


Abstract
RNA-small molecule binding is a key regulatory mechanism which can stabilize 3D structures and activate molecular functions. The discovery of RNA-targeting compounds is thus a current topic of interest for novel therapies. Our work is a first attempt at bringing the scalability and generalization abilities of machine learning methods to the problem of RNA drug discovery, as well as a step towards understanding the interactions which drive binding specificity. Our tool, RNAmigos, builds and encodes a network representation of RNA structures to predict likely ligands for novel binding sites. We subject ligand predictions to virtual screening and show that we are able to place the true ligand in the 71st–73rd percentile in two decoy libraries, showing a significant improvement over several baselines, and a state of the art method. Furthermore, we observe that augmenting structural networks with non-canonical base pairing data is the only representation able to uncover a significant signal, suggesting that such interactions are a necessary source of binding specificity. We also find that pre-training with an auxiliary graph representation learning task significantly boosts performance of ligand prediction. This finding can serve as a general principle for RNA structure-function prediction when data is scarce. RNAmigos shows that RNA binding data contains structural patterns with potential for drug discovery, and provides methodological insights for possible applications to other structure-function learning tasks. The source code, data and a Web server are freely available at http://rnamigos.cs.mcgill.ca.
INTRODUCTION
Recent studies have identified small organic molecules as important non-covalent regulators of RNA function (1). These discoveries contribute to a better understanding of pathways present in all organisms, but also pose RNA molecules as a large class of promising novel drug targets. For example, Ribocil, which has recently been uncovered through a phenotypic assay to target the FMN riboswitch, is currently undergoing clinical trials as a novel antibiotic (2). Various other small molecule-activated RNA systems are also being proposed (3–5). Notable among these is the application to CRISPR activation regulation (6). The list of possible therapies is likely to expand given the observations of KD Warner et al. that only a small fraction of the genome is translated into protein (1.5%) while the vast majority is transcribed into potentially druggable non-coding RNA (70%) (7).
RNA structural organization
RNAs possess multiple levels of structural organization which together determine function, and by extension, ligand binding ability. At the simplest level, RNA is a string of monomers 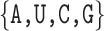 linked by a chain of covalent bonds known as the backbone. This is commonly known as as the primary structure of RNA. Non-covalent pairwise interactions between nucleotides (bases) in the chain give rise to the secondary and tertiary structure. Canonical pairs (i.e. A–U, C–G) give rise to the secondary structure. Notably, these pairs form loops and stacks (helices), assembling a stable scaffold for the full structure (8). The experimental determination of binding energies for these pairs (9) prompted a boom of algorithms for sequence to secondary structure prediction such as RNAfold, (10). In seminal work, Leontis and Westhof identified 11 additional types of base pairing occurring in 3D structures (11,12), known as non-canonical base pairs. These interactions can occur between any pair of nucleotides and are defined by the relative orientations of three faces of the interacting bases in 3D. By considering all combinations of faces and a cis and trans orientation, we arrive at 12 possible base pairing geometries. Whereas canonical pairs form stable helices, non-canonical pairs are typically found in loops (i.e. regions without canonical pairs) and create more complex structural patterns (13,14). These pairings fine-tune RNA function by defining structure at the 3D level (15). Interestingly, non-canonical pairs were also found to be enriched in ligand binding sites (16,17), which corroborates with the observation that increased structural complexity is associated with binding specificity (7).
linked by a chain of covalent bonds known as the backbone. This is commonly known as as the primary structure of RNA. Non-covalent pairwise interactions between nucleotides (bases) in the chain give rise to the secondary and tertiary structure. Canonical pairs (i.e. A–U, C–G) give rise to the secondary structure. Notably, these pairs form loops and stacks (helices), assembling a stable scaffold for the full structure (8). The experimental determination of binding energies for these pairs (9) prompted a boom of algorithms for sequence to secondary structure prediction such as RNAfold, (10). In seminal work, Leontis and Westhof identified 11 additional types of base pairing occurring in 3D structures (11,12), known as non-canonical base pairs. These interactions can occur between any pair of nucleotides and are defined by the relative orientations of three faces of the interacting bases in 3D. By considering all combinations of faces and a cis and trans orientation, we arrive at 12 possible base pairing geometries. Whereas canonical pairs form stable helices, non-canonical pairs are typically found in loops (i.e. regions without canonical pairs) and create more complex structural patterns (13,14). These pairings fine-tune RNA function by defining structure at the 3D level (15). Interestingly, non-canonical pairs were also found to be enriched in ligand binding sites (16,17), which corroborates with the observation that increased structural complexity is associated with binding specificity (7).
These observations, together with the well-studied role of secondary structure in RNA ligand binding (18), led us to hypothesize that studying RNA structures at the augmented base-pairing level (i.e. including non-canonical pairs) holds useful spatial and chemical information about ligand binding. However, studying RNA at this level of structure comes with major algorithmic challenges, such as the lack of binding energies and more complex interaction patterns. For these reasons, non-canonical interactions are typically modeled with statistical methods, and represented using more general data structures such as graphs (19). In practice, this means that a graph using vertices to represent nucleotides and multi-relational edges to encode base-pairing interactions could offer a signature for RNA ligand binding sites (see Figure 1 for an example of a binding site and its associated base pairing network). We call this graphical representation of RNA sites annotated with canonical and non-canonical interactions an Augmented Base Pairing Network (ABPN) since we consider base pairs beyond the canonicals. Indeed, similar representations of RNA base pairing networks have been exploited in various tools (14,19–21) for their ability to capture RNA-specific interactions in an interpretable manner. This paradigm distinguishes RNA from protein–ligand interactions where surface-cavity topologies tend to drive binding preferences (22), hence direct use of atomic coordinates can be more appropriate.
Figure 1.

(A) Binding site atomic coordinates. (B) Graph encoding of binding site as an augmented base pairing network (ABPN). RNA structure representation of the THF riboswitch binding site (PDB: 4LVV) as atomic coordinates using UCSF Chimera (23)(left) and resulting augmented base pairing network (ABPN) (right). We superpose the ABPN in the 3D visualization. Nodes are drawn as white spheres, backbone connections are in white, and canonical and non-canonical base pairs are green and red tubes respectively. We color the edges simply to guide the eye to the corresponding base pairs but note that edge color has no special meaning to our graphs. We annotate the graph representation with the standard Leontis-Westhof nomenclature for pairing type symbols. In this case, the binding site has three canonical interactions denoted (•), and three non canonicals of types (□○, ▹, □▹).
Structure-based drug discovery and RNA base pairing networks
The central aim of structure-based drug discovery is to identify compounds with high affinity to a given site or set of binding sites. A natural problem to address in this context is the prediction of binding affinity from a binding site–ligand pair. Machine learning models which solve this task can be used as alternatives to computationally expensive docking simulations to screen ligand databases for strong binders (24). And in some cases have shown superior performance to methods built on explicit chemicophysical knowledge (25). This setting is quite feasible in the protein domain as affinities and drug screens are abundant, hence various methods have been proposed (26). Recently, some repositories of RNA small molecule data have been made public (27) however, only a handful of binding affinities are known. Given a pose for a ligand inside a binding site, various scoring approaches have been proposed; DrugScoreRNA, and LigandRNA (28,29), SPA-LN (30) which are built on a priori chemical knowledge and rely on accurate docking. While RNA docking methods which search for the optimal pose in a binding site are still showing limited success (22,31).
