

Abstract
The expression of synthetic receptors in primary T cells enables the programming of user-defined responses when designing T-cell therapies. Chimeric antigen receptors (CARs) are synthetic receptors that have demonstrated efficacy in cancer therapy by targeting immobilized antigens on the surface of malignant cells. We have recently shown they can also rewire T-cell responses to soluble ligands. In contrast to other synthetic receptors, CARs are not only readily engineered by rational design, but also clinically translatable with robust function in primary human T cells. This protocol discusses design principles for CARs responsive to soluble ligands and delineates steps for producing T cells expressing synthetic receptors. Functional assays for quantifying the ability of CAR-T cells to sense and respond to soluble ligands are also presented. This protocol provides a framework for proficient immune-cell researchers to test novel T-cell therapies targeting soluble ligands in under two weeks.
Keywords: Chimeric antigen receptor, primary human T cells, soluble factors
INTRODUCTION
Chimeric antigen receptors (CARs) are modular, synthetic receptors that have demonstrated remarkable promise in the field of cancer immunotherapy, where they have been used to reprogram T-cell responses to surface markers presented by malignant cells. Multiple clinical trials have shown complete remission achieved in >80% of patients with relapsed or refractory B-cell leukemias when treated with T cells expressing a CAR targeting CD19, a B-cell antigen1–5. With tumor targeting as their primary purpose, CARs have conventionally been designed to recognize immobilized, surface-bound antigens. However, we recently demonstrated that CARs can also be engineered to target soluble factors6. Here, we describe a protocol for the generation and functional validation of CAR-T cells with rewired responses to soluble ligands.
Development of the protocol
A method to rewire cellular responses to soluble ligands can confer cell-based therapies with new ways to interface with immunosuppressive cytokines and growth factors commonly found in the tumor microenvironment. Given the modularity of the CAR and its well-established ability to program T-cell function against membrane-bound antigens, we reasoned that the CAR would provide an attractive platform for engineering T-cell responses to soluble factors. The design and functional validation of CARs responsive to soluble factors is highly analogous to the process for conventional, tumor antigen-targeting CARs, and is therefore readily accessible for those who are already familiar with CAR-T–cell engineering. Instead of target cell lysis, the main functional readouts for CAR signaling in response to soluble factors are activation-marker upregulation and cytokine production. Assays can also be performed at low cell densities in order to verify that soluble ligand—and not ligand bound to the surface of neighboring CAR-T cells—is sufficient for CAR-T–cell activation. Our lab has reported on this novel class of soluble-ligand–responsive CARs, demonstrating robust and tunable CAR-mediated T-cell activation6. A follow-up study subsequently demonstrated the potential anti-tumor applications of one such CAR when paired with tumor-targeting T-cell receptors (TCRs) or CARs7.
Applications
Although CARs targeting soluble factors do not directly program tumor-cell killing, they expand the toolbox of synthetic receptors that can be used to program novel T-cell functions. For instance, CARs responsive to soluble ligands can be used to convert typically immunosuppressive cytokines, such as transforming growth factor-beta (TGF-β), into potent T-cell stimulants. Indeed, instead of becoming dysfunctional in the presence of TGF-β, T cells engineered to express a TGF-β–responsive CAR proliferate and produce Th1 cytokines such as IFN-γ, TNF-α, and IL-26. Furthermore, TGF-β CAR-T cells can protect neighboring, tumor-specific T cells from TGF-β–mediated immune suppression7. CARs targeting soluble ligands may also serve as reporters for the presence of their cognate ligand in the in vivo milieu, but this will likely be contingent on proper T-cell trafficking to the site(s) of interest and has yet to be experimentally validated.
Alternative methods
Other novel synthetic receptor constructs targeting soluble ligands have been described in the literature8. Switch receptors, which are chimeras between different cytokine receptors, have been demonstrated to function in primary human T cells, effectively reprogramming T-cell responses to inhibitory cytokines such as IL-49–11. Although the construction of such receptors is straightforward, it requires the existence of endogenous receptors for the ligand of interest. Furthermore, because ligand-binding domains are obtained from endogenous receptors, binding affinity—and consequently, signaling strength—cannot be easily tuned. Lastly, though in theory one could mix-and-match domains from different receptors, it is likely that not all combinations will work, and that trial-and-error will be necessary to obtain switch receptors with proper protein folding, surface expression, and signal transduction. In contrast, the CAR architecture has been shown to be highly modular and tunable, and easily adaptable to a variety of target ligands12.
Although not yet validated in primary human T cells, the modular extracellular sensor architecture (MESA), dCas9 synthetic receptor (dCas9-synR), and generalized extracellular molecule sensor (GEMS) platforms have been used to program responses to soluble ligands in immortalized cell lines13–15. Whereas dCas9 synthetic receptors leverage the extracellular domain of endogenous ligand receptors, MESA and GEMS receptors, like CARs, utilize synthetic ligand-binding domains. A notable feature distinguishing the MESA, dCas9-synR, and GEMS receptors from CARs is the full programmability over transcriptional output upon ligand binding. Although the complexity of these outputs is currently constrained by the size of transgenes that can be efficiently introduced into T cells, continued advances in genetic engineering promise to alleviate some of these limitations.
Advantages and limitations
Our approach has several key advantages over alternative methods for engineering cellular responses to soluble factors. First and foremost, the modularity of the CAR lends itself to facile and intuitive design by simply swapping the extracellular ligand-binding domain for any given ligand of interest, with little to no tuning of the signaling domains required. Another main advantage of CARs is their clinical translatability, as they can robustly reprogram not only immortalized cell lines, but also primary human T cells. However, the signaling output is hard-wired by the intracellular signaling domains that have been used in CARs (e.g., CD3ζ, CD28, 4–1BB, etc.), limiting the ability to control both receptor input and output simultaneously. In addition, the design space of CARs responsive to soluble ligands is limited by the fact that signal transduction requires ligand-mediated receptor dimerization. Nevertheless, given the trade-off that currently exists between complexity and programmability of signaling outputs encoded by other types of synthetic receptors, CARs are an attractive tool for cancer immunotherapy, where an array of functional outputs may be required to achieve the most potent anti-tumor effect.
Overview of the procedure
Using well-established molecular cloning and virus production methods, CARs targeting a soluble ligand of interest can be cloned into lentiviral vectors for production of lentiviruses encoding the desired CAR constructs16–18. We present a procedure for isolation of T cells from whole blood, followed by activation, lentiviral transduction, and expansion of T cells, to generate a primary T-cell line expressing synthetic constructs of choice (steps 1–42). Alternatives to and variations of this protocol (e.g., using different transgene vectors, buffers, T-cell stimulation conditions, cytokines, cell-line purification procedures, time in culture, etc.) are discussed elsewhere19–22. Validation of CAR signaling upon soluble ligand binding is achieved by monitoring activation marker upregulation and cytokine production (steps 43–48; 49–51). These methods involve assaying T-cell properties after exposure to CAR ligand, can be applied to CARs targeting either soluble or immobilized ligands, and should be familiar to those experienced with CAR-T–cell research. One might also be interested in assaying CAR-T–cell degranulation by CD107a staining, where distinct patterns can be observed upon exposure to soluble versus immobilized ligand (steps 52–57). To specifically verify that interactions with soluble ligands are sufficient to trigger downstream receptor signaling, we also describe two approaches where cells are seeded at low densities to minimize cell-cell contacts (steps 58–65; 66–74).
Experimental design
CAR components.
A CAR that responds to soluble ligands does not deviate from the basic CAR architecture: an antigen-binding extracellular domain is linked via a spacer and transmembrane domain to intracellular signaling domains12. CARs that respond to soluble ligand are expected to exhibit many of the same modular properties as CARs that respond to immobilized ligands, and several specific domains have been reported for each part6,23 (Fig. 1a).
Fig. 1. CAR structure and mechanisms for response to soluble ligands.
