

Abstract
Inhibition of neuronal nitric oxide synthase (nNOS), an enzyme implicated in neurodegenerative disorders, is an attractive strategy for treating or preventing these diseases. We previously developed several classes of 2-aminoquinoline-based nNOS inhibitors, but these compounds had drawbacks including off-target promiscuity, low activity against human nNOS, and only modest selectivity for nNOS over related enzymes. In this study, we synthesized new nNOS inhibitors based on 7-phenyl-2-aminoquinoline, and assayed them against rat and human nNOS, human eNOS, and murine and (in some cases) human iNOS. Compounds with a meta-relationship between the aminoquinoline and a positively charged tail moiety were potent and had up to nearly 900-fold selectivity for human nNOS over human eNOS. X-ray crystallography indicates that the amino groups of some compounds occupy a water-filled pocket surrounding an nNOS-specific aspartate residue (absent in eNOS). This interaction was confirmed by mutagenesis studies, making 7-phenyl-2-aminoquinolines the first aminoquinolines to interact with this residue.
Graphical Abstract

Introduction
Neurodegenerative diseases such as Alzheimer’s, Huntington’s, Parkinson’s, and amyotrophic lateral sclerosis (ALS), are characterized by the gradual loss of neuronal integrity and are responsible for a wide range of neurological deficiencies. Neuronal damage or death associated with stroke, ischemic events, and cerebral palsy (as well as acute or chronic brain injuries) has also been linked to similarly debilitating motor, cognitive, and psychological impairments. The overproduction of the vital secondary messenger nitric oxide (NO), produced by neuronal nitric oxide synthase (nNOS) in tissues of the central (CNS) and peripheral nervous system (PNS), is directly implicated in these disorders.1,2 Because NO plays a key role in these diseases, rational control of NO levels in neuronal tissues via nNOS-specific inhibition is therapeutically desirable.
Nitric oxide synthases (NOS) are a family of homodimeric enzymes that are responsible for the biosynthesis of NO. Functional regulation of NO is differentiated by subcellular localization, tissue distribution, and regulatory gene expression of three isoforms of NOS: endothelial NOS (eNOS), inducible NOS (iNOS), and neuronal NOS (nNOS), which are responsible for regulating blood pressure and vascular tone, immune activation, and normal neuronal communication, respectively.3 Functional NOS is a homodimer. Each NOS monomer contains a reductase domain and an oxygenase domain, separated by a flexible region where calmodulin binds when activated by calcium ions. The reductase domain contains binding sites for flavin adenine dinucleotide (FAD), flavin mononucleotide (FMN), and reduced nicotinamide adenine dinucleotide phosphate (NADPH), whereas the oxygenase domain contains binding sites for (6R)-5,6,7,8-tetrahydrobiopterin (H4B), the metallocofactor heme, and the substrate L-arginine. Electron flow proceeds from one monomer’s reductase domain, sequentially through NADPH, FAD, and FMN, to the opposite monomer’s oxygenase domain, where the electron is transferred between FMN and heme, by which L-arginine is oxidized to L-citrulline and NO.4
Most compounds initially investigated for nNOS inhibition were designed as competitive mimics of L-arginine. These inhibitors have high basicity and polarity, a large total polar surface area (tPSA), and an overabundance of hydrogen bond donors, and as a result, suffer from poor bioavailability and blood-brain barrier (BBB) penetrability. Furthermore, many promising L-arginine-mimetic inhibitors are not nNOS-selective, owing to the high sequence similarity and nearly identical active-site architecture between the three NOS isoforms. High nNOS selectivity is crucial; non-selective inhibitors have the potential for dangerous side effects. For example, iNOS inhibition could impair immune system activation, while eNOS inhibition can lead to severe hypertension or other cardiovascular complications.5
We have been investigating 2-aminoquinoline-based scaffolds as isoform-selective arginine bioisosteres with more favorable pharmacokinetic properties. Since 2014, we have reported several generations of aminoquinolines that are modestly potent and selective towards nNOS (Figure 1). The first generation of aminoquinolines (such as 16), were found to be potent and selective nNOS inhibitors with improved pharmacokinetics. Unfortunately, 1 was found to have high rat nNOS (rnNOS) over human nNOS (hnNOS) selectivity, low hnNOS over human eNOS (heNOS) selectivity and caused toxic side effects, possibly because of its off-target promiscuity.4 The second generation of NOS inhibitors, (e. g., phenyl ether 27) reduced off-target binding while preserving potency and selectivity against rnNOS. However, these compounds suffered from decreased Caco-2 permeability, low hnNOS activity, and similarly low hn/heNOS selectivity. Newer generation inhibitors, such as 3 and 4, improved upon their respective parent series by incorporating elements such as the quinoline 4-methyl group and cyano-containing tail moieties.8,9 These compounds have greatly enhanced hnNOS potency, hn/heNOS selectivity, and improved cellular permeability and off-target profiles.
Figure 1.
Representative aminoquinoline nNOS inhibitors
Because of some of the drawbacks associated with previous inhibitor generations, we have been investigating alternative aminoquinoline-containing scaffolds. Interestingly, the 7-phenylquinoline compound 5 appears in the literature as part of a Glaxo-SmithKline screening library and was recently employed in several high-throughput screening studies.10,11 However, there are no articles, patents, or other reports of what research program this compound may have belonged to originally, but it appears to now be part of an “open source” drug discovery program.
Because of its distinctively nNOS inhibitor-like structure but with fewer rotatable bonds than earlier series, a docking study with 5 in a nNOS crystal structure was conducted. Consequently, 5 was predicted to bind in an nNOS inhibitor-like mode, in which the aminoquinoline forms a salt-bridge with Glu592/Glu597 (rnNOS/hnNOS) and the phenethylamine tail portion faces out toward the regions of the active site. To this end, lead compound 5 and related compounds 6–9 (modified at the amine portion (Figure 2) were designed, synthesized, and assayed against purified NOS isoforms to test the hypothesis that 5 and analogues could act as nNOS inhibitors. Satisfyingly, these compounds possessed encouraging nNOS inhibitory activity and good isoform selectivity, and we chose to undertake a more thorough structure-activity relationship (SAR) study. First, we investigated whether meta- or ortho-substitution of the central phenyl ring (compounds 10–13) might be more effective than the para-substitution of the parent compound. This early optimization revealed that meta-substituted analogue 12 displayed good inhibitory potency against rat and human nNOS, excellent hn/heNOS selectivity and n/i selectivity, as well as good solubility and desirable properties (few rotatable bonds and low tPSA).
Figure 2.

Initial derivatives of 5 prepared in this study
Encouraged by both the inhibitory constants and the agreement between X-ray crystallography and our docking results, 12 was used as a launching point for further optimization. Efforts were made to develop a set of compounds with modifications made at the 5-position of the central phenyl ring (e.g., nitrile 14 and pyridine 15) to investigate whether additional interactions could be made with the heme propionate or another active site residue.
Because of 12’s high n/eNOS selectivity, we hypothesized that the flexible tail amino group might be contacting (directly or otherwise) a specific aspartate residue (Asp597/Asp602 in rnNOS/hnNOS, respectively). This residue is missing in eNOS isoforms, replaced by asparagine. Consequently, contact (H-bonding or electrostatic) between an inhibitor and this residue can impart very high n/eNOS selectivity (1000-fold or more). We hypothesized that a second set of compounds could be designed to solidify any existing contacts with Asp597/Asp602 by incorporating the tail amino group functionality into a rigid ring system, thereby reducing its overall flexibility and locking the interaction in place. As the amino group of 5 is quite flexible, both meta-/para- and ortho-/meta-constrained derivatives (isoindoline 16 and the two isomeric racemic indanylamines 17 and 18) were prepared.
Additional docking studies indicated that a variety of groups might be accommodated at the 4-position of the central phenyl ring of 12, which could form van der Waals interactions with Met336/Met341 (rnNOS/hnNOS), a residue that was previously implicated in high n/eNOS selectivity for 2-aminoquinoline-based inhibitors7 as it is absent in eNOS isoforms (replaced by a smaller valine).12 To this end, 3,4-substituted compounds 19–37 (Figure 3), possessing a variety of steric, electronic, and H-bonding substituents at the 4-position were investigated to determine if this substitution pattern could make extra contacts with the isoform-specific residues Met336/Met341 and/or the hnNOS-specific residue His342.
Figure 3.

Optimization of 12 by 5-substitution, amino group constraint, and 4-substitution
All synthesized compounds were assayed against rnNOS, and selected compounds were also assayed against hnNOS. Murine iNOS and human eNOS were used to determine selectivity, and selected compounds were also assayed against human iNOS.
Results and Discussion
Chemistry.
To prepare compound 5, we envisioned that the quinoline-aryl bond could be constructed via Suzuki coupling. To this end, we sought to install the boron-containing moiety on the quinoline, taking advantage of a large and diverse set of available aryl halides (which are less expensive and easier to synthesize than an analogous series of boronates or boronic esters). Using versatile 7-bromoquinoline 38,8 a 7-BPin moiety was first installed via Miyaura borylation (Scheme 1). This intermediate was not isolated but rather converted to trifluoroborate 39, which was readily purified because of its insolubility in most organic solvents.
Scheme 1a.

Synthesis of lead 5
To prepare the halide precursor, commercially available 4-bromophenethylamine 40 was Boc-protected to yield 41. Many Suzuki conditions were screened for the coupling of 39 and 41, but the strong protic bases usually required for reductive elimination and activation of 39 often led to deacetylation of the quinoline and decomposition. The use of NaHCO3 as the base13 in a mixed aqueous solvent was more successful, and microwave irradiation of this mixture yielded phenylquinoline 42 within 25 minutes at 120 ˚C without substantial deacetylation. The intermediate protected phenylquinolines were not extensively characterized but were isolated and immediately deprotected. Deprotection was accomplished stepwise, first with K2CO3 in refluxing methanol to cleave the acetyl group, followed by treatment of the free aminoquinoline with methanolic HCl to remove the Boc group and provide 5 as its water-soluble dihydrochloride salt.6
To prepare the initial set of derivatives with different para-aminoalkyl tail portions (6–9), the halides were first prepared. Compound 41 was methylated to yield 43 (Scheme 2A). Commercially available iodobenzylamine 44 was Boc-protected to yield 45, which was also methylated to yield 46 (Scheme 2B). For the (S)-alpha-methyl-phenethylamine group of 9, the Ellman auxiliary method14 was used (Scheme 2C). Ketone 47 was condensed with (S)-tert-butylsulfinamide, and the intermediate sulfinyl imine was reduced at low temperature to afford (S,S)-48 in a good d.r. of ~7:1.
Scheme 2a.

Preparation of precursor p-, o-, and m-substituted halides
Desulfinylation under acidic conditions and Boc-protection subsequently afforded derivative 49. For ortho- and meta-substituted derivatives 10/11 and 12/13, respectively, the commercially available benzylamines (50, 54) and phenethylamines (51, 55) were Boc-protected to yield o-substituted (52, 53) and m-substituted (56, 57) bromides, respectively (Schemes 2D and 2E). Suzuki coupling between 39 and these halides under the conditions described above (Scheme 3) was facile and displayed a high substrate tolerance, affording protected phenylquinolines 5–65 in good yields. Generally, the only impurity isolated was a small amount (<10%) of proto-deborylated acetamidoquinoline. Deprotection of 58–66 (Scheme 3) afforded analogues 6–13.
Scheme 3.

Assembly and deprotection of p-, o-, and m-substituted phenylquinolines.
To synthesize 5-cyano derivative 14, bromobenzene 66 was prepared as previously described. Treatment of 66 with the anion derived from Boc2NH (Scheme 4A) yielded, surprisingly, mono-Boc protected amine 67, indicating that one Boc group was cleaved during the reaction or workup. Suzuki coupling with 39 yielded 68, which was then deprotected to provide amine 14. In contrast, heating Boc2NH and pyridine 69 under basic conditions (Scheme 4B) afforded the N,N-di-Boc compound (70).15 Likewise, the major product (71) isolated upon coupling of 70 and 39 contained both Boc groups intact. During deacetylation, a longer period of heating (4.5 h) was employed to remove both the acetyl group and one Boc group, and the second Boc group was then removed with HCl to yield 15.
Scheme 4.

Synthesis of 14 and 15.
Synthesis of isoindoline derivative 16 (Scheme 5A) commenced with commercially available 4-bromoisoindoline salt 72, which was converted to the free base and Boc-protected to yield 73. Coupling with 39 afforded 74, which then yielded 16 upon deprotection. Indanylamine derivatives 17 and 18 (Scheme 5B) were prepared from the 5- and 6-bromoindanones (75 and 76), respectively. The Ellman method (as in Scheme 2C)14 was used to install the amino group as its racemate. However, yields of sulfinamides 77 and 78 were fairly low, and large amounts of insoluble ketone condensation by-products were obtained. Nonetheless, 77 and 78 were desulfinylated and Boc-protected, and carbamates 79 and 80 were readily amenable to Suzuki coupling with 39, affording 81 and 82, which were deprotected to yield, respectively, 17 and 18.
Scheme 5a.

Synthesis of conformationally constrained derivatives 16-18
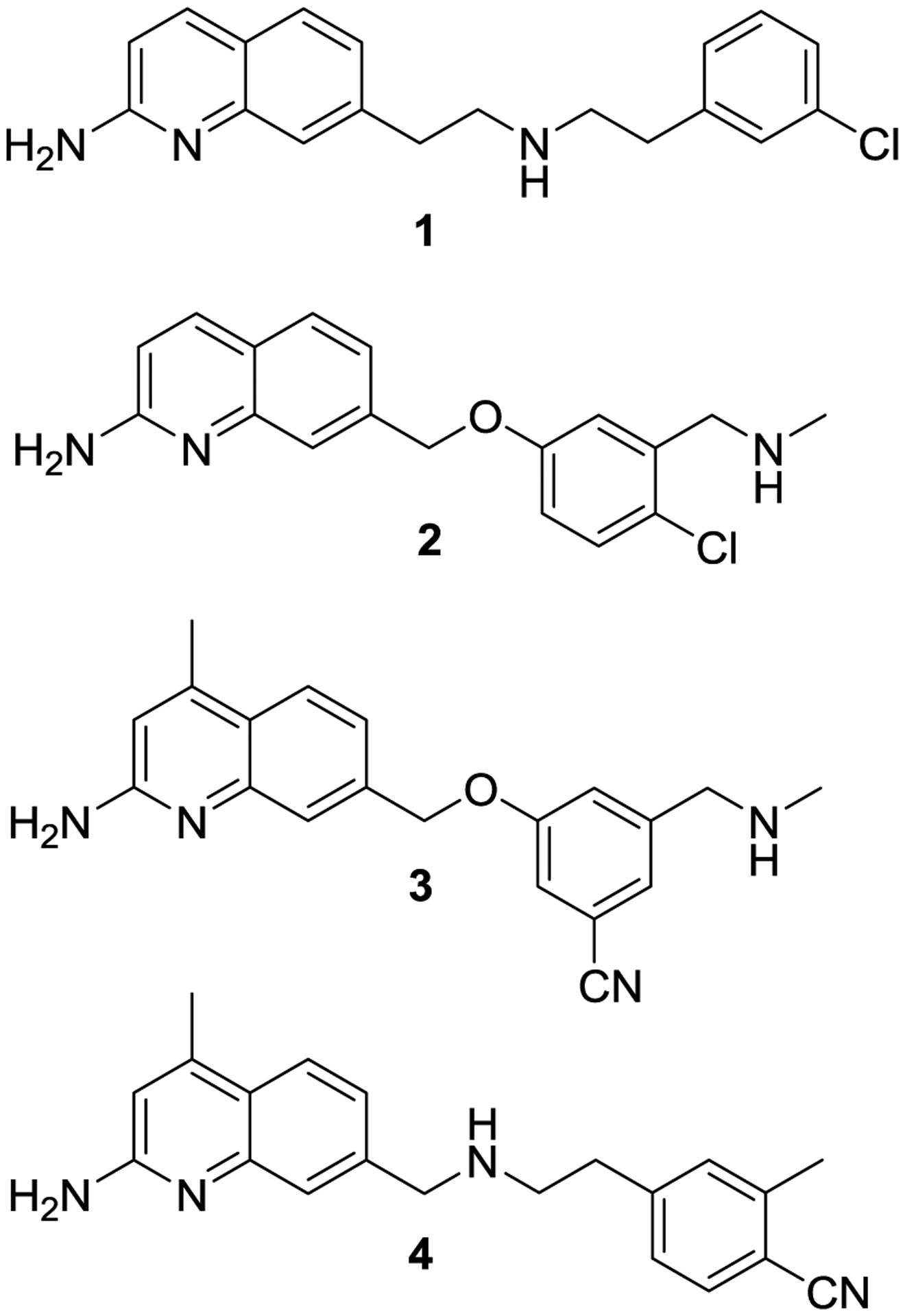
We envisioned that 4-substituted derivatives 19-24 could be accessed from commercially available benzaldehydes or toluene derivatives via conversion to the benzyl halides. To this end, fluorotoluene 83 and chlorotoluene 84 (for 19 and 20) were converted to benzyl bromides 85 and 86. A Delépine reaction, involving treatment with hexamethylenetetramine and acidic hydrolysis of the resulting hexaminium adduct, and subsequent Boc-protection of the amine yielded 87 and 88 (Scheme 6A). For the trifluoromethyl derivative en route to 21, commercially available nitrile 90 was reduced with BH3-DMS,16 and the isolated amine was protected to yield 91 (Scheme 6B). 2-Ethybenzaldehyde (91, for 22) was complexed with AlCl317 and brominated to yield the major regioisomer (92b) as an inseparable 3:1 mixture with 92a. Following borohydride reduction, the isomers were separated, and the major isomer (93) was chlorinated to yield 94. Delépine reaction and Boc protection provided bromobenzene derivative 95 (Scheme 6C).
Scheme 6a.
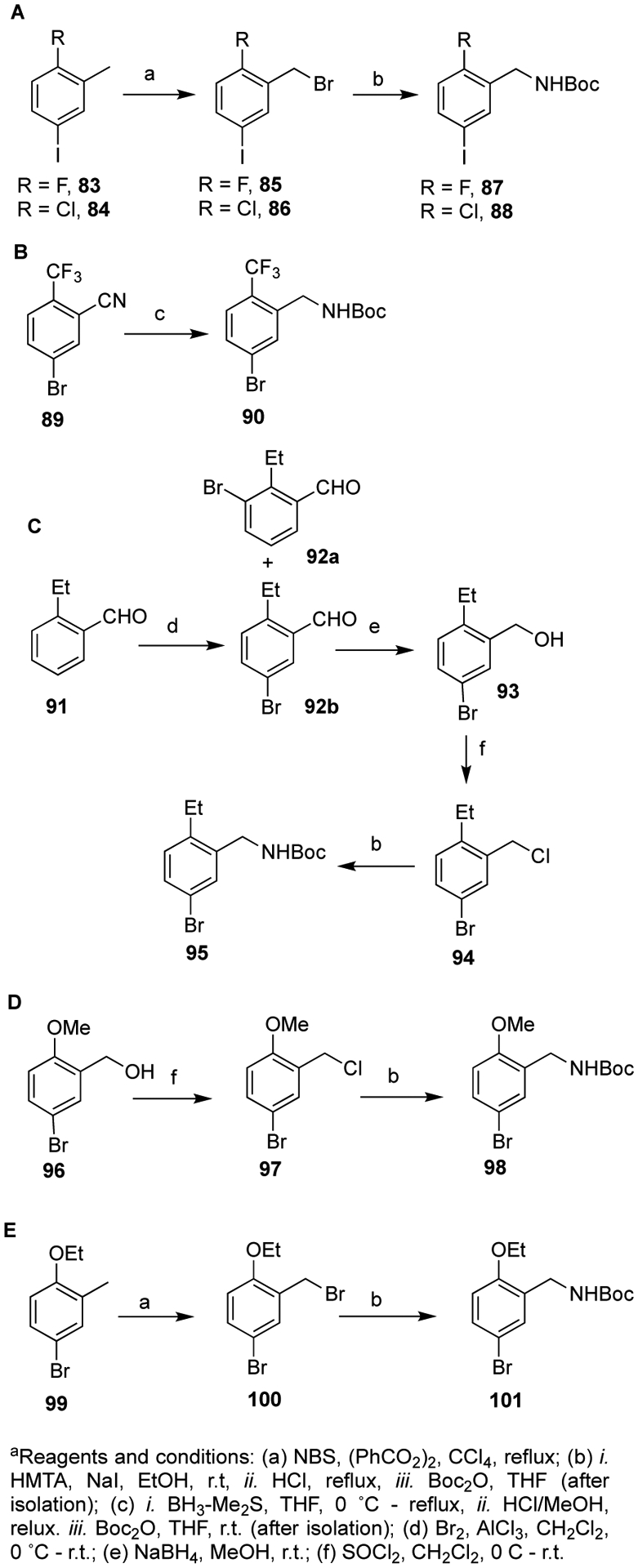
Preparation of intermediate carbamates for the preparation of 4-substituted derivatives
Methoxybenzyl alcohol 96 (for 23) was prepared as previously described (Scheme 6D) and elaborated via 97 as described above to yield 98. Finally, ethoxylated toluene 99 was brominated to yield 100 (Scheme 6E). Delépine reaction and Boc protection afforded carbamate 101.
As the multiple steps of this route would make the preparation of many similar analogues time consuming, and the Boc2NH method of Scheme 4 was unpredictable, a slightly different strategy was used to prepare 4-ether halide derivatives 25–37 (Scheme 7). In this route, protected amine 103 was first prepared via reduction of 102, and then the ether functionality was installed by deprotonation of the phenol and treatment with alkyl or benzyl halides.
Scheme 7a.

