

Abstract
Isothermal, cell-free, synthetic biology-based approaches to pathogen detection leverage the power of tools available in biological systems, such as highly active polymerases compatible with lyophilization, without the complexity inherent to live-cell systems, of which nucleic acid sequence based amplification (NASBA) is well known. Despite the reduced complexity associated with cell-free systems, side reactions are a common characteristic of these systems. As a result, these systems often exhibit false positives from reactions lacking an amplicon. Here we show that the inclusion of a DNA duplex lacking a promoter and unassociated with the amplicon fully suppresses false positives, enabling a suite of fluorescent aptamers to be used as NASBA tags (Apta-NASBA). Apta-NASBA has a 1 pM detection limit and can provide multiplexed, multicolor fluorescent readout. Furthermore, Apta-NASBA can be performed using a variety of equipment, for example, a fluorescence microplate reader, a qPCR instrument, or an ultra-low-cost Raspberry Pi-based 3D-printed detection platform using a cell phone camera module, compatible with field detection.
Keywords: NASBA, T7 RNA polymerase, fluorescent aptamer, isothermal amplification, pathogen detection
INTRODUCTION
Low-cost, rapid pathogen detection methods are of high interest for field diagnostics and for use in the developing world, both for disease detection and pathogen monitoring (e.g., in water supplies or agricultural products). Several nucleic acid amplification techniques have been developed to identify disease-causing pathogens, of which the polymerase chain reaction (PCR) is the best-known (Garibyan and Avashia 2013). PCR is robust, specific and widely used, and it constitutes the basis for a wide range of diagnostic tests. However, the thermocyclers required to perform PCR are energy intensive and expensive (∼$5–10K), making them cost-prohibitive for field diagnostics or use in low-resource environments. Furthermore, the typical readout of PCR, electropherograms, requires trained technicians and additional equipment. It is possible to monitor PCR reactions in real time, but this requires the coupling of a fluorescence detection system to the thermocycler, increasing the cost of the instrument by as much as an order of magnitude. These reactions are typically monitored by expensive oligonucleotide probes or nonspecific DNA intercalating dyes (Arya et al. 2005).
Isothermal nucleic acid amplification techniques are an attractive means to mitigate the costs and complexity associated with thermal cycling. Several isothermal techniques have been reported, such as nucleic acid sequence based amplification (NASBA), loop mediated amplification, recombinase polymerase amplification (RPA), helicase dependent amplification, and SHERLOCK (Notomi et al. 2000; Deiman et al. 2002; An et al. 2005; Piepenburg et al. 2006; Gootenberg et al. 2017). In each case, these reactions use strand displacement, nucleases, or recombinases to generate double-stranded DNA, either as a product or as an intermediate that is transcribed to an RNA output. Reactions that generate RNA outputs are of high interest, given that a number of RNA polymerases can readily be overexpressed, lyophilized (negating the need for a cold chain), and used to generate large quantities of RNA in these reactions. Furthermore, unlike dsDNA, RNA can exhibit diverse functional behaviors, including binding of fluorogenic dyes and enzymatic activity, enabling a wide range of potential readouts (Abdolahzadeh et al. 2019).
A common pitfall associated with isothermal amplification reactions is nonspecific amplification, which cannot be mitigated by thermal cycling, as in PCR. Investigators have used a range of techniques to mitigate this pitfall, including engineered thermally stabilized polymerases and two-step nested reactions targeting different regions of the amplicon of interest (including another fluorescent aptamer-based method, Nested Mango NASBA). These strategies, while they enable high sensitivity and specificity, necessarily add complexity to the reaction (Abdolahzadeh et al. 2019; Wu et al. 2020).
One potential source of nonspecific amplification in reactions using viral polymerases, such as T7 RNA polymerase, is unpromoted transcription. This latent behavior of T7 RNA polymerase was recently shown to be the reason transcripts produced by this enzyme can exhibit innate immune activation. This occurs because the enzyme produces a low level of antisense transcript, which hybridizes with the main sense transcript, producing dsRNA and causing innate immune activation (Mu et al. 2018). We speculated that unpromoted transcription could be operative in NASBA reactions, generating a primer dimer that codes for the fluorescent aptamer, which can go on to seed the reaction. Here, we demonstrate that the inclusion of a competitor duplex that does not contain a promoter sequence can fully suppress nonspecific NASBA amplification. Using this technique, we demonstrate multicolor tagging of NASBA amplicons with fluorescent aptamers of diverse sequence and structure, including in multiplexed, one-pot reactions with real-time readout.
