swiss National Science Foundation (Project Funding to A.C.) and Swiss Cancer League - Krebsliga (to C.N. and A.C.). P.S. is grateful to the UZH BioEntrepreneurship program for financial support.
The authors declare no competing financial interest.
Notes
The PDB codes of ATAD2 in complex with the following compounds are: 1, 5F36; 2, 6EPU; 3, 6EPX; 4, 6HI3; 5, 6EPV; 6, 6EPJ; 7, 6HI4; 8, 6HI5; 9, 6HI6; 10, 6HI7; 11, 6HI8; 12, 6EPT; 13, 6HIA; 14, 6HIB; 15, 6HIC; 16, 6HID; 17, 6HIE. The authors will release the atomic coordinates and experimental data upon article publication.
Supplementary Material
References
- Blundell T. L. Structure-Based Drug Design. Nature 1996, 384, 23–26. 10.1016/B978-012518170-9/50007-6. [DOI] [PubMed] [Google Scholar]
- Arkin M. R.; Randal M.; DeLano W. L.; Hyde J.; Luong T. N.; Oslob J. D.; Raphael D. R.; Taylor L.; Wang J.; McDowell R. S.; et al. Binding of Small Molecules to an Adaptive Protein-Protein Interface. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 1603–1608. 10.1073/pnas.252756299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells J. A.; McClendon C. L. Reaching for High-Hanging Fruit in Drug Discovery at Protein-Protein Interfaces. Nature 2007, 450, 1001–1009. 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- Śledź P.; Caflisch A. Protein Structure-Based Drug Design: From Docking to Molecular Dynamics. Curr. Opin. Struct. Biol. 2018, 48, 93–102. 10.1016/j.sbi.2017.10.010. [DOI] [PubMed] [Google Scholar]
- Durrant J. D.; McCammon J. A. Molecular Dynamics Simulations and Drug Discovery. BMC Biol. 2011, 9, 71. 10.1186/1741-7007-9-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganesan A.; Coote M. L.; Barakat K. Molecular Dynamics-Driven Drug Discovery: Leaping Forward with Confidence. Drug Discovery Today 2017, 22, 249–269. 10.1016/j.drudis.2016.11.001. [DOI] [PubMed] [Google Scholar]
- Zhao H.; Caflisch A. Molecular Dynamics in Drug Design. Eur. J. Med. Chem. 2015, 91, 4–14. 10.1016/j.ejmech.2014.08.004. [DOI] [PubMed] [Google Scholar]
- Su J.; Liu X.; Zhang S.; Yan F.; Zhang Q.; Chen J. Insight Into Selective Mechanism of Class of I-BRD9 Inhibitors Toward BRD9 Based on Molecular Dynamics Simulations. Chem. Biol. Drug Des. 2019, 93, 163–176. 10.1111/cbdd.13398. [DOI] [PubMed] [Google Scholar]
- Su J.; Liu X.; Zhang S.; Yan F.; Zhang Q.; Chen J. A Theoretical Insight Into Selectivity of Inhibitors Toward Two Domains of Bromodomain-Containing Protein 4 Using Molecular Dynamics Simulations. Chem. Biol. Drug Des. 2018, 91, 828–840. 10.1111/cbdd.13148. [DOI] [PubMed] [Google Scholar]
- Huang D.; Rossini E.; Steiner S.; Caflisch A. Structured Water Molecules in the Binding Site of Bromodomains Can Be Displaced by Cosolvent. ChemMedChem 2014, 9, 573–579. 10.1002/cmdc.201300156. [DOI] [PubMed] [Google Scholar]
- Magno A.; Steiner S.; Caflisch A. Mechanism and Kinetics of Acetyl-Lysine Binding to Bromodomains. J. Chem. Theory Comput. 2013, 9, 4225–4232. 10.1021/ct400361k. [DOI] [PubMed] [Google Scholar]
- Steiner S.; Magno A.; Huang D.; Caflisch A. Does Bromodomain Flexibility Influence Histone Recognition?. FEBS Lett. 2013, 587, 2158–2163. 10.1016/j.febslet.2013.05.032. [DOI] [PubMed] [Google Scholar]