The fundamental commonality in these tools is that they all require a binding site and ligand as input. Therefore, identifying a binder consists of docking and scoring all combinations of RNA and small molecules from a desired library in all putative poses, which can be prohibitive. In this work, we ask whether base pairing patterns, which creates a scaffold for the 3D structures and is easier to obtain, can be used to accelerate these searches. Or in other words, if a coarse-grained representation of RNA structures provides sufficient information about potential ligands.
To our knowledge, the closest contribution to this work is a template-based approach named Inforna (32). Inforna searches through an input sequence and secondary structure for motifs that are similar to those found in a library of small-molecule structural binding motifs, and return candidate ligands. Here, we propose two major innovations to such approaches. First, we take the first step towards learning a generalizable RNA binding landscape that can be used to infer compounds which are not explicitly present in compound libraries. Previous contributions have shown success in using protein 3D structures information to reach this objective in proteins (25,33–35), but this is to our knowledge the first attempt to apply a similar strategy to RNAs. Next, because we also aim to leverage the specificity of the RNA structural organization, we investigate the impact of higher-order base pair interactions (beyond classical secondary structure), which has yet to be explored.
Contribution
RNAmigos brings together domain knowledge of RNA structure, currently available crystal structure data, and graph neural networks, to show that base pairing networks can be used to automatically predict ligands for RNA structures. Importantly, we propose the use of Augmented Base Pairing (ABPNs) networks, an enriched alphabet of base pairing interactions, and demonstrate that they are a necessary component for capturing binding signatures. Molecular fingerprints predicted by RNAmigos serve as ligand search tools across diverse ligand classes and show strong performance in two different ligand screens, as well as compared to a state of the art method, Inforna (32). Additionally, we explore the use of an unsupervised graph representation learning scheme for boosting model performance in this low-data setting. The implications of our work are 2-fold (i) we show for the first time that we can learn from non-canonical interaction data to make predictions about RNA function and (ii) our ability to enrich for actives in compound libraries shows potential for RNAmigos as an upstream filtering step for more fine-grained drug discovery tools such as docking. The core implementation of RNAmigos is built in Pytorch (36) and DGL (37) and is available as an open source Python 3.6 software package.
MATERIALS AND METHODS
Model overview
Our model (RNAmigos) seeks to identify possible ligands for a given coarse-grained representation of an RNA binding site (see Figure 2). More precisely, our input is an ABPN modelling the RNA structure, from which we predict a molecular fingerprint for a potential ligand. This fingerprint can be used to search a library of compounds for active binders. We train RNAmigos on RNA-ligand pairs found in the RCSB PDB Data Bank (38), and use graph neural networks (39) to learn the relationship between RNA structure and ligand binding preferences.
Figure 2.
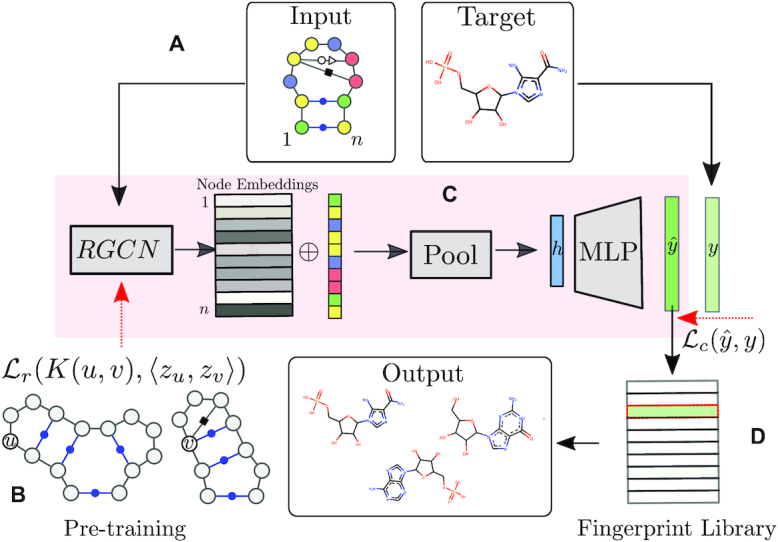
Outline of the RNAmigos pipeline. A base pairing network is passed as input to RNAmigos. In training mode, it is paired with a native ligand (Target) from which a target fingerprint y is constructed. The embedding network (RGCN) produces a matrix of node embeddings of dimension n × d where n is the number of nodes in the graph, and d is a fixed embedding size. This is followed by a pooling step which reduces node embeddings to a single graph-level vector. Finally, the graph representation is fed through a multi-layer perceptron (MLP) to produce a predicted fingerprint 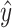 that minimizes the distance
that minimizes the distance 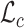 to the native fingerprint y. The fingerprint is then used to search for similar ligands to the prediction in a ligand screen and thus enriches the probability of identifying an active compound. The RGCN network is pre-trained using an unsupervised node embedding framework which allows us to leverage structural patterns from a large dataset of RNA structures. This network is trained to generate embeddings which minimize the distance (
to the native fingerprint y. The fingerprint is then used to search for similar ligands to the prediction in a ligand screen and thus enriches the probability of identifying an active compound. The RGCN network is pre-trained using an unsupervised node embedding framework which allows us to leverage structural patterns from a large dataset of RNA structures. This network is trained to generate embeddings which minimize the distance (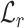 ) between kernel similarities k(u,
) between kernel similarities k(u, 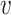 ) and embedding similarities 〈
) and embedding similarities 〈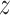 u,
u, 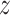
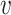 〉.
〉.
Dataset preparation
We begin by collecting a set of RNA-small molecule complexes from the PDB Data Bank (38). We download all crystal structures (90% identity threshold) which contain RNA and at least one ligand. This results in 2993 PDB structures. We omit ions such as magnesium (Mg+) from the set of valid ligands as they vastly outnumber organic ligands and likely require customized models. (40) We choose a maximum allowable distance between any ligand atom and any RNA atom of 10 Angstroms according to David-Eden et al. (16) which statistically characterized ribosome antibiotic binding sites. The number of valid sites is further reduced when we remove binding sites with fewer than five RNA residues and remove binding sites containing a large proportion of protein residues, (See Supplementary Figure S1). The final training set consists of 773 binding sites associated to 270 unique ligands.
