

Abstract

Pan-BET inhibitors have shown profound efficacy in a number of in vivo preclinical models and have entered the clinic in oncology trials where adverse events have been reported. These inhibitors interact equipotently with the eight bromodomains of the BET family of proteins. To better understand the contribution of each domain to their efficacy and to improve from their safety profile, selective inhibitors are required. This Letter discloses the profile of GSK973, a highly selective inhibitor of the second bromodomains of the BET proteins that has undergone extensive preclinical in vitro and in vivo characterization.
Keywords: Bromodomain, BET, constrain, selectivity, dihydrobenzofuran, fluorine
The bromodomain and extra-terminal (BET) family of proteins are known as epigenetic readers of chromatin. Indeed, the two tandem bromodomains (BD1 and BD2) of each of the four proteins (BRD2,3,4, and T) recognize and bind to specific acetylated-lysine residues of histone tails and non-histone proteins and play a major role in the regulation of gene transcription (ref (1)). Pan-BET inhibitors have shown profound efficacy in a number of preclinical in vitro and in vivo models (ref (2)) with several molecules progressed into the clinic. A number of adverse events have been reported in oncology trials to date (ref (3)), in particular thrombocytopenia. Various strategies (dosing holidays, combination therapy) have been put in place in an attempt to circumvent these safety concerns and maximize the potential of this mechanism.
Pan-BET inhibitors interact equipotently with the eight bromodomains of the BET family. In order to elucidate the functional contribution of each bromodomain to the phenotype observed with pan-BET inhibitors and with the ambition of exploring whether the efficacy and toxicology effects observed could be dissociated by selective bromodomain targeting, we aimed to identify selective small molecule inhibitors of BD2s. The high homology between the 4 BD1s and the 4 BD2s (see the Supporting Information) makes the identification of isoform selective inhibitors (interacting only with one or two BD of a given protein) difficult. Indeed, an HTS performed at GSK only identified hits either equipotent at all BDs or biased against all BD1 or all BD2 BDs (ref (4)).
We (refs (4−6)) like others (refs (7 and 8)) have recently disclosed the identification of drug-like selective pan-BD2 inhibitors. In particular, we have disclosed the profile of a series of pyridone derivatives with excellent selectivity for BD2s over BD1s (ref (4)). The pyridone series was characterized by a methyl amide KAc mimetic, stabilized in its bound conformation by an internal hydrogen bond; a benzylic substituent projected toward the BET WPF shelf and a secondary amide (cyclopropyl amide in the case of 1, Figure 1) which proved to increase both potency and selectivity thanks to an additional hydrogen bond with Asn429 (BRD4 BD2 numbering) and better shape complementary for BD2 compared to BD1. This series delivered drug-like molecules with good potency and BD2/BD1 selectivity (in most cases >100-fold). However, to fully interrogate the properties of domain selectivity, we were interested in identifying novel series with similar potency (FRET BRD4 BD2 pIC50 > 7) but increased level of BD2/BD1 selectivity (>1000-fold). Crystallographic data suggested that constraining the WPF shelf substituent by cyclization of the benzylic position should mimic the bound conformation of 1, which may have a positive impact on both potency and selectivity. In order to keep the internal H-bond known to provide enhanced potency, bicyclic cores A were considered (Figure 1).
Figure 1.

Strategy to increase potency and selectivity.
Dihydrobenfurans (DBFs) could be readily accessed to test this hypothesis (Figure 2). Alkylation of 2, Claisen rearrangement, and amide formation led to 3 which could be cyclized using refluxing TFA to afford racemic compounds 4–6 in low yields (6–16%) (6 is obtained via a Wagner–Meerwein rearrangement). Among the three derivatives isolated, compound 5 appeared to be the most interesting in terms of both potency and selectivity and compared favorably with the profile of fragment 7, which had led to 1.
Figure 2.

Synthesis and comparison of substituted dihydrobenzofuran cores. Reagents and conditions: (a) Cinnamyl chloride, K2CO3, KI, acetone, reflux, 88%; (b) N,N-dimethylaniline, reflux, 55%; (c) MeNH2, THF, room temperature, 91%; (d) TFA, reflux, 6–16%.