Preparation of ether-substituted carbamates and final assembly of 4-substituted analogues 19–37
By this method, the n-propyl (104), isopropyl (105), isobutyl (106), methylcyclobutyl (107), methylcyclopropyl (108), 3-fluorobenzyl (109), 4-cyanobenzyl (110), (5-methylisoxazol-3-methyl (113), 4- and 5-methyl thiazoles (114/116), and oxazol-4-methyl (115) ethers were prepared. As the thiazol-5-methyl chloride and pyridylmethyl bromides are only commercially available as the HCl and HBr salts, respectively, these salts were converted to their free base immediately prior to the formation of the 2-pyridylmethyl (111), 3-pyridylmethyl (112), and thiazol-5-methyl (116) ethers. All of these ether-containing halides were subjected to Suzuki coupling with 39 to yield protected phenylquinolines 117–135 in moderate to excellent yields, and stepwise deprotection as described above yielded final analogues 19–37.
nNOS Inhibitory Assay and Crystallography.
The hemoglobin capture assay (see Experimental Section) was used to determine the inhibition constants (Ki) of synthesized compounds 5–37.18,19 All compounds were assayed against purified rnNOS as a prescreen, and eighteen of the most potent compounds against rnNOS were further assayed against purified human nNOS (hnNOS), murine iNOS (miNOS), and human eNOS (heNOS) to determine isoform selectivity. Table 1 summarizes the apparent Ki values and isoform selectivities for 5–37. Values for compounds 1–4 are included for comparison. The rnNOS and miNOS isoforms were used to approximate n/i isoform selectivity, because they are the easiest to express and purify, and those are the species used for crystallography. Furthermore, for preclinical purposes, it is essential to prove efficacy and selectivity in lower animals prior to advancement to clinical trials. Recent advances have made it possible to obtain and crystallize both hnNOS and heNOS, which were used to support our human isoform SAR development. Because high-resolution structures of murine and human iNOS were not available until more recently, the majority of the structural discussion will focus on nNOS and eNOS; discussion of a comparison between murine and human iNOS inhibition follows that.
Table 1.
Inhibition of NOS enzymes by synthesized compounds 5–37.a
| Ki (μM)a | Selectivity | ||||||
|---|---|---|---|---|---|---|---|
| compd | rnNOS | hnNOS | miNOS | heNOS | rn/mi | hn/he | |
| 1 | 0.066 | 0.440 | 28.4 | 11.8 | 431 | 27 | |
| 2 | 0.058 | 0.295 | 27.7 | 7.41 | 478 | 25 | |
| 3 | 0.033 | 0.031 | 6.7 | 5.63 | 203 | 181 | |
| 4 | 0.025 | 0.030 | 4.83 | 5.76 | 193 | 192 | |
| 5 | 0.105 | 0.122 | 21.7 | 23.3 | 207 | 191 | |
| 6 | 0.246 | - | - | - | - | - | |
| 7 | 0.045 | 0.088 | 16.3 | 18.7 | 362 | 212 | |
| 8 | 0.090 | 0.149 | - | - | - | - | |
| 9 | 0.140 | - | 27.5 | - | 197 | - | |
| 10 | 0.119 | 0.151 | 2.86 | 41.9 | 24 | 277 | |
| 11 | 0.935 | - | - | - | - | - | |
| 12 | 0.055 | 0.060 | 24.2 | 52.6 | 440 | 877 | |
| 13 | 0.107 | - | - | - | - | - | |
| 14 | 1.03 | - | - | - | - | - | |
| 15 | 0.285 | - | - | - | - | - | |
| 16 | 0.254 | - | - | - | - | - | |
| 17 | 0.159 | - | - | - | - | - | |
| 18 | 0.180 | - | - | - | - | - | |
| 19 | 0.108 | - | - | - | - | - | |
| 20 | 0.235 | - | - | - | - | - | |
| 21 | 1.42 | - | - | - | - | - | |
| 22 | 0.390 | - | - | - | - | - | |
| 23 | 0.106 | - | - | - | - | - | |
| 24 | 0.058 | 0.111 | 24.2 | 18.2 | 418 | 164 | |
| 25 | 0.072 | 0.058 | 23.9 | 12.9 | 333 | 223 | |
| 26 | 0.071 | 0.066 | 25.5 | 20.2 | 360 | 307 | |
| 27 | 0.062 | 0.072 | 9.66 | 5.65 | 156 | 78 | |
| 28 | 0.049 | 0.096 | 10.4 | 22.8 | 212 | 238 | |
| 29 | 0.039 | 0.046 | 32.8 | 21.0 | 841 | 457 | |
| 30 | 0.083 | - | - | - | - | - | |
| 31 | 0.055 | 0.114 | 13.5 | 8.31 | 246 | 73 | |
| 32 | 0.052 | 0.076 | 45.7 | 23.7 | 879 | 312 | |
| 33 | 0.044 | 0.045 | 8.18 | 7.73 | 186 | 172 | |
| 34 | 0.056 | 0.106 | - | - | - | - | |
| 35 | 0.043 | 0.031 | 4.32 | 6.20 | 100 | 200 | |
| 36 | 0.043 | 0.063 | 16.4 | 27.3 | 382 | 433 | |
| 37 | 0.053 | 0.046 | 21.0 | 20.0 | 397 | 435 | |
The compounds were assayed for in vitro inhibition against four purified NOS isoforms: rat nNOS (rnNOS), human nNOS (hnNOS), murine iNOS (miNOS), and human eNOS (heNOS) using known literature methods (see Experimental Section for details), and Ki values are calculated directly from IC50 values. IC50 values are the average of at least two replicates from 6–9 data points; all experimental standard error values (for the LogIC50) are less than 10%, and all correlation coefficients are good (r2> 0.87). Selectivity values are ratios of respective Ki values.
Initial Inhibitory and Structural Analysis of Modified Amine Tail Analogues.
The initial lead 7-phenyl-2-aminoquinoline (5) has good rat and human nNOS inhibitory activity (105 nM and 122 nM, respectively) with moderately high n/iNOS and n/eNOS selectivity of 207-fold and 191-fold, respectively. The X-ray crystal structure of 5 bound to rnNOS, hnNOS, and heNOS (Figure 4A–C) revealed the structural basis for inhibitory potency. The quinoline portion of 5 mimics arginine and forms a bifurcated hydrogen bond system with the main chain carbonyl of Trp587/Trp592 (rnNOS/hnNOS) and the side chain carboxylate of Glu592/Glu602 (rnNOS/hnNOS). This is identical to the structural details observed for other nNOS inhibitors containing a 2-aminoquinoline.6,7,8,9 All three crystal structures clearly reveal that the central phenyl ring resides between heme propionates A and D. In the hnNOS-5 structure, the tail phenethylamine moiety makes a direct H-bond interaction with the H4B and propionate A, displacing the water there, while the rnNOS-5 structure only makes H-bonding contacts with the carbonyl of the H4B and is unable to displace the water molecule bridging propionate A and H4B.
Figure 4.

X-ray crystal structures of 5 bound in the active sites of (A) rnNOS, (B) hnNOS, and (C) heNOS at 1.80 Å, 1.95 Å, and 2.19 Å resolution, respectively. In this and all of the following figures the Polder Fo – Fc omit maps are contoured at 3.5 σ for the bound inhibitors. Major hydrogen bonds are depicted with dashed lines. Two heme propionates are labeled as A and D. Figures were made with PyMol.67
In heNOS there are two molecules of 5 bound. Ligand A binds in the active site in a manner very similar to the nNOS structures, while Ligand B displaces H4B, with the aminoquinoline positioned in the pterin binding pocket. As a result, the tail ethylamine of 5 bound to heNOS is oriented in the opposite direction to what we observed in the nNOS structures, and instead interacts with propionate D. Ligand B is stabilized by aromatic stacking interactions with both Trp447 as well as Trp74 and Phe460 from the opposite chain at the dimer interface. Two-site binding is not unique to 5 and has been observed in several NOS-inhibitor structures.20,21 In all four molecules of the asymmetric unit of heNOS, the aminoquinoline and phenyl rings of 5 are clearly defined. The electron density is weaker for the tail ethylamines, with slightly more density observed near propionate D, indicating a potential interaction with the heme propionate. Interestingly, the ability of 5 and other compounds to bind in both sites has little correlation with inhibitory potency, indicating that active site binding (and not H4B displacement) determines potency.
With the goal of improving hnNOS activity and isoform selectivity, we made efforts to optimize the conformational positioning of the tail amine of 5. To this end, homologation, chain shortening, and isomerization of lead molecule 5 resulted in compounds 6–13. Methylation of the tail nitrogen atom of 5 (compound 6) resulted in over a 2-fold loss in rnNOS activity, while shortening the ethylene linker between the central ring and the terminal nitrogen atom by one carbon (compound 7) resulted in a 2.3-fold increase in rnNOS activity. Moreover 7 showed increased hnNOS inhibitory activity, as well as improvements in both rn/miNOS and hn/heNOS selectivity ratios, relative to parent molecule 5. The X-ray crystal structures of 7 bound to rnNOS and hnNOS (Figure 5) reveal well-defined density at the aminoquinoline and central ring regions that closely overlaps with lead 5. However, the orientation of the tail aminomethyl group in both structures could not be determined because of poor density, even at a low contour levels, suggesting free rotation of the aminomethyl occurs toward either propionate A and the H4B site water, as modeled in Figure 5A and B, or toward propionate D and Tyr706/Tyr711 (rnNOS/hnNOS). In both nNOS structures, the position of the central phenyl ring of 7 forces heme propionate D into a downward conformation. As observed for 5 and 6, methylation of the tail nitrogen atom of 7 to yield 8 resulted in a loss of potency against both rat and human nNOS, suggesting that a primary amino group at this position is important for achieving maximal inhibitory activity against nNOS isoforms.
Figure 5.

X-ray crystal structures of 7 bound in the active sites of (A) rnNOS and (B) hnNOS at 1.75 Å, and 2.05 Å resolution, respectively
It appears that some parts of the phenylaminoquinoline SAR overlap with that of the previously reported phenyl ether compounds.7 For example, phenyl ether compounds with one methylene between the amino group and the aryl ring (benzyl) are more potent and selective than those with two (phenethyl) or more methylenes, and the same trend is observed here (cf. 7 and 8 vs. 5 and 6); compound 9 (Ki (rnNOS) = 140 nM) also has slightly less potency than 5. Nonetheless, other parts of the SAR are distinct from the previous aminoquinoline SARs. For instance, N-methylated compounds (6, 8) are less potent than the desmethyl analogues (5, 7), which is the reverse of what was generally observed for benzyl and phenethyl ether compounds.
The o-substituted isomers (11 and 10) possess less inhibitory potency (11, Ki (rnNOS) = 935 nM; 10, Ki (rnNOS) = 119 nM) than their corresponding para-substituted derivatives 5 and 7, respectively. Furthermore, 10 displays a similar loss in hnNOS potency and a sharp decrease in rn/miNOS selectivity. However, poor binding affinity to heNOS (10, Ki (heNOS) = 41,900 nM) results in a gain in hnNOS selectivity over heNOS compared to 7. In contrast to o-substitution, m-substituted isomers 13 and 12 exhibited comparable potencies to 5 and 7, respectively [13, Ki (rnNOS) = 107 nM; 12, Ki (rnNOS) = 55 nM]. Moreover, 12 also has very good nNOS selectivity over iNOS (rn/miNOS = 440) and outstanding selectivity over eNOS (hn/heNOS = 877). The assay data for 12 suggest that placing the aminoalkyl tail portion meta- to the quinoline favors binding to the human nNOS isoform, which prompted examination of the crystal structures of both rnNOS-12 and hnNOS-12. As shown in Figures 6A and 6B, the tail amino group in the meta-position of the benzene ring may participate in maximal binding interactions with a water-filled polar pocket composed of the carboxylate of propionate A, the side chains of Gln478/Gln483 and Arg481/Arg486, and, most notably, Asp597/Asp602 (rnNOS/hnNOS, respectively), resulting in reduced flexibility in the tail aminomethyl group.
Figure 6.

X-ray crystal structures of 12 bound in the active sites of (A) rnNOS, (B) hnNOS, and (C) heNOS at 2.05 Å, 2.15 Å, and 2.05 Å resolution, respectively
In sharp contrast, the tail aminomethyl group in the heNOS-12 structure (Fig. 6C) shows a different orientation, making H-bonds with heme propionate A. It is important to note that there is a major difference in the active site of human eNOS, namely, Asn366 replaces Asp597/Asp602 (rnNOS/hnNOS) in the polar pocket noted for nNOS. In earlier studies we found that this Asp/Asn difference makes substantial contributions to isoform selectivity.22 This is the likely the basis for the high selectivity observed for 12, because the aminomethyl group is oriented toward Asp597/Asp602 in the rnNOS/hnNOS structures.
We proposed that homologated analogue 13 could have improved activity because the longer chain might allow it to potentially displace one of the structural waters near the nNOS-specific Asp residue. However, the nNOS inhibitory activity of 13 is weaker, comparable to para-substituted derivative 5 (and about two-fold lower than 12), indicating that either a) this water molecule is not displaced, or b) any energy gained from water displacement does not offset entropic costs of chain elongation, internal torsion, or other negative interactions between the enzyme and inhibitor. The crystal structures of rnNOS-13 and hnNOS-13 (Figure 7) support the former hypothesis; the tail amino group does not occupy this polar pocket, but rather only displaces the water molecule bridging propionate A and the H4B. Consequently, the aminoethyl group can only reach the closer heme propionate A for an electrostatic interaction rather than the farther Asp597/Asp602 site (rnNOS/hnNOS).
Figure 7.

X-ray crystal structures of 13 bound in the active sites of (A) rnNOS and (B) hnNOS at 1.95 Å and 2.40 Å resolution, respectively
Previously, installation of a nitrile at the 5-position of phenyl ether-linked aminoquinolines greatly improved potency, as the nitrile fit into a small, previously undiscovered auxiliary pocket, where it formed a H-bond with a deep structural water and stabilized binding;8 likewise, 1,3,5-trisubstituted nitrile-containing aminopyridine derivatives23 exert augmented potency and selectivity via differential interactions with the Asp site (vs. the Asn site in eNOS). For the phenylquinoline scaffold, however, the nitrile is a very deleterious modification [14, Ki (rnNOS) = 10,300 nM]. Examination of the crystal structure of 12 (Figure 6A) indicates that the 5-position nitrile of the more compact and rigid phenylquinoline (compared to the more flexible phenyl ether molecule) cannot reach this auxiliary pocket. Instead, the position of the phenyl ring of 14 (just like 12) would force heme propionate D into a downward conformation; thus, the 5-position nitrile would cause serious clashes with nearby residues. This is another area where the SARs of phenyl ether-linked aminoquinolines and phenylquinolines drastically diverge.
On the basis of the 12-bound NOS crystal structures (Figure 6), pyridine 15 was expected to be a potent inhibitor. In addition to having an identical binding mode to 12, the pyridine nitrogen atom forms a hydrogen bond with heme propionate D in all three NOS structures (Figure 8). The aminomethyl group of 15 approaches Gln478 in rnNOS, and is disordered but may point to Asp602 in hnNOS, and heads to the water molecule near heme propionate A in heNOS, where a second molecule of 15 is bound in the pterin site. Nonetheless, the pyridine analogue is approximately 5-fold less potent than 12 in rnNOS, contradicting both the crystallographic observations and previous results for pyridine-containing 2-aminopyridine and 2-aminoquinoline compounds.7 The unpredicted effect may be electronic, or the pyridine may influence interactions of the aminomethyl group with nearby water molecules.
Figure 8.

X-ray crystal structures of 15 bound in the active sites of (A) rnNOS, (B) hnNOS, and (C) heNOS at 2.10 Å, 2.10 Å and 2.15 Å resolution, respectively
Constrained Amine Analogues 16–18.
See Supporting Information for details.
4-Alkoxy-3-aminomethyl Analogues 19–29.
Docking studies with 12 also indicated that a variety of groups might be accommodated at position 4 of the phenyl ring (Supporting Information Figure S1). Substituents at this position could form favorable van der Waals interactions with Met336/Met341 or Leu337/His342 (rnNOS/hnNOS), residues that have previously been implicated in high n/eNOS selectivity for 2-aminoquinoline-based inhibitors,7 and which are replaced by Val104 and Phe105 in heNOS, respectively.12 To this end, 19–29, which have a variety of steric, electronic, and H-bonding properties that could provide crucial SAR information, were prepared and assayed for NOS inhibitory potency. In general, small substituent modifications at the 4-position of lead 12, such as fluorine (19), chlorine (20), ethyl (22), and methoxyl (23) do not add additional good contacts in the rat nNOS active site, resulting in a loss of potency. Compound 21 is also a less potent inhibitor given that the trifluoromethyl group of 21 is likely too bulky to fit. However, changing the 4-position methoxyl group (23) to an ethoxyl group (24) restored the potency in rnNOS. The crystal structure of 24 bound to rnNOS (Figure 9A) shows strong density for the aminoquinoline and central phenyl ring as seen in parent compound 12. While the aminomethyl moiety occupies the water site between H4B and propionate A, the ethoxyl group is large enough to establish some contacts with Met336. It seems that as a minimum a 3-atom, nonpolar moiety at the 4-position fits better into rnNOS.
Figure 9.

X-ray crystal structures of 24 bound in the active sites of (A) rnNOS, (B) hnNOS, and (C) heNOS, at 2.23 Å, 2.10 Å, and 2.20 Å resolution, respectively
In the hnNOS-24 structure, the central phenyl ring of 24 flips 180° relative to the orientation seen in rnNOS. The aminomethyl moiety interacts with Asn574 (Figure 9B) and the ethoxyl group still can reach Met341 for contact. It might be that the polar nature of His342 pushes the ethoxyl group away and causes the central phenyl ring to flip. The binding mode of 24 in heNOS (Figure 9C) is almost identical to that observed in rnNOS (Figure 9A). The better nonbonded contacts between the ethoxyl moiety of 24 and the bulky Phe105 side chain makes the inhibitor more ordered in structure and leads to rather low n/e selectivity (164).
Discussion of the small-4-alkoxy-3-aminomethyl and 4-cycloalkoxy analogues (25–28) is in the Supporting Information.
A further improvement for rnNOS inhibition was observed by the addition of a small cycloalkyl group (28 and 29). The addition of a cyclobutyl tail moiety allows 28 to form van der Waals interactions in the Met336-Leu337-Tyr706 pocket, resulting in good potency with rnNOS (Ki (rnNOS) = 49 nM). The slightly less bulky cyclopropane analogue 29 was found to have even greater rnNOS potency [Ki (rnNOS) = 39 nM], making it the most potent rnNOS inhibitor in the entire series. The X-ray crystal structure of 29 bound to rnNOS (Figure 10A) reveals that the tail cyclopropane ring fits nicely into the hydrophobic pocket surrounded by Met336, Leu337, and Tyr706, with the side chain of Tyr706 occupying two alternate conformations. Propionate D is pushed by 29 into the downward conformation. However, the orientation of the aminomethyl group shows two alternate directions that form H-bonds to either the H4B site water (as shown in Figure 10A) or with the side chain of Arg481 via water bridging. Compound 29 has a nearly 2-fold increase in rn/miNOS selectivity compared to 12 (29, rn/miNOS = 841; 12, rn/miNOS = 441).
Figure 10.