Finally, we build an ABPN from the 3D structure of each binding site identified in the previous step. In the ABPN, each node corresponds to a residue in the binding site, and links/edges are formed between nodes if they form a backbone or base pair interaction. Node and edge annotations are taken from the BGSU RNA 3D Motif Atlas (14) database which maintains base pairing annotations of all PDBs with Leontis-Westhof and backbone interaction types computed by the software FR3D (41). In this manner, each ABPNs stores the nucleotide identity (A, U, C, G) of each of its residues as a node attribute, and each base pairing interaction corresponds to an edge with one of 13 different types (backbone + 12 base pairing geometries). The resulting graphs are on average 15.76 nodes in size. At this point, the ligand is removed from the structure so that the graph contains only RNA base-pairing information. While atomic coordinates are the current source of data, we highlight that a key feature of taking ABPNs as input is that we can eventually learn from many other sources of ABPN data which are easier to obtain than crystal structures. A promising example comes from recent developments in predicting base pairing networks from RNA sequences in high-throughput (21,42). Our model would then be able to directly use such predictions once they are linked to a functional label (such as a ligand in this case). For full details on binding site extraction and graph construction, see Supplementary Material S1.1.
Fingerprint prediction
Given a binding site, our model predicts a set of chemical features which can be used to identify a ligand. This set of features is typically known as a molecular fingerprint (43). Many approaches to compute fingerprints from chemical structures have been developed; all with the common aim of numerically encoding chemicals (44–46). Such encodings greatly facilitate searches for similar compounds in databases and screens. In this work, we use a common fingerprint implementation known as the MDL Molecular Access Keys (MACCS) fingerprint (47) which has the advantage of providing compact and interpretable entries. For a given chemical compound c, the MACCS fingerprint fc is a 166 bit binary vector where each entry indicates the presence or absence of a chemical property. For the ith chemical property, fc[i] is set to 1 if the chemical property is present and is 0 otherwise. We use the set of 166 predefined chemical properties from the Pybel (48) implementation as a target vector for our model. We emphasize that the computation of the fingerprint depends only on the chemical composition of the ligand and not on the RNA binding site. The main objective of our model is to predict the set of chemical features (fingerprint) that is close to that of the co-crystallized ligand using only RNA base pairing networks. For convenience, we call the ligand co-crystallized with a given site its native ligand.
Model architecture
Since a key feature of our ABPNs is the fact that we encode base pairing geometry as an edge category (or relation type) in a graph, we use a Relational Graph Convolutional Network (RGCN) (39) as the core of the fingerprint prediction model (see Figure 2). An RGCN is a specialized neural network which acts directly on graphs, allowing us to naturally model ABPN structures. Here, a nucleotide is associated with a node and a base pair interaction represented by an edge. At a high level, the RGCN computes an encoding for each node, known as a node embedding. Formally, we denote a node embedding for node i as a d-dimensional real-valued vector, 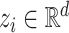 . The notion of a node embedding can be understood in a similar manner to molecular fingerprints. Each entry of the vector numerically encodes a feature of the node and its neighbourhood (i.e. the nucleotide).
. The notion of a node embedding can be understood in a similar manner to molecular fingerprints. Each entry of the vector numerically encodes a feature of the node and its neighbourhood (i.e. the nucleotide).
We can choose the embeddings such that they maximize performance on some classification task (supervised; analogous to image classification), or to capture structural similarity relationships (unsupervised; analogous to dimensionality reduction, and molecular fingerprints). More formally, a supervised task is one where each training point has an associated external label (i.e. the feature we want to predict). In our case, the native ligand acts as a label for the binding site. On the other hand, an unsupervised task is one where we only have the input data but no ground truth; and the task becomes to compute the best possible classification of the data points. We will therefore additionally train a model to recognize structurally similar RNA neighbourhoods via unsupervised node embedding techniques.
In this work, we propose a pipeline that combines supervised and unsupervised node embedding methods to best represent ABPN structure and maximize predictive performance. Figure 2 provides an overview of our system. Given an ABPN, (Figure 2A) we use an RGCN to compute an embedding for each node, to which add the identity of the corresponding nucleotide. In this manner, node embeddings represent structure and sequence identity. The node embedding RGCN is pre-trained using an unsupervised structure encoding task (Figure 2B). Since our task is to associate the entire ABPN with a molecular fingeprint, we use a pooling process (Figure 2C), which aggregates node-level embeddings into a graph-level (binding site) representation. The final graph-level representation (the vector h in Figure 2C), is fed through a simple neural network to output the final fingerprint. The entire network is trained to minimize the difference between the predicted fingerprint  and the native fingerprint y using a standard binary cross-entropy loss. Finally, we evaluate our predictions by using the predicted fingerprint to identify the native ligand from a compound library (Figure 2D). See Supplemental Material S2 for a full description of the neural network.
and the native fingerprint y using a standard binary cross-entropy loss. Finally, we evaluate our predictions by using the predicted fingerprint to identify the native ligand from a compound library (Figure 2D). See Supplemental Material S2 for a full description of the neural network.
Unsupervised pre-training: ABPN node embeddings
Since RNA-small molecule binding events are relatively infrequent in the set of RNA 3D structures, the number of training points for ligand prediction (supervised learning) is limited. However, we are still able to leverage the full set of RNA 3D structures (3,972 full RNA structures versus 773 binding sites) using unsupervised pre-training, which is known to boost performance when labeled data is scarce (49). Recent methods have been developed for unsupervised learning on network data (50,51). As described in the preceding section, node embeddings can be trained to maximize performance on a prediction task (e.g. ligand prediction), or an unsupervised task (encoding similarity relationships, e.g. molecular fingerprints). In our case, we would train an RGCN to simply produce similar embeddings for RNA nodes with similar local structures. This would define a learning task on RNA structures for which we don’t have a label (native ligand).
This process is analogous to molecular fingerprint building, where we wish to numerically encode structural similarity relationships. Once the RGCN has learned to encode the local structure of each node, the downstream task of ligand prediction becomes less prone to overfitting and more likely to learn general patterns (49). In the unsupervised setting, we train a model to produce embeddings for a pair nodes such that the similarity between the embeddings 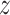 u and
u and 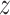
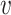 is proportional to a user-defined similarity measure K which compares nodes u and
is proportional to a user-defined similarity measure K which compares nodes u and 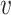 in the graph.
in the graph.
We are free to choose the pairwise node similarity function K: (u, 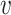 ) → [0, 1] according to the application domain.
) → [0, 1] according to the application domain.
Here, we adapt the node similarity function proposed in struc2vec (52) which allows us to capture local structural similarity across graphs. Other node similarity functions such as the ones used in GraphClust for RNA 2D structures (53) are only able to compare nodes within the same graph and are affected by the distance between nodes which is not necessarily related to structural identity. Our similarity function addresses these limitations by comparing the counts of edge types in the local neighbourhood of u and 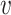 . We provide an example of a comparison between a pair of two nodes on simplified graphs in Figure 3 and show the result on a sample ABPNs in Supplementary Figure S3.
. We provide an example of a comparison between a pair of two nodes on simplified graphs in Figure 3 and show the result on a sample ABPNs in Supplementary Figure S3.
Figure 3.
