

Abstract
Background
Nanopore sequencing enables portable, real-time sequencing applications, including point-of-care diagnostics and in-the-field genotyping. Achieving these outcomes requires efficient bioinformatic algorithms for the analysis of raw nanopore signal data. However, comparing raw nanopore signals to a biological reference sequence is a computationally complex task. The dynamic programming algorithm called Adaptive Banded Event Alignment (ABEA) is a crucial step in polishing sequencing data and identifying non-standard nucleotides, such as measuring DNA methylation. Here, we parallelise and optimise an implementation of the ABEA algorithm (termed f5c) to efficiently run on heterogeneous CPU-GPU architectures.
Results
By optimising memory, computations and load balancing between CPU and GPU, we demonstrate how f5c can perform ∼3-5 × faster than an optimised version of the original CPU-only implementation of ABEA in the Nanopolish software package. We also show that f5c enables DNA methylation detection on-the-fly using an embedded System on Chip (SoC) equipped with GPUs.
Conclusions
Our work not only demonstrates that complex genomics analyses can be performed on lightweight computing systems, but also benefits High-Performance Computing (HPC). The associated source code for f5c along with GPU optimised ABEA is available at https://github.com/hasindu2008/f5c.
Keywords: Nanopore, Signal alignment, Event alignment, Methylation, GPU, GPU acceleration, Optimisation, SoC, Nanopolish, f5c
Background
Advances in genomic technologies have improved the feasibility and accessibility of rapid species identification, accurate clinical diagnostics, and specialised therapeutics, amongst other applications. The latest generation (third generation) of sequencing technologies generate data in the order of terabytes. Oxford Nanopore Technologies’ (ONT) pocket-sized MinION device generates ∼1 TB of raw signal data during a typical sequencing run, while their high-throughput PromethION device can generate >50TB of data in <60h. Computational analysis of such massive data currently poses a challenge.
Nanopore sequencing measures characteristic disruptions in the electric current (referred to hereafter as raw signal) when DNA passes through a nanopore (Fig. 1). The instantaneous current measured in the R9.4.1 pore model depends on 5-6 contiguous bases [1]. The measured signal also presents stochastic noise due to a number of factors [2]. Additionally, the speed of the DNA strand moving through the pore can vary, causing the signal to warp in the time domain [2]. The raw signal is converted to nucleotide strings (reads) through a process called base-calling (Fig. 1). Despite recent improvements, nanopore base-calling often introduces errors (∼3-5% at the time of writing) given the use of probabilistic methods to infer biological sequences from often noisy raw signal [3]. To overcome base-calling errors, raw signal can be revisited to improve the reconstitution of the base-called sequence a posteriori (Fig. 1). This process, termed ‘polishing’, can correct base-calling errors by aligning the raw signal to a biological reference sequence [4, 5], thus identifying idiosyncrasies in the raw signal by comparing observed signal levels to the expected levels at all aligned positions. Polishing can also reveal base substitutions (i.e. mutations) or base modifications such as 5-methylcytosine (5mC), a dynamic biochemical modification of DNA that is associated with genetic activity and regulation [6]. Detecting 5mC bases is important for the study of DNA methylation in the field of epigenetics [7].
Fig. 1.

Nanopore DNA sequencing and associated data analysis. A consumable flowcell containing an array of hundreds or thousands of such nanopores is loaded into the sequencing device (e.g. MinION). Ionic current (in pico amperes) is measured when DNA strands pass through nanopores to produce the raw signal, which is eventually basecalled. The base-called reads are then aligned to a reference genome. The raw signal is then revisited during the polishing step. Images of nanopore devices are reproduced with permission from ONT
A crucial algorithmic component of polishing is the alignment of raw signal—a time series of electric current—to a biological reference sequence. One of the first and most popular raw nanopore signal alignment algorithms is implemented in Nanopolish [6], which employs a dynamic programming strategy referred to as Adaptive Banded Event Alignment (ABEA). ABEA is one of the most time consuming steps during the process of analysing raw nanopore data. For instance, in-house profiling revealed that when performing methylation detection with Nanopolish, the ABEA step alone consumes ∼70% of the total CPU time. Considering the increasing amount of data generated by high-throughput nanopore sequencers, solutions are required to accelerate ABEA and reduce the turnaround time of certain nanopore sequencing applications, such as real-time polishing or methylation detection.