X-ray crystal structures of 29 bound in the active sites of (A) rnNOS, (B) hnNOS, and (C) heNOS at 1.90 Å, 1.95 Å and 1.76 Å resolution, respectively
Not only does 29 have high inhibition potency against rnNOS, but it also maintains equally high potency against hnNOS, making it a potent dual rnNOS and hnNOS inhibitor. Similar to the binding mode found in the rnNOS-29 structure, the central phenyl ring of 29 bound to hnNOS presses into heme propionate D. The polar nature of His342 forces the tail cyclopropyl group to move away from the imidazole side chain, making contacts with Met341 instead. The binding conformation of the tail cyclopropyl group closely overlaps with the tail cyclobutyl group of 28 in the hnNOS-28 structure, suggesting the placement of the aminomethyl substituent gives rise to the difference in hnNOS activity between 28 and 29. In hnNOS-29, the density for the aminomethyl group is weak. However, there is no sign that the tail amino group interacts with the H4B site water as is the case in the hnNOS-28 structure. Rather, there is electron density to support H-bonding with water near Gln483, which is very similar to the binding conformation of the aminomethyl moiety of 12 bound to hnNOS. The positive ammonium group of 29 faces a water-filled pocket noted above for 12 that is influenced by the negative charge of Asp602. In heNOS, again the bulky Phe105 pushes the cyclopropyl group toward heme propionate A, allowing the aminomethyl moiety to make H-bonds with both the propionate and H4B. The aminomethyl group would not point to the water-filled pocket because there is no negatively charged residue lining the pocket in heNOS. Rather, Asn366 is part of this pocket and is very likely the origin of the 457-fold hn/heNOS selectivity.
4-Phenyloxymethylaryl Analogues 30–37.
Thus far, it appears that considerably larger alkoxyl substituents can be accommodated at the 4-position. Aryl substituents are also tolerated; for example, compound 30, with its bulky 3-fluorobenzyl group at position 4, is only slightly less potent than 24, although it is considerably less potent than an aminopyridine-pyrrolidine compound (18 nM) after which it is modeled.24 This trend is also observed with the benzonitrile ring of 31; previously, 4-cyanoaryl compounds had good hnNOS activity, but lower hn/heNOS selectivity (generally ~30-fold),9 but in this phenylquinoline scaffold, the hn/heNOS selectivity is higher with the 4-cyanoaryl group present. However, the hn/heNOS selectivity of 31 remains inferior to leads 5, 12, and 24 because of increased binding to heNOS (8300 nM). The unusual crystallographic binding behavior of 31 is discussed in the Supporting Information.
Given the lower hn/heNOS selectivity for 31, as well as the disfavored binding to the human nNOS active site, we decided to reduce the overall length of the inhibitors to better fit into the hnNOS active site, while maintaining H-bond accepting capability in the tail ring. The 2- and 3-pyridinylmethyl ether groups of 32 and 33, respectively, were introduced to provide contact with either Leu337 in rnNOS or His342 in hnNOS. Relative to 12, both pyridinyl modifications provided slight improvements in rnNOS potency (32, Ki = 52 nM; 33, Ki = 44 nM). The crystal structure of rnNOS-32 with compound 33 overlaid (Figure 11A) shows nearly identical binding positions for the aminoquinoline and tail pyridine, with the pyridinyl nitrogen facing away from Leu337 in both cases (to allow hydrophobic interactions). The difference lies in the orientation of the central phenyl ring. As a result, the central-ring aminomethyl group of 33 displaces the H4B site water, whereas this moiety points away from the existing water in 32.
Figure 11.

X-ray crystal structures of 32 (yellow) bound in the active sites of (A) rnNOS with 33 (magenta) overlaid, (B) hnNOS, and (C) heNOS, at 1.70 Å, 1.81 Å, and 1.95 Å resolution, respectively
In the hnNOS-32 structure (Figure 11B), the overall binding remains largely the same, although the orientation of the middle phenyl ring is different in the two nNOS cases. Here, the ring does not press sufficiently against heme propionate D, and the latter is not distorted as seen in the rnNOS-32 structure. The differences might be the result of the Leu337 (rat) and His342 (human) variation, where the bulkier His342 side chain in human nNOS pushes the tail pyridine of 32 away so that the central phenyl ring does not make such close contacts with heme propionate D. Because of the 2-position of the pyridine nitrogen, a H-bond with the His342 side chain is not possible. Additionally, the position of the aminomethyl group on the phenyl ring has some uncertainty, although some weak electron density supports a likely interaction with Asn574. These deleterious interactions lead to a decreased binding affinity of 32 to hnNOS (Ki = 76 nM). However, by virtue of substantially diminished binding potency towards miNOS and heNOS, the rn/miNOS and hn/heNOS selectivities of 32 remain quite high (rn/miNOS = 879, hn/heNOS = 312).
Compound 32 is unique in that previously reported aminoquinolines substituted with pyridines3 generally had only 10–20-fold n/eNOS selectivity. However, 32 exhibits fairly good hn/heNOS selectivity (312-fold). The main difference is that in the heNOS-32 crystal structure (Figure 11C) the orientation of the central phenyl ring is perpendicular to the aminoquinoline ring rather than parallel as in hnNOS-32. As a result, the amino group of 32 in heNOS is about 2 Å farther from heme propionate D than it is in hnNOS-32. In addition, the amino group in hnNOS-32 H-bonds with Asn574. These interactions are possible reasons for the good hn/heNOS selectivity.
The 3-pyridylmethyl ether of 33 moderately increases hnNOS potency (Ki = 45 nM for 33 vs. 60 nM for 12). This is not the first instance that the 3-pyridyl moiety has improved binding to hnNOS,9,25 although it is the first example where such a compound has equal potency for both the rat and human enzymes. The hnNOS-33 X-ray structure (Figure 12B) shows good density for the aminoquinoline and central phenyl rings. The tail pyridine portion shows signs of disorder, but a potential H-bond between the pyridine and His342 is more feasible for 33 than for 32. In the rnNOS-33 structure, however, the hydrophobic portions of the pyridine about the nonpolar residues Leu337 and Met336 (Figure 12A), suggesting that the pyridine can be either a hydrophobic or a H-bonding moiety depending on the nNOS isoform it is bound to. The aminomethyl moiety of 33 still interacts with Asn574 of hnNOS (Figure 12B), while in rnNOS-33 it makes an electrostatic interaction with heme propionate A (Figure 12A). Both interactions are favorable, and the results taken together may suggest a reason for equal potency against rat and human enzymes.
Figure 12.

X-ray crystal structures of 33 bound in the active sites of (A) rnNOS, (B) hnNOS, and (C) heNOS at 1.84 Å, 1.81 Å and 2.20 Å resolution, respectively
In heNOS-33, the aminomethyl moiety H-bonds heme propionate A (Figure 12C), rather than pointing out toward Gln247 as in the case of 32 (Figure 11C). How could the pyridinyl nitrogen atom position make such a large difference? The pyridine ring nitrogen atom of 33 can make an extra H-bond with a water molecule next to the H4B, which pulls the inhibitor closer to heme propionate A and results in better interactions. This may explain the 3-fold improvement in binding affinity of 33 (7700 nM) versus 32 (23,700 nM) toward heNOS, which leads to the poorer n/eNOS selectivity observed for 33 (172-fold).
The incorporation of heterocyclic H-bond acceptors leads to favorable increases in both potency and selectivity. Utilizing heterocycles with the dual ability to form hydrophobic interactions in rnNOS while also forming H-bonding interactions with isoform-specific His342 in hnNOS may be advantageous for in vivo studies (where activity in rats is necessary). Considering the importance of the heterocycle, we also sought to exchange the bulky pyridine for smaller heterocycles (as in analogues 34–37). These compounds all demonstrated high potencies against rnNOS, with Ki values all below 60 nM.
The use of isoxazole (as in 34) resulted in lost activity against hnNOS. However, 4-(methyloxy)-1,3-thiazole (35) resulted in a Ki of 31 nM against hnNOS, making 35 the most potent hnNOS inhibitor in this series. Comparing the X-ray crystal structures of rnNOS-35 (Figure 13A) and hnNOS-35 (Figure 13B) reveals a common binding orientation of the aminoquinoline ring, but with differences in the central phenyl ring resulting from different interactions between the thiazole ring and the enzyme. In the rnNOS structure, the thiazole is positioned near Leu337 with its carbon atom making the closest contact. In hnNOS-35, the thiazole is near His342, with its S atom facing toward His342 and its N atom H-bonding with a water molecule. The bulkier His342 in hnNOS pushes 35 slightly farther away from the heme than its position in rnNOS. Therefore, the aminomethyl group on the central phenyl ring in hnNOS H-bonds with Asn574, whereas the same moiety points toward Gln478 in rnNOS, and the phenyl ring flips almost 180°. Unfortunately, single digit micromolar inhibitory activity of 35 against miNOS and heNOS results in low rn/mi and hn/heNOS selectivities relative to 32.
Figure 13.

X-ray crystal structures of 35 bound in the active site of (A) rnNOS and (B) hnNOS at 1.88 Å and 1.95 Å resolution, respectively
The use of oxazole (36) results in a slight loss in hnNOS potency (Ki = 63 nM). However, reduced inhibitory activity against miNOS and heNOS led to excellent selectivities (rn/miNOS = 382, hn/heNOS = 433). Crystallographic details of 36 and 37 are discussed in the Supporting Information.
Human iNOS Inhibition Study.
Recently, we successfully expressed and purified human iNOS protein21 that showed robust activity. Human iNOS (hiNOS) assay data were collected for eight compounds, including the three simple aminomethyl compounds, 7, 10, and 12, as well as a sampling of the more potent compounds from the latter series (29, 32, 36, and 37) to determine the SAR for the human system. Ki values obtained from rat and human nNOS, as well as murine and human iNOS, were used to approximate the cross-species selectivity between lower (rn/mi) and higher order species (hn/hi) (vide infra). The major difference between murine and human iNOS that might affect inhibitor binding is that Asn115 in miNOS is Thr121 in hiNOS. The tail part of many inhibitors might reach the site based on observations in the available nNOS and eNOS structures. Without an iNOS-inhibitor structure, we can only speculate on potential interactions by superimposing the iNOS structures onto the known nNOS-inhibitor structures.
The general inhibition trends in Table 2 are more or less consistent (<2-fold) between murine and human iNOS, with only 7 and 33 as the principal exceptions. Compound 7 is a compact inhibitor that makes weaker contacts with the Asn115/Thr121 (miNOS/hiNOS) site. The two-fold difference in binding affinity might result from the variation in a water-mediated H-bonding network or another unknown reason. Contrarily, overlaying the structure of miNOS or hiNOS on hnNOS-33 (Figure 14) reveals that the pyridine nitrogen atom of 33 is capable of making a direct H-bond with either Asn115 or Thr121. The three-fold weaker binding affinity to hiNOS might indicate a weaker interaction with Thr121 in hiNOS (than with Asn115 in miNOS). In the same overlay, another variation site, Ser256/Ala262 (miNOS/hiNOS) is more than 5.0 Å from the potential position of the aminomethyl moiety of 33, likely too far away to influence inhibitor binding.
Table 2.
Inhibition of rat and human nNOS compared to murine and human iNOS by selected compoundsa
| Ki (μM) | Selectivity | ||||||
|---|---|---|---|---|---|---|---|
| compd | rnNOS | hnNOS | miNOS | hiNOS | rn/mi | hn/hi | |
| 7 | 0.045 | 0.088 | 16.3 | 6.94 | 362 | 79 | |
| 10 | 0.119 | 0.151 | 2.86 | 3.55 | 24 | 24 | |
| 12 | 0.055 | 0.060 | 24.2 | 29.9 | 441 | 498 | |
| 29 | 0.039 | 0.046 | 32.8 | 21.1 | 841 | 459 | |
| 32 | 0.052 | 0.076 | 45.7 | 43.9 | 879 | 578 | |
| 33 | 0.044 | 0.045 | 8.18 | 29.4 | 186 | 653 | |
| 36 | 0.043 | 0.063 | 16.4 | 24.6 | 382 | 390 | |
| 37 | 0.053 | 0.046 | 21.0 | 34.8 | 397 | 757 | |
Compounds 7, 10, 12, 29, 32, 33, 36, and 37 were assayed for in vitro inhibition against purified human iNOS (hiNOS) using known literature methods, and Ki values were calculated directly from IC50 values using the Cheng–Prusoff equation. IC50 values are the average of at least two replicates from 6–9 data points; all experimental standard error values (for the LogIC50) are less than ± 0.10. Ki values for isoforms: rat nNOS (rnNOS), human nNOS (hnNOS), and murine iNOS (miNOS) were included for comparison. Selectivity values are ratios of respective Ki values.
Figure 14.