RESULTS AND DISCUSSION
Apta-NABSA entails the use of two primers: The first is a conventional NASBA primer, which installs the T7 RNA polymerase promoter sequence. The second binds the amplicon and additionally contains the antisense sequence for a functional RNA (here, a fluorogenic aptamer). Thus, as the amount of RNA amplicon transcribed increases, the amount of RNA aptamer increases. The aptamer can then fold and bind its corresponding small molecule, generating a specific, real-time fluorescence signal (Fig. 1). The use of primers coding for a fluorescent aptamer is particularly appealing, since this enables the specificity typically associated with a dual-labeled probe (e.g., TaqMan, molecular beacons), coupled with the low cost associated with a small molecule dye (e.g., SYBR Green) (Arya et al. 2005). We designed primers for an enteroaggregative E. coli virulence gene, aggR, encoding a transcriptional activator of aggregative adherence fimbriae I using primers previously developed for real-time PCR (Hidaka et al. 2009; Morin et al. 2013). The primer lacking the T7 RNA polymerase promoter sequence contained the coding sequence for the Broccoli aptamer, acting as a genetically encoded, green fluorescent RNA tag when complexed with the fluorogenic dye DFHBI-1T (Filonov et al. 2014).
FIGURE 1.

Apta-NASBA reaction. Schematic of the reaction. Promoter-containing primer (PP) binds the amplicon of interest. Reverse transcriptase (RT) extends PP, generating a DNA-RNA hybrid comprised of the amplicon target sequence (TS) and an antisense DNA copy of the amplicon with an overhang containing the T7 RNA polymerase (T7 RNAP) promoter. RNase H degrades the original amplicon RNA of interest. Coding primer (CP) binds. RT extends CP, generating a DNA duplex corresponding to the amplicon, with a T7 RNAP promoter (red) and an aptamer coding sequence (blue). T7 RNAP generates an RNA fusion construct that contains the sequence antisense to TS, fused at the 3′ end with the aptamer corresponding to the sequence in CP (green). CP binds this RNA, which again enters the RT-RNase H-T7 RNAP cycle. At each step, RNA is generated with multiple turnover, enabling exponential amplification. Images on the right are representative Apta-NASBA reactions using aptamers that span the fluorescence spectrum.
As expected, fluorogenic aptamer-encoding primers gave exponential amplification curves (Fig. 2A). However, as others have observed in NASBA reactions, untemplated amplification occurred in template-free controls (Fig. 2A–E; Abdolahzadeh et al. 2019). T7 RNA polymerase is well-known to catalyze a range of reactions beyond its canonical promoter-driven DNA-dependent RNA polymerase activity, including RNA-dependent RNA polymerase activity (Konarska and Sharp 1989) and unpromoted DNA-dependent polymerization, as was recently reported (Mu et al. 2018). We speculated the latter could have been the source of the observed false positives. Given that either the sense or antisense RNA strand is sufficient to seed a NASBA reaction, and that our NASBA primers contain the T7 RNA polymerase promoter in one primer and the aptamer coding sequence in the other, a small amount of primer dimer could cause a false positive. In each case, false positives were smaller than the expected amplicon and stained with DFHBI, consistent with primer-derived RNA products (Fig. 2B,E; Supplemental Fig. S1).
FIGURE 2.

NASBA is highly prone to false positives, which can be suppressed by modulation of reaction conditions. Apta-NASBA was performed using the enteroaggregative E. coli gene, aggR. Typical NASBA reactions exhibit fluorescence developing over ∼2 h, with frequent false positives. These false positives can be suppressed by inclusion of Duplex40 as observed by fluorescence spectroscopy (A) and PAGE (B). (A) Samples containing template = green closed markers, samples lacking template = black open markers, samples containing Duplex40 = circles, samples lacking Duplex40 = squares. Shading represents S.E.M., N = 3. (B, left) SYBR Gold stained ladder, (right) DFHBI stained gel. Arrow indicates size of expected full-length amplicon-aptamer fusion product (144 nt); false positives are of lower size, consistent with a primer-derived product and not template contamination. (C) Images of Apta-NASBA reactions with and without Duplex40. False positives can also be suppressed by decreased Mg2+ concentration (without inclusion of Duplex40), which has previously been reported to suppress unpromoted transcription (Mu et al. 2018), as observed by fluorescence spectroscopy (D) and PAGE (E). (D) Samples containing template = closed markers, samples lacking template = open markers. Samples containing 12 mM Mg2+ (blue diamonds) exhibited false positives, while 10 mM Mg2+ (green triangles) still exhibited NASBA with ∼40% reduction in signal intensity, and false positives were not detected by fluorescence spectroscopy, but they were detected by PAGE. Samples containing 8 mM Mg2+ (orange squares) did not exhibit detectable product by fluorescence spectroscopy, but product was detectable by PAGE. 5 mM Mg2+ did not support NASBA amplification (red circles). (E, left) SYBR Gold stained ladder, (right) DFHBI stained gel. Arrow indicates size of expected full-length amplicon-aptamer fusion product (144 nt); false positives are of lower size, indicating a primer-derived product, and not template contamination. (F) Optimized Apta-NASBA enables specific sensitive detection of E. coli virulence gene aggR at an amplicon concentration as low as 1 pM. Shading represents S.E.M., N = 3.
