

Abstract
PURPOSE
To investigate cancer treatment plus pathogenic germline mutations (PGMs) in DNA repair genes (DRGs) for identification of childhood cancer survivors at increased risk of subsequent neoplasms (SNs).
METHODS
Whole-genome sequencing was performed on blood-derived DNA from survivors in the St Jude Lifetime Cohort. PGMs were evaluated in 127 genes from 6 major DNA repair pathways. Cumulative doses of chemotherapy and body region–specific radiotherapy (RT) were abstracted from medical records. Relative rates (RRs) and 95% CIs of SNs by mutation status were estimated using multivariable piecewise exponential models.
RESULTS
Of 4,402 survivors, 495 (11.2%) developed 1,269 SNs. We identified 538 PGMs in 98 DRGs (POLG, MUTYH, ERCC2, and BRCA2, among others) in 508 (11.5%) survivors. Mutations in homologous recombination (HR) genes were significantly associated with an increased rate of subsequent female breast cancer (RR, 3.7; 95% CI, 1.8 to 7.7), especially among survivors with chest RT ≥ 20 Gy (RR, 4.4; 95% CI, 1.6 to 12.4), or with a cumulative dose of anthracyclines in the second or third tertile (RR, 4.4; 95% CI, 1.7 to 11.4). Mutations in HR genes were also associated with an increased rate of subsequent sarcoma among those who received alkylating agent doses in the third tertile (RR, 14.9; 95% CI, 4.0 to 38.0). Mutations in nucleotide excision repair genes were associated with subsequent thyroid cancer for those treated with neck RT ≥ 30 Gy (RR, 12.9; 95% CI, 1.6 to 46.6) with marginal statistical significance.
CONCLUSION
Our study provides novel insights regarding the contribution of genetics, in combination with known treatment-related risks, for the development of SNs. These findings have the potential to facilitate identification of high-risk survivors who may benefit from genetic counseling and/or testing of DRGs, which may further inform personalized cancer surveillance and prevention strategies.
INTRODUCTION
The population of childhood cancer survivors is growing and estimated to exceed 500,000 in the United States by 2020.1 Childhood cancer survivors experience a substantial burden of long-term chronic health conditions, including a high incidence of subsequent neoplasms (SNs).2 Although the development of SNs among survivors is largely considered therapy related,3 pathogenic germline mutations in cancer predisposition genes also contribute to this elevated risk.4
CONTEXT
Key Objective
We aimed to characterize germline pathogenic mutations in DNA repair genes and evaluate the risk of developing subsequent neoplasms on the basis of mutation status among childhood cancer survivors in conjunction with genotoxic cancer treatment exposures.
Knowledge Generated
By analyzing 4,402 survivors from the St Jude Lifetime Cohort, we identified 3 groups with elevated risk: (1) female survivors who had mutations in homologous recombination genes and received high doses of chest irradiation or anthracyclines have 4-fold increased risk of subsequent breast cancer; (2) survivors who had mutations in homologous recombination genes and received high doses of alkylating agents have 15-fold increased risk of subsequent sarcoma; and (3) survivors who had mutations in nucleotide excision repair genes and received high doses of neck irradiation have 13-fold increased risk of subsequent thyroid cancer.
Relevance
Identifying survivors at the highest risk of subsequent neoplasms and implementing personalized cancer surveillance and prevention strategies may reduce morbidity and mortality.
Using the St Jude Lifetime Cohort (SJLIFE),5 we previously characterized the prevalence and spectrum of pathogenic/likely pathogenic (P/LP) mutations in 60 genes associated with well-established autosomal dominant cancer predisposition syndromes and found that carriers of P/LP mutations have a greater risk of SNs.4 Notably, some cancer predisposition genes are also DNA repair genes (DRGs), suggesting that pathogenic mutations in DNA repair processes may play a role in the development of SNs among childhood cancer survivors.
DNA repair is essential for the maintenance of genome integrity. The various forms of DNA damage caused by genetic (ie, germline mutations) and/or environmental (ie, genotoxic agents) factors6 can be repaired through 6 major DNA repair pathways: base excision repair (BER), Fanconi anemia (FA), homologous recombination (HR), mismatch repair (MMR), nucleotide excision repair (NER), and nonhomologous end joining (NHEJ). Defects in some DRGs elevate risks for hereditary cancers or syndromes associated with specific malignancies.7,8 Most of these syndromes, such as Bloom syndrome, Nijmegen breakage syndrome, and FA, are characterized by autosomal recessive inheritance of DRG defects with low penetrance in the general population and have a limited contribution to the overall cancer burden.8 Nevertheless, monoallelic pathogenic mutations of these DRGs have been reported to be associated with increased risk of cancers. For instance, monoallelic mutation carriers of BLM,9 an HR gene, and monoallelic mutation carriers of MUTYH,10 a BER gene, have increased risk of developing colorectal cancer; and monoallelic FANCC mutations in the FA pathway increase risk of breast cancer.11 Moreover, specific DNA repair pathways are responsible for repairing DNA damages caused by certain treatment exposures cancer survivors had received.12-16 For example, DNA double-strand breaks caused by ionizing radiation or anthracyclines can be repaired by HR or NHEJ pathway.12 Germline deficiencies in these DNA repair pathways may be exacerbated by these treatment exposures and lead to oncogenic genomic instability and hence increase SN risk. Therefore, we hypothesized that pathogenic mutations in DRGs of specific pathways, regardless of the mode of inheritance of the associated syndromes, may lead to suboptimal DNA repair proficiency resulting in the accumulation of specific DNA errors and contribute to the risk of SNs among childhood cancer survivors, especially those treated with high doses of certain radiotherapy (RT) and/or chemotherapeutic agents.