The structure of hnNOS-33 (yellow) with the variant residues of miNOS (Asn115 and Ser256, gray, PDB code 1NOD) and hiNOS (Thr121 and Ala262, magenta, PDB code 4NOS) overlaid
Directed Mutagenesis Supports Water-mediated Interaction of Inhibitors with Asp597.
Previous studies indicated that Asp597 in rnNOS electrostatically stabilizes cationic inhibitors and can account for much of the e/nNOS selectivity (as this residue is Asn in heNOS). A second difference, Met336 in rnNOS (Val in heNOS), can provide better nonpolar contacts with inhibitors and impart selectivity. To test the importance of these differences further, we determined the Ki values for certain compounds against various rnNOS mutants (Table 3).
Table 3.
Inhibition data for wild-type and mutant NOS enzymes by selected compounds (7, 10, 12, 29, 32, and 36)a
| Ki (μM) | Selectivity | ||||||
|---|---|---|---|---|---|---|---|
| Compd | WT-rnNOS | heNOS | D597N rnNOS | M336V/D597N rnNOS | WT/SM | WT/DM | |
| 7 | 0.045 | 18.7 | 0.273 | 0.215 | 6 | 5 | |
| 10 | 0.119 | 41.9 | 2.11 | 1.57 | 18 | 13 | |
| 12 | 0.055 | 52.6 | 0.416 | 1.60 | 8 | 29 | |
| 29 | 0.039 | 21.0 | 0.246 | 0.189 | 6 | 5 | |
| 32 | 0.052 | 23.7 | 0.384 | 0.159 | 7 | 3 | |
| 36 | 0.043 | 27.3 | 0.575 | 0.114 | 13 | 3 | |
The compounds were assayed for in vitro inhibition against purified NOS isoforms: rat nNOS (rnNOS, and human eNOS (heNOS), as well as single mutant (SM) (D597N) and double mutant (DM) (M336V/D597N) of rat nNOS using known literature methods, and Ki values were calculated directly from IC50 values using the Cheng–Prusoff equation. IC50 values are the average of at least two replicates from 6–9 data points; all experimental standard error values (for the LogIC50) are less than ± 0.10. Selectivity values are ratios of respective Ki values.
All tested compounds exhibited decreased potency against the single mutant enzyme (D597N) (perhaps, as expected), but against the double mutant, all compounds had potency comparable to the single mutant (except for 12, where it was greatly reduced). Compound 36, actually displayed 5-fold increased potency against the double mutant (versus the single mutant). The rnNOS-36 structure shows that the tail end of the inhibitor contacts Met336 and Leu337. These two residues are Val104 and Phe105 in heNOS, and contacts made with the tail end of the inhibitor may involve more than the Met/Val difference. It is possible that the local environment near the Val336 and Leu337 residues in the M336V/D597N double mutant enables better contacts with the tail end of the inhibitor than the same area in the D597N single mutant does, thus improving the potency against the double mutant versus the single mutant. This trend is reflected in the behaviors of 29, 32, and 36, which all have a long tail moiety that fits in this region of the enzyme. On the other hand, more compact inhibitors might be less sensitive to the Met/Val mutation site.
We conclude by focusing on 12, since this inhibitor exhibits the best hn/heNOS selectivity (877-fold). The D597N mutant decreases potency 8-fold, which further drops to 29-fold against the M336V/D597N mutant – even though this inhibitor does not appear to make contact with the Met/Val site. It is perhaps more instructive to consider the change in ΔGbind obtained from the Ki values, where ΔGbind = -RTlnKi. ΔGbind for WT hnNOS, WT heNOS, and the hnNOS D597N mutant are −9.9, −4.5, and −7.3 kcal/mol, respectively. Thus the Asp/Asn difference accounts for about half of ΔΔGbind between heNOS and hnNOS, leaving about −2.6 kcal/mol of binding affinity unexplained. This underscores the limitation of quantitatively explaining selectivity (against isoforms or mutants) by a few simple amino acid differences and a comparison of static X-ray structures. There are clear examples of differences in active site dynamics and the ability of even conserved side chains to adjust to inhibitor binding that could also contribute to selectivity (anchored plasticity) in cases like this.26
Off-Target Profiling.
Four structurally diverse compounds from this series (12, 29, 32, and 33) were screened by the National Institute of Mental Health’s Psychoactive Drug Screening Program (PDSP, Table 4). In this assay,27 compounds were screened against a panel of 45 pharmacologically relevant CNS targets and receptors using a radioligand displacement assay. Initially, the assay used a primary high dose (10 μM) and then a secondary Ki determination was performed for compounds showing >50% binding in the primary assay. We classify off-target binding using the following rubric: concerning (Ki < 100 nM, or <~2 × nNOS Ki value), moderate (100–300 nM, or ~2–5 × nNOS Ki value), weak (>300 nM, or > ~5 × nNOS Ki value, typically ~1 μM), and insignificant (<50% at 10 μM). The off-target profiles of previous aminoquinolines 1 and 4 have been included for comparison. Although not as effective as 4, a slight decrease in the fraction of concerning or moderate hits for 32 is observed (11/45 for 32 compared to 15/45 for 1), while this fraction decreases further to 8/45 for 33. Unfortunately, most of the flagged targets for 32 and 33 are serotonin receptors, suggesting that the heteroaryl-alkyl tails may resemble a GPCR-ligand-like pharmacophore.28 Conversely, the off-target profiles for 12 and 29 reveal the cleanest CNS counterscreening observed for 2-aminoquinolines to date, flagging only the H2 and H3 receptors as concerning for 12 and 29, respectively, which is unsurprising as these receptors are known to bind cationic amidine groups (e.g., ranitidine), while 29 only flagged an additional 4/45 targets for moderate binding.29 For 12, 21/45 targets were classified as weak (27/45 for 29), while 23/45 (12) and 13/45 (29) were classified as insignificant. These results indicate that reducing the tail group’s size (as in 29) or eliminating it completely (as in 12) are both effective strategies to reduce off-target CNS binding, which may translate to improved safety in vivo.
Table 4.
PDSP Binding Summary for Selected Compoundsa
| Compd | Concerning | moderate | weak | insignificant | Total |
|---|---|---|---|---|---|
| 1 | 8 | 7 | 22 | 8 | 45 |
| 4 | 3 | 6 | 17 | 22 | 45 |
| 12 | 1 | 0 | 21 | 23 | 45 |
| 29 | 1 | 4 | 27 | 13 | 45 |
| 32 | 4 | 7 | 22 | 12 | 45 |
| 33 | 3 | 5 | 17 | 20 | 45 |
Off-target binding is classified into four categories: concerning (Ki < 100 nM, or < ~2× nNOS Ki value), moderate (100–300 nM, or ~2– 5× nNOS Ki value), weak (>300 nM, or > ~5× nNOS Ki value, typically ~1 μM), and insignificant (<50% bound at 10 μM), for a total of 45 receptors as assayed by the PDSP’s “comprehensive screen” (see ref 28).
Membrane Permeability and Microsome Stability.
As our ultimate goal is to utilize these compounds as CNS drugs, membrane permeability for compounds 7, 12, 29, 33 and 37 was determined using the parallel artificial membrane permeability for the blood–brain barrier (PAMPA-BBB) assay. In this assay, an artificial membrane containing BBB phospholipids is used to assess permeability. A compound is predicted to have good BBB penetration and is classified as a “CNS (+)” molecule if its effective permeability (Pe) in this assay is larger than 4.0 × 10–6 cm s–1.30,31,32,33 Additionally, the web tool SwissADME was used to make in-silico passive-BBB penetrability predictions based on their validated BOILED-Egg (Brain Or IntestinaL EstimateD permeation) predictive model.34,35 Table 5 contains Pe values of three commercial drug standards and selected nNOS inhibitors 7, 12, 29, 33 and 37. All of the selected nNOS inhibitors are predicted “CNS (+)” with Pe values up to 15.5 × 10–6 cm/s. Compounds 33 and 37 display the lowest permeability among the selected compounds (Pe = 8.09 ± 0.67 × 10−6 cm s−1 and 7.04 ± 2.43 × 10−6 cm s−1, respectively), indicating that the presence of the heterocyclic tail portion may reduce the permeability of these compounds. However, while the Pe values for 33 and 37 were high enough to score as “CNS (+)”, the BOILED-Egg permeant model predicted they would not have BBB penetration because of their higher tPSA values of 87.05 Å2 and 115.29 Å2, respectively, although these two compounds, along with all the tested compounds, have predicted LogD and LogP values that are favorable for BBB penetration. Cyclopropyl compound 29, which lacks 33 and 37’s polar heterocycles, has a lower tPSA (74.16 Å2), a two-fold higher Pe, and is predicted to be BBB (+). Eliminating the substitution at the 4-position gave mixed results. Although not as high as 29, compound 7 maintained a high Pe value (11.3 ± 1.64 × 10−6 cm s−1) relative to 33 and 37, although compound 12 was found to have the highest Pe out of all compounds assayed. Additional cellular pharmacokinetic assays are in progress.
Table 5.
Effective Permeability (Pe) of Five Commercial Drugs and nNOS Inhibitors in the PAMPA–BBB Assaya
| Compd | log Db | log Pb | TPSA (Å2)c | reported Pe (10−6 cm s−1)d | determined Pe (10−6 cm s−1)e | BBB permeant predictionc | prediction |
|---|---|---|---|---|---|---|---|
| verapamil | 2.29 | 4.55 | 63.95 | 16 | 21.3 ± 1.5f 18.5 ± 1.9 | BBB (+) | CNS (+) |
| chlorpromazine | 2.76 | 4.56 | 31.78 | 6.5 | 8.04 ± 0.41f 8.90 ± 0.68 | BBB (+) | CNS (+) |
| dopamine | −1.50 | 0.03 | 66.48 | 0.2 | 0.12 ± 0.41f 0.125 ± 0.14 | BBB (−) | CNS (−) |
| 7 | 1.36 | 3.32 | 64.93 | 11.3 ± 1.64 | BBB (+) | CNS (+) | |
| 12 | 1.27 | 3.18 | 64.93 | 15.5 ± 2.32 | BBB (+) | CNS (+) | |
| 29 | 2.37 | 3.78 | 74.16 | 14.6 ± 0.97 | BBB (+) | CNS (+) | |
| 33 | 2.13 | 3.53 | 87.05 | 8.09 ± 0.67 | BBB (−) | CNS (+) | |
| 37 | 1.94 | 3.35 | 115.29 | 7.04 ± 2.43 | BBB (−) | CNS (+) |
All assays were performed over 17 h at a concentration of 200 μM; see Experimental Section for details.
Log D (pH = 7.4) and log P values of the free-base species were predicted using ChemAxon software.
TPSA calculations and BBB permeation was predicted using the free web tool SwissADME.
Effective permeability values from literature.32
Effective permeability values obtained in-house.
Experimental Pe values reported previously by Do et al.68
Additionally, stability in the presence of human liver microsomes (HLM) was determined for 12 and positive control terfenadine. The results of this study (Table 6) show that 12 displays high stability relative to the positive control, as indicated by a half-life (t1/2) >60 minutes. However, 12 did show a sign of degradation in buffer control samples (Supporting Information, Table S3).
Table 6.
Metabolic stability of 12 and positive control in human liver microsome.a
| 12 | 87 | 67 | >60 | 8 |
| terfenadined | 101 | 100 | 23 | 108 |
All assays were performed in 50 mM Kphos buffer (pH 7.4) containing HLM (0.714 mg/mL) over 60 min at 1.428 μM drug concentration. Parent compound peak disappearance were monitored by LC-MS/MS.
t1/2: half-life.
CL’int: in vitro intrinsic clearance.
Positive control.
Conclusions
In summary, we prepared a series of novel 7-phenyl-2-aminoquinolines possessing tail amines designed to target nNOS-specific aspartate residues Asp597/Asp602 (rnNOS/hnNOS), and thereby result in higher n/eNOS selectivity. Initially, screening compounds 5–13 revealed a preference for meta-substituted benzylamines, such as 12, which shows excellent potency and outstanding selectivity for nNOS over the other isozymes. A number of modifications to 12 were made, which included reducing or constraining amino group flexibility, substituting the 4-position of the phenyl ring, and using a 1,3,5-tri-substituted phenyl core. While the amine cannot be constrained effectively, and 1,3,5-tri-substitutions clash deleteriously with heme propionates in the enzyme, some 4-position additions enhance potency and maintain high isoform selectivity via favorable interactions with isoform-specific residues on the far end of the substrate access channel, namely, Leu337/His342 (rnNOS/hnNOS). Crystal structures indicate that these compounds act as competitive arginine mimics, where the aminoquinoline forms hydrogen bonds with the active-site glutamate residue, but also that substitutions at the 4-position can reduce phenyl ring rotation to favor interactions with the Asp (Asp597/Asp602)/H4B/propionate A site over the propionate D site. Mutagenesis studies confirmed the influence of the Asp site on inhibitor potency, making this class of aminoquinolines the first with the ability to target this site in nNOS, although preference for this site varies from compound to compound. Finally, with these inhibitors, the hn/heNOS selectivity derives more from a weakening of affinity to heNOS rather than an increase in affinity for hnNOS; inhibitors more complex than 12 tend to be less selective because of an increased affinity for heNOS. Apparently, more complex substituents provide additional contacts that improve binding to heNOS relative to hnNOS. On the basis of both their good potency and selectivity, clean CNS counterscreening (PDSP) profiles, and excellent permeability in our PAMPA-BBB assay, the most promising compounds, 12 and 29, are being advanced into preclinical studies.
Experimental Section
General Procedures.
Anhydrous solvents (THF, CH2Cl2, MeOH, Et3N, and DMF) were distilled prior to use. All other solvents, reactants, and reagents were purchased from commercial vendors and were used without further purification. Methanolic HCl (3 M, for ammonium hydrochloride salt formation and Boc-deprotection) was prepared fresh by the reaction of acetyl chloride and anhydrous MeOH at 0 ˚C. Melting points were determined in capillary tubes using a Buchi melting point B-540 apparatus and are uncorrected. 1H-NMR spectra were recorded at 500 MHz, using a Bruker Avance III 500 (direct cryoprobe), and 13C-NMR spectra were obtained at 126 MHz using the same instrument. High-resolution mass spectral data were obtained at the Integrated Molecular Structure Education and Research Center (IMSERC, Northwestern University) on an Agilent 6210A TOF mass spectrometer in positive ion mode coupled to an Agilent 1200 series HPLC system. Data were processed using MassHunter software version B.04.00. Flash column chromatography was performed using an Agilent 971-FP automated flash purification system with a Varian column station and SiliCycle cartridges (12–80 g, both normal and High Performance). Analytical HPLC was performed using an Agilent Infinity 1260 HPLC system with injection volumes of 5–10 μL. A Phenomenex Luna 5 μm C-8(2) 100 Å column, 50 × 4.60 mm, was used for all HPLC experiments, using a 10-min gradient of 95% H2O/5% acetonitrile + 0.05% TFA to 95% acetonitrile/5% H2O + 0.05% TFA, at 1.5 mL/min. The purity of all final target compounds was found to be ≥95% by HPLC. Analytical thin-layer chromatography was performed on Silicycle extra-hard 250 μm TLC plates. Compounds were visualized with short-wavelength UV light, and with ninhydrin and CAM stains, where appropriate. The preparation of quinoline precursors and assembly of final compounds is described below, while the preparation of other precursors is discussed in the Supporting Information. Compounds 388, 4136, 4337, 4538, 5239, 5340, 5641, 578, 6642, 6943, 7015, 8644, 8945, 9040, and 9646 were prepared by literature procedures or are known, and their spectral or analytical data are identical to those reported.
General Procedure 1: Suzuki Coupling between 39 and Aryl Halides.
Compound 39 (1 eq.), the requisite aryl halide (1–1.2 eq.), Pd(dppf)Cl2 (5 mol%) and NaHCO3 (3.5–4 eq.) were combined in dimethoxyethane/H2O (3:1, 4 mL solvent per 0.25 mmol 39 is sufficient) in a 20 mL sealable microwave vial. The mixture was briefly sparged with argon, sealed, and heated to 120 ˚C under microwave irradiation with stirring (720 rpm) for 20–25 minutes. After cooling, the mixture was partitioned between EtOAc and H2O (10 mL each), the layers were separated, and the aqueous layer was extracted with EtOAc (3 × 30 mL), and the organic layer was washed with 5% aq. NaCl (2 × 50 mL) and sat. aq. NaCl (50 mL), dried over anhydrous sodium sulfate and concentrated. The crude residue was purified as listed below under subheadings for individual compounds.
General Procedure 2: Deprotection of Aminoquinolines.
The protected intermediate was immediately diluted with MeOH (9–10 mL/0.2 mmol of protected quinoline), and K2CO3 (2 eq.) was added. The mixture was heated at reflux for 2–2.5 h, cooled, and concentrated, and the residue was partitioned between EtOAc (10 mL) and H2O/sat. aq. NaCl (1:1, 10 mL). The layers were separated, the aqueous phase was extracted with EtOAc (3 × 20 mL), and the organic layers were combined, washed with sat. aq. NaCl (20 mL), dried over anhydrous sodium sulfate and concentrated. Purification, if necessary, is described below under subheadings for individual compounds. The resulting free 2-aminoquinoline was dissolved in MeOH or ether/MeOH (10–15 mL), filtered to remove particulate matter, and treated with methanolic HCl (~3 M, ~1.5–2 mL). The mixture was stirred overnight at r.t. and workup (as described below) afforded the deprotected compounds as hydrochloride salts.
7-(4-(2-Aminoethyl)phenyl)-4-methylquinolin-2-amine Dihydrochloride (5).
Compounds 39 (0.050 g, 0.163 mmol) and 41 (0.050 g, 0.167 mmol) were coupled using General Procedure 1. Purification by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 30% EtOAc in CH2Cl2, afforded protected phenylquinoline 42 as a white solid (0.052 g, 76%). This compound was immediately deprotected using General Procedure 2. After workup, the free aminoquinoline was diluted in ether/MeOH (1:1, 20 mL), heated gently to affect solution, filtered, and cooled. After treatment with methanolic HCl, 5 was obtained as a cream-colored solid (0.037 g, 86% from 42) after triturating with ether and drying in vacuo: 1H-NMR (500 MHz; DMSO-d6): δ 14.09 (s, 1 H), 8.07–7.94 (m, 5 H), 7.81 (dd, J = 8.5, 1.6 Hz, 1 H), 7.75 (d, J = 8.2 Hz, 2 H), 7.46 (d, J = 8.2 Hz, 2 H), 6.93 (s, 1 H), 3.12–3.08 (m, 2 H), 2.96 (t, J = 7.8 Hz, 2 H), 2.65 (s, 3 H); the aminoquinoline –NH protons are mostly broadened into the baseline at 8.1 and 8.9 ppm; 13C-NMR (126 MHz; DMSO-d6): δ 154.3, 152.6, 143.8, 138.6, 137.4, 136.9, 130.2 (2 C), 127.8 (2 C), 126.7, 123.9, 120.9, 115.4, 112.9, 33.1, 19.4; one of the aliphatic carbon signals is obscured by the solvent peak; HRMS calcd for C18H20N3+: 278.1652; found, 278.1661.
4-Methyl-7-(4-(2-(methylamino)ethyl)phenyl)quinolin-2-amine Dihydrochloride (6).
Compounds 39 (0.075 g, 0.245 mmol) and 43 (0.079 g, 0.251 mmol) were coupled using General Procedure 1. Purification by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 30% EtOAc in CH2Cl2, afforded protected phenylquinoline 58 as a yellow foam (0.070 g, 70%). This compound was immediately deprotected using General Procedure 2. After workup, the free aminoquinoline was passed through a short SiO2 plug, eluting with 1% MeOH in EtOAc. The filtrate was concentrated, washed with hexanes, and diluted in ether/MeOH (8:1, 20 mL). After treatment with methanolic HCl, 6 was obtained as a flocculent cream-colored solid (0.057 g, 76% from 58) after filtering, precipitating from MeOH (1 mL) with ether (5 mL) triturating with ether, and drying in vacuo: mp 307–308.3 ˚C (dec). 1H-NMR (500 MHz; DMSO-d6): δ 14.09 (s, 1 H), 8.86–8.85 (m, 2 H), 8.06 (d, J = 8.5 Hz, 1 H), 7.94 (d, J = 1.7 Hz, 1 H), 7.82 (dd, J = 8.5, 1.7 Hz, 1 H), 7.77–7.75 (m, 2 H), 7.46 (d, J = 8.3 Hz, 2 H), 6.93 (d, J = 1.0 Hz, 1 H), 3.22–3.16 (m, 2 H), 3.02 (t, J = 8.0 Hz, 2 H), 2.66 (d, J = 0.9 Hz, 3 H), 2.59 (t, J = 5.1 Hz, 3 H); the aminoquinoline –NH protons are mostly broadened into the baseline at 8.1 and 8.9 ppm; 13C-NMR (126 MHz; DMSO-d6): δ 154.33, 152.50, 143.75, 138.36, 137.40, 136.98, 130.14 (2 C), 127.79 (2 C), 126.66, 123.84, 120.89, 115.42, 112.93, 49.36, 32.93, 31.57, 19.41; HRMS calcd for C19H22N3+: 292.1808; found, 292.1822.