We reasoned that the presence of a competitor duplex of a sequence unrelated to the amplicon of interest and lacking both the T7 RNA polymerase promoter and the aptamer-encoding sequence could suppress this phenomenon. We screened a range of concentrations of a 40 bp duplex Duplex40 (Supplemental Fig. S2) and observed that Duplex40, when present at 10 µM duplex, fully suppressed untemplated amplification with a minimal diminution in final fluorescence intensity relative to reactions lacking Duplex40 (Fig. 2A–C). A longer 100 bp duplex, Duplex100, also sufficed to suppress false positives, with similar effectiveness at a lower concentration of 0.316 µM duplex (Supplemental Fig. S3).
The required concentration of competitor duplex DNA (in base pairs) to suppress false positives decreased with increasing length, with a minimal concentration of 400 µM bp for Duplex40 and 31.7 µM bp for Duplex100. We thus speculated that T7 RNA polymerase could be associated with nucleic acids for a duration proportional to their length, and that very long nucleic acids could act as ultra-potent inhibitors of false positives. However, this was not the case; genomic (calf thymus) DNA did not suppress false positives (Supplemental Fig. S4). We also screened heparin, another highly negatively charged polyelectrolyte and inhibitor of T7 RNA polymerase, at submaximal levels using a Spinach-tagged Apta-NASBA construct (Supplemental Fig. S5; Sastry and Ross 1997; Capila and Linhardt 2002; Paige et al. 2011). Heparin reduced the maximum signal seen from both the positive and false positive signals by approximately the same extent.
We also screened Mg2+ concentrations, previously reported to suppress unpromoted transcription (Mu et al. 2018). Reactions not containing Duplex40 and with decreasing Mg2+ from 12 mM to 10 mM also diminished false positive signal, albeit with a decrease in signal in positive reactions (Fig. 2D). A further decrease to 8 mM Mg2+ fully suppressed unpromoted transcription with a further diminution in final fluorescence intensity (Fig. 2D) resulting in the positive reaction only being detectable by a weak band on PAGE (Fig. 2E). T7 RNA polymerase exhibits differing confirmations when initiating transcription vs. during elongation. High Mg2+ concentrations promote the elongation-associated conformation of T7 RNA polymerase, allowing for unpromoted transcription to occur on the primers in the Apta-NASBA reactions, as previously speculated (Mu et al. 2018).
We performed further optimization of the reaction using Duplex40 as the false-positive inhibitor. Optimizations included removing chloride (Supplemental Fig. S6; Chamberlin and Ring 1973) altering primer concentration (Supplemental Fig. S7), adding osmolytes (Supplemental Figs. S8–S13), altering potassium concentration (Supplemental Fig. S14), and adding inorganic pyrophosphatase (Supplemental Fig. S15; Cunningham and Ofengand 1990). Optimized reaction conditions resulted in a limit of detection of 1 pM (Fig. 2F), with no signal from negative reactions (i.e., those lacking template). Concentrations lower than 1 pM failed to amplify.
Having observed that the Apta-NASBA system functions with Broccoli, a green aptamer (λex = 472 nm, λem = 507 nm), we sought to expand Apta-NASBA into different colors, using Corn, a yellow aptamer (λex = 505 nm, λem = 545 nm) (Song et al. 2017; Warner et al. 2017). We designed primers for two additional pathogen-associated genes, estP (observed using Corn) and estH (observed using Broccoli) by modifying primers previously developed for real-time PCR. estP and estH produce two different variants of heat-stable enterotoxins found in enterotoxigenic E. coli, and enterotoxigenic E. coli is a major cause of diarrhea in travelers and infants (Grate and Wilson 1999; Plourde 2003; Hidaka et al. 2009). Additionally, we designed another primer for aggR to observe using malachite green aptamer, a red aptamer (λex = 630 nm, λem = 650 nm) (Grate and Wilson 1999; Babendure et al. 2003; Hidaka et al. 2009). Reactions were successful using Corn to detect estP and malachite green aptamer to detect aggR (Fig. 3A; Supplemental Fig. S16). Consistent with the high degree of specificity we found Duplex40 to afford, amplicons only exhibited signal when amplified with their cognate primer set (Supplemental Fig. S17).
FIGURE 3.