The synthetic route to access functionalized analogues of 5 is shown in Scheme 1. Epoxide 9 could be obtained from 8 using the same Claisen rearrangement as that for 3, followed by epoxidation using m-CPBA. Interestingly, 9 was obtained as a 1:1 mixture of diastereoisomers, but the cyclization to 10 under mild basic conditions led only to the more thermodynamically stable trans-DBF (dr > 95:5) thanks to an in situ epimerization of the dibenzylic position. From 10, a carbonylation led to ester 11. It was also possible to substitute the primary hydroxyl of 10 by a fluorine (12) or reduce it to the corresponding alkane (13). Saponification of the ester followed by amide coupling provided compounds 14–36. Separation of the enantiomers by chiral chromatography could be done at any stage once 10 was obtained. One enantiomer was always significantly more potent than the other and when cocrystallized with the bromodomain construct always had the stereochemistry as drawn in the tables and scheme. We therefore considered that the most potent BD2 activities always resided in this isomer. Data provided below are for single enantiomers unless otherwise stated.
Scheme 1. Synthesis of Functionalized DBFs.

Reagents and conditions: (a) Cinnamyl chloride, K2CO3, KI, acetone, reflux, 100%; (b) N,N-dimethylaniline, reflux; (c) MeNH2, THF, room temperature, 56% (two steps); (d) m-CPBA, CH2Cl2, 0 °C, 98%; (e) KOH, DMSO/H2O, 0 °C, 67%; (f) Deoxofluor, CH2Cl2, 0–40 °C, 71%; (g) Pd(OAc)2, Xantphos, CO, MeOH, DMF, 70 °C, 49%; (h) I2, PPh3, imidazole, CH2Cl2, rt, 79%; (i) NEt3, Pd/C, H2 (1 atm), MeOH, rt, 56%; (j) LiOH or NaOH, MeOH, THF, H2O, rt, >95%; (k) RNH2, HATU, NEt3, rt, 13–92%.
As can be seen in Table 1, the DBF series was more potent and selective than the pyridone series: For most pairs presented (14–21), the DBF analogue was at least 3-fold more potent against BRD4 BD2 than its pyridone equivalent with excellent ligand efficiency (LE > 0.4) for both series when lipophilic amines were used. The selectivity was superior in the DBF series by virtue of not only higher potency against BD2 but also lower potency against BD1. Excellent selectivity (>1000-fold) could be achieved with small amide substituents (compounds 19 and 21), but this was to the detriment of lipophilicity which was systematically increased by at least 1.1 log units compared to the equivalent pyridone. Introducing polarity in the amide substituent was detrimental to activity (compounds 22–25).
Table 1. Comparison of the Pyridone and DBF Series Activities.

| BRD4
pIC50a |
||||||
|---|---|---|---|---|---|---|
| Cpd | seriesb | R | BD2 (LE) | BD1 | fold selectivity | ChromLogD @ pH 7.4 |
| 14 | pyridone | H | 6.5 (0.42) | 5.1 | 25 | 2.03 |
| 15 | DBF | 7 (0.42) | 4.8 | 160 | 3.28 | |
| 16 | pyridone | methyl | 6.9 (0.43) | 5.1 | 60 | 2.38 |
| 17 | DBF | 7.5 (0.43) | 4.9 | 400 | 3.55 | |
| 18 | pyridone | ethyl | 6.9 (0.41) | 4.8 | 125 | 2.99 |
| 19 | DBF | 7.4 (0.41) | 4.4 | 1000 | 4.11 | |
| 20 | pyridone | c-Pr | 7.1 (0.41) | 4.8 | 200 | 3.07 |
| 21 | DBF | 7.9 (0.42) | 4.7 | 1600 | 4.24 | |
| 22 | pyridone | CH2CH2OH | 6.2 (0.35) | 4.7 | 30 | 1.9 |
| 23 | DBF | 6.8 (0.36) | <4.3 | >320 | 3.12 | |
| 24 | pyridone | CH2CH2OCH3 | 6.2 (0.34) | 5.1 | 13 | 2.74 |
| 25 | DBF | 6.7 (0.34) | 4.4 | 200 | 3.81 | |
BRD4 potencies are representative of the potencies against the BET proteins (see the Supporting Information for full profiling).
See ref (4) for all data on the pyridone series.