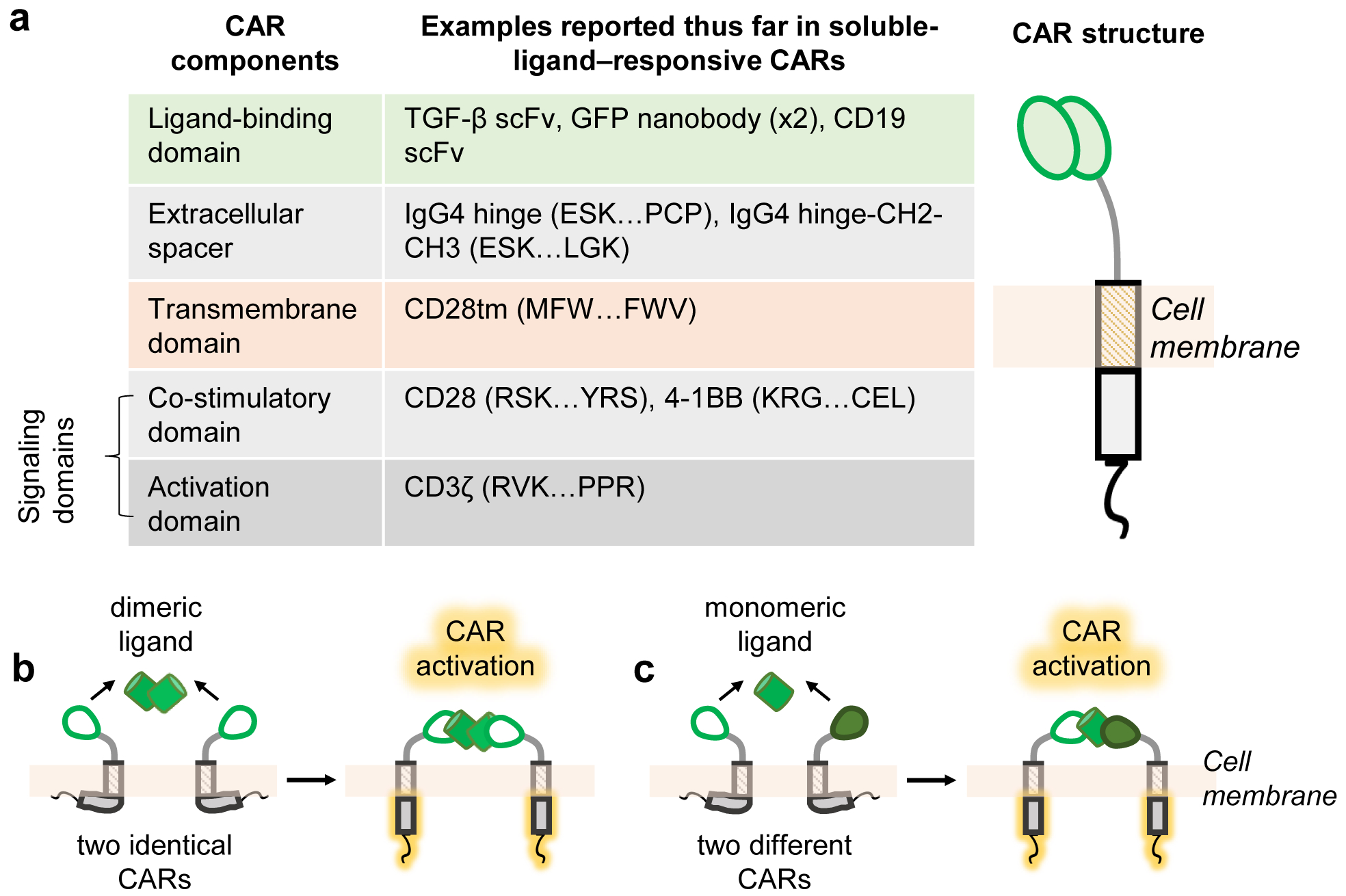
a, A CAR presents an extracellular ligand-binding domain linked via an extracellular spacer and transmembrane domain to intracellular signaling domains. The indicated specific domains have been shown to function in the context of responding to natural and synthetic soluble ligands. For common domains sourced from native proteins, the specific amino acid stretches we have used are demarcated by the first 3 amino acids and last 3 amino acids6,23. The full amino acid sequence of each native protein can be obtained from public databases such as NCBI and Uniprot. b, CARs signal in response to soluble ligand if the cognate ligand contains two binding epitopes that can cause CAR dimerization. c, For ligands of interest that do not contain multiple binding epitopes, ligand responsiveness can be conferred by expressing two CARs capable of simultaneously binding separate epitopes on the same ligand. In b-c, the proposed models shown assume that CAR cytoplasmic tails interact with the plasma membrane in the inactive state, based on the membrane association observed for CD28 and CD3ζ cytoplasmic domains in their native contexts32–34.
Mechanistic considerations.
Previous work has demonstrated that ligand-mediated CAR dimerization is a critical feature for responding to soluble ligands6. Thus, cells engineered with a single CAR can only respond to a soluble ligand if that ligand contains two or more epitopes for the CAR to bind (Fig. 1b). For ligands that lack redundant epitopes, receptor activation can be achieved by co-expressing two CARs that can simultaneously bind the same ligand at separate epitopes (Fig. 1c).
The CAR’s signaling strength in response to a soluble ligand is influenced by CAR structure, with evidence suggesting that tighter mechanical coupling between extracellular and intracellular domains results in increased signaling at lower thresholds of ligand6,24. Extracellular binding affinity has also been shown to impact CAR signaling thresholds in the context of immobilized ligand25,26. Thus, depending on the application, CARs with varying ligand-binding affinities or structural properties can be tested to obtain one with the desired responsiveness threshold and signaling intensity.
Demonstrating soluble-ligand–mediated CAR activation.
Standard bulk assays for CAR-T cell activation generally utilize cells seeded at high densities. In this context, it is possible for T-cell activation to result from dimeric ligands being cross-presented by one cell to another, as opposed to individual T cells being activated by freely soluble ligands. We use two assays that avoid this confounding possibility. Microscopy can be used to observe activation events in the demonstrable absence of any cell-cell contact, particularly with time-lapse images. However, this approach may be limited by throughput, technical ability, or instrument accessibility. An alternative approach involves assessing CAR-T cell activation across different cell densities, where the probability of cell-cell contact events varies. Critical to this assay is a low–cell-density regime, where cell-cell contacts are rare and soluble-ligand–mediated T-cell activation is quantifiably independent of cell density. Samples in this experiment with the lowest cell densities will have few cells per well. Therefore, to observe a statistically reliable number of events, a large number of wells can be seeded and subsequently combined for analysis. To reduce labor intensity, we use NFAT reporter Jurkat T-cell lines, which allows cell activation assessment without the need for antibody staining and avoids the cell attrition associated with the staining process.
Sorting for pure populations.
To obtain a pure transduced (i.e., 100% CAR+) population, we recommend using magnetism-activated cell sorting (MACS) as an efficient and gentle method to sort large cell samples. This method labels antigen-expressing cells with antibody-coated magnetic beads, and labeled cells are subsequently captured onto, and eluted from, magnetic columns. Cells should not be labelled by direct antibody binding to the CAR molecule, as such labeling is likely to result in CAR crosslinking, which can cause cell activation and perturb cell biology. Instead, the CAR-encoding transgene can be co-expressed with a transduction marker that is presented to the cell surface for affinity sorting. In our lentiviral constructs, we use a 2A “self-cleaving” peptide to incorporate a truncated epidermal growth factor receptor as a sortable marker27. Although MACS sorting is relatively gentle compared to other methods such as fluorescence-activated cell sorting, shear stress exerted during elution of cells from magnetic columns can still be detrimental to T-cell viability. In our hands, T cells that have been sufficiently well-transduced (i.e., >60% CAR+) often do not require MACS sorting to be useful in downstream experiments. We therefore recommend sorting for a pure CAR+ population as an optional step dependent on the CAR transduction efficiency and the planned subsequent use of the CAR-T cell line.
Assays to assess CAR activity.
Although the procedure details the generation of CAR-expressing primary human T cells, immortalized Jurkat T-cell lines can also be transduced with CAR lentivirus. For higher throughput and/or faster turnaround times, one might consider first assaying CAR activity in Jurkat cells prior to functional validation in primary human T cells. The use of Jurkat T-cells can be particularly useful in initial testing of multiple CAR variants.
We describe two different assays for cytokine production. In our hands, the intracellular cytokine staining protocol described, which employs formaldehyde fixation and ice-cold methanol permeabilization, has consistently worked well for TNF-ɑ, but is less sensitive for other cytokines (Supplementary Fig. 1). Consequently, ELISA and/or cytometric bead array assays are typically performed as confirmatory tests. Alternatively, commercially available fixation and permeabilization buffers can be considered for intracellular staining. We advise those interested in quantifying multiple cytokines at once to perform cytometric bead array assays for their ease of use. However, since bead-based kits are typically more costly than ELISA kits, those interested only in a limited set of cytokines may find it preferable to perform ELISA for cytokine quantification.
Controls.
As primary T cells derived from different donors can vary in quality, mock-transduced T cells cultured alongside CAR-transduced T cells from the same donor serve as informative controls for T-cell behavior. Assays for CAR function can be further controlled with comparison to a second CAR-T–cell product generated from the same donor’s cells but targeted to an irrelevant antigen. The off-target CAR does not respond to the ligand of choice, but is otherwise functional (such as one of the well-characterized CD19 CARs, if appropriate, or an scFv-less CAR that lacks the ligand-binding domain). A positive control for CAR activity involves exposure to immobilized cognate ligand, whether presented by beads or cells, or seeded on the surface of a culture well. Unspecific stimulation with 10 ng/mL PMA and 2.2 μM ionomycin can also serve as a positive control, in the event that the CAR fails to respond to both soluble and immobilized ligand. For degranulation assays, positive controls can involve exposure to immobilized ligand, immobilized OKT3 for spatially polarized unspecific stimulation, or 10 ng/mL PMA and 2.2 μM ionomycin for spatially nonpolarized unspecific stimulation.
MATERIALS
Biological materials
CAUTION: For work with materials derived from human subjects, appropriate approvals from the researcher’s institution are required.
Human blood
NFAT (RRID:CVCL_YB71) and/or NFκB (RRID:CVCL_XF35) fluorescent reporter Jurkat T-cell lines28,29
Reagents
Lentivirus encoding CAR of choice
Using well-documented methods of molecular cloning, construct a lentiviral plasmid vector encoding the CAR16,17. With pure plasmid, generate lentivirus that will deliver DNA encoding the CAR18.
Rosette Sep CD4+ T cell enrichment cocktail (STEMCELL Technologies, catalog no. 15062)
Rosette Sep CD8+ T cell enrichment cocktail (STEMCELL Technologies, catalog no. 15063)
RPMI-1640 with 25 mM HEPES and L-Glutamine (Lonza, catalog no. 12–115Q)
Heat-inactivated fetal bovine serum (HI-FBS; Gibco, catalog no. 16140–071)
Ficoll-Paque PLUS (GE Healthcare, catalog no. 17144002)
Phosphate Buffered Saline (1X), without Calcium and Magnesium (PBS; Lonza, catalog no. 17–516F)
Dynabeads™ Human T-Activator CD3/CD28 for T Cell Expansion and Activation (Gibco, catalog no. 11132D)
Recombinant human IL-2 (Gibco, catalog no. PHC0023)
Recombinant human IL-15 (Miltenyi Biotec, catalog no. 130-095-765)
Polybrene (Sigma Aldrich, catalog no. H9268)
Dimethyl Sulfoxide (DMSO; Sigma Aldrich, catalog no. D2650)
Fluo-4 AM (Invitrogen, catalog no. F14201)
Pluronic™ F-127 (20% Solution in DMSO) (Invitrogen, catalog no. P3000MP)
Phorbol 12-myristate 13-acetate (PMA; Sigma Aldrich, catalog no. P8139)
Ionomycin (Sigma Aldrich, catalog no. I3909)
Brefeldin A Solution (1,000X) (BioLegend, catalog no. 420601)
Monensin Solution (1,000X) (BioLegend, catalog no. 420701)
Methanol (Fisher Scientific, catalog no. A412)
CAUTION: Methanol is flammable; keep away from open flame.
Formaldehyde (Sigma Aldrich, catalog no. F8775)
CAUTION: Formaldehyde is carcinogenic.
