

Abstract
Background
Esophageal carcinoma (ESCA) is a health challenge with poor prognosis and limited treatment options. Our aim is to screen for hub genes and pathways associated with ESCA pathology as diagnostic or therapeutic targets.
Material/Methods
We downloaded 2 ESCA-related datasets from the Gene Expression Omnibus (GEO) database. Subsequently, differentially expressed genes (DEGs) of ESCA were determined by statistical analysis. Both Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of DEGs were performed using online analytic tools. Network analysis was employed to construct a protein-protein interaction (PPI) network and to filter hub genes. We evaluated the expression level and impact of hub genes on survival of ESCA patients using the OncoLoc webserver.
Results
A total of 210 DEGs were identified. The GO analysis showed that the DEGs were enriched in cell division. The KEGG pathway analysis showed DEGs that were enriched in cell cycle regulation, known cancer pathways, the PI3K-Akt signaling pathway, and the cGMP-PKG signaling pathway. The top 10 hub genes were markedly upregulated in ESCA tissue compared with normal esophageal tissue. Moreover, the expression level of the hub genes was different at different pathological stages of ESCA. Further prognostic analysis identified that the top 10 hub genes were related to late survival of ESCA patients, while exhibiting few associations with early survival time.
Conclusions
The signaling pathways involving the DEGs probably represent the pathological mechanism underlying ESCA. The hub genes were associated with survival of ESCA patients, and as such have the potential to serve as diagnostic indicators and therapeutic targets.
MeSH Keywords: Computational Biology, Esophageal Neoplasms, Gene Expression
Background
Esophageal carcinoma (ESCA) is the eighth most common cancer and the sixth most common cancer leading to death, with more than 456 000 sufferers worldwide [1,2]. Previous studies have documented that the overall 5-year survival of ESCA patients is approximately 15% to 25% [2].
ESCA includes two major subtypes: esophageal squamous cell carcinoma (ESCC), the predominant form, and esophageal adenocarcinoma (EAC), which affects a growing percentile of patients [3]. There are distinct differences between the pathological risks underlying these histological subtypes that affect their incidence. The mechanism underlying ESCC is complicated and embraces a broad spectrum of hazards contributing to its rapidly increasing incidence. There are two primary types of risk factors: inheritable and environmental factors. There are several environmental risks, including alcohol and tobacco consumption, low vegetable and fruit intake, and low socioeconomic status [4]. As for EAC, almost all cases are complicated by precancerous lesions called Barrett’s oesophagus, which mainly derive from gastroesophageal reflux disease, a common condition throughout the human population [5]. Despite this, the risk of progression from this kind of chronic premalignant condition into EAC is low; approximately 0.12%, which makes it difficult to perform predictions of EAC from Barrett’s oesophagus. Fortunately, there are some mechanistic connections between heritable elements and EAC. A meta-analysis demonstrated that single nucleotide polymorphisms (SNPs) located in the LPA, TPPP, and CEP72 genes were associated with high risk of both EAC and its premalignant precursor. SNPs near HTR3C and ABCC5 implicated this region as a specific locus for EAC [6].
Given its extremely invasive nature and poor prognosis among gastrointestinal malignancies, a majority of people die from ESCA, placing a heavy burden on the international economy [7]. When it comes to treatment, ESCA demands multidisciplinary knowledge, covering endoscopic procedures, neoadjuvant therapy with chemotherapy, chemoradiotherapy, surgery, and other procedures [8]. If early-stage, mild symptoms are neglected by patients and clinical workers, treatment will be even more difficult. Studies have demonstrated that early diagnosis of dysplasia allows for decisive therapy and ultimately lengthens survival time [9]. In addition, immuno-oncology therapies with molecular targets have emerged, promising early results among ESCA patients [10]. Pursuit of available biomarkers therefore has the potential to exert positive effects via early diagnosis and treatment.
At present, exploring pathological mechanisms of diseases based on bioinformatics theories has becoming an increasingly important and effective method [11]. With bioinformatics analysis, researchers can gain comprehensive knowledge regarding the studied diseases from molecular data. More crucially, it can provide novel insight leading to early diagnosis, definitive treatment, and survival prediction [12]. Previous research in the bioinformatics field has yielded some achievements in ESCA knowledge, such as identification of associated genes and pathways and altered methylation associated with ESCA pathology [13,14]. However, analytical ability for ESCA has been limited due to insufficient sample size and a lack of in-depth evaluation of key genes which play a dominant role in the malignant development of ESCA.
Learning from our predecessors, we added the expression data from two microarrays to our research to enlarge the sample size, in order to reduce errors. We aimed to seek out hub genes and pathways involved in ESCA using integrated analysis of the chosen microarray data. Moreover, we aimed to conduct further expression and survival assessments of hub genes, which may provide reference and direction for early diagnosis and practical therapeutic strategies for ESCA. To our knowledge, the present study represents the first assessment of key hub genes associated with ESCA using bioinformatics methods to obtain information on altered expression of these genes at different stages, as well as their prognostic impact for diagnosed patients.
Material and Methods
Acquisition of datasets
The appropriate datasets were obtained from the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo). Inclusion criteria were as follows: (1) sample from human esophageal tissue (cell-line and animal experiments excluded); (2) normalized expression data; (3) coverage of diverse types of ESCA, including ESCC, EAC, and rare types of ESCA; (4) control group from non-carcinoma oesophageal tissue; (5) inclusion of mRNA expression level.
After an overall scrutiny, the datasets of GSE100942 [15] and GSE111044 [16] were selected as they suited the inclusion standards we set up. Moreover, both of them had been created recently, in the past 5 years, which met the need for timeliness. GSE100942 consisted of ten samples, including five samples from ESCC and five samples from normal esophageal tissue. GSE111044 was made up of six samples, including three samples from small cell esophageal carcinoma (SCEC) and three samples from corresponding normal tissue.
Identification of DEGs
GEO2R (http://www.ncbi.nlm.nih.gov/geo/geo2r/), an online web tool affiliated with GEO, already provided access to the data set and collection. Further analysis was applied to the prepared datasets using GEO2R. The selected datasets were divided into two groups: the normal tissue group and the cancer group. Subsequently, direct statistical analysis of each dataset was conducted with cutoff values of: p<0.01 and 2 |log FC| ≥3. The intersection of the two filtered datasets, representing DEGs, was identified by a web-based tool named Venny v2.1. (https://bioinfogp.cnb.csic.es/tools/venny/).
Enrichment analysis
Gene ontology (GO) enrichment analysis was conducted using the online platform Metascape [17] (http://metascape.org). A set of functional classifications and annotations are included in Metascape, such as biological processes (BP), cellular components (CC), and molecular functions (MF). The norm for statistical significance of p<0.05 was determined as the threshold value for our statistical analysis, to deeply screen enriched GO terms.
The Kyoto Encyclopedia of Genes and Genomes (KEGG) Orthology Based Annotation System, third version (KOBAS v3.0) [18] (http://kobas.cbi.pku.edu.cn/kobas3) was used to perform the KEGG pathway enrichment analysis. A corrected p value <0.05 was considered to represent statistical significance. Moreover, we used the remaining pathways to draw a bubble chart of the top 20 pathways as a visualization, using OmicShare tools (https://www.omicshare.com.html).
Construction of the PPI network
The screened DEGs were imported into the Search Tool for the Retrieval of Interacting Genes (STRING) [19] (http://string.embl.de/) to begin network analysis. STRING possesses a potent ability to process multiple proteins or amino acid sequences input by users, which are then used to construct a protein-protein interaction (PPI) network. Conveniently, STRING can provide various types of exported files, including tsv, PNG, and tables. In our research, we downloaded the requisite files (tsv and PNG) to prepare for validation of hub genes and further research.
Expression analysis of hub genes
In light of degree level, hub genes were identified by cytohubba, an analytic tool attached to Cytoscape software [20] (https://cytoscape.org/). Cytohubba can rank nodes in a network by their network features in order to find a requested objective from imported elements of a PPI network, at levels of degree, closeness, and so on.
We successfully used Gene Expression Profiling Interactive Analysis (GEPIA2) [21] (http://gepia2.cancer-pku.cn) to compare mRNA expression levels between ESCA tissue and paired normal tissue. Additionally, differential expression levels in patients at each stage of ESCA were compared in GEPIA.