Compound 21 was considered as the lead molecule and further profiled. As can be seen in Table 2, the low molecular weight and moderate lipophilicity were properties affording good passive permeability and consequently good cell potency, as seen by inhibition of monocyte chemoattractant protein-1 (MCP-1) in a lipopolysaccharide (LPS)-stimulated human peripheral blood mononuclear cell (PBMC) assay. Moderate protein binding aided the retention of high potency for inhibition of MCP-1 release in an LPS-stimulated human whole blood assay. High throughput solubility was moderate. Unfortunately, the compound showed moderate turnover in vitro in rat and human hepatocytes. In vivo, in the rat, this translated to high blood clearance. In vivo clearance was assumed to be metabolic as renal clearance was minimal. The compound was distributed into tissues, but the high clearance resulted in a short elimination half-life unacceptable for further development as an oral therapeutic.
Table 2. Characterization of Compounds 21 and 26.
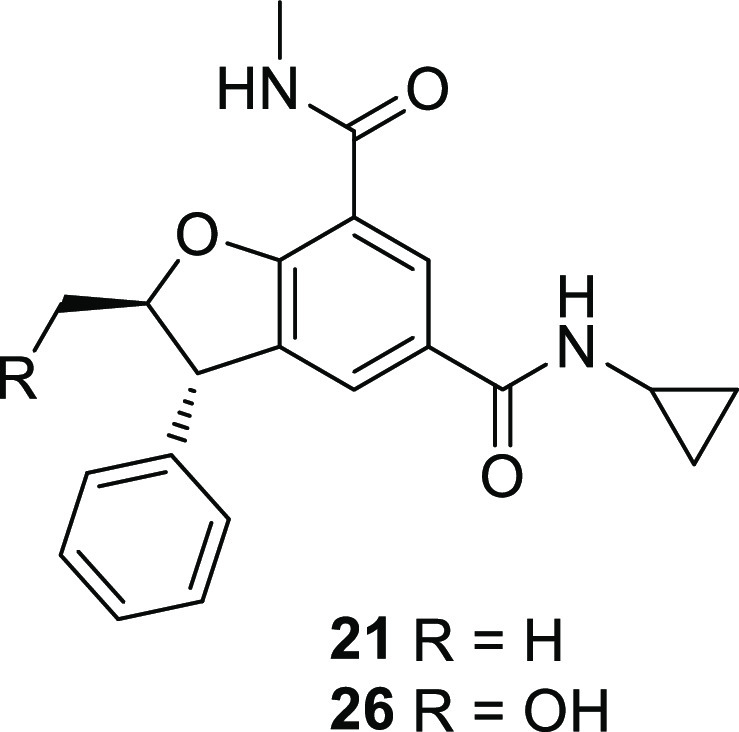
| compound | 21 | 26 | |
|---|---|---|---|
| MW, cLogP, ChromLogD | 350.4; 2.88; 4.24 | 366.4; 1.41; 2.53 | |
| AMP permeability (nM/s) | 440 | 266 | |
| CLND solubility (μg/mL) | 137 | >177 | |
| HSA binding (%) | 89 | 79 | |
| h PBMC pIC50 (MCP-1) | 8.2 | ||
| h whole blood pIC50 (MCP-1) | 6.7 | 6.1 | |
| hepatocyte Cli (rat, human, mL/min/g tissue) | 2.48; 1.42 | <0.8; <0.45 | |
| rat PK (n = 1, 1 mg/kg IV) | CL (mL/min/kg) (% LBF) | 90 (>100%) | 74 (65%) |
| Clrenal (mL/min/kg) | 0.2 | 23 | |
| Vss (L/kg) | 2.3 | 1.4 | |
| T1/2 (h) | 0.4 | 0.4 | |
| rat PK (n = 3, 3 mg/kg po) | Fpo (%) | 2 | |
We considered that reducing the lipophilicity in this series could improve not only pharmacokinetics but also have a beneficial impact on other developability properties such as solubility. However, retaining low molecular weight and ligand efficiency was essential. As compounds 23 and 25 (Table 1) had shown that small polar substituents were poorly tolerated in the amide region and as chemistry enabled further functionalization of the C2 position, we assessed the impact on the potency and lipophilicity of a small polar substituent in this position. Hydroxyl and fluorine were considered, knowing that the latter also reduces lipophilicity when attached to an alkyl group (refs (9−11)).