MilliQ water
APC anti-DYKDDDDK [clone L5] (BioLegend Cat# 637308, RRID:AB_2561497)
PE anti-human CD4 [clone RPA-T4] (BioLegend Cat# 300508, RRID:AB_314076)
APC anti-human CD8α [clone RPA-T8] (BioLegend Cat# 301049, RRID:AB_2562054)
FITC anti-human CD3 [clone UCHT1] (BioLegend Cat# 300406, RRID:AB_314060)
Pacific Blue™ anti-human CD69 [clone FN50] (BioLegend Cat# 310920, RRID:AB_493667)
APC anti-human IL-2 [clone MQ1–17H12] (BioLegend Cat# 500310, RRID:AB_315097)
PE anti-human TNF-α [clone MAb11] (BioLegend Cat# 502909, RRID:AB_315261)
Pacific Blue™ anti-human IFN-γ [clone 4S.B3] (BioLegend Cat# 502522, RRID:AB_893525)
Pacific Blue™ anti-human CD107a [clone H4A3] (BioLegend Cat# 328623, RRID:AB_2134615)
OKT3 (Thermo Fisher Scientific Cat# 16-0037-81, RRID:AB_468854)
Erbitux (Bristol-Myers Squibb, NDC 66733-948-23)
ELISA MAX™ Deluxe Set Human IL-2 (BioLegend, catalog no. 431804)
ELISA MAX™ Deluxe Set Human TNF-α (BioLegend, catalog no. 430204)
ELISA MAX™ Deluxe Set Human IFN-γ (BioLegend, catalog no. 430104)
Cytometric Bead Array, Human Th1/Th2 Cytokine Kit II (BD Biosciences, catalog no. 551809)
Anti-Biotin MicroBeads (Miltenyi Biotec, catalog no. 130-090-485)
Equipment
Polypropylene centrifuge tubes (15 mL and 50 mL; Olympus, catalog nos. 28–103 and 28–108)
Falcon® Round-Bottom Polystyrene Tubes (5 mL flow tube; Corning, catalog no. 352058)
Pipette tips (10 μL, 200 μL, and 1000 μL; Olympus, catalog nos. 24–120RS, 24–151RS, 24–165RS)
Filter tips (10 μL, 200 μL, and 1000 μL; Olympus, catalog nos. 24–401, 24–412, 24–430); for handling lentivirus and cells exposed to lentivirus (steps 21–29)
Serological pipettes (5 mL, 10 mL, and 25 mL; Olympus, catalog nos. 12–102, 12–104, 12–106)
Pipettes (2 μL, 20 μL, 200 μL, 1000 μL; Gilson, catalog nos. FA10001M, FA10003M, FA10005M, FA10006M)
Multichannel pipette (300 μL, 12 channels; Eppendorf, catalog no. 3125000060)
Pipette aid (Drummond, catalog no. 4-000-110-TC)
Tissue-culture well plates (12-well, 24-well, 48-well, 96-well U-bottom; Olympus, catalog nos. 25–106, 25–107, 25–108, 25–221)
Glass-bottom well plates (48-well; MatTek, catalog no. P48G-1.5–6-F)
Tissue-culture flask (25 cm2; Olympus, catalog no. 25–213)
Cryovials (2 mL; Fisher Scientific, catalog no. 10-500-26)
Transfer pipettes (5 mL; Olympus, catalog no. 30-206S)
Reagent reservoir (25 mL; Olympus, catalog no. 28-121)
Mr. Frosty™ Freezing Containers (Nalgene, catalog no. 5100–0001)
DynaMag-5 (Invitrogen, catalog no. 12303D) or DynaMag-15 (Invitrogen, catalog no. 12301D)
MACS Cell Separation
MACS Multistand (Miltenyi Biotec, catalog no. 130-042-303)
OctoMACS Separator (Miltenyi Biotec, catalog no. 130-042-109) or QuadroMACS Separator (Miltenyi Biotec, catalog no. 130-090-976)
MS Columns (Miltenyi Biotec, catalog no. 130-042-201) or LS Columns (Miltenyi Biotec, catalog no. 130-042-401)
Liquid nitrogen cryo tank
−80°C freezer
Biosafety cabinet
37°C water bath
Humidified tissue-culture incubator (37°C, 5% CO2)
Eppendorf 5810R centrifuge
Flow cytometer
Confocal microscope
Reagent Setup
T-cell media In a sterile environment, mix 111 mL of HI-FBS and 1000 mL of RPMI-1640 with 25 mM HEPES and L-Glutamine. Store the media at 4°C for up to 3 months.
Fluo-4-AM stock solutions In a sterile environment, reconstitute 50 μg lyophilized Fluo-4-AM in 50 μL DMSO. Dilute 5-fold in DMSO to get ~9 mM stock solutions and freeze at −20°C in single-use aliquots for up to 6 months. Protect from light.
5% (w/v) Pluronic F-127 solution In a sterile environment, dilute Pluronic F-127 (20% stock solution in DMSO) four-fold in PBS and store at room temperature.
IL-2 stock solution In a sterile environment, reconstitute lyophilized IL-2 in 100 mM acetic acid to reach a concentration of 1 mg/mL. Using the lot-specific ED50, determine the specific activity as defined by the formula (units/mg) = 106/ED50. Based on the calculation, further dilute the 1 mg/mL solution of IL-2 to 1000 units/μL using RPMI-1640 + 10% HI-FBS. This is the concentrated stock solution. Dilute 500 μL of the concentrated stock solution with 9.5 mL RPMI-1640 + 10% HI-FBS to obtain a working stock concentration of 50 units/μL (1,000X). Both concentrated stock solutions and working stock solutions can be stored at −20°C for up to 1 year.
IL-15 stock solution In a sterile environment, reconstitute lyophilized IL-15 in 0.1% BSA to reach a concentration of 10 ng/μL. This is a 10,000X working stock. Store at −20°C for up to 1 year.
FACS buffer (PBS + 2% (v/v) HI-FBS) In a sterile environment, mix 10 mL of HI-FBS and 500 mL of PBS. Store the solution at 4°C for up to 6 months.
Polybrene stock solution Dissolve polybrene to final concentration of 4 mg/mL in MilliQ water. Sterile filter solution (0.22 μm) and store ~1 mL aliquots at −20°C for up to 1 year.
PMA stock solution In a sterile environment, reconstitute in DMSO to 10 ng/μL. Solutions can be stored at −20°C for up to six months.
PROCEDURE
Isolate primary human T cells from donor blood (Timing: 2 hours)
-
1
Obtain human blood and aliquot 15 mL into 50-mL conical tubes.
-
2
Use the RosetteSep CD4+ enrichment kit and/or the CD8+ enrichment kit (STEMCELL Technologies) according to experimental needs. Add RosetteSep cocktail to the sample (50 μL/mL of blood), mix, and incubate at room temperature (22°C) for 20 min.
-
3
Dilute samples with 15 mL room-temperature PBS.
-
4
Underlay 14 mL Ficoll, a density gradient medium, into the conical tube.
CRITICAL STEP: Use a 10-mL serological pipette and draw up the maximum possible volume of Ficoll. Plant serological pipette at the base of the conical tube and gently release the Ficoll slowly from the pipette. The Ficoll solution should not mix with the blood solution and should slowly displace the blood solution upwards. This step is more easily performed with a well-charged serological pipette with a low-speed setting. An alternative method, which can be chosen purely by preference, is to overlay blood on top of Ficoll that is already placed in the conical tube.
-
5
Spin tubes at 1200 x g for 20 min at room temperature, with the brakes off. At the end of the spin, be sure to gently handle the samples when removing spin buckets from the centrifuge and placing samples in racks in the biosafety cabinet.
-
6
Use an eye-dropper pipette to gently harvest the buffy coat layer to a new tube. This cell-containing layer lies at the interface between the straw-colored plasma above and the Ficoll below. At the base of the tube will be a maroon-colored pellet.
-
7
Wash the collected cells three times. For each wash, fill the tube with room-temperature PBS, centrifuge at 300 x g for 10 minutes at room temperature and low brakes, then discard supernatant.
-
8
Count cell yield with a hemocytometer. Assess the isolation efficiency with flow cytometry to determine the fraction that is CD3+CD4+ and/or CD3+CD8+.
-
9
Resuspend cells in T-cell culture medium and incubate at 37°C in 5% CO2 incubator.
PAUSE POINT: T cells can be stored as a freezer stock in a liquid nitrogen cryo tank. Generate freezer stock by collecting cells and resuspending in T-cell culture media + 10% DMSO to the desired cell density (we recommend a range between 1 × 107 and 2 × 107 cells/mL). Aliquot cell suspension into 2-mL cryovials labeled with cell line ID, cell count, and date. Place cryovials in Mr. Frosty containers (filled with isopropanol) for 24 hours at −80°C, then transfer cryovials to a liquid nitrogen cryo tank. When ready to use cells, first warm T-cell media to 37°C. Bring cryovial of cells out of the liquid nitrogen tank and immediately swish continuously in a 37°C water bath to thaw cell solution as rapidly as possible. Clean the outside of the cryovial with 70% ethanol, then transfer cells into warm T-cell media.
Activate T cells (Timing: 30 min, followed by 10 min/day for 2 days for routine culture maintenance)
-
10
Prepare CD3/CD28 Dynabeads (Gibco) by resuspending in the original vial and transfer 25 μL of Dynabead solution for every 1 × 106 T cells that will be activated into a tube that will fit in the DynaMag, usually 15-mL conical tubes (for DynaMag-15) or 5-mL flow tubes (for DynaMag-5).
-
11
Add 100 μL PBS for every 25 μL of Dynabead solution to the flow tube and pulse vortex 3 times.
-
12
Place tube on a DynaMag magnet for 1 min and aspirate the supernatant.
-
13
Remove tube from the magnet and resuspend the washed Dynabeads in 25 μL T-cell culture medium for every 1 × 106 T cells that will be activated (i.e., the same volume that was drawn in step 10).
-
14
Plate T cells at 1 × 106 T cells/2 mL T-cell culture media for each 24-well plate well.
-
15
Add 25 μL Dynabead solution/1 × 106 T cells to get a 1:1 cell:bead ratio and mix by pipetting up and down.
-
16
Supplement cell culture with 50 U/mL IL-2 and 1 ng/mL IL-15, and continue to do so every 2–3 days.