7-(4-(Aminomethyl)phenyl)-4-methylquinolin-2-amine Dihydrochloride (7).
Compounds 39 (0.075 g, 0.245 mmol) and 45 (0.082 g, 0.245 mmol) were coupled using General Procedure 1. Purification by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 40% EtOAc in CH2Cl2, afforded protected phenylquinoline 59 as a yellow solid (0.062 g, 63%). This compound was immediately deprotected using General Procedure 2. After workup, the free aminoquinoline was precipitated from EtOAc (1 mL) with hexanes (9 mL), triturated with hexanes, and diluted in ether/MeOH (5:1). After treatment with methanolic HCl, 7 was obtained as a cream-colored solid (0.043 g, 84% from 59) after filtering, precipitating twice from hot MeOH (2 mL) with ether (10 mL), washing with ether, and drying in vacuo: mp >300 ˚C (darkens slowly), >360 ˚C (melts or decomposes). 1H-NMR (500 MHz; DMSO-d6): δ 14.10 (s, 1 H), 9.06 (br s, 1 H), 8.43 (s, 3 H), 8.30 (br s, 1 H), 8.09 (d, J = 8.5 Hz, 1 H), 7.98 (d, J = 1.7 Hz, 1 H), 7.86–7.84 (m, 3 H), 7.67 (d, J = 8.4 Hz, 2 H), 6.95 (d, J = 1.0 Hz, 1 H), 4.13–4.10 (m, 2 H), 2.67 (d, J = 0.9 Hz, 3 H); 13C-NMR (126 MHz; DMSO-d6): δ 154.3, 152.6, 143.5, 138.9, 136.7, 135.1, 130.3 (2 C), 127.7 (2 C), 126.7, 124.0, 121.0, 115.5, 113.1, 42.3, 19.4; HRMS calcd for C17H18N3+: 264.1495; found, 264.1503.
4-Methyl-7-(4-((methylamino)methyl)phenyl)quinolin-2-amine Dihydrochloride (8).
Compounds 39 (0.075 g, 0.245 mmol) and 46 (0.085 g, 0.245 mmol) were coupled using General Procedure 1. Purification by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 40% EtOAc in CH2Cl2, afforded protected phenylquinoline 60 as a yellow solid (0.072 g, 70%). This compound was immediately deprotected using General Procedure 2. After workup, the free aminoquinoline was precipitated from EtOAc (1 mL) with hexanes (9 mL). The solid was collected and diluted in ether/MeOH (3:1). After treatment with methanolic HCl, ether (10 mL) was added, and 8 was obtained as a cream-colored solid (0.047 g, 78% from 60) after filtering, precipitating from hot MeOH (2 mL) with ether (10 mL), washing with ether, and drying in vacuo. 1H-NMR (500 MHz; DMSO-d6): δ 14.06 (s, 1 H), 9.20 (s, 2 H), 9.00 (br s, 1 H), 8.20 (br s, 1 H), 8.08 (d, J = 8.5 Hz, 1 H), 7.97 (d, J = 1.7 Hz, 1 H), 7.86–7.82 (m, 2.0 Hz, 3 H), 7.70 (d, J = 8.3 Hz, 2 H), 6.94 (d, J = 1.0 Hz, 1 H), 4.21–4.19 (m 2 H), 2.66 (d, J = 0.9 Hz, 3 H), 2.58–2.56 (m, 3 H); 13C-NMR (126 MHz; DMSO-d6): δ 154.3, 152.6, 143.3, 139.4, 136.8, 133.1, 131.3 (2 C), 127.8 (2 C), 126.7, 124.0, 121.1, 115.6, 113.1, 51.2, 32.6, 19.4; HRMS calcd for C18H20N3+: 278.1652; found, 278.1664.
(S)-7-(4-(2-Aminopropyl)phenyl)-4-methylquinolin-2-amine Dihydrochloride (9).
Compounds 39 (0.075 g, 0.245 mmol) and 49 (0.077 g, 0.245 mmol) were coupled using General Procedure 1. Purification by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 35% EtOAc in CH2Cl2, afforded protected phenylquinoline 61 as a pale-yellow foam (0.092 g, 87%). This compound was immediately deprotected using General Procedure 2. After workup, the free aminoquinoline was washed with 10:1 hexanes/CH2Cl2 (10 mL). The solid was collected and diluted in ether/MeOH (3:1). After treatment with methanolic HCl, the mixture was concentrated, and the residue was azeotroped with toluene twice. After precipitation of the residue from MeOH (1 mL) with ether (10 mL), 9 was obtained as a tan hygroscopic powder (0.058, 75% from 61) after washing with ether and drying in vacuo: mp >260 ˚C (darkens slowly), 281–282 ˚C (dec). 1H-NMR (500 MHz; DMSO-d6): δ 14.27 (s, 1 H), 9.00 (br s, 1 H), 8.20 (br s, 1 H), 8.14–8.13 (m, 3 H), 8.06 (d, J = 8.5 Hz, 1 H), 7.96 (d, J = 1.5 Hz, 1 H), 7.82 (dd, J = 8.5, 1.5 Hz, 1 H), 7.76 (d, J = 8.2 Hz, 2 H), 7.45 (d, J = 8.2 Hz, 2 H), 6.94 (s, 1 H), 3.50–3.45 (m, 1 H), 3.10 (dd, J = 13.4, 5.3 Hz, 1 H), 2.78 (dd, J = 13.4, 8.8 Hz, 1 H), 2.66 (s, 3 H), 1.17 (d, J = 6.5 Hz, 3 H). 13C-NMR (126 MHz; DMSO-d6): δ 154.3, 152.5, 143.8, 138.1, 137.3, 136.8, 130.7 (2 C), 127.7 (2 C), 126.7, 123.9, 120.8, 115.3, 112.9, 48.4, 19.4, 18.2; one of the aliphatic carbons is obscured by the solvent peak; HRMS calcd for C19H22N3+: 292.1808; found, 292.1818.
7-(2-(Aminomethyl)phenyl)-4-methylquinolin-2-amine Dihydrochloride (10).
Compounds 39 (0.075 g, 0.245 mmol) and 52 (0.070 g, 0.245 mmol) were coupled using General Procedure 1. Purification by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 35% EtOAc in CH2Cl2, afforded protected phenylquinoline 62 as pale-yellow crystals (0.098 g, 99%). This compound was immediately deprotected using General Procedure 2. After workup, the free aminoquinoline was diluted in ether/MeOH (5:1). After treatment with methanolic HCl, the mixture was concentrated, and 10 was obtained as a white solid (0.070 g, 86% from 62) after precipitating twice from hot MeOH (1 mL) with ether (10 mL), washing with ether, and drying in vacuo: mp 290–291.5 ˚C. 1H-NMR (500 MHz; DMSO-d6): δ 14.21 (s, 1 H), 9.00 (br s, 1 H), 8.42 (s, 3H), 8.30 (br s, 1 H), 8.07 (d, J = 8.4 Hz, 1 H), 7.74 (d, J = 7.6 Hz, 1 H), 7.67 (s, 1 H), 7.59–7.51 (m, 3 H), 7.39–7.38 (m, 1 H), 6.97 (s, 1 H), 3.97 (br d, J = 4.0 Hz, 2 H), 2.67 (s, 3 H); 13C-NMR (126 MHz; DMSO-d6): δ 154.4, 143.7, 140.4, 132.0, 130.5, 129.29, 129.18, 129.09, 126.4, 126.0, 120.9, 118.7, 113.3, 19.5; two of the quinoline carbons are not visible due to baseline broadening; one of the aliphatic carbon signals is obscured by the solvent peak; HRMS calcd for C17H18N3+: 264.1495; found, 264.1503.
7-(2-(2-Aminoethyl)phenyl)-4-methylquinolin-2-amine Dihydrochloride (11).
Compounds 39 (0.075 g, 0.245 mmol) and 53 (0.077 g, 0.257 mmol) were coupled using General Procedure 1. Purification by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 35% EtOAc in CH2Cl2, afforded protected phenylquinoline 63 as pale-yellow crystals (0.093 g, 90%). This compound was immediately deprotected using General Procedure 2. After workup, the free aminoquinoline was sonicated with hexanes, and the solid was collected and diluted in ether/MeOH (2:1). After treatment with methanolic HCl, the mixture was concentrated, and 11 was obtained as an off-white powder (0.064 g, 83% from 63) after precipitating twice from hot MeOH (2 mL) with ether (10 mL), washing with ether, and drying in vacuo: mp >300 ˚C (darkens slowly), 323–325 ˚C (dec). 1H-NMR (500 MHz; DMSO-d6): δ 14.37 (s, 1 H), 8.10 (br s, 1 H), 8.10 (br s, 1 H), 8.07 (d, J = 8.4 Hz, 1 H), 7.92 (s, 3 H), 7.68 (d, J = 1.0 Hz, 1 H), 7.49–7.39 (m, 4 H), 7.30 (d, J = 7.4 Hz, 1 H), 6.98 (s, 1 H), 2.89–2.86 (m, 4 H), 2.67 (s, 3 H); 13C-NMR (126 MHz; DMSO-d6): δ 154.2, 152.6, 144.9, 140.5, 136.1, 135.1, 130.40, 130.22, 129.1, 127.6, 126.4, 126.1, 120.7, 118.1, 113.1, 30.7, 19.5; one of the aliphatic carbon signals is obscured by the solvent peak; HRMS calcd for C18H20N3+: 278.1652; found, 278.1664.
7-(3-(2-Aminoethyl)phenyl)-4-methylquinolin-2-amine Dihydrochloride (12).
Compounds 39 (0.075 g, 0.245 mmol) and 56 (0.070 g, 0.245 mmol) were coupled using General Procedure 1. Purification by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 40% EtOAc in CH2Cl2, afforded protected phenylquinoline 64 as a yellow foam (0.089 g, 90%). This compound was immediately deprotected using General Procedure 2. After workup, the free aminoquinoline was precipitated from EtOAc (2 mL) with hexanes (20 mL), and diluted in ether/MeOH (5:1). After treatment with methanolic HCl, 12 was obtained as a cream-colored flocculent solid (0.053 g, 72% from 64) after filtering, precipitating twice from hot MeOH (2 mL) with ether (10 mL), washing with ether, and drying in vacuo: mp 305.5–306.5 ˚C. 1H-NMR (500 MHz; DMSO-d6): δ 14.01 (s, 1H), 9.00 (br s, 1 H), 8.41 (s, 3H), 8.25 (br s, 1 H), 8.11 (d, J = 8.5 Hz, 1 H), 7.95–7.93 (m, 2 H), 7.84 (dd, J = 8.4, 1.2 Hz, 1 H), 7.78 (d, J = 7.6 Hz, 1 H), 7.62–7.59 (m, 2 H), 6.94 (s, 1 H), 4.15 (q, J = 5.3 Hz, 2 H), 2.67 (s, 3 H); 13C-NMR (126 MHz; DMSO-d6): δ 154.4, 143.7, 139.2, 135.6, 130.1, 129.7, 128.3, 127.6, 126.8, 124.0, 121.1, 115.5, 113.2, 42.7, 19.4; two of the quinoline carbons are not visible due to baseline broadening; HRMS calcd for C17H18N3+: 264.1495; found, 264.1504.
7-(3-(2-Aminoethyl)phenyl)-4-methylquinolin-2-amine Dihydrochloride (13).
Compounds 39 (0.075 g, 0.245 mmol) and 57 (0.077 g, 0.257 mmol) were coupled using General Procedure 1. Purification by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 45% EtOAc in CH2Cl2, afforded protected phenylquinoline 65 as a yellow syrup (0.090 g, 88%). This compound was immediately deprotected using General Procedure 2. After workup, the free aminoquinoline was diluted in ether/MeOH (5:1), and after treatment with methanolic HCl, ether (5 mL) was added and 13 was obtained as a tan hygroscopic semisolid (0.053 g, 71% from 65) after filtering, precipitating twice from hot MeOH (2 mL) with ether (10 mL), washing with ether, and drying in vacuo: 1H-NMR (500 MHz; DMSO-d6): δ 14.17 (s, 1 H), 9.00 (br s, 1 H), 8.08–8.06 (m, 5 H), 7.95 (d, J = 1.4 Hz, 1 H), 7.84 (dd, J = 8.5, 1.5 Hz, 1 H), 7.66–7.64 (m, 2 H), 7.53 (t, J = 7.8 Hz, 1 H), 7.39 (d, J = 7.6 Hz, 1 H), 6.94 (s, 1 H), 3.17–3.09 (m, 2 H), 3.01 (t, J = 7.8 Hz, 2 H), 2.66 (s, 3 H); 13C-NMR (126 MHz; DMSO-d6): δ 154.3, 152.6, 144.1, 139.2, 139.0, 136.7, 130.1, 129.6, 128.0, 126.6, 126.1, 124.1, 120.9, 115.5, 113.0, 33.5, 19.4; HRMS calcd for C18H20N3+: 278.1652; found, 278.1662.
3-(2-Amino-4-methylquinolin-7-yl)-5-(aminomethyl)benzonitrile Dihydrochloride (14).
Compounds 39 (0.075 g, 0.245 mmol) and 67 (0.083 g, 0.269 mmol) were coupled using General Procedure 1. Purification by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 35% EtOAc in CH2Cl2, afforded protected phenylquinoline 68 as a yellow foam (0.082 g, 78%). This compound was immediately deprotected using General Procedure 2. After workup, the free aminoquinoline was purified by flash column chromatography, eluting with a gradient of EtOAc to 5% MeOH in EtOAc to yield a white semisolid that was diluted in ether/MeOH (10:1). After treatment with methanolic HCl, ether (5 mL) was added, and 14 was obtained as a cream-colored solid (0.069 g, 68% from 68) after filtering, precipitating from hot MeOH (2 mL) with ether (10 mL), washing with ether, and drying in vacuo: mp >320 ˚C (darkens) >360 ˚C (melts or decomposes). 1H-NMR (500 MHz; DMSO-d6): δ 14.15 (s, 1 H), 9.10 (br s, 1 H), 8.63 (s, 3 H), 8.40 (br s, 1 H), 8.34 (s, 1 H), 8.29 (s, 1 H), 8.14 (d, J = 8.5 Hz, 1 H), 8.08 (s, 1 H), 7.99 (d, J = 1.1 Hz, 1 H), 7.91 (dd, J = 8.5, 1.2 Hz, 1 H), 6.99 (s, 1 H), 4.21 (s, 2 H), 2.67 (s, 3 H). 13C-NMR (126 MHz; DMSO-d6): δ 154.6, 152.5, 141.6, 140.2, 137.2, 136.6, 133.36, 133.19, 131.1, 127.0, 124.1, 121.6, 118.8, 115.9, 113.6, 112.8, 41.9, 19.5; HRMS calcd for C18H17N4+: 289.1448; found, 289.1459.
7-(5-(Aminomethyl)pyridin-3-yl)-4-methylquinolin-2-amine Trihydrochloride (15).
Compounds 39 (0.075 g, 0.245 mmol) and 70 (0.114 g, 0.294 mmol) were coupled using General Procedure 1. Purification by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 70% EtOAc in CH2Cl2, afforded protected phenylquinoline 71 as a white solid (0.080 g, 65%). 1H-NMR confirmed the presence of two Boc groups. This compound was immediately deprotected using General Procedure 2, and after 3 h, an additional equivalent of K2CO3 was added to remove one of the Boc groups. The mixture was heated a total of 4.5 h, and after workup, the free aminoquinoline was precipitated from EtOAc (0.5 mL) with hexanes (5 mL), and diluted in ether/MeOH (1:1). After treatment with methanolic HCl, ether (20 mL) was added and 15 was obtained as a tan solid (0.053 g, 90% from 71) after filtering, precipitating from hot MeOH (5 mL) with ether (10 mL), washing with ether, and drying in vacuo: mp 290–292 ˚C. 1H-NMR (500 MHz; DMSO-d6): δ 14.18 (s, 1 H), 9.03 (d, J = 2.0 Hz, 1 H), 8.81 (d, J = 1.6 Hz, 1 H), 8.62 (s, 3 H), 8.52 (s, 1 H), 8.17 (d, J = 8.5 Hz, 1 H), 8.01 (d, J = 1.5 Hz, 1 H), 7.91 (dd, J = 8.5, 1.7 Hz, 1 H), 6.99 (s, 1 H), 4.22 (q, J = 5.7 Hz, 2 H), 2.68 (s, 3 H); the pyridinium –NH proton is broadened into residual water around 4 ppm; 13C-NMR (126 MHz; DMSO-d6): δ 154.5, 152.7, 149.8, 147.5, 140.4, 137.0, 136.5, 134.4, 131.1, 127.2, 124.1, 121.5, 115.7, 113.6, 19.5; one of the aliphatic carbon signals is obscured by the solvent peak; HRMS calcd for C16H17N4+: 265.1459; found, 265.1464.
7-(Isoindolin-4-yl)-4-methylquinolin-2-amine Dihydrochloride (16).
Compounds 39 (0.075 g, 0.245 mmol) and 73 (0.077 g, 0.257 mmol) were coupled using General Procedure 1. Purification by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 35% EtOAc in CH2Cl2, afforded protected phenylquinoline 74 as a yellow gum (0.093 g, 91%). This compound was immediately deprotected using General Procedure 2. After workup, the free aminoquinoline was purified by flash column chromatography, eluting with EtOAc to yield a pale yellow solid. The solid was diluted in ether/MeOH (2:1). After treatment with methanolic HCl, the mixture was concentrated, and the residue was diluted in MeOH (3 mL) and filtered through a small pad of Celite. Reconcentration afforded a residue that was then precipitated three times from hot MeOH (3 mL) with ether (10 mL) to yield 16 as a pale tan solid (0.040 g, 52% from 74) after washing with ether and drying in vacuo: mp 220–224 ˚C (softens), 233–235 ˚C (dec). 1H-NMR (500 MHz; DMSO-d6): δ 13.98 (s, 1 H), 9.87 (s, 2 H), 9.00 (br s, 1 H), 8.20 (br s, 1 H), 8.09 (d, J = 8.5 Hz, 1 H), 7.78 (s, 1 H), 7.63 (d, J = 8.3 Hz, 1 H), 7.59–7.53 (m, 3 H), 6.95 (s, 1 H), 4.64 (s, 2 H), 4.61 (s, 2 H), 2.66 (s, 3 H); 13C-NMR (126 MHz; DMSO-d6): δ 154.4, 152.3, 142.5, 137.1, 136.8, 135.5, 133.5, 129.9, 128.9, 126.6, 124.9, 123.7, 121.1, 117.2, 113.4, 50.5, 50.2, 19.4; HRMS calcd for C18H18N3+: 276.1495; found, 276.1508.
7-(1-Amino-2,3-dihydro-1H-inden-5-yl)-4-methylquinolin-2-amine Dihydrochloride (17).
Compounds 39 (0.075 g, 0.245 mmol) and 79 (0.076 g, 0.245 mmol) were coupled using General Procedure 1. Purification by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 35% EtOAc in CH2Cl2, afforded protected phenylquinoline 81 as a yellow foam (0.091 g, 86%). This compound was immediately deprotected using General Procedure 2. After deprotection, the free aminoquinoline was precipitated from EtOAc (1 mL) with hexanes (20 mL), and the gel-like solid was filtered and diluted in ether/MeOH (2:1). After treatment with methanolic HCl, ether (8 mL) was added, and 17 was obtained as a white solid (0.059 g, 78% from 81) after precipitating from hot MeOH (5 mL) with ether (10 mL), washing with ether, and drying in vacuo: mp: >300 ˚C (darkens slowly), >340 ˚C (chars). 1H-NMR (500 MHz; DMSO-d6): δ 13.98 (s, 1 H), 9.00 (br s, 1 H), 8.49 (s, 3 H), 8.10 (br s, 1 H), 8.07 (d, J = 8.5 Hz, 1 H), 7.94 (s, 1 H), 7.82 (dd, J = 8.5, 1.3 Hz, 1 H), 7.76–7.69 (m, 3 H), 3.20–3.14 (m, 1 H), 3.02–2.95 (m, 1 H), 2.57–2.54 (m, 1 H), 2.10–2.03 (m, 1 H); 13C-NMR (126 MHz; DMSO-d6): δ 154.3, 145.8, 143.9, 140.5, 139.9, 126.7, 126.32, 126.18, 124.10, 124.01, 121.0, 115.8, 113.1, 54.9, 31.0, 30.4, 19.4; two of the quinoline carbons are not visible due to baseline broadening; HRMS calcd for C19H20N3+: 290.1652; found, 290.1663.
7-(3-Amino-2,3-dihydro-1H-inden-5-yl)-4-methylquinolin-2-amine Dihydrochloride (18).
Compounds 39 (0.075 g, 0.245 mmol) and 80 (0.076 g, 0.245 mmol) were coupled using General Procedure 1. Purification by flash column chromatography, eluting with a gradient of 2% EtOAc in CH2Cl2 to 35% EtOAc in CH2Cl2, afforded protected phenylquinoline 82 as a yellow semisolid (0.093 g, 88%). This compound was immediately deprotected using General Procedure 2. After deprotection, the free aminoquinoline was purified by flash column chromatography, eluting with EtOAc. Concentration afforded a residue that was diluted in MeOH. After treatment with methanolic HCl, the mixture was concentrated, azeotroped with toluene twice, and 18 was obtained as a white solid (0.064 g, 82% from 82) after precipitating twice from hot MeOH (2 mL) with ether (10 mL), washing with ether, and drying in vacuo: mp 250–256 ˚C (darkens, then chars). 1H-NMR (500 MHz; DMSO-d6): δ 14.15 (s, 1 H), 9.00 (br s, 1 H), 8.61 (s, 3 H), 8.20 (br s, 1 H), 8.11 (t, J = 4.2 Hz, 2 H), 7.90 (d, J = 1.5 Hz, 1 H), 7.79 (dd, J = 8.5, 1.6 Hz, 1 H), 7.74 (dd, J = 7.9, 1.5 Hz, 1 H), 7.51 (d, J = 7.9 Hz, 1 H), 6.94 (s, 1 H), 4.83–4.80 (m, 1 H), 3.14 (ddd, J = 19.1, 7.1, 4.3 Hz, 1 H), 2.98–2.92 (m, 1 H), 2.58–2.52 (m, 1 H), 2.10–2.03 (m, 1 H); 13C-NMR (126 MHz; DMSO-d6): δ 154.5, 145.3, 144.1, 141.1, 137.6, 136.8, 128.4, 126.9, 126.2, 124.3, 123.9, 120.9, 115.2, 113.0, 55.1, 31.0, 30.2, 19.4; HRMS calcd for C19H20N3+: 290.1652; found, 290.1664.
7-(3-(Aminomethyl)-4-fluorophenyl)-4-methylquinolin-2-amine Dihydrochloride (19).
Compounds 39 (0.075 g, 0.245 mmol) and 87 (0.099 g, 0.282 mmol) were coupled using General Procedure 1. Purification by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 35% EtOAc in CH2Cl2, afforded protected phenylquinoline 117 as a dark yellow foam (0.088 g, 85%). This compound was immediately deprotected using General Procedure 2. After workup, the free aminoquinoline was diluted in ether/MeOH (5:1). After treatment with methanolic HCl, 19 was obtained as a cream-colored flocculent solid (0.060 g, 81% from 117) after precipitating from hot MeOH (1 mL) with ether (10 mL), washing with ether, and drying in vacuo: mp 300–302 ˚C (dec). 1H-NMR (500 MHz; DMSO-d6): δ 14.08 (s, 1 H), 9.05 (br s, 1 H), 8.52 (s, 3 H), 8.25 (br s, 1 H), 8.12–8.08 (m, 2 H), 7.92 (d, J = 1.4 Hz, 1 H), 7.86–7.82 (m, 2 H), 7.49 (t, J = 9.2 Hz, 1 H), 6.95 (s, 1 H), 4.18 (br d, J = 3.2 Hz, 2 H), 2.67 (s, 3 H); 13C-NMR (126 MHz; DMSO-d6): δ (162.1 + 160.1, 1 C), 154.5, 152.6, 142.8, 136.6, (135.34 + 135.31, 1 C), (130.90 + 130.87), 1 C), (129.99 + 129.92, 1 C), 126.9, 124.0, (122.47 + 122.35, 1 C), (117.05 + 116.87, 1 C), 115.3, 113.2, (36.17 + 36.14, 1 C), 19.5; HRMS calcd for C17H17FN3+: 282.1401; found, 282.1408.
7-(3-(Aminomethyl)-4-chlorophenyl)-4-methylquinolin-2-amine Dihydrochloride (20).