METHODS
Study Participants
The SJLIFE study is a retrospective cohort with prospective clinical follow-up and ongoing enrollment of survivors of childhood cancer treated at St Jude Children’s Research Hospital.5 For the current analysis, 4,402 survivors completed SJLIFE clinical assessment, provided a blood sample for DNA sequencing, and were followed until December 31, 2016. Procedures of SJLIFE have been approved by the St Jude Institutional Review Board, and participants provided written informed consent. Genetic findings from this study are strictly used for research purpose only, as indicated in the signed consent form.
Treatment Exposures and Characterization of Subsequent Neoplasms
Region-specific RT exposures used radiation oncology treatment records.17 Chemotherapy exposures, including cumulative doses of anthracyclines, alkylating agents (cyclophosphamide equivalent dose18), and epipodophyllotoxins, were abstracted from medical records. Reported SNs were verified pathologically and/or radiologically, through clinical notes, or from National Death Index reports. The locations of SNs were reviewed in conjunction with RT records and categorized as in or out of the radiation field. The occurrence of any SN and that of each common specific SN diagnosis (nonmelanoma skin cancer [NMSC], meningioma, thyroid cancer, female breast cancer, and sarcoma) were studied.
DNA Sequencing
DNA was isolated from blood samples. Whole-genome sequencing (WGS) and whole-exome sequencing (WES) were performed on the initial 3,006 survivors using the Illumina (San Diego, CA, USA) HiSeq X Ten and HiSeq 4000, respectively.4 The second set of 1,396 survivors was sequenced with Illumina NovaSeq (WGS, average genome-wide coverage of 38.7×; and WES, average coverage of coding exons of 234.1×).
Selection of DNA Repair Pathway Genes
We selected 127 genes in 6 major DNA repair pathways on the basis of information from the MD Anderson DNA repair gene website19-23 and a literature review. The gene list included 42 HR genes (40 core genes described previously,24 POLQ as a radiosensitivity HR gene,25 and RECQL as an HR gene implicated in breast cancer risk26), 12 MMR genes, 33 NER genes, 20 FA genes, 9 NHEJ genes, and 22 BER genes (Fig 1).
FIG 1.
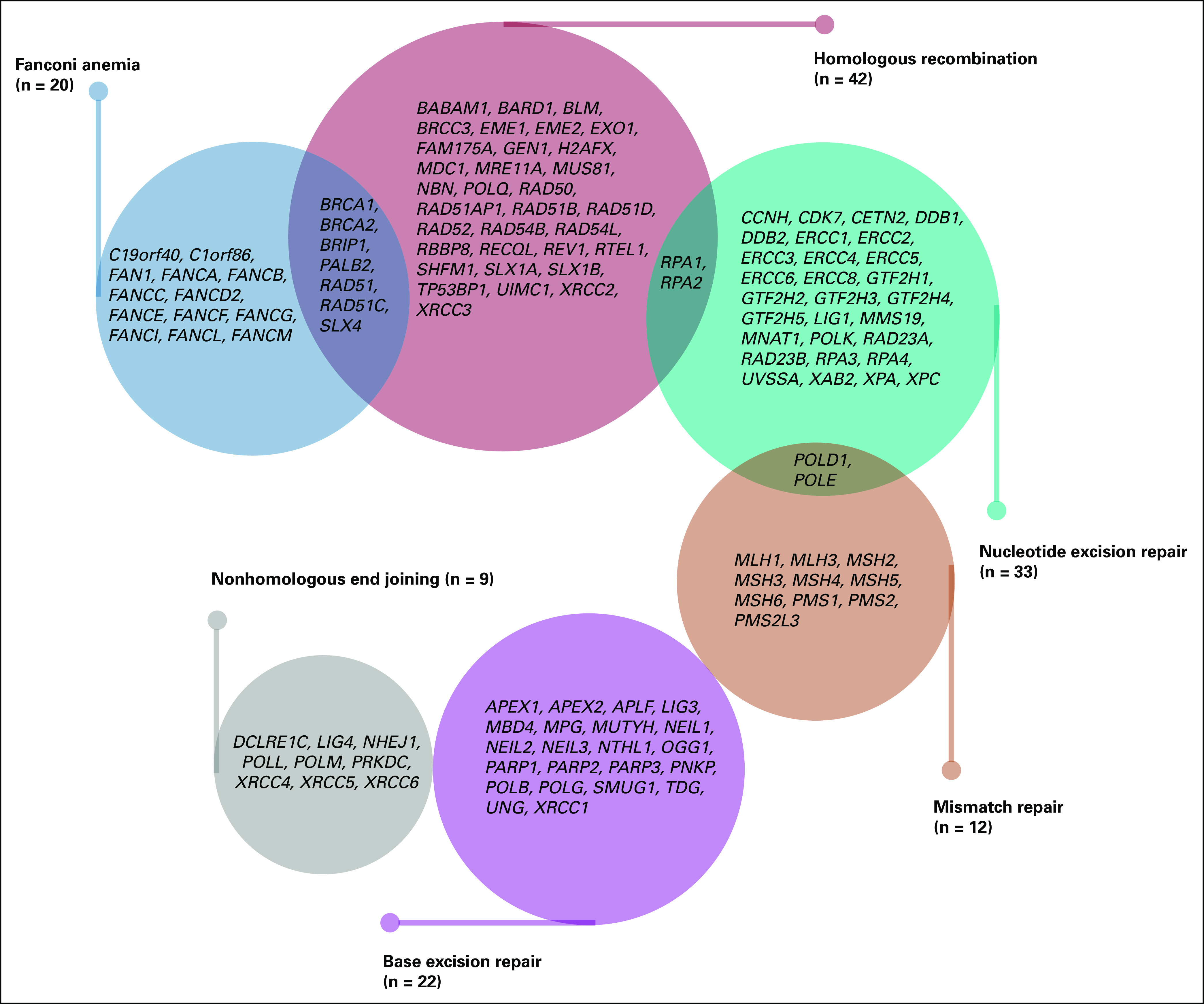
Summary information for 127 genes from 6 major DNA repair pathways selected for germline mutation analysis.