Survival analysis of hub genes
OncoLnc [22] (http://www.oncolnc.org/) was used to analyze predictive hub gene data. OncoLnc is an efficient online analytic box, which can supply survival data correlated with mRNA expression level based on The Cancer Genome Atlas (TCGA) data. We set 50 as the cutoff to divide paired data into high- and low-expression groups, for a Kaplan plot of input genes related to ESCA.
Results
Confirmation of DEGs
After GEO2R analysis of the two screened datasets, the criterion p<0.01 and 2 |log FC| ≥3 was utilized for statistical analysis. A total of 3170 and 318 gene fragments were selected from the GSE111044 and GSE100942 datasets. Genes found to be present in both datasets were selected as DEGs. Consequently, a total of 210 DEGs were screened from the two genetic datasets (Table 1, Figure 1).
Table 1.
| Source | Total | Elements |
|---|---|---|
| Intersection of GSE111044 and GSE100942 | 210 | SLMAP, SAMD4A, KCNMA1, LDB3, LINC01279, TPX2, EPB41L4B, WISP2, IGF2BP3, CCNB1, MSRB3, ANP32E, NFIA, CNN1, COL1A1, ANLN, BIRC5, GHR, BCHE, DNMT3B, SMAD9, LRRK2, HLF, ADAMTS2, AURKA, KIF14, FHL1, SORBS2, ADRB1, CSRP1, TCF21, KIF4A, KIT, RAD54B, C1QTNF7, NEGR1, FAM72A, KCNAB1, MELK, CDH11, NDC80, CCNA2, GTSE1, NUF2, KRT78, PDK4, MMP1, MYOCD, EPCAM, ANGPTL1, DAAM2, P2RY14, ECT2, KIF23, DEPDC1, EMCN, LPAR3, OGN, POSTN, NBEA, MYLK, CYSLTR1, CCNB2, PRC1, IGFBP6, TNXB, CEP55, MYOC, CCNE2, MCM2, MCM4, CGNL1, ADIRF, HEY1, SRPX, IL1RN, DLGAP5, RAD51AP1, SMTN, TMEM108, SCN7A, CCDC69, CDCA2, MYL9, LRRN4CL, RHPN2, CDH19, PGM5, SOBP, C2orf40, AOX1, COL1A2, LAPTM4B, GULP1, CFD, RNASE4, NEK2, ABLIM1, KIF26B, LPP, FGL2, CHRDL1, HSPB8, RAP1A, DEPDC1B, CILP, ITIH5, HMGB3, TMEM110, NETO2, SYNPO2, SOX4, PLN, MRVI1, EZH2, TNFSF10, CD36, CAPN14, CFH, MYOT, MT1M, RASEF, EMP1, PGM5-AS1, FAM149A, STEAP4, AOC3, LMOD1, ASPA, TMEM35A, CCL14, MYH11, KIF2C, EBF1, EREG, AGTR1, CDCA7, TNS1, CDC20, RBPMS2, C7, BUB1, CCL2, PRR11, IL6, CPED1, RGS2, COL3A1, DPT, SCARA5, PTPRN2, ATP1A2, CDCA3, IFI6, ATAD2, KNL1, EXOSC7, ADAMTS1, ANOS1, SORBS1, FGF7, STIL, UBE2C, STXBP6, MTURN, CCDC80, CLU, CXCL12, IL36A, SLC35F6, TOP2A, AVPR1A, COL14A1, HELLS, FANCI, IRX2, NEXN, SPC25, KIF18A, BUB1B, DKK1, HJURP, AQP1, LMO3, SOCS2, DTL, ACTG2, ADH1B, MEST, ARHGAP6, HMMR, CAB39L, FAM107A, GPX3, KIF20A, SHISA2, ENC1, PTGS1, CASQ2, CTHRC1, UHRF1, SGO2, MAMDC2, TTK, CDKN3, NCAPG, FXYD3, CENPF, NUSAP1, CRCT1 |
Figure 1.
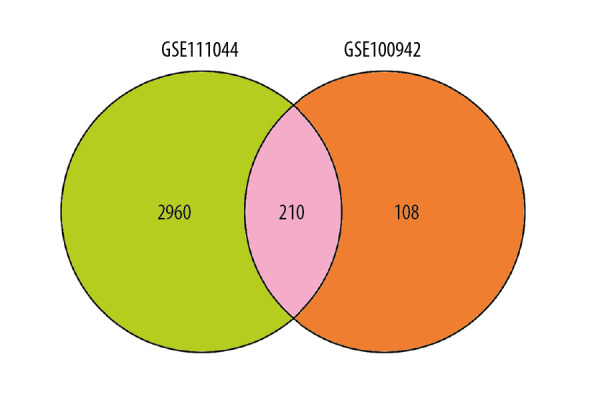
Venn diagram of filtered differentially expressed genes (DEGs) shared by the GSE111044 and GSE100942 datasets. The overlapping area, which contained 210 items, was considered to contain DEGs for ESCA.
GO enrichment analysis
In total, 114 GO terms mediated by the DEGs were identified as enriched, spanning the categories of molecular function (MF), cellular component (CC), and biological process (BP) (Table 2). The MF results implicated DEGs that were profoundly enriched in actin binding, extracellular matrix structural constituents, and structural constituents of muscle (Figure 2). The CC results indicated that DEGs were enriched in chromosome, centromere, spindle, extracellular matrix, and stress fibers (Figure 3). With respect to BP, DEGs were noticeably enriched in processes related to cell division, mitotic cell cycle phase transition, muscle system process, and muscle structural development (Figure 4). We then obtained the top 20 terms of the GO functional enrichment analysis (Figure 5). The results indicated that ESCA-related DEGs were mainly involved in cell division, muscle system process, development and attachment of spindle microtubules to the kinetochore at the midbody, and extracellular matrix.
Table 2.
GO enrichment analysis of ESCA-related DEGs.
