

Abstract
Clinical interpretation of DNA sequence variants is a critical step in reporting clinical genetic testing results. Application of next-generation sequencing technology in molecular genetic testing has facilitated diagnoses of genetic disorders in clinical practice. However, the large number of DNA sequence variants detected in clinical specimens, many of which have never been seen before, make clinical interpretation challenging. Recommendations by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP) have been widely adopted by clinical laboratories around the world to guide clinical interpretation of sequence variants. The ClinGen Sequence Variant Interpretation Working Group and various disease-specific variant curation expert panels have also developed specifications for the ACMG/AMP recommendations. Despite these efforts to standardize variant interpretation in clinical practice, different laboratories may subjectively use professional judgment to determine which criteria are applicable when classifying a variant. In addition, clinicians and researchers who are not familiar with the variant interpretation process may have difficulty understanding clinical genetic reports and communicating the clinical significance of genetic testing results. Here we provide a step-by-step protocol for clinical interpretation of sequence variants, including practical examples. By following this protocol, clinical laboratory geneticists can interpret the clinical significance of sequence variants according to the ACMG/AMP recommendations and ClinGen framework. Furthermore, this article will help clinicians and researchers to understand variant classification in clinical genetic testing reports and evaluate the quality of the reports.
Basic Protocol
Interpreting the clinical significance of sequence variants
Support Protocol
Reevaluating the clinical significance of sequence variants
Keywords: ACMG/AMP recommendations, ClinGen, clinical genetic testing, sequence variant interpretation, variant reevaluation
INTRODUCTION
Clinical use of next-generation sequencing (NGS) has improved the ability to diagnose rare genetic disorders and increased the number of sequence variants encountered in clinical settings. Typically, 200,000–400,000 or 3,900,000 sequence variants are identified per patient following whole-exome or whole-genome sequencing, respectively (Lionel et al., 2018; Yang et al., 2013). Hundreds of clinical variants with possible clinical significance remain even after multistage filtering, and it is challenging to identify the disease-causing variants among them. Proper interpretation of the clinical significance of these variants is key to high-quality clinical genetic reporting (Current Protocols article Song, Duzkale, & Shen, 2018).
In 2015, the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) published updated recommendations for standards and guidelines in sequence variant interpretation (Richards et al., 2015). These recommendations were intended to help clinical laboratories determine the clinical significance of a sequence variant and avoid associating variants with disease without sufficient evidence. They defined a five-tier framework for classifying variants as “pathogenic,” “likely pathogenic,” “uncertain significance,” “likely benign” or “benign” according to criteria that address levels of evidence for pathogenicity. Each criterion was assigned a code consisting of two to three letters and a number. The first letter indicates a direction, pathogenic (P) or benign (B); the following letter(s) denotes the level of strength: very strong (VS), strong (S), moderate (M), supporting (P), and stand-alone (A); the number acts as a serial number to distinguish criteria of the same strength. Combinations of different evidence are used to determine the classification of a sequence variant (Richards et al., 2015).
Richards et al. (2015) recognized that further specifications are required to ensure that the guidelines are suitable for each particular disorder. The guidelines are being further developed through the ClinGen Sequence Variant Interpretation (SVI) Working Group (referred to as SVI) and various disease-specific Variant Curation Expert Panel (VCEP) Working Groups in the US. The SVI aims to support the refinement and evolution of the guidelines and develop quantitative approaches to variant interpretation. They have proposed general recommendations for ACMG/AMP criteria, including PVS1 (Abou Tayoun et al., 2018), BA1 (Ghosh et al., 2018), PS3/BS3 (Brnich et al., 2019), PP5/BP6 (Biesecker, Harrison, & ClinGen SVI, 2018), PS2/PM6, and PM3, and have approved 26 disease-specific VCEPs that are establishing disease/gene-specific guidelines (Current Protocols article Harrison, Biesecker, & Rehm, 2019; Gelb et al., 2018; Kelly et al., 2018; Lee et al., 2018; Luo et al., 2019; Mester et al., 2018; Oza et al., 2018; Zastrow et al., 2018). Although standardized efforts are being made, the subjectivity of criterion selection can still lead to inconsistent classification of variants across clinical laboratories (Amendola et al., 2016; Balmana et al., 2016; Harrison et al., 2017; Pepin et al., 2016).
The ACMG/AMP recommendations have been widely adopted in clinical practice worldwide. In the UK, the Association for Clinical Genomic Science (ACGS) recommended adoption of the ACMG/AMP recommendations in 2016 and has updated their specifications annually ever since (Ellard et al., 2020). In China, The ACMG/AMP recommendations were translated into Chinese by the Chinese Board of Genetic Counseling (CBGC) in 2017, but no detailed specifications have been released since publication of the translated recommendations.