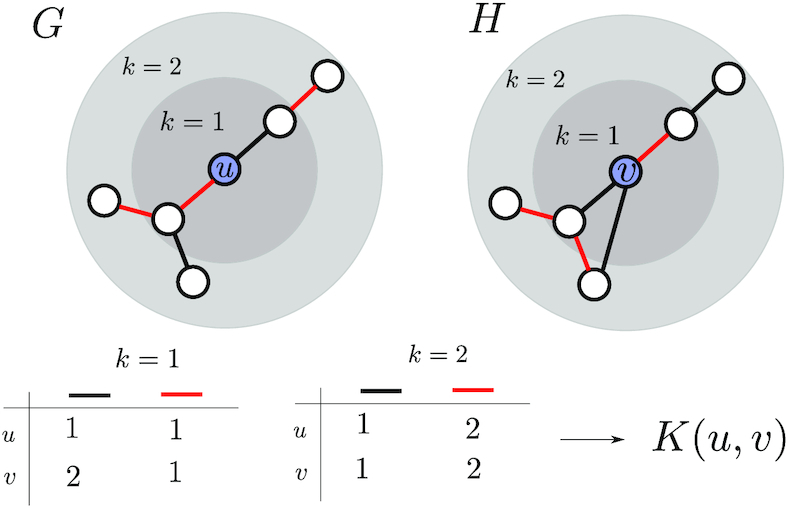
Here, we compare the local neighbourhoods of node u in graph G and node 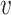 in graph H. In this simple example, graphs only have one of two possible edge types, red and black. We compare the distributions of edge labels at each distance from the source nodes (u and
in graph H. In this simple example, graphs only have one of two possible edge types, red and black. We compare the distributions of edge labels at each distance from the source nodes (u and 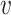 ) to obtain the final similarity value K(u,
) to obtain the final similarity value K(u, 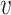 ).
).
Therefore, in the first training phase, our network tries to learn node embeddings on a large data set which are aware of general RNA structural patterns. Once this phase has converged, the model is then asked to predict ligand fingerprints.
Ligand screen
Here, we propose a test to interpret the usefulness of our model by measuring its performance in a ligand screen setting. In a ligand screen, we are given a set of compounds and we seek to identify the most promising one. For validation, we know a native binder and we hide it in a set of inactive compounds, also known as as decoys. The model is asked to find back the active. Given a binding site, our model produces a predicted fingerprint. We then rank all compounds of the decoy set according to distance to the predicted fingerprint. We normalize this score by the size of the set. Thus, a successful predictor will rank the native ligand as closest to its prediction (normalized rank close to 1), while a random predictor will result in an average rank of 0.5.
Considering that the distribution of RNA ligands appears to cluster to specific sub-regions (see Supplementary Figure S2), this evaluation method also ensures that a classifier does not obtain a good score by simply predicting the average ligand as it would when only considering the absolute distance between the predicted and the native fingerprints.
We construct two decoy sets for our experiments. Since there are currently no experimentally validated data sets of active and inactive binders for a given RNA site (such as DUDE for protein (54)), our first set consists of all RNA-binding ligands in the PDB (270 ligands). The second decoy set is constructed using DecoyFinder (55) on default settings, which samples a list of 36 decoys for each compound such that generic chemical properties are preserved while potentially disturbing binding potential. Of course, this test assumes that chemical dissimilarity between an active compound implies inactivity which is not always the case (56). However, the current aim of our work is simply to determine whether ABPNs retain significant amount of information about its observed ligand preferences, for which this test is sufficient.
RESULTS
We report resulting rank over the list of all RNA-small molecule pairs as well as the set of all decoys for each ligand, following the two decoy benchmark process.
Due to the limited size of our labeled data set, we performed a 10-fold cross-validation to include all training pairs in the evaluation and provide a more accurate measure of performance. All results are reported from the held-out sets in our validation, hence the model is never trained on the same binding sites that are being predicted on.
Node embeddings are computed using a three-layer RGCN, each layer consisting of 16 dimensional inputs and outputs, a graph attention layer computing a 16-dimensional graph embedding and a fully-connected layer, which outputs a 166-dimensional vector. See Supplementary Table S1 for full model architecture and hyperparameters. Variations of the architecture used did not have strong effects on performance, so no extensive hyper-parameter search was conducted. We leave the exploration of other architecture choices for future work.
Augmented RNA base pairing networks encode binding preferences
Setting
The first hypothesis to test is that the proposed framework (ABPN) is able to retrieve some information about ligand binding. To explore this question, we compute the rank and distance metrics on ablated data. We compare this performance to three baselines:
random consists of a synthetic label set where each binding site is assigned a uniformly random 166-dimensional binary vector (fingerprint).
swap is designed to account for imbalances in the data (some ligands are more frequent than others): each binding site is assigned a fingerprint selected at random from the set of observed fingerprints. The overall distribution of ligand fingerprints thus remains the same but the input-output correlations are broken.
majority is a constant ligand annotation computed as a majority vote over all fingerprints at each index. This is to be compared to the swap to check that the only thing that can be learnt on swapped data is over-representation of some ligands within the experiment.
The distributions of performance over each binding site–ligand pair is visualized for all experiments in Figure 4 as a box plot. Summary statistics can be found in Table 1 with accompanying standard deviations in Supplementary Table S2, and Euclidean distances from the native ligand in Supplementary Figure S5. We also assessed the statistical significance of the difference of the means in a pairwise Wilcoxon rank test which is shown in Table 2.
Figure 4.
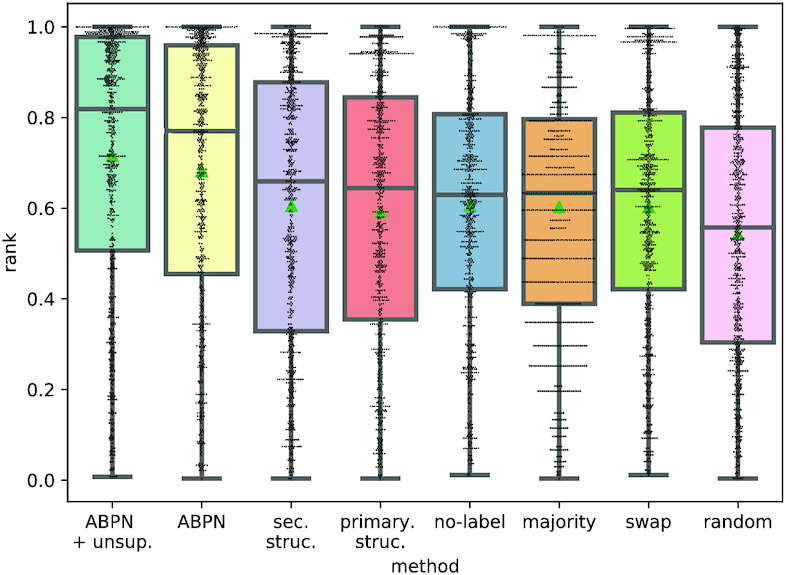
Distribution of rank achieved on ligand screening. All points are from test set data on a 10-fold cross validation. The median is denoted with a dashed line and the mean with a green triangle. Each point is the normalized rank of one binding site’s native ligand when searching for it using our network’s predicted fingerprint.
Table 1.