Compound 26 (Table 2) is a representative example of the C2-hydroxyl derivatives that were prepared. It is less potent (BD2 pIC50 = 7.4) than 21 but remains selective (BD1 pIC50 = 4.5, selectivity 800-fold). Lipophilicity is significantly reduced, but despite increased free fraction, 26 is significantly less potent in human whole blood (pIC50 of 6.1 versus 6.7 for 21). Surprisingly, despite the low turnover in rat hepatocytes, the compound was still rapidly cleared in vivo in rats. Metabolic clearance was somewhat reduced, in line with lower hepatocyte clearance, but an increased renal clearance was observed. This was likely a resulting effect of the reduced ChromLogD altering filtration and/or secretion mechanisms. The elimination half-life remained short and oral bioavailability very low. Overall, the drop in potency and the poor in vivo pharmacokinetics of 26 despite low ChromLogD led us to put work on this subseries on hold and focus on the C2-fluoro derivatives.
The profile of a number of representative examples of C2-fluoro inhibitors is presented in Table 3. The chemical complexity of introducing a substituent on the C3 phenyl ring to tune intrinsic properties led us to focus on modifying the C5-amide. We also knew from the pyridone series that substituting the C3-phenyl ring with electron withdrawing or low LogD substituents was usually detrimental to potency (ref (4)).
Table 3. Profile of C2-Fluoro Inhibitors.
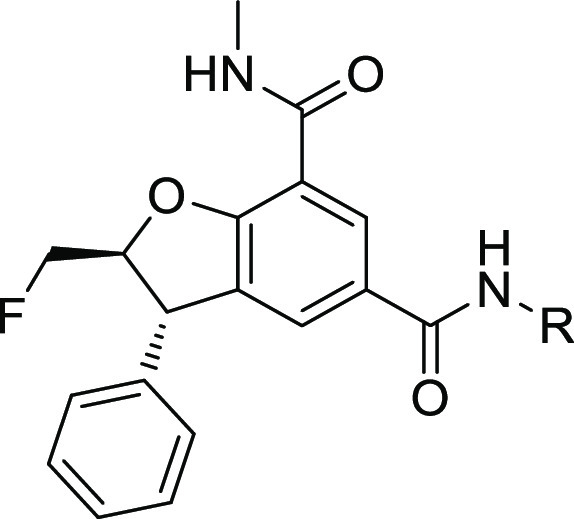
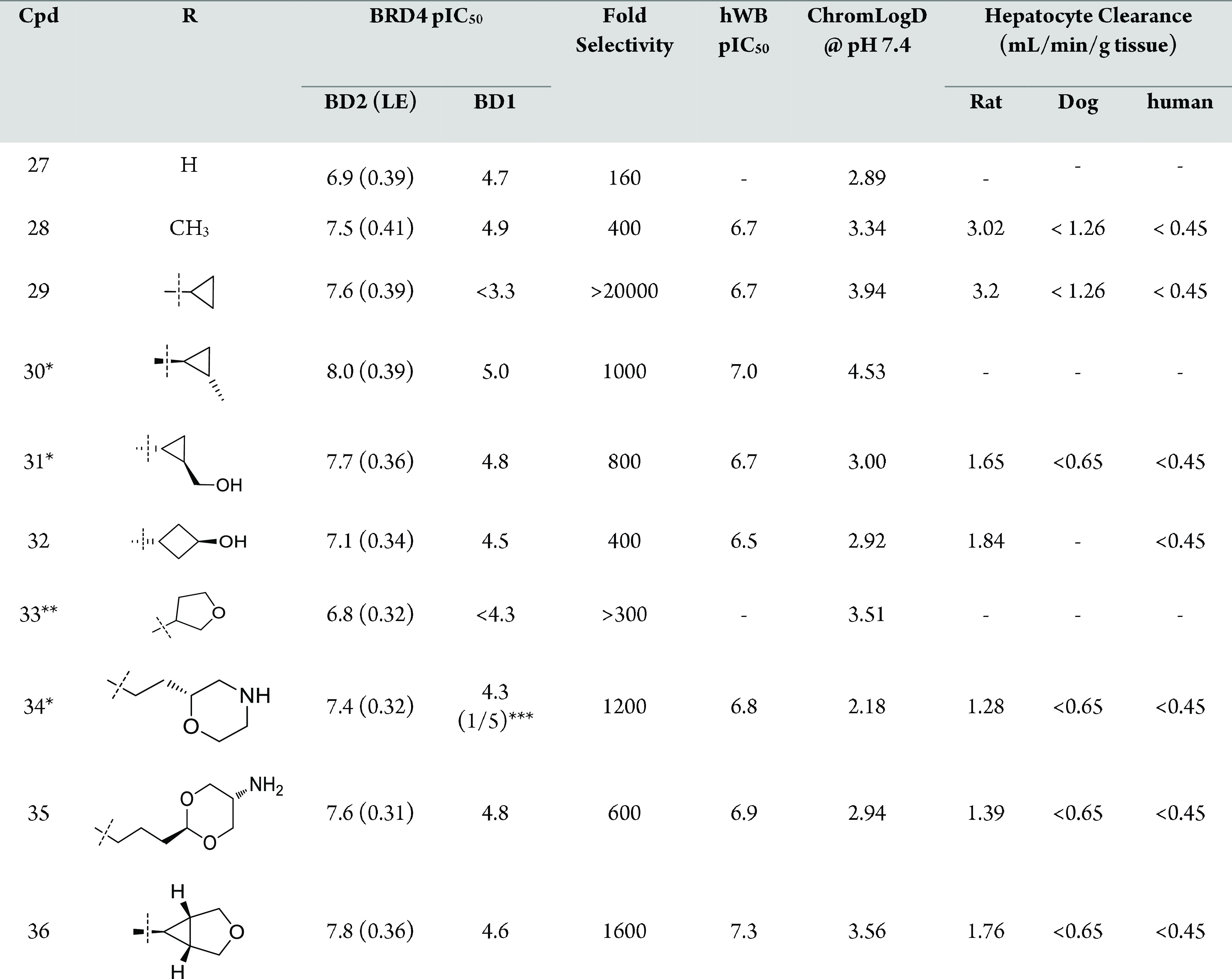
The R group is a single enantiomer.
The R group is a 1:1 mixture of epimers.
The C2-fluoro derivatives were potent and selective with the cyclopropyl derivative 29 providing excellent selectivity to the detriment of lipophilicity (compare 29 with 27 and 28, Table 3). Overall, the ChromLogD values of the C2-fluorine derivatives were lower than their C2-methyl analogue (compare 29 and 21 as examples). Crystallographic data (not shown) suggested that the introduction of a specific methyl substituent on the cyclopropyl ring of 29 could increase van der Waals interactions with the protein, and this proved indeed the case with compound 30 being significantly more potent than 29. It was possible to reduce the lipophilicity by