New evidence emerges constantly over time. New cases with sequence variants are identified, relevant databases are updated, and new scientific discoveries about genes are published every day. It is therefore important to periodically reevaluate clinical significance and reclassify variants using updated information, especially for variants without a definitive classification. In this article, we provide two protocols. A Basic Protocol describes step-by-step procedures for interpreting sequence variants in a clinical setting. Practical examples are provided using updated information according to the ACMG/AMP and ClinGen recommendations. A Support Protocol describes step-by-step procedures for reevaluating the clinical significance of sequence variants.
STRATEGIC PLANNING
Sequence variant interpretation is a complicated aspect of clinical genetic testing. Strategic planning is, therefore, essential. The workflow for sequence variant interpretation is divided into five sections: verification of variant nomenclature, evaluation of population data, evaluation of case data, variant-type-specific analysis, and computational predictions (Fig. 1). An interpretation is made if the evidence is sufficient to classify the variant. It is not necessary to follow all steps in the workflow.
Figure 1. Strategic planning workflow for sequence variant interpretation.
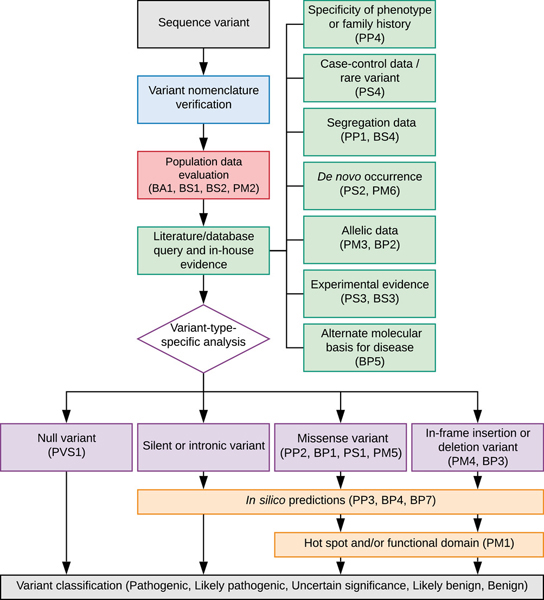
Basic Protocol: INTERPRETATING THE CLINICAL SIGNIFICANCE OF SEQUENCE VARIANTS
Proper variant interpretation is critical for generating an accurate genetic testing report, which guides the diagnosis and management of patients with a genetic disorder. The following protocol describes the step-by-step procedures for sequence variant interpretation according to the ACMG/AMP and ClinGen recommendations. Practical examples and notes are provided for better usage. Users should be able to interpret the clinical significance of sequence variants as practiced in clinical genetic testing laboratories by following this protocol.
Materials
Hardware: Any internet-enabled computer.
Software: Up-to-date web browsing software (e.g., Chrome, Safari, Internet Explorer).
Files: variants to be interpreted and raw data from the laboratory (may be in various formats)
- Variant nomenclature verification tools:
- ○
-
○VariantValidator (https://variantvalidator.org/) (Freeman, Hart, Gretton, Brookes, & Dalgleish, 2018)
-
○Human Genome Variation Society (HGVS) recommendations for sequence variant nomenclature (https://varnomen.hgvs.org/) (den Dunnen et al., 2016)
- Integrated databases:
-
○VarSome (https://varsome.com/) (Kopanos et al., 2019)
-
○VarCards (http://varcards.biols.ac.cn/) (Li et al., 2018)
-
○Mastermind Genomic Search Engine (https://mastermind.genomenon.com/)
-
○wANNOVAR (http://wannovar.wglab.org/) (Yang & Wang, 2015)
-
○
- Population databases:
-
○Genome Aggregation Database (gnomAD) (https://gnomad.broadinstitute.org/)
-
○Bravo (https://bravo.sph.umich.edu/)
-
○Chinese Millionome Database (CMDB) (https://db.cngb.org/cmdb/)
-
○
- Variant databases:
-
○ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/)
-
○Human Gene Mutation Database (HGMD) (license required for professional version) (http://www.hgmd.org/) (Stenson et al., 2017)
-
○Leiden Open Variation Database (LOVD) (https://www.lovd.nl/) (Fokkema et al., 2011)
-
○ClinGen Evidence Repository (https://erepo.clinicalgenome.org/evrepo/)
- ○
-
○
- Literature search tools:
-
○PubMed (https://pubmed.ncbi.nlm.nih.gov/)
-
○Google Scholar (https://scholar.google.com/)
- ○
-
○variant2literature (https://variant2literature.taigenomics.com/) (Lin et al., 2019)
-
○Variant Information Search Tool (VIST) (https://vist.informatik.hu-berlin.de/) (Seva et al., 2019)
-
○
- Gene-disease association databases:
-
○ClinGen (https://www.clinicalgenome.org/)
-
○OMIM (https://www.omim.org/)
-
○GeneReviews (https://www.ncbi.nlm.nih.gov/books/NBK1116/)
-
○ClinGen Dosage Sensitivity Map (https://dosage.clinicalgenome.org/)
-
○DECIPHER (https://decipher.sanger.ac.uk/)