In this study, we describe and dissect the ABEA algorithm in detail to optimise and parallelise its execution to exploit heterogeneous CPU-GPU architectures, commonplace in mainstream computing systems. We demonstrate the utility of our GPU-optimised ABEA by incorporating a completely re-engineered version of the popular methylation detection tool Nanopolish. First, we modified the original Nanopolish methylation detection tool to efficiently utilise existing CPU resources, which we refer to as f5c. Then, we incorporated a GPU-optimised ABEA algorithm into f5c. We demonstrate how f5c enables DNA methylation detection using nanopore sequencers in real-time (i.e. on-the-fly processing of the output) by using a lightweight embedded computer system equipped with a GPU (e.g., NVIDIA Jetson TX2). We also demonstrate how f5c benefits a wide range of computing devices, from embedded systems and laptops to workstations and high performance servers. f5c is available at https://github.com/hasindu2008/f5c.
The ABEA algorithm
ABEA was first introduced in the raw nanopore signal analysis package Nanopolish [6]. The origin of the ABEA algorithm can be tracked to the Smith-Waterman (SW) dynamic programming sequence alignment algorithm that was first described in 1981. The original SW algorithm has a computational complexity of O(n2) and is most practical when the sequences are very short. Several optimisations to SW have since been introduced. Heuristic approaches, such as banded SW, attempt to reduce the search space by limiting computation along the diagonal of the dynamic programming table [8]. While the banded approach is suitable for fast alignment of second-generation sequencing data—which are composed of relatively short reads—it is less so for third generation long reads, as significantly longer width is required to contain the alignment within the band. The more recent Suzuki-Kasahara (SK) algorithm [9] uses a heuristic optimisation to banded SW that allows the band to adapt and move during the alignment, thus containing the optimal alignment within the band while allowing large gaps in the alignment. The SK algorithm is well-suited for aligning long and error-prone third generation reads in base-space (nucleotide sequences). The SK alignment algorithm was later modified and extended to ABEA in Nanopolish to enable signal-space alignment of time series signal data instead of nucleotide sequences. A simplified example of the ABEA algorithm and a representative dynamic programming table is shown in Fig. 2. Algorithm 1 summarises the ABEA algorithm and the reader may refer to Supplementary Materials for a detailed explanation.
Fig. 2.

Nanopore raw signal, events and ABEA algorithm. In aevents are the result of the event detection step (time series segmentation of the raw signal based on abrupt changes — detailed in Supplementary Materials) and true annotation is the expected output of ABEA. In b, c and d, vertical axis represents the events and horizontal axis represents the ref k-mers (k-mers within the base-called read). The dynamic programming table (DP table) is for 13 events, indexed from e0−e12 vertically, and the ref k-mers, indexed from k0−k5 horizontally. For computational and memory efficiency, only the diagonal bands (marked using blue rectangles) with a band width of W (typically W=100 for nanopore signals) are computed. The bands are computed along the diagonal from top-left (b0) to bottom-right (b17). Each cell score is computed in function of five factors: scores from the three neighbouring cells (up, left and diagonal); the corresponding ref k-mer; and, the event (shown for the cell e6, k3 via red arrows in c). Observe that all the cells in the nth band can be computed in parallel as long as the n−1th and n−2th bands are computed beforehand. To contain the optimal alignment, the band adapts by moving down or to the right as shown using blue arrows. The adaptive band movement is determined by the Suzuki-Kasahara heuristic rule [9]

Methods
CPU-GPU optimisations
f5c employs a fork-join multi-threading model (with work stealing) implemented using C POSIX threads.
Implementing the ABEA algorithm for GPU execution is not a straightforward task due to three main factors: (i) inefficient memory access patterns, which are not ideal for GPUs with relatively less powerful and smaller caches (compared to CPUs), resulting in frequent instruction stalls; (ii) read lengths of the input vary significantly (from ∼100 bases to >1M bases), requiring millions to billions of dynamic memory allocations—an expensive operation in GPUs; and (iii) non uniform distribution of read lengths in the input causes irregular utilisation of GPU cores. These challenges were overcome by: (i) tailoring the algorithm and GPU user-managed cache to exploit cache friendly memory access patterns; (ii) employing a custom heuristic-based memory allocation scheme; and (iii) using a heuristic-based work partitioning and load balancing scheme between CPU and GPU.
The GPU implementation of ABEA algorithm was performed using CUDA C. A brief summary of our optimisations is listed below.
