

There is long-standing interest in therapies targeting full-length CD33 (CD33FL) for acute myeloid leukemia (AML).1–3 Longer survival with gemtuzumab ozogamicin (GO) in some patients validates this approach but GO does not benefit many patients with CD33+ leukemias.4 Numerous efforts to develop more effective CD33-directed therapeutics are therefore ongoing.5
The immune-dominant epitopes recognized by existing CD33 therapeutics are located within the membrane-distal V-set domain of CD33FL (Supplementary Figure 1).5 However, alternative splicing of CD33 results in the transcription of several shorter isoforms in AML cells, with one variant missing exon 2 (CD33ΔE2) being of particular interest.6 CD33ΔE2 is predicted to contain only the membrane-proximal C2-set domain in its extracellular portion and is, at the mRNA level, present in myeloblasts from all AML patients.6 A single nucleotide polymorphism (SNP), rs12459419, modulates exon 2 splicing efficiency.7–9 The minor (T) allele is associated with reduced CD33FL expression on AML cells and, conversely, increased CD33ΔE2 transcription.9 Data from the pediatric COG-AAML0531 trial, showing improved outcome with GO add-on to intensive chemotherapy was limited to patients homozygous for the major C allele,9 suggest CD33 splicing events may be clinically important. This observation has prompted interest in targeting CD33ΔE2 with antibodies recognizing the C2-set domain, perhaps particularly for the 50% of patients with rs12459419 CT or TT genotypes. Human AML cells can be generated that display cell surface CD33ΔE2 when expression is forced via lentiviral transduction6 but whether CD33ΔE2 is a viable therapeutic target is unknown. Since commercial antibodies specifically recognizing CD33ΔE2 do not exist, rigorous testing to what degree endogenous CD33ΔE2 mRNA is translated, post-translationally modified, and transported to the cell surface of AML cells has so far not been possible.
To examine expression and localization of CD33ΔE2 in human AML cell lines and primary AML blasts, we therefore first raised antibodies that bind human CD33ΔE2 but not CD33FL (Supplementary Figure 1). Briefly (see Online Supplement for detailed methods), immunogens consisting of the murine Fc domain and either the entire extracellular domain of human CD33FL or the entire extracellular domain of human CD33ΔE2 were generated, expressed in HEK293 cells, and purified (Supplementary Figure 2). BALB/c, CD1, and F1 mice were injected with a mixture of both immunogens. We identified two hybridomas, 11D5 and 13E11, showing binding to CD33ΔE2 but not CD33FL in early screening assays (data not shown). 11D5 and 13E11 were then sequenced and purified monoclonal antibodies generated (Supplementary Figure 2).10
To confirm the specificity of 11D5 and 13E11 as recombinant antibodies, we used parental CD33neg human acute lymphoblastic leukemia (ALL) REH cells and sublines engineered to overexpress either CD33FL or CD33ΔE2.6 As shown by flow cytometry (Figure 1A) and immunofluorescence microscopy (Supplementary Figure 3), 11D5 and 13E11 only bound cells expressing CD33ΔE2 but neither parental cells nor cells expressing CD33FL, indicating the V-set domain (present in CD33FL) hinders access of both antibodies to CD33. Testing of both antibodies with additional CD33neg human ALL cell lines (RS4;11, RCH-ACV) and a CD33low human AML cell line (OCI-AML3) engineered to overexpress CD33FL or CD33ΔE2 confirmed their specificity for CD33ΔE2 (Supplementary Figure 4), demonstrating both 11D5 and 13E11 recognize epitopes within the membrane-proximal C2-set domain of CD33 that are only present/accessible in the absence of the V-set domain (“CD33ΔE2-specific antibody”).
Figure 1. Cell surface characterization of human acute leukemia cells with anti-CD33ΔE2 antibodies.