Mean ligand screen ranks and L2 (Euclidean) distance achieved on held-out binding sites for each experiment and decoy set
| Experiment | Ranks | L2 | ||
|---|---|---|---|---|
| DecoyFinder | RNA | DecoyFinder | RNA | |
| Random | 0.611 | 0.542 | 0.502 | 0.502 |
| Majority | 0.621 | 0.603 | 0.175 | 0.179 |
| Swap | 0.617 | 0.603 | 0.177 | 0.179 |
| No-label | 0.628 | 0.606 | 0.176 | 0.180 |
| Primary | 0.624 | 0.592 | 0.181 | 0.186 |
| Secondary | 0.631 | 0.605 | 0.178 | 0.182 |
| ABPN | 0.695 | 0.681 | 0.155 | 0.160 |
| ABPN + unsup. | 0.735 | 0.715 | 0.145318 | 0.150189 |
Table 2.
Wilcoxon rank test for all pairs of training conditions. Each entry in the table is the P-value for testing the hypothesis that the ranks resulting from a pair of experiments come from the same distribution. These are performed on the RNA decoy set. We provide the test results for the DecoyFinder decoy set in Supplementary Table S3 material and show consistent results.
| Experiment 2 | aBPN | Secondary | Primary | No-label | Majority | Swap | Random |
|---|---|---|---|---|---|---|---|
| Experiment 1 | |||||||
| ABPN + unsup. | 2.9e–06 | 5.1e–26 | 1.4e–22 | 2.1e–21 | 9.3e–25 | 7.2e–26 | 2.3e–18 |
| ABPN | – | 1.7e–11 | 5.6e–11 | 1.5e–08 | 4.3e–10 | 6.4e–12 | 2.0e–08 |
| Secondary | – | 3.2e–01 | 7.7e–01 | 1.3e–01 | 2.8e–02 | 1.7e–01 | |
| Primary | – | 4.3e–01 | 2.7e–01 | 2.4e–02 | 3.2e–01 | ||
| No-label | – | 5.5e–01 | 1.5e–02 | 1.8e–01 | |||
| Majority | – | 3.7e–01 | 3.3e–01 | ||||
| Swap | – | 5.5e–01 |
Performance
In the RNA ligands setting, our full model achieves a rank of 0.68 and an mean-squared error (MSE) of 0.150 to the native fingerprint. The random, swap and majority experiments respectively yield ranks of 0.542, 0.603 and 0.603 and mean squared errors (MSEs) of 0.5, 0.18 and 0.18. This confirms that this model retrieves signal for the data and outperforms baselines. This conclusion is statistically significant based on a Wilcoxon P-value of at most 7e−18 between the model and the randomized results. As expected, the majority scheme is statistically equivalent to the swapped one and superior to the random one. These results are similar in the DecoyFinder (See Supplementary Figure S4) setting where the mean rank of the model is 0.69 compared to 0.62 in the majority setting. This shows that the full model successfully retrieves some signal and outperforms the baselines (Wilcoxon test results for DecoyFinder are shown in Supplementary Table S3).
Augmented base pairing networks encode ligand binding information
Next, we test the hypothesis that robust descriptors in the form of ABPNs from RNA domain knowledge are key to retrieve this signal. The question is whether the non canonical interactions encode information that lower levels of structure (secondary, primary) do not. We answer this question by performing three ablation experiments on our training set:
primary encodes the binding sites as graphs that only contain node sequence and backbone interactions.
secondary uses only information from the secondary structure which includes canonical pairs and backbones.
no-label preserves all the interactions (and thus graph structure) in the graph (including non canonical) but do not distinguish between different edge types (i.e. edges only have one label).
In all these conditions, we find that performance is no better than the randomized baselines, indicating that non canonical interactions are essential for encoding specificity in ligand binding.
Indeed, the best performing model is no-label which has a Wilcoxon P-value of 0.55 with the majority experiment and of 1.7e−18 with the ABPN. This finding is in agreement with biological literature on RNA binding sites and the importance of complex structural motifs for determining functional specificity (7,16). This is a major validation of the hypothesis that these are the correct representation for RNA structure for this task.
Unsupervised pre-training boosts performance
As explained in ‘Unsupervised Pre-Training: ABPN Node Embeddings’, one major limitation for this supervised task is the paucity of data. We investigated the possibility of using unsupervised learning by pre-training on an unsupervised task, and denote this experiment as ABPN unsup. The use of unsupervised pre-training of the node embedding network provides a significant performance boost over a network trained only on fingerprint reconstruction (MSE = 0.68 versus MSE = 0.715), with a P-value of 2.9e−6 This is a methodological insight that can have applications for various other RNA-related tasks for which labeled data is typically scarce.
Our model can predict diverse ligand classes
Next, we ask whether the positive results can be explained by a small set of ligands, or whether it is able to achieve high scores on a diverse set of ligands. To get a better view of performance, we plot the same prediction scores but averaged over ligand types (270 unique ligands) against a hierarchical clustering dendrogram of each ligand (shown in Figure 5).
Figure 5.

RNAmigos performance by ligand class. Hierarchical clustering dendrogram of the ligands, classifying ligand families by similarity. Each cell in the horizontal grid is the average score for binding sites containing a given ligand. Ligands belonging to the same tree are grouped together by the clustering procedure. Colored-in sub-trees denote tight clusters which contain ligands within 0.25 Jaccard distance.
Colored-in subtrees indicate groups of ligands that are similar, (i.e. within 0.25 Jaccard distance of each other) which would indicate strong clustering. In this manner, we are able to assess the performance across ‘classes’ of similar ligands. We first observe that successful classifications are not restricted to a single class of ligands and instead show good predictions for diverse ligands. Interestingly, the class that is most consistently predicted accurately corresponds to the aminoglycosides (highlighted in the green cluster in the middle). Aminoglycosides are a class of antibiotics binding to bacterial RNA with well-defined binding sites (57), and are quite abundant in the dataset. Nucleic acid-like compounds, many of which bind riboswitches, also form a large family of binders (green) however results were less consistent than for aminoglycosides. A possible explanation for strong performance on aminoglycosides, apart for the larger number of examples obtained, is that these are typically large polysaccharide-like structures with a large number of interactions with the RNA. On the other hand, riboswitches bind much smaller molecules with a limited number of interactions. As a result, binding site requirements are much more complex and specific with aminoglycosides and the large number of interactions can only be fulfilled by a limited number of molecules. We leave this question for future work, as with the current dataset size, we are unable to provide quantitative evidence of such phenomena. Finally, ligands clustered on the left of the dendrogram show the weakest performance. Since these groups show little branching in the dendrogram, we can conclude that they represent sparsely populated ligand classes for which we have few examples and thus, obtaining more data in these regions could improve performance.
Comparison with a secondary structure-based tool
Finally, we compare the performance of RNAmigos with the closest related tool we could find, Inforna (32). Inforna accepts as input a RNA sequence with a secondary structure and returns a list of candidate ligands, based on sequence and structural similarities with motifs stored in a database. Although the input is not strictly identical, it is quite close to the one of RNAmigos. Similarly for the output, Inforna provides direct ligand information. In this benchmark, we provide to Inforna secondary structures computed with Forgi (58) directly from the PDB files, which is the most accurate input available.