introduction of a hydroxyl (compound 31). All other attempts to introduce substituents balancing potency and selectivity in a reasonable LogD space were unsuccessful (see compounds 32 and 33 as examples). Another opportunity to lower lipophilicity was to introduce basicity on the chain, which was feasible as long as the basic nitrogen was far enough from the amide to be solvent exposed (compounds 34 and 35) (ref (12)). From the cyclopropyl motif, it was also possible to increase selectivity and lower lipophilicity with appropriate ether derivatives (compound 36) (ref (13)). Overall, in vitro potency translated into good human whole blood potency (hWB) with 36 being one of the most potent and selective compounds made.
Regarding in vitro PK, the series showed a marked difference between species with most compounds being more metabolically stable in dog and human hepatocytes than rat. Table 4 describes the in vivo rat PK data for a set of optimized compounds. As can be seen, in most cases, high in vitro clearance translated to high in vivo clearance and, as a result, limited oral bioavailability in the rat when tested. Compound 36 was deemed worth progressing further thanks to its excellent in vitro profile and acceptable exposure in rat. Indeed, it showed excellent pharmacokinetics in dog with low blood clearance (in agreement with the hepatocyte data), good oral bioavailability, and a moderate half-life. The correlation between in vitro and in vivo metabolic clearance provided confidence that low metabolic clearance could be achieved in humans.
Table 4. In Vivo Pharmacokinetics of a Set of C2-Fluoro DBFs.
| compound | 31 | 35 | 36 | ||
|---|---|---|---|---|---|
| AMP (permeability, nM/s) | 73 | 45 | 165 | ||
| CLND solubility (μg/mL) | 114 | 174 | 173 | ||
| species | rata | rata | ratb | dogc | |
| IV PK | CL (mL/min/kg) (% LBF) | 98 (120%) | 73 (81%) | 73 (89%) | 3 (5%) |
| Clrenal (mL/min/kg) | 4 | 10 | 4 | 0.3 | |
| Vss (L/kg) | 1.7 | 2.1 | 2.1 | 1.3 | |
| T1/2 (h) | 0.5 | 0.6 | 0.6 | 4.8 | |
| oral PK | Fpo (%) | 15 | 3 | 48 | 87d |
n = 1, 1 h IV infusion at 1 mg/kg, n = 3 PO gavage at 3 mg/kg.
n = 3, 1 h IV infusion at 1 mg/kg, n = 3 PO gavage at 3 mg/kg.
n = 3, 1 h IV infusion at 1 mg/kg, n = 2 PO gavage at 2 mg/kg.