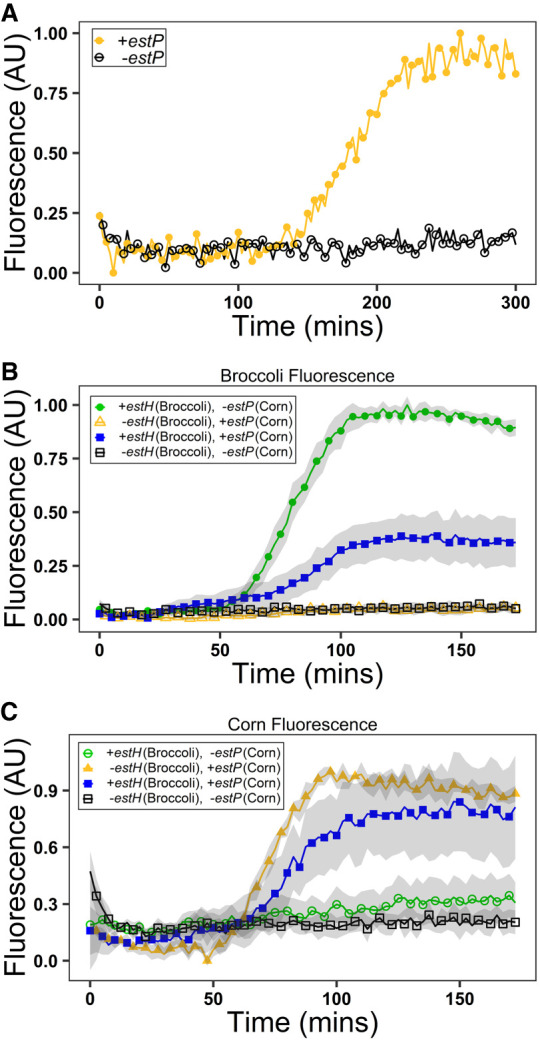
Apta-NASBA functions with multiple fluorescent aptamers. (A) Apta-NASBA of enteroinvasive E. coli estP gene with Corn (λex = 505 nm, λem = 545 nm) detecting 218 pM template RNA performed with SpectraMax Gemini XS plate reader. The ability to use different fluorescent aptamers allows for multiplexing. Multiplexed Apta-NASBA reactions using Broccoli to detect E. coli estH gene and E. coli estP gene with Corn (B,C). (B) Multiplexed Apta-NASBA reactions detected using Broccoli. (C) Multiplexed Apta-NASBA reactions detected using Corn. Samples where signal should be seen = closed symbols, samples where no signal should be seen = open symbol. Green symbols represent reactions containing template detected with Broccoli, blue symbols represent reactions containing both template detected with Broccoli and template detected with Corn, yellow symbols represent reactions containing template detected with Corn, black symbols represent reactions containing no template. Shading represents S.E.M., N = 3 (B,C).
Given the strict primer-to-amplicon relationship, we sought to use these orthogonal primer pairs in a multiplexed detection system. Here we show detection of estP and estH in a single reaction using Corn and Broccoli, respectively (Fig. 3B,C). We have also demonstrated dual-detection estP and aggR using combinations of Broccoli, Corn and the malachite green aptamer for readout (Supplemental Figs. S18, S19).
The isothermal nature of Apta-NASBA eliminates the need for energy intensive thermocyclers. To further Apta-NASBA's utility in field diagnostics and low resource areas we coupled a lyophilized Apta-NASBA reaction with a low-cost Raspberry Pi-based detection platform (Hoog et al. 2019) using a cell phone camera module (Fig. 4C). A short Python script was written to control the image acquisition of fluorescent reactions and fluorescence intensity was determined based on pixel value (Hoog et al. 2019). The fluorescence curves obtained from Apta-NASBA on more expensive plate readers and qPCR-based fluorescence detection platforms are functionally identical to those observed on the Raspberry Pi-based detection platform (Fig. 4A–C). We show, with the addition of a cryoprotectant (trehalose), Apta-NASBA reactions are viable post lyophilization, allowing for transportation to remote locations without a cold chain (Fig. 4C; Supplemental Fig. S20; Supplemental Video S1).
FIGURE 4.
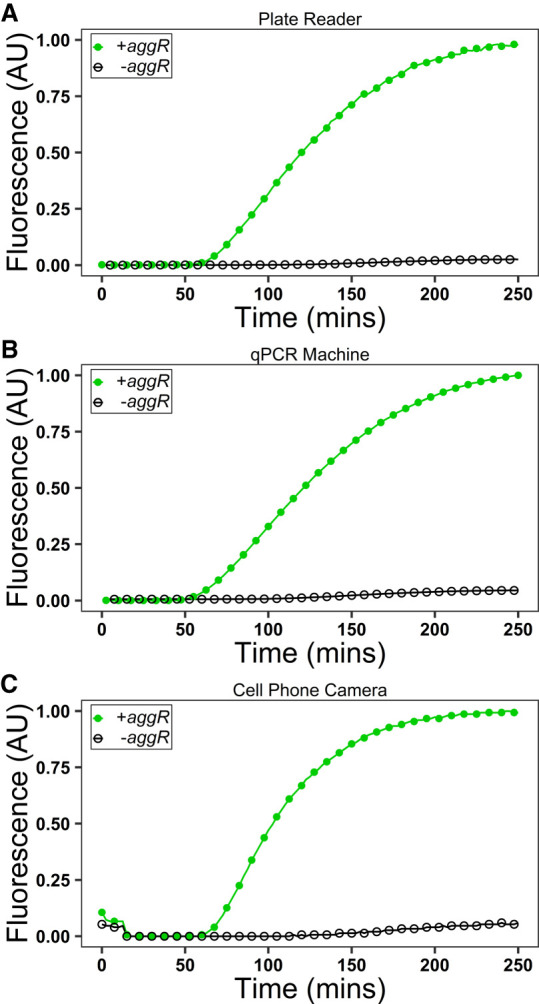
Apta-NASBA can be lyophilized, enabling transportation to remote locations, and the fluorescence signal generated can be imaged using a Raspberry Pi-based apparatus that is compatible with fieldwork. (A) Lyophilized Apta-NASBA reaction of aggR gene performed in a fluorescence plate reader (SpectraMax Gemini XS). (B) Lyophilized Apta-NASBA reaction of aggR gene performed in qPCR machine (ChaiBio Open qPCR). (C) Lyophilized Apta-NASBA reaction of aggR gene performed with the Raspberry Pi-based fluorescence imaging apparatus. All detection platforms show a characteristic template-dependent sigmoidal amplification curve.