For each chain in each RNA PDB associated with a ligand in the PDB databank set, we query the Inforna web server and obtain a list of candidate ligands that we use to search for the native ligand (see Supplementary Table S4). Importantly, in contrast to RNAmigos for which we directly provide the binding site, Inforna scans the whole input structure for candidate sites. To address this discrepancy, we only take the maximum score returned by Inforna for each structure.
We show the results of our benchmark in Figure 6 (see Supplementary Figure S6 for corresponding distance comparisons). We were able to obtain predictions for 176 unique RNA chains corresponding to 82 unique ligands. The reduction from the RNAmigos set is maily due to excluding PDBs which contain protein and some secondary structure extraction failures. In this context, Inforna achieves an average enrichment at the level of our random model (mean rank 0.43, and distance 0.48), and to some extent also consistent with the performance of RNAmigos using only secondary structure information.
Figure 6.
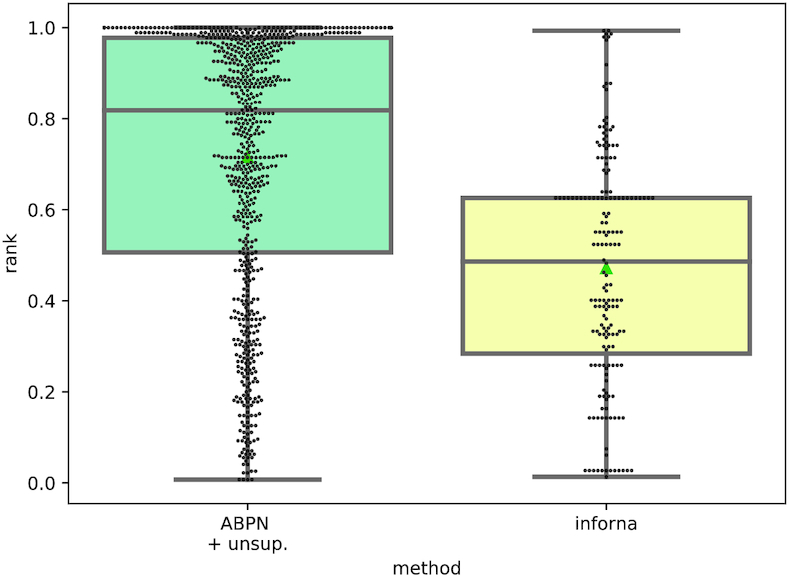
Distribution of native ligand rank achieved on ligand screening in RNAmigos and Inforna.
We analyzed the performance across ligand classes (see Supplementary Figure S7) and observed that the accuracy of the predictions appears to be stronger in well-known classes such as aminoglycosides and riboswitch ligands, but the performance decreases sharply outside these classes. This phenomenon could highlight a shortcoming of non-generalizable models, and thus a benefit of our approach. Looking at ligand classes where both tools made predictions (Supplementary Table S4), we observe that RNAmigos outperforms Inforna in nearly all classes (Inforna outperformed RNAmigos on 10 of 66 ligands tested on Inforna by a margin larger than 0.1.). It suggests that the richer structural representation leveraged by RNAmigos is an important source of specificity. Since both tools work with differing levels of representation (2D versus augmented 2D) and at different scales (binding site versus full sequence), we stress that this benchmark do not intend to be a direct comparison but rather a demonstration that higher-level interactions are a crucial source of information.
DISCUSSION
We have developed a unique computational platform, RNAmigos, to show that augmented RNA base pairing networks contain useful ligand binding information. The significance of our results is 2-fold.
We show for the first time that ABPNs encode sufficient information for a classification task, and establish an initial methodological primitive for such a task. To date, the majority of computational methods which leverage ABPNs have focused on sequence to structure (21,59) prediction and motif identification (14,20). While these tasks involve some degree of learning, the relevance of higher-order interactions lies ultimately in their potential to specify function, which until now has been left unexplored. Interestingly, these findings come at a time when information of the type our model uses is becoming more widely available. Computational prediction tools such as (21,59) promise to yield large amounts of higher-order RNA pairwise interaction data without need for costly crystallography experiments. This opens the door to applying such data in other important biological problems such as RNA binding protein prediction (60) and ion binding (40). Furthermore, the promising results obtained from the unsupervised pre-training provide a methodological building block for assisting in supervised learning on complex RNA structures.
Second, our findings take a first step towards learning-based methods for systematically identifying drugs binding to RNA, and pinpoint ABPNs as essential tools for this task. The finding that only ABPN representations of binding sites was able to produce a significant signal in the task indicates that richer representations are necessary for successful classification when complex interactions are at play. Since our prediction is a fingerprint vector (chemical descriptor) and not a simple classification of ligands (i.e directly selecting a single ligand as output, or predicting an affinity), the fingerprint itself can be used to search large ligand databases, and can eventually be applied to direct molecule generation (25). While performance was strong across different ligand classes, it is apparent that classes for which data is more abundant received more consistently positive predictions. Therefore, as more examples of RNA–ligand complexes are characterized by experimental and computational techniques, we believe that the performance of our platform will improve. Additional data will also allow us to account for properties desired in medical applications such as synthesizability, and drug-likeness (61). Our choice of graphs for binding site representation reflects this consideration, as graphs can natively hold additional information such as evolutionary or chemical properties without requiring changes to the pipeline. Furthermore, recent advances in graph neural networks would provide the ability to model binding site flexibility (62). Eventually, computational predictions of ABPNs from sequence (21) combined with our methods will enable large-scale searches for binding sites.
We hope that this work will motivate further investigation of the links between ABPNs and RNA function, and eventually facilitate efforts in RNA targeted drug discovery.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Mathieu Blanchette and Jacques Boitreaud for helpful feedback and discussions.
Contributor Information
Carlos Oliver, School of Computer Science, McGill University, Montreal H3A 0E9, Canada; Mila - Quebec Artificial Intelligence Institute, H2S 3S1, Canada.
Vincent Mallet, Institut Pasteur, Structural Bioinformatics Unit, Paris, F-75015, France; MINES ParisTech, PSL Research University, CBIO - Centre for Computational Biology, F-75006 Paris, France.
Roman Sarrazin Gendron, School of Computer Science, McGill University, Montreal H3A 0E9, Canada.
Vladimir Reinharz, Department of Computer Science, Université du Québec à Montréal, Montreal H2X 3Y7, Canada.
William L Hamilton, School of Computer Science, McGill University, Montreal H3A 0E9, Canada; Mila - Quebec Artificial Intelligence Institute, H2S 3S1, Canada.
Nicolas Moitessier, Department of Chemistry, McGill University, Montreal H3A 0B8, Canada.
Jérôme Waldispühl, School of Computer Science, McGill University, Montreal H3A 0E9, Canada.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Natural Sciences and Engineering Research Council of Canada; Canadian Institutes of Health Research; Genome Canada. Funding for open access charge: NSERC Discovery grant.
Conflict of interest statement. None declared.