Variant Detection and Classification
Germline single-nucleotide variants, small insertions and deletions, and copy number variations (CNVs) were detected as previously described.27 Details for the classification of “pathogenic mutations” are provided in the Data Supplement.
Statistical Analyses
Multivariable piecewise-exponential models,28 defined as generalized linear models with a logarithmic link function and Poisson error distribution, were used to assess the effect of carrying pathogenic mutations in each DNA repair pathway on the rate of each SN outcome (overall and specific SN types) among survivors of childhood cancer, from which relative rates (RRs) and 95% CIs were estimated. The at-risk follow-up started from the fifth anniversary from childhood cancer diagnosis and ended at the earliest of the occurrence of a specific SN type, last date of contact, or at death. Each survivor’s follow-up time was divided into age-specific segments, where each segment’s age-specific rate of SN was modeled by explanatory variables. Additional details of the modeling approach are provided in the Data Supplement. When estimating the cumulative incidence of each SN by age, death was considered a competing risk event. Gray’s method29 was used to evaluate statistical significance of the differences in cumulative incidence curves between each pair of groups and across all groups. Analyses were performed using SAS 9.4 (SAS Institute, Cary, NC), and figures were generated using R 3.5.1.30 All tests were 2-sided. Benjamini-Hochberg’s method was used to control the false discovery rate (FDR).
RESULTS
Study Participants
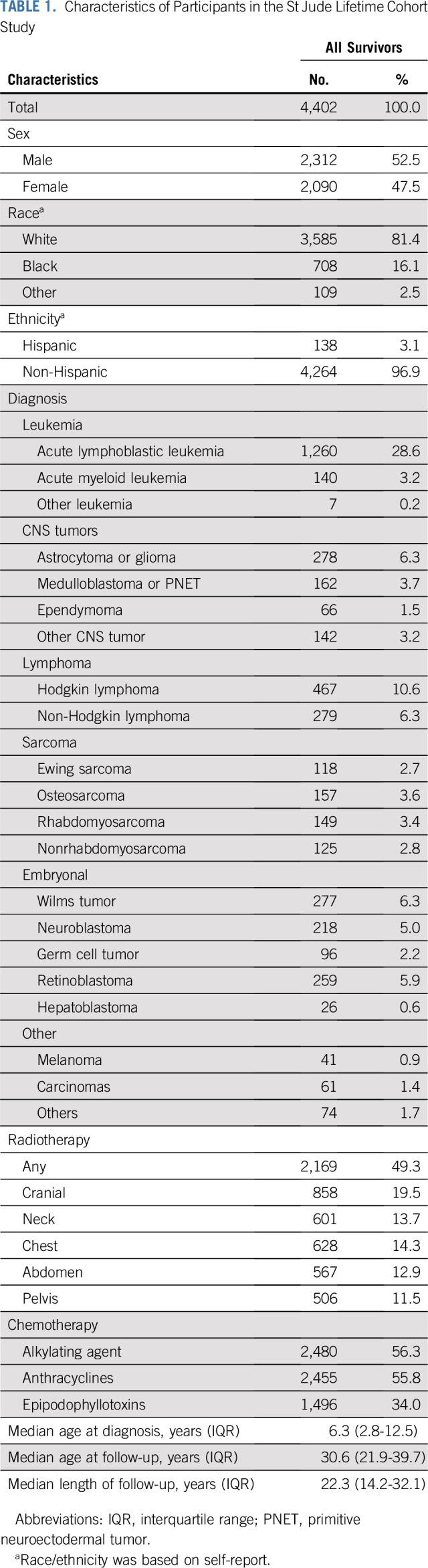
Of 4,402 survivors (Table 1), 52.5% were males, and 81.4% were white. The primary diagnoses included leukemia (32.0%), CNS malignancies (14.7%), lymphoma (16.9%), and other solid tumors (36.4%). Approximately half (49.3%) were treated with RT, 55.8% with anthracyclines, 56.3% with alkylating agents, and 34.0% with epipodophyllotoxins. The median ages at childhood cancer diagnosis and at last follow-up were 6.3 (interquartile range [IQR], 2.8-12.5) years and 30.6 (IQR, 21.9-39.7) years, respectively, with a median follow-up of 22.3 (IQR, 14.2-32.1) years.
TABLE 1.
Characteristics of Participants in the St Jude Lifetime Cohort Study
SNs
A total of 1,269 SNs were diagnosed among 495 (11.2%) survivors. Median time from childhood cancer diagnosis to the first SN was 26.5 (IQR, 19.1-33.9) years, with the median age at first SN of 35.9 (IQR, 29.0-43.2) years. The most common SNs were NMSC (4.3%; 686 in 191 survivors), meningioma (2.7%; 272 in 120 survivors), female breast cancer (2.7%; 64 in 57 survivors), thyroid cancer (1.6%; 73 in 72 survivors), and sarcoma (0.7%; 30 in 30 survivors).