| Category | Term | Annotation | Count | P Value | FDR |
|---|---|---|---|---|---|
| MF | GO: 0042393 | Histone binding | 5 | 0.040956 | 43.57063 |
| GO: 0004672 | Protein kinase activity | 9 | 0.037858 | 41.02474 | |
| GO: 0017048 | Rho GTPase binding | 3 | 0.035287 | 38.83208 | |
| GO: 0008083 | Growth factor activity | 6 | 0.029217 | 33.3507 | |
| GO: 0003678 | DNA helicase activity | 3 | 0.026484 | 30.73641 | |
| GO: 0042826 | Histone deacetylase binding | 5 | 0.023265 | 27.53669 | |
| GO: 0019901 | Protein kinase binding | 10 | 0.019069 | 23.15887 | |
| GO: 0016887 | ATPase activity | 7 | 0.013453 | 16.91675 | |
| GO: 0048407 | Platelet-derived growth factor binding | 3 | 0.005775 | 7.618473 | |
| GO: 0005201 | Extracellular matrix structural constituent | 5 | 0.005574 | 7.362384 | |
| GO: 0005524 | ATP binding | 28 | 0.004315 | 5.745381 | |
| GO: 0008574 | ATP-dependent microtubule motor activity, plus-end-directed | 4 | 7.15E-04 | 0.97331 | |
| GO: 0008017 | Microtubule binding | 10 | 3.85E-04 | 0.525754 | |
| GO: 0008201 | Heparin binding | 9 | 3.10E-04 | 0.423032 | |
| GO: 0003777 | Microtubule motor activity | 7 | 2.05E-04 | 0.280292 | |
| GO: 0005515 | Protein binding | 118 | 1.98E-04 | 0.271172 | |
| GO: 0008307 | Structural constituent of muscle | 6 | 7.85E-05 | 0.107306 | |
| GO: 0003779 | Actin binding | 15 | 1.63E-06 | 0.002223 | |
| CC | GO: 0005814 | Centriole | 5 | 0.034311 | 36.49999 |
| GO: 0097149 | Centralspindlin complex | 2 | 0.032082 | 34.56688 | |
| GO: 0005694 | Chromosome | 5 | 0.0272 | 30.14128 | |
| GO: 0070062 | Extracellular exosome | 42 | 0.0257 | 28.72761 | |
| GO: 0005680 | Anaphase-promoting complex | 3 | 0.025342 | 28.38631 | |
| GO: 0005584 | Collagen type i trimer | 2 | 0.021504 | 24.62943 | |
| GO: 0016363 | Nuclear matrix | 5 | 0.021056 | 24.17972 | |
| GO: 0051233 | Spindle midzone | 3 | 0.017618 | 20.64245 | |
| GO: 0005925 | Focal adhesion | 11 | 0.010247 | 12.53825 | |
| GO: 0015629 | Actin cytoskeleton | 8 | 0.009682 | 11.8868 | |
| GO: 0005654 | Nucleoplasm | 44 | 0.008794 | 10.8543 | |
| GO: 0005576 | Extracellular region | 29 | 0.008174 | 10.12583 | |
| GO: 0048471 | Perinuclear region of cytoplasm | 15 | 0.007728 | 9.598831 | |
| GO: 0005813 | Centrosome | 12 | 0.006822 | 8.519486 | |
| GO: 0000922 | Spindle pole | 6 | 0.006621 | 8.277421 | |
| GO: 0015630 | Microtubule cytoskeleton | 7 | 0.003771 | 4.795012 | |
| GO: 0001725 | Stress fiber | 5 | 0.002747 | 3.514485 | |
| GO: 0045120 | Pronucleus | 3 | 0.002356 | 3.021411 | |
| GO: 0005737 | Cytoplasm | 77 | 0.001617 | 2.083603 | |
| GO: 0005829 | Cytosol | 54 | 0.00154 | 1.984823 | |
| GO: 0031012 | Extracellular matrix | 11 | 0.001477 | 1.904168 | |
| GO: 0005876 | Spindle microtubule | 5 | 0.001282 | 1.655013 | |
| GO: 0031262 | Ndc80 complex | 3 | 6.88E-04 | 0.890813 | |
| GO: 0000942 | Condensed nuclear chromosome Outer kinetochore | 3 | 6.88E-04 | 0.890813 | |
| GO: 0005874 | Microtubule | 12 | 5.90E-04 | 0.765086 | |
| GO: 0030018 | Z disc | 8 | 2.97E-04 | 0.385939 | |
| GO: 0005581 | Collagen trimer | 8 | 6.25E-05 | 0.081295 | |
| GO: 0000776 | Kinetochore | 8 | 2.74E-05 | 0.035668 | |
| GO: 0005871 | Kinesin complex | 7 | 2.22E-05 | 0.028929 | |
| GO: 0005615 | Extracellular space | 33 | 2.02E-05 | 0.026247 | |
| GO: 0005578 | Proteinaceous extracellular matrix | 14 | 7.32E-06 | 0.009524 | |
| GO: 0005819 | Spindle | 10 | 6.68E-06 | 0.008694 | |
| GO: 0000775 | Chromosome, centromeric region | 8 | 2.59E-06 | 0.003375 | |
| GO: 0030496 | Midbody | 11 | 1.40E-06 | 0.001819 | |
| GO: 0000777 | Condensed chromosome kinetochore | 11 | 3.37E-08 | 4.38E-05 | |
| BP | GO: 0007015 | Actin filament organization | 4 | 0.048188 | 55.68998 |
| GO: 0051436 | Negative regulation of ubiquitin-protein ligase activity involved in mitotic cell cycle | 4 | 0.046554 | 54.41983 | |
| GO: 0001666 | Response to hypoxia | 6 | 0.046026 | 54.00186 | |
| GO: 0046777 | Protein autophosphorylation | 6 | 0.046026 | 54.00186 | |
| GO: 0044344 | Cellular response to fibroblast growth factor stimulus | 3 | 0.045033 | 53.20648 | |
| GO: 2000660 | Negative regulation of interleukin-1-mediated signaling pathway | 2 | 0.044501 | 52.77484 | |
| GO: 0010574 | Regulation of vascular endothelial growth factor production | 2 | 0.044501 | 52.77484 | |
| GO: 0043154 | Negative regulation of cysteine-type endopeptidase activity involved in apoptotic process | 4 | 0.043372 | 51.8463 | |
| GO: 0097421 | Liver regeneration | 3 | 0.042339 | 50.98273 | |
| GO: 0051384 | Response to glucocorticoid | 4 | 0.037348 | 46.59797 | |
| GO: 0060326 | Cell chemotaxis | 4 | 0.037348 | 46.59797 | |
| GO: 0007049 | Cell cycle | 7 | 0.037147 | 46.41426 | |
| GO: 0045840 | Positive regulation of mitotic nuclear division | 3 | 0.034645 | 44.07219 | |
| GO: 0071173 | Spindle assembly checkpoint | 2 | 0.033564 | 43.03115 | |
| GO: 0060414 | Aorta smooth muscle tissue morphogenesis | 2 | 0.033564 | 43.03115 | |
| GO: 0006977 | DNA damage response, signal transduction by p53 class mediator resulting in cell cycle arrest | 4 | 0.033134 | 42.61195 | |
| GO: 0000070 | Mitotic sister chromatid segregation | 3 | 0.032215 | 41.70664 | |
| GO: 0006306 | DNA methylation | 3 | 0.032215 | 41.70664 | |
| GO: 0042787 | Protein ubiquitination involved in ubiquitin-dependent protein catabolic process | 6 | 0.030104 | 39.57493 | |
| GO: 0051966 | Regulation of synaptic transmission, glutamatergic | 3 | 0.02757 | 36.92013 | |
| GO: 0051439 | Regulation of ubiquitin-protein ligase activity involved in mitotic cell cycle | 3 | 0.02757 | 36.92013 | |
| GO: 0043547 | Positive regulation of GTPase activity | 13 | 0.027349 | 36.68291 | |
| GO: 0070301 | Cellular response to hydrogen peroxide | 4 | 0.026698 | 35.9813 | |
| GO: 1903779 | Regulation of cardiac conduction | 4 | 0.0255 | 34.66984 | |
| GO: 0008283 | Cell proliferation | 10 | 0.023601 | 32.53933 | |
| GO: 0048146 | Positive regulation of fibroblast proliferation | 4 | 0.023193 | 32.07322 | |
| GO: 0000022 | Mitotic spindle elongation | 2 | 0.022502 | 31.27789 | |
| GO: 0001501 | Skeletal system development | 6 | 0.019827 | 28.11186 | |
| GO: 0010881 | Regulation of cardiac muscle contraction by regulation of the release of sequestered calcium ions | 3 | 0.019192 | 27.33961 | |
| GO: 0006939 | Smooth muscle contraction | 3 | 0.017299 | 24.99359 | |
| GO: 0051726 | Cell cycle regulation | 6 | 0.013422 | 19.96503 | |
| GO: 0031145 | Anaphase-promoting complex-dependent catabolic process | 5 | 0.012345 | 18.5125 | |
| GO: 0006957 | Complement activation, alternative pathway | 3 | 0.009152 | 14.06012 | |
| GO: 0051310 | Metaphase plate congression | 3 | 0.007801 | 12.10953 | |
| GO: 0055119 | Relaxation of cardiac muscle | 3 | 0.007801 | 12.10953 | |
| GO: 0048565 | Digestive tract development | 4 | 0.007152 | 11.1566 | |
| GO: 0008284 | Positive regulation of cell proliferation | 13 | 0.006888 | 10.76681 | |
| GO: 0030574 | Collagen catabolic process | 5 | 0.005947 | 9.363414 | |
| GO: 0034501 | Protein localization to kinetochore | 3 | 0.005399 | 8.535467 | |
| GO: 0032060 | Bleb assembly | 3 | 0.005399 | 8.535467 | |
| GO: 0007019 | Microtubule depolymerization | 3 | 0.005399 | 8.535467 | |
| GO: 0006268 | DNA unwinding involved in DNA replication | 3 | 0.005399 | 8.535467 | |
| GO: 0009612 | Response to mechanical stimulus | 5 | 0.00445 | 7.087078 | |
| GO: 0071549 | Cellular response to dexamethasone stimulus | 4 | 0.004196 | 6.694943 | |
| GO: 0019221 | Cytokine-mediated signaling pathway | 7 | 0.003769 | 6.034147 | |
| GO: 0032355 | Response to estradiol | 6 | 0.00373 | 5.973802 | |
| GO: 0001558 | Regulation of cell growth | 6 | 0.00213 | 3.453634 | |
| GO: 0000910 | Cytokinesis | 5 | 0.002095 | 3.397158 | |
| GO: 0007094 | Mitotic spindle assembly checkpoint | 4 | 0.00141 | 2.299434 | |
| GO: 0000086 | G2/M transition of mitotic cell cycle | 8 | 9.49E-04 | 1.552707 | |
| GO: 0007080 | Mitotic metaphase plate congression | 5 | 7.83E-04 | 1.283031 | |
| GO: 0007155 | Cell adhesion | 15 | 7.52E-04 | 1.232171 | |
| GO: 0007018 | Microtubule-based movement | 7 | 3.11E-04 | 0.511889 | |
| GO: 0030199 | Collagen fibril organization | 6 | 7.41E-05 | 0.122099 | |
| GO: 0006936 | Muscle contraction | 9 | 2.91E-05 | 0.047888 | |
| GO: 0007052 | Mitotic spindle organization | 6 | 1.99E-05 | 0.032833 | |
| GO: 0000281 | Mitotic cytokinesis | 6 | 1.68E-05 | 0.027614 | |
| GO: 0007059 | Chromosome segregation | 8 | 1.15E-05 | 0.018962 | |
| GO: 0007062 | Sister chromatid cohesion | 12 | 2.35E-08 | 3.87E-05 | |
| GO: 0007067 | Mitotic nuclear division | 20 | 5.66E-11 | 9.33E-08 | |
| GO: 0051301 | Cell division | 25 | 1.55E-12 | 2.55E-09 |
Figure 2.

Top 16 enriched molecular function GO terms. The horizontal axis is a logarithmic calculation of p value, while GO terms are listed on the y axis. The length of each bar represents lower p value (higher significance). Actin binding was the term with the highest significance level.
Figure 3.

Top 16 enriched cellular component GO terms. The horizontal axis is a logarithmic calculation of p value, while GO terms are listed on the y axis. The length of each bar represents lower p value (higher significance). Chromosome and centromeric region was the term with the highest significance level.
Figure 4.

Top 20 enriched biological process GO terms. The horizontal axis is a logarithmic calculation of p value, while GO terms are listed on the y axis. The length of each bar represents lower p value (higher significance). Cell division was the term with the highest significance level.
Figure 5.

Top 20 results from GO functional enrichment analysis. The size of the filled circles indicates statistical significance. Larger circle size indicates lower p value. Circle color indicates type of GO term, as listed in the legend (lower left corner).
Enrichment analysis of the KEGG pathway
In total, 107 pathways connected with DEGs were procured through KOBAS v3.0. The corrected p value <0.05 was set as the statistical cutoff, and 60 pathways without statistical difference were rejected. The remaining 47 pathways with statistical significance are presented in Table 3. Among these remaining pathways, we chose the top 20 enriched pathways using OmicShare tools (Figure 6). These pathways revealed that a broad spectrum of DEGs participated in the cell cycle, pathways in cancer, the phosphatidylinositol-3-kinase (PI3K)/protein kinase B (AKT) signaling pathway, the cGMP-protein kinase G (PKG) signaling pathway, cytokine-cytokine receptor interaction, and vascular smooth muscle contraction.
Table 3.
KEGG pathway enrichment analysis of ESCA-related DEGs.
| Term | Number | Corrected p value | Genes |
|---|---|---|---|
| Cell cycle | 10 | 1.19E-08 | CCNB2, CCNB1, CCNA2, BUB1B, CCNE2, CDC20, TTK, MCM4, MCM2, BUB1 |
| cGMP-PKG signaling pathway | 9 | 1.51E-06 | MRVI1, MYLK, KCNMA1, ADRB1, ATP1A2, RGS2, PLN, AGTR1, MYL9 |
| Vascular smooth muscle contraction | 8 | 2.68E-06 | MYH11, MRVI1, AVPR1A, ACTG2, MYLK, KCNMA1, AGTR1, MYL9 |
| AGE-RAGE signaling pathway in diabetic complications | 6 | 0.000105714 | CCL2, IL6, COL1A2, COL1A1, COL3A1, AGTR1 |
| Pathways in cancer | 11 | 0.00016471 | CXCL12, CCNE2, BIRC5, KIT, IL6, HLF, FGF7, LPAR3, AGTR1, HEY1, MMP1 |
| PI3K-Akt signaling pathway | 9 | 0.000181898 | GHR, CCNE2, KIT, IL6, COL1A2, EREG, COL1A1, FGF7, LPAR3 |
| Platelet activation | 6 | 0.000181898 | PTGS1, MYLK, RAP1A, COL1A2, COL1A1, COL3A1 |
| Calcium signaling pathway | 7 | 0.000181898 | AVPR1A, CASQ2, MYLK, ADRB1, PLN, AGTR1, CYSLTR1 |
| Oocyte meiosis | 6 | 0.000181898 | CCNB2, CCNB1, BUB1, CCNE2, CDC20, AURKA |
| Cytokine-cytokine receptor interaction | 8 | 0.000256568 | CCL2, IL1RN, TNFSF10, GHR, DTL, IL6, IL36A, CXCL12 |
| Protein digestion and absorption | 5 | 0.000372719 | COL3A1, COL14A1, COL1A2, ATP1A2, COL1A1 |
| Rheumatoid arthritis | 5 | 0.000372719 | IL6, CCL2, CXCL12, DTL, MMP1 |
| Progesterone-mediated oocyte maturation | 5 | 0.000505745 | CCNB2, CCNB1, CCNA2, AURKA, BUB1 |
| p53 signaling pathway | 4 | 0.001955985 | CCNE2, CCNB2, CCNB1, GTSE1 |
| Human T-cell leukemia virus 1 infection | 6 | 0.001955985 | CCNB2, CCNA2, BUB1B, CCNE2, CDC20, IL6 |
| Adrenergic signaling in cardiomyocytes | 5 | 0.002613273 | ADRB1, AGTR1, SCN7A, ATP1A2, PLN |
| Neuroactive ligand-receptor interaction | 7 | 0.002678945 | AVPR1A, GHR, P2RY14, ADRB1, LPAR3, AGTR1, CYSLTR1 |
| ECM-receptor interaction | 4 | 0.003021888 | HMMR, COL1A2, CD36, COL1A1 |
| Cellular senescence | 5 | 0.003023275 | IL6, CCNB2, CCNB1, CCNE2, CCNA2 |
| Jak-STAT signaling pathway | 5 | 0.003030745 | IL6, AOX1, FHL1, GHR, SOCS2 |
| Tyrosine metabolism | 3 | 0.003030745 | AOX1, AOC3, ADH1B |
| Amoebiasis | 4 | 0.003552075 | IL6, COL3A1, COL1A2, COL1A1 |
| Viral protein interaction with cytokine and cytokine receptor | 4 | 0.004092655 | IL6, CCL2, CXCL12, TNFSF10 |
| Malaria | 3 | 0.005771037 | IL6, CCL2, CD36 |
| Intestinal immune network for IgA production | 3 | 0.005771037 | IL6, CXCL12, DTL |
| Focal adhesion | 5 | 0.005771037 | MYL9, COL1A2, MYLK, RAP1A, COL1A1 |
| cAMP signaling pathway | 5 | 0.007342941 | ADRB1, MYL9, ATP1A2, RAP1A, PLN |
| Regulation of actin cytoskeleton | 5 | 0.007342941 | FGF7, CFD, CXCL12, MYLK, MYL9 |
| Relaxin signaling pathway | 4 | 0.008337936 | COL3A1, COL1A1, COL1A2, MMP1 |
| FoxO signaling pathway | 4 | 0.008510363 | IL6, CCNB2, CCNB1, TNFSF10 |
| Renin secretion | 3 | 0.011964411 | ADRB1, AGTR1, KCNMA1 |
| Phospholipase D signaling pathway | 4 | 0.011964411 | LPAR3, AGTR1, AVPR1A, KIT |
| PPAR signaling pathway | 3 | 0.015020846 | SORBS1, CD36, MMP1 |
| Hepatitis B | 4 | 0.015736213 | IL6, CCNE2, BIRC5, CCNA2 |
| Complement and coagulation cascades | 3 | 0.015736213 | CLU, C7, CFH |
| Tight junction | 4 | 0.017269759 | MYH11, EPB41L4B, RAP1A, MYL9 |
| Salivary secretion | 3 | 0.021192296 | ADRB1, KCNMA1, ATP1A2 |
| MicroRNAs in cancer | 5 | 0.021964309 | DNMT3B, CCNE2, EZH2, KIF23, SOX4 |
| IL-17 signaling pathway | 3 | 0.021964309 | IL6, CCL2, MMP1 |
| Hematopoietic cell lineage | 3 | 0.023983878 | IL6, CD36, KIT |
| Pancreatic secretion | 3 | 0.024052267 | KCNMA1, RAP1A, ATP1A2 |
| Human papillomavirus infection | 5 | 0.029816413 | CCNE2, CCNA2, COL1A1, COL1A2, HEY1 |
| Rap1 signaling pathway | 4 | 0.029816413 | FGF7, LPAR3, RAP1A, KIT |
| Prion diseases | 2 | 0.030030738 | IL6, C7 |
| DNA replication | 2 | 0.03062247 | MCM4, MCM2 |
| Leukocyte transendothelial migration | 3 | 0.03062247 | CXCL12, RAP1A, MYL9 |
| AMPK signaling pathway | 3 | 0.035976653 | CAB39L, CCNA2, CD36 |
Figure 6.

The top 20 KEGG pathways involving the differentially expressed genes (DEGs). Rich factor is represented on the x axis. KEGG pathway annotation is represented on the y axis. The size of the filled circles symbolizes the number of genes involved in the pathways. The color of the filled circles denotes significance, with gradation from red to blue symbolizing increasing p value.
Construction of the PPI network and identification of hub genes
The PPI network exported by STRING comprised 202 nodes and 1571 edges (Figure 7). The modular interaction network was viewed as statistically enriched, due to the enrichment p value <1.0e-16 (<0.05). The top 10 genes in the PPI network, in terms of degree ranking, were regarded as hub genes (Figure 8); these genes probably play crucial roles in the incidence and development of ESCA. TOP2A was in the highest rank among all the DEGs, followed by UBE2C, BUB1, KIF20A, TTK, CCNB2, KIF2C, TPX2, CCNA2, and CCNB1.
Figure 7.

PPI network of ESCA-related differentially expressed genes (DEGs). The number of nodes was 202 (with 8 untouched genes). The number of edges was 1571; much higher than the expected 298 edges. The average node degree was 15.6. The average local clustering coefficient was 0.582. The PPI enrichment p value was less than 1.0e-16.
Figure 8.
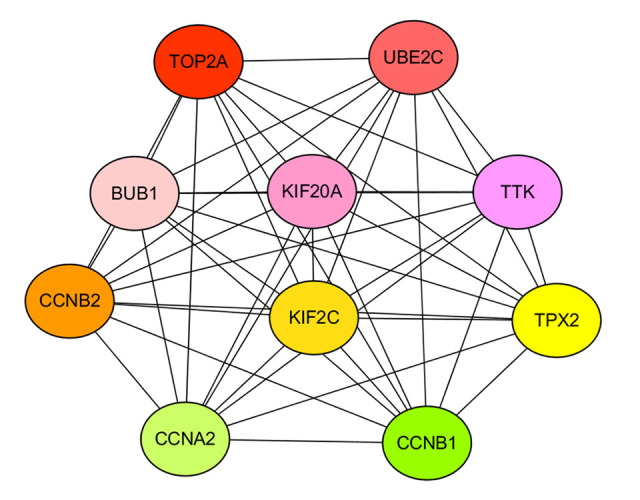
Topological diagram of the top 10 hub genes. In accordance with degree ranking, the top 10 differentially expressed genes (DEGs) were accepted as hub genes. Among all the DEGs, TOP2A was in the highest degree rank, followed by UBE2C, BUB1, KIF20A, TTK, CCNB2, KIF2C, TPX2, CCNA2, and CCNB1.
Expression analysis of hub genes
The expression boxplots of the hub genes, conveyed on the basis of TCGA and GTEx data, demonstrated that these hub genes were upregulated in ESCA tissue compared with normal esophageal tissue (Figure 9). Further, we evaluated the expression level of the hub genes at different pathological stages among ESCA patients (stage I, stage II, stage III, and stage IV patients). The stage-plots were scored using GEPIA (Figure 10). As is shown below, the expression level of the hub genes differed at different stages.
Figure 9.

Differences in expression level of the top 10 hub genes between ESCA tissue and matched normal tissue. The plots are based on TCGA and GTEx data, and reveal that these hub genes are significantly upregulated in ESCA tissue compared with normal tissue (p<0.01). The red * denotes statistically significant difference.
Figure 10.

GEPIA stage-plots for the top 10 hub genes in patients at different stages of ESCA. The stage-plot is considered statistically significant when Pr >F. Greater F values represent increasing significance.
Survival analysis for the hub genes
The survival curve output by OncoLnc showed that all the hub genes with high expression, in comparison with those with low expression, improved the percentage of late survival, after the 2000-day timepoint (>2000 on the x axis) (Figure 11). Moreover, when the surviving percentile of ESCA patients was stationary, high expression of the hub genes prolonged survival time. However, the hub genes exhibited few associations with early survival.
Figure 11.

Effects of the top 10 hub genes on prognosis of ESCA patients, as calculated by OncoLnc. All of the hub genes improved late survival time of ESCA patients (x>2000), while few exhibited any association with early survival time.
Discussion
ESCA is a major global health challenge with multifactorial etiology, including both genetic and environmental components. Efforts to detect inchoate changes have attenuated the development of malignant cancer, and have even had some success in prevention [9]. Therefore, it is of vital importance to seek out predictive indicators and therapeutic markers for ESCA.
In our research, 210 DEGs were screened from two genetic expression datasets, via the GEO bioinformatics portal. Then, GO function enrichment analysis and KEGG pathway enrichment analysis were applied to filter the DEGs. A PPI network was constructed using STRING to obtain hub genes that are likely to be central to the ESCA pathological process. Most previous studies in this field stopped at this point, resulting in limited clinical usefulness. Therefore, we implemented further analysis targeting the crucial hub genes identified in the first steps of our study. We focused on the assessment of screened hub genes with greater possibility as new therapeutic targets for treating ESCA. We conducted expression analysis of these hub genes in patients at different stages of ESCA, as well as prognostic analysis. These analyses showed the potential of these hub genes as biomarkers for appraisal in the process of therapy and post-therapy.
The results of GO functional enrichment analysis identified several DEGs exhibiting a distinction from genes expressed in healthy esophageal tissue. The DEGs were principally targeted to the midbody, with functions related to cell division and attachment of spindle microtubules to the kinetochore. The results suggested that multitudes of DEGs were closely associated with nuclear activities, especially cell division. Cell division as a functional category includes mechanisms to properly orient and position the mitotic spindle, which is important because incorrect activity related to the spindle contributes to disease, even carcinogenesis [23,24]. Echoing the KEGG pathway enrichment analysis, the DEGs were strongly related to the cell cycle. Dysregulation of the cell cycle leading to endless proliferation of cells has been implicated in tumorigenesis [25]. Jing Wen et al. [26] found that, in vitro and in vivo, transcriptional activation of miR-424 elevated proliferation of ESCC cell lines and led to poor survival in ESCC patients; this activation facilitates both G1/S and G2/M cell cycle transitions. However, inhibition of cell cycle progression alleviates the development of malignancy. Chinese researchers have demonstrated that a novel Notch inhibitor called FLI-06 exerts antitumor activity in a dose-dependent manner via cell-cycle arrest and proliferation suppression in ESCC cells [27]. Above all, therapeutic interventions targeting cell division may generate antitumor activity.