-
17
Label cell culture with today’s date as Day 0 of culture and incubate at 37°C for 2 days.
Transduce T cells with lentivirus
Timing: 7 hours (with 4-hour break) on the day of transduction, then 0–30 min for next 6 days.
-
18
On Day 2 post T-cell activation, examine cells under light microscope to confirm cluster formation. Cells should have settled on the bottom of the culture well and gathered into scattered asymmetric clumps of beads and dividing cells. To increase cell concentration (and avoid the need for an extra centrifugation step to concentrate the cells), reduce the volume in the T-cell culture well by gently removing 1.4 mL media from a 24-well plate well without disturbing the T-cell/Dynabead mix settled at the bottom of the well. Resuspend cells and count them.
-
19
Depending on cell counts and desired number of transduction groups, seed 0.5–1 × 106 T cells in 500 μL warm T-cell culture media in each well of a 24-well plate. One can also consider seeding 0.2–0.5 × 106 T cells in 250 μL per well in a 48-well plate. Let cells settle at 37°C for 0.5–1 hour.
-
20
Thaw lentivirus at room temperature for 20 min. Vortex the virus for 10–20 sec and spin the tube at maximum speed for 2 minutes. If cryovials do not fit conveniently in centrifuge, one could place them in 50-mL conical tubes, then spin the conical tubes in a centrifuge.
-
21
Add the lentivirus to the T cell culture plates. We recommend an MOI of 1.5–3 based on virus titers obtained using H9 cells as transduction targets.
-
22
Add polybrene to a final concentration of 5 μg/mL (1:800 dilution of 4 mg/mL stock).
-
23
Add IL-2 at 50 U/mL.
-
24
Gently swirl plate in cross-wise fashion to distribute the transduction mix.
-
25
Place T-cell culture plate in bucket with aerosol-tight lid and centrifuge for 30 min at 800 x g, 32°C, lowest acceleration, and no brake.
-
26
Incubate at 37°C for 4 hours.
-
27
Add 1 mL pre-warmed T-cell culture media to each 24-well plate well.
-
28
Incubate for 48 hours at 37°C.
-
29
Wash and resuspend cells in fresh T-cell culture media pre-warmed to 37°C. Plate cells in a fresh well and supplement with 50 U/mL IL-2 and 1 ng/mL IL-15.
-
30
Continue to culture T cells at 37°C, supplementing with 50 U/mL IL-2 and 1 ng/mL IL-15 every 2–3 days. Split cells when they appear confluent at the bottom of the culture vessels and media changes color to yellow-orange over a 24-hour period. Maintain cultures at a cell density of around 1 × 106 cells/mL. Advance cells from well plates to culture flasks as necessary. When split is performed on a non-feeding day, supplement the new media with 25 U/mL IL-2 and 0.5 ng/mL IL-15.
Remove Dynabeads from culture
Timing: 45 min
-
31
On Day 9 of culture, gently transfer culture vessels from the tissue-culture incubator to the biosafety cabinet, taking care not to disturb the cells. Remove half to two-thirds of the culture supernatant from the top, and then resuspend cells in the reduced culture volume.
-
32
Transfer cell solution to tubes that fit in the DynaMag, usually 15-mL conical tubes (for DynaMag-15) or flow tubes (for DynaMag-5). Let sit for 1 minute at room temperature.
-
33
Remove cell solution from tubes and continue to culture. Take a small aliquot to analyze for CAR surface expression by flow cytometry. If employing a transduction marker, then transduction-marker staining allows for quantification of transduction efficiency, assuming the marker efficiently presents at the cell surface. When testing the CAR for the first time, stain for both the CAR and the transduction marker together. Direct CAR staining alone quantifies CAR surface expression, which may be lower than actual transduction efficiency if the CAR does not localize to the cell surface efficiently. ?Troubleshooting
PAUSE POINT: Transduced T cells can be stored as a freezer stock for at least several years in a liquid nitrogen cryo tank after Dynabead removal. Generate freezer stocks and revive cell lines as detailed above in the pause point after step 9.
OPTIONAL Sort for transduced cells
Timing: 2.5–3 hours
-
34
If a pure receptor-positive cell line is desired, sort for the transduced cells on Day 10. We use the magnetic bead-based sorting system MACS Cell Separation (Miltenyi Biotec) to positively select for transduced cells.
-
35
Count cells to determine the scale of the sort. Prepare an appropriate column, such as the MS column for sorting a 50% transduced population of 107 cells.
-
36
Prepare a sorting buffer of PBS + 2% (v/v) FBS. Mount the column on a magnetic stand, such as the OctoMACS Separator on a MACS Multistand and add sorting buffer to equilibrate the column (500 μL buffer for an MS column).
-
37
Stain cells with appropriate antibodies and MicroBeads and resuspend in 500 μL sorting buffer.
-
38
Apply cell solution to the column and collect the flow-through.
-
39
Wash column three times (with 500 μL buffer for an MS column) and collect each flow-through separately.
-
40
Remove column from the stand and place in a collection tube. Add appropriate volume of sorting buffer (1 mL for an MS column) and flush immediately with the plunger.
-
41
Take small aliquots of each flow-through and the final eluent to stain and analyze by flow cytometry to determine the sorting efficiency.
-
42
Depending on your sorting efficiency, need for purity, and cell numbers, consider performing a second round of sorting by repeating steps 35–40. ?Troubleshooting
PAUSE POINT: Sorted T cells can be stored as a freezer stock for at least several years in a liquid-nitrogen cryo tank after Dynabead removal. Generate freezer stocks and revive cell lines as detailed above in the pause point after step 9.
Assays to assess CAR activity
OPTIONAL Assay for NFAT, NFκB, and the early activation marker CD69
Timing: 1 hour for set-up, 4–24 hour incubation period, 1 hour to assay results
-
43
Use the NFAT and NFκB fluorescent reporter Jurkat T-cell lines expressing the CAR to assess NFAT and NFκB transcriptional activity. CD69 upregulation can be assessed in both Jurkat cells and primary human T cells.
-
44
Determine the number of samples needed for the experiment of interest. A typical experiment may include triplicate wells of the cell line with or without exposure to the soluble target ligand. A positive control can involve exposure to immobilized ligand, or unspecific stimulation with 10 ng/mL PMA and 2.2 μM ionomycin. A negative control can employ the parental cell line expressing an off-target CAR. These controls are particularly important as current limitations in protein engineering can result in unpredictable CAR signaling properties30,31.
-
45
Make solutions of 0.5 × 106/mL CAR-expressing cells in T-cell culture media for each cell line that will be used. If using a multichannel pipette and a trough to seed samples, prepare 5–10% extra sample.
-
46
Seed 100 μL cell solution per well in 96-well U-bottom plates.
-
47
Apply ligand of choice to each well, mix by pipetting up and down 3 times, and let the samples sit in the tissue-culture incubator at 37°C until the experiment is finished. We recommend incubation for at least 4 hours, but not exceeding 24 hours when first assessing CD69 upregulation. NFAT or NFκB reporter activity can be assessed after 8–24 hours.
-
48
At the end of the experiment, pellet cells by spinning the 96-well plate at 300 x g at room temperature for 2 minutes. Aspirate supernatant and resuspend samples in 100 μL FACS buffer. NFAT or NFκB reporter cell lines can be directly analyzed for fluorescent reporter expression using flow cytometry. CD69 expression can be assessed with surface staining using fluorescently labeled anti-CD69 antibodies, followed by flow cytometry. ?Troubleshooting
OPTIONAL Assay for IFN-γ, TNF-ɑ, and IL-2 cytokine production
Timing: 1 hour for set-up, 24 hour incubation period, 2 hours to assay results with intracellular staining, 5 hours to assay results with ELISA or cytometric bead array kits.
-
49
Determine the number of samples needed for the experiment. A typical experiment may include triplicate wells of the cell line with or without exposure to the soluble target ligand.
-
50
Make solutions of 0.5 × 106/mL CAR-T cells in T-cell culture media for each cell line that will be used. If using a multichannel pipette and a trough to seed samples, prepare 5–10% extra sample.
-
51Assess cytokine production by either: (A) detecting intracellular cytokine expression in cells where cytokine release is blocked through inhibition of vesicle formation; or (B) measuring cytokine levels in the supernatant. Option A allows for the capture of cell-to-cell variability in cytokine production. However, it employs a toxic small molecule that likely perturbs multiple cell processes in addition to cytokine secretion, and may require optimization of a number of cell-fixation and cell-permeabilization steps. Option B measures the total cytokine actually secreted by a bulk cell population into supernatant, but is typically more resource- and time-intensive.
- Intracellular staining for cytokine expression:
- Add 5 μg/mL Brefeldin A to cell solution and mix well by pipetting up and down.
- Seed 100 μL cell solution per well in 96-well U-bottom plate wells.
- Apply ligand of choice to each well, mix with pipetting up and down 3 times, and let samples sit in the tissue-culture incubator at 37°C for 24 hours.
- Pellet cells by spinning the 96-well plate at 300 x g at room temperature for 2 minutes and wash in PBS.
- Incubate samples in 1.5% (v/v) formaldehyde in PBS for 15 minutes at room temperature.
- Pellet cells by spinning the 96-well plate at 300 x g at room temperature for 2 minutes.
- Aspirate supernatant, then permeabilize cells with ice-cold methanol on ice for 30 minutes.
- Pellet cells by spinning the 96-well plate at 300 x g at room temperature for 2 minutes. Wash samples in FACS buffer once.
- Stain samples with antibodies to IFN-γ, TNF-ɑ, and IL-2, and analyze using a flow cytometer. ?Troubleshooting
- Assess cytokine production with an ELISA or cytometric bead array:
- Seed 100 μL cell solution per well in 96-well U-bottom plate wells.
- Apply ligand of choice to each well, mix by pipetting up and down 3 times, and let the samples sit in the tissue-culture incubator at 37°C for 18–24 hours.
- Pellet cells by spinning the 96-well plate at 300 x g at room temperature for 2 minutes and carefully remove 50 μL supernatant from each well.
- Assess for cytokine levels in the supernatant using method of choice (ELISA or cytometric bead array).