Parallelisation and computational optimisations: To achieve fast performance on GPUs, their thousands of tiny computing cores must be sufficiently occupied. For this, thousands of parallel threads must be launched, which requires thousands of parallel tasks. This is achieved by processing a batch of reads in parallel and concurrently computing all the cells of a dynamic programming matrix band (Fig. 2b, lines ??-?? in Algorithm 1). As the bandwidth is 100 cells, a read batch of a few hundred can sufficiently occupy thousands of GPU cores. GPU core utilisation is further enhanced by improving memory access latency by using the GPU’s fast cache memory (shared memory) and a technique called memory coalescing. The current, previous and 2nd previous bands, which are frequently accessed by hundreds of threads in parallel, are kept in the shared memory. Data arrays such as the score array, trace array, reference k-mers and events that are in slow DRAM (global memory) are placed (laid out) such that contiguous threads access contiguous memory locations. This facilitates memory coalescing (one memory access can fetch data required by a large number of threads), consequently reducing the number of accesses to DRAM.
Memory optimisation: Dynamic memory allocations in the GPU memory are expensive and must be minimised for fast performance. We significantly reduced the number of dynamic memory allocations by employing a lightweight heuristic-based custom memory allocation scheme. In brief, large chunks of contiguous memory are pre-allocated when initiating the program to accommodate a batch of reads, which are then reused throughout the execution of the program. The sizes of these large chunks are determined by the available GPU memory and a heuristically determined value for the average number of events per base (i.e. average value of the number of events divided by the read length).
Heterogeneous processing: If all queried reads were of similar length, GPU threads that process the reads would complete approximately at the same time, and thus GPU cores will be equally busy throughout the execution. However, nanopore read length distributions can include reads which are significantly longer than the average read length. When the GPU threads process reads in parallel, longer reads cause all other GPU threads to wait until processing of the longest read is completed. These waiting threads lead to underutilisation of GPU cores. This issue is remedied by employing heterogeneous processing, where the CPU processes these very long reads while the GPU is processing the rest of the reads in parallel. CPU cores have a higher clock frequency than the GPU cores, therefore such very long reads can be independently and quickly processed by the CPU while the remaining reads are processed by GPU cores in parallel.
A detailed breakdown of these optimisations, experimental evidence that justify design and optimisation decisions—including a section describing the fundamentals of GPU architecture and programming—can be found in Supplementary Materials.
Biological data analysis
Comparative performance benchmarking was performed using the publicly available NA12878 (human genome) “Nanopore WGS Consortium” sequencing data [4]. The datasets used for the experiments, their statistics (number of reads, total bases, mean read length and maximum read length) and their source are listed in Table 1. Dsmall, a small subset, was used for testing a wide range of systems (all systems in Table 2, i.e. embedded system, low-end and high-end laptops, workstation and a high-performance server). Two complete nanopore MinION data sets (Dligation and Drapid) are only tested on three systems due to the larger run-time and incidental access to the other two systems. Dligation and Drapid represent the two existing nanopore sample preparation methods (ligation and rapid [10]) that affects the read length distribution.
Table 1.
Information of the datasets
| Dataset | Number of reads | Number of bases (Gbases) | Mean read length (Kbases) | Max read length (Kbases) | Source / SRA accession |
|---|---|---|---|---|---|
| Dsmall | 19275 | 0.15 | 7.7 | 196 | [11] |
| Dligation | 451020 | 3.62 | 8.0 | 1500 | ERR2184733 |
| Drapid | 270189 | 2.73 | 10.0 | 386 | ERR2184734 |
Table 2.
Different systems used for experiments
| System Name | Info | CPU | CPU cores/ threads | RAM (GB) | GPU | GPU mem (GB) | GPU arch |
|---|---|---|---|---|---|---|---|
| SoC | NVIDIA Jetson TX2 embedded module | ARMv8 Cortex-A57 + NVIDIA Denver2 | 6 / 6 | 8 | Tegra | shared with RAM | Pascal / 6.2 |
| lapL | Acer F5-573G laptop | i7-7500U | 2/4 | 8 | Geforce 940M | 4 | Maxwell / 5.0 |
| lapH | Dell XPS 15 laptop | i7-8750H | 6/12 | 16 | Geforce 1050 Ti | 4 | Pascal / 6.1 |
| ws | HP Z640 workstation | Xeon E5-1630 | 4/8 | 32 | Tesla K40 | 12 | Kepler / 3.5 |
| HPC | Dell PowerEdge | Xeon Silver 4114 | 20/40 | 376 | Tesla V100 | 16 | Volta / 7.0 |
| C4140 |
For “Speedup of ABEA algorithm”, time measurements were obtained by inserting gettimeofday timestamp function invocations directly into the C source code. Total execution time and the peak RAM usage in “Comparative performance of f5c with Nanopolish” sections were measured by running the GNU time utility with the verbose option.