Wet Beadmilled Spray Dried material (EXP109376) formulated in 1.5% HPMC PHARMACOAT 603:0.15% (w/v) sodium lauryl sulfate (aq) (1.5: v/v).
Compound 36 (GSK973) was further profiled. It showed acceptable FaSSIF solubility from crystalline material (80 μg/mL) and no cardiac liability in vitro (hERG pIC50 < 4.3), and it was negative in the AMES and mouse lymphoma assays (MLA), a glutathione (GSH) trapping, and CYP3A4 time dependent inhibition (TDI) assays. The selectivity profile against the other bromodomains was assessed using BROMOscan (Figure 3, details in the Supporting Information) and showed no significant cross-reactivity (the lowest selectivity was against TAF1 BD2 and was 200-fold). The BD2/BD1 selectivity was confirmed in this assay format as well as by SPR (with Kd against BRD4 BD2 and BD1 of 34 nM and >3000 nM; see the Supporting Information). As a further assessment of possible off-target activity, 36 was screened and proved inactive against an internal panel of 40 targets considered as potential liabilities (data not shown). The enantiomer of GSK973, with BD1/BD2 pIC50’s of <4.3 and 5.1, represents a good negative control (ref (14)).
Figure 3.
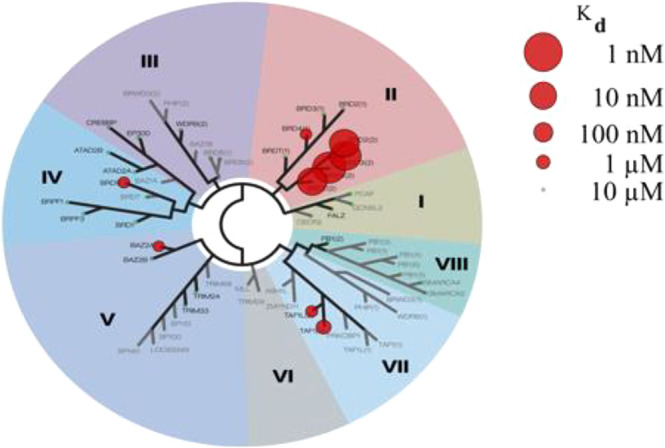
BROMOscan data for compound 36.
A crystal structure of compound 36 bound to BRD4 BD2 superimposed with 1 is shown in Figure 4. As intended, both bind to the KAc pocket in a similar way. The carbonyl of the C7 amide of GSK973 hydrogen-bonds to the conserved asparagine Asn429 and via a bridging water molecule to Tyr386. In addition, the NH of the C5 amide makes a second interaction to the carbonyl group of Asn429. The phenyl group in the C3 position sits on the WPF shelf and is involved in three-way edge-to-face aromatic interactions between Trp370 and His433. The fluoromethyl group off the C2 position projects into the ZA channel, between Typ370 and Leu381.
Figure 4.
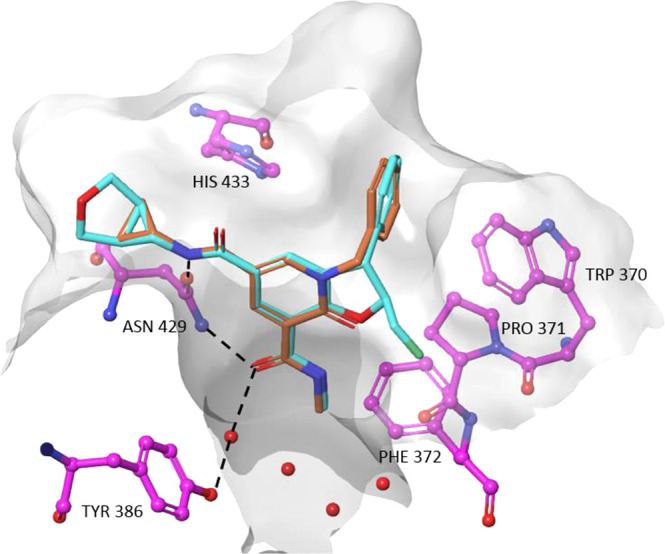
Compounds 1 (blue, PDB: 6zb1) and 36 (brown, PDB 6z8p) bound to BRD2 BD2.