Prevalence and Spectrum of Mutations
A total of 538 pathogenic mutations in 98 DRGs were identified in 508 survivors (prevalence, 11.5%; 95% CI, 10.6% to 12.5%), including 192 HR gene mutations in 185 survivors (prevalence, 4.2%; 95% CI, 3.6% to 4.8%), 36 MMR gene mutations in 36 survivors (prevalence, 0.8%; 95% CI, 0.6% to 1.1%), 98 NER gene mutations in 97 survivors (prevalence, 2.2%; 95% CI, 1.8% to 2.7%), 143 FA gene mutations in 140 survivors (prevalence, 3.2%; 2.7% to 3.7%), 26 NHEJ gene mutations in 26 survivors (prevalence, 0.6%; 95% CI, 0.4% to 0.9%), and 112 BER gene mutations in 110 survivors (prevalence, 2.5%; 95% CI, 2.1% to 3.0%). The proportions (with 95% CIs) of mutation carriers in each DNA repair pathway were reported among survivors without SN, with any SN, or with a specific SN (Data Supplement). Of the 538 pathogenic mutations, 70 were missense mutations, 175 were nonsense mutations, 196 were frameshift mutations, one was an in-frame protein deletion, 78 were splicing mutations, and 18 were CNVs. Of the 508 mutation carriers, no one had > 1 pathogenic mutation in the same gene, 11 carried ≥ 2 pathogenic mutations in the same DNA repair pathway, and 16 had > 1 pathogenic mutation in different pathways (Data Supplement).
The most frequently altered DRGs were POLG (n = 30), MUTYH (n = 27), ERCC2 (n = 25), BRCA2 (n = 20), FANCA (n = 16), FAN1 (n = 15), FANCC (n = 15), BRCA1 (n = 14), BLM (n = 13), ERCC3 (n = 13), PALB2 (n = 13), POLQ (n = 13), LIG4 (n = 12), RECQL (n = 10), and ERCC6 (n = 10; Data Supplement). Thirty-three mutations in 27 DRGs (eg, BRCA1, BRCA2, ERCC2, FAN1) with a low variant allele frequency (range, 0.13-0.30) were considered as germline mosaicism (Data Supplement).
Associations Between Mutation Status and Subsequent Neoplasms
Any SN and SN-specific baseline clinical models are shown in Table 2. We observed significantly increased rates of any SN among carriers of mutations in HR (RR, 1.5; 95% CI, 1.1 to 2.2), NER (RR, 1.6; 95% CI, 1.1 to 2.5), or MMR genes (RR, 2.1; 95% CI, 1.2 to 3.7; Table 3). The subsequent breast cancer rate was significantly increased among female survivors carrying pathogenic mutations in HR (RR, 3.7; 95% CI, 1.8 to 7.7) or FA genes (RR, 3.7; 95% CI, 1.3 to 10.6; Table 3). We also observed a higher rate of subsequent sarcoma (RR, 5.0; 95% CI, 1.4 to 12.8) among survivors carrying pathogenic mutations in HR genes, and higher rates of subsequent thyroid cancer (RR, 2.7; 95% CI, 1.1 to 6.9) and NMSC (RR, 2.9; 95% CI, 1.1 to 7.9) among survivors carrying pathogenic mutations in NER genes, but these associations were not statistically significant after controlling FDR (Table 3). We found no significant association for subsequent meningioma (Table 3). For HR, FA, and MMR pathways, we performed additional analyses by excluding 61 mutation carriers of 7 well-established cancer predisposition genes (14 in BRCA1, 20 in BRCA2, 13 in PALB2, 8 in MSH6, and 6 in PMS2; no carrier of mutations in MLH1 or MSH2) and found that the estimated RRs ranged from 1.35-3.15, although they were not statistically significant (Data Supplement).
TABLE 2.
Baseline Nongenetic Multivariable Model, Including Diagnostic and Treatment Variables, of Relative Rates of Any and Specific Subsequent Neoplasms

TABLE 3.
Adjusted Associations Between Pathogenic Mutations in DNA Repair Genes and Rates of Subsequent Neoplasms Among Childhood Cancer Survivors

Associations Between Mutation Status and Subsequent Neoplasms by Treatment Exposures
When stratified by treatment exposures, the associations of mutations in HR or FA genes with increased rates of subsequent breast cancer were slightly stronger among female survivors treated with chest RT ≥ 20 Gy (HR genes: RR, 4.4; 95% CI, 1.6 to 12.4; Pfdr = 0.02; FA genes: RR, 5.5; 95% CI, 1.5 to 14.2; Pfdr = 0.03) and those who received anthracycline doses in the second or third tertile (≥ 98 mg/m2; HR genes: RR, 4.4; 95% CI, 1.7 to 11.4; Pfdr = 0.01; FA genes: RR, 4.2; 95% CI, 1.4 to 12.3; Pfdr = 0.03; Fig 2A; Data Supplement). The association of HR gene mutations with subsequent sarcoma was substantially stronger among survivors treated with alkylating agent doses in the third tertile (≥ 10,451 mg/m2; RR, 14.9; 95% CI, 4.0 to 38.0; Pfdr = 0.001; Fig 2B). NER gene mutations were associated with a higher rate of subsequent thyroid cancer among those treated with neck RT ≥ 30 Gy (RR, 12.9; 95% CI, 1.6 to 46.6; Pfdr = 0.06; Fig 2C) and an increased rate of subsequent NMSC occurring in the RT field (RR, 3.2; 95% CI, 1.1 to 9.2; Pfdr = 0.06; Data Supplement), both with marginal statistical significance after accounting for multiple comparisons.
FIG 2.

Adjusted associations of pathogenic mutations in DNA repair genes with rates of subsequent neoplasms by treatment exposures. (A) The associations of mutations in homologous recombination (HR) genes and the rate of subsequent female breast cancer. (B) The association of mutations in HR genes and the rate of subsequent sarcoma. (C) The association of mutations in nucleotide excision repair (NER) genes and the rate of subsequent thyroid cancer. The relative rate is denoted as not applicable (NA) if the model did not converge. RT, radiotherapy.