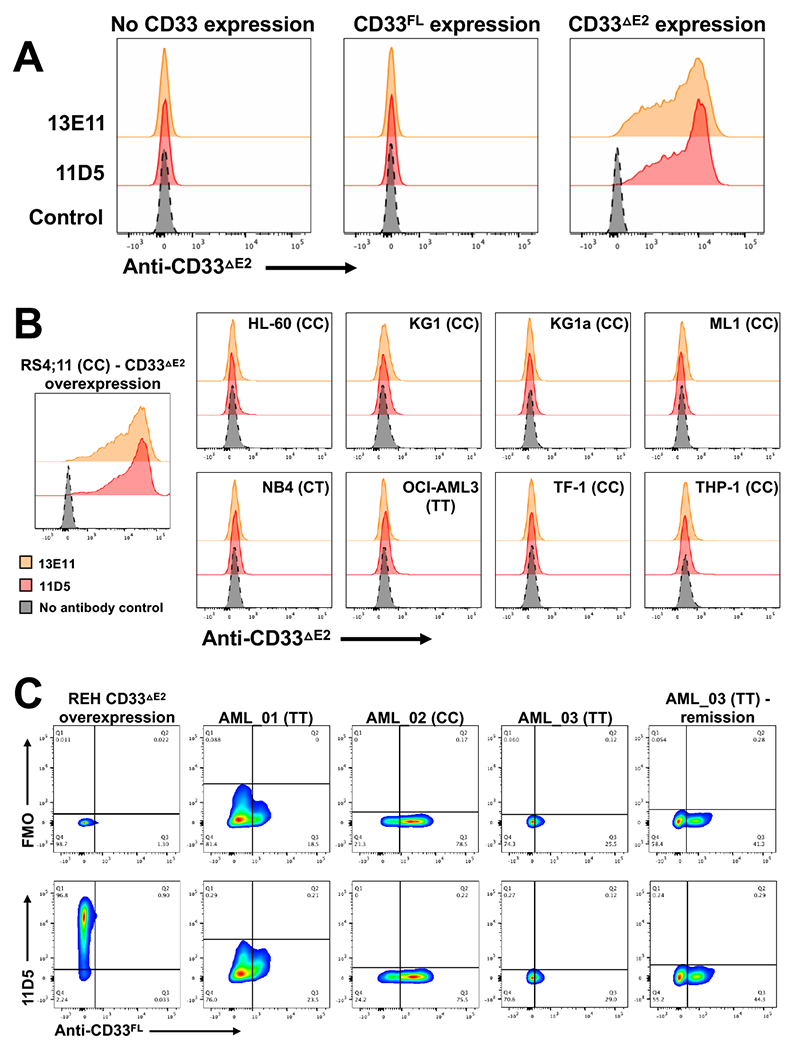
Anti-CD33ΔE2 antibody clones 11D5 and 13E11 were tested by flow cytometry against (A) parental REH cells (human acute lymphoblastic cell line, endogenously CD33neg) and sublines engineered to overexpress CD33FL and CD33ΔE2 as well as (B) a panel of human AML cell lines and, as a positive control, RS4;11 (human acute lymphoblastic cell line, endogenously CD33neg) cells engineered to overexpress CD33ΔE2. A no-primary-antibody control was included. Data are representative of three separate experiments. CD33 rs12459419 genotypes are shown in parentheses. (C) The anti-CD33ΔE2 antibody 11D5 and standard anti-CD33FL antibody clone p67.6 were used to co-stain three frozen/thawed samples from AML patients with active disease (AML_01, AML_02, AML_03) as well as a corresponding remission sample from AML_03. As a positive control, engineered REH cells overexpressing CD33ΔE2 were included. Both full stain and fluorescence minus one (FMO, −11D5) are shown. CD33 rs12459419 genotypes are shown in parentheses. Abbreviations: CD33FL, full length CD33 isoform; CD33ΔE2, CD33 isoform arising from transcript missing exon 2.