- Bacci M.; Langini C.; Vymětal J.; Caflisch A.; Vitalis A. Focused Conformational Sampling in Proteins. J. Chem. Phys. 2017, 147, 195102. 10.1063/1.4996879. [DOI] [PubMed] [Google Scholar]
- Gay J. C.; Eckenroth B. E.; Evans C. M.; Langini C.; Carlson S.; Lloyd J. T.; Caflisch A.; Glass K. C. Disulfide Bridge Formation Influences Ligand Recognition by the ATAD2 Bromodomain. Proteins: Struct., Funct., Genet. 2019, 87, 157–167. 10.1002/prot.25636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain A. K.; Barton M. C. Bromodomain Histone Readers and Cancer. J. Mol. Biol. 2017, 429, 2003–2010. 10.1016/j.jmb.2016.11.020. [DOI] [PubMed] [Google Scholar]
- Muller S.; Filippakopoulos P.; Knapp S. Bromodomains as Therapeutic Targets. Expert Rev. Mol. Med. 2011, 13, e29 10.1017/S1462399411001992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferri E.; Petosa C.; McKenna C. E. Bromodomains: Structure, Function and Pharmacology of Inhibition. Biochem. Pharmacol. 2016, 106, 1–18. 10.1016/j.bcp.2015.12.005. [DOI] [PubMed] [Google Scholar]
- Hwang H. W.; Ha S. Y.; Bang H.; Park C. K. ATAD2 as a Poor Prognostic Marker for Hepatocellular Carcinoma after Curative Resection. Cancer Res. Treat. 2015, 47, 853–861. 10.4143/crt.2014.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morozumi Y.; Boussouar F.; Tan M.; Chaikuad A.; Jamshidikia M.; Colak G.; He H.; Nie L.; Petosa C.; De Dieuleveult M.; et al. ATAD2 Is a Generalist Facilitator of Chromatin Dynamics in Embryonic Stem Cells. J. Mol. Cell Biol. 2016, 8, 349–362. 10.1093/jmcb/mjv060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciró M.; Prosperini E.; Quarto M.; Grazini U.; Walfridsson J.; McBlane F.; Nucifero P.; Pacchiana G.; Capra M.; Christensen J.; et al. ATAD2 Is a Novel Cofactor for MYC, Overexpressed and Amplified in Aggressive Tumors. Cancer Res. 2009, 69, 8491–8498. 10.1158/0008-5472.CAN-09-2131. [DOI] [PubMed] [Google Scholar]
- Zhang M. J.; Zhang C. Z.; Du W. J.; Yang X. Z.; Chen Z. P. ATAD2 Is Overexpressed in Gastric Cancer and Serves as an Independent Poor Prognostic Biomarker. Clin. Transl. Oncol. 2016, 18, 776–781. 10.1007/s12094-015-1430-8. [DOI] [PubMed] [Google Scholar]
- Hou M.; Huang R.; Song Y.; Feng D.; Jiang Y.; Liu M. ATAD2 Overexpression Is Associated with Progression and Prognosis in Colorectal Cancer. Jpn. J. Clin. Oncol. 2016, 46, 222–227. 10.1093/jjco/hyv195. [DOI] [PubMed] [Google Scholar]
- Caron C.; Lestrat C.; Marsal S.; Escoffier E.; Curtet S.; Virolle V.; Barbry P.; Debernardi A.; Brambilla C.; Brambilla E.; et al. Functional Characterization of ATAD2 as a New Cancer/Testis Factor and a Predictor of Poor Prognosis in Breast and Lung Cancers. Oncogene 2010, 29, 5171–5181. 10.1038/onc.2010.259. [DOI] [PubMed] [Google Scholar]