Results
Speedup of ABEA algorithm
We initially compared the optimised GPU version with the optimised CPU version of the ABEA algorithm (not the unoptimised CPU version in the original Nanopolish, see below) by executing them on publicly available raw nanopore genome sequencing data. The CPU version was run with maximum supported threads on the tested systems. The optimised CPU version will be henceforth referred to as C-opti and the optimised GPU version will be referred to as G-opti.
First we benchmarked on five different systems (Table 2) over Dsmall dataset. Speedups (including all the overheads) observed for G-opti compared to C-opti are: ∼4.5× on the low-end-laptop and the workstation; ∼4× on Jetson TX2 SoC; and ∼3× on high-end-laptop and HPC (Fig. 3). Note that only a ∼3× speedup was observed on high-end-laptop and HPC (versus >=4 × on other systems) due to the CPU on those particular systems having a comparatively higher amount of CPU cores (12 and 40 respectively).
Fig. 3.
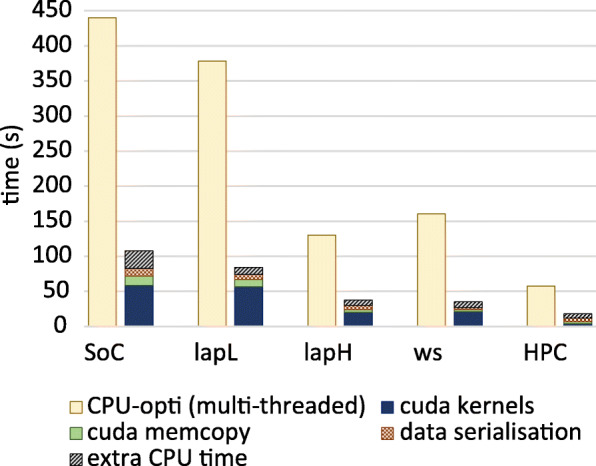
Performance comparison of ABEA on CPU vs GPU over a wide range of systems. Runtime for C-opti (left bars) and the G-opti (right bars). Runtime for the GPU has been broken down into: compute kernel time; different overheads (memory copying to/from the GPU, data serialisation time), and the extra CPU time due to CPU processing of the reads. The compute kernel time includes the sum of time for all GPU kernels. The extra CPU time is the additional time spent by the CPU to process very long reads and ultra long reads (see Supplementary materials) assigned to the CPU (excluding the processing time that overlaps with the GPU execution, i.e. only the extra time which the GPU has to wait after the execution is included)
We next benchmarked on two larger datasets (Drapid and Dligation). A speedup up of ∼3× was observed for all three systems for the two big datasets— Dligation and Drapid (Fig. 4). Due to more ultra long reads (>100kb) in Dligation and Drapid than in Dsmall, the overall speedup for SoC is limited to around ∼3× compared to ∼4× for Dsmall.
Fig. 4.
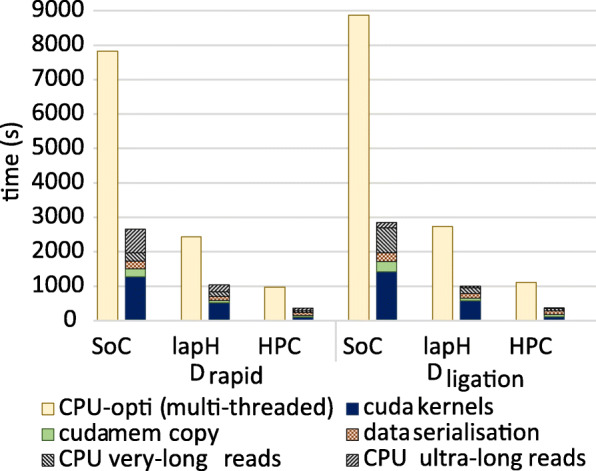
Performance comparison of ABEA on CPU vs GPU across over large datasets Runtime for C-opti (left bars) and the G-opti (right bars). Runtime for the GPU has been broken down into: compute kernel time; different overheads (memory copying to/from the GPU, data serialisation time), extra CPU time due to very long reads and ultra long reads (see Supplementary materials)
It is noteworthy to mention that comparing performance to the unoptimised CPU version in Nanopolish is not straightforward, as the time for individual components (e.g. ABEA) cannot be accurately measured because each read executes on its own code path (detailed in Supplementary Materials). We nonetheless estimated the runtime of unoptimised ABEA by injecting timestamp (gettimeofday) functions into the original Nanopolish code, directly before and after the ABEA component to measure runtimes for individual reads. Nanopolish was launched with multiple threads and the runtimes were averaged by the number of threads to get a reasonable estimate for ABEA. When evaluated using the Dsmall dataset, the optimised ABEA CPU version in f5c was ∼1.3-1.7 × times faster than the unoptimised ABEA in the original Nanopolish program (∼1.4× speedup on Jetson TX2, workstation and HPC, ∼1.7× on low-end-laptop and ∼1.3× on high-end-laptop).