The predicted canonical signal peptide of CD33 consists of 16 amino acids, with 12 amino acids encoded by exon 1 and 4 amino acids encoded by exon 2, and cleavage predicted to occur after the 16th residue (Supplementary Figure 5). Since CD33ΔE2 retains exon 1 but lacks exon 2, the predicted signal peptide cleavage site present in CD33FL is lost. Computational models identified only 2 low-probability cleavage sites in the 5’ portion of exon 3 (Supplementary Figure 5). Thus, we considered the possibility that the amino acids encoded by exon 1 (which are cleaved in CD33FL) are retained in the mature CD33ΔE2 protein. Consistent with this possibility, mass spectrometry detected the signal peptide as part of the CD33ΔE2 immunogen (Supplementary Figure 6). To test whether the signal peptide served as binding epitope for 11D5 and/or 13E11, we generated two variant CD33 constructs with modified signal peptide sequences. Specifically, we generated a CD33ΔE2 variant in which the N-terminal amino acids of exon 2 that comprise the signal peptide for CD33FL were included (Supplementary Figure 5, “CD33ΔE2 + SPFL”). Additionally, we generated a CD33FL variant in which the N-terminal amino acids encoded by exon 2 were replaced with the N-terminal amino acids of exon 3 to mimic the structure of the signal peptide in CD33ΔE2 (Supplementary Figure 5, “CD33FL + SPΔE2”). As depicted in Supplementary Figure 7, 13E11 but not 11D5 bound CD33FL + SPΔE2, whereas 11D5 but not 13E11 bound CD33ΔE2 + SPFL. Together, these findings suggest 13E11 and 11D5 bind non-overlapping epitopes on CD33ΔE2, with 13E11 but not 11D5 recognizing the signal peptide retained in CD33ΔE2.
We previously reported universal expression of CD33ΔE2 at the mRNA transcript level in human AML cell lines and primary bone marrow and peripheral blood samples from patients with AML.6 With 13E11 and 11D5 now available, we tested whether CD33ΔE2 could be detected as protein on the cell surface of human AML cells. In a panel of human AML cell lines shown to express CD33ΔE2 mRNA (Supplementary Figure 8), neither 13E11 nor 11D5 detected cell surface CD33ΔE2 (Figure 1B). Moreover, in 2 primary human AML specimens (both of which expressed CD33ΔE2 mRNA; Supplementary Figure 8), flow cytometric analyses with 11D5 similarly did not demonstrate any CD33ΔE2 protein on AML blasts (Figure 1C). We also found no evidence of CD33ΔE2 in 4 healthy donor bone marrow specimens which all expressed CD33FL (Supplementary Figure 9A). Of note, all three rs12459419 genotypes were represented in our panel of cell lines, primary AML cells, and healthy donor samples. To study the potential expression of CD33ΔE2 on primary AML cells in more detail, we performed multiparameter flow cytometric analyses of 21 residual clinical samples positive for AML or myelodysplastic syndrome in which directly-labeled 11D5 was integrated in a multi-tube reagent panel. In all 21 samples, no convincing evidence of expression of CD33ΔE2 was identified above background.
Consistent with our findings, recent studies have indicated CD33ΔE2 protein is not expressed on the cell surface of healthy donor neutrophils and monocytes.11 Rather, some evidence suggested CD33ΔE2 was retained intracellularly, accumulating in peroxisomes.11 To address this possibility in AML, we conducted flow cytometric studies with 11D5 on permeabilized AML cells that we found to express CD33ΔE2 mRNA. In REH cells lentivirally-transduced to overexpress CD33ΔE2, staining was retained following permeabilization, demonstrating that the target antigen can still be detected after the permeabilization process. Likewise, 11D5 bound ML-1 cells in which endogenous CD33 was deleted via CRISPR/Cas9 and CD33ΔE2 introduced via lentiviral mediated gene transfer (ML-1del33+CD33ΔE2) (Figure 2A). Neither REHCD33ΔE2 nor ML-1del33+CD33ΔE2 cells showed increased 11D5 staining following permeabilization. Similarly, neither HL-60 or U937 cells nor primary AML patient cells stained with 11D5 after permeabilization. An ML-1 cell subline with CRISPR/Cas9-mediated deletion of exon 2 with resulting exclusive expression of CD33ΔE2 mRNA12 likewise did not show cell surface or intracellular CD33ΔE2 protein expression (Figure 2A), arguing against the possibility that heterodimer formation between CD33FL and CD33ΔE2 could prevent antibody access to endogenously-expressed CD33ΔE2. These cells, however, stained positive with an antibody against myeloperoxidase used as a positive control for detection of an intracellular antigen. These findings suggested the lack of CD33ΔE2 cell surface expression in AML is not due to sequestration of CD33ΔE2 protein in intracellular compartments. To test further whether CD33ΔE2 protein might be retained in intracellular compartments, we performed immunoprecipitations using commercial antibodies raised against C-terminal CD33 peptides to enrich for both CD33FL and CD33ΔE2. As shown in Figure 2B, we found CD33 protein with the predicted size of CD33ΔE2 in human ALL cell lines forced to overexpress CD33ΔE2. In contrast, a similar protein band was not found in any of the parental human AML cell lines or 4 primary AML patient samples studied, again suggesting that CD33ΔE2 protein is not present at a level detectable by antibody staining in human AML cells. It is plausible but speculative that retention of a non-cleaved signal peptide could alter intracellular trafficking and target CD33ΔE2 for degradation, as has been demonstrated for other proteins (e.g. references13,14).
Figure 2. Whole cell characterization of human acute leukemia cells with anti-CD33ΔE2 antibodies.
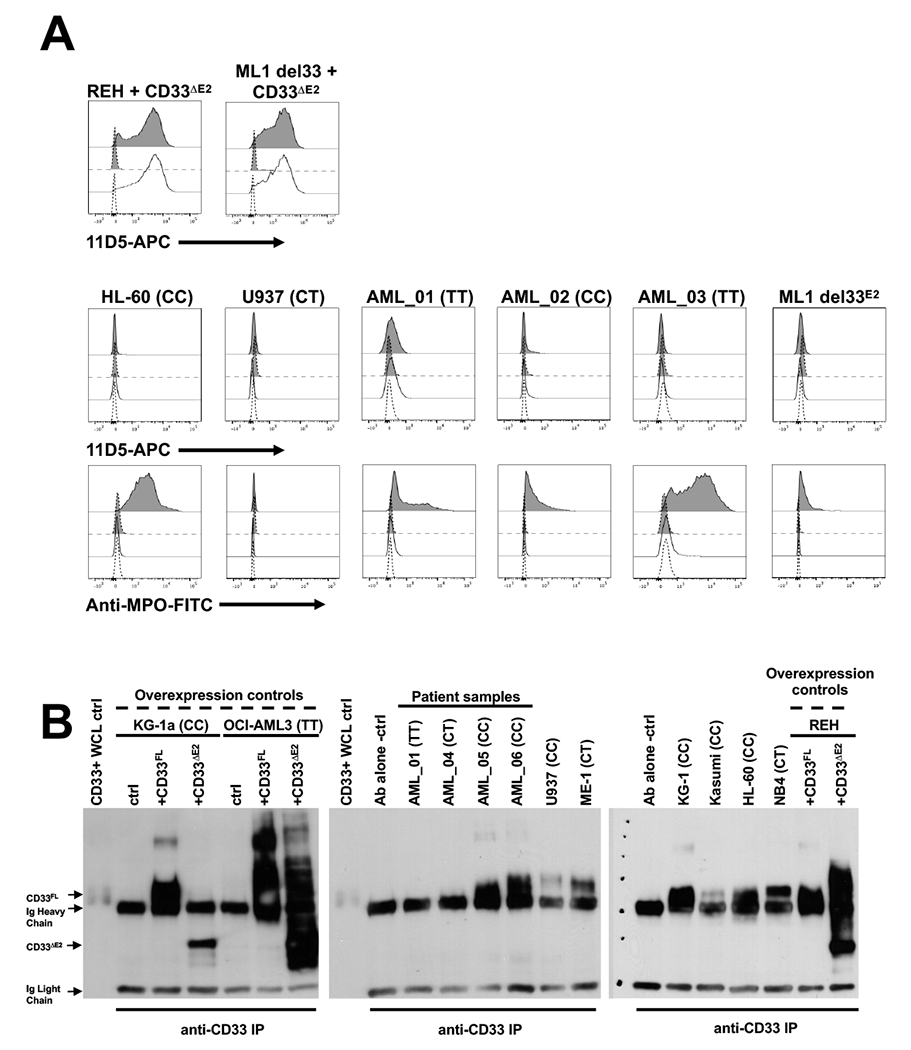
(A) Engineered REH cells overexpressing CD33ΔE2, ML-1 cells with deletion of CD33 (del CD33) via CRISPR/Cas9 and subsequent overexpression of CD33ΔE2, HL-60 cells, U937 cells, three primary AML patient specimens (AML_01, AML_02, AML_03) and an ML-1 cell subline with genomic deletion of CD33 exon 2 via CRISPR/Cas9 (del33E2) with resulting exclusive expression of CD33ΔE2 transcript12 were included. All cell lines and patient specimens were stained with 11D5-APC with (gray histograms) or without (white histograms) permeabilization to detect intracellular antigens. Anti-myeloperoxidase (MPO) antibody was used as a positive control for intracellular antigen detection. Dotted lines represent isotype control antibody. Results are representative of N=3 separate experiments. CD33 rs12459419 genotypes are shown for non-engineered cell lines and patient specimens in parentheses. (B) Antibodies against the C-terminal portion of CD33 (present in both CD33FL and CD33ΔE2) were used for immunoprecipitation and subsequent immunoblotting to enrich for, and detect, CD33 isoforms. KG-1a, OCI-AML3, and REH cells engineered to overexpress either CD33FL or CD33ΔE2 are shown as positive/negative controls together with a panel of parental AML cells lines and thawed AML patient samples. CD33 rs12459419 genotypes are shown in parentheses. Ab, antibody; ctrl, control; IP, immunoprecipitation; WCL, whole cell lysate.
Together, in our studies conducted with newly-developed CD33ΔE2-specific antibodies, we were unable to detect CD33ΔE2 on human AML cell lines or primary blast cells from a smaller cohort of AML patients, which contained cells with CC, CT, and TT rs12459419 genotypes. Different from what has been reported previously in normal neutrophils and monocytes,11 we could not find CD33ΔE2 protein accumulations in intracellular compartments of AML cells or maturing healthy donor myeloid cells. While we cannot exclude that CD33ΔE2 might be expressed on the cell surface of myeloblasts in a smaller subset of AML patients, or might be expressed at earlier differentiation stages in AML, our data do not provide evidence for the value of CD33ΔE2 as a therapeutic target in AML regardless of the CD33 rs12459419 genotype of the patient.
Supplementary Material
ACKNOWLEDGMENTS
Research reported in this publication was supported by the Leukemia & Lymphoma Society (Translational Research Program, grant 6489-16) and the National Institutes of Health/National Cancer Institute (NIH/NCI) (R21-CA234203, P30-CA015704, and P50-CA100632 [MD Anderson Cancer Center Leukemia SPORE]). C.D.G. is supported by a fellowship training grant from the NIH/National Heart, Lung, and Blood Institute (NHLBI; T32-HL007093), an institutional K12 grant from the NIH/NCI (K12-CA076930) an American Society of Clinical Oncology/Conquer Cancer Foundation Young Investigator Award and an Alex’s Lemonade Stand Young Investigator Grant.
Conflict of interest: H.P.K. is a consultant to and has ownership interests with Rocket Pharma and Homology Medicines and is a consultant to CSL Behring and Magenta Therapeutics. R.B.W. received laboratory research grants and/or clinical trial support from Agios, Amgen, Aptevo Therapeutics, Arog, BioLineRx, Jazz, Pfizer, Seattle Genetics, and Selvita; has ownership interests with Amphivena Therapeutics; and is (or has been) a consultant to Agios, Amphivena Therapeutics, Astellas, BiVictrix, Boehringer Ingelheim, Covagen, Emergent Biosolutions/Aptevo Therapeutics, Jazz, Kite, Pfizer, and Seattle Genetics. The other authors declare no competing financial interests.