REFERENCES
- 1. Donlic A., Hargrove A.E.. Targeting RNA in mammalian systems with small molecules. WIRES RNA. 2018; 9:e1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Howe J.A., Wang H., Fischmann T.O., Balibar C.J., Xiao L., Galgoci A.M., Malinverni J.C., Mayhood T., Villafania A., Nahvi A. et al.. Selective small-molecule inhibition of an RNA structural element. Nature. 2015; 526:672. [DOI] [PubMed] [Google Scholar]
- 3. Wagner T.E., Becraft J.R., Bodner K., Teague B., Zhang X., Woo A., Porter E., Alburquerque B., Dobosh B., Andries O. et al.. Small-molecule-based regulation of RNA-delivered circuits in mammalian cells. Nat. Chem. Biol. 2018; 14:1043. [DOI] [PubMed] [Google Scholar]
- 4. Porter E.B., Polaski J.T., Morck M.M., Batey R.T.. Recurrent RNA motifs as scaffolds for genetically encodable small-molecule biosensors. Nat. Chem. Biol. 2017; 13:295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rauch S., Jones K.A., Dickinson B.. Small molecule-inducible RNA-targeting systems for temporal control of RNA regulation in vivo. RNA. 2020; 12:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kundert K., Lucas J.E., Watters K.E., Fellmann C., Ng A.H., Heineike B.M., Fitzsimmons C.M., Oakes B.L., Qu J., Prasad N. et al.. Controlling CRISPR-Cas9 with ligand-activated and ligand-deactivated sgRNAs. Nat. Commun. 2019; 10:2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Warner K.D., Hajdin C.E., Weeks K.M.. Principles for targeting RNA with drug-like small molecules. Nat. Rev. Drug. Discov. 2018; 17:547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tinoco Jr I., Bustamante C.. How RNAfolds. J. Mol. Biol. 1999; 293:271–281. [DOI] [PubMed] [Google Scholar]
- 9. Freier S.M., Kierzek R., Jaeger J.A., Sugimoto N., Caruthers M.H., Neilson T., Turner D.H.. Improved free-energy parameters for predictions of RNA duplex stability. Proc. Natl. Acad. Sci. U.S.A. 1986; 83:9373–9377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lorenz R., Bernhart S.H., Zu Siederdissen C.H., Tafer H., Flamm C., Stadler P.F., Hofacker I.L.. ViennaRNA Package 2.0. Algorithm Mol. Biol. 2011; 6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Leontis N.B., Westhof E.. Geometric nomenclature and classification of RNA base pairs. Rna. 2001; 7:499–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Leontis N.B., Westhof E.. Conserved geometrical base-pairing patterns in RNA. Q. Rev. Biophys. 1998; 31:399–455. [DOI] [PubMed] [Google Scholar]
- 13. Leontis N.B., Westhof E.. Analysis of RNAmotifs. Curr. Opin. Struc. Biol. 2003; 13:300–308. [DOI] [PubMed] [Google Scholar]
- 14. Petrov A.I., Zirbel C.L., Leontis N.B.. Automated classification of RNA 3D motifs and the RNA 3D motif atlas. RNA. 2013; 19:1327–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Leontis N.B., Lescoute A., Westhof E.. The building blocks and motifs of RNA architecture. Curr. Opin. Struc. Biol. 2006; 16:279–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. David-Eden H., Mankin A.S., Mandel-Gutfreund Y.. Structural signatures of antibiotic binding sites on the ribosome. Nucleic Acids Res. 2010; 38:5982–5994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kligun E., Mandel-Gutfreund Y.. Conformational readout of RNA by small ligands. RNA Biol. 2013; 10:981–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Thomas J.R., Hergenrother P.J.. Targeting RNA with small molecules. Chem. Rev. 2008; 108:1171–1224. [DOI] [PubMed] [Google Scholar]
- 19. Cruz J.A., Westhof E.. Sequence-based identification of 3D structural modules in RNA with RMDetect. Nat. Methods. 2011; 8:513. [DOI] [PubMed] [Google Scholar]
- 20. Reinharz V., Soulé A., Westhof E., Waldispühl J., Denise A.. Mining for recurrent long-range interactions in RNA structures reveals embedded hierarchies in network families. Nucleic Acids Res. 2018; 46:3841–3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sarrazin-Gendron R., Reinharz V., Oliver C.G., Moitessier N., Waldispühl J.. Automated, customizable and efficient identification of 3D base pair modules with BayesPairing. Nucleic Acids Res. 2019; 47:3321–3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Luo J., Wei W., Waldispühl J., Moitessier N.. Challenges and current status of computational methods for docking small molecules to nucleic acids. Eur. J. Med. Chem. 2019; 168:414–425. [DOI] [PubMed] [Google Scholar]
- 23. Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E.. UCSF Chimera—a visualization system for exploratory research and analysis. J. Computat. Chem. 2004; 25:1605–1612. [DOI] [PubMed] [Google Scholar]
- 24. Kitchen D.B., Decornez H., Furr J.R., Bajorath J.. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat. Rev. Drug. Discov. 2004; 3:935–949. [DOI] [PubMed] [Google Scholar]
- 25. Gómez-Bombarelli R., Wei J.N., Duvenaud D., Hernández-Lobato J.M., Sánchez-Lengeling B., Sheberla D., Aguilera-Iparraguirre J., Hirzel T.D., Adams R.P., Aspuru-Guzik A.. Automatic chemical design using a data-driven continuous representation of molecules. ACS Central Sci. 2018; 4:268–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jiménez J., Skalic M., Martinez-Rosell G., De Fabritiis G.. K deep: protein–ligand absolute binding affinity prediction via 3d-convolutional neural networks. J. Chem. Inf. Model. 2018; 58:287–296. [DOI] [PubMed] [Google Scholar]
- 27. Wang K., Jian Y., Wang H., Zeng C., Zhao Y.. RBind: computational network method to predict RNAbinding sites. Bioinformatics. 2018; 34:3131–3136. [DOI] [PubMed] [Google Scholar]
- 28. Pfeffer P., Gohlke H.. DrugScoreRNA knowledge-based scoring function to predict RNA-ligand interactions. J. Chem. Inf. Model. 2007; 47:1868–1876. [DOI] [PubMed] [Google Scholar]
- 29. Philips A., Milanowska K., Łach G., Bujnicki J.M.. LigandRNA: computational predictor of RNA–ligand interactions. RNA. 2013; 19:1605–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yan Z., Wang J.. SPA-LN: a scoring function of ligand–nucleic acid interactions via optimizing both specificity and affinity. Nucleic Acids Res. 2017; 45:e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sun L.-Z., Zhang D., Chen S.-J.. Theory and modeling of RNA structure and interactions with metal ions and small molecules. Ann. Rev. Biophys. 2017; 46:227–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Disney M.D., Winkelsas A.M., Velagapudi S.P., Southern M., Fallahi M., Childs-Disney J.L.. Inforna 2.0: a platform for the sequence-based design of small molecules targeting structured RNAs. ACS Chem. Biol. 2016; 11:1720–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mallet V., Oliver C.G., Moitessier N., Waldispuhl J.. Leveraging binding-site structure for drug discovery with point-cloud methods. 2019; arXiv doi:28 May 2019, preprint: not peer reviewedhttps://arxiv.org/abs/1905.12033.