Associations Between Mutation Status and Subsequent Neoplasms by Age
When stratified by age during follow-up (Data Supplement), the associations of HR or FA gene mutations and increased rates of specific SNs appeared stronger during the follow-up period corresponding to age ≤ 35years, including any SN (HR genes: RR, 2.1; 95% CI, 1.3 to 3.3; FA genes: RR, 1.9; 95% CI, 1.0 to 3.5), subsequent female breast cancer (HR genes: RR, 6.4; 95% CI, 2.3 to 18.1; FA genes: RR, 8.7; 95% CI, 2.4 to 22.2), and subsequent sarcoma (HR genes: RR, 9.2; 95% CI, 2.5 to 23.5). In contrast, the associations of NER gene mutations with the rates of any SN (RR, 2.0; 95% CI, 1.2 to 3.4), or subsequent NMSC (RR, 3.5; 95% CI, 1.4 to 8.5) and the association of MMR gene mutations with the rate of any SN (RR, 2.3; 95% CI, 1.0 to 5.1) appeared stronger during the follow-up period corresponding to age > 35years.
Cumulative Incidence of Second Neoplasms by Mutation Status and Treatment Exposures
Cumulative incidence curves of subsequent female breast cancer at age 20-55 years and that of subsequent sarcoma and thyroid cancer at age 10-55 years by mutation status and treatment modality are shown in Figure 3 and the Data Supplement. Among the 4 groups in Figure 3A, cumulative incidence of subsequent breast cancer varied greatly, being highest among female survivors who carried HR gene mutations and received chest RT ≥ 20 Gy: at age 45 years, it was 49.2%, 13.9%, 10.4%, and 2.5%, for the 4 subgroups. Among the 4 subgroups in Figure 3B, cumulative incidence of subsequent breast cancer was the highest among female survivors who carried HR gene mutations and received moderate to high cumulative doses of anthracyclines (the second or third tertile); at age 45 years, it was 36.9%, 8.6%, 8.5%, and 3.8%, for the 4 subgroups. Cumulative incidence of subsequent sarcoma was the highest among survivors who carried HR gene mutations and received high doses of alkylating agents (the third tertile); at age 45 years it was 24.6%, 1.6%, 0.8%, and 0.0%, for the 4 subgroups (Fig 3C). Cumulative incidence of subsequent thyroid cancer was the highest among survivors who carried NER-gene mutations and received high-dose neck RT (≥ 30 Gy); at age 45 years it was 36.2%, 6.4%, 6.3%, and 3.4%, for the 4 subgroups (Fig 3D).
FIG 3.
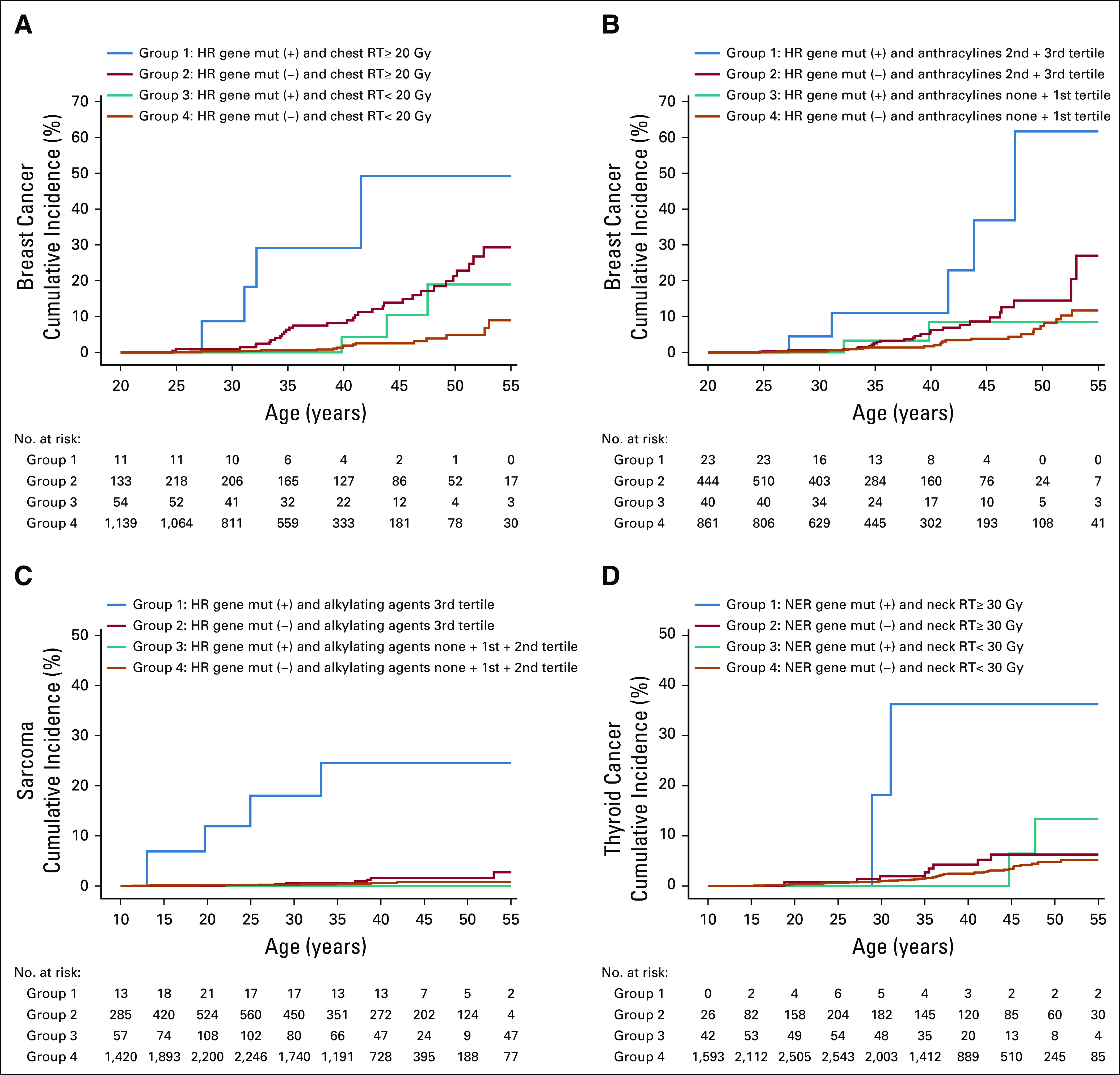
Cumulative incidence of subsequent neoplasms by mutation (mut) status of DNA repair genes and treatment exposures. (A) Subsequent female breast cancer by mutation status of homologous recombination (HR) genes and dose of chest radiotherapy (RT). (B) Subsequent female breast cancer by mutation status of HR genes and dose of anthracyclines. (C) Subsequent sarcoma by mutation status of HR genes and dose of alkylating agents. (D) Subsequent thyroid cancer by mutation status of nucleotide excision repair (NER) genes and dose of neck RT. The cumulative incidence curves are statistically different (P < .001) in all 4 panels. Additional statistical tests between group pairs and across the 3 groups except Group 1 (blue line) are included in the Data Supplement. A similar set of cumulative incidence curves with years since diagnosis as the x-axis is provided in the Data Supplement.