Comparative performance of f5c with Nanopolish
The overall performance of the GPU-accelerated ABEA algorithm was evaluated through a DNA methylation (5-methylcytosine) detection work-flow. We compared the total runtime for methylation calling using the original Nanopolish against f5c (both CPU-only and GPU-accelerated versions) by running on two publicly available nanopore datasets (see “Methods” section).
We refer to the original Nanopolish (version 0.9) as nanopolish-unopti, f5c run only on the CPU as f5c-C-opti and GPU accelerated f5c as f5c-G-opti. We executed nanopolish-unopti, f5c-C-opti and f5c-G-opti on the full datasets Drapid and Dligation. Note that all execution instances were performed with the maximum number of CPU threads available on each system.
f5c-C-opti on the Drapid dataset was: ∼2× faster than nanopolish-unopti on SoC and lapH and ∼4× faster on HPC. On Dligation, nanopolish-unopti crashed on SoC (limited by 8GB RAM) and lapH (16GB RAM) due to the Linux Out Of Memory (OOM) killer [12] (Fig. 5). On Dligation, f5c-C-opti on HPC was not only 6 × faster than nanopolish-unopti, but also consumed only ∼15 GB RAM, as opposed to >100 GB used by nanopolish-unopti (both with 40 compute threads). Hence, it is evident that CPU optimisations alone can do significant improvements.
Fig. 5.
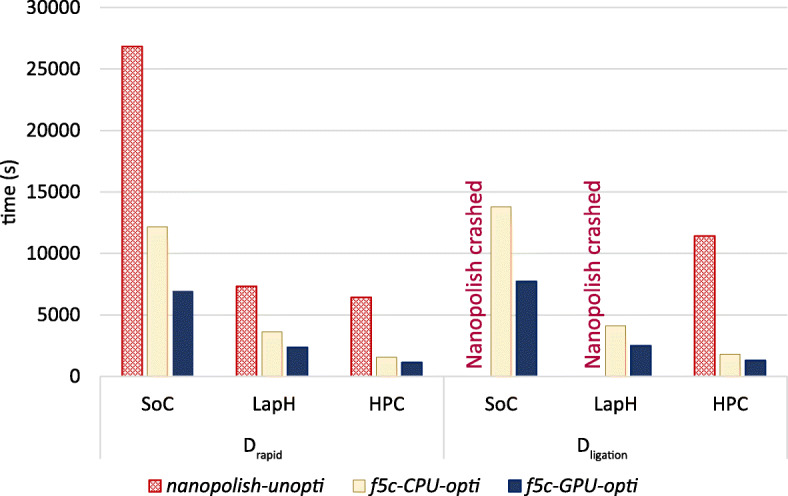
Comparison of f5c to Nanopolish. The reported run-times are for the complete methylation calling and also include disk I/O time
When comparing the total execution time (including disk I/O) of the entire methylation calling process with different hardware acceleration options in f5c, f5c-G-opti was 1.7 × faster than f5c-C-opti on SoC, 1.5-1.6 × on lapH and <1.4× on HPC (Fig. 5). On HPC, the speedup was limited to <1.4× due to file I/O being the bottleneck. N.B. only the ABEA algorithm step utilises the GPU acceleration.
For the Drapid dataset, the execution time of f5c-G-opti versus nanopolish-unopti was ∼4×, ∼3× and ∼6× faster on SoC, lapH and HPC, respectively (Fig. 5). On the Dligation dataset on HPC, f5c-G-opti was a remarkable ∼9× faster.