REFERENCES
- 1.Grossbard ML, Press OW, Appelbaum FR, Bernstein ID, Nadler LM. Monoclonal antibody-based therapies of leukemia and lymphoma. Blood 1992; 80(4): 863–878. [PubMed] [Google Scholar]
- 2.Walter RB, Appelbaum FR, Estey EH, Bernstein ID. Acute myeloid leukemia stem cells and CD33-targeted immunotherapy. Blood 2012; 119(26): 6198–6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Laszlo GS, Estey EH, Walter RB. The past and future of CD33 as therapeutic target in acute myeloid leukemia. Blood Rev 2014; 28(4): 143–153. [DOI] [PubMed] [Google Scholar]
- 4.Godwin CD, Gale RP, Walter RB. Gemtuzumab ozogamicin in acute myeloid leukemia. Leukemia 2017; 31(9): 1855–1868. [DOI] [PubMed] [Google Scholar]
- 5.Walter RB. Investigational CD33-targeted therapeutics for acute myeloid leukemia. Expert Opin Investig Drugs 2018; 27(4): 339–348. [DOI] [PubMed] [Google Scholar]
- 6.Laszlo GS, Harrington KH, Gudgeon CJ, Beddoe ME, Fitzgibbon MP, Ries RE. et al. Expression and functional characterization of CD33 transcript variants in human acute myeloid leukemia. Oncotarget 2016; 7(28): 43281–43294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Malik M, Simpson JF, Parikh I, Wilfred BR, Fardo DW, Nelson PT. et al. CD33 Alzheimer’s risk-altering polymorphism, CD33 expression, and exon 2 splicing. J Neurosci 2013; 33(33): 13320–13325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Raj T, Ryan KJ, Replogle JM, Chibnik LB, Rosenkrantz L, Tang A. et al. CD33: increased inclusion of exon 2 implicates the Ig V-set domain in Alzheimer’s disease susceptibility. Hum Mol Genet 2014; 23(10): 2729–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lamba JK, Chauhan L, Shin M, Loken MR, Pollard JA, Wang YC. et al. CD33 splicing polymorphism determines gemtuzumab ozogamicin response in de novo acute myeloid leukemia: report from randomized phase III Children’s Oncology Group trial AAML0531. J Clin Oncol 2017; 35(23): 2674–2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Correnti CE, Laszlo GS, de van der Schueren WJ, Godwin CD, Bandaranayake A, Busch MA. et al. Simultaneous multiple interaction T-cell engaging (SMITE) bispecific antibodies overcome bispecific T-cell engager (BiTE) resistance via CD28 co-stimulation. Leukemia 2018; 32(5): 1239–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Siddiqui SS, Springer SA, Verhagen A, Sundaramurthy V, Alisson-Silva F, Jiang W. et al. The Alzheimer’s disease-protective CD33 splice variant mediates adaptive loss of function via diversion to an intracellular pool. J Biol Chem 2017; 292(37): 15312–15320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Humbert O, Laszlo GS, Sichel S, Ironside C, Haworth KG, Bates OM. et al. Engineering resistance to CD33-targeted immunotherapy in normal hematopoiesis by CRISPR/Cas9-deletion of CD33 exon 2. Leukemia 2019; 33(3): 762–808. [DOI] [PubMed] [Google Scholar]
- 13.Stewart RS, Drisaldi B, Harris DA. A transmembrane form of the prion protein contains an uncleaved signal peptide and is retained in the endoplasmic Reticulum. Mol Biol Cell 2001; 12(4): 881–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamamoto K, Hayashishita M, Minami S, Suzuki K, Hagiwara T, Noguchi A. et al. Elimination of a signal sequence-uncleaved form of defective HLA protein through BAG6. Sci Rep 2017; 7(1): 14545. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.