Proportions of Mutation Carriers Among High-Risk Groups
For female survivors who developed subsequent breast cancer and were exposed to high-dose chest RT (≥ 20 Gy) or moderate to high cumulative doses of anthracyclines (the second or third tertile), the proportions of HR gene mutation carriers were 11.1% (95% CI, 4.4% to 25.3%) and 16.1% (95% CI, 7.1% to 32.6%), which were 2- to 3-fold higher than that observed in all survivors. Among survivors exposed to high cumulative doses of alkylating agents (the third tertile) who developed subsequent sarcoma, the prevalence of HR gene mutation carriers was 30.8% (95% CI, 12.7% to 57.6%), more than 7-fold higher than that observed in all survivors. Finally, for survivors who developed subsequent thyroid cancer after high-dose neck RT (≥ 30 Gy) exposure, the prevalence of NER gene mutation carriers was 20.0% (95% CI, 5.7% to 51.0%), more than 9-fold higher than that in all survivors (Data Supplement).
Genetic Heterogeneity of DNA repair Gene Mutations in Subsequent Neoplasms
We observed substantial genetic heterogeneity and pleiotropic effects (Data Supplement). Although survivors who developed the same type of SN carry mutations in different DRGs (genetic heterogeneity), carriers of mutations in the same DRG developed different types of SNs (pleiotropy). For example, subsequent breast cancers developed in female survivors who carried mutations in HR genes (BRCA1, BRCA2, NBN, BRIP1, EME2, RECQL, and XRCC3), FA genes (BRCA1, BRCA2, and BRIP1), and NER genes (ERCC3 and GTF2H4), as well as 1 BER gene (POLG); subsequent sarcomas developed in those who carried mutations in HR genes (BRCA2, PALB2, BRIP1, POLQ, and RAD54L) and FA genes (BRCA2, PALB2, and BRIP1), as well as 1 MMR gene (PMS1), NER gene (XPC), and NHEJ gene (LIG4); subsequent thyroid cancer occurred in survivors who carried mutations in HR genes (BRCA2, EME1, RAD50, RAD54L, and UIMC1), FA genes (BRCA2 and FAN1), and NER genes (ERCC2, ERCC3, POLK, CDK7, and RPA3), as well as 1 MMR gene (MSH5), BER gene (POLG), and NHEJ gene (POLM).
DISCUSSION
Within this cohort of 4,402 SJLIFE survivors, approximately 11.5% were carriers of a pathogenic mutation in ≥ 1 DRG. These mutations were significantly associated with increased rates of subsequent female breast cancer, sarcoma, thyroid cancer, and NMSC among survivors, particularly within treatment- and age-specific subgroups. This finding is of great interest to the research and clinical communities, as it not only provides novel insights into genetic susceptibility underlying SN development but also identifies populations with substantially higher probability of carrying a pathogenic mutation in a DRG and populations at significantly increased SN risk.
Although DNA damage caused by genotoxic agents for cancer therapy cures primary childhood cancers by killing tumor cells, it also produces harmful genetic lesions within normal cells, which may lead to SNs.31 Thus, survivors with a pathogenic DRG mutation are at elevated risk for therapy-related SNs. The Data Supplement summarizes cancer therapy–associated genotoxicity with well-known clinical presentations.12-16 Mechanistically, the end products of DNA damage associated with specific anticancer therapies and their correction through specific repair pathways are consistent with and support our findings of increased SN rates among survivors who carry mutations in a specific DNA repair pathway and received specific genotoxic treatment.
Given the increased SN risk experienced by childhood cancer survivors, efforts to define those survivors at highest risk have potential to further inform clinical care recommendations and cancer surveillance practices. On the basis of our previous finding of a prevalence of 5.8% for P/LP mutations in 60 genes involved in autosomal dominant cancer predisposition syndromes with moderate to high penetrance, we recommended that all childhood cancer survivors be referred for genetic counseling.4 The associations observed within SJLIFE between pathogenic DRG mutations and SN risk add substantial new information regarding genetic contribution along with known treatment-related risks. Within the context of genetic counseling and potential clinical genetic testing of childhood cancer survivors, consideration should be given to assessing DRG mutations, especially among those survivors with treatment exposures known to increase the risk of female breast cancer, subsequent sarcomas, NMSC, and thyroid cancer. Establishing the presence of a genetic mutation in cancer predisposition and/or DRGs has potential to inform future precision medicine approaches for the treatment of high-risk patients, for example by avoiding or limiting exposure to radiation therapy.32 Moreover, mutation status may inform precision prevention/intervention strategies designed to reduce risk and/or provide early detection of SNs, beyond what is recommended in current clinical guidelines.33 Last, survivors’ genetic information may provide family members with valuable information about potential cancer risk to consider in decision-making about personal genetic testing and participation in strategies that facilitate early detection/prevention.