-
○Monarch Disease Ontology (MONDO) (https://www.ebi.ac.uk/ols/ontologies/mondo/) (Mungall et al., 2017)
-
○Orphanet (https://www.orpha.net/)
-
○
- Genome browsers and sequence databases:
-
○UCSC Genome Browser (https://genome.ucsc.edu/)
-
○RefSeq (https://www.ncbi.nlm.nih.gov/refseq/)
-
○Locus Reference Genomic (LRG) (https://www.lrg-sequence.org/)
-
○Ensembl Genome Browser (https://www.ensembl.org/)
-
○
- Computational prediction tools:
-
○Protein Variation Effect Analyzer (PROVEAN) (http://provean.jcvi.org/) (Choi & Chan, 2015)
-
○MutationTaster (http://mutationtaster.org/) (Schwarz, Cooper, Schuelke, & Seelow, 2014)
-
○PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) (Current Protocols article Adzhubei, Jordan, & Sunyaev, 2013)
-
○Human Splicing Finder (http://www.umd.be/HSF3/index.html/) (Desmet et al., 2009)
- ○
- ○
- ○
- ○
-
○Pathogenic Variant Enriched Regions (PER) viewer (http://per.broadinstitute.org/) (Perez-Palma et al., 2020)
-
○Constrained Coding Regions (CCRs) Browser (http://www.rebrand.ly/ccrregions/) (Havrilla, Pedersen, Layer, & Quinlan, 2019)
-
○MISCAST (http://miscast.broadinstitute.org/) (Iqbal et al., 2020)
-
○Rare Exome Variant Ensembl Learner (REVEL) (https://sites.google.com/site/revelgenomics/) (Ioannidis et al., 2016)
- ○
-
○
- Other useful tools:
-
○alleleFrequencyApp (https://www.cardiodb.org/allelefrequencyapp/) (Whiffin et al., 2017)
-
○Automatic PVS1 (AutoPVS1) interpretation tool (http://autopvs1.genetics.bgi.com/) (Xiang, Peng, & Peng, 2019)
- ○
-
○InterVar (http://wintervar.wglab.org/) (Li & Wang, 2017)
-
○
- Sequence variant interpretation resources:
-
○ACMG/AMP recommendations for standards and guidelines for sequence variant interpretation (Richards et al., 2015). A list of ACMG/AMP evidence criteria can be found in Table 3, and rules for combining criteria to classify variants are found in Table 5.
-
○ClinGen General Sequence Variant Curation Process SOP (https://clinicalgenome.org/docs/variant-curation-sop/)
- ○
-
○ACGS Best Practice Guidelines for Variant Classification in Rare Disease (https://www.acgs.uk.com/quality/best-practice-guidelines/#VariantGuidelines)
-
○
Verify variant nomenclature
-
Obtain raw variant data.
Raw sequence variant data may be in various formats. The minimum requirement is the ability to uniquely define a sequence variant. This requires a human reference sequence, the position coordinate corresponding to the selected reference sequence, and the change in the sequence.
-
Check variant nomenclature. Go to Mutalyzer website and click the “Name Check” tool, or go to VarSome or VariantValidator and enter the variant.
Free user registration is required to use VarSome. VarSome uses a user-friendly search engine that can recognize variants in many different formats. A full list of acceptable formats can be found at https://varsome.com/about/examples/.
Outputs of the above tools include many different formats describing the same variant, including many forms of standard variant nomenclature according to Human Genome Variation Society (HGVS) recommendations (den Dunnen et al., 2016).
It is important to make sure there are no errors. Errors often arise from typos, incorrect reference accession ID or version, or incorrect position coordinates. Correct any error(s) before proceeding to the next step.
-
Identify the clinically relevant transcript of the gene by checking ClinVar or HGMD. If a reference transcript for the gene is not defined in ClinVar or HGMD, it is necessary to curate clinically relevant transcripts.
Clinical interpretation of sequence variants depends on using consistent nomenclature that uniquely defines a sequence variant. There are often multiple transcripts of many genes. It is important to annotate variants using clinically relevant transcripts. Canonical transcripts, often defined as ones with the longest coding sequence, are commonly chosen for variant annotation; however, the “canonical” transcript is not always clinically relevant. The systematic strategy for curