

Abstract
With roughly 2 billion people infected, the neurotropic protozoan Toxoplasma gondii remains one of the most pervasive and infectious parasites. Toxoplasma infection is the 2nd leading cause of death due to foodborne illness in the US, causes severe disease in immunocompromised patients, and is correlated with several cognitive and neurological disorders. Currently, no therapies exist capable of eliminating the persistent infection in the central nervous system (CNS). In this study we report the identification of triazine nitrile inhibitors of Toxoplasma cathepsin L (TgCPL) from a high throughput screen, and their subsequent optimization. Through rational design, we improved inhibitor potency to as low as 5 nM, identified pharmacophore features that can be exploited for isoform selectivity (up to 7-fold for TgCPL versus human isoform), and improved metabolic stability (t1/2 > 60 min in mouse liver microsomes) guided by a metabolite ID study. We demonstrated this class of compounds is capable of crossing the blood-brain barrier in mice (1:1 Brain:Plasma at 2 hr). Importantly, we also show for the first time that treatment of T. gondii bradyzoite cysts in vitro with triazine nitrile inhibitors reduces parasite viability with efficacy equivalent to a TgCPL genetic knockout.
Keywords: Toxoplasma gondii, antiparasitics, toxoplasmosis, cathepsin L, triazine nitrile, protease, infectious disease, antiprotazoal agents, blood-brain barrier, central nervous system
Graphical Abstract:

INTRODUCTION
Estimates suggest over 30% of the global human population is currently infected with Toxoplasma gondii, with approximately 60 million infected in the United States alone.1–3 Capable of infecting nearly all warm-blooded mammals, this single-celled apicomplexan is the second leading cause of death due to foodborne illness in the US.4 As a member of the phylum Apicomplexa, Toxoplasma gondii exhibits a complex infection cycle in humans.5 Through consumption of contaminated food, or direct exposure to cat fecal matter, the parasite enters an initial tachyzoite infection stage, in which the parasite undergoes rapid asexual replication and disseminates throughout the host tissues, including the central nervous system (CNS).6 Upon activation of the host immune response, T. gondii transforms into its chronic bradyzoite phase, in which the parasite exists as lifelong tissue cysts within the muscle and CNS. If the host maintains a healthy immune system, this chronic infection generally presents few symptoms, although recently there is a growing body of evidence suggesting that this chronic stage infection may significantly contribute to a wide array of neuropsychiatric symptoms including schizophrenia, bipolar disorder, obsessive compulsive disorder, and various other mood alterations.7–10
A more obvious impact on human health from this parasite is revealed when the host immune system is compromised. The unchecked parasite cysts reactivate into the acute tachyzoite form, causing tissue inflammation and more serious complications such as: encephalitis, blindness, and death.11 Treatment options for toxoplasmosis reactivation are generally pyrimethamine in combination with sulfadiazine, or clindamycin.12 However, these options are prone to adverse side effects and only act upon the parasite in the peripheral system. Currently, no approved treatment options exist to clear the latent and chronic Toxoplasma infection in the CNS, therefore it is critical to develop new and alternative treatment strategies. A recent review by Deng et al13 summarizes current approaches to targeting enzymes critical to the life cycle of the parasite, including calcium-dependent protein kinase 1 (TgCDPK1), thymidylate synthase-dihydrofolate reductase, enoyl-acyl carrier protein reductase, and cathepsin L. TgCDPK1 inhibitors in particular have shown promise for treating the chronic form of T. gondii.14
Cathepsins L and B are the major cysteine proteases found in protozoans, and are emerging as viable targets for anti-parasitic agents due to their essential roles in parasite survival.15–17 For example, Rhodesain (T. brucei), cruzain (T. cruzi), and falcipain (P. falciparum) express cathepsin L-like proteases that have gained a considerable amount of attention for the potential treatment of African sleeping sickness, Chagas disease, and malaria respectively.18 Toxoplasma gondii expresses five members of the C1 family of cysteine proteases, including TgCPL, TgCPB, and TgCPC1–3.19–22 TgCPL is localized in the plant like-vacuole (PLV) vacuolar compartment (VAC, used hereafter), and is also responsible for the maturation and activation of TgCPB.20, 23 Although inhibition or disruption of TgCPL in the parasite has been shown to moderately impede parasite invasion and growth in the tachyzoite stage of infection, TgCPL is not critical to the acute stage.21, 23 Interestingly, parasites in the cystic bradyzoite stage express heightened levels of TgCPL and TgCPB, suggesting proteolysis in the VAC plays an important role in the chronic infection.24 Accordingly, bradyzoites deficient in TgCPL die after forming cysts in both culture and in the neuronal cells of infected mice. Upon restoration of catalytically active TgCPL, bradyzoite viability is re-established. In contrast, expression of a catalytically inactive TgCPL fails to restore cyst viability. Taken together, these strongly implicate TgCPL as a viable target for the treatment of this chronic stage infection. The parasite dependence on TgCPL has been further validated in vitro, as well as through the treatment of cysts with the covalent, irreversible cathepsin inhibitor LHVS (1, Figure 1).25 This disruption of the proteolytic activity in the lysosomal VAC ultimately results in parasite death.26 However, LHVS is not a viable pharmaceutical lead as it failed to reduce neural cysts in infected mice, due to its poor drug-like properties (e.g., fails to cross the blood-brain barrier).27
Figure 1.
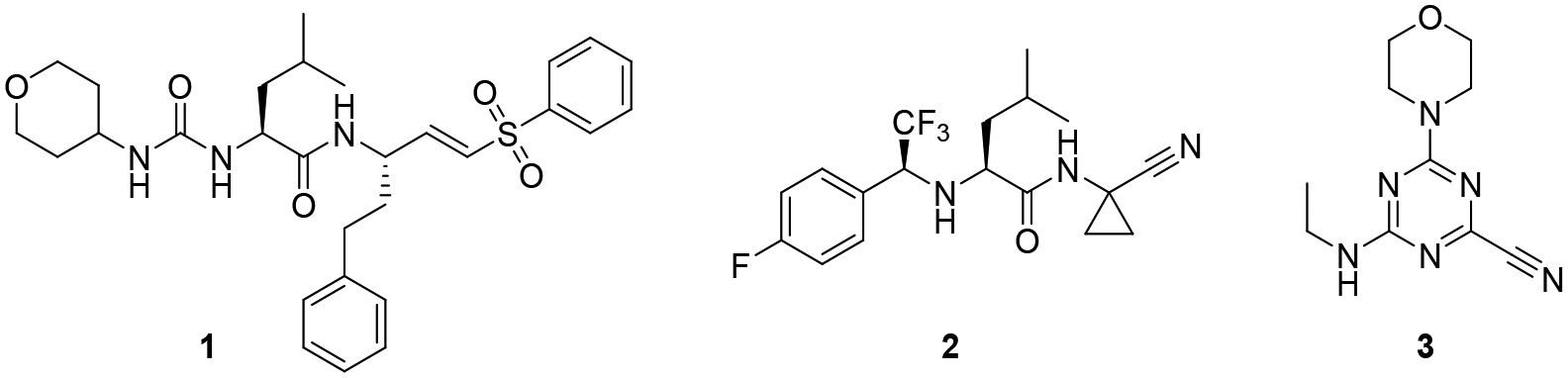
Inhibitors of Toxoplasma gondii cathepsin L
Recently, we reported the first lead optimization study for the inhibition of TgCPL with a series of dipeptide nitriles.28 While we were able to gain insight into the pharmacophore necessary for TgCPL inhibition and significantly improve the potency and selectivity over human isoforms, we were unable to carry forward the dipeptide series due to the inadequate pharmacokinetic (PK) properties of optimized lead 2. Despite being potent in vitro and demonstrably CNS penetrant, the drug was rapidly cleared in vivo. While the dipeptide-nitriles are synthetically convenient for investigating SAR in the cathepsin pockets, they have less than desirable physiochemical properties for gaining robust blood-brain barrier (BBB) permeability, such as several rotational bonds, extensive hydrogen bonding, and high topological polar surface area (TPSA).29 We thus began searching for alternative scaffolds that might exhibit better physiochemical properties to improve upon the in vivo PK, while maintaining the pharmacophoric elements necessary for potent TgCPL inhibition.
A high throughput screen for TgCPL inhibitors was conducted at the Center for Chemical Genomics at the University of Michigan on over 150,000 small molecules. From this we identified several inhibitor classes that demonstrated promising inhibitory activity against TgCPL, reasonable synthetic approachability, and physiochemical profiles optimal for use as a lead scaffold in a CNS targeted campaign. To minimize potential PK and off-target toxicity issues in later development, we chose to exclude compounds that were peptidic in structure, or irreversibly covalent. After our triage process had identified several potential lead molecules, we searched the literature to determine if any of these chemotypes had any previously demonstrated activity against cysteine proteases. An extensive amount of study has been done on inhibitors of human cathepsin L (HsCPL).30 Since Toxoplasma gondii cathepsin L shares high homology with HsCPL, we considered chemotypes with precedent for inhibiting HsCPL as good potential leads. The overall sequence similarity between the two is about 50 %. However, for residues within 4.5 A of the triazine, the similarity is about 72 %. Only 5 out of 18 residues in the binding site are different in the two structures (Figure 2). Among the several potential lead molecules discovered in our screen, we selected a modestly active triazine nitrile 3 (TgCPL IC50 = 2.5 μM, HsCPL IC50 = 2.3 μM) as a lead candidate (Figure 1), given its fragment-like size and solid literature precedent as a cathepsin inhibitor class. Several recent publications have reported triazine nitrile-based inhibitors of human cathepsins, and additionally demonstrated potent inhibition of rhodesain and falcipain.31–33 The activity reported against these analogous cysteine proteases from the related apicomplexan parasites P. falciparum and T. brucei further supports the potential of this chemotype as an antiparasitic agent. Additional triazine nitriles, pyrazolopyrimidine nitriles, and related heterocylic-nitrile motifs have also been reported as inhibitors of cysteine proteases.34–37
Figure 2:
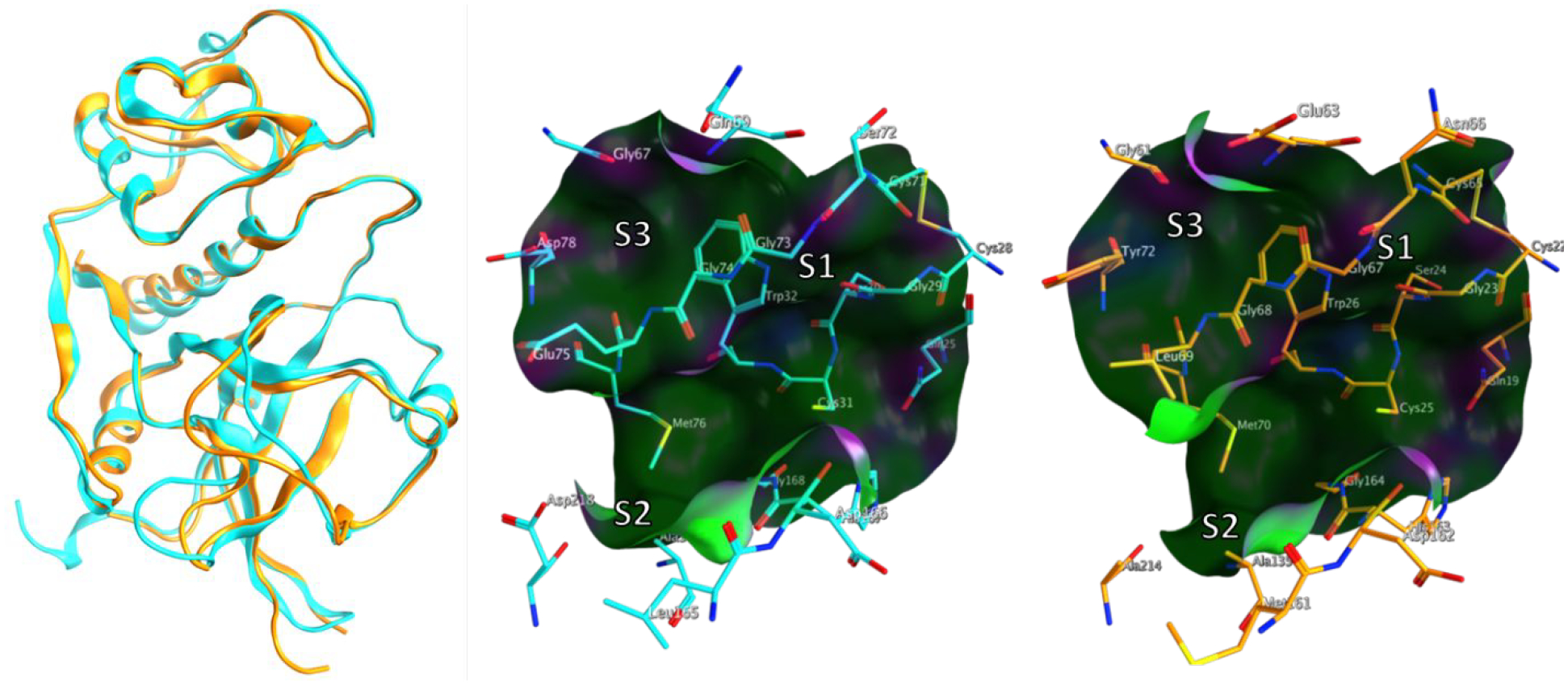
Structural overlay and active sites of apo TgCPL (Cyan; PDB-3F75) and apo HsCPL (Orange PDB-5MAJ)
Although these recent studies have established the potential of this chemotype as a clinically relevant antiparasitic agent, Toxoplasma gondii, as well as other protozoan parasites such as T. brucei, establish a chronic infection in the CNS. Compounds intended for CNS targets have a somewhat constrained physiochemical profile as compared to peripheral drugs. Key among these are the minimization of both the number of hydrogen bond donors and rotatable bonds, which can increase the rate of passive BBB penetration. Additionally, CNS drugs exhibit relatively low MW (<450), high lipophilicity (logP 2–5), and exclusion of acidic moieties. The triazinyl-nitrile scaffold was particularly promising in that it could readily incorporate the structural motifs necessary for binding in the S2 and S3 pockets of TgCPL (Figure 2), which we elucidated in our previous dipeptide work. This scaffold also has a potentially improved pharmacokinetic profile and CNS permeability by reducing the rotational degrees of freedom, eliminating a hydrolyzable amide and removing two hydrogen bond donors (HBD). Molecular docking and energy minimization of the dipeptide nitrile (2) and proposed triazine nitrile (4) in the active site of TgCPL showed good structural agreement into the pharmacophore of TgCPL inhibition (Figure 3). Altogether this encouraged us to attempt a scaffold-hop from the optimized dipeptide to a triazine nitrile. In this paper we report our success at this transformation, and our initial SAR work at enhancing selectivity for the parasite enzyme vs human cathepsin L. Furthermore, we demonstrate for the first time the ability of this class of compounds to inhibit bradyzoite viability in vitro and to penetrate into the brain in vivo, two key milestones for developing an effective therapeutic for the chronic stage of T. gondii.
Figure 3. Predicted scaffold hop from optimized dipeptide nitrile to a triazine nitrile.

A) Predicted projection of common vectors into the S2 and S3 sites of TgCPL based on computational docking of the dipeptide scaffold 2 and a corresponding triazine nitrile scaffold 4 into the crystal structure of TgCPL derived from PDB: 3F75. B) Molecular overlay of dipeptide nitrile (2, yellow) and triazine nitrile (4, teal) demonstrating good structural alignment.; HBD= Hydrogen bond donor; Rot B= rotatable bonds;
RESULTS AND DISCUSSION
Synthesis of TgCPL inhibitors
Under anhydrous conditions, the desired aryl aldehyde or acetophenone Int-1a was condensed with the desired alkylamine overnight, at which point the formed imine was reduced with sodium borohydride or sodium triacetoxyborohydride (Scheme 1). Next, the reaction between the secondary amine Int-2a and cyanuric chloride provided intermediates Int-3a. Subsequent SNAr with morpholine afforded intermediates Int-4a. In the case of 50, oxetanamine was used as the P1 solubilizing group in place of morpholine. Finally, cyanation of the scaffold with potassium cyanide in DMSO provided the desired final compounds.
Scheme 1.

General synthesis of triazine nitrile inhibitors
Reagents and conditions: a) R2NH2, NaBH4, DCM, rt, or Titanium (IV) isopropoxide, Na(OAc)3BH, DCM, 0°C-rt, O/N; b) cyanuric chloride, DIPEA, DCM, −10°C, 1–2 hr; c) morpholine, DIPEA, DCM, 0°C-rt, 4 hr; d) KCN, DMSO:H2O (9:1), 80°C, O/N.
To expedite rapid production of diverse analogs, a synthetic strategy analogous to that reported by Giroud et. al. for the synthesis of triazine-nitrile HsCPL inhibitors was employed (Scheme 2).31 First, cyanuric chloride underwent SNAr reactions with the desired benzyl or alkyl amines Int-1b to set the various S2 and S3 vectors for intermediates Int-2b. A second SNAr step was employed to install a morpholine as a P1 solubilizing group. Cyanation of the intermediate was performed with potassium cyanide in DMSO, to afford Int-3b. Subsequently, the anion of intermediates Int-3b was generated using sodium hydride, and the desired alkyl/benzyl halide was added to provide analogs Int-4b. In the case of analogs bearing a Boc-protected amine side chain, TFA:DCM deprotection was employed to provide the desired final compounds.
Scheme 2.

Alternative general synthesis of triazine nitrile inhibitors
Reagents and conditions: a) i. Cyanuric chloride, DIPEA, DCM, −10°C, 1–2 hr; ii. Morpholine, DIPEA, DCM, 0°C-rt, 4 hr; b) KCN, DMSO:H2O (9:1), 80°C, O/N; c) NaH, DMF, 0°C 30 min, then ArCH2X when X=Br, Cl, Ts, or Ms; d) TFA:DCM (1:2), rt, 0.5–1 hr.
Docking of the triazine scaffold into TgCPL and HsCPL:
The IC50 for the triazine nitrile we identified in our HTS (3) was benchmarked at 3.5 μM and 2.3 μM against TgCPL and HsCPL, respectively (Table 1). Considering this lead bears an ethyl as the only potential P2/3 vector, it left obvious room for improvement. As previously discussed (Figure 3), we elected to first synthesize the triazine nitrile 4 analogous to an optimum dipeptide nitrile from our previous work. Figure 4 presents a comparison of models of 4 docked into the active sites of TgCPL and HsCPL. The triazine nitrile replaces the P1–P2 amide bond and retains the reversibly covalent nitrile interaction with the active site cysteine. In the P2 position, we previously found that leucine is the optimal dipeptide amino acid, so in the respective position on the triazine, which we hypothesized would project into P2, we placed an isoamyl amine to achieve the same length into the pocket. In P3, our most active compound previously bore a 4-fluorobenzyl, and therefore we placed the same motif in the hypothetical P3 position for the triazine nitrile scaffold. An additional feature found both in our HTS lead and known literature analogs was the P1 morpholine. While the S1 pocket in TgCPL is absent, the morpholine in the P1 position projects into solvent and has been shown to improve the solubility of this chemotype (Figure 4).
Table 1.
SAR of P3 vectors
| No. | R | TgCPLa IC50 (nM) | HsCPLb IC50 (nM) | Selectivity for TgCPLc |
|---|---|---|---|---|
| HTS Lead 3 | see Fig 1 | 3,500 | 2,300 | 0.7 |
| 4 |  |
34 | 32 | 0.9 |
| 5 |  |
47 | 20 | 0.4 |
| 6 |  |
53 | 19 | 0.4 |
| 7 |  |
356 | 33 | 0.09 |
| 8 | 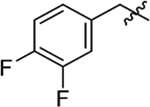 |
53 | 34 | 0.6 |
| 9 | 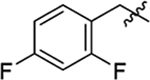 |
48 | 35 | 0.7 |
| 10 |  |
101 | 62 | 0.6 |
| 11 |  |
110 | 86 | 0.8 |
| 12 |  |
230 | 35 | 0.2 |
| 13 |  |
61,457 | 12 | 0.0002 |
| 14 |  |
15,200 | 2 | 0.001 |
| 15 |  |
82 | 33 | 0.4 |
| 16 | 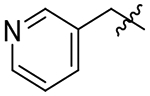 |
39 | 27 | 0.7 |
| 17 | 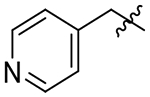 |
107 | 41 | 0.4 |
| 18 |  |
230 | 87 | 0.4 |
| 19 |  |
95 | 28 | 0.3 |
| 20 |  |
137 | 296 | 2.2 |
| 21 | 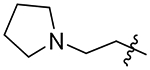 |
304 | 492 | 1.6 |
| 22 |  |
32 | 6 | 0.2 |
| 23 |  |
95 | 36 | 0.4 |
| 24 |  |
107 | 14 | 1.3 |
| 25 |  |
64 | 22 | 0.3 |
| 26 |  |
19 | 5 | 0.3 |
| 27 |  |
37 | 41 | 1.1 |
| 28 |  |
17 | 47 | 2.8 |
| 29 |  |
17 | 3 | 0.2 |
| 30 |  |
47 | 6 | 0.1 |
| 31 |  |
27 | 96 | 3.6 |
| 32 | 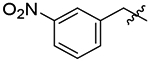 |
42 | 14 | 0.3 |
| 33 |  |
59 | 51 | 0.9 |
| 34 |  |
105 | 56 | 0.5 |
| 35 | 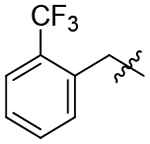 |
740 | 266 | 0.4 |
| 36 |  |
123 | 91 | 0.7 |
| 37 |  |
970 | 67 | 0.07 |

IC50 for Toxoplasma gondii Cathepsin L (TgCPL) or human Cathepsin L (HsCPL). Values are mean of at least 3 independent experiments with SEM typically < 30%.
Selectivity for Toxoplasma gondii Cathepsin L as defined by the ratio HsCPL IC50/TgCPL IC50
Figure 4.

Comparison of TgCPL and HsCPL binding sites. A) Covalent docking of 4 into TgCPL active site and electrostatic surface mapping. Red= polar negative, White = neutral, Blue= polar positive. (Derived from PDB: 3F75); B) Covalent docking of 4 into the active site of HsCPL (PDB: 5MAJ). Pocket view; Red= polar negative, White= neutral, Green= lipophilic
The installment of these two pendants in our first triazine analog 4 indeed provided over a 100-fold improvement in potency over the HTS lead 3, affording an IC50 of 34 nM and 32 nM for TgCPL and HsCPL, respectively (Table 1). These results supported our docking model, and validated our hypothesis that we could successfully scaffold-hop from the dipeptides to the triazine, suggesting that much of our dipeptide SAR should translate directly to the triazine series. The active site of TgCPL contains four residues (Figure 4A) that are either non-conserved across the human cathepsin isoforms A-X, or unique to TgCPL. Notably, the S3 pocket of TgCPL tends to be significantly more polar as compared to HsCPL, with Gln69, Asp78, and Glu75 making up the key pocket residues. The human cathepsin L (Figure 4B) is somewhat more lipophilic with the S3 pocket residues Glu63, Tyr72 and Leu69, respectivly. While this presents potential for differences in ligand selectivity, the S3 pocket is shallow and somewhat promiscuous with its preferred residues. We anticipated that the majority of our potential selectivity would come from the S2 pocket, which tends to be deeper and somewhat more defined. Importantly, the parasitic cathepsin bears an Asp218 in the S2 pocket, while the human isoform contains an Ala214 in the corresponding position. We therefore expected that selectivity for TgCPL could be achieved by exploiting the differences in the overall topology and unique residues. As such, we chose to explore various P2 and P3 vectors to probe these diffences and elucidate the pharmacophore for selective and potent inhibition of TgCPL.
SAR of P3 Vectors
We began our SAR investigation by retaining the isoamyl P2 vector and varying the P3 position (Table 1). A scan of fluorine mono-substitution of the P3 benzyl (5 and 6) showed retention of activity, but no significant improvement in either overall potency or selectivity. A similar outcome was seen with the P3 di-fluoro analogs 7, 8, and 9. The non-fluorinated P3 benzyl pendant 10 exhibited roughly a 3-fold loss of potency versus the 4-fluoro lead 4. Homologation of the linker from methyl to ethyl (11) or propyl (12), while better tolerated for the human isoform, resulted in the same reduced potency for TgCPL. An even greater loss of potency for the parasitic enzyme was seen when large bulky pendants like the quinoline or naphthyl of 13 and 14 were installed. Taken together, this demonstrates that the parasitic isoform does not productively accomodate large lipophilic vectors in the P3 position, which is consistent with the topologically more polar nature of the S3 pocket defined by Asp78 and Glu75 (Figure 4A).
With this in mind, we tried the 2-, 3-, and 4-pyridyl pendants (15, 16, and 17) in P3, with the objective of improving our selectivity for the parasite, as well as potentially improving our metabolic profile by decreasing the clogP. Of these, the 3-pyridyl (16) retained potency, but unfortunately did not offer any improvement in selectivity over the human isoform. Anilines 18, 19, and 20 were also evaluated. We had hoped to gain a meaningful interaction with Asp78, Gln69, or Glu75 in TgCPL (Figure 4A), but none of the anilines were very potent for TgCPL. However, analog 20 did afford a small measure of selectivity toward TgCPL (HsCPL IC50/TgCPL IC50 = 2.2). Analog 21 was tested to determine if a basic amine might afford greater TgCPL selectivity, but resulted in nearly a 9-fold loss in potency versus parent compound 4.
As a comparison to our 4-fluorobenzyl pendant, we tested the 4-chlorobenzyl analog 22 to determine the impact of a larger halogen. While equally effective against TgCPL to the 4-fluoro, this change afforded an order of magnitude improvement in potency for HsCPL. Analog 23 was synthesized to assess the effects of a moderately electron withdrawing acetophenone, but afforded a mild reduction in potency against TgCPL. A methyl scan was performed to probe the space around the ring and determine if any of the substitution patterns might lead toward some selectivity for TgCPL. While 24 and 25 did not offer any improvement in potency, 26 was nearly twice as potent (TgCPL IC50 = 19 nM) as the parent scaffold 4, but more strongly improved HsCPL inhibition (HsCPL IC50= 5 nM), similar to what we observed with the 4-chloro analog 23. It is worth noting that the o-methyl benzyl analog 24, while less potent than the 4-flurobenzyl, showed mild selectivity for TgCPL. Dimethyl analogs 27 and 28 provided more potent inhibitors and were consistent with the trend that an ortho substitution (28) may favor selectivity for TgCPL. Undesirably, these also increased overall molecular weight and introduced potential metabolic hotspots, and therefore were not further pursued. The electron withdrawing 3-trifluoromethyl 29 provided one of the more potent TgCPL inhibitors (IC50 = 17 nM), but exhibited selectivity in favor of HsCPL. The increased MW versus 4 was also undesirable in trying to remain within the calculated properties for BBB permeability. The corresponding 3-methoxy analog 30 was also tested to assess the effects of electron donation into the ring; however, this did not seem to offer any significant improvement in potency or selectivity as compared to 29. Interestingly, a nitro scan in analogs 31, 32, and 33 revealed that the o-nitrobenzyl P3 vector of 31 exhibited a 3-fold selectivity over the human isoform (TgCPL IC50 = 27 nM, HsCPL IC50 = 96 nM), consistent with the modest selectivity we observed with ortho-methyl analogs 24 and 28. As this was the most TgCPL-selective analog to date, additional ortho-substituted analogs 34, 35, 36, and 37 were synthesized in an effort to better explain the observed selectivity. The nitrile (34), trifluoromethyl (35), and methyl ester (35) vectors were tested as isosteres of the nitro. Unfortunately, none of these substituents reproduced the selectivity we saw with the o-nitro vector. The o-methoxy analog 37 was also synthesized to determine if the observed effects were due to the electron withdrawing nature of the nitro, or a conformational change due to sterics. While tolerated in HsCPL, the potency significantly decreased against TgCPL.
Based on observations in our modeling (Figure 5), we hypothesize that the P3 pendant may have the ability to bind outside of the S3 pocket and interact with the backbone of Asp166, giving rise to a higher tolerance for vectors as compared to that of S2. This concept is supported by the overall lack of robust SAR trends for the S3 pocket and the reltively high substrate tolerance in the P3 position observed in analogs 4–47. Furthermore, a recent publication observed a similar trend in triazine nitrile inhibitors of human cathepsin L.31
Figure 5.

Potential alternative binding mode for triazine nitriles (e.g. 4). Derived from PDB: 3F75
SAR Analysis of P2 Vectors:
We then turned our attention to evaluating the SAR among the postulated S2 vectors. In general, the papain family of proteases achieve their substrate specificity primarily from interactions in the S2 pocket.38 Based on our previous SAR studies with the dipeptide nitriles, we had determined that leucine was the optimal amino acid for the P2 position. As already noted with 4, the isoamyl sidechain in the respective position for the triazine nitriles afforded a huge improvement in potency to HTS lead 3 (TgCPL IC50 = 34 nM). We wanted to first determine if the previous dipeptide SAR trends we found tracked in a similar fashion with this new chemotype. Initially, we evaluated the effects of shorter, aliphatic vectors. The n-propyl, n-butyl, and n-butenyl (38, 39, and 40) were tolerated in HsCPL, but all decreased in potency for TgCPL. Consistent with our previous SAR, the shorter isobutyl S2 vector in 42, which closely mimics a valine sidechain, slightly decreased activity for TgCPL to 67 nM, but enhanced HsCPL inhibition (IC50 = 2 nM). Interestingly, the dehydroleucine analog 41 retained nearly equivalent potency (TgCPL IC50 = 46 nM) to 4, a trend that was not observed in the previous dipeptide series. This indicated to us that, while similar, the SAR between the dipeptide and triazine chemotypes was not identical and that binding of the triazine nitrile series may be somewhat better accomodated by the enzyme. Extension of the S2 sidechain of 4 by one carbon in 43 resulted in a significant loss of potency. We believe this may be due to a clash in the back of the S2 pocket, consistant with the S2 size constraints observed previously.28 Compound 44 was made to determine if the S2 pocket could tolerate a less lipophilic group thereby reducing overall clogP and improving compound solubility. Unfortunately, this resulted in a large decrease in potency for both enzymes. Given that TgCPL bears a unique Asp218 versus the Ala214 in HsCPL (Figure 4), we rationalized that inclusion of a basic group in the S2 position might afford some selectivity, as well as improve compound solubility. As such, compounds 45-48 were synthesized. We had hoped that the stepwise increase in side chain length might elucidate the ideal distance required to gain a productive interaction with Asp218. Unfortunately, these changes were not well tolerated and resulted in a drastic drop in potency down to the low micromolar range for both enzymes, indicating that perhaps these compounds are not binding as predicted, or that Asp218 is not oriented the way it appears in our model.
P2 Vectors for Improved Metabolic Profile and Selectivity:
The previously developed dipeptide nitrile 2 exhibited a fair level of stability toward mouse liver microsomes (MLM) with a half-life of t1/2 = 22 min. While the scaffold hop from dipeptide 2 to the triazine nitrile 4 resulted in an improvement in both potency and predicted CNS profile, it also imparted a significant drop in metabolic stability (MLM t1/2 = 7 min). The metabolic liabilities predicted by SmartCyp (Figure 6A) led us to install the α-methyl and exchange the P1 solublizing group with 3-amino-oxetane in analog 50 (Table 3). Unfortunately, this did not offer any improvement to the microsomal stability profile (MLM t1/2 = 5 min). Therefore, a metabolite ID study was performed to experimentally determine sites of oxidative metabolism. As shown in Figure 6B oxidation of 4 occurs predominantly on the P2 isoamyl vector and the P3 benzylic methylene. Similar oxidation of S2 isoamyl sidechains has been reported in the development of odanacatib for the inhibition of cathepsin K, and it was overcome by replacement of the sidechain with fluoroleucine.39 With this guidance in mind, analogs were designed to retain the physiochemical profile of a CNS drug and simultaneously circumvent these potential metabolic liabilities (Table 3).
Figure 6.

Predicted and actual metabolism of 4 by CYPs. A) Top-ranked metabolic sites predicted by SmartCYP for CYP3A4. B) Metabolite ID results (incubation with mouse liver microsomes) identifying the isoamyl S2 vector as the primary site of oxidation.
Table 3.
P2 Vectors for Improved Metabolic profile and Selectivity
| No. | R1 | R2 | TgCPLa IC50 (nM) | HsCPLb IC50 (nM) | Selectivity for TgCPLc | MLM t1/2 (min) |
|---|---|---|---|---|---|---|
| 4 | 4-Fluorobenzyl |  |
34 | 32 | 0.94 | 7 |
| 49 | 4-Fluoro-α-methylbenzyl |  |
26 | 15 | 0.58 | |
| 50* | 4-Fluoro-α-methylbenzyl | 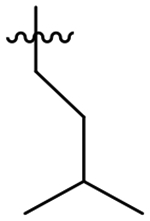 |
92 | 23 | 0.25 | 5 |
| 51 | 4-Fluorobenzyl | 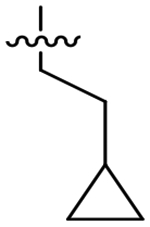 |
310 | 190 | 0.61 | |
| 52 | 4-Fluoro-α-methylbenzyl |  |
143 | 103 | 0.72 | 4.4 |
| 53 | 4-Fluorobenzyl | 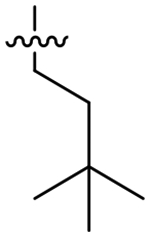 |
68 | 160 | 2.4 | |
| 54 | 4-Fluorobenzyl |  |
17 | 26 | 1.5 | 3.5 |
| 55 | 4-Fluorobenzyl | 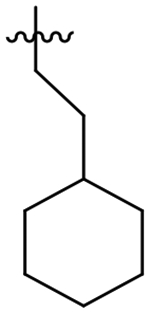 |
66,800 | 87,500 | 1.3 | |
| 56 | 4-Fluorobenzyl |  |
26 | 95 | 3.7 | 15 |
| 57 | 4-Fluorobenzyl |  |
3,570 | 66 | 0.018 | |
| 58 | 4-Fluorobenzyl |  |
110 | 77 | 0.70 | |
| 59 | 4-Fluorobenzyl |  |
74 | 60 | 0.81 | |
| 60 | 4-Fluorobenzyl | 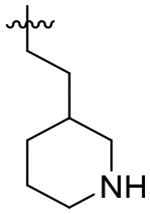 |
1,900 | 1,190 | 0.63 | |
| 61 | 4-Fluorobenzyl |  |
326 | 969 | 2.9 | |
| 62 | 4-Fluorobenzyl | 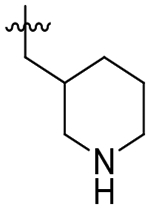 |
181 | 1,200 | 6.7 | >60 |
| 63 | 4-Fluorobenzyl | 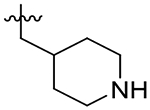 |
1,200 | 960 | 0.80 | |
| 64 | 4-Fluorobenzyl |  |
400 | 94 | 0.25 | |
| 65 | 4-Fluorobenzyl |  |
49 | 170 | 3.5 | 3.5 |
| 66 | 4-Fluorobenzyl |  |
630 | 370 | 0.58 | |
| 67 | 2-Nitrobenzyl |  |
3,970 | 1,240 | 0.3 | |
| 68 | 4-Fluorobenzyl |  |
1,600 | 517 | 0.33 |

IC50 for Toxoplasma gondii Cathepsin L (TgCPL) or human Cathepsin L (HsCPL). Values are mean of at least 3 independent experiments with SEM typically < 30%.
Selectivity for Toxoplasma gondii Cathepsin L as defined by the ratio HsCPL IC50/TgCPL IC50.
S1 morpholine group replaced with oxetan-3-amine.
Analog 49 was prepared first to evaluate the impact of the alpha-methylbenzyl on potency since we had previously observed a loss of potency with oxetane analog 50. In fact this single change was well tolerated and provided a small improvement in overall potency; however, the potency improvement was 2-fold greater for HsCPL. Presumably due to the added MW and lipophilicity, compound solubility significantly decreased versus the parent compound (thermodynamic aqueous solubility: 4 = 42 μM, 49 = 16 μM). In an effort to resolve this, we tried replacing the S1 morpholine group with oxetanamine in analog 50. As noted previously, the potency decreased to 92 nM for TgCPL, but solubility improved to 42 μM. Analogs 51, 53, and 54 were all synthesized with the intent of blocking oxidation of the isoamyl CH.39–40 The cyclopropyl leucine mimic (51) lost an order of magnitude in potency (TgCPL IC50 = 310 nM) compared to 4, and the t-butyl analog, 53, while 2-fold selective for TgCPL, decreased to 68 nM against TgCPL. It should be noted, however, that these respective changes in the dipeptide nitrile series were not tolerated nearly as well, giving us some confidence that the triazine nitrile may be a superior scaffold with regards to potential for improving PK properties. Furthermore, this data suggests these two chemotypes are not binding in precisely the same manner, consistent with what we had observed earlier in our SAR. Compound 52 was synthesized to determine if the cyclopropyl and α-methylbenzyl combination in P2 and P3 respectively would be sufficient to slow metabolism. Unfortunately, this actually decreased the MLM t1/2 to 4.4 min, indicating a likely switch to another metabolic site, perhaps N-dealkylation of the P2 sidechain or oxidation of the P1-morpholine. Compound 54, bearing a fluoroleucine in S2 (a vector also not tolerated in the dipeptide series), demonstrated excellent potency for TgCPL (IC50 = 17 nM) and HsCPL (IC50 = 26 nM), although MLM stability was decreased to t1/2 = 3.5 min, further indicating that blocking the primary metabolic site alone is insufficient for improving stability. The ethyl-cyclohexyl vector of 55 resulted in a significant decrease in potency for both cathepsins, likely due to the same steric clash in the S2 pocket observed for compound 43. Interestingly, shortening the pendant by one carbon to the methyl-cyclohexyl (56) both retained potency for TgCPL (IC50 = 26 nM) and achieved nearly 4-fold selectivity for the parasite isoform over human. Furthermore, it doubled the MLM t1/2 of lead 4 to 15 min. The P2 cyclopentyl in 59 is comparable to the cyclohexyl in terms of potency, but not as selective. The smaller ring sizes in analogs 57 and 58 significantly decreased potency for TgCPL and were not further pursued.
Given the favorable TgCPL selectivity imparted by the P2 cyclohexyl ring for 56, we also explored incorporation of a basic amine with the objective of enhancing this selectivity through a favorable ionic interaction with Asp218. As such, S2 piperidinylmethyl compounds 62 and 63 were synthesized. Interestingly, the 4-piperidine of 63 resulted in sharply reduced activity against both enzymes, while the 3-piperidine 62 demonstrated good selectivity (7-fold) for TgCPL. Despite a decrease in overall potency to 0.180 μM, 62 exhibited a much improved MLM stability with t1/2 > 60 minutes. The drastic improvement in stability seen for compound 62 is likely due to the added charge of the basic amine impeding CYP binding. The addition of an extra carbon in the sidechain for analog 60 decreased potency to 1.9 μM for TgCPL, consistent with the poor activity of the homologated cyclohexane analog 55. The ethyl 4-piperidine P2 vector 61 showed a slightly improved potency as compared to analog 60, however it was still significantly lower than parent analog 56. The respective tetrahydropyran analogs 64 and 65 were made to determine if the basic amine was important for this observed selectivity. These displayed a similar potency and selectivity profile, with the heteroatom being better tolerated at the 3-position (65, TgCPL IC50 = 49 nM) than the 4-position (64, TgCPL IC50 = 400 nM). Unfortunately, compound 65 was not stable to MLM. Reduction of the piperidine ring size to a pyrrolidine in analog 66 abolished the selectivity and potency profile. Finally, we attempted to combine our most selective P2 (3-piperidinylmethyl) and P3 (o-nitro benzyl) vectors with analog 67. Unfortunately, these effects were not synergistic and resulted in a loss of both selectivity and potency, strongly suggesting that the two congeners are not binding the same way.
Pharmacokinetics:
Compound 4 was evaluated in a mouse PK study to benchmark the CNS profile of the triazine nitrile chemotype and compare it to that of the dipeptide nitrile 2 (Table 4). We were pleased to find that 4 does in fact access the CNS with a brain AUC/plasma AUC = 0.61. However, the compound levels decreased rather quickly, likely due to a combination of high volume of distribution and metabolic instability. Nonetheless, this was a promising result indicating that the triazine nitrile scaffold has the physiochemical profile needed to cross the blood-brain barrier.
Table 4.
Pharmacokinetic studies in mice*
| No. | [Plasma] (ng/mL) |
[Brain] (ng/g) |
AUCBrain (ng*hr/g) |
AUCPlasma (ng*hr/mL) |
AUCBrain/AUCPlasma | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.5 | 2 | 4 | 7 | 0.5 | 2 | 4 | 7 | ||||
| 2 | 110 | 138 | 10.2 | - | 53.6 | - | - | - | 53.6 | 376 | 0.14 |
| 4 | 141 | 17.3 | 7.4 | 3.1 | 90.8 | 16.1 | - | - | 119 | 195 | 0.61 |
| 50 | 1,530 | 57.7 | 4.7 | 11.4 | 671 | 82.5 | 12.5 | 13.8 | 868 | 1660 | 0.52 |
| 62 | 473 | 200 | 65.9 | 14.3 | 36.1 | 32 | 16.6 | 12.4 | 152 | 1010 | 0.15 |
Compounds were administered as a single IP dose at 10 mg/kg.
We hoped that by increasing the metabolic stability of the scaffold, we might gain a better pharmacokinetic profile. We attempted to evaluate compound 56, as this analog exhibited an improved half-life in vitro, good potency, and demonstrated some selectivity for TgCPL, but unfortunately its poor solubility precluded in vivo dosing. As mentioned above, the replacement of the P1 morpholine of 49 with a 3-amino-oxetane in compound 50 offered an improvement in solubility (16 μM to 42 μM respectively). We decided to perform a PK study on 50 to determine if the additional hydrogen bond donor of the oxetanamine would impede our CNS penetrance, or if this could be a viable strategy to improve the solubility of future compounds (Table 4). We were very pleased to find that in addition to retaining CNS access, the overall exposure levels were significantly increased compared to 4. Given that the 3-piperidine P2 vector in 62 significantly improved the metabolic stability, we also evaluated its pharmacokinetic profile. While the exposure levels were improved in plasma versus 4, this compound had markedly reduced BBB penetrance. Basic amines are often associated with efflux by Pgp, so the difluoro-3-piperidine compound 68 was synthesized with the intent of attenuating the basicity of this sidechain (Table 3). Unfortunately, this change was not well tolerated and potency was reduced to low micromolar for this compound (TgCPL IC50 = 1.6 μM).
Because of their markedly divergent brain:plasma ratios observed in the PK studies, compounds 50 and 62 were assessed in vitro to determine whether they are potential substrates of P-glycoprotein (P-gp). Evaluation in an MDR1/MDCK permeability assay indicated that compound 50 has high passive permeability and an efflux ratio <1, and thus does not appear to be a Pgp substrate (Table 5). This would be expected to translate to good CNS penetrance, which is in agreement with our PK results. 62 also demonstrated high passive permeability, but has an efflux ratio of over 17, indicating this compound is likely a P-gp substrate, explaining its poor brain exposure in vivo. As noted above, this is very likely to be due to the presence of the basic amine in 62.
Table 5.
MDR1/MDCK Assay results
| Sample | Pappa (cm/sec) | Efflux Ratio (B-A)/(A-B) |
|---|---|---|
| 50 (A-B) | 3.55 × 10−5 | 0.223 |
| 50 (B-A) | 7.92 × 10−6 | |
| 62 (A-B) | 2.59 × 10−5 | 17.2 |
| 62 (B-A) | 4.47 × 10−4 |
Values shown are the average of two sample measurements of the acceptor wells from three replicate assays.
In vitro efficacy studies:
Based on their potencies vs TgCPL, five compounds were selected for evaluation of their effects on plaque formation in a T. gondii bradyzoite cyst qPCR/plaque viability assay in vitro (Figure 7). This assay involves isolating bradyzoites after treatment and quantifying viability by applying them to a new monolayer of host cells. Viable bradyzoites invade the monolayer, differentiate to tachyzoites, and form a visible plaque after 12 days of growth. Viability is calculated as the number of plaques/1000 genomes of input bradyzoites (measure by qPCR) and expressed as a percentage of solvent (DMSO) treated bradyzoite cysts. Positive controls in the assay included a TgCPL knockout strain of T. gondii (Δcpl), as well as the irreversible TgCPL inhibitor LHVS (1)25. We were pleased to find that the triazine nitriles 4 and 50 exhibited strong efficacy at 5 μM that was equivalent to the genetic knockout and irreversible inhibitor LHVS at 1 μM, with zero plaques formed in any biological replicate. To our knowledge, this is the first example of a reversible inhibitor of TgCPL achieving in vitro efficacy against the chronic (bradyzoite) stage of T. gondii. Triazine nitrile analogs 56, 65, and 62, on the other hand, showed variable efficacy. 56 and 65 were slightly less efficacious than 4 and 50 at the concentration tested, reducing cyst viability by ~80% across three biological replicates. Only one biological replicate of compound 62 reduced viability (<20%), suggesting that it was comparatively ineffective. Dipeptide nitrile lead (2) demonstrated only modest activity against the bradyzoite cysts, exemplifying the improved potential of the triazine nitrile as a therapeutic.
Figure 7.

Efficacy of triazine nitrile compounds for treatment of in vitro bradyzoite cysts within human foreskin fibroblast (HFF) host cells. Cultures were treated with 1 μM LHVS (1) or 5 μM nitrile compounds daily for 7 days.
To confirm that the observed reduction in bradyzoite viability was not due to human foreskin fibroblast (HFF) host cell viability being indirectly compromised, MTS assays were also conducted to assess cell cytotoxicity (CC). The relatively high CC50 values indicate that none of the compounds would be expected to cause significant host cell cytotoxicity at the concentration used in the qPCR/plaque bradyzoite cyst viability assays (5 μM).
CONCLUSIONS AND PERSPECTIVES
Throughout this project, significant advances have been made in developing small molecule inhibitors of TgCPL. We have elucidated key SAR for the inhibition of TgCPL, developed potent and CNS penetrant inhibitors, and demonstrated efficacy against bradyzoite cysts in vitro. We started by conducting a high throughput screen to identify new chemotypes capable of the inhibition of Toxoplasma gondii cathepsin L as potential new therapeutics for toxoplasmosis. The low μM triazine nitrile scaffold (3) was selected based on the previously established literature precedent of this chemotype for inhibition of human cathepsins, its low molecular weight, and physical properties consistent with CNS-active compounds. Translation of the pharmacophore previously developed with our dipeptide nitrile series to the triazine nitrile scaffold resulted in an immediate 100-fold gain in potency (TgCPL IC50 = 34 nM), but reduced metabolic stability relative to the corresponding dipeptide. A TgCPL covalent docking model was developed using the crystal structure of TgCPL and the crystal structure(s) of human cathepsin (HsCPL) in complex with various dipeptide and triazine nitrile inhibitors. Using this we were able to identify key distinctions between the parasite and human isoforms of cathepsin L. In particular, the smaller S2 pocket and the four unique residues (Asp218, Glu75, Asp78, and Gln69) were targeted with the goal of gaining selectivity for TgCPL. 68 analogs in this series were synthesized with varying S2 and S3 vectors to optimize metabolic stability, CNS permeability, and selectivity over human isoforms. In general, we found that variations in the P3 position had little impact on potency or selectivity. As expected, the bulk of the potency and selectivity seems to be imparted by efficient binding in the S2 position. The S2 pocket across the human cathepsins tends to be more uniformly lipophilic than TgCPL, and we predicted that inclusion of a basic residue in the P2 position might interact favorably with non-conserved Asp218 in TgCPL. It is worth noting that out of the nine analogs bearing an amine in P2, all exhibited IC50s for HsCPL of >0.5 μM, supporting that the human isoform does not tolerate a basic amine in S2. TgCPL, conversely, appears to be able to better accommodate either a nitrogen or oxygen in S2, likely due to the polar Asp218 residue in the back of the S2 pocket, and this may represent an avenue for further TgCPL selectivity enhancement. Work in our laboratory is underway to exploit these features and further explore the SAR trends with various basic and polar groups in P2.
Overall, we improved potency against TgCPL to as low as 5 nM and identified features that can be exploited to gain selectivity vs HsCPL (up to 5–7 fold), and significantly improved metabolic stability (up to t1/2 >60 min in MLM). Importantly, we were able to demonstrate significant brain penetration with several analogs in vivo in mice. Although our most Tg-selective analog 62 bearing a basic amine in the P2 pendant proved to be highly susceptible to efflux in MDR1-MDCK cells in vitro and demonstrated low brain exposure in vivo, an analog lacking this basic amine (50) was shown not to be a P-gp substrate and achieved good exposure in the brains of mice after IP dosing, suggesting the triazine nitrile chemotype has promise as an in vivo probe for chronic T. gondii infection studies. Synthesis of new analogs to further improve PK and isoform selectivity are underway. Finally, we demonstrated for the first time that in vitro treatment of T. gondii bradyzoite cysts with non-peptidic triazine nitrile inhibitors reduces parasite viability with efficacy equivalent to a TgCPL genetic knockout, in contrast to one of our previously developed dipeptide nitriles, highlighting the increased therapeutic potential of the triazine template. This is the first example of a CNS penetrant, reversibly covalent TgCPL inhibitor showing efficacy in an in vitro model of bradyzoite stage parasites. Current studies are underway to advance the triazine nitrile series into an in vivo model of latent Toxoplasma infection.
METHODS
Chemistry General Information:
All reagents were used without further purification as received from commercial sources unless noted otherwise. 1H NMR spectra were taken in DMSO-d6, MeOD, or CDCl3 at room temperature on Varian Inova 400 MHz or Varian Inova 500 MHz instruments. Reported chemical shifts for the 1H NMR and 13C NMR spectra were recorded in parts per million (ppm) on the δ scale from an internal standard of residual tetramethylsilane (0 ppm). Mass spectrometry data were obtained on either a Micromass LCT or Agilent Q-TOF. An Agilent 1100 series HPLC with an Agilent Zorbax Eclipse Plus–C18 column was used to determine purity of biologically tested compounds. Unless otherwise noted, all tested compounds were determined to be >95% pure using a 6 minute gradient of 10–90% acetonitrile in water followed by a 2 minute hold at 90% acetonitrile with detection at 254 nm. Flash chromatographic purifications were performed using a Teledyne ISCO Combiflash RF with Redisep Gold RF columns.
General Procedure A1: Reductive Amination
To a dry round bottom flask with DCM was added aryl aldehyde or aryl ketone Int-1a (1 eq), followed by the addition of the desired primary amine (1 eq), final reaction concentration of 0.1–0.3 mM. The vessel was then stirred under a nitrogen atmosphere at room temperature overnight. Sodium triacetoxyborohydride (3 eq) was then added and reaction was stirred at room temperature for 1–3 hr. The reaction was quenched slowly with water then poured into water and extracted 3x with DCM. The combined organic layer was dried over sodium sulfate and concentrated in-vacuo. The crude product was further purified by column chromatography (0–10% MeOH in DCM + 0.1% TEA) to afford the desired secondary amine Int-2a.
General Procedure A2: SNAr
Under a dry nitrogen atmosphere, cyanuric chloride (1 eq) was added a roundbottom flask along with DCM (final conc. = 0.1–0.3 mM). The vessel was cooled to −10°C (Ice/Brine) and the desired secondary amine (1 eq) and DIPEA (1 eq) were then added. Reaction was stirred at −10°C for 1 h. The reaction was poured into water and extracted 3x with DCM. Combined organic layers were dried over MgSO4 and concentrated in vacuo. The crude product was further purified by column chromatography (0–100% EtOAc in Hexanes gradient) to afford the desired intermediate Int-3a.
General Procedure A3: SNAr
Under a dry nitrogen atmosphere, dichloro triazine intermediate (1 eq) was added a roundbottom flask along with DCM to a final reaction concentration of 0.1 mM. The vessel was cooled to −10°C (Ice/Brine) and DIPEA (1 eq) and morpholine (1 eq) were then added. Reaction was stirred and allowed to warm to room temperature over 6–12 h. The reaction was poured into water and extracted 3x with DCM. Combined organic layers were dried over MgSO4 and concentrated in vacuo. The crude product was further purified by column chromatography (0–100% EtOAc in Hexanes gradient) to afford the desired intermediate Int-4a.
General Procedure A4: Cyanation
A dry pressure vessel was charged with the appropriate triazinyl chloride (1.0 eq), and suspended in DMSO/H2O 9:1. KCN (1.1 eq.) and 1,4-diazabicyclo[2.2.2]octane (DABCO, 2.0 eq.) were added and the reaction was heated to 80°C. Reaction was left to stir at this temperature until LC/MS showed completion of the reaction (6–12 h). Reaction was cooled to room temperature, diluted with EtOAc, and washed thoroughly with brine (3–5 x). The organic layer was separated, dried over MgSO4, filtered, and evaporated. The crude product was further purified by column chromatography (0–100% EtOAc in Hexanes gradient) to afford the desired product.
General Procedure B1: One-pot double SNAr
Under a dry nitrogen atmosphere, cyanuric chloride (1 eq) was added a roundbottom flask along with DCM (final conc. = 0.1–0.3 mM). The vessel was cooled to −10°C (Ice/Brine) and DIPEA (1 eq) followed by the desired primary amine Int-1b (1eq) were then added. Reaction was stirred at for 1–3 hr, until TLC/HPLC indicated substitution was complete. Next, DIPEA (1 eq) was added followed by morpholine (1 eq) and reaction was allowed to stir and warm to rt over 4–12h until TLC/HPLC indicated substitution was complete. The reaction was poured into water and extracted 3x with DCM. Combined organic layers were dried over MgSO4 and concentrated in vacuo. The crude product was further purified by column chromatography (0–100% EtOAc in Hexanes gradient) to afford the desired intermediate Int-2b.
General Procedure B2: Cyanation
A dry pressure vessel was charged with the appropriate triazinyl chloride (1.0 eq), and suspended in DMSO/H2O 9:1. KCN (1.1 eq) and 1,4-diazabicyclo[2.2.2]octane (DABCO, 2.0 eq) were added and the reaction was heated to 80°C. Reaction was left to stir at this temperature until LC/MS showed completion of the reaction (6–12 h). Reaction was cooled to room temperature, diluted with EtOAc and washed thoroughly with brine (3–5 x). The organic layer was separated, dried over MgSO4, filtered, and evaporated. The crude product was further purified by column chromatography (0–100% EtOAc in Hexanes gradient) to afford the desired secondary amine Int-3b.
General Procedure B3: Alkylation with NaH and aryl/alkyl X/Ms/Ts
The triazinyl-nitrile (1 eq) was dissolved in DMF and cooled to −10°C (Ice/Brine bath). 60% NaH in mineral oil (1–1.5 eq) was added and reaction was stirred for 30 min. The appropriate alkyl or aryl Br/Cl/Ms/Ts (1–2 eq) was then added. The reaction was allowed to slowly warm to room temperature and left to stir (2–12 hr) until HPLC indicated reaction was complete. The solution was poured into EtOAc, washed 3x with brine, dried over NaSO4, and concentrated. Crude residue was purified by flash chromatography EtOAc:Hexanes 0–100% gradient to give the pure tertiary amine Int-4b.
General Procedure B4: Boc-Deprotection
The Boc-protected amine (1 eq) was dissolved into a 1:2 mixture of TFA:DCM (~0.1–0.3 mM) and allowed to stir at room temperature for 0.5–2h, until HPLC or TLC indicated reaction was complete. Solvent was removed in vacuo and compound was purified by reverse phase chromatography (C18, 10–100% ACN in water + 0.1% TFA gradient) and concentrated to afford desired product as the TFA salt.
Computational Modeling:
To construct a model for TgCatL-triazine complex, the X-ray structure of TgCPL in complex with its propeptide (PDB: 3F75) was superposed with the X-ray structure of HsCPL (PDB: 5MAJ) using The Molecular Operating Environment (MOE), version 2008.10, Chemical Computing Group Inc., Montreal, Quebec, Canada. Then, the HsCPL enzyme from the 5MAJ and the propeptide of TgCPL from the 3F75 were removed and the TgCPL enzyme and the triazine compound are saved as a model for TgCPL-triazine complex. All the water molecules from the 3F75 and water molecules outside the binding site from 5MAJ were removed. Covalent docking of the triazine compound in the TgCPL was performed with induced fit option instead of rigid receptor for refinement. The covalent docking protocol in MOE was developed by creating a reaction profile for this particular virtual reaction and saving in the MarvinSketch 17.14.0 (http://www.chemaxon.com).
TgCPL and HsCPL Inhibition Assay:
Compound potency and selectivity was evaluated in vitro for both TgCPL and HsCPL activity in a fluorescence-based assay by monitoring the hydrolysis of Cbz-Leu-Arg-aminomethylcoumarin (Z-L-R-AMC). Protein was expressed and purified as described previously.25 The substrate hydrolysis results in the release of fluorescent 7-amino-4-methylcoumarin (AMC) that can be monitored spectrophotometrically with linear kinetics for up to 60 min. Inhibitors were serial diluted in DMSO in a 1-to-3 dilution, spanning at least 10 concentration points in either duplicate or triplicate. The enzyme was pre-incubated with the inhibitory compounds for 5 min at 23°C, followed by the addition of the AMC substrate. The assay final conditions had a total volume of 200 μL, consisting of 90 μL enzyme (conc.=50 ng/mL), 100 μL Z-L-R-AMC substrate (conc.= 80 μM), and 10 μL inhibitor or DMSO. The relative fluorescence of the AMC generation is measured over the course of 5 min (Excitation: 380 nm, Emission: 460 nm). LHVS (1) and DMSO were used as positive and negative controls, respectively. All dose response data was obtained with at least three independent replicates (n=3). Graphpad Prism software was used to visualize inhibition curves and calculate IC50 values from the reaction mean ν. Assay final concentrations in each well: 40 μM ZLR-AMC, TgCPL or HsCPL (final conc= 0.0225 ng/μL). Assay Buffer: 100 mM NaAc, 2 mM EDTA, 900 mM NaCl, 50 mM DTT. Substrate: Cbz-Leu-Arg-AMC (Bachem, purchased as HCl salt).
High throughput library screen:
We utilized compound libraries provided by the University of Michigan Center for Chemical Genomics (CCG) to screen for inhibitors against TgCPL. The HTS primary screen was conducted on ~150,000 candidates, representing a wide diversity of structures and sources. The libraries were composed of small molecule compounds and clinically tested bioactive drugs obtained from a variety of sources include ChemBridge, ChemDiv, Maybridge, MicroSource, TimTec, NCI, and the NIH Clinical Collection. Reactions of the aforementioned Z-Leu-Arg-AMC assay were conducted in a 384-well plate at the scale of 10 μL per well. Each reaction was initiated by the addition of 50 nL of 2 mM stocks of the compounds to each well using a Caliper Sciclone ALH3000 with a V&P pintool, resulting in a final concentration of 10 μM for each compound. A Thermo Multidrop Combi Liquid Dispenser was used to add 5 μL of recombinant TgCPL (0.5 μg/mL) and 5 μL of Z-Leu-Arg-AMC (80 μM), both diluted in activity assay buffer. After 10 min incubation at RT, 10 μL of E64 (100 μM) diluted in in assay buffer was added with the multidrop reagent dispenser. A Perkin Elmer Envision Multimode Plate Reader measured the fluorescence after 30 min at RT with excitation at 355 and the emission at 460 nm. Active compounds were triaged using computational and experimental results to eliminate compounds likely to interfere with the assay or to present development issues. Computational evaluation of primary screen hits was first conducted in light of toxicity, cysteine reactivity, promiscuity, and inhibitor level. Primary screen hits available after this attribute triage were then subjected to confirmation screen to compose the experimental evaluation. Assay conditions from the primary screen were repeated, but conducted in triplicate for each candidate (using a TTP Labtech Mosquito X1 for compound addition). Confirmed hits were retested to establish a concentration response curve of activity in a secondary screen. Assay conditions were repeated on the 384-well plates, and conducted in duplicate for each compound. Compounds found active after the concentration response secondary screen were prioritized based on chemical properties, and given appropriate rankings by consulting medicinal chemists. High and medium ranked hits were prioritized, and fresh powders of the library stocks were obtained from Sigma Aldrich. The available compounds were reconstituted in DMSO to 10 μM. All assay conditions of the concentration response screen were repeated for the repurchased compounds against both TgCPL and HsCPL.
Metabolic Stability in Mouse Liver Microsomes:
The metabolic stability was assessed using CD-1 mouse liver microsomes. One micromolar of each compound was incubated with 0.5 mg/mL microsomes and 1.7 mM cofactor β-NADPH in 0.1 M phosphate buffer (pH = 7.4) containing 3.3 mM MgCl2 at 37°C. The DMSO concentration was less than 0.1% in the final incubation system. At 0, 5, 10, 15, 30, 45, and 60 min of incubation, 40 μL of reaction mixture were taken out, and the reaction is quenched by adding 3-fold excess of cold acetonitrile containing 100 ng/mL of internal standard for quantification. The collected fractions were centrifuged at 15000 rpm for 10 min to collect the supernatant for LC–MS/ MS analysis, from which the amount of compound remaining was determined. The natural log of the amount of compound remaining was plotted against time to determine the disappearance rate and the half-life of tested compounds.
Pharmacokinetic Studies in Mice:
All animal experiments in this study were approved by the University of Michigan Committee on Use and Care of Animals and Unit for Laboratory Animal Medicine. The abbreviated pharmacokinetics for compounds was determined in female CD-1 mice following intraperitoneal (ip) injection at 10 mg/kg. Compound was dissolved in the vehicle containing 15% (v/v) DMSO, 15–20% (v/v) PEG-400, and 70% (v/ v) PBS. Four blood samples (50 μL) were collected over 7 h (at 0.5h, 2h, 4h, and 7h), centrifuged at 3500 rpm for 10 min, and plasma was frozen at −80°C for later analysis. Plasma concentrations of the compounds were determined by the LC–MS/MS method developed and validated for this study. The LC–MS/MS method consisted of a Shimadzu HPLC system, and chromatographic separation of tested compound which was achieved using a Waters Xbridge-C18 column (5 cm × 2.1 mm, 3.5 μm). An AB Sciex QTrap 4500 mass spectrometer equipped with an electrospray ionization source (ABI-Sciex, Toronto, Canada) in the positive-ion multiple reaction monitoring (MRM) mode was used for detection. All pharmacokinetic parameters were calculated by non-compartmental methods using WinNonlin, version 3.2 (Pharsight Corporation, Mountain View, CA, USA).
MDR1/MDCK Assay:
12-well transwell plates were seeded with MDCKII-MDR1 cells (0.5 million/well) and cultured for 24h. Cells were washed with DMEM 3 times (both sides). 0.5 ml of 1μM test compound in DMEM was added to apical side (for A to B measurement) or basolateral side (for B to A) (donor side) and 0.5ml of DMEM+0.1% DMSO to the receiving side. Cells were incubated for 4h and 2 × 200μL was sampled from the receiving side and stored at −20°C for future use. For calibration standard, compound standards were dissolved in DMSO then further diluted in acetonitrile to a concentration of 10 μg/mL. Calibration standards were prepared from this stock with internal standard (5 nM CE302). Samples were prepared by protein precipitation from media followed by addition of internal standard. Samples were analyzed by LC–MS/MS. The LC–MS/MS method consisted of a Shimadzu HPLC system, and chromatographic separation of tested compound which was achieved using a Waters Xbridge-C18 column (5 cm × 2.1 mm, 3.5 μm). An AB Sciex QTrap 4500 mass spectrometer equipped with an electrospray ionization source (ABI-Sciex, Toronto, Canada) in the positive-ion multiple reaction monitoring (MRM) mode was used for detection.
Bradyzoite Viability Assay:
Bradyzoite viability was assessed by combining plaque assay and quantitative polymerase chain reaction (qPCR) analysis of genome number, as previously performed by Di Cristina et al, 2017.26 Briefly, human foreskin fibroblast (HFF) cells were infected with T. gondii tachyzoites in six-well plates. Following host cell invasion by parasites, tachyzoites underwent differentiation to bradyzoites by being maintained at 37°C/0% CO2 in alkaline media (RPMI 1540 w/o NaHCO3, 50 mM HEPES, 3% FBS, Pen/Strep, pH 8.2), resulting in the generation of in vitro tissue cysts. Differentiation was carried out over the course of seven days, replacing the alkaline media daily. Samples were subsequently treated with the test compounds for seven days, replacing them daily. A DMSO-treated vehicle control sample was also incorporated into each assay. Following the treatment period, the culture media in each well was replaced with 2 mL HBSS and cysts were liberated from the infected HFF monolayers by mechanical extrusion, by lifting cells with a cell scraper and syringing several times through 26G needles. Then, 2 mL of pre-warmed 2× pepsin solution (0.026% pepsin in 170 mM NaCl and 60 mM HCl, final concentration) was added to each sample and sample were left to incubate at 37°C for 30 min. Reactions were stopped by adding 94 mM Na2CO3, removing the supernatant after centrifugation at 1,500g for 10 min at room temperature and re-suspending pepsin-treated parasites in 1 mL of DMEM without serum. Parasites were enumerated and 1000 parasites per well were added to six-well plates containing confluent monolayers of HFFs in D10 media, in triplicate. To allow for the formation of plaques, parasites were left to grow undisturbed for 12 days. After this period, the number of plaques in each well was determined by counting plaques with the use of a light microscope. Five hundred microlitres of the initial 1 mL of pepsin-treated parasites was used for genomic DNA purification, performed using the DNeasy Blood & Tissue Kit (Qiagen). Genomic DNA was eluted in a final volume of 100 μL. To determine the number of parasite genomes per microliter, 10 μL of each gDNA sample was analysed by qPCR, in duplicate, using the tubulin primers TUB2.RT.F and TUB2.RT.R. Quantitative PCR analyses were performed using Brilliant II SYBR Green QPCR Master Mix (Agilent) and a Stratagene Mx3000PQ-PCR machine. The number of plaques that formed per genome was then calculated and expressed as a percentage of control (DMSO) treated parasites.
HFF Cell Viability by MTS Assay:
MTS assays were performed to evaluate the cytotoxicity of test compounds on HFF cells in vitro. HFF cells were plated onto 96-well plates and allowed to become fully confluent at 37°C/5% CO2, prior to the addition of the test compounds. Test compounds were diluted to 100 μM in conversion media (the same culture media used in bradyzoite viability assays) and applied to HFF cells in triplicate. Subsequently, test compounds underwent two-fold serial dilution across the plate to a concentration of 0.195 μM. Plates were incubated for a further 7 days at 37°C/5% CO2, replacing the media daily to apply fresh compounds. Following the 7-day treatment, 20 μl of CellTiter 96 Aqueous One Solution reagent (Promega) was added to each well and left to incubate for 2 hours at 37°C. Absorbance was then measured at 490nm using a Synergy H1 Hybrid Multi-Mode Reader (BioTek). MTS assays were performed in triplicate. Untreated HFF cells were used as a negative control. HFF cell viability was calculated as a percentage by dividing the measured A490 of test samples by the mean A490 of the untreated control samples. The 50% toxicity dosage (TD50) of each test compound was established by plotting the % HFF cell viability against compound concentration and reading the concentration at 50% HFF cell viability.
Supplementary Material
Table 2.
Screen of various P2 vectors
| No. | R | TgCPLa IC50 (nM) | HsCPLb IC50 (nM) | Selectivity for TgCPLc |
|---|---|---|---|---|
| 4 | 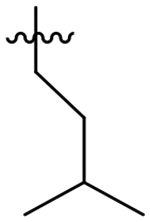 |
34 | 32 | 0.94 |
| 38 |  |
246 | 67 | 0.27 |
| 39 |  |
117 | 26 | 0.22 |
| 40 |  |
144 | 64 | 0.44 |
| 41 |  |
46 | 32 | 0.70 |
| 42 |  |
67 | 2 | 0.030 |
| 43 |  |
220 | 328 | 1.5 |
| 44 |  |
2,260 | 759 | 0.33 |
| 45 | 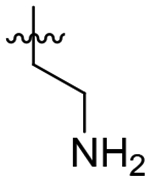 |
6,490 | 3,430 | 0.52 |
| 46 | 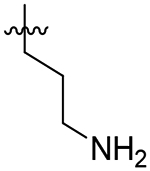 |
3,590 | 4,090 | 1.1 |
| 47 |  |
1,410 | 561 | 0.40 |
| 48 |  |
1,990 | 2,050 | 1.1 |

IC50 for Toxoplasma gondii Cathepsin L (TgCPL) or human Cathepsin L (HsCPL). Values are mean of at least 3 independent experiments with SEM typically < 30%.
Selectivity for Toxoplasma gondii Cathepsin L as defined by the ratio HsCPL IC50/TgCPL IC50
Table 6.
MTS assay results in HFF cells.
| Compound | CC50 (μM)a |
|---|---|
| 4 | 65.19 (±16.69) |
| 50 | 29.05 (±2.6) |
| 56 | >100 |
| 62 | 61.05 (±2.08) |
| 65 | 34.58 ±1.33) |
All values are the mean of at least three independent experiments.
Funding Sources:
This work was supported by NIAID of the National Institutes of Health under award number R21/R33AI127492 (V.B.C. and S.D.L.). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. V.B.C. and S.D.L. were also supported by a grant from the Center for Discovery of New Medicines, University of Michigan. J.D.Z. acknowledges support from the University of Michigan Pharmacological Sciences Training Program (PSTP) training grant (T32-GM007767), and the Rackham Merit Fellowship (RMF).
Footnotes
Supporting Information
Synthetic procedures for and characterization of all compounds.
The authors declare no competing financial interest.
References
- 1.Dubey JP; Jones JL, Toxoplasma gondii infection in humans and animals in the United States. Int J Parasitol 2008, 38 (11), 1257–78. [DOI] [PubMed] [Google Scholar]
- 2.Jones JL; Kruszon-Moran D; Elder S; Rivera HN; Press C; Montoya JG; McQuillan GM, Toxoplasma gondii Infection in the United States, 2011–2014. Am J Trop Med Hyg 2018, 98 (2), 551–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoffmann S; Batz MB; Morris JG Jr., Annual cost of illness and quality-adjusted life year losses in the United States due to 14 foodborne pathogens. J Food Prot 2012, 75 (7), 1292–302. [DOI] [PubMed] [Google Scholar]
- 4.Scallan E; Griffin PM; Angulo FJ; Tauxe RV; Hoekstra RM, Foodborne illness acquired in the United States--unspecified agents. Emerg Infect Dis 2011, 17 (1), 16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Innes EA, A brief history and overview of Toxoplasma gondii. Zoonoses Public Health 2010, 57 (1), 1–7. [DOI] [PubMed] [Google Scholar]
- 6.Dubey JP, History of the discovery of the life cycle of Toxoplasma gondii. Int J Parasitol 2009, 39 (8), 877–82. [DOI] [PubMed] [Google Scholar]
- 7.Fuglewicz AJ; Piotrowski P; Stodolak A, Relationship between toxoplasmosis and schizophrenia: A review. Adv Clin Exp Med 2017, 26 (6), 1031–1036. [DOI] [PubMed] [Google Scholar]
- 8.Afonso C; Paixao VB; Costa RM, Chronic Toxoplasma infection modifies the structure and the risk of host behavior. PLoS One 2012, 7 (3), e32489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nascimento FS; Dantas CD; Netto MP; Mella LFB; Suzuki LA; Banzato CEM; Rossi CL, Prevalence of antibodies to Toxoplasma gondii in patients with schizophrenia and mood disorders. Schizophr Res 2012, 142 (1–3), 244–245. [DOI] [PubMed] [Google Scholar]
- 10.Pearce BD; Kruszon-Moran D; Jones JL, The Relationship Between Toxoplasma Gondii Infection and Mood Disorders in the Third National Health and Nutrition Survey. Biol Psychiat 2012, 72 (4), 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lanjewar DN; Surve KV; Maheshwari MB; Shenoy BP; Hira SK, Toxoplasmosis of the central nervous system in the acquired immunodeficiency syndrome. Indian J Pathol Microbiol 1998, 41 (2), 147–51. [PubMed] [Google Scholar]
- 12.Dedicoat M; Livesley N, Management of toxoplasmic encephalitis in HIV-infected adults--a review. S Afr Med J 2008, 98 (1), 31–2. [PubMed] [Google Scholar]
- 13.Deng Y; Wu T; Zhai SQ; Li CH, Recent progress on anti-Toxoplasma drugs discovery: Design, synthesis and screening. Eur J Med Chem 2019, 183, 111711. [DOI] [PubMed] [Google Scholar]
- 14.Rutaganira FU; Barks J; Dhason MS; Wang Q; Lopez MS; Long S; Radke JB; Jones NG; Maddirala AR; Janetka JW; El Bakkouri M; Hui R; Shokat KM; Sibley LD, Inhibition of Calcium Dependent Protein Kinase 1 (CDPK1) by Pyrazolopyrimidine Analogs Decreases Establishment and Reoccurrence of Central Nervous System Disease by Toxoplasma gondii. J Med Chem 2017, 60 (24), 9976–9989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sajid M; McKerrow JH, Cysteine proteases of parasitic organisms. Mol Biochem Parasitol 2002, 120 (1), 1–21. [DOI] [PubMed] [Google Scholar]
- 16.Ang KKH; Ratnam J; Gut J; Legac J; Hansell E; Mackey ZB; Skrzypczynska KM; Debnath A; Engel JC; Rosenthal PJ; McKerrow JH; Arkin MR; Renslo AR, Mining a Cathepsin Inhibitor Library for New Antiparasitic Drug Leads. Plos Neglect Trop D 2011, 5 (5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sajid M; McKerrow JH, Cysteine proteases of parasitic organisms (vol 120, pg 1, 2002). Mol Biochem Parasit 2002, 121 (1), 159–159. [DOI] [PubMed] [Google Scholar]
- 18.Chung JY; Bae YA; Na BK; Kong Y, Cysteine protease inhibitors as potential antiparasitic agents. Expert Opin Ther Pat 2005, 15 (8), 995–1007. [Google Scholar]
- 19.Dou Z; Carruthers VB, Cathepsin proteases in Toxoplasma gondii. Adv Exp Med Biol 2011, 712, 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dou Z; Coppens I; Carruthers VB, Non-canonical maturation of two papain-family proteases in Toxoplasma gondii. J Biol Chem 2013, 288 (5), 3523–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dou Z; McGovern OL; Di Cristina M; Carruthers VB, Toxoplasma gondii ingests and digests host cytosolic proteins. MBio 2014, 5 (4), e01188–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang R; Que XC; Hirata K; Brinen LS; Lee JH; Hansell E; Engel J; Sajid M; Reed S, The cathepsin L of Toxoplasma gondii (TgCPL) and its endogenous macromolecular inhibitor, toxostatin. Mol Biochem Parasit 2009, 164 (1), 86–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parussini F; Coppens I; Shah PP; Diamond SL; Carruthers VB, Cathepsin L occupies a vacuolar compartment and is a protein maturase within the endo/exocytic system of Toxoplasma gondii. Mol Microbiol 2010, 76 (6), 1340–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pittman KJ; Aliota MT; Knoll LJ, Dual transcriptional profiling of mice and Toxoplasma gondii during acute and chronic infection. BMC Genomics 2014, 15, 806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Larson ET; Parussini F; Huynh MH; Giebel JD; Kelley AM; Zhang L; Bogyo M; Merritt EA; Carruthers VB, Toxoplasma gondii Cathepsin L Is the Primary Target of the Invasion-inhibitory Compound Morpholinurea-leucyl-homophenyl-vinyl Sulfone Phenyl. Journal of Biological Chemistry 2009, 284 (39), 26839–26850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Di Cristina M; Dou Z; Lunghi M; Kannan G; Huynh MH; McGovern OL; Schultz TL; Schultz AJ; Miller AJ; Hayes BM; van der Linden W; Emiliani C; Bogyo M; Besteiro S; Coppens I; Carruthers VB, Toxoplasma depends on lysosomal consumption of autophagosomes for persistent infection. Nat Microbiol 2017, 2, 17096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barclay J; Clark AK; Ganju P; Gentry C; Patel S; Wotherspoon G; Buxton F; Song C; Ullah J; Winter J; Fox A; Bevan S; Malcangio M, Role of the cysteine protease cathepsin S in neuropathic hyperalgesia. Pain 2007, 130 (3), 225–34. [DOI] [PubMed] [Google Scholar]
- 28.Zwicker JD; Diaz NA; Guerra AJ; Kirchhoff PD; Wen B; Sun D; Carruthers VB; Larsen SD, Optimization of dipeptidic inhibitors of cathepsin L for improved Toxoplasma gondii selectivity and CNS permeability. Bioorg Med Chem Lett 2018, 28 (10), 1972–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Di L; Rong H; Feng B, Demystifying brain penetration in central nervous system drug discovery. Miniperspective. J Med Chem 2013, 56 (1), 2–12. [DOI] [PubMed] [Google Scholar]
- 30.Li YY; Fang J; Ao GZ, Cathepsin B and L inhibitors: a patent review (2010 - present). Expert Opin Ther Pat 2017, 27 (6), 643–656. [DOI] [PubMed] [Google Scholar]
- 31.Giroud M; Ivkovic J; Martignoni M; Fleuti M; Trapp N; Haap W; Kuglstatter A; Benz J; Kuhn B; Schirmeister T; Diederich F, Inhibition of the Cysteine Protease Human Cathepsin L by Triazine Nitriles: AmideHeteroarene pi-Stacking Interactions and Chalcogen Bonding in the S3 Pocket. ChemMedChem 2017, 12 (3), 257–270. [DOI] [PubMed] [Google Scholar]
- 32.Ehmke V; Winkler E; Banner DW; Haap W; Schweizer B; Rottmann M; Kaiser M; Freymond C; Schirmeister T; Diederich F, Optimization of Triazine Nitriles as Rhodesain Inhibitors: StructureActivity Relationships, Bioisosteric Imidazopyridine Nitriles, and X-ray Crystal Structure Analysis with Human CathepsinL. Chemmedchem 2013, 8 (6), 967–975. [DOI] [PubMed] [Google Scholar]
- 33.Giroud M; Harder M; Kuhn B; Haap W; Trapp N; Schweizer WB; Schirmeister T; Diederich F, Fluorine Scan of Inhibitors of the Cysteine Protease Human Cathepsin L: Dipolar and Quadrupolar Effects in the pi-Stacking of Fluorinated Phenyl Rings on Peptide Amide Bonds. Chemmedchem 2016, 11 (10), 1042–1047. [DOI] [PubMed] [Google Scholar]
- 34.Cai J; Fradera X; van Zeeland M; Dempster M; Cameron KS; Bennett DJ; Robinson J; Popplestone L; Baugh M; Westwood P; Bruin J; Hamilton W; Kinghorn E; Long C; Uitdehaag JC, 4-(3-Trifluoromethylphenyl)-pyrimidine-2-carbonitrile as cathepsin S inhibitors: N3, not N1 is critically important. Bioorg Med Chem Lett 2010, 20 (15), 4507–10. [DOI] [PubMed] [Google Scholar]
- 35.Cai J; Bennett DJ; Rankovic Z; Dempster M; Fradera X; Gillespie J; Cumming I; Finlay W; Baugh M; Boucharens S; Bruin J; Cameron KS; Hamilton W; Kerr J; Kinghorn E; McGarry G; Robinson J; Scullion P; Uitdehaag JC; van Zeeland M; Potin D; Saniere L; Fouquet A; Chevallier F; Deronzier H; Dorleans C; Nicolai E, 2-Phenyl-9H-purine-6-carbonitrile derivatives as selective cathepsin S inhibitors. Bioorg Med Chem Lett 2010, 20 (15), 4447–50. [DOI] [PubMed] [Google Scholar]
- 36.Altmann E; Cowan-Jacob SW; Missbach M, Novel purine nitrile derived inhibitors of the cysteine protease cathepsin K. J Med Chem 2004, 47 (24), 5833–6. [DOI] [PubMed] [Google Scholar]
- 37.Schroder J; Noack S; Marhofer RJ; Mottram JC; Coombs GH; Selzer PM, Identification of semicarbazones, thiosemicarbazones and triazine nitriles as inhibitors of Leishmania mexicana cysteine protease CPB. PLoS One 2013, 8 (10), e77460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Turk V; Stoka V; Vasiljeva O; Renko M; Sun T; Turk B; Turk D, Cysteine cathepsins: from structure, function and regulation to new frontiers. Biochim Biophys Acta 2012, 1824 (1), 68–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gauthier JY; Chauret N; Cromlish W; Desmarais S; Duong LT; Falgueyret JP; Kimmel DB; Lamontagne S; Leger S; LeRiche T; Li CS; Masse F; McKay DJ; Nicoll-Griffith DA; Oballa RM; Palmer JT; Percival MD; Riendeau D; Robichaud J; Rodan GA; Rodan SB; Seto C; Therien M; Truong VL; Venuti MC; Wesolowski G; Young RN; Zamboni R; Black WC, The discovery of odanacatib (MK-0822), a selective inhibitor of cathepsin K. Bioorg Med Chem Lett 2008, 18 (3), 923–8. [DOI] [PubMed] [Google Scholar]
- 40.Talele TT, The “Cyclopropyl Fragment” is a Versatile Player that Frequently Appears in Preclinical/Clinical Drug Molecules. J Med Chem 2016, 59 (19), 8712–8756. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.