OPTIONAL Assay for degranulation
Timing: 1 hour for set up, 5 hours of incubation, 1 hour to assay results.
-
52
A typical experiment may include triplicate wells of the cell line with or without exposure to the soluble target ligand.
-
53
Make solutions of 1 × 106/mL CAR-T cells in T-cell culture media for each cell line that will be used. If using a multichannel pipette and a trough to seed samples, prepare 5–10% extra sample.
-
54
Seed 60 μL of the appropriate cell line to each well of a 96-well U-bottom plate.
-
55
Apply ligand of choice to each well, mix by pipetting up and down 3 times, and let samples sit in the tissue-culture incubator at 37°C for 1 hour.
-
56
Remove plate from incubator and add monensin at 2 μM final concentration and anti-CD107a antibody at appropriate concentration in 40 μL volumes. For our particular antibody product, antibody is added to achieve a 1:50 dilution in the final cell mixture. Incubate plate at 37°C for 4 hours.
CRITICAL STEP: To reduce pipetting error, dilute antibody and monensin in warm culture buffer and add larger volumes of solution (> 10 μL if pipetting error can be assumed to be < 1 μL) to each well. As specified above, we typically seed T cells in 60 μL volumes and apply the monensin and antibody in a 40 μL volume to reach a final volume of 100 μL.
-
57
Remove plate from incubator, wash twice with FACS buffer chilled at 4°C, and analyze samples with flow cytometry.
OPTIONAL Assess activation across various cell densities
Timing: 1.5 hours for set-up, 8 hours incubation, 1.5 hours to assay results
-
58
Using a CAR-expressing NFAT reporter Jurkat T-cell line, determine the various cell seeding densities to be used in the experiment. Cell densities should span several orders of magnitude, such as seeding at 150, 500, 1000, 2000, 5000, 104, and 3 × 104 cells per well in 96-well flat-bottom plates.
CRITICAL STEP: For each replicate, plan to seed multiple wells such that a minimum of 1500 events will be counted when determining the extent of NFAT reporter upregulation by flow cytometry. For samples seeded at 150 cells/well, 16 wells should be combined to reliably reach 1500 events when samples are washed and assessed with flow cytometry.
-
59
Prepare a solution of 0.6 × 105 cells/mL in T-cell media. This solution can be used to seed 3 × 104 cells/50 μL/well. Starting with this mixture, perform serial dilutions to prepare cell solutions that can be used to seed more dilute wells.
CRITICAL STEP: Seed extra replicate wells at each seeding density to immediately check the actual seeding densities in the experiment. This will correct for any pipetting errors in the serial dilution steps and determine the actual cell seeding densities used in the experiment.
-
60
After all cells have been seeded, add 50 μL T-cell media with or without ligand to each well and mix by pipetting up and down 3 times. A multichannel pipette can be used for convenience.
-
61
Incubate samples in the tissue-culture incubator at 37°C for 8 hours. In the interim, the extra replicate wells seeded in step 59 can be counted by flow cytometry.
-
62
Remove samples from the incubator and pellet at 300 x g at room temperature for 5 minutes.
-
63
Aspirate and resuspend in 100 μL FACS buffer. For samples that need to be combined to reach a reliable number of events, add 100 μL to only one well and then transfer to each additional well to collect all cells into one solution.
-
64
Pellet at 300 x g at room temperature for 5 minutes, aspirate, and resuspend in 100 μL FACS buffer.
-
65
Analyze NFAT reporter expression by flow cytometry, running the samples on a chilled rack.
OPTIONAL Imaging CAR-T cell activation in the absence of any cell-cell contacts using Ca2+ flux
Timing: 2.5 hours
-
66
Dilute 1 μL 9 mM Fluo-4-AM into 900 μL PBS and add 3.6 μL 5% (w/v) Pluronic F-127 detergent to make the 10 μM Fluo-4-AM staining solution
-
67
Collect cells to be stained into a centrifuge tube and pellet at 300 x g at room temperature for 5 minutes.
-
68
Wash cells once with PBS.
-
69
Load cells with Fluo-4-AM by incubating cells at 1 × 106 cells/mL 10 μM Fluo-4-AM staining solution for 20 minutes at room temperature.
-
70
Wash cells twice with T-cell culture media, pelleting at 300 x g at room temperature for 5 minutes in between.
-
71
Seed cells in T-cell culture media in 50 μL volumes at 0.4 × 106 cells/mL in 48-well glass-bottom plate wells. Seed extra wells to allow for repeat attempts at the experiment. If performing the experiment for the first time, also prepare extra wells that can be used to establish microscope settings.
-
72
To set microscope settings, add ligand into a well and mix to generate activated cells. View cells under an objective lens that allows for individual cells to be distinguished. Higher power allows for increased detail, but reduces the number of cells that can be observed at one time. Set laser power, exposure time, and gain settings that are sufficient to visualize the Fluo-4 AM dye in activated cells. Afterward, image a well with Fluo-4-AM–loaded cells but no ligand to determine the background signal at your settings. We use a confocal microscope for this experiment, but a fluorescence microscope could also be used.
CRITICAL STEP: Exposure time and laser power must be high enough to reliably detect Ca2+ flux in the majority of cells. However, a lower exposure time and lower laser power will reduce phototoxicity and reduce the likelihood of observing artifactual Ca2+ fluxes.
-
73
Under the microscope, identify a region of interest in a fresh well where multiple cells are present but not in contact with each other. Begin recording time-lapse images every 20 sec in brightfield and green fluorescence channels. Imaging under brightfield helps ensure that no cells are touching the cells of interest outside the plane of focus. ?Troubleshooting
-
74
Gently pipette a small volume of ligand in one corner of the well and continue imaging while waiting for the ligand to reach the particular region of the well that is being imaged. The experiment ends when the ligand has diffused into the region of interest and Ca2+ flux events are observed under the microscope. ?Troubleshooting
TROUBLESHOOTING
| Step | Problem | Possible Reason(s) | Possible Solution(s) |
|---|---|---|---|
| 33 | CAR does not express | Improper protein folding and/or trafficking of receptor to cell membrane | Test multiple scFv sequences or ligand-binding domains (Fig. 2a). |
| 48 | CAR-T cell does not signal | ||
| Poor T-cell product | Control for ability of T cell to produce an effector response using PMA/ionomycin stimulation (Fig. 2c). | ||
| 8–33 | Cell line with low viability | ||
| Improper maintenance of T-cell cultures | Maintain cultures at 1 × 106 cells per mL; supplement cultures with 50 U/mL IL-2 and 1 ng/mL IL-15 every 2–3 days. | ||
| 34–42 | Poor yield from cell sorting | ||
| Positively transduced cells inadequately labeled | Increase labeling time with antibody. | ||
| 51A | Poor cell viability | Brefeldin A toxicity | Reduce incubation period in Brefeldin A. Depending on the exact measurement desired in the experiment, this can be performed by either shortening the incubation in 51Aiii to 4–12 hours, or initiating the experiment without Brefeldin A and adding Brefeldin A to cells 4–12 hours prior to completing the incubation. |
| 73 | Cells touching when attempting to image without cell-cell contacts | Cells are too dense | Test various cell densities. |
| 74 | Cells flow out of view after adding ligand to well when imaging | Addition of ligand causes too much disturbance to wells | Use higher concentration of ligand at smaller volume; apply ligand further away from region being imaged. |
Fig. 2. Troubleshooting CAR expression and effector activity.
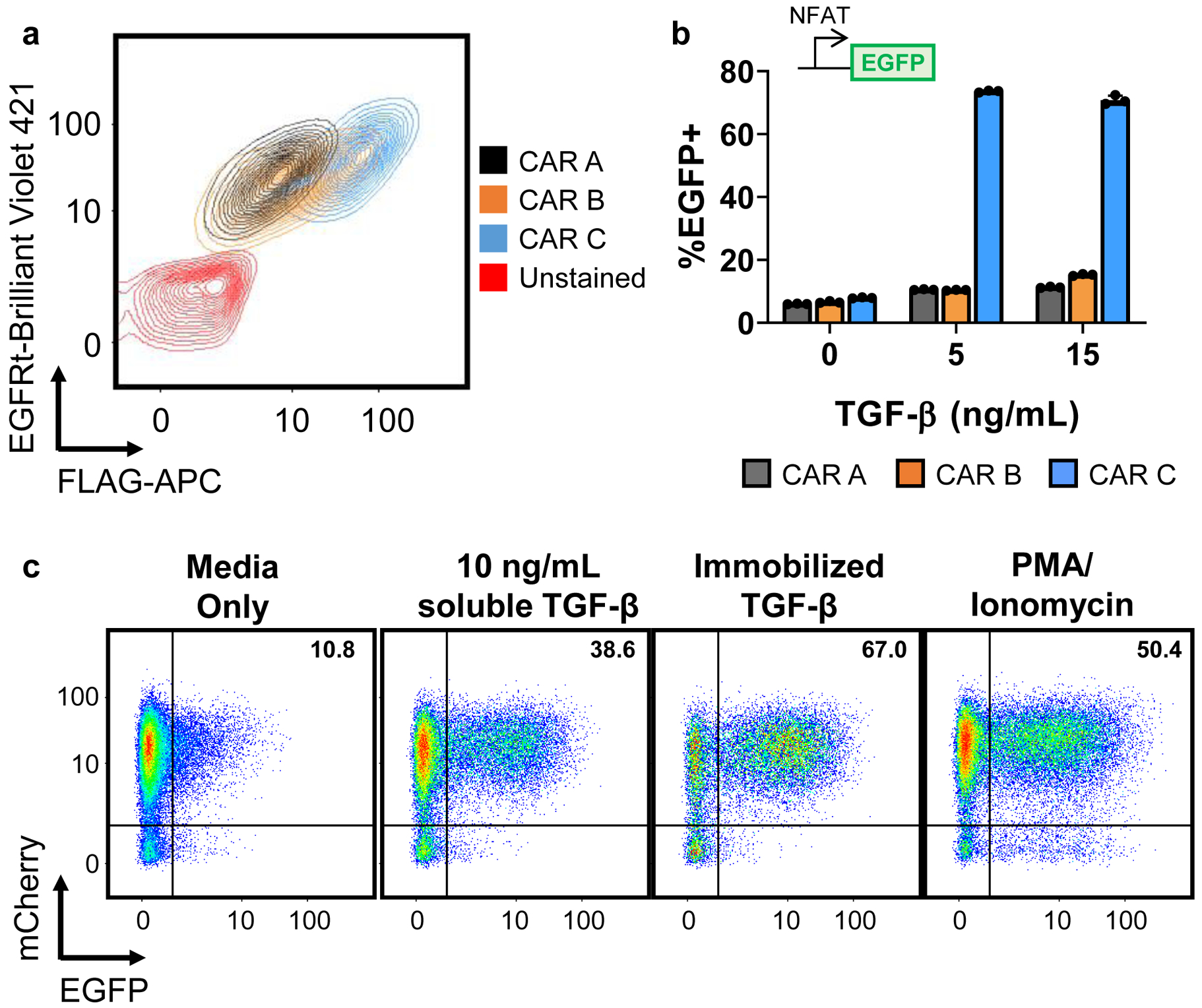
a, Different TGF-β scFvs with increasing TGF-β–binding affinities were used to construct CARs A, B, and C, respectively. CARs were fused to an N-terminal FLAG tag, and connected via a T2A “self-cleaving” peptide to truncated EGFR (EGFRt). As such, FLAG staining allows quantification of CAR surface expression while EGFRt staining indicates transduction efficiency. When expressed in Jurkat cells, all three CAR designs transduced well and expressed on the cell surface, albeit with varying surface-localization efficiencies. b, When tested in Jurkat NFAT reporter cell lines, only CAR C produced a clear response at the tested levels of TGF-β concentration. Data points from n = 3 samples are shown with means ± 1 standard deviation. c, NFAT reporter cells transduced to express an mCherry-fused TGF-β CAR were exposed to media alone, 10 ng/mL soluble TGF-β, immobilized TGF-β, or 50 ng/mL PMA and 1 μM ionomycin. TGF-β was immobilized in cell-culture wells by covering the well with 100 ng/mL TGF-β at room temperature for 30 min, washing once with PBS, then allowing the well to dry.