Although parameters that may affect biological accuracy were untouched, we did observe subtle variations in the output as a consequence of hardware-based fluctuations in the treatment of floating point units. We assessed the impact of these subtle variations on the measurement of relative methylation frequencies by comparing results for Nanopolish, f5c-C-opti and f5c-G-opti on the Dsmall dataset, which encompasses 5M bases of human chromosome 20 with an average read coverage of 30 ×. Of the ∼32,000 surveyed CpG sites, f5c-C-opti and f5c-G-opti produced different methylation frequencies for only 6 (∼0.02%) and 65 (∼0.2%) positions, with an average position-specific difference in methylation frequency values of ∼1.5% and ∼0.4%, respectively. Both variants of f5c yielded overall Pearson correlation values of 0.99999 with Nanopolish. Moreover, the overall correlation between Nanopolish and bisulfite sequencing data from NA12878 is 0.88723, while the correlation for f5c-C-opti and f5c-G-opti is 0.88723 and 0.88724, respectively. The impact of hardware-based differences in the calculation of methylation frequencies is therefore negligible.
Discussion and future work
High-throughput nanopore data analysis is a relatively new field that emerged with the release of the first ONT sequencing device (MinION) in 2014. Numerous nanopore data analysis algorithms have since been developed by biologists and bioinformaticians. However, work that explores computational bottlenecks, acceleration and parallisation techniques for such algorithms are limited, especially for those that exploit raw nanopore signal data, such as the ABEA algorithm.
There are a handful of methods that have been developed to accelerate the analysis of nanopore data. The proprietary base-calling software Guppy developed by ONT exploits NVIDIA GPUs for fast and accurate processing of raw nanopore data via deep neural networks [3]. Although the design details of Guppy are not publicly disclosed, they are likely to have benefited by a plethora of work focusing on GPU optimisations for neural networks. Another example of highly optimised software for third generation sequencing data is minimap2, a popular open source sequence aligner for long reads (including nanopore reads) that has recently been accelerated with the simultaneous use of GPUs and Intel Xeon Phi co-processors [13]. However, alignment in base-space is considerably different from signal-space, which is explored in this work. Recently, the NVIDIA corporation has shown an interest in developing open source libraries such as Clara Genomics[14] for accelerating long read data analysis on their GPUs. The Clara Genomics library contributes to nanopore data analysis domain through the acceleration of core algorithmic components such as all-vs-all read mapping and partial order alignments for genome assembly. Nonetheless, none of these algorithms focus on accelerating signal-space alignment.
A number of GPU accelerated versions of SW alignment have previously been reported [15–17]. However, differences between SW and ABEA significantly affect the efficient mapping of the algorithm and data structures to GPU architectures. For instance, band movement in ABEA during execution and random memory accesses to the pore-model in ABEA affect data dependencies (thus, the parallelism) and the memory layout (thus, the memory access patterns). Therefore, the GPU acceleration solutions proposed in these reports are ill-suited for ABEA. In addition, the above-mentioned works were developed for short, static read-lengths. Third generation sequencers produce variable long read lengths that vary significantly over a given dataset. Consequently, the strategies we disclose herein for efficient GPU memory allocation and load balancing are novel and significant improvements for ABEA.
Moreover, we demonstrate that a complete DNA methylation analysis of a human genome using raw Oxford Nanopolish sequencing data can be executed on an embedded system (e.g., a SoC equipped with ARM processor and an NVIDIA GPU) as shown in Fig. 6. The data processing speed is sufficient to keep up with data generated in real-time by four Oxford Nanopore MinION devices in parallel, or a GridION sequencer. GPU-enabled f5c can process such data using a single NVIDIA TX2 SoC, at a speed of >600 Kbases per second to keep up with the sequencing output (∼600 Kbases per second [18]), as shown in Fig. 6. Conversely, if the original Nanopolish was executed on the NVIDIA TX2 SoC, the processing speed is limited to ∼256 Kbases per second. The base-space alignment speed of ∼715 Kbases/s in Fig. 6 was obtained by running Minimap2 [19] on the Jetson TX2 with only 8GB using the partitioned-index approach we previously presented in [20].
Fig. 6.
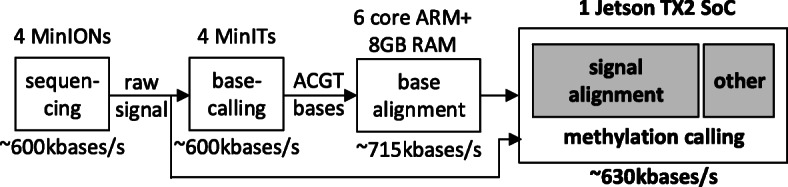
Human genome processing on-the-fly
It is also noteworthy to mention that we used Nanopolish v0.9 for the performance comparisons as subsequent releases of the software incorporated some of our optimisations, excluding those that did not require major code refactoring, such as GPU implementation, threading models and I/O processing interleaving. Furthermore, the optimisations we performed were focused on restructuring and fine-tuning the implementation of the algorithm to conform with computer hardware. Parameters that might affect the biological accuracy were untouched. Still, we extensively compared results of f5c with that of Nanopolish and verified that they are almost identical, greatly surpassing variation observed using alternative experimental approaches (i.e, bisulfite sequencing for 5mC detection).