- Bamborough P.; Chung C. W.; Demont E. H.; Furze R. C.; Bannister A. J.; Che K. H.; Diallo H.; Douault C.; Grandi P.; Kouzarides T.; et al. A Chemical Probe for the ATAD2 Bromodomain. Angew. Chem., Int. Ed. 2016, 55, 11382–11386. 10.1002/anie.201603928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Montalván A. E.; Berger M.; Kuropka B.; Koo S. J.; Badock V.; Weiske J.; Puetter V.; Holton S. J.; Stöckigt D.; Ter Laak A.; et al. Isoform-Selective ATAD2 Chemical Probe with Novel Chemical Structure and Unusual Mode of Action. ACS Chem. Biol. 2017, 12, 2730–2736. 10.1021/acschembio.7b00708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamborough P.; Chung C. W.; Furze R. C.; Grandi P.; Michon A. M.; Watson R. J.; Mitchell D. J.; Barnett H.; Prinjha R. K.; Rau C.; et al. Aiming to Miss a Moving Target: Bromo and Extra Terminal Domain (BET) Selectivity in Constrained ATAD2 Inhibitors. J. Med. Chem. 2018, 61, 8321–8336. 10.1021/acs.jmedchem.8b00862. [DOI] [PubMed] [Google Scholar]
- Bamborough P.; Chung C. W.; Demont E. H.; Bridges A. M.; Craggs P. D.; Dixon D. P.; Francis P.; Furze R. C.; Grandi P.; Jones E. J.; et al. A Qualified Success: Discovery of a New Series of ATAD2 Bromodomain Inhibitors with a Novel Binding Mode Using High-Throughput Screening and Hit Qualification. J. Med. Chem. 2019, 62, 7506–7525. 10.1021/acs.jmedchem.9b00673. [DOI] [PubMed] [Google Scholar]
- Sunseri J.; Koes D. R. Pharmit: Interactive Exploration of Chemical Space. Nucleic Acids Res. 2016, 44, W442–W448. 10.1093/nar/gkw287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baell J. B.; Holloway G. A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) From Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53, 2719–2740. 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- A recent study on 2-AT containing fragments and HTS hits revealed that not all 2-AT molecules are problematic and that acylation of the 2-amino group, as in our case, reduces substantially the risk of pan hitting:Devine S. M.; Mulcair M. D.; Debono C. O.; Leung E. W. W.; Nissink J. W. M.; Lim S. S.; Chandrashekaran I. R.; Vazirani M.; Mohanty B.; Simpson J. S.; et al. Promiscuous 2-Aminothiazoles (PrATs): A FrequentHitting Scaffold. J. Med. Chem. 2015, 58, 1205–1214. 10.1021/jm501402x. [DOI] [PubMed] [Google Scholar]
- Langini C.; Caflisch A.; Vitalis A. The ATAD2 Bromodomain Binds Different Acetylation Marks on the Histone H4 in Similar Fuzzy Complexes. J. Biol. Chem. 2017, 292, 16734–16745. 10.1074/jbc.M117.786350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford T. D.; Tsui V.; Flynn E. M.; Wang S.; Taylor A. M.; Côté A.; Audia J. E.; Beresini M. H.; Burdick D. J.; Cummings R.; Dakin L. A.; Duplessis M.; Good A. C.; Hewitt M. C.; Huang H.-R.; Jayaram H.; Kiefer J. R.; Jiang Y.; Murray J.; Nasveschuk C. G.; Pardo E.; Poy F.; Romero F. A.; Tang Y.; Wang J.; Xu Z.; Zawadzke L. E.; Zhu X.; Albrecht B. K.; Magnuson S. R.; Bellon S.; Cochran A. G. Diving into the Water: Inducible Binding Conformations for BRD4, TAF1(2), BRD9, and CECR2 Bromodomains. J. Med. Chem. 2016, 59, 5391–5402. 10.1021/acs.jmedchem.6b00264. [DOI] [PubMed] [Google Scholar]
- Batiste L.; Unzue A.; Dolbois A.; Hassler F.; Wang X.; Deerain N.; Zhu J.; Spiliotopoulos D.; Nevado C.; Caflisch A. Chemical Space Expansion of Bromodomain Ligands Guided by in Silico Virtual Couplings (AutoCouple). ACS Cent. Sci. 2018, 4, 180–188. 10.1021/acscentsci.7b00401. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.