TIMING
Stably expressing synthetic receptors in primary T cells
Steps 1–9, 2 hours
Steps 10–17, 30 min followed by 10 min/day for 2 days
Steps 18–30, 7 hours followed by 0–30 min over the next 6 days
Steps 31–33, 45 min
Steps 34–41, 2.5 hours
Step 42 (optional), 30 min
Assays to assess CAR activity
Steps 43–48, 1 hour for set-up, 4–24 hour incubation, 1 hour to assay results
Steps 49–51, 1 hour for set-up, 24 hours incubation, 2–5 hours to assay results
Steps 52–57, 1 hour for set-up, 5 hours incubation, 1 hour to assay results
Steps 58–65, 1.5 hours for set-up, 8 hours incubation, 1.5 hours to assay results
Steps 66–74, 2.5 hours
ANTICIPATED RESULTS
The above protocols were used previously to generate cell lines expressing a TGF-β CAR and demonstrate the CAR’s activity6. Evaluation of steps in cell line production (steps 1–42) are presented in Fig. 3. Isolation of primary human T cells from donor blood using Rosette Sep CD4+ or CD8+ enrichment kits (STEMCELL Technologies) routinely yields >80% CD3+CD4+ or CD3+CD8+ T cells, respectively (Fig. 3a). These T cells are then transduced with lentivirus at varying efficiencies and can typically be sorted, when desired, to isolate a 95–99% pure CAR+ fraction (Fig. 3bc).
Fig. 3. Steps in the generation of CAR+ primary human T cells from human whole blood.
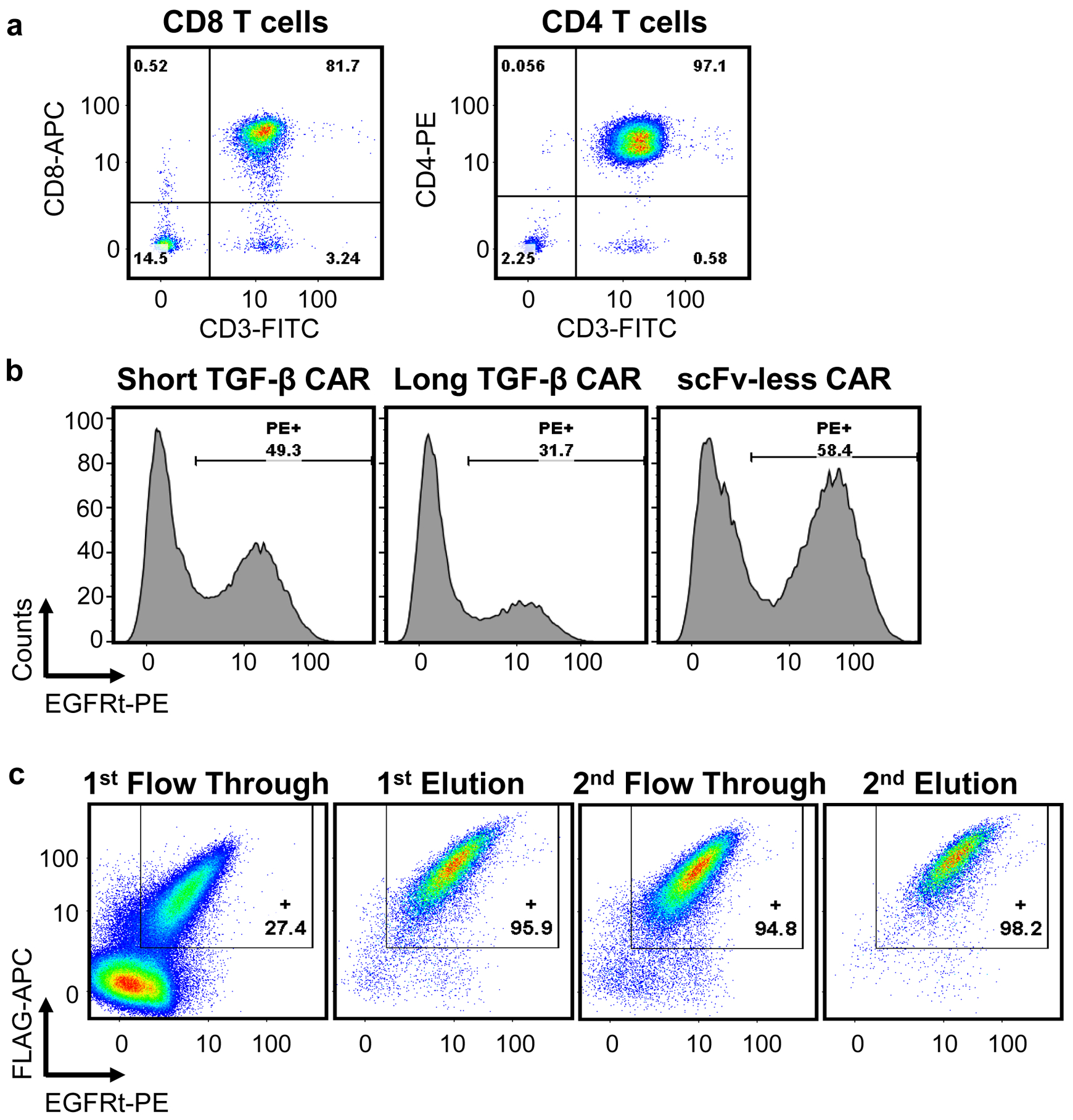
a, Cells isolated from human donor whole blood using a CD8+ or CD4+ enrichment kit, as in steps 1–7, were stained with antibodies for CD3 and CD8 (left) or CD3 and CD4 (right). b, CD4+ T cells were transduced with lentivirus encoding a TGF-β CAR with a short extracellular spacer domain, a TGF-β CAR with a long extracellular spacer domain, or an scFv-less CAR (as in steps 10–33). Each CAR was linked via a self-cleaving peptide to truncated EGFR (EGFRt). On Day 9 of culture, transduction efficiency was evaluated by staining for EGFRt. c, EGFRt-expressing cells were tagged with Erbitux-Biotin, then Anti-Biotin MicroBeads, and sorted through magnetic columns twice (steps 34–42). The sorting process was monitored by staining for both EGFRt and FLAG-tagged CARs in the flow-through and elution fractions.
Data representing the activity of the TGF-β CAR as assayed according to steps 43–74 are shown in Figs. 4 and 5. Jurkat-T cells stably expressing the TGF-β CAR exhibit robust activation of NFAT and NFκB in the presence of TGF-β (Fig. 4ab). Primary human T cells expressing the TGF-β CAR upregulate the early activation marker CD69 and secrete Th1 cytokines in the presence of TGF-β (Fig. 4cd). To elucidate whether CAR activation is a result of receptor signaling in response to soluble ligand as opposed to ligand cross-presented by neighboring CAR-T cells, TGF-β CAR-expressing NFAT-reporter Jurkat-T cells were seeded at varying densities in the absence or presence of TGF-β. At the lowest cell densities tested, signaling occurs in a density-independent manner, suggesting that ligand-receptor interactions in individual cells can in fact trigger T-cell activation (Fig. 4e). This is further confirmed by microscopy with Fluo-4-AM–labeled Jurkat T cells, where calcium flux in response to the addition of TGF-β is observed in the absence of cell-cell contacts (Fig. 4f). A comparison of how the TGF-β CAR upregulates NFAT or triggers degranulation in response to soluble vs. immobilized TGF-β demonstrates how CAR-T cells may act differently in response to different ligand modalities (Figs. 2c and 5). Examples of raw flow-cytometry data and its analysis are shown in Supplementary Fig. 3.
Fig. 4. Assessing functionality of the TGF-β CAR.