The interpretation of our findings should be considered with the following limitations. First, because WGS data were available only for participants who were alive at SJLIFE recruitment, mutations associated with increased mortality, either from a childhood cancer or SN, may be underrepresented. Thus, the mutation prevalence and associations between mutation carrier status and SN risk are likely underestimated in our analysis. Second, the increased SN risk among mutation carriers was associated with mutations in multiple genes. The substantial genetic heterogeneity and limited sample size prevented us from conducting more granular analyses of specific SN-gene associations and differentiating the contributions of mutations in genes associated with autosomal dominant syndromes with relatively higher penetrance versus those with autosomal recessive syndromes with relatively lower penetrance. Although excluding mutation carriers of 7 well-established cancer predisposition genes in HR, FA, and MMR pathways attenuated the associations of these pathways with specific SN rates, the directions of association remained consistent, suggesting that other DRGs also contributed but to a lesser extent. Third, our findings of higher SN rates among survivors with defined dose exposures were not based on a rigorous investigation of dose categories and their interactions with DRG mutations; additional research with larger populations is warranted to better determine the thresholds of treatment doses with strength of these genetic associations. Finally, because SJLIFE survivors are relatively young, with a median age of 36 years, SN risk in DRG mutation carriers may change with increasing age and extended follow-up.
Collectively, our findings provide compelling evidence of increased SN risk among childhood cancer survivors with DRG mutations and prior genotoxic treatment exposures. Thus, the results have the potential for informing future clinical recommendations for the broad range of medical professionals who provide health care for this growing population. Identifying survivors at the highest SN risk and implementing personalized cancer surveillance and prevention strategies may reduce the substantial morbidity and mortality associated with these outcomes.
ACKNOWLEDGMENT
We thank all the individuals who participated in this study; we also thank Dr Angela J. McArthur (St Jude Children’s Research Hospital) for scientific editing of this manuscript.
SUPPORT
Supported by funding from the American Lebanese Syrian Associated Charities and by Grants No. CA021765, CA195547, and CA216354 from the National Institutes of Health to St Jude Children’s Research Hospital. The funders of the study had no role in the design and conduct of the study: collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; or decision to submit manuscript for publication.
AUTHOR CONTRIBUTIONS
Conception and design: Zhaoming Wang, James R. Downing, Melissa M. Hudson, Jinghui Zhang, Leslie L. Robison, Yutaka Yasui
Financial support: Melissa M. Hudson, Leslie L. Robison
Administrative support: Zhaoming Wang, Melissa M. Hudson, Jinghui Zhang, Leslie L. Robison
Provision of study material or patients: Melissa M. Hudson, Leslie L. Robison
Collection and assembly of data: Zhaoming Wang, Carmen L. Wilson, Kyla Shelton, John Easton, Heather Mulder, Dennis Kennetz, Melissa M. Hudson, Jinghui Zhang, Leslie L. Robison
Data analysis and interpretation: Na Qin, Zhaoming Wang, Qi Liu, Nan Song, Carmen L. Wilson, Matthew J. Ehrhardt, Dennis Kennetz, Michael N. Edmonson, Michael C. Rusch, Melissa M. Hudson, Kim E. Nichols, Leslie L. Robison, Yutaka Yasui
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Pathogenic Germline Mutations in DNA Repair Genes in Combination With Cancer Treatment Exposures and Risk of Subsequent Neoplasms Among Long-Term Survivors of Childhood Cancer
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/authors/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
John Easton
Patents, Royalties, Other Intellectual Property: Co-inventor of patented technology–Primary Template Directed Amplification: https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019148119 (Inst)
Melissa M. Hudson
Consulting or Advisory Role: Oncology Research Information Exchange Network, Princess Máxima Center
Kim E. Nichols
Research Funding: Incyte/Novartis, Alpine Immune Sciences
No other potential conflicts of interest were reported.
REFERENCES
- 1.Robison LL, Hudson MM. Survivors of childhood and adolescent cancer: Life-long risks and responsibilities. Nat Rev Cancer. 2014;14:61–70. doi: 10.1038/nrc3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhakta N, Liu Q, Ness KK, et al. The cumulative burden of surviving childhood cancer: An initial report from the St Jude Lifetime Cohort Study (SJLIFE) Lancet. 2017;390:2569–2582. doi: 10.1016/S0140-6736(17)31610-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Turcotte LM, Liu Q, Yasui Y, et al. Temporal trends in treatment and subsequent neoplasm risk among 5-year survivors of childhood cancer, 1970-2015. JAMA. 2017;317:814–824. doi: 10.1001/jama.2017.0693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang Z, Wilson CL, Easton J, et al. Genetic risk for subsequent neoplasms among long-term survivors of childhood cancer. J Clin Oncol. 2018;36:2078–2087. doi: 10.1200/JCO.2018.77.8589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hudson MM, Ehrhardt MJ, Bhakta N, et al. Approach for classification and severity grading of long-term and late-onset health events among childhood cancer survivors in the St. Jude Lifetime Cohort. Cancer Epidemiol Biomarkers Prev. 2017;26:666–674. doi: 10.1158/1055-9965.EPI-16-0812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Torgovnick A, Schumacher B. DNA repair mechanisms in cancer development and therapy. Front Genet. 2015;6:157. doi: 10.3389/fgene.2015.00157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jeggo PA, Pearl LH, Carr AM. DNA repair, genome stability and cancer: A historical perspective. Nat Rev Cancer. 2016;16:35–42. doi: 10.1038/nrc.2015.4. [DOI] [PubMed] [Google Scholar]