Additionally, the KEGG pathway enrichment analysis showed that the DEGs were enriched in the PI3K-Akt signaling pathway, the cGMP-PKG signaling pathway, cytokine-cytokine receptor interaction, and vascular smooth muscle contraction. Intriguingly, those findings differed from those of a similar study using bioinformatics methods conducted by He et al. [25]. Their findings indicated that DEGs related to ECA were mainly enriched in complement and coagulation cascades, mature- onset diabetes in the young, and retinol metabolism. Given that their sample was extracted from ECA patients and ours from patients diagnosed with ESCC and SCEC, it was inevitable for the two studies to find differences. Still, the evidence supporting our scientific results are as follows:
The PI3K-Akt signaling pathway is implicated in several cancers, including breast cancer, colorectal carcinoma, hepatocellular cancer, and others [28–30]. Invariably, this pathway plays an important role in metastasis and prognosis among ESCA patients [31,32]. Ni Shi et al. [33] showed that MK2206/BEZ235 enhances apoptosis and inhibits tumor growth targeted at AKT phosphorylation in a mouse xenograft model of ESCC and in vitro. Moreover, the combination of MK2206 and BEZ235 boost antitumor effects because of dual PI3K and AKT inhibition. Another study demonstrated that Osthole, extracted from the herb Cnidium monnieri (L.) Cuss, reduced expression of PI3K and phosphorylated AKT (p-AKT), and thereby decreased ESCC proliferation [34]. Hence, treatments targeting the PI3K-Akt signaling pathway provide a promising approach to prevent ESCA and/or to slow its progression. The cGMP-PKG signaling pathway modulates massive physiological and pathological parameters [35]. Correlation between inflammation and the cGMP-PKG signaling pathway has been identified, involving mechanisms of neutrophil migration, mitochondrial permeability transition, and oscillations of calcium ions [36,37]. Interestingly, the mechanism responsible for dysplasia-induced (especially by Barrett’s esophagus) early EAC included a substantial promotion in the levels of inflammatory cytokines [38]. This implies that the cGMP-PKG signaling pathway is possibly connected with ESCA pathology in the early stages. Although studies testing the potential role of pharmacological modulation of the cGMP-PKG signaling pathway as an anticancer therapy are scant, we still hold a positive belief in their potential.
Tumor events are elicited by the collective expression of multiple genes, while single genes function as carcinogens particularly associated with severity [39]. As revealed in the PPI network, coordinated DEGs mediated the occurrence and development of ESCA. To select the critical genes for the interlaced construction, we input information for all DEGs to Cytoscape to screen the top 10 nodes in terms of degree. As a consequence, the degree of TOP2A ranked the highest, followed by UBE2C, BUB1, KIF20A, TTK, CCNB2, KIF2C, TPX2, CCNA2, and CCNB1. These nodes were regarded as hub genes for ESCA. In contrast, He et al. [25] identified IL8, IVL, TIMP1, FN1, SERPINE1, SERPINA1, CFTR, SPP1, COL1A1, and AGT as hub genes for ESCA. They focused on EAC while we focused on ESCC and SCEC. It has been established that there are substantial differences between EAC and ESCC, especially in terms of pathology [3], so it is not surprising to obtain distinct results. Taken together, all 10 hub genes we identified were linked to cell proliferation, involving the cell cycle or chromosome activity. Details about key hub genes are demonstrated as follows:
CCNA2 controls both the G1/S and the G2/M transition phases of the cell cycle, via the formation of specific serine/threonine protein kinase holoenzyme complexes with the cyclin-dependent protein kinases CDK1 or CDK2 [40]. Emerging evidence indicates that miR-219-5p mitigates ESCC cell proliferation by decreasing expression levels of CCNA2. Knockdown of CCNA2 enhances the impacts of miR-219-5p in terms of cell proliferation and cell cycle process [41]. CCNB2 and CCNB1 are elementary parts of the G2/M (mitosis) transition. One of the reasons why the overexpression of erythrocyte membrane protein band 4.1 like 3 (EPB41L3) noticeably improved overall survival rates among ESCA patients was its activation of CCNB1 signaling to induce G2/M cell cycle arrest [42]. A number of studies have found the high expression of CCNB2 to be related to tumor growth and poor prognosis in several cancers, including non-small cell lung carcinomas (NSCLC), hepatocellular carcinoma, and gastric cancer [43–45]. Without laboratory data on ESCA, the role of CCNB2 is still controversial. Given that CCNB2, CCNA2, and CCNB1 are essential for cell cycle regulation, and are subsidiary to the cyclin family, they have potential as therapeutic targets for ESCA.
KIF20A and KIF2C are implicated in chromosome activity. They belong to the kinesin superfamily, and probably possess the potential to ameliorate malignancy. The kinesin family plays a significant role in microtubule motor activity of several cellular and extracellular structures, including Golgi membranes, chromosomes, associated vesicles along microtubules, and the central spindle [46]. KIF20A is a member of the kinesin-6 subfamily, and is crucial for cytokinesis and spindle assembly. Its expression pattern is linked to tumor development and prognosis [47]. KIF2C is responsible for the majority of microtubule plus-end depolymerizing activity in mitotic cells. It is required for chromosome segregation and congression during mitosis, and regulates the turnover and conversion of microtubules [48]. An immunohistochemical analysis conducted in ESCA tissue and adjacent non-cancerous tissues revealed that higher KIF2C expression led to higher pathologic tumor status and poorer tumor differentiation, but only for male patients [49].
TOP2A controls topological states of DNA, a crucial role for proper segregation of daughter chromosomes during mitosis and meiosis [50]. One clinical study suggested that high TOP2A could serve as a biomarker driving medical therapy [51]. Another study showed that the knockdown of the long noncoding RNA (lncRNA) DDX11-AS1 reversed paclitaxel (PTX) resistance by means of inhibiting the expression level of TOP2A [52]. Both of these results imply that targeting the expression of TOP2A may have promise as a pharmacological treatment.
BUB1 is identified as a mitotic checkpoint that is important for spindle-assembly checkpoint signaling and correct chromosome alignment [53]. The transcriptional level of BUB1 in Barrett’s patients, leading to early chromosomal instability (CI), can be helpful in EAC [54]. For the time being, we can hypothesize that treatment modifying BUB1 expression may benefit ESCA patients.
Meanwhile, the expression of UBE2C, TTK, and TPX2 is involved in ESCA pathology and prognosis. As a ubiquitin-conjugating enzyme, UBE2C promotes the degradation of several cell cycle-regulated proteins that control progression through mitosis [55]. Knockdown of UBE2C significantly inhibits cell proliferation via cell-cycle regulation, in in-vitro ESCA cell models, particularly in TE-1 cell lines [56]. TTK is associated with cell proliferation and chromosome alignment [57]. Previous clinical trials found that immunotherapies using epitope peptides derived from TTK potentiated cell response and immunity against ESCA [58,59]. TPX2 is required for normal assembly of microtubules as well as mitotic spindles during apoptosis. As detected by RT-qPCR, the relative expression of TPX2 is upregulated in ESCA tumor tissue, and in lymph node metastasis [60]. Moreover, low expression of miR-491 aimed at TPX2 promotes cell invasion in ESCA clinical samples, affecting carcinogenesis and cancer development [61].
Collectively, we identified 210 DEGs and implemented GO analysis, KEGG pathway enrichment analysis, and PPI network construction to understand their possible roles in ESCA carcinogenesis. The enriched KEGG pathway implicated pathological mechanisms in ESCA, including the cell cycle and the PI3K-Akt and cGMP-PKG signaling pathways. Furthermore, we identified 10 hub genes and assessed their prognostic value. The 10 hub genes were TOP2A (highest ranking in terms of degree), UBE2C, BUB1, KIF20A, TTK, CCNB2, KIF2C, TPX2, CCNA2, and CCNB1. These genes showed marked connections with late survival of ESCA patients, indicating their value in diagnosis and survival improvement.
Our research provides new insights for ESCA early diagnosis and survival evaluation, at the level of genes, pathways, and molecular functions. Even though we analyzed two microarray datasets containing data from patients diagnosed with ESCA, the chosen samples in our research are still inadequate for definitive conclusions, especially in light of the scarcity of experimental evidence related to our findings. It will be necessary to validate our research findings through practical experiments, such as immunohistochemistry on tumor tissue and paired healthy control tissue.