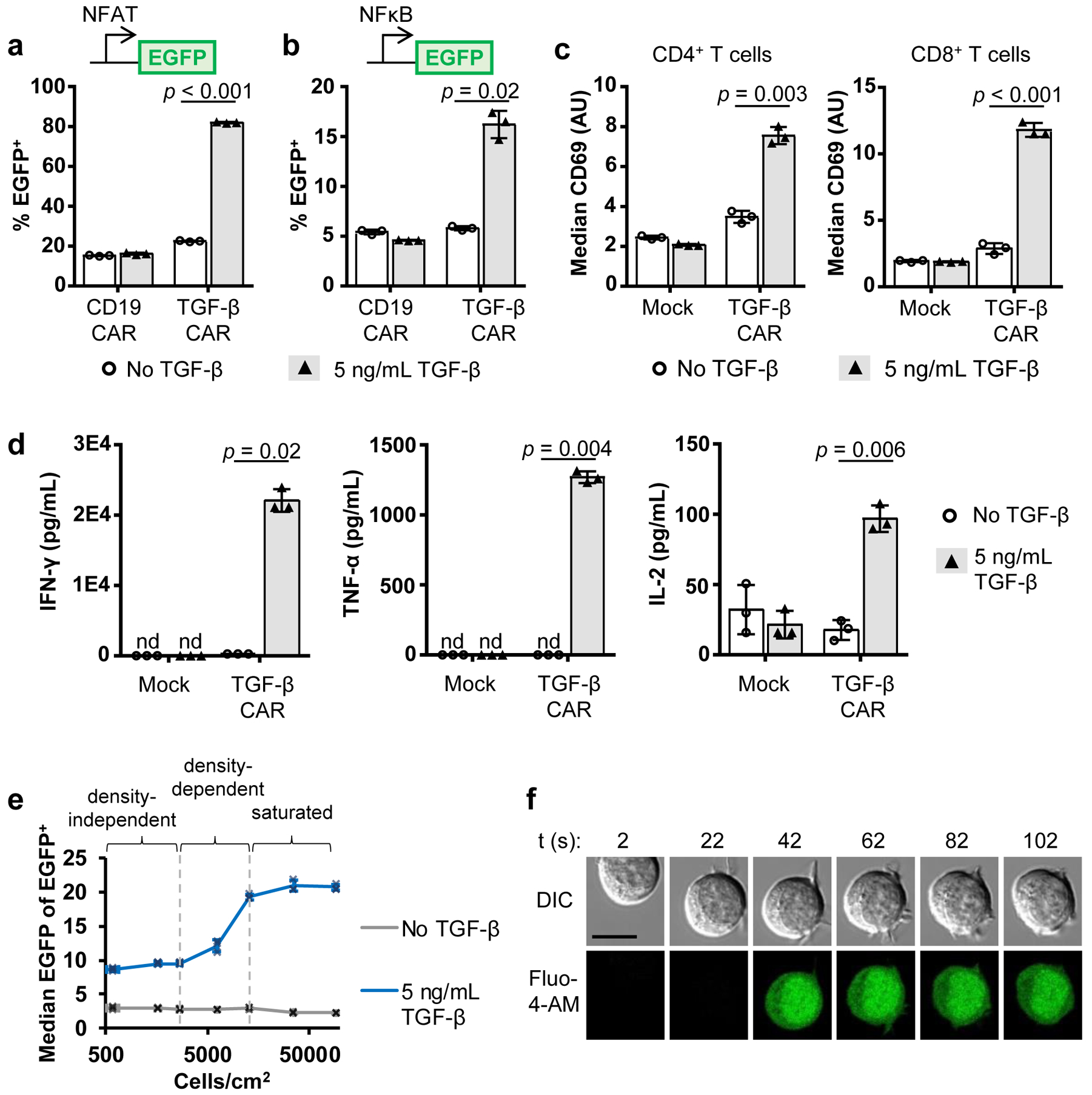
Data presented are reprinted with permission from a previously published study that employed the protocols mentioned in this article6. a, An NFAT reporter Jurkat T-cell line demonstrates that TGF-β stimulates NFAT signaling in TGF-β CAR-expressing Jurkat-T cells. b, Similarly, an NFκB reporter Jurkat T-cell line demonstrates that TGF-β stimulates NFκB signaling in TGF-β CAR-expressing Jurkat-T cells. c, Exposure of TGF-β CAR-expressing primary human CD4+ or CD8+ T cells to TGF-β triggers CD69 upregulation. Data in a-c are from experiments described in steps 43–48. d, Primary human CAR-T cell lines were assessed for cytokine production in the presence or absence of TGF-β as described in steps 49–51 (using a cytometric bead array). e, TGF-β CAR-expressing NFAT reporter Jurkat T cells were incubated with or without cognate ligand at varying cell densities as described in steps 58–65. Flow cytometry analysis showed the presence of a density-independent regime at low cell densities, a density-dependent regime at intermediate cell densities, and a density-independent regime at high cell densities where the signal output is likely saturated. The presence of a density-independent regime at low cells densities is consistent with the hypothesis that the TGF-β CAR can be activated by soluble TGF-β ligand independent of cell-to-cell interfaces. f, Microscopy of a TGF-β CAR-expressing Jurkat-T cell loaded with the Ca2+ flux indicator Fluo-4 AM was performed as described in steps 66–74. TGF-β ligand was added at time 0, and an activation event in the absence of cell-cell contact was observed. Scale bar represents 10 μm. In all graphs, data points from n = 3 samples are shown with means ± 1 standard deviation. Two-tailed Student’s t-tests with unequal variances were used to generate the p-values presented in a-d.
Fig. 5. Degranulation in response to soluble and immobilized target ligand.
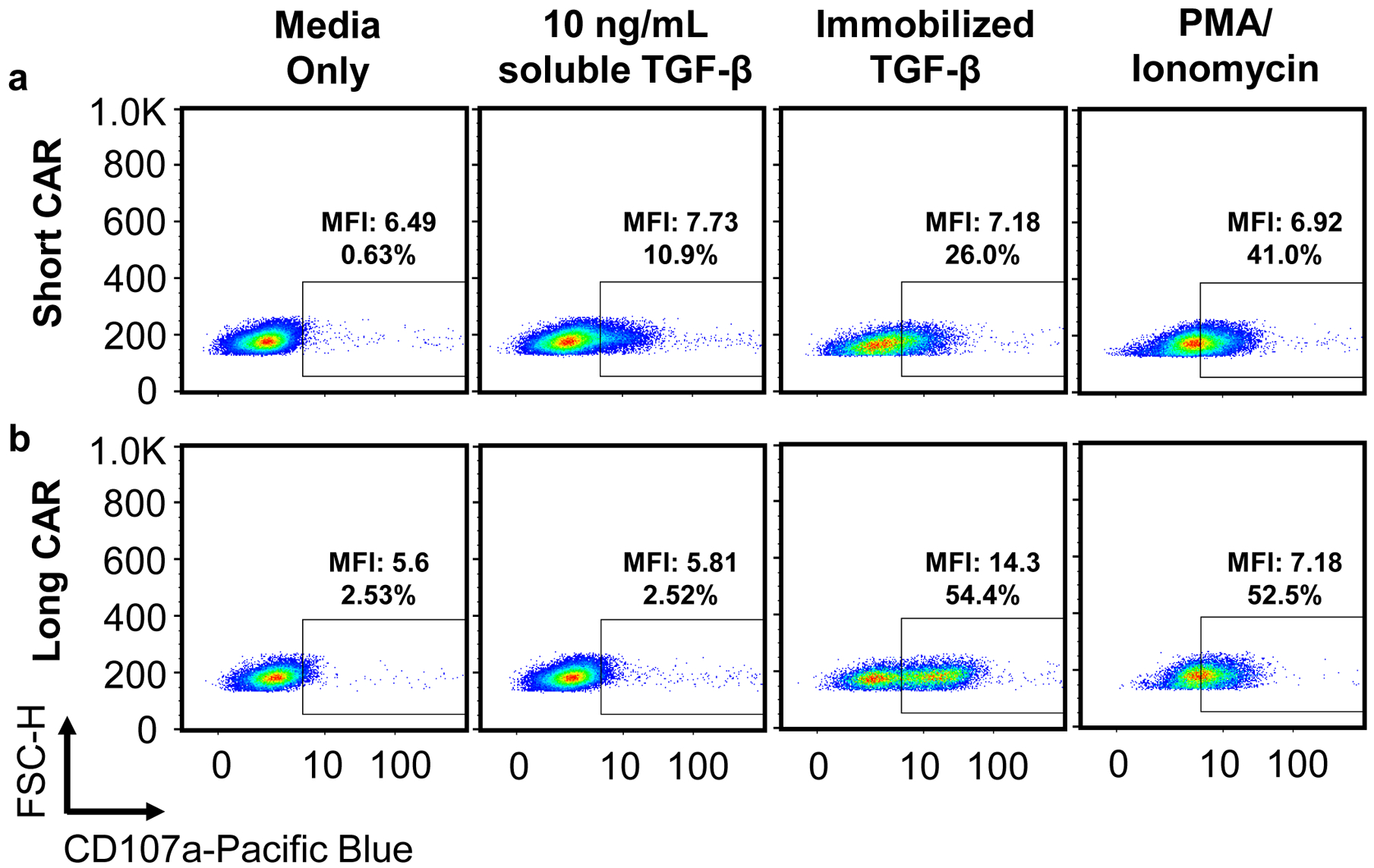
T cells expressing a TGF-β CAR with an IgG4 hinge spacer (a, short CAR) and IgG4-CH2-CH3 spacer (b, long CAR) were exposed to media alone, 10 ng/mL soluble TGF-β, immobilized TGF-β, or 50 ng/mL PMA and 1 μM ionomycin. TGF-β was immobilized in cell-culture wells by covering the well with 100 ng/mL TGF-β at room temperature for 30 min, washing once with PBS, then allowing the well to dry. Analysis of degranulation according to steps 52–57 showed that when targeting TGF-β immobilized on the bottom of culture wells, only the long-CAR cell line exhibits a distinct CD107ahi population consistent with that of a full degranulation response, perhaps because only the long CAR provides appropriate T-cell–to–target spacing. In contrast, for both cell lines, stimulation with either soluble TGF-β or soluble PMA/ionomycin resulted in a broadening of the CD107a expression distribution into the CD107a+ gate, but no distinct CD107ahi degranulated population. Scatter plots are representative of n = 3 samples.
Supplementary Material
Supplementary Fig. 1 Comparison of quantifying cytokine secreted into supernatant and intracellular cytokine staining.