In conclusion, our strategy of constraining the WPF shelf substituent of the pyridone series using a DBF template in order to obtain more potent and selective pan-BD2 BET inhibitors proved successful. Compound 36 is a drug-like inhibitor with exceptional potency and selectivity. Its in vitro profile and good pharmacokinetics in preclinical species make it a useful tool for further probing the separate contributions of the BDs to BET-associated functional phenotypes.
Acknowledgments
Thanks to Dr Richard Upton for NMR support, Mr Adam Flinders for generating the SPR data, and Mrs Emily Lowdnes for crystallization support.
Glossary
Abbreviations
- AMP
artificial membrane permeability
- BET
bromodomain and extra-terminal
- BD
bromodomain
- DBF
dihydrobenzofuran
- LBF
liver blood flow
- LE
ligand efficiency
- FaSSIF
fasted state simulated intestinal fluid
- hWB
human whole blood
- TAF1
transcription initiation factor TFIID subunit 1.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00247.
Assay and pharmacokinetic study protocols, protein production and crystallographic information, compound synthesis and characterization, and BROMOscan data for 36 (PDF)
Author Present Address
∥ S.T.: DMPK, Drug Discovery Services, Pharmaron, Hertford Road, Hoddesdon, Hertfordshire EN11 9BU, U.K.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Financial support for this research was provided by GSK.
The authors declare no competing financial interest.
Notes
PDB: The authors will release the atomic coordinates and experimental data upon article publication.
Supplementary Material
References
- Filippakopoulos P.; Knapp S. Targetting bromodomains: epigenetic readers of lysine acetylation. Nat. Rev. Drug Discovery 2014, 13, 337–356. 10.1038/nrd4286. [DOI] [PubMed] [Google Scholar]
- Tough D. F.; Tak P. P.; Tarakhovsky A.; Prinjha R. K. Epigenetic drug discovery: breaking through the immune barrier. Nat. Rev. Drug Discovery 2016, 15, 835–853. 10.1038/nrd.2016.185. [DOI] [PubMed] [Google Scholar]
- Pervaiz M.; Mishra P.; Gunther S. Bromodomain Drug Discovery – The Past, the Present, and the Future. Chem. Rec. 2018, 18, 1808–1817. 10.1002/tcr.201800074. [DOI] [PubMed] [Google Scholar]
- Seal J.; Atkinson S. J.; Aylott H.; Bamborough P.; Chung C.-w.; Copley R. C. B.; Gordon L.; Grandi P.; Gray J. R. J.; Harrison L. A.; Hayhow T. G.; Lindon M.; Messenger C.; Michon A.-M.; Mitchell D. J.; Preston A.; Prinjha R. K.; Rioja I.; Taylor S.; Wall I.; Watson R. J.; Woolven J.; Demont E. H.. The Optimisation of a Novel, Weak Bromo and Extra Terminal Domain (BET) Bromodomain Fragment Ligand to a Potent and Selective Second Bromodomain (BD2) Inhibitor. J. Med. Chem., submitted for publication, 2020. [DOI] [PubMed] [Google Scholar]
- Gilan O.; Rioja I.; Knezevic K.; Bell M. J.; Yeung M. M.; Harker N. R.; Lam E. Y. N.; Chung C.-w.; Bamborough P.; Petretich M.; Urh M.; Atkinson S. J.; Bassil A. K.; Roberts E. J.; Vassiliadis D.; Burr M. L.; Preston A. G. S.; Wellaway C.; Werner T.; Gray J. R.; Michon A.-M.; Gobbetti T.; Kumar V.; Soden P. E.; Haynes A.; Vappiani J.; Tough D. F.; Taylor S.; Dawson S.-J.; Bantscheff M.; Lindon M.; Drewes G.; Demont E. H.; Daniels D. L.; Grandi P.; Prinjha R. K.; Dawson M. A. Selective targeting of BD1 and BD2 of the BET proteins in cancer and immuno-inflammation. Science 2020, 368, 387–394. 10.1126/science.aaz8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preston A.; Atkinson S. J.; Seal J.; Mitchell D. J.; Watson R. J.; Gray J. R. J.; Taylor S.; Woolven J. M.; Wall I.; Chung C.-w.; Bamborough P.; Rianjongdee F.; Grandi P.; Michon A.-M.; Rioja I.; Prinjha R. K.; Lindon M. J.; Demont E. H.. The Design and Synthesis of a Highly Selective and in Vivo Capable Inhibitor of the Second Bromodomain (BD2) of the Bromodomain and Extra Terminal Domain (BET) Family of Proteins. J. Med. Chem., submitted for publication, 2020. [DOI] [PubMed] [Google Scholar]