Our work can not only reduce the hardware and bandwidth requirements for analysing raw Nanopore data, but can also improve the turnaround time for performing reference-guided raw nanopore signal processing, an analytic process that is used for base-calling and detecting non-standard nucleotides. In addition to embedded systems, our work benefits all computational systems, with or without GPU. For instance, our work enables methylation calling on laptops with <16GB of RAM. Furthermore, we have demonstrated that a posteriori methylation calling execution with f5c on high performance computers also benefits from a significant speedup.
Conclusions
ABEA is a prominent bioinformatics algorithm for raw nanopore signal analysis. Although this algorithm is not massively parallel, we present a highly efficient implementation of ABEA that includes the (optional) use of GPUs. Through a number of memory optimisations and a heterogeneous processing strategy that uses both CPU and GPU, we were able to overcome several inherent challenges, such as prominent variations in sequencing read lengths. Our optimisations yield around 3-5 × performance improvement on a CPU-GPU system when compared to CPU only. We demonstrate that these optimisations are sufficient for the execution and completion of a DNA methylation detection workflow on an embedded SoC equipped with a hexa-core ARM processor and NVIDIA GPU (256 cores) in real-time. This work not only benefits embedded SoCs, but also a wide range of systems equipped with GPUs, from laptops to servers, as highlighted by a 9 × speedup and 6-fold memory reduction when performing methylation detection on a high-performance computing server. The source code of f5c is made available at https://github.com/hasindu2008/f5c.
Supplementary information
Additional file 1 Supplementary materials. PDF file that details all the supplementary materials.
Acknowledgements
We thank NVIDIA for providing the Jetson TX2 board (to University of New South Wales) and Tesla K40 GPU (to University of Peradeniya) through the GPU donation programme. We thank Dr. Roshan Ragel and Dr. Swarnalatha Radhakrishnan at University of Peradeniya who assisted the research. We thank our colleagues who provided support, especially, James Ferguson and Shaun Carswell at Garvan Institute of Medical Research; Thomas Daniell, Hassaan Saadat and Darshana Jayasinghe at University of New South Wales; Geesara Pratap at Innopolis University; and, Pim Schravendijk. We also thank the Data Intensive Computer Engineering (DICE) team at Data Sciences Platform, Garvan Institute of Medical Research.
Abbreviations
- ABEA
Adaptive banded event alignment
- CPU
Central processing unit
- GPU
Graphics processing unit
- SoC
System on chip
- HPC
High-performance computer/computing
- ONT
Oxford nanopore technologies
- SW
Smith-waterman
- SK
Suzuki-kasahara
- POSIX
Portable operating system interface
- CUDA
Compute unified device architecture
- DRAM
Dynamic random access memory
- RAM
Random access memory
- GNU
GNU’s not unix
- OOM
Out Of memory
Authors’ contributions
H.G. and S.P. conceived the study. H.G., C.W.L., H.S. and G.J. re-engineered, wrote, modified and optimised software. H.G. designed, developed and optimised the GPU implementation. H.G. and C.W.L. conducted the experiments and benchmarks. M.A.S., J.T.S and S.P. provided strategic oversight for the work. H.G, M.A.S. and S.P wrote and revised the manuscript. J.T.S. critically revised the manuscript. All author(s) read and approved the manuscript.
Funding
J.T.S. is supported by the Ontario Institute for Cancer Research through funds provided by the Government of Ontario and the Government of Canada through Genome Canada and Ontario Genomics (OGI-136).
Availability of data and materials
All data generated or analysed during this study are included in this published article and its supplementary information files. Source code of f5c is available in the GitHub repository, https://github.com/hasindu2008/f5c. A reproducible Code Ocean compute capsule is available at 10.24433/CO.3078978.v1
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
H.G. received a travel bursary from Oxford Nanopore Technologies to present at conferences. J.T.S. receives research funding from ONT and has received travel support to attend and speak at meetings organized by ONT. M. A. S. received travel and accommodation expenses to speak at Oxford Nanopore Technologies conferences and are listed as inventors on patent PCT/AU2018/050265 relating to signal alignment.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Martin A. Smith and Sri Parameswaran contributed equally to this work.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s12859-020-03697-x.