- 8.Romero-Laorden N, Castro E. Inherited mutations in DNA repair genes and cancer risk. Curr Probl Cancer. 2017;41:251–264. doi: 10.1016/j.currproblcancer.2017.02.009. [DOI] [PubMed] [Google Scholar]
- 9.de Voer RM, Hahn MM, Mensenkamp AR, et al. Deleterious germline BLM mutations and the risk for early-onset colorectal cancer. Sci Rep. 2015;5:14060. doi: 10.1038/srep14060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Croitoru ME, Cleary SP, Di Nicola N, et al. Association between biallelic and monoallelic germline MYH gene mutations and colorectal cancer risk. J Natl Cancer Inst. 2004;96:1631–1634. doi: 10.1093/jnci/djh288. [DOI] [PubMed] [Google Scholar]
- 11.Thompson ER, Doyle MA, Ryland GL, et al. Exome sequencing identifies rare deleterious mutations in DNA repair genes FANCC and BLM as potential breast cancer susceptibility alleles. PLoS Genet. 2012;8:e1002894. doi: 10.1371/journal.pgen.1002894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shibata A. Regulation of repair pathway choice at two-ended DNA double-strand breaks. Mutat Res. 2017;803-805:51–55. doi: 10.1016/j.mrfmmm.2017.07.011. [DOI] [PubMed] [Google Scholar]
- 14.Fu D, Calvo JA, Samson LD. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat Rev Cancer. 2012;12:104–120. doi: 10.1038/nrc3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spencer DM, Bilardi RA, Koch TH, et al. DNA repair in response to anthracycline-DNA adducts: A role for both homologous recombination and nucleotide excision repair. Mutat Res. 2008;638:110–121. doi: 10.1016/j.mrfmmm.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 16.Figueiredo N, Chora A, Raquel H, et al. Anthracyclines induce DNA damage response-mediated protection against severe sepsis. Immunity. 2013;39:874–884. doi: 10.1016/j.immuni.2013.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stovall M, Donaldson SS, Weathers RE, et al. Genetic effects of radiotherapy for childhood cancer: Gonadal dose reconstruction. Int J Radiat Oncol Biol Phys. 2004;60:542–552. doi: 10.1016/j.ijrobp.2004.03.017. [DOI] [PubMed] [Google Scholar]
- 18.Green DM, Nolan VG, Goodman PJ, et al. The cyclophosphamide equivalent dose as an approach for quantifying alkylating agent exposure: A report from the Childhood Cancer Survivor Study. Pediatr Blood Cancer. 2014;61:53–67. doi: 10.1002/pbc.24679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wood RD, Mitchell M, Lindahl T. Human DNA repair genes. doi: 10.1126/science.1056154. https://www.mdanderson.org/documents/Labs/Wood-Laboratory/human-dna-repair-genes.html. [DOI] [PubMed]
- 20.Wood RD, Mitchell M, Sgouros J, et al. Human DNA repair genes. Science. 2001;291:1284–1289. doi: 10.1126/science.1056154. [DOI] [PubMed] [Google Scholar]
- 21.Wood RD, Mitchell M, Lindahl T. Human DNA repair genes, 2005. Mutat Res. 2005;577:275–283. doi: 10.1016/j.mrfmmm.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 22.Lange SS, Takata K, Wood RD. DNA polymerases and cancer. Nat Rev Cancer. 2011;11:96–110. doi: 10.1038/nrc2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Friedberg EC, Walker GC, Siede W, et al: DNA Repair and Mutagenesis, (ed 2) Washington, DC, ASM Press, 2006. [Google Scholar]
- 24.Riaz N, Blecua P, Lim RS, et al. Pan-cancer analysis of bi-allelic alterations in homologous recombination DNA repair genes. Nat Commun. 2017;8:857. doi: 10.1038/s41467-017-00921-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ceccaldi R, Liu JC, Amunugama R, et al. Homologous-recombination-deficient tumours are dependent on Polθ-mediated repair. Nature. 2015;518:258–262. doi: 10.1038/nature14184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tervasmäki A, Mantere T, Hartikainen JM, et al. Rare missense mutations in RECQL and POLG associate with inherited predisposition to breast cancer. Int J Cancer. 2018;142:2286–2292. doi: 10.1002/ijc.31259. [DOI] [PubMed] [Google Scholar]
- 27.Zhang J, Walsh MF, Wu G, et al. Germline mutations in predisposition genes in pediatric cancer. N Engl J Med. 2015;373:2336–2346. doi: 10.1056/NEJMoa1508054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Friedman M. Piecewise exponential models for survival-data with covariates. Ann Stat. 1982;10:101–113. [Google Scholar]
- 29.Gray RJ. A class of K-sample tests for comparing the cumulative incidence of a competing risk. Ann Stat. 1988;16:1141–1154. [Google Scholar]
- 30. R Foundation: The R project for statistical computing. http://www.R-project.org.
- 31.Pearce MS, Salotti JA, Little MP, et al. Radiation exposure from CT scans in childhood and subsequent risk of leukaemia and brain tumours: A retrospective cohort study. Lancet. 2012;380:499–505. doi: 10.1016/S0140-6736(12)60815-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kerns SL, Ostrer H, Rosenstein BS. Radiogenomics: Using genetics to identify cancer patients at risk for development of adverse effects following radiotherapy. Cancer Discov. 2014;4:155–165. doi: 10.1158/2159-8290.CD-13-0197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Landier W, Bhatia S, Eshelman DA, et al. Development of risk-based guidelines for pediatric cancer survivors: The Children’s Oncology Group Long-Term Follow-Up Guidelines from the Children’s Oncology Group Late Effects Committee and Nursing Discipline. J Clin Oncol. 2004;22:4979–4990. doi: 10.1200/JCO.2004.11.032. [DOI] [PubMed] [Google Scholar]