Compounds 39 (0.075 g, 0.245 mmol) and 88 (0.105 g, 0.282 mmol) were coupled using General Procedure 1. Purification by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 40% EtOAc in CH2Cl2, afforded protected phenylquinoline 118 as a yellow foam (0.095 g, 88%). This compound was immediately deprotected using General Procedure 2. After workup, the free aminoquinoline was diluted in ether/MeOH (5:1). After treatment with methanolic HCl, 20 was obtained as a cream-colored powder (0.057 g, 71% from 118) after precipitating four times from hot MeOH (2 mL) with ether (15 mL), washing with ether, and drying in vacuo: mp 249–250 ˚C (softens slightly), 255–257 ˚C (melts). 1H-NMR (500 MHz; DMSO-d6): δ 14.13 (s, 1 H), 8.65 (s, 3 H), 8.13–8.11 (m, 2 H), 7.95 (d, J = 1.5 Hz, 1 H), 7.87 (dd, J = 8.5, 1.1 Hz, 1 H), 7.81 (dd, J = 8.4, 2.2 Hz, 1 H), 7.72 (d, J = 8.4 Hz, 1 H), 6.95 (s, 1 H), 4.24 (s, 2 H), 3.39–3.30 (m, 3 H), 2.66 (s, 3 H); the aminoquinoline –NH protons are not visible as discrete peaks, but are broadened into the baseline around 8.4 ppm; 13C-NMR (126 MHz; DMSO-d6): δ 154.7, 152.5, 142.3, 138.1, 133.8, 132.9, 130.8, 130.0, 129.1, 126.9, 123.8, 121.4, 115.8, 113.3, 19.4; one of the quinoline carbons is not visible due to baseline broadening; one of the aliphatic carbon signals is obscured by the solvent peak; HRMS calcd for C17H17ClN3+: 298.1106; found, 298.1114.
7-(3-(Aminomethyl)-4-trifluoromethylphenyl)-4-methylquinolin-2-amine Dihydrochloride (21).
Compounds 39 (0.075 g, 0.245 mmol) and 90 (0.100 g, 0.282 mmol) were coupled using General Procedure 1. Purification by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 35% EtOAc in CH2Cl2, afforded protected phenylquinoline 119 as a yellow solid (0.102 g, 88%). This compound was immediately deprotected using General Procedure 2. After workup, the free aminoquinoline was purified by flash column chromatography, eluting with EtOAc to yield a pale yellow gum that was diluted in ether/MeOH (10:1). After treatment with methanolic HCl, ether (5 mL) was added, and 21 was obtained as a cream-colored powder (0.054 g, 62% from 119) after precipitating from hot MeOH (2 mL) with ether (10 mL), washing with ether, and drying in vacuo: mp 327–329 ˚C. 1H-NMR (500 MHz; DMSO-d6): δ 14.09 (s, 1H), 9.00 (br s, 1 H), 8.30 (br s, 1 H), 8.76 (s, 3 H), 8.31 (s, 1 H), 8.17 (d, J = 8.5 Hz, 1 H), 8.03 (d, J = 1.5 Hz, 1 H), 7.99–7.95 (m, 3 H), 6.98 (s, 1 H), 4.29 (s, 2 H), 2.68 (s, 3 H); 13C-NMR (126 MHz; DMSO-d6): δ 154.6, 152.4, 143.0, 141.9, 136.8, 133.6, 130.2, 127.82, (127.77 + 127.73 + 127.68 + 127.64 + 127.59, 1 C), (127.49 + 127.3, 1 C), (125.6 + 123.4, 1 C), 127.0, 124.2, 121.7, 116.1, 113.7, 39.14, 39.13, 19.5; the splitting of the signals for the trifluoromethyl group and nearby carbons is partially obscured by other signals; HRMS calcd for C18H17F3N3+: 332.1369; found, 332.1383.
7-(3-(Aminomethyl)-4-ethylphenyl)-4-methylquinolin-2-amine Dihydrochloride (22).
Compounds 39 (0.060 g, 0.196 mmol) and 95 (0.065 g, 0.206 mmol) were coupled using General Procedure 1. Purification by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 35% EtOAc in CH2Cl2, afforded protected phenylquinoline 120 as a cream-colored solid (0.074 g, 87%). This compound was immediately deprotected using General Procedure 2. After workup, the free aminoquinoline was triturated with 5% EtOAc in hexanes and the solid was collected, washed with hexanes, diluted in EtOAc, filtered, and concentrated. The residue was diluted in ether/MeOH (6:1). After treatment with methanolic HCl, the mixture was concentrated, and 22 was obtained as a an off-white solid (0.035 g, 56% from 120) after precipitating three times from hot MeOH (1.5 mL) with ether (10 mL), washing with ether, and drying in vacuo: mp 307–309 ˚C. 1H-NMR (500 MHz; DMSO-d6): δ 14.06 (s, 1 H), 9.05 (br s, 1 H), 8.48 (s, 3 H), 8.25 (br s, 1 H), 8.10 (d, J = 8.5 Hz, 1 H), 7.94 (t, J = 2.2 Hz, 2 H), 7.88 (dd, J = 8.5, 1.6 Hz, 1 H), 7.73 (dd, J = 8.0, 1.8 Hz, 1 H), 7.46 (d, J = 8.1 Hz, 1 H), 6.94 (d, J = 0.6 Hz, 1 H), 4.16–4.13 (m, 2 H), 2.76 (q, J = 7.5 Hz, 2 H), 2.65 (s, 3 H), 1.22 (t, J = 7.5 Hz, 3 H); 13C-NMR (126 MHz; DMSO-d6): δ 154.4, 152.7, 143.71, 143.55, 136.65, 136.54, 133.1, 130.0, 128.6, 127.6, 126.7, 124.0, 120.9, 115.0, 113.0, 39.5, 25.1, 19.5, 15.4; HRMS calcd for C19H22N3+: 292.1808; found, 292.1815.
7-(3-(Aminomethyl)-4-methoxyphenyl)-4-methylquinolin-2-amine Dihydrochloride (23).
Compounds 39 (0.075 g, 0.245 mmol) and 98 (0.085 g, 0.269 mmol) were coupled using General Procedure 1. Purification by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 40% EtOAc in CH2Cl2, afforded protected phenylquinoline 121 as a yellow foam (0.077 g, 72%). This compound was immediately deprotected using General Procedure 2. After workup, the free aminoquinoline was triturated with hexanes. The solid was collected and diluted in ether/MeOH (8:1). After treatment with methanolic HCl, 23 was obtained as a yellow amorphous solid (0.057 g, 88% from 121) after precipitating from hot MeOH (5 mL) with ether (10 mL), washing with ether, and drying in vacuo: mp 232–234 ˚C. 1H-NMR (500 MHz; DMSO-d6): δ 14.16 (s, 1 H), 9.00 (br s, 1 H), 8.34–8.30 (2 br s, 4 H), 8.07 (d, J = 8.6 Hz, 1 H), 7.91 (d, J = 2.2 Hz, 1H), 7.90 (d, J = 1.4 Hz, 1 H), 7.82 (ddd, J = 8.5, 4.7, 2.0 Hz, 2 H), 7.26 (d, J = 8.7 Hz, 1 H), 6.93 (s, 1 H), 4.08 (q, J = 5.3 Hz, 2 H), 3.92 (s, 3 H), 2.65 (s, 3 H); 13C-NMR (126 MHz; DMSO-d6): δ 158.3, 154.4, 152.7, 143.5, 136.7, 130.8, 129.7, 129.3, 126.7, 123.7, 123.1, 120.5, 114.5, 112.7, 112.4, 56.4, 38.1, 19.4; HRMS calcd for C18H20N3O+: 294.1601; found, 294.1611.
7-(3-(Aminomethyl)-4-ethoxyphenyl)-4-methylquinolin-2-amine Dihydrochloride (24).
Compounds 39 (0.075 g, 0.245 mmol) and 101 (0.089 g, 0.269 mmol) were coupled using General Procedure 1. Purification by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 40% EtOAc in CH2Cl2, afforded protected phenylquinoline 122 as a cream-colored solid (0.072 g, 65%). This compound was immediately deprotected using General Procedure 2. After workup, the free aminoquinoline was triturated with hexanes. The solid was collected and diluted in ether/MeOH (2:1). After treatment with methanolic HCl, 24 was obtained as a yellow glassy solid (0.060 g, 99% from 122) after washing with 2% MeOH in ether, diluting in MeOH and reconcentrating, washing with ether, and drying in vacuo: mp 250 ˚C softens), 265–267 ˚C (bubbles, melts). 1H-NMR (500 MHz; DMSO-d6): δ 14.23 (s, 1 H), 9.07 (br s, 1 H), 8.39 (s, 3 H), 8.25 (br s, 1 H), 8.07 (d, J = 8.6 Hz, 1 H), 7.92 (d, J = 2.1 Hz, 1 H), 7.89 (d, J = 1.2 Hz, 1 H), 7.82 (dd, J = 8.5, 1.4 Hz, 1 H), 7.78 (dd, J = 8.6, 2.2 Hz, 1 H), 7.24 (d, J = 8.7 Hz, 1 H), 6.93 (s, 1 H), 4.18 (br q, J = 6.9 Hz, 2 H), 4.07 (d, J = 5.4 Hz, 2 H), 2.65 (s, 3 H), 1.42 (t, J = 6.9 Hz, 3 H); 13C-NMR (126 MHz; DMSO-d6): δ 157.5, 154.4, 152.6, 143.5, 136.7, 130.6, 129.6, 129.2, 126.7, 123.7, 123.1, 120.5, 114.4, 113.0, 112.7, 64.5, 37.9, 19.4, 15.0; HRMS calcd for C19H22N3O+: 308.1757; found, 308.1772.
7-(3-(Aminomethyl)-4-propoxyphenyl)-4-methylquinolin-2-amine Dihydrochloride (25).
Compounds 39 (0.075 g, 0.245 mmol) and 104 (0.093 g, 0.269 mmol) were coupled using General Procedure 1. Purification by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 35% EtOAc in CH2Cl2, afforded protected phenylquinoline 123 as an off-white solid (0.083 g, 73%). This compound was immediately deprotected using General Procedure 2. After deprotection, the free aminoquinoline was triturated with 5% EtOAc in hexanes (15 mL), and the obtained solid was diluted in ether/MeOH (2:1). After treatment with methanolic HCl, the mixture was concentrated, the residue was azeotroped twice with toluene, and 25 was obtained as a yellow glassy solid (0.044 g, 62% from 123) after precipitating twice from hot MeOH (2 mL) with ether (10 mL), and drying in vacuo: mp 220 ˚C (softens), 264–266 ˚C (bubbles, melts). 1H-NMR (500 MHz; DMSO-d6): δ 14.06 (s, 1H), 9.00 (br s, 1 H), 8.32 (s, 4 H), 8.07 (d, J = 8.5 Hz, 1 H), 7.91 (d, J = 2.2 Hz, 1 H), 7.89 (d, J = 1.4 Hz, 1 H), 7.82 (dd, J = 8.6, 1.2 Hz, 1 H), 7.78 (dd, J = 8.6, 2.3 Hz, 1 H), 7.25 (d, J = 8.7 Hz, 1 H), 6.91 (s, 1 H), 4.09 (br t, J = 6.5 Hz, 4 H), 2.63 (t, J = 1.8 Hz, 3 H), 1.83 (sextet, J = 7.0 Hz, 2 H), 1.03 (t, J = 7.4 Hz, 3 H); 13C-NMR (126 MHz; DMSO-d6): δ 157.6, 154.4, 143.51, 130.7, 129.4, 129.2, 126.7, 123.6, 123.2, 120.6, 114.6, 113.1, 112.7, 70.2, 37.8, 22.4, 19.4, 10.9; two of the quinoline carbons are not visible due to baseline broadening; HRMS calcd for C20H24N3O+: 322.1914; found, 322.1927.
7-(3-(Aminomethyl)-4-isopropoxyphenyl)-4-methylquinolin-2-amine Dihydrochloride (26).
Compounds 39 (0.075 g, 0.245 mmol) and 105 (0.093 g, 0.269 mmol) were coupled using General Procedure 1. Purification by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 35% EtOAc in CH2Cl2, afforded protected phenylquinoline 124 as an off-white solid (0.083 g, 73%). This compound was immediately deprotected using General Procedure 2. After deprotection, the free aminoquinoline was precipitated from EtOAc (1 mL) with hexanes (10 mL) and the obtained solid was diluted in ether/MeOH (4:1). After treatment with methanolic HCl, the mixture was concentrated and azeotroped twice with toluene, and 26 was obtained as an off-white solid (0.0097 g, 14% from 124) after precipitating twice from hot MeOH (1 mL) with ether (10 mL), washing with ether, and drying in vacuo: mp 237–239 ˚C (softens), 245–246 ˚C (melts). 1H-NMR (500 MHz; DMSO-d6): δ 13.97 (s, 1 H), 9.00 (br s, 1 H), 8.26 (br s, 4 H), 8.07 (d, J = 8.6 Hz, 1 H), 7.89 (d, J = 9.5 Hz, 2 H), 7.81 (d, J = 8.3 Hz, 1 H), 7.77 (d, J = 8.7 Hz, 1 H), 7.28 (d, J = 8.4 Hz, 1 H), 6.91 (s, 1 H), 4.81–4.75 (m, 1 H), 4.06 (d, J = 4.9 Hz, 2 H), 2.66 (s, 3 H), 1.37 (d, J = 6.0 Hz, 6 H; 13C-NMR (126 MHz; DMSO-d6): δ 156.5, 154.5, 152.5, 143.5, 136.9, 130.4, 129.5, 129.0, 126.7, 123.80, 123.61, 120.5, 114.5, 114.2, 112.7, 71.0, 37.9, 22.2 (2 C), 19.4; HRMS calcd for C20H24N3O+: 322.1914; found, 322.1933.
7-(3-(Aminomethyl)-4-isobutoxyphenyl)-4-methylquinolin-2-amine Dihydrochloride (27).
Compounds 39 (0.050 g, 0.163 mmol) and 106 (0.058 g, 0.160 mmol) were coupled using General Procedure 1. Purification by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 35% EtOAc in CH2Cl2, afforded protected phenylquinoline 125 as a yellow (0.056 g, 72%). This compound was immediately deprotected using General Procedure 2. After deprotection, the free aminoquinoline was diluted in MeOH (10 mL), filtered, and diluted with 4 mL ether. After treatment with methanolic HCl, the mixture was concentrated and azeotroped with toluene twice to yield 27 as a cream-colored solid (0.043 g, 90% from 125) after precipitating from MeOH (2 mL) with ether (15 mL), washing with ether, and drying in vacuo: mp: 283–285 ˚C (softens, bubbles), 308 ˚C (chars). 1H-NMR (500 MHz; DMSO-d6): δ 13.85 (s, 1 H), 8.26 (s, 3 H), 8.08 (d, J = 8.5 Hz, 1 H), 7.89 (dd, J = 8.8, 1.6 Hz, 2 H), 7.82–7.78 (m, 2 H), 7.26 (d, J = 8.7 Hz, 1 H), 6.90 (s, 1 H), 4.11 (q, J = 5.4 Hz, 2 H), 3.91 (d, J = 6.5 Hz, 2 H), 2.66 (s, 3 H), 2.16–2.10 (m, 1 H), 1.04 (d, J = 6.7 Hz, 6 H); the aminoquinoline –NH protons are broadened into the baseline around 8.5 ppm; 13C-NMR (126 MHz; DMSO-d6): δ 157.5, 154.4, 152.7, 143.5, 130.7, 129.23, 129.12, 126.7, 123.6, 123.2, 120.6, 114.5, 113.1, 112.7, 74.8, 37.7, 28.1, 19.60, 19.43; one of the aminoquinoline carbons is not visible due to baseline broadening; HRMS calcd for C21H26N3O+: 336.2070; found, 336.2080.
7-(3-(Aminomethyl)-4-(cyclobutylmethoxy)phenyl)-4-methylquinolin-2-amine Dihydrochloride (28).
Compounds 39 (0.082 g, 0.270 mmol) and 107 (0.110 g, 0.300 mmol) were coupled using General Procedure 1. Purification by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 80% EtOAc in CH2Cl2, afforded protected phenylquinoline 126 as a tan foam (0.052 g, 40%). A portion of this compound (0.04 g) was immediately deprotected using General Procedure 2. After workup, the free aminoquinoline was taken up in MeOH (1.0 mL) and treated with methanolic HCl for 18h. The mixture was concentrated to minimal solvent and triturated with ether (10 mL) 3 × to afford 28 as an off-white solid (0.031 g, 88% from 126) after filtering, washing with ether, and drying in vacuo: mp 83–84 ˚C (softens) >209.5 ˚C (darkens) >241.2 ˚C (dec) 1H-NMR (500 MHz, DMSO-d6) δ 14.38 (s, 1 H), 9.20 (bs, 1 H), 8.58 (s, 3 H), 8.28 (bs, 1 H), 8.05 (d, J = 8.4 Hz, 1 H), 7.97 (d, J = 2.3 Hz, 1H), 7.88 (d, J = 1.7 Hz, 1H), 7.84 (dd, J = 8.6, 1.7 Hz, 1 H), 7.75 (dd, J = 8.6, 2.4 Hz, 1 H), 7.24 (d, J = 8.6 Hz, 1 H), 6.96 (s, 1 H), 4.09 (d, J = 6.4 Hz, 2 H), 4.06 (d, J = 5.3 Hz, 2 H), 2.81 (hept, J = 7.0 Hz, 1 H), 2.64 (s, 3 H), 2.18 – 2.05 (m, 2 H), 1.99 – 1.82 (m, 4 H). 13C-NMR (126 MHz, DMSO-d6) δ 156.97, 153.88, 151.95, 143.00, 135.99, 130.05, 128.66, 128.37, 126.08, 123.12, 122.73, 119.86, 113.68, 112.59, 112.08, 71.86, 36.84, 33.78, 24.15, 18.85, 18.07; one of the aminoquinoline carbons is not visible due to baseline broadening; HRMS calcd for C22H26N3O+: 348.2076; found, 348.2073.
7-(3-(Aminomethyl)-4-(cyclopropylmethoxy)phenyl)-4-methylquinolin-2-amine Dihydrochloride (29).
Compound 108 (0.078 g, 0.219 mmol), tetrahydroxydiboron (0.059 g, 0.77 mmol), KOAc (0.064 g, 0.66 mmol), XPhos-Pd-G3 (2.4 mg, 2.8 μmol), and XPhos (2.6 mg, 5.5 μmol) were added to an oven-dried glass vial equipped with a Teflon-lined cap.47 EtOH (2.2 mL, degassed) was then added to the reaction vessel and a nitrogen stream was allowed to bubble through the solution at room temperature for 15 min with stirring. The vessel was then sealed, and the reaction mixture was heated to 80 °C for 2 h. The reaction was then allowed to cool to ambient temperatures before 3 equiv (0.364 mL, 0.66 mmol) of 1.8 M degassed aqueous K2CO3 was added via syringe followed by compound 38 (0.055 g, 0.20 mmol). The reaction vial was degassed with argon, resealed with a Teflon-lined cap, and left to stir at 80 °C for an additional 15 h. The reaction mixture was cooled to room temperature and partitioned between EtOAc and H2O (20 mL each). The layers were separated, the aqueous layer was extracted with EtOAc (3 × 20 mL), the organic layer was washed with 5% aq. NaCl (2 × 20 mL) and sat. aq. NaCl (20 mL), dried over anhydrous sodium sulfate, and concentrated. Purification by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 90% EtOAc in CH2Cl2, afforded protected phenylquinoline 127 as a colorless solid (0.073 g, 78%). A portion of this compound (0.069 g) was immediately deprotected using General Procedure 2. After workup, the free aminoquinoline was taken up in MeOH (3.0 mL) and treated with methanolic HCl for 18 h, and concentrated to dryness. The title compound (29) was obtained as a yellow-colored solid (0.029 g, 49% from 127) after reverse phase flash column chromatography, eluting through a 15.5 g C18 RediSep RF Gold cartridge column with a gradient of 100% water to 100% acetonitrile, azeotropic removal of water with toluene, and drying in vacuo: mp >193.5 ˚C (foam dec) 1H-NMR (500 MHz, DMSO-d6) δ 14.30 (s, 1 H), 8.50 (s, 3 H), 8.06 (d, J = 8.5 Hz, 1 H), 7.95 (d, J = 2.4 Hz, 1 H), 7.89 (d, J = 1.7 Hz, 1 H), 7.83 (dd, J = 8.6, 1.8 Hz, 1 H), 7.75 (dd, J = 8.7, 2.4 Hz, 1 H), 7.23 (d, J = 8.7 Hz, 1 H), 6.99 – 6.87 (m, 1 H), 4.09 (d, J = 5.0 Hz, 2 H), 3.99 (d, J = 6.9 Hz, 2 H), 2.65 (s, 3 H), 1.33 (tt, J = 7.3, 5.0 Hz, 1 H), 0.61 (td, J = 7.7, 6.1, 4.5 Hz, 2 H), 0.41 (q, J = 4.9 Hz, 2 H); 13C NMR (126 MHz, DMSO-d6) δ 156.93, 153.87, 151.99, 142.97, 136.11, 130.08, 128.75, 128.43, 126.09, 123.08, 122.71, 119.91, 113.79, 112.80, 112.10, 72.70, 37.08, 18.84, 9.93, 3.01; HRMS calcd for C21H24N3O+: 334.1914; found, 334.1910.
7-(3-(Aminomethyl)-4-((3-fluorobenzyl)oxy)phenyl)-4-methylquinolin-2-amine Dihydrochloride (30).
Compounds 39 (0.060 g, 0.196 mmol) and 109 (0.090 g, 0.216 mmol) were coupled using General Procedure 1. Purification by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 35% EtOAc in CH2Cl2, afforded protected phenylquinoline 128 as a brown foam (0.078 g, 76%). This compound was immediately deprotected using General Procedure 2. After deprotection, the free aminoquinoline was purified by passing through a short plug of SiO2, washing with EtOAc and then 10% MeOH in EtOAc. Concentration afforded a residue that was diluted in ether/MeOH (2:1). After treatment with methanolic HCl, the mixture was concentrated, and 30 was obtained as a white solid (0.057 g, 84% from 128) after precipitating twice from MeOH (2 mL) with ether (10 mL), and drying in vacuo: mp 242–243.5 ˚C. 1H-NMR (500 MHz; DMSO-d6): δ 14.00 (s, 1 H), 9.00 (br s, 1 H), 8.35 (br s, 4 H), 8.07 (d, J = 8.5 Hz, 1 H), 7.94 (d, J = 2.2 Hz, 1 H), 7.89 (d, J = 1.4 Hz, 1 H), 7.81 (ddd, J = 12.4, 8.8, 1.8 Hz, 2 H), 7.50–7.46 (m, 1 H), 7.43–7.38 (m, 2 H), 7.29 (d, J = 8.7 Hz, 1 H), 7.19 (td, J = 8.7, 2.2 Hz, 1 H), 6.91 (s, 1 H), 4.16 (br q, J = 5.5 Hz, 2 H), 2.65 (s, 3 H); 13C-NMR (126 MHz; DMSO-d6): δ (163.8 +161.8, 1 C) 157.0, 154.4, 152.8, 143.4, (140.16 + 140.10, 1 C), 131.19, (131.06 + 130.99, 1 C), 129.7, 129.2, 126.7, 123.88, (123.86 + 123.67, 1 C), 123.5, 120.6, (115.31 + 115.15, 1 C), (114.74 + 114.57, 1 C), 113.6, 112.8, 69.4, 37.9, 19.4; three of the quinoline and aryl ring carbons are not visible because of baseline broadening; HRMS calcd for C24H23FN3O+: 388.1820; found, 388.1828.
4-((4-(2-Amino-4-methylquinolin-7-yl)-2-(aminomethyl)phenoxy)methyl)benzonitrile Dihydrochloride (31).
Compounds 39 (0.075 g, 0.245 mmol) and 110 (0.102 g, 0.245 mmol) were coupled using General Procedure 1. After workup, the crude residue was suspended in EtOAc/ether (1:1) and filtered through a small pad of Celite overlaid with silica. The filtrate was concentrated to yield a residue that was washed with 5% hexanes/EtOAc (30 mL) to afford 129 as a white solid (0.073 g, 56%) after washing with hexanes. This compound was immediately deprotected using General Procedure 2. After deprotection, the free aminoquinoline was purified by flash column chromatography, eluting with a gradient of EtOAc to 2% MeOH in EtOAc to yield a white solid that was diluted in ether/MeOH (2:1). After treatment with methanolic HCl, the mixture was concentrated, the residue was azeotroped with toluene, and 31 was obtained as a white solid (0.054 g, 84% from 129) after precipitating twice from MeOH (3 mL) with ether (15 mL), and drying in vacuo: mp 280–282 ˚C. 1H-NMR (500 MHz; DMSO-d6): δ 14.03 (s, 1 H), 8.37 (s, 3 H), 8.06 (d, J = 8.4 Hz, 1 H), 7.94–7.88 (m, 4 H), 7.80–7.75 (m, 4 H), 7.27 (d, J = 8.5 Hz, 1 H), 6.90 (s, 1 H), 5.40 (s, 2 H), 4.17 (s, 2 H), 2.64 (s, 3 H); the aminoquinoline –NH protons are not visible as discrete peaks, but are broadened into the baseline around 8.4 and 7.9 ppm; 13C-NMR (126 MHz; DMSO-d6): δ 19.38, 37.79, 69.30, 111.09, 112.77, 113.52, 115.19, 119.23, 120.72, 123.49, 126.60, 128.52, 129.11, 129.80, 131.43, 132.93, 143.01, 143.16, 154.74, 156.78; three of the aminoquinoline and aryl carbons are not visible because of baseline broadening; HRMS calcd for C25H23N4O+: 395.1866; found, 395.1872.
7-(3-(Aminomethyl)-4-(pyridin-2-ylmethoxy)phenyl)-4-methylquinolin-2-amine Trihydrochloride (32).