The computer-aided design of multidomain RNAs containing fluorescent tags can be problematic, given that a significant number of known fluorescent RNAs fold into G-Quadruplex structures. We observed that, despite predicting an incorrect fold, the use of folding prediction software was generally successful in predicting whether a domain of a given RNA would fold independently or induce misfolding (Supplemental Fig. S21).
The RNA constructs developed in this paper provide proof-of-concept for design of multidomain RNAs that exhibit diverse folds, including those not accommodated by secondary structure prediction algorithms—particularly G-Quadruplexes (Supplemental Fig. S21; Huang et al. 2014; Warner et al. 2014). These appear to be ubiquitous in fluorescent aptamers selected in vitro. For example, folding prediction algorithms showed Spinach, the first published G-Quadruplex-containing fluorescent RNA aptamer, to be a stem–loop structure (Paige et al. 2011). However, Spinach is a G-Quadruplex not captured by existing folding prediction algorithms (Huang et al. 2014; Warner et al. 2014). The presence of G-Quadruplexes in Apta-NASBA readouts is the reasoning behind an increase in potassium leading to a greater signal. Monovalent cations are required for proper G-Quadruplex folding and stability (Supplemental Fig. S14). Diverse and novel folds have also been observed in several other fluorescent aptamers. The work in this paper provides important proof of concept for design and deployment of arbitrary RNAs, including those forming noncanonical secondary structures not accounted for by structure prediction software.
Apta-NASBA represents an attractive platform for pathogen detection with the capability to be performed in remote, low resource areas. Given its modularity and compatibility with one-pot, multicolor sequence detection, it promises to be a highly extensible platform. In the current work, the readout used is a fluorescent aptamer. Given that the functional RNA is installed using the second primer, the Apta-NASBA platform is highly versatile, and the possibility exists to install arbitrary nucleic acids, including other functional RNAs, such as aptamers for modulation of a cell-free protein expression system, or small transcription activating RNAs (STARs) (Chappell et al. 2015).
MATERIALS AND METHODS
Oligonucleotides were obtained from Integrated DNA Technologies desalted and used as received. Sequences of oligonucleotides used are given in Supplemental Table S1. Enzymes were from New England Biolabs (Ipswitch), except for T7 RNAP and mMuLV, which were overexpressed in-house as described below.
NASBA reaction
NASBA was performed based on extensive modifications to a procedure originally reported by Sooknanan, van Gemen, and Malek (Sooknanan et al. 1995). Reactions were run with a total volume of 25 µL. A 5× Buffer stock was prepared containing 200 mM Tris-Acetate, 420 mM KOAc, 60 mM MgOAc, 5 mM each of dATP, dTTP, dGTP and dCTP, 10 mM each of ATP, UTP, GTP, and CTP. An enzyme mix was prepared containing T7 RNAP, Moloney Murine Leukemia Virus reverse transcriptase (mMuLV), RNase H (New England Biolabs Inc., M0297) and bovine serum albumin (New England Biolabs Inc., B9000S). Final reaction concentrations/amounts of enzymes present were 1 µM T7 RNAP, 0.25 µM mMuLV, 0.2 U RNase H and 2.6 µg bovine serum albumin per 25 µL reaction. 5× Buffer was added to a final concentration of 1× to a solution containing 1 µM Primer 1, 1 µM Primer 2, 100 µM cognate dye, 1 mM DTT, 166 mM KOAc, 10 µM Duplex40, 400 mM trehalose, 50 µg inorganic pyrophosphatase (Bayou Biolabs) as described previously (Heili et al. 2018) and 4 mM MgOAc. Reactions not containing enzyme mix were heated for 5 min at 65°C followed by 5 min at 40°C. Enzyme mix was then added to the reaction and reactions were incubated at 40°C. All aptamers were used below their melting temperature. For lyophilized reactions, the full reaction except amplicon and dye was flash-frozen in liquid nitrogen and lyophilized overnight using a LABCONCO FreeZone 1 liter benchtop freeze dry system, then resuspended in a solution of amplicon and dye. No 65°C step was performed for lyophilized reactions. Reactions without addition of template were used as negative controls. Fluorescence spectroscopy has been normalized in each graph by subtracting the smallest value from each measurement followed by dividing by the largest value to normalize for instrumental response. Template used was transcribed in vitro as described below. Primer sequences can be found in Supplemental Table S1. Primers were adapted from previously published primers from Hidaka et al. (2009). Geneious 10.1.3 was used to assist in primer design.