- Faivre E. J.; McDaniel K. F.; Albert D. H.; Mantena S. R.; Plotnik J. P.; Wilcox D.; Zhang L.; Bui M. H.; Sheppard G. S.; Wang L.; Sehgal V.; Lin X.; Huang X.; Lu X.; Uziel T.; Hessler P.; Lam L. T.; Bellin R. J.; Mehta G.; Fidanze S.; Pratt J. K.; Liu D.; Hasvold L. A.; Sun C.; Panchal S. C.; Nicolette J. J.; Fossey S. L.; Park C. H.; Longenecker K.; Bigelow L.; Torrent M.; Rosenberg S. H.; Kati W. M.; Shen Y. Selective inhibition of the BD2 bromodomain of BET proteins in prostate cancer. Nature 2020, 578 (7794), 306–310. 10.1038/s41586-020-1930-8. [DOI] [PubMed] [Google Scholar]
- Sheppard G. S.; Wang L.; Fidanze S. D.; Hasvold L. A.; Liu D.; Pratt J. K.; Park C. H.; Longenecker K. L.; Qiu W.; Torrent M.; Kovar P.; Bui M.; Faivre E. J.; Huang X.; Lin X.; Wilcox D.; Zhang L.; Shen Y.; Albert D. H.; Magoc T. J.; Rajaraman G.; Kati W. M.; McDaniel K. F. Discovery of N-Ethyl-4-[2-(4-fluoro-2,6-dimethyl-phenoxy)-5-(1-hydroxy-1-methyl-ethyl)phenyl]-6-methyl-7-oxo-1H-pyrrolo[2,3-c]pyridine-2-carboxamide (ABBV-744), a BET Bromodomain Inhibitor with Selectivity for the Second Bromodomain. J. Med. Chem. 2020, 63, 5585–5623. 10.1021/acs.jmedchem.0c00628. [DOI] [PubMed] [Google Scholar]
- Gillis E. P.; Eastman K. J.; Hill M. D.; Donnelly D. J.; Meanwell N. A. Applications of fluorine in medicinal chemistry. J. Med. Chem. 2015, 58, 8315–8359. 10.1021/acs.jmedchem.5b00258. [DOI] [PubMed] [Google Scholar]
- Bamborough P.; Chung C.-w.; Demont E.; Furze R.; Bannister A.; Che K.; Diallo H.; Douault C.; Paola Grandi P.; Tony Kouzarides T.; Michon A.-M.; Mitchell D.; Prinjha R.; Rau C.; Robson S.; Sheppard R.; Upton R.; Watson R. A chemical probe for the ATAD2 bromodomain. Angew. Chem., Int. Ed. 2016, 55, 11382–11386. 10.1002/anie.201603928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meanwell N. A. Fluorine and fluorinated motifs in the design and application of bioisosteres for drug design. J. Med. Chem. 2018, 61, 5822–5880. 10.1021/acs.jmedchem.7b01788. [DOI] [PubMed] [Google Scholar]
- The amine of 35 is known to be stable and of low pKa. See:Ndubaku C. O.; Crawford J. J.; Drobnick J.; Aliagas I.; Campbell D.; Dong P.; Dornan L. M.; Duron S.; Epler J.; Gazzard L.; Heise C. E.; Hoeflich K. P.; Jakubiak D.; La H.; Lee W.; Lin B.; Lyssikatos J. P.; Maksimoska J.; Marmorstein R.; Murray L. J.; O’Brien T.; Oh A.; Ramaswamy S.; Wang W.; Zhao X.; Zhong Y.; Blackwood E.; Rudolph J. Design of selective PAK1 inhibitor G-5555: improving properties by employing an unorthodox low-pKa polar moiety. ACS Med. Chem. Lett. 2015, 6, 1241–1246. 10.1021/acsmedchemlett.5b00398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The detail of the synthesis of the amines present in compounds 35 and 36 can be found in the Supporting Information.
- Bunnage M. E.; Piantniski Chekler E. L.; Jones L. H. Target validation using chemical probes. Nat. Chem. Biol. 2013, 9, 195–199. 10.1038/nchembio.1197. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.