Compounds 39 (0.075 g, 0.245 mmol) and 111 (0.096 g, 0.245 mmol) were coupled using General Procedure 1. Purification by flash column chromatography, eluting with a gradient of 10% EtOAc in CH2Cl2 to 85% EtOAc in CH2Cl2, afforded protected phenylquinoline 130 as a white powder (0.091 g, 72%) after precipitating from EtOAc (1 mL) with hexanes (20 mL). This compound was immediately deprotected using General Procedure 2. After deprotection, the free aminoquinoline was purified by flash column chromatography, eluting with a gradient of EtOAc to 15% MeOH in EtOAc. Concentration afforded a residue that was diluted in MeOH. After treatment with methanolic HCl, the mixture was concentrated, the residue was azeotroped three times with toluene, and 32 was obtained as a white powder (0.063 g, 74% from 130) after precipitating three times from MeOH (5 mL) with ether (10 mL), washing with ether, and drying in vacuo: mp 231–233 ˚C (dec). 1H-NMR (500 MHz; DMSO-d6): δ 14.08 (s, 1 H), 9.04 (br s, 1 H), 8.66 (d, J = 4.8 Hz, 1 H), 8.46 (s, 3 H), 8.22 (br s, 1 H), 8.08 (d, J = 8.6 Hz, 1 H), 7.99 (t, J = 7.3 Hz, 1 H), 7.94 (d, J = 2.3 Hz, 1 H), 7.89 (d, J = 1.6 Hz, 1 H), 7.82 (dd, J = 8.6, 1.7 Hz, 1 H), 7.79 (dd, J = 8.6, 2.3 Hz, 1 H), 7.72 (d, J = 7.8 Hz, 1 H), 7.49 (t, J = 6.1 Hz, 1 H), 7.33 (d, J = 8.7 Hz, 1 H), 6.92 (s, 1 H), 4.21 (d, J = 5.7 Hz, 2 H); the pyridinium –NH proton is broadened into residual water around 4.3 ppm; 13C-NMR (126 MHz; DMSO-d6): δ 157.1, 156.0, 154.4, 152.7, 148.7, 143.4, 139.0, 136.6, 131.5, 130.0, 129.3, 126.8, 124.2, 123.73, 123.67, 122.8, 120.6, 114.6, 113.8, 112.8, 70.4, 38.3, 19.4; HRMS calcd for C23H23N4O+: 371.1866; found, 371.1880.
7-(3-(Aminomethyl)-4-(pyridin-3-ylmethoxy)phenyl)-4-methylquinolin-2-amine Trihydrochloride (33).
Compounds 39 (0.075 g, 0.245 mmol) and 112 (0.096 g, 0.245 mmol) were coupled using General Procedure 1. As the product was poorly soluble in EtOAc, the aqueous layer was extracted with 200 mL of EtOAc during the workup. After concentration, the residue was washed with 50% hexanes/EtOAc (30 mL) to afford 131 as an off-white solid (0.100 g, 80%) after washing with hexanes. This compound was immediately deprotected using General Procedure 2. After deprotection, the free aminoquinoline was purified by flash column chromatography, eluting with a gradient of 3% MeOH in EtOAc to 15% MeOH in EtOAc to yield a colorless foam. This was diluted with ether/MeOH (1:1). After treatment with methanolic HCl, the mixture was concentrated, and 33 was obtained as a pale orange solid (0.062 g, 66% from 131) after precipitating twice from hot MeOH (1 mL) with ether (10 mL), washing with ether, and drying in vacuo: mp 238–240 ˚C (softens) 248–250 ˚C (melts). 1H-NMR (500 MHz; DMSO-d6): δ 14.07 (s, 1H), 9.00 (br s, 1 H), 8.94 (s, 1 H), 8.72 (d, J = 4.7 Hz, 1 H), 8.39 (s, 3 H), 8.30 (d, J = 6.9 Hz, 1 H), 8.25 (br s, 1 H), 8.08 (d, J = 8.6 Hz, 1 H), 7.95 (d, J = 2.1 Hz, 1 H), 7.90 (d, J = 1.3 Hz, 1 H), 7.84–7.81 (m, 2 H), 7.74–7.72 (m, 1 H), 7.35 (d, J = 8.7 Hz, 1 H), 6.93 (s, 1 H), 5.40 (s, 2 H), 4.15 (q, J = 5.5 Hz, 3 H), 2.66 (s, 3 H); the pyrdinium –NH proton is broadened into residual water around 4.0 ppm; 13C-NMR (126 MHz; DMSO-d6): δ 156.9, 154.4, 152.8, 146.9, 143.4, 136.6, 134.4, 131.4, 130.0, 129.3, 126.8, 125.4, 123.74, 123.55, 120.6, 114.5, 113.6, 112.8, 67.5, 37.9, 19.4; one of the quinoline carbons is not visible due to baseline broadening; 13C-NMR (126 MHz; DMSO-d6): δ 157.1, 156.0, 154.4, 152.7, 148.6, 143.4, 139.0, 136.6, 131.5, 130.0, 129.3, 126.8, 124.2, 123.73, 123.67, 122.8, 120.6, 114.6, 113.8, 112.8, 70.4, 38.3, 19.4; HRMS calcd for C23H23N4O+: 371.1866; found, 371.1878.
7-(3-(Aminomethyl)-4-((5-methylisoxazol-3-yl)methoxy)phenyl)-4-methylquinolin-2-amine Dihydrochloride (34).
Compounds 39 (0.085 g, 0.276 mmol) and 113 (0.132 g, 0.33 mmol) were coupled using General Procedure 1. Purification by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 70% EtOAc in CH2Cl2, afforded protected phenylquinoline 133 as a white solid (0.032 g, 22%). This compound was immediately deprotected using General Procedure 2. After workup, the free aminoquinoline was taken up in MeOH (1.0 mL) and treated with methanolic HCl (1 mL) for 18 h, resulting in the formation of a white precipitate, which was concentrated to dryness. The title compound (34) was obtained as a tan solid (0.012 g, 43% from 132) after recrystallization overnight from methanol that was slowly diffused with ether, followed by removal of mother liquor, washing with 1:20 methanol-ether, and drying in vacuo: mp >267 ˚C (browns slowly) 272–274 ˚C (melts/gels) >277 ˚C (foams and decomp.). 1H NMR (500 MHz, DMSO-d6) δ 14.29 (s, 1 H), 8.52 (s, 3 H), 8.10 – 8.01 (m, 1 H), 8.01 – 7.93 (m, 1 H), 7.90 (s, 1 H), 7.83 (dd, J = 8.5, 2.0 Hz, 1 H), 7.79 (dt, J = 8.8, 2.4 Hz, 1 H), 7.36 (d, J = 8.6 Hz, 1 H), 6.94 (d, J = 4.6 Hz, 1 H), 6.49 (d, J = 4.4 Hz, 1 H), 5.32 (s, 2 H), 4.10 (d, J = 5.6 Hz, 2 H), 2.65 (s, 3 H), 2.44 (s, 3 H); 13C NMR (126 MHz, DMSO-d6) δ 169.98, 164.79, 160.32, 156.15, 153.88, 142.80, 130.94, 129.19, 128.56, 126.44, 126.14, 123.17, 122.96, 120.01, 114.03, 112.93, 112.20, 101.47, 61.85, 37.13, 18.84, 11.76; HRMS calcd for C22H23N4O2+: 375.1821; found, 375.1816.
7-(3-(Aminomethyl)-4-(thiazol-4-ylmethoxy)phenyl)-4-methylquinolin-2-amine Trihydrochloride (35).
Compounds 39 (0.087 g, 0.282 mmol) and 114 (0.122 g, 0.304 mmol) were coupled using General Procedure 1. Purification by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 80% EtOAc in CH2Cl2, afforded protected phenylquinoline 133 as a cream-colored solid (0.056 g, 38%). A portion of this compound (0.047 g) was immediately deprotected using General Procedure 2. After workup, the free aminoquinoline was treated with methanolic HCl (2 mL), and sonicated for 10 minutes before stirring at room temperature for 18 h. The resulting grey precipitate was collected to afford 35 as a grey solid (0.044 g, 99% from 133) after filtering, washing with ether, and drying in vacuo. Because of the NMR spectral overlap of differing HCl species of 35, the precipitate was further purified using reverse phase chromatography, eluting through a 15.5 g C18 RediSep RF Gold cartridge column with a gradient of 100% water to 100% acetonitrile to afford 35 as a yellow solid (0.009 g, 38%) which was used for characterization: mp 210.8–212 ˚C (softens) >315 ˚C (dec); 1H-NMR (500 MHz, DMSO-d6) δ 14.20 (s, 1 H), 9.18 (d, J = 1.9 Hz, 1 H), 8.43 (s, 4 H), 8.05 (d, J = 8.5 Hz, 1 H), 7.94 (d, J = 2.4 Hz, 1 H), 7.92 (d, J = 2.0 Hz, 1 H), 7.89 (d, J = 1.8 Hz, 1 H), 7.84 – 7.75 (m, 2 H), 7.42 (d, J = 8.6 Hz, 1 H), 6.91 (s, 1 H), 5.42 (s, 2 H), 4.12 (s, 2 H), 2.64 (s, 3 H); 13C-NMR (126 MHz, 500 MHz, DMSO-d6) δ 156.45, 154.96, 154.03, 152.12, 151.95, 142.98, 136.00, 130.73, 129.20, 128.44, 126.18, 123.27, 123.18, 119.99, 118.35, 113.82, 113.36, 112.24, 65.99, 37.17, 18.97. HRMS calcd for C21H21N4OS+: 377.1436; found, 377.1429.
7-(3-(Aminomethyl)-4-(oxazol-4-ylmethoxy)phenyl)-4-methylquinolin-2-amine Trihydrochloride (36).
Compounds 39 (0.089 g, 0.289 mmol) and 115 (0.133 g, 0.347 mmol) were coupled using General Procedure 1. Purification by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 80% EtOAc in CH2Cl2, afforded protected phenylquinoline 134 as a tan powder (0.027 g, 19%). A portion of this compound (0.024 g) was immediately deprotected using General Procedure 2. After workup, the free aminoquinoline was taken up in MeOH (1.0 mL) and treated with methanolic HCl (1.5 mL) for 18h, and concentrated to dryness. The title compound (36) was obtained as a tan solid (0.014 g, 65% from 134) in 90% purity after recrystallizing once from 1:3 methanol-ethanol followed by an additional overnight recrystallization from methanol that was slowly diffused with ether, followed by removal of mother liquor, washing with 1:20 methanol-ether, and drying in vacuo. Additional purification of the crude product was achieved after reverse phase flash column chromatography, eluting through a 15.5 g C18 RediSep RF Gold cartridge column with a gradient of 100% water to 100% acetonitrile to afford pure 36 as a cream-colored solid (0.011 g, 47% from 135) after azeotropic removal of water with toluene and drying in vacuo: mp >151 ˚C (slowly browns) 253–254 ˚C). 1H-NMR (500 MHz, DMSO-d6) δ 8.45 (s, 1 H), 8.30 (s, 1 H), 8.22 – 7.91 (m, 1 H), 7.88 (d, J = 2.5 Hz, 1 H), 7.82 (d, J = 8.4 Hz, 1 H), 7.75 (dd, J = 8.6, 2.5 Hz, 1 H), 7.72 (d, J = 1.9 Hz, 1 H), 7.50 (dd, J = 8.5, 2.1 Hz, 1 H), 7.35 (dd, J = 8.7, 2.8 Hz, 1 H), 6.61 (s, 1 H), 6.33 (s, 2 H), 5.18 (s, 2 H), 4.05 (s, 2 H), the aminoquinoline -NH proton is broadened into baseline and the benzylic CH3 protons are obscured by the solvent peak around 2.5; 13C NMR (126 MHz, DMSO-d6) δ 158.23, 155.33, 152.46, 148.39, 143.80, 139.52, 137.92, 135.55, 132.62, 128.25, 127.67, 124.30, 123.67, 122.30, 121.93, 119.46, 113.05, 112.08, 62.21, 37.52, 18.08. HRMS calcd for C21H21N4O2+: 361.1659; found, 361.1655.
7-(3-(Aminomethyl)-4-(thiazol-5-ylmethoxy)phenyl)-4-methylquinolin-2-amine Trihydrochloride (37).
Compounds 39 (0.081 g, 0.265 mmol) and 116 (0.127 g, 0.32 mmol) were coupled using General Procedure 1. Purification by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 70% EtOAc in CH2Cl2, afforded protected phenylquinoline 135 as a tan solid (0.036 g, 26%). A portion of this compound (0.032 g) was immediately deprotected using General Procedure 2. After workup, the free aminoquinoline was taken up in MeOH (1.0 mL) and treated with methanolic HCl (1 mL) for 18 h, resulting in the formation of a white precipitate, which was concentrated to dryness. The pure title compound (37) was obtained as a brown crystalline solid (0.018 g, 60% from 135) after recrystallizing once from 1:5 methanol-ethanol and then from methanol, washing with 1:20 methanol-ether, and drying in vacuo: mp >238 ˚C (darkens) 252–255 ˚C (slowly softens) >256 ˚C (decomposes into brown foam). 1H NMR (500 MHz, DMSO-d6) δ 14.33 (s, 1 H), 9.18 (s, 1 H), 8.54 (d, J = 6.1 Hz, 3 H), 8.12 (s, 1 H), 8.06 (d, J = 8.5 Hz, 1 H), 7.98 (d, J = 2.4 Hz, 1 H), 7.90 (d, J = 1.8 Hz, 1 H), 7.84 (dd, J = 8.6, 1.8 Hz, 1 H), 7.79 (dd, J = 8.6, 2.3 Hz, 1 H), 7.42 (d, J = 8.7 Hz, 1 H), 6.96 (s, 1 H), 5.57 (s, 2 H), 4.06 (q, J = 5.8 Hz, 3 H), 2.65 (s, 4 H); 13C NMR (126 MHz, DMSO) δ 155.94, 155.51, 153.86, 152.02, 142.93, 142.82, 135.97, 133.64, 130.85, 129.06, 128.42, 126.13, 123.18, 123.04, 119.97, 115.67, 113.86, 113.19, 112.18, 62.40, 37.04, 18.85; HRMS calcd for C21H21N4OS+: 377.1436 found, 377.1431.
Potassium 2-Acetamido-4-methylquinoline-7-trifluoroborate (39).
Compound 38 (0.368 g, 1.32 mmol), B2Pin2 (0.503 g, 1.98 mmol), anhydrous KOAc (0.388 g, 3.96 mmol), and Pd(dppf)Cl2 (5 mol%, 0.048 g) were combined in a sealable vial and diluted with anhydrous dioxane (5 mL). The mixture was purged with argon, sealed, and heated at 75 ˚C for 19 h. The mixture was then cooled, diluted with EtOAc (20 mL) and filtered through a pad of Celite overlaid with a thin layer of SiO2. The pad was washed with EtOAc (200 mL), and the combined filtrate was washed with 5% aq. NaCl and sat. aq. NaCl (100 mL each). The organic layer was dried over anhydrous sodium sulfate and concentrated to yield a brown foam containing the crude 7-BPin quinoline, which was immediately diluted in THF/H2O (5 mL/1.5 mL). KHF2 (0.412 g, 5.28 mmol) was added, and the mixture was stirred at r.t. for 3 h. THF (2 mL) and H2O (1 mL) were then added, and the mixture was sonicated vigorously for 5 min and concentrated. The residue was azeotroped three times with toluene, diluted with acetone, and filtered. The filter cake was extracted with boiling acetone (6 × 60 mL), and the combined acetone filtrates were concentrated. The residue was washed with 10% hexanes in CH2Cl2 to yield 39 as a light tan powder (0.371 g, 92%): mp > 260 ˚C (darkens), 290–292 ˚C (dec). 1H-NMR (500 MHz; acetone-d6): δ 9.33 (s, 1 H), 8.12 (s, 1 H), 7.91 (s, 1 H), 7.71 (d, J = 8.2 Hz, 1 H), 7.66 (d, J = 8.2 Hz, 1 H), 2.63 (d, J = 0.8 Hz, 3 H), 2.23 (s, 3 H); 13C-NMR (126 MHz; DMSO-d6): δ 169.4, 150.2, 146.0, 145.1, 129.46, 129.45, 129.42, 123.9, 121.0, 112.7, 24.0, 18.7; this compound does not ionize well by ESI-MS or LC-TOF.
Purified NOS Enzyme Assays.
Rat and human nNOS, rat nNOS mutants, murine macrophage iNOS, human iNOS, and human eNOS were recombinant enzymes (expressed in E. coli and purified as reported previously).21,48,49,50 To test for NOS inhibition, the hemoglobin capture assay was used to measure nitric oxide production. The assay was performed at 37 ˚C in HEPES buffer (100 mM with 10% glycerol, pH 7.4) in the presence of 10 μM L-arginine, used because a) it is close to the Km values all three isoforms, where detection of competitive inhibitors is most sensitive, and b) significant NOS uncoupling does not occur at this concentration. Also included were 100 μM NADPH, 0.83 mM CaCl2, approximately 320 units/mL of calmodulin, 10 μM H4B, and human oxyhemoglobin (3 μM). For iNOS, the CaCl2 and calmodulin were omitted and replaced with HEPES buffer (as neither are required for activation of iNOS). The assay was performed in 96-well plates using a Synergy 4 BioTek hybrid reader. The dispensing of NOS enzyme and hemoglobin was automated, and after 30 sec (maximum delay), NO production was read by monitoring the absorbance at 401 nm (resulting from the conversion of oxyhemoglobin to methemoglobin). Kinetic readouts were performed for 5 min. Each compound was assayed at least in duplicate, and six to nine concentrations (500 μM-50 nM or 100 μM-10 nM for eNOS and iNOS; 50 μM to 5 nM for rat and human nNOS) were used to construct concentration-response curves. IC50 values were calculated by nonlinear regression (variable slope, four parameters) using GraphPad Prism software (standard error is reported for the LogIC50), and Ki values were obtained from IC50 values using the Cheng-Prusoff51 equation [Ki = IC50/(1+[S]/Km)] with the following Km values: 1.3 μM (rat nNOS), 1.6 μM (human nNOS), 8.2 μM (murine macrophage iNOS), 8 μM (human iNOS),52 3.9 μM (human eNOS),53 1.7 μM (rat nNOS D597N single mutant),54 and 1.9 μM (rat nNOS D597N/M336V double mutant).48
PAMPA-BBB Assay.
The PAMPA-BBB assay was performed following a protocol described previously.21 Briefly, the assay was done in 10 mM phosphate-buffered saline (PBS) buffer (pH 7.5), and compounds were tested at a concentration of 200 μM. The donor plate was first coated with 4 μL of porcine brain lipid (20 mg/mL in dodecane), followed by addition of 250 μL of a test compound. The acceptor plate was filled with 250 μL of PBS, and the donor plate was carefully placed on top of the acceptor plate to make a “sandwich”. The plate was incubated at 25 °C for 17 h in a saturated humidity atmosphere with orbital agitation at 100 rpm. Verapamil, chlorpromazine, and dopamine were used as a positive, intermediate, and negative control, respectively. After incubation, 150 μL of test solution was taken from each well from both sides (donor and acceptor) and transferred to the UV plate for measurement. The effective permeability (Pe) was calculated using the following equation55: , where Pe is the effective permeability (cm/s), VA and VD are the volume of the acceptor and donor well (0.25 cm3), respectively, CA (t) is the concentration of the acceptor well at time t, CD (0), CD (t) is the concentration of the donor well at t0 and t, respectively, A is the filter well area (0.21 cm2). t is the incubation time (s). τss is the time to reach a steady state (usually very short compared to the incubation time). R is the retention membrane factor and was calculated using the following equation: . Pe was reported as an average of triplicate measurements with a standard deviation.
Human Liver Microsome Stability Assay.
A 1120 μL aliquot of potassium phosphate buffer (50 mM, pH 7.4) containing liver microsomes (0.714 mg/mL) was added to individual 2 mL tubes (final concentration 0.5 mg/mL). Positive control (terfenadine) and 12 (1 mM DMSO stocks) were directly spiked into respective tubes to prepare a concentration of 1.428 μM (final concentration 1 μM). From the above mixture, 70 μL was added to individual wells of 96-well reaction plates and preincubated at 37 ˚C for 5 min. All reactions were initiated by adding 30 μL of 3.33 mM NADPH (1 mM final concentration). Reactions without NADPH and buffer controls (minus NADPH) at 0 min and 60 min were also incubated to rule out non-NADPH metabolism or chemical instability in the incubation buffer. All reactions were terminated using 100 μL of ice-cold acetonitrile containing internal standard (glipizide) at 0, 5, 15, 30, and 60 min. The plates were centrifuged at 4000 rpm for 15 min and 100 μL aliquots were analysis for parent compound disappearance in multiple-reaction monitoring (MRM) mode using LC-MS/MS. The percent remaining of test compounds and positive controls in each sample was determined by considering peak area ratio in the 0-minute sample as 100%. The half-life (t1/2 in min), and the in vitro intrinsic clearance (CL’int units in mL/min/kg) were calculated according to the following equations, where k is the gradient of line determined from a plot of peak area ratio (compound peak area / internal standard peak area) against time:
For liver microsomes, a scaling factor used was 45 mg microsomal protein per g liver.
Inhibitor Complex Crystal Preparation.
The sitting drop vapor diffusion method was used to grow crystals at 4 °C for the heme domains of rnNOS (8 mg/mL containing 20 mM histidine), the hnNOS K301R/R354A/G357D mutant (10 mg/mL containing 20 mM histidine), and heNOS (6 mg/mL). The crystal growth conditions were as described previously.8 The only exception is that the pH for the heNOS crystal growth is 7.5 rather than 6.5 as mistakenly reported there. Fresh crystals were first passed stepwise through cryoprotectant solutions. The pH of the final soaking solution for rnNOS was adjusted from 5.8 through 6.5, 7.0 (in MES) to 7.5 (in HEPES) and that for hnNOS from pH 7.2 to 7.5 (in HEPES), for heNOS the BIS-TRIS buffer at pH 7.5 was unchanged. At pH 7.5, crystals were soaked with 5–10 mM inhibitor for 2–4 h at 4 °C before being flash cooled with liquid nitrogen and stored until data collection.
X-ray Diffraction Data Collection, Data Processing, and Structural Refinement.
The cryogenic (100 K) X-ray diffraction data were collected remotely at the Stanford Synchrotron Radiation Lightsource (SSRL) or Advanced Light Source (ALS) through the data collection control software Blu-Ice56 and a crystal-mounting robot. When a CCD detector was used, 100–125° of data were typically collected with 0.5° per frame. If a Pilatus pixel array detector was used, 140–200° of fine-sliced data were collected with 0.2° per frame. More data frames were collected for heNOS which has a low symmetry (P21). Raw CCD data frames were indexed, integrated, and scaled using iMOSFLM,57 but the pixel array data were preferably processed with XDS58 and scaled with Aimless.59 The binding of inhibitors was detected by initial difference Fourier maps calculated with REFMAC.60 The inhibitor molecules were then modeled in Coot61 and refined using REFMAC or PHENIX.62 The symmetry of heNOS crystals was changed from the orthorhombic P212121 reported previously63 to monoclinic P21, with a β angle only 0.6–0.7° off compared to the original 90°. Therefore, a molecular replacement calculation with PHASER-MR64 was needed to solve the structure. In the P21 space group, there are two heNOS dimers in the asymmetric unit. Disordering in portions of inhibitors bound in the NOS active sites was often observed, sometimes resulting in poor density quality. However, partial structural features were usually still visible if the contour level of the sigmaA weighted 2m|Fo| − D|Fc| map was dropped to 0.5 σ, which afforded the building of reasonable models into the disordered regions. Water molecules were added in PHENIX and visually checked by Coot. The TLS65 protocol was implemented in the PHENIX refinements with each subunit as one TLS group. The omit Fo − Fc density maps were calculated by the Polder map facility in PHENIX for the bound inhibitors.66 The refined structures were validated in Coot before deposition in the Protein Data Bank. The X-ray diffraction data collection and structure refinement statistics are summarized in Table S1 in Supporting Information.