T7 RNAP and mMuLV-RT overexpression and purification
An amount of 10 mL LB containing 100 µg/µL carbenicillin was inoculated with E. coli DH5α containing either pT7-911Q (T7 RNAP) (Ichetovkin et al. 1997) or pET-MRT (mMuLV-RT) (Yasukawa et al. 2010). Cultures were grown overnight at 37°C, then used to inoculate an additional 1 L of LB containing 100 µg/µL carbenicillin and grown at 37°C to an OD600 between 0.5 and 1. Cultures were then induced with 1 mM IPTG, and grown at 37°C for 3 h. Culture was cooled on ice for 20 min and pelleted at 3700 RPM for 15 min. Pellets were flash frozen in liquid nitrogen and frozen at −80°C overnight. Pellets were held in a cold room for 30 min, then dissolved in 20 mL lysis buffer (50 mM HEPES-KOH pH 7.6, 1 M NH4Cl, 10 mM MgCl2, 7 mM BME). Pellets were incubated in lysis buffer for 30 min followed by tip sonication. Sonication was performed at 50% power in 15 sec intervals until 2 kJ total energy had been applied, then the sample was allowed to cool for 5 min. This was repeated a total of four times. Pellets were then centrifuged for 45 min at 15,000g at 4°C.
The supernatant was applied to 0.6 mL Ni-NTA agarose beads (GoldBio, H-350-50) and incubated on a rocker in a cold room for 1 h. Washing and elution steps were done in batch method. Beads were washed with 10 mL wash buffer for 10 min then washed again with 10 mL wash buffer (50 mM HEPES pH 7.6, 1 M NH4Cl, 10 mM MgCl2, 15 mM imidazole, 7 mM BME) for 15 min. An amount of 3 mL elution buffer (50 mM HEPES-KOH pH 7.6, 100 mM KCl, 10 mM MgCl2, 300 mM imidazole, 7 mM BME) was applied to beads and incubated on a rocker for 12 min in a 4°C cold room. Elution was dialyzed against 500 mL 2× storage buffer (100 mM Tris-HCl pH 7.6, 200 mM KCl, 20 mM MgCl2, 14 mM BME) using Slide-A-Lyzer Dialysis Cassette, 2000 MWCO (Thermo Fisher Scientific, 66203) overnight, followed by dialysis against an additional 500 mL 2× storage buffer for 3 h. Enzymes intended for lyophilization were prepared in the same storage buffer with the omission of glycerol.
Proteins were quantified using the calculated A280 using a NanoDrop ND-1000. Protein activity was assessed by in vitro transcription of Broccoli aptamer and kinetic monitoring on a fluorescence plate reader (T7 RNAP) or reverse transcription of estH gene and visualization on a SYBR Gold-stained urea-PAGE gel (mMuLV-RT).
Template transcription
One millileter transcriptions were performed to obtain template for NASBA reactions. Reactions contained 40 mM Tris-acetate, 24 mM Mg-glutamate, 100 mM K-glutamate, 2 mM spermidine, 1 mM DTT, 4 mM ATP, 4 mM GTP, 4 mM UTP, 4 mM CTP, 1 µM overexpressed T7 RNA polymerase (conversely New England Biolabs T7 RNA Polymerase M0251L can be used, but it does not give as great a yield), 1X Pyrophosphatase as used previously, and double-stranded DNA templates were used according to Supplemental Table S1 (Heili et al. 2018). Optimal template concentration was determined empirically and was typically ∼100 nM for the amplicons used here. Reactions were incubated for 8 h at 37°C. For transcript purification, reactions were first treated with 1 U/µg Turbo DNase (Thermo Fisher Scientific, S11494) for 15 min at 37°C followed by ethanol precipitation. The precipitated RNA was resuspended in 1× loading buffer (1× TBE, 8 M Urea) and separated on a 10% Urea-PAGE gel; the full-length product band was excised via UV shadowing on a fluorescent TLC plate. The gel slice was physically disturbed by forcing it through a 21 Ga needle in 1 to 3 mL 1× TBE, followed by 3 freeze-thaw cycles alternating between liquid nitrogen and a 55°C water bath. The TBE containing the RNA was decanted, and an ethanol precipitation was performed. The final product was suspended in RNase-free water, and concentration was determined by A260 using a NanoDrop ND-1000.