References
- 1.Lu H, Giordano F, Ning Z. Oxford nanopore minion sequencing and genome assembly. Genomics Proteomics Bioinforma. 2016;14(5):265–79. doi: 10.1016/j.gpb.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rang FJ, Kloosterman WP, de Ridder J. From squiggle to basepair: computational approaches for improving nanopore sequencing read accuracy. Genome Biol. 2018;19(1):90. doi: 10.1186/s13059-018-1462-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wick RR, Judd LM, Holt KE. Performance of neural network basecalling tools for oxford nanopore sequencing. Genome Biol. 2019;20(1):129. doi: 10.1186/s13059-019-1727-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jain M, Koren S, Miga KH, Quick J, Rand AC, Sasani TA, Tyson JR, Beggs AD, Dilthey AT, Fiddes IT, et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat Biotechnol. 2018;36(4):338. doi: 10.1038/nbt.4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Loman NJ, Quick J, Simpson JT. A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat Methods. 2015;12(8):733. doi: 10.1038/nmeth.3444. [DOI] [PubMed] [Google Scholar]
- 6.Simpson JT, Workman RE, Zuzarte P, David M, Dursi L, Timp W. Detecting dna cytosine methylation using nanopore sequencing. Nat Methods. 2017;14(4):407. doi: 10.1038/nmeth.4184. [DOI] [PubMed] [Google Scholar]
- 7.Bird A. Dna methylation patterns and epigenetic memory. Genes Dev. 2002;16(1):6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 8.Chao K-M, Pearson WR, Miller W. Aligning two sequences within a specified diagonal band. Bioinformatics. 1992;8(5):481–7. doi: 10.1093/bioinformatics/8.5.481. [DOI] [PubMed] [Google Scholar]
- 9.Suzuki H, Kasahara M. Introducing difference recurrence relations for faster semi-global alignment of long sequences. BMC Bioinformatics. 2018;19(1):45. doi: 10.1186/s12859-018-2014-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Technologies ON. Ligation sequencing kit 1D or rapid sequencing kit. 2017. https://store.nanoporetech.com/media/Ligation_Sequencing_Kit_1D_or_Rapid_Sequencing_Kit_v5_Feb2017.pdf. Accessed 29 July 2020.
- 11.Simpson J. Stats and analysis. 2017. https://nanopolish.readthedocs.io/en/latest/quickstart_call_methylation.html. Accessed 29 July 2020.
- 12.Chase R. How to configure the linux out-of-memory killer. 2013. https://www.oracle.com/technical-resources/articles/it-infrastructure/dev-oom-killer.html. Accessed 29 July 2020.
- 13.Feng Z, Qiu S, Wang L, Luo Q. Proceedings of the 48th International Conference on Parallel Processing. Kyoto: ACM; 2019. Accelerating long read alignment on three processors. [Google Scholar]
- 14.NVIDIA. Clara genomics. 2020. https://developer.nvidia.com/Clara-Genomics. Accessed 20 Jan 2020.
- 15.Liu Y, Huang W, Johnson J, Vaidya S. International Conference on Computational Science. Berlin: Springer; 2006. Gpu accelerated smith-waterman. [Google Scholar]
- 16.Manavski SA, Valle G. Cuda compatible gpu cards as efficient hardware accelerators for smith-waterman sequence alignment. BMC Bioinformatics. 2008;9(2):10. doi: 10.1186/1471-2105-9-S2-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Y, Maskell DL, Schmidt B. CUDASW++: optimizing Smith-Waterman sequence database searches for CUDA-enabled graphics processing units. BMC Res Notes. 2009;2(1):73. doi: 10.1186/1756-0500-2-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Technologies ON. MinIT is out - an analysis and device control accessory to enable powerful, real-time DNA/RNA sequencing by anyone, anywhere. 2018. https://nanoporetech.com/about-us/news/minit-launch. Accessed 29 July 2020.
- 19.Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34(18):191. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gamaarachchi H, Parameswaran S, Smith MA. Featherweight long read alignment using partitioned reference indexes. Sci Rep. 2019;9(1):4318. doi: 10.1038/s41598-019-40739-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1 Supplementary materials. PDF file that details all the supplementary materials.
Data Availability Statement
All data generated or analysed during this study are included in this published article and its supplementary information files. Source code of f5c is available in the GitHub repository, https://github.com/hasindu2008/f5c. A reproducible Code Ocean compute capsule is available at 10.24433/CO.3078978.v1