Conclusion
In this research, a total of 210 ESCA-related DEGs were found, and their enriched functions and pathways were analyzed using bioinformatics methods. Taking the mechanistic link between enriched pathways and ESCA into consideration, our results may provide helpful information for understanding ESCA pathology and ESCA treatment. The PPI network we constructed from the DEGs revealed that these genes mediate the occurrence and development of ESCA through multi-gene interactions. From these, we selected the top 10 hub genes: TOP2A (highest ranking of degree), UBE2C, BUB1, KIF20A, TTK, CCNB2, KIF2C, TPX2, CCNA2, and CCNB1. These genes are closely associated with the etiology and prognosis of ESCA, and have potential as therapeutic or early-diagnostic biomarkers. Nevertheless, there are some limitations in our research. The absence of laboratory evidence imposes restrictions on the conclusions that can be drawn from our results. In addition, there were no more than 10 ESCA patients in each sample; this sample size may be insufficient. Therefore, in order to verify our findings, further experimental research, such as immunohistochemistry studies and large clinical studies, is needed.
Acknowledgements
We express our cordial thanks to all those who participated in this study.
Footnotes
Conflict of interest
None.
Source of support: Scientific Research Fund of Hunan Provincial Education Department (No. 19B436); Hunan Provincial Innovation Foundation For Postgraduates (No. CX2018B515); Open Fund of the Domestic First-Class Discipline Construction Project of Integrated Traditional Chinese and Western Medicine of Hunan University of Chinese Medicine (No. 2018ZXYJH35); First-Class Discipline Construction Project of Basic Medicine in 13th Five-Year Plan of Hunan University of Chinese Medicine (06)
References
- 1.Abnet CC, Arnold M, Wei WQ. Epidemiology of esophageal squamous cell carcinoma. Gastroenterology. 2018;154(2):360–73. doi: 10.1053/j.gastro.2017.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pennathur A, Gibson MK, Jobe BA, et al. Oesophageal carcinoma. Lancet. 2013;381(9864):400–12. doi: 10.1016/S0140-6736(12)60643-6. [DOI] [PubMed] [Google Scholar]
- 3.Smyth EC, Lagergren J, Fitzgerald RC, et al. Oesophageal cancer. Nat Rev Dis Primers. 2017;3:17048. doi: 10.1038/nrdp.2017.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sardana RK, Chhikara N, Tanwar B, et al. Dietary impact on esophageal cancer in humans: A review. Food Funct. 2018;9(4):1967–77. doi: 10.1039/c7fo01908d. [DOI] [PubMed] [Google Scholar]
- 5.Coleman HG, Xie SH, Lagergren J. The epidemiology of esophageal adenocarcinoma. Gastroenterology. 2018;154(2):390–405. doi: 10.1053/j.gastro.2017.07.046. [DOI] [PubMed] [Google Scholar]
- 6.Gharahkhani P, Fitzgerald RC, Vaughan TL, et al. Genome-wide association studies in oesophageal adenocarcinoma and Barrett’s oesophagus: A large-scale meta-analysis. Lancet Oncol. 2016;17(10):1363–73. doi: 10.1016/S1470-2045(16)30240-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Domper Arnal MJ, Ferrández Arenas Á, Lanas Arbeloa Á. Esophageal cancer: Risk factors, screening and endoscopic treatment in Western and Eastern countries. World J Gastroenterol. 2015;21(26):7933–43. doi: 10.3748/wjg.v21.i26.7933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lagergren J, Smyth E, Cunningham D, et al. Oesophageal cancer. Lancet. 2017;390(10110):2383–96. doi: 10.1016/S0140-6736(17)31462-9. [DOI] [PubMed] [Google Scholar]
- 9.Alsop BR, Sharma P. Esophageal cancer. Gastroenterol Clin North Am. 2016;45(3):399–12. doi: 10.1016/j.gtc.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 10.Wang C, Wang J, Chen Z, et al. Immunohistochemical prognostic markers of esophageal squamous cell carcinoma: A systematic review. Chin J Cancer. 2017;36(1):65. doi: 10.1186/s40880-017-0232-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hernández M, Quijada NM, Rodríguez-Lázaro D, et al. [Bioinformatics of next generation sequencing in clinical microbiology diagnosis]. Rev Argent Microbiol. 2019 doi: 10.1016/j.ram.2019.06.003. [Online ahead of print] [DOI] [PubMed] [Google Scholar]
- 12.Keerthikumar S. An introduction to proteome bioinformatics. Methods Mol Biol. 2017;1549:1–3. doi: 10.1007/978-1-4939-6740-7_1. [DOI] [PubMed] [Google Scholar]
- 13.Peng D, Guo Y, Chen H, et al. Integrated molecular analysis reveals complex interactions between genomic and epigenomic alterations in esophageal adenocarcinomas. Sci Rep. 2017;7:40729. doi: 10.1038/srep40729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He F, Ai B, Tian L. Identification of genes and pathways in esophageal adenocarcinoma using bioinformatics analysis. Biomed Rep. 2018;9(4):305–12. doi: 10.3892/br.2018.1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ming XY, Zhang X, Cao TT, et al. RHCG suppresses tumorigenicity and metastasis in esophageal squamous cell carcinoma via inhibiting NF-κB Signaling and MMP1 expression. Theranostics. 2018;8(1):185–98. doi: 10.7150/thno.21383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu D, Xu X, Wen J, et al. Integrated genome-wide analysis of gene expression and DNA copy number variations highlights stem cell-related pathways in small cell esophageal carcinoma. Stem Cells Int. 2018;2018 doi: 10.1155/2018/3481783. 3481783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou Y, Zhou B, Pache L, et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019;10(1):1523. doi: 10.1038/s41467-019-09234-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xie C, Mao X, Huang J, et al. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011;9:W316–22. doi: 10.1093/nar/gkr483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szklarczyk D, Gable AL, Lyon D, et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47:D607–13. doi: 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shannon P, Markiel A, Ozier O, et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tang Z, Kang B, Li C, et al. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019;47:W556–60. doi: 10.1093/nar/gkz430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anaya J. OncoLnc: Linking TCGA survival data to mRNAs, miRNAs, and lncRNAs. Peer J Computer Science. 2016;2:e67. [Google Scholar]
- 23.Canman JC, Cabernard C. Mechanics of cell division and cytokinesis. Mol Biol Cell. 2018;29(6):685–86. doi: 10.1091/mbc.E17-11-0671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.López-Lázaro M. The stem cell division theory of cancer. Crit Rev Oncol Hematol. 2018;123:95–113. doi: 10.1016/j.critrevonc.2018.01.010. [DOI] [PubMed] [Google Scholar]
- 25.Kar S. Unraveling cell-cycle dynamics in cancer. Cell Syst. 2016;2(1):8–10. doi: 10.1016/j.cels.2016.01.007. [DOI] [PubMed] [Google Scholar]
- 26.Wen J, Hu Y, Liu Q, et al. miR-424 coordinates multilayered regulation of cell cycle progression to promote esophageal squamous cell carcinoma cell proliferation. EBioMedicine. 2018;37:110–24. doi: 10.1016/j.ebiom.2018.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu Z, Ren Y, Zhang M, et al. FLI-06 suppresses proliferation, induces apoptosis and cell cycle arrest by targeting LSD1 and Notch pathway in esophageal squamous cell carcinoma cells. Biomed Pharmacother. 2018;107:1370–76. doi: 10.1016/j.biopha.2018.08.140. [DOI] [PubMed] [Google Scholar]
- 28.Costa RLB, Han HS, Gradishar WJ. Targeting the PI3K/AKT/mTOR pathway in triple-negative breast cancer: a review. Breast Cancer Res Treat. 2018;169(3):397–406. doi: 10.1007/s10549-018-4697-y. [DOI] [PubMed] [Google Scholar]