Calf thymus DNA
Calf thymus DNA (Sigma D1501-100MG) was diluted in 10 mM Tris-Acetate pH 8, 1 mM EDTA to 2 mg/mL, and rotated until homogeneous. In order to minimize shearing, no vortexing was performed. To determine the amount of DNA used, solution concentration was determined to be 2.32 mM by A260 using a NanoDrop ND-1000.
Gel staining
Ten percentage of Urea-PAGE gels (1× TBE, 8 M urea) were incubated in two changes of folding buffer (100 mM Tris-Acetate pH 8, 10 mM MgCl2, 500 mM KCl) for 20 min each time to remove urea from gels and to supply K+ and Mg2+ to support aptamer folding. Gels were then incubated in folding buffer spiked with 10 µM DFHBI for 15 min. If the fluorescence background on a gel was high, it was destained for 5 min in folding buffer. Gels were imaged on a gel doc (Omega Lum G, Aplegen) using the orange filter. For visualization of non-aptamer containing sequences, gels were stained in 1× SYBR Gold for an additional 15 min and reimaged under the same conditions.
3D printed cell phone camera module/raspberry Pi imaging device
The Raspberry Pi imaging device and codes were used as described previously (Hoog et al. 2019).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Peter Unrau and members of the Engelhart laboratory for helpful discussions. This work was supported by NASA Contract 80NSSC18K1139 under the Center for Origin of Life (to A.E.E. and K.P.A.).
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.075192.120.
REFERENCES
- Abdolahzadeh A, Dolgosheina EV, Unrau PJ. 2019. RNA detection with high specificity and sensitivity using nested fluorogenic Mango NASBA. RNA 25: 1806–1813. 10.1261/rna.072629.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- An L, Tang W, Ranalli TA, Kim H-J, Wytiaz J, Kong H. 2005. Characterization of a thermostable UvrD helicase and its participation in helicase-dependent amplification. J Biol Chem 280: 28952–28958. 10.1074/jbc.M503096200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arya M, Shergill IS, Williamson M, Gommersall L, Arya N, Patel HRH. 2005. Basic principles of real-time quantitative PCR. Expert Rev Mol Diagn 5: 209–219. 10.1586/14737159.5.2.209 [DOI] [PubMed] [Google Scholar]
- Babendure JR, Adams SR, Tsien RY. 2003. Aptamers switch on fluorescence of triphenylmethane dyes. J Am Chem Soc 125: 14716–14717. 10.1021/ja037994o [DOI] [PubMed] [Google Scholar]
- Capila I, Linhardt RJ. 2002. Heparin–protein interactions. Angew Chem Int Ed 41: 391–412. [DOI] [PubMed] [Google Scholar]
- Chamberlin M, Ring J. 1973. Characterization of T7-specific ribonucleic acid polymerase. II. Inhibitors of the enzyme and their application to the study of the enzymatic reaction. J Biol Chem 248: 2245–2250. [PubMed] [Google Scholar]
- Chappell J, Takahashi M, Lucks J. 2015. Creating small transcription activating RNAs. Nat Chem Biol 11: 214–220. 10.1038/nchembio.1737 [DOI] [PubMed] [Google Scholar]
- Cunningham PR, Ofengand J. 1990. Use of inorganic pyrophosphatase to improve the yield of in vitro transcription reactions catalyzed by T7 RNA polymerase. BioTechniques 9: 713–714. [PubMed] [Google Scholar]
- Deiman B, van Aarle P, Sillekens P. 2002. Characteristics and applications of nucleic acid sequence-based amplification (NASBA). Mol Biotechnol 20: 163–179. 10.1385/MB:20:2:163 [DOI] [PubMed] [Google Scholar]
- Filonov GS, Moon JD, Svensen N, Jaffrey SR. 2014. Broccoli: rapid selection of an RNA mimic of green fluorescent protein by fluorescence-based selection and directed evolution. J Am Chem Soc 136: 16299–16308. 10.1021/ja508478x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garibyan L, Avashia N. 2013. Polymerase chain reaction. J Invest Dermatol 133: 1–4. 10.1038/jid.2013.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gootenberg JS, Abudayyeh OO, Lee JW, Essletzbichler P, Dy AJ, Joung J, Verdine V, Donghia N, Daringer NM, Freije CA, et al. 2017. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 356: 438–442. 10.1126/science.aam9321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grate D, Wilson C. 1999. Laser-mediated, site-specific inactivation of RNA transcripts. Proc Natl Acad Sci 96: 6131–6136. 10.1073/pnas.96.11.6131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heili JM, Gomez-Garcia J, Gaut NJ, Cash BW, Aufdembrink LM, Heffron BA, Shirley JD, Carlson EE, Adamala KP, Engelhart AE. 2018. Real-time visualization of in vitro transcription of a fluorescent RNA aptamer: an experiment for the upper-division undergraduate or first-year graduate laboratory. J Chem Educ 95: 1867–1871. 10.1021/acs.jchemed.7b00735 [DOI] [Google Scholar]