Supplementary Material
Acknowledgments
The authors thank the National Institutes of Health (R01 GM049725 and R35 GM131788 to R.B.S.; GM057353 and GM131920 to T.L.P.) for their support of this work. We thank the beamline staff at SSRL and ALS for their assistance during the remote X-ray diffraction data collections. H. L. would like to thank Carla Plaza for her efforts in protein preparations, which provided samples for both the structure determinations and the inhibitory assays. M. A. C. would like to thank Mr. Saman Shafaie and Dr. S. Habibi Goudarzi for assistance with HRMS experiments. This work made use of IMSERC at Northwestern University, which has received support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF NNCI-1542205), the State of Illinois, and the International Institute for Nanotechnology (IIN).
Abbreviations Used.
- NO
nitric oxide
- nNOS
neuronal nitric oxide synthase
- iNOS
inducible nitric oxide synthase
- eNOS
endothelial nitric oxide synthase
- rnNOS
rat neuronal nitric oxide synthase
- hnNOS
human neuronal nitric oxide synthase
- heNOS
human endothelial nitric oxide synthase
- miNOS
murine inducible nitric oxide synthase
- hiNOS
human inducible nitric oxide synthase
- FMN
flavin mononucleotide
- H4B
(6R)-5,6,7,8-tetrahydrobiopterin
- tPSA
total polar surface area
- Bis-Tris
bis(2-hydroxyethyl)amino-tris(hydroxymethyl)-methane
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- PAMPA-BBB
parallel artificial membrane permeability for the blood–brain barrier
- PDSP
Psychoactive Drug Screening Program
- WT
wild-type
Footnotes
Supporting Information. Crystallographic data collection and refinement statistics for rat and human nNOS and human eNOS structures. Results and Discussion pertaining to constrained amine analogues (16–18) and small 4-alkyloxy-3-aminomethyl analogues 25–28, and binding behavior of 31, 36, and 37. Relevant X-ray structures for 25, 26, 28, 31, 36, 37 and protein secondary structure displacement (Figures S1–S6), molecular docking protocol, synthesis and analytical data for relevant precursor compounds 46–49, 67, 73, 77–80, 85, 87–88, 90, 92–95, 98, 100–116, and full PDSP secondary binding assay results. A Molecular Formula Strings table (.csv) is also available.
PDB ID Codes. PDB codes for X-ray crystal structures described in this study have been deposited in the Protein Data Bank under the following accession codes (See Table S1 in SI for details): 6PMV, 6PMW, 6PMX, 6PMY, 6PMZ, 6PN0, 6PN1, 6PN2, 6PN3, 6PN4, 6PN5, 6PN6, 6PN7, 6PN8, 6PN9, 6PNA, 6PNB, 6PNC, 6PND, 6PNE, 6PNF, 6PNG, 6PNH, 6PO5, 6PO7, 6PO8, 6PO9, 6POA, 6POB, 6POC, 6POT, 6POU, 6POV, 6POW, 6POX, 6POY, 6POZ, 6PP0, 6PP1, 6PP2, 6PP3, 6PP4.
References Cited
- (1).Torreilles F; Salman-Tabcheh S; Guérin M; Torreilles J Neurodegenerative disorders: the role of peroxynitrite. Brain Res. Rev 1999, 30, 153–163. [DOI] [PubMed] [Google Scholar]
- (2).Uehara T; Nakamura T; Yao D; Shi Z; Gu Z; Ma Y; Masliah E; Nomura Y; Lipton SA S-Nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature 2006, 441, 513–517. [DOI] [PubMed] [Google Scholar]
- (3).Trippier PC; Jansen Labby K; Hawker DD; Mataka JJ; Silverman RB Target- and mechanism-based therapeutics for neurodegenerative diseases: strength in numbers. J. Med. Chem 2013, 56, 3121–3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Campbell MG; Smith BC; Potter CS; Carragher B; Marletta MA Molecular architecture of mammalian nitric oxide synthase. Proc. Natl. Acad. Sci. U. S. A 2014, 111, E3614–E3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Gamboa A; Shibao C; Diedrich A; Choi L; Pohar B; Jordan J; Paranjape S; Farley G; Biaggioni I Contribution of endothelial nitric oxide to blood pressure in humans. Hypertension 2007, 49, 170–177. [DOI] [PubMed] [Google Scholar]
- (6).Cinelli MA; Li H; Chreifi G; Martasek P; Roman LJ; Poulos TL; Silverman RB Simplified 2-aminoquinoline-based scaffold for potent and selective neuronal nitric oxide synthase inhibition. J. Med. Chem 2014, 57, 1513–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Cinelli MA; Li H; Pensa AV; Kang S; Roman LJ; Martasek P; Poulos TL; Silverman RB Phenyl ether- and aniline-containing 2-aminoquinolines as potent and selective inhibitors of neuronal nitric oxide synthase. J. Med. Chem 2015, 58, 8694–8712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Cinelli MA; Li H; Chreifi G; Poulos TL; Silverman RB Nitrile in the hole: discovery of a small auxiliary pocket in neuronal nitric oxide synthase leading to the development of potent and selective 2-aminoquinoline inhibitors. J. Med. Chem 2017, 60, 3958–3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Pensa AV; Cinelli MA; Li H; Chreifi G; Mukherjee P; Roman LJ; Martasek P; Poulos TL; Silverman RB Hydrophilic, potent, and selective 7-substituted 2-aminoquinolines as improved human neuronal nitric oxide synthase inhibitors. J. Med. Chem 2017, 60, 7146–7165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Ballell L; Bates RH; Young RJ; Alvarez-Gomez D; Alvarez-Ruiz E; Barroso V; Blanco D; Crespo B; Escribano J; González R; Lozano S; Huss S; Santos-Villarejo A; Martín-Plaza JJ; Mendoza A; Rebollo-Lopez MJ; Remuiñan-Blanco M; Lavandera JL; Pérez-Herran E; Gamo-Benito FJ; García-Bustos JF; Barros D; Castro JP and Cammack N Fueling open-source drug discovery: 177 small-molecule leads against tuberculosis. ChemMedChem 2013, 8, 313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Mugumbate G; Abrahams KA; Cox JAG; Papadatos G; van Westen G; Lelièvre J; Calus ST; Loman NJ; Ballell L; Barros D; Overington JP; Besra GS Mycobacterial dihydrofolate reductase inhibitors identified using chemogenomic methods and in vitro validation. PLOS ONE 2015, 10, e0121492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Xue F; Li H; Fang J; Roman LJ; Martásek P; Poulos TL; Silverman RB Peripheral but crucial: A hydrophobic pocket (Tyr706, Leu337, and Met336) for potent and selective inhibition of neuronal nitric oxide synthase. Bioorg. Med. Chem. Lett 2010, 20, 6258–6261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Duquenne C; Johnson N; Knight SD; Lafrance L; Miller WH; Newlander K; Romeril S; Rouse MB; Tian X; Verma SK Indazoles. WO2011140325, A1 November 10, 2011. [Google Scholar]
- (14).Borg G; Cogan DA; Ellman JA; One-pot asymmetric synthesis of tert-butanesulfinyl-protected amines from ketones by the in situ reduction of tert-butanesulfinyl ketimines. Tetrahedron Lett. 1999, 40, 6709–6712. [Google Scholar]
- (15).Lind KE; Cao K; Lin EY-S; Nguyen TB; Tangonan BT; Erlanson DA; Guckian K; Simmons RC; Lee W-C; Sun; Hansen S; Pathan N; Zhang. Pyridinonyl pdk1 Inhibitors. WO 2008005457, January 10, 2008. [Google Scholar]
- (16).Heil M; Heilmann EK; Sudau A; Kapferer T; Muhlthau FA; Jeschke P; Voerste R; Gorgens U; Raming K; Ebbinghaus-Kinstscher U; Drewes M; Adamczewski M; Becker A Halogenalkyl-Substituted Amides Used as Insecticides and Acaricides. WO2011054436, July 7, 2010. [Google Scholar]
- (17).Brunton SA; Guicherit OM; Kruse LI; Haydar SN; and Springer DM Small Organic Molecule Regulators of Cell Proliferation. WO2008057469, May 15, 2008. [Google Scholar]
- (18).Labby KJ; Xue F; Kraus JM; Ji H; Mataka J; Li H; Martásek P; Roman LJ; Poulos TL; Silverman RB Intramolecular hydrogen bonding: A potential strategy for more bioavailable inhibitors of neuronal nitric oxide synthase. Bioorg. Med. Chem 2012, 20, 2435–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Hevel JM; Marletta MA [25] Nitric-oxide synthase assays. Methods in Enzymology 1994, 233, 250–258. [DOI] [PubMed] [Google Scholar]
- (20).Delker SL; Xue F; Li H; Jamal J; Silverman RB; Poulos TL Role of zinc in isoform-selective inhibitor binding to neuronal nitric oxide synthase. Biochemistry 2010, 49, 10803–10810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Do HT; Li H; Chreifi G; Poulos TL; Silverman RB Optimization of blood-brain barrier permeability with potent and selective human heuronal nitric oxide synthase inhibitors having a 2-aminopyridine scaffold. J. Med. Chem 2019, 62, 2690–2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Poulos TL; Li H Structural basis for isoform-selective inhibition in nitric oxide synthase. Acc. Chem. Res 2013, 46, 390–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Huang H; Li H; Martasek P; Roman LJ; Poulos TJ; Silverman RB Structure-guided design of selective inhibitors of neuronal nitric oxide synthase. J. Med. Chem 2013, 56, 3024–3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Delker SL; Ji H; Li H; Jamal J; Fang J; Xue F; Silverman RB; Poulos TL Unexpected binding modes of nitric oxide synthase inhibitors effective in the prevention of cerebral palsy. J. Am. Chem. Soc 2010, 132, 5437–5442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Mukherjee P; Li H; Sevrioukova I; Chreifi G; Martásek P; Roman LJ; Poulos TL; Silverman RB Novel 2,4-disubstituted pyrimidines as potent, selective, and cell-permeable inhibitors of neuronal nitric oxide Synthase. J. Med. Chem 2015, 58, 1067–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Garcin ED; Arvai AS; Rosenfeld RJ; Kroeger MD; Crane BR; Andersson G; Andrews G; Hamley PJ; Mallinder PR; Nicholls DJ; St-Gallay SA; Tinker AC; Gensmantel NP; Mete A; Cheshire DR; Connolly S; Stuehr DJ; Aberg A; Wallace AV; Tainer JA; Getzoff ED Anchored plasticity opens doors for selective inhibitor design in nitric oxide synthase. Nat. Chem. Biol 2008, 4, 700–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Besnard J; Ruda GF; Setola V; Abecassis K; Rodriguiz RM; Huang X; Norval S; Sassano MF; Shin AI; Webster LA; Simeons FRC; Stojanovski L; Prat A; Seidah NG; Constam DB; Bickerton GR; Read KD; Wetsel WC; Gilbert IH; Roth BL; Hopkins AL Automated design of ligands to polypharmacological profiles. Nature 2012, 492, 215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Basith S; Cui M; Macalino SJY; Park J; Clavio NAB; Kang S; Choi S Exploring G-protein-coupled Receptors (GPCRs) ligand space via cheminformatics approaches: impact on rational drug design. Front. Pharmacol 2018, 9, 128, doi: 10/3389/fphar.2018.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Yanagisawa I; Hirata Y; Ishii Y Studies on histamine H2 receptor antagonists. 2. Synthesis and pharmacological activities of N-sulfamoyl and N-sulfonyl amidine derivatives. J. Med. Chem 1987, 30, 1787–1793. [DOI] [PubMed] [Google Scholar]
- (30).Rankovic Z CNS drug design: balancing physicochemical properties for optimal brain exposure J. Med. Chem 2015, 58, 2584–2608. [DOI] [PubMed] [Google Scholar]
- (31).Di L; Kerns EH; Fan K; McConnell OJ; Carter GT High throughput artificial membrane permeability assay for blood– brain barrier. Eur. J. Med. Chem 2003, 38, 223–232. [DOI] [PubMed] [Google Scholar]
- (32).Di L; Kerns EH; Bezar IF; Petusky SL; Huang Y Comparison of blood–brain barrier permeability assays: in situ brain perfusion, MDR1-MDCKII and PAMPA-BBB J. Pharm. Sci 2009, 98, 1980–1991. [DOI] [PubMed] [Google Scholar]
- (33).Könczöl Á; Müller J; Földes E; Béni Z; Végh K; Kéry Á; Balogh GT Applicability of a blood–brain barrier specific artificial membrane permeability assay at the early stage of natural product-based CNS drug discovery J. Nat. Prod 2013, 76, 655–663 [DOI] [PubMed] [Google Scholar]
- (34).Daina A, Michielin O & Zoete V SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep 7, 42717 (2017) doi: 10.1038/srep42717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Daina A, Zoete V, A BOILED-Egg to predict gastrointestinal absorption and brain penetration of small molecules. ChemMedChem 2016, 11, 1117–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Dialer LO; Selivanova SV; Muller CJ; Muller A; Stellfeld T; Graham K; Dinkelborg LM; Kramer SD; Schibli R; Reiher M, Ametamey SM Studies toward the development of new silicon-containing building blocks for the direct (18)F-labeling of peptides. J. Med. Chem 2013, 56, 7552–7563. [DOI] [PubMed] [Google Scholar]
- (37).Demont EH; Garton NS; Gosmini RLM; Hayhow TGC; Seal J; Wilson DM; Woodrow MD Tetrahydroquinoline Derivatives and their Pharmaceutical Use. WO2011054841, May 12, 2011. [Google Scholar]
- (38).Gunbas DD and Brouwer AM Degenerate molecular shuttles with flexible and rigid spacers. J. Org. Chem 2012, 77, 5724–5735. [DOI] [PubMed] [Google Scholar]
- (39).Brown AD; Bunnage ME; Glossop PA; James K; Lane CAL; Lewthwaite RA; Moses IA; Price DA; Thomson NM Sulfonamide Derivatives for the Treatment of Diseases. US Patent 20100273758, October 28, 2010. [Google Scholar]
- (40).Jarboe SG; Terrazas, and Beak, P. The endocyclic restriction test: the geometries of nucleophilic substitutions at sulfur (VI) and sulfur (II). J. Org. Chem 2008, 73, 9627–9632. [DOI] [PubMed] [Google Scholar]
- (41).Ballatore C; Soper JH; Piscitelli F; James M; Huang L; Atasoylu O; Huryn DM; Trojanowski JQ; Lee VM; Brunden KR; Smith AB Cyclopentane-1,3-dione: a novel isostere for the carboxylic acid functional group. Application to the design of potent thromboxane (A2) receptor antagonists. J. Med. Chem 2011, 54, 6969–6983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Fisher TH; Dershem S,M; Prewitt ML Meta-substituent effects on benzyl free-radical stability. J. Org. Chem 1990, 55, 1040–1043. [Google Scholar]
- (43).Hoyt SB; Petrilli WL; London C; Xiong Y; Taylor JA; Ali A; Lo M; Henderson TJ; Hu Q; Hartmann R; Yin L; Heim R; Bey E; Saxena R; Samanta SK; Kulkarni BA Aldosterone Synthase Inhibitors. WO2012148808, November 1, 2012. [Google Scholar]
- (44).Finkelstein BL; Taggi AE; Long JK; Sharpe PL; Tseng C-P; McCann SF; Ding AX; Swann SL Jr Substituted Benzene Fungicides. WO 2008124092, December 18, 2008. [Google Scholar]
- (45).Nielsen SF; Larsen M; Boesen T; Schonning K; Kromann H Cationic chalcone antibiotics. Design, synthesis, and mechanism of action. J. Med. Chem 2005, 48, 2667–2677. [DOI] [PubMed] [Google Scholar]
- (46).Andreini M; Gabellieri E; Guba W; Marconi G; Narquizian R; Power E; Travagli M; Woltering T; Wostl W 4,5-Dihydro-oxazol-2-yl Amine Derivatives. US Patent 10090209529, August 20, 2009. [Google Scholar]
- (47).Molander GA; Trice SLJ; Kennedy SM Scope of the two-step, one-pot palladium-catalyzed borylation/suzuki cross-coupling reaction utilizing bis-boronic acid. J. Org. Chem 2012, 77, 8678–8688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Roman LJ; Sheta EA; Martásek P; Gross SS; Liu Q; Masters BSS High-level expression of functional rat neuronal nitric oxide synthase in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A 1995, 92, 8428–8432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Hevel JM; White KA; Marletta MA Purification of the inducible murine macrophage nitric oxide synthase: identification as a flavoprotein. J. Biol. Chem 1991, 266, 22789–22791. [PubMed] [Google Scholar]
- (50).Gerber NC; Ortiz de Montellano PR Neuronal nitric oxide synthase: expression in Escherichia coli, irreversible inhibition by phenyldiazene, and active site topology. J. Biol. Chem 1995, 270, 17791–17796. [DOI] [PubMed] [Google Scholar]
- (51).Cheng Y-C; Prusoff WH Relationship between the inhibition constant (Ki) and the concentration of the inhibitor which causes 50 per cent inhibition (IC50) of an enzymatic reaction. Biochem. Pharmacol 1973, 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
- (52).Fossetta JD; Niu XD; Lunn CA; Zavodny PJ; Narula SK; Lundell D Expression of human inducible nitric oxide synthase in Escherichia coli. FEBS Letters 1996, 379, 135–138. [DOI] [PubMed] [Google Scholar]
- (53).Leber A; Hemmens B; Klosch B; Goessler W; Raber G; Mayer B; Schmidt K Characterization of recombinant human endothelial nitric-oxide synthase purified from the yeast pichia pastoris. J. Biol. Chem 1999, 274, 37658–37665. [DOI] [PubMed] [Google Scholar]
- (54).Li H; Wang HY; Kang S; Silverman RB; Poulos TL Electrostatic control of isoform selective inhibitor binding in nitric oxide synthase. Biochemistry 2016, 55, 3702–3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Müller J; Esső K; Dargó G; Könczoö Á; Balogh GT Tuning the predictive capacity of the PAMPA-BBB model. Eur. J. Pharm. Sci 2015, 79, 53–60. [DOI] [PubMed] [Google Scholar]
- (56).McPhillips TM; McPhillips SE; Chiu H-J; Cohen AE; Deacon AM; Ellis PJ; Garman E; Gonzalez A; Sauter NK; Phizackerley RP; Soltis SM; Kuhn P Blu-Ice and the Distributed Control System: software for data acquisition and instrument control at macromolecular crystallography beamlines. J. Synchrotron Radiat 2002, 9, 401–406. [DOI] [PubMed] [Google Scholar]
- (57).Battye TGG; Kontogiannis L; Johnson O; Powell HR; Leslie AGW iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr., Sect. D: Biol. Crystallogr 2011, 67, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Kabsch W XDS. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Evans P Scaling and assessment of data quality. Acta Crystallogr., Sect. D: Biol. Crystallogr 2006, 62, 72–82. [DOI] [PubMed] [Google Scholar]
- (60).Murshudov GN; Vagin AA; Dodson EJ Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., Sect. D: Biol. Crystallogr 1997, 53, 240–255. [DOI] [PubMed] [Google Scholar]
- (61).Emsley P; Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr 2004, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- (62).Adams PD; Afonine PV; Bunkoczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung L-W; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Li H; Jamal J; Plaza C; Pineda SH; Chreifi G; Jing Q; Cinelli MA; Silverman RB; Poulos TL Structures of human constitutive nitric oxide synthases. Acta Crystallogr., Sect. D: Biol. Crystallogr 2014, 70, 2667–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ Phaser crystallographic software. J. Appl. Crystallogr 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Winn MD; Isupov MN; Murshudov GN Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallog., Sect. D: Biol. Crystallogr 2001, 57, 122–133. [DOI] [PubMed] [Google Scholar]
- (66).Liebschner D; Afonine PV; Moriarty NW; Poon BK; Sobolev OV; Terwilliger TC; Adams PD Polder maps: improving OMIT maps by excluding bulk solvent. Acta Crystallog., Sect. D: Biol. Crystallogr 2017, 73, 148–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Schrodinger LLC. The PyMOL Molecular Graphics System, Version 1.8, 2015.
- (68).Do HT; Wang H-Y; Li H; Chreifi G; Poulos TL; Silverman RB Improvement of Cell Permeability of Human Neuronal Nitric Oxide Synthase Inhibitors Using Potent and Selective 2-Aminopyridine-Based Scaffolds with a Fluorobenzene Linker. J. Med. Chem 2017, 60, 9360–9375. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.