- 29.Slattery ML, Mullany LE, Sakoda LC, et al. Associations of miRNAs with dysregulated gene expression in colorectal cancer. Mol Carcinog. 2018;57(2):243–61. doi: 10.1002/mc.22752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang SS, Chen YH, Chen N, et al. Hydrogen sulfide promotes autophagy of hepatocellular carcinoma cells through the PI3K/Akt/mTOR signaling pathway. Cell Death Dis. 2017;8(3):e2688. doi: 10.1038/cddis.2017.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li B, Xu WW, Lam AKY, et al. Significance of PI3K/AKT signaling pathway in metastasis of esophageal squamous cell carcinoma and its potential as a target for anti-metastasis therapy. Oncotarget. 2017;8(24):38755–66. doi: 10.18632/oncotarget.16333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu N, Du Z, Zhu Y, et al. The expression and prognostic impact of the PI3K/AKT/mTOR signaling pathway in advanced esophageal squamous cell carcinoma. Technol Cancer Res Treat. 2018;17 doi: 10.1177/1533033818758772. 1533033818758772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shi N, Yu H, Chen T. Inhibition of esophageal cancer growth through the suppression of PI3K/AKT/mTOR signaling pathway. Onco Targets Ther. 2019;12:7637–47. doi: 10.2147/OTT.S205457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu X, Li Z, Li T, et al. Osthole inhibits the PI3K/AKT signaling pathway via activation of PTEN and induces cell cycle arrest and apoptosis in esophageal squamous cell carcinoma. Biomed Pharmacother. 2018;102:502–9. doi: 10.1016/j.biopha.2018.03.106. [DOI] [PubMed] [Google Scholar]
- 35.Li ZH, Cui D, Qiu CJ, et al. Cyclic nucleotide signaling in sensory neuron hyperexcitability and chronic pain after nerve injury. Neurobiol Pain. 2019;6:100028. doi: 10.1016/j.ynpai.2019.100028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spiller F, Oliveira Formiga R, Fernandes da Silva Coimbra J, et al. Targeting nitric oxide as a key modulator of sepsis, arthritis and pain. Nitric Oxide. 2019;89:32–40. doi: 10.1016/j.niox.2019.04.011. [DOI] [PubMed] [Google Scholar]
- 37.Inserte J, Garcia-Dorado D. The cGMP/PKG pathway as a common mediator of cardioprotection: translatability and mechanism. Br J Pharmacol. 2015;172(8):1996–2009. doi: 10.1111/bph.12959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tanţău M, Laszlo M, Tanţău A. Barrett’s esophagus – state of the art. Chirurgia (Bucur) 2018;113(1):46–60. doi: 10.21614/chirurgia.113.1.46. [DOI] [PubMed] [Google Scholar]
- 39.Kim H, Kim YM. Pan-cancer analysis of somatic mutations and transcriptomes reveals common functional gene clusters shared by multiple cancer types. Sci Rep. 2018;8(1):6041. doi: 10.1038/s41598-018-24379-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wood DJ, Endicott JA. Structural insights into the functional diversity of the CDK-cyclin family. Open Biol. 2018;8(9):180112. doi: 10.1098/rsob.180112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ma Q. MiR-219-5p suppresses cell proliferation and cell cycle progression in esophageal squamous cell carcinoma by targeting CCNA2. Cell Mol Biol Lett. 2019;24:4. doi: 10.1186/s11658-018-0129-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zeng R, Liu Y, Jiang ZJ, et al. EPB41L3 is a potential tumor suppressor gene and prognostic indicator in esophageal squamous cell carcinoma. Int J Oncol. 2018;52(5):1443–54. doi: 10.3892/ijo.2018.4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qian X, Song X, He Y, et al. CCNB2 overexpression is a poor prognostic biomarker in Chinese NSCLC patients. Biomed Pharmacother. 2015;74:222–27. doi: 10.1016/j.biopha.2015.08.004. [DOI] [PubMed] [Google Scholar]
- 44.Li R, Jiang X, Zhang Y, et al. Cyclin B2 overexpression in human hepatocellular carcinoma is associated with poor prognosis. Arch Med Res. 2019;50(1):10–17. doi: 10.1016/j.arcmed.2019.03.003. [DOI] [PubMed] [Google Scholar]
- 45.Shi Q, Wang W, Jia Z, et al. ISL1, a novel regulator of CCNB1, CCNB2 and c-MYC genes, promotes gastric cancer cell proliferation and tumor growth. Oncotarget. 2016;7(24):36489–500. doi: 10.18632/oncotarget.9269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lucanus AJ, Yip GW. Kinesin superfamily: Roles in breast cancer, patient prognosis and therapeutics. Oncogene. 2018;37(7):833–38. doi: 10.1038/onc.2017.406. [DOI] [PubMed] [Google Scholar]
- 47.Wu WD, Yu KW, Zhong N, et al. Roles and mechanisms of Kinesin-6 KIF20A in spindle organization during cell division. Eur J Cell Biol. 2019;98(2–4):74–80. doi: 10.1016/j.ejcb.2018.12.002. [DOI] [PubMed] [Google Scholar]
- 48.Gilbert SP, Guzik-Lendrum S, Rayment I. Kinesin-2 motors: Kinetics and biophysics. J Biol Chem. 2018;293(12):4510–18. doi: 10.1074/jbc.R117.001324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Duan H, Zhang X, Wang FX, et al. KIF-2C expression is correlated with poor prognosis of operable esophageal squamous cell carcinoma male patients. Oncotarget. 2016;7(49):80493–507. doi: 10.18632/oncotarget.11492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bush NG, Evans-Roberts K, Maxwell A. DNA topoisomerases. EcoSal Plus. 2015;6(2):10. doi: 10.1128/ecosalplus.esp-0010-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Purim O, Beny A, Inbar M, et al. Biomarker-driven therapy in metastatic gastric and esophageal cancer: Real-life clinical experience. Target Oncol. 2018;13(2):217–26. doi: 10.1007/s11523-017-0548-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang S, Jiang H, Xu Z, et al. The resistance of esophageal cancer cells to paclitaxel can be reduced by the knockdown of long noncoding RNA DDX11-AS1 through TAF1/TOP2A inhibition. Am J Cancer Res. 2019;9(10):2233–48. [PMC free article] [PubMed] [Google Scholar]
- 53.Raaijmakers JA, van Heesbeen RGHP, Blomen VA, et al. BUB1 is essential for the viability of human cells in which the spindle assembly checkpoint is compromised. Cell Rep. 2018;22(6):1424–38. doi: 10.1016/j.celrep.2018.01.034. [DOI] [PubMed] [Google Scholar]
- 54.Doak SH, Jenkins GJ, Parry EM, et al. Differential expression of the MAD2, BUB1 and HSP27 genes in Barrett’s oesophagus-their association with aneuploidy and neoplastic progression. Mutat Res. 2004;547(1–2):133–44. doi: 10.1016/j.mrfmmm.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 55.Nicolau-Neto P, Palumbo A, De Martino M, et al. UBE2C is a transcriptional target of the cell cycle regulator FOXM1. Genes (Basel) 2018;9(4):188. doi: 10.3390/genes9040188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li L, Li X, Wang W, et al. UBE2C is involved in the functions of ECRG4 on esophageal squamous cell carcinoma. Biomed Pharmacother. 2018;98:201–6. doi: 10.1016/j.biopha.2017.12.066. [DOI] [PubMed] [Google Scholar]
- 57.Pachis ST, Kops GJPL. Leader of the SAC: Molecular mechanisms of Mps1/TTK regulation in mitosis. Open Biol. 2018;8(8):180109. doi: 10.1098/rsob.180109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mizukami Y, Kono K, Daigo Y, et al. Detection of novel cancer-testis antigen-specific T-cell responses in TIL, regional lymph nodes, and PBL in patients with esophageal squamous cell carcinoma. Cancer Sci. 2008;99(7):1448–54. doi: 10.1111/j.1349-7006.2008.00844.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Iwahashi M, Katsuda M, Nakamori M, et al. Vaccination with peptides derived from cancer-testis antigens in combination with CpG-7909 elicits strong specific CD8+ T cell response in patients with metastatic esophageal squamous cell carcinoma. Cancer Sci. 2010;101(12):2510–17. doi: 10.1111/j.1349-7006.2010.01732.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sui C, Song Z, Yu H, et al. Prognostic significance of TPX2 and NIBP in esophageal cancer. Oncol Lett. 2019;18(4):4221–29. doi: 10.3892/ol.2019.10747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Niu H, Gong L, Tian X, et al. Low expression of miR-491 promotes esophageal cancer cell invasion by targeting TPX2. Cell Physiol Biochem. 2015;36(6):2263–73. doi: 10.1159/000430190. [DOI] [PubMed] [Google Scholar]