- Hidaka A, Hokyo T, Arikawa K, Fujihara S, Ogasawara J, Hase A, Hara-Kudo Y, Nishikawa Y. 2009. Multiplex real-time PCR for exhaustive detection of diarrhoeagenic Escherichia coli. J Appl Microbiol 106: 410–420. 10.1111/j.1365-2672.2008.04043.x [DOI] [PubMed] [Google Scholar]
- Hoog TG, Pawlak MR, Aufdembrink LM, Bachan BR, Galles MB, Bense NB, Adamala KP, Engelhart AE. 2019. DNA transistors switched by the Hofmeister effect. bioRxiv 784561 10.1101/784561 [DOI] [Google Scholar]
- Huang H, Suslov NB, Li N-S, Shelke SA, Evans ME, Koldobskaya Y, Rice PA, Piccirilli JA. 2014. A G-quadruplex-containing RNA activates fluorescence in a GFP-like fluorophore. Nat Chem Biol 10: 686–691. 10.1038/nchembio.1561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichetovkin IE, Abramochkin G, Shrader TE. 1997. Substrate recognition by the leucyl/phenylalanyl-tRNA-protein transferase. Conservation within the enzyme family and localization to the trypsin-resistant domain. J Biol Chem 272: 33009–33014. 10.1074/jbc.272.52.33009 [DOI] [PubMed] [Google Scholar]
- Konarska MM, Sharp PA. 1989. Replication of RNA by the DNA-dependent RNA polymerase of phage T7. Cell 57: 423–431. 10.1016/0092-8674(89)90917-3 [DOI] [PubMed] [Google Scholar]
- Morin N, Santiago AE, Ernst RK, Guillot SJ, Nataro JP. 2013. Characterization of the AggR regulon in enteroaggregative Escherichia coli. Infect Immun 81: 122–132. 10.1128/IAI.00676-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu X, Greenwald E, Ahmad S, Hur S. 2018. An origin of the immunogenicity of in vitro transcribed RNA. Nucleic Acids Res 46: 5239–5249. 10.1093/nar/gky177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notomi T, Okayama H, Masubuchi H, Yonekawa T, Watanabe K, Amino N, Hase T. 2000. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res 28: E63 10.1093/nar/28.12.e63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paige JS, Wu KY, Jaffrey SR. 2011. RNA mimics of green fluorescent protein. Science 333: 642–646. 10.1126/science.1207339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piepenburg O, Williams CH, Stemple DL, Armes NA. 2006. DNA detection using recombination proteins. PLoS Biol 4: e204 10.1371/journal.pbio.0040204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plourde PJ. 2003. Travellers’ diarrhea in children. Paediatr Child Health 8: 99–103. 10.1093/pch/8.2.99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastry SS, Ross BM. 1997. Nuclease activity of T7 RNA polymerase and the heterogeneity of transcription elongation complexes. J Biol Chem 272: 8644–8652. 10.1074/jbc.272.13.8644 [DOI] [PubMed] [Google Scholar]
- Song W, Filonov GS, Kim H, Hirsch M, Li X, Moon JD, Jaffrey SR. 2017. Imaging RNA polymerase III transcription using a photostable RNA-fluorophore complex. Nat Chem Biol 13: 1187–1194. 10.1038/nchembio.2477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sooknanan R, Malek LT, van Gemen B. 1995. Nucleic acid sequence-based amplification. In Molecular methods for virus detection (ed. Wiedbrauk DL, Farkas DH), pp. 261–285. Academic Press, San Diego. [Google Scholar]
- Warner KD, Chen MC, Song W, Strack RL, Thorn A, Jaffrey SR, Ferré-D'Amaré AR. 2014. Structural basis for activity of highly efficient RNA mimics of green fluorescent protein. Nat Struct Mol Biol 21: 658–663. 10.1038/nsmb.2865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner KD, Sjekloća L, Song W, Filonov GS, Jaffrey SR, Ferré-D'Amaré AR. 2017. A homodimer interface without base pairs in an RNA mimic of red fluorescent protein. Nat Chem Biol 13: 1195–1201. 10.1038/nchembio.2475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MZ, Asahara H, Tzertzinis G, Roy B. 2020. Synthesis of low immunogenicity RNA with high-temperature in vitro transcription. RNA 26: 345–360. 10.1261/rna.073858.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasukawa K, Mizuno M, Konishi A, Inouye K. 2010. Increase in thermal stability of Moloney murine leukaemia virus reverse transcriptase by site-directed mutagenesis. J Biotechnol 150: 299–306. 10.1016/j.jbiotec.2010.09.961 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.