CD4 TGF-β CAR-T cells were stimulated with 5 ng/mL TGF-β for 24 hours. Side-by-side samples were prepared for analysis of supernatant or intracellular cytokine staining. a, Quantification of IFN-γ and TNF-α levels secreted into supernatant using a cytometric bead array showed comparable IFN-γ and TNF-α production. Data points from n = 3 (without TGF-β) or n = 2 (with TGF-β) samples are shown with means ± 1 standard deviation or total ranges, respectively. b, In contrast, intracellular cytokine staining detected more robust TNF-α production than IFN-γ production, indicating lower sensitivity for IFN-γ staining using the protocol described.
Supplementary Fig. 2 Cell sorting results are impacted by transduction efficiency of starting material.
Cells lines A and B were generated by transduction with a synthetic construct linked via a T2A “self-cleaving” peptide to truncated EGFR (EGFRt). The poorly transduced cell line (Cell Line A) and well-transduced cell line (Cell Line B) were then sorted according to steps 34–41. The poorly transduced starting material yielded an enriched product that was only 68.5% pure, while the well-transduced cell line yielded a 97.1% pure population.
Supplementary Fig. 3 Gating strategy for flow cytometry experiments.
a, An example gating scheme for all flow cytometry experiments is shown. Initial FSC-Area/SSC-Area gates are drawn to remove debris and dead cells. A subsequent FSC-Area/FSC-Height gate demarcates singlet events. b–d, Histograms are shown for flow cytometry of TGF-β CAR-expressing cell lines, gated for viable singlets, demonstrating upregulation of (b) NFAT-driven EGFP, (c) NFκB-driven EGFP, and (d) CD69. Plots in b–d are representative of data used to generate the bar graphs in Fig. 4a–c.
ACKNOWLEDGEMENTS
Several protocols presented reflect discussion and contribution from present and past members of the Yvonne Chen lab, notably Eugenia Zah, Patrick Ho, Meng-yin Lin, and Michael Lorenzini, as well as members of the Michael Jensen lab (Seattle Children’s Research Institute). The NFAT reporter Jurkat T-cell line was a gift of Arthur Weiss (University of California, San Francisco). The NFκB reporter Jurkat T-cell line was a gift of Xin Lin (University of Texas MD Anderson Cancer Center). This work was supported by the National Institutes of Health (DP5OD012133, grant to Y.Y.C.; UC CAI Grant U54HL119893; and UCLA CTSI Grant Number UL1TR001881). Z.L.C. was supported by an NIH F30 Fellowship (F30CA183528). A.J.H. was supported by the NIH Biotechnology Training in Biomedical Sciences and Engineering Program (T32 GM067555).
Footnotes
DATA AVAILABILITY
The majority of data analyzed to generate what is shown in this manuscript are compiled in the Supplemental Spreadsheet file. Any additional data is available from the corresponding author upon reasonable request. Data from Fig. 4 and Supplementary Fig. 3 have been used in prior publication6.
COMPETING INTERESTS
Y.Y.C. and Z.L.C. declare competing financial interests in the form of a patent application whose value may be affected by the publication of this work.
REFERENCES
- 1.Maude SL et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N. Engl. J. Med 371, 1507–1517 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maude SL et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med 378, 439–448 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Porter DL et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med 7, 303ra139 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brentjens RJ et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood 118, 4817–4828 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kochenderfer JN et al. Chemotherapy-Refractory Diffuse Large B-Cell Lymphoma and Indolent B-Cell Malignancies Can Be Effectively Treated With Autologous T Cells Expressing an Anti-CD19 Chimeric Antigen Receptor. J. Clin. Oncol 33, 540–549 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang ZL et al. Rewiring T-cell responses to soluble factors with chimeric antigen receptors. Nat. Chem. Biol 14, 317–324 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hou AJ, Chang ZL, Lorenzini MH, Zah E & Chen YY TGF‐β–responsive CAR‐T cells promote anti‐tumor immune function. Bioeng. Transl. Med 3, 75–86 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen LC & Chen YY Outsmarting and outmuscling cancer cells with synthetic and systems immunology. Curr. Opin. Biotechnol 60, 111–118 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Leen AM et al. Reversal of Tumor Immune Inhibition Using a Chimeric Cytokine Receptor. Mol. Ther 22, 1211–1220 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mohammed S et al. Improving Chimeric Antigen Receptor-Modified T Cell Function by Reversing the Immunosuppressive Tumor Microenvironment of Pancreatic Cancer. Mol. Ther 25, 249–258 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilkie S et al. Selective expansion of chimeric antigen receptor-targeted T-cells with potent effector function using interleukin-4. J. Biol. Chem 285, 25538–25544 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang ZL & Chen YY CARs: Synthetic Immunoreceptors for Cancer Therapy and Beyond. Trends Mol. Med 23, 430–450 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwarz KA, Daringer NM, Dolberg TB & Leonard JN Rewiring human cellular input-output using modular extracellular sensors. Nat. Chem. Biol 13, 202–209 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baeumler TA, Ahmed AA & Fulga TA Engineering Synthetic Signaling Pathways with Programmable dCas9-Based Chimeric Receptors. Cell Rep. 20, 2639–2653 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scheller L, Strittmatter T, Fuchs D, Bojar D & Fussenegger M Generalized extracellular molecule sensor platform for programming cellular behavior. Nat. Chem. Biol 14, 723 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Green MR & Sambrook J Molecular Cloning: A Laboratory Manual. (Cold Spring Harbor Press, 2012). [Google Scholar]
- 17.Gibson DG et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 (2009). [DOI] [PubMed] [Google Scholar]
- 18.Tiscornia G, Singer O & Verma IM Production and purification of lentiviral vectors. Nat. Protoc 1, 241–245 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Liu L et al. Inclusion of Strep-tag II in design of antigen receptors for T-cell immunotherapy. Nat. Biotechnol 34, 430–434 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ghassemi S et al. Reducing Ex Vivo Culture Improves the Antileukemic Activity of Chimeric Antigen Receptor (CAR) T Cells. Cancer Immunol. Res 6, 1100–1109 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hollyman D et al. Manufacturing validation of biologically functional T cells targeted to CD19 antigen for autologous adoptive cell therapy. J. Immunother. Hagerstown Md 1997 32, 169–180 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vormittag P, Gunn R, Ghorashian S & Veraitch FS A guide to manufacturing CAR T cell therapies. Curr. Opin. Biotechnol 53, 164–181 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Chang ZL et al. ASGCT 22nd Annual Meeting Abstracts: 832. Rewiring T-Cell Signaling Responses to Extracellular Soluble Cues with Chimeric Antigen Receptors. Mol. Ther 27, 384 (2019). [Google Scholar]
- 24.Zah E, Lin M-Y, Silva-Benedict A, Jensen MC & Chen YY T Cells Expressing CD19/CD20 Bispecific Chimeric Antigen Receptors Prevent Antigen Escape by Malignant B Cells. Cancer Immunol. Res 4, 498–508 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Caruso HG et al. Tuning Sensitivity of CAR to EGFR Density Limits Recognition of Normal Tissue While Maintaining Potent Antitumor Activity. Cancer Res. 75, 3505–3518 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu X et al. Affinity-tuned ErbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against tumors in mice. Cancer Res. 75, 3596–3607 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang X et al. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood 118, 1255–1263 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wei P et al. Bacterial virulence proteins as tools to rewire kinase pathways in yeast and immune cells. Nature 488, 384–388 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang D et al. A requirement for CARMA1 in TCR-induced NF-κB activation. Nat. Immunol 3, 830–835 (2002). [DOI] [PubMed] [Google Scholar]
- 30.Long AH et al. 4–1BB Costimulation Ameliorates T Cell Exhaustion Induced by Tonic Signaling of Chimeric Antigen Receptors. Nat. Med 21, 581–590 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frigault MJ et al. Identification of chimeric antigen receptors that mediate constitutive or inducible proliferation of T cells. Cancer Immunol. Res 3, 356–367 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aivazian D & Stern LJ Phosphorylation of T cell receptor ζ is regulated by a lipid dependent folding transition. Nat. Struct. Biol 7, 1023–1026 (2000). [DOI] [PubMed] [Google Scholar]
- 33.Zhang H, Cordoba S-P, Dushek O & Merwe PA van der. Basic residues in the T-cell receptor ζ cytoplasmic domain mediate membrane association and modulate signaling. PNAS 108, 19323–19328 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dobbins J et al. Binding of the cytoplasmic domain of CD28 to the plasma membrane inhibits Lck recruitment and signaling. Sci. Signal 9, ra75–ra75 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. 1 Comparison of quantifying cytokine secreted into supernatant and intracellular cytokine staining.
CD4 TGF-β CAR-T cells were stimulated with 5 ng/mL TGF-β for 24 hours. Side-by-side samples were prepared for analysis of supernatant or intracellular cytokine staining. a, Quantification of IFN-γ and TNF-α levels secreted into supernatant using a cytometric bead array showed comparable IFN-γ and TNF-α production. Data points from n = 3 (without TGF-β) or n = 2 (with TGF-β) samples are shown with means ± 1 standard deviation or total ranges, respectively. b, In contrast, intracellular cytokine staining detected more robust TNF-α production than IFN-γ production, indicating lower sensitivity for IFN-γ staining using the protocol described.
Supplementary Fig. 2 Cell sorting results are impacted by transduction efficiency of starting material.
Cells lines A and B were generated by transduction with a synthetic construct linked via a T2A “self-cleaving” peptide to truncated EGFR (EGFRt). The poorly transduced cell line (Cell Line A) and well-transduced cell line (Cell Line B) were then sorted according to steps 34–41. The poorly transduced starting material yielded an enriched product that was only 68.5% pure, while the well-transduced cell line yielded a 97.1% pure population.
Supplementary Fig. 3 Gating strategy for flow cytometry experiments.
a, An example gating scheme for all flow cytometry experiments is shown. Initial FSC-Area/SSC-Area gates are drawn to remove debris and dead cells. A subsequent FSC-Area/FSC-Height gate demarcates singlet events. b–d, Histograms are shown for flow cytometry of TGF-β CAR-expressing cell lines, gated for viable singlets, demonstrating upregulation of (b) NFAT-driven EGFP, (c) NFκB-driven EGFP, and (d) CD69. Plots in b–d are representative of data used to generate the bar graphs in Fig. 4a–c.