- 34. Aumentado-Armstrong T. Latent molecular optimization for targeted therapeutic design. 2018; arXiv doi:05 September 2018, preprint: not peer reviewedhttps://arxiv.org/abs/1809.02032.
- 35. Torng W., Altman R.B.. Graph convolutional neural networks for predicting drug-target interactions. J. Chem. Inf. Model. 2019; 59:4131–4149. [DOI] [PubMed] [Google Scholar]
- 36. Paszke A., Gross S., Massa F., Lerer A., Bradbury J., Chanan G., Killeen T., Lin Z., Gimelshein N., Antiga L. et al.. Wallach H., Larochelle H., Beygelzimer A., d’Alché-Buc F., Fox E., Garnett R.. PyTorch: An Imperative Style, High-Performance Deep Learning Library. Advances in Neural Information Processing Systems 32. 2019; Curran Associates, Inc; 8024–8035. [Google Scholar]
- 37. Wang M., Yu L., Zheng D., Gan Q., Gai Y., Ye Z., Li M., Zhou J., Huang Q., Ma C. et al.. Deep graph library: towards efficient and scalable deep learning on graphs. 2019; arXiv doi:03 September 2019, preprint: not peer reviewedhttps://arxiv.org/abs/1909.01315v1.
- 38. Berman H., Westbrook J., Feng Z., Gilliland G., Bhat T., Weissig H., Shindyalov I., Bourne P.. The Protein Data Bank, Nucleic Acids Res. 2000; 28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schlichtkrull M., Kipf T.N., Bloem P., Van Den Berg R., Titov I., Welling M.. Modeling relational data with graph convolutional networks. European Semantic Web Conference. 2018; Springer; 593–607. [Google Scholar]
- 40. Philips A., Milanowska K., Lach G., Boniecki M., Rother K., Bujnicki J.M.. MetalionRNA: computational predictor of metal-binding sites in RNA structures. Bioinformatics. 2012; 28:198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sarver M., Zirbel C.L., Stombaugh J., Mokdad A., Leontis N.B.. FR3D: finding local and composite recurrent structural motifs in RNA 3D structures. J. Math Biol. 2008; 56:215–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Roll J., Zirbel C.L., Sweeney B., Petrov A.I., Leontis N.. JAR3D Webserver: scoring and aligning RNA loop sequences to known 3D motifs. Nucleic Acids Res. 2016; 44:W320–W327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cereto-Massagué A., Ojeda M.J., Valls C., Mulero M., Garcia-Vallvé S., Pujadas G.. Molecular fingerprint similarity search in virtual screening. Methods. 2015; 71:58–63. [DOI] [PubMed] [Google Scholar]
- 44. Glen R.C., Bender A., Arnby C.H., Carlsson L., Boyer S., Smith J.. Circular fingerprints: flexible molecular descriptors with applications from physical chemistry to ADME. IDrugs. 2006; 9:199. [PubMed] [Google Scholar]
- 45. Duvenaud D.K., Maclaurin D., Iparraguirre J., Bombarell R., Hirzel T., Aspuru-Guzik A., Adams R.P.. Convolutional networks on graphs for learning molecular fingerprints. Advances in neural information processing systems. 2015; 2224–2232. [Google Scholar]
- 46. Rogers D., Hahn M.. Extended-connectivity fingerprints. J. Chem. Inf. Model. 2010; 50:742–754. [DOI] [PubMed] [Google Scholar]
- 47. Durant J.L., Leland B.A., Henry D.R., Nourse J.G.. Reoptimization of MDL keys for use in drug discovery. J. Chem. Inf. Comp. Sci. 2002; 42:1273–1280. [DOI] [PubMed] [Google Scholar]
- 48. O’Boyle N.M., Morley C., Hutchison G.R.. Pybel: a Python wrapper for the OpenBabel cheminformatics toolkit. Chem. Cent. J. 2008; 2:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Erhan D., Bengio Y., Courville A., Manzagol P.-A., Vincent P., Bengio S.. Why does unsupervised pre-training help deep learning?. J. Mach. Learn. Res. 2010; 11:625–660. [Google Scholar]
- 50. Hamilton W.L., Ying R., Leskovec J.. Representation learning on graphs: methods and applications. Bulletin of the Technical Committee on Data Engineering. 2017; 40:52–75. [Google Scholar]
- 51. Sun F.-Y., Hoffmann J., Tang J.. InfoGraph: unsupervised and semi-supervised graph-level representation learning via mutual information maximization. 2019; arXiv doi:17 January 2020, preprint: not peer reviewedhttps://arxiv.org/abs/1908.01000.
- 52. Ribeiro L.F., Saverese P.H., Figueiredo D.R.. struc2vec: learning node representations from structural identity. Proceedings of the 23rd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. 2017; 385–394. [Google Scholar]
- 53. Heyne S., Costa F., Rose D., Backofen R.. GraphClust: alignment-free structural clustering of local RNA secondary structures. Bioinformatics. 2012; 28:i224–i232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mysinger M.M., Carchia M., Irwin J.J., Shoichet B.K.. Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking. J. Med. Chem. 2012; 55:6582–6594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Adrià C.M., Garcia-Vallvé S., Pujadas G.. DecoyFinder, a tool for finding decoy molecules. J, Cheminformatics. 2012; 4:1. [Google Scholar]
- 56. Bantscheff M., Scholten A., Heck A.J.. Revealing promiscuous drug–target interactions by chemical proteomics. Drug Discov. Today. 2009; 14:1021–1029. [DOI] [PubMed] [Google Scholar]
- 57. Walter F., Vicens Q., Westhof E.. Aminoglycoside–RNAinteractions. Curr. Opin. Chem. Biol. 1999; 3:694–704. [DOI] [PubMed] [Google Scholar]
- 58. Thiel B.C., Beckmann I.K., Kerpedjiev P., Hofacker I.L.. 3D based on 2D: Calculating helix angles and stacking patterns using forgi 2.0, an RNA Python library centered on secondary structure elements. F1000Research. 2019; 8:doi:10.12688/f1000research.18458.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zirbel C.L., Roll J., Sweeney B.A., Petrov A.I., Pirrung M., Leontis N.B.. Identifying novel sequence variants of RNA 3D motifs. Nucleic Acids Res. 2015; 43:7504–7520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Uhl M., Tran V.D., Heyl F., Backofen R.. GraphProt2: a novel deep learning-based method for predicting binding sites of RNA-binding proteins. 2019; bioRxiv doi:07 April 2019, preprint: not peer reviewed 10.1101/850024. [DOI]
- 61. Lipinski C.A. Lead-and drug-like compounds: the rule-of-five revolution. Drug Discov. Today: Technol. 2004; 1:337–341. [DOI] [PubMed] [Google Scholar]
- 62. Pareja A., Domeniconi G., Chen J., Ma T., Suzumura T., Kanezashi H., Kaler T., Leisersen C.E.. Evolvegcn: evolving graph convolutional networks for dynamic graphs. AAAI. 2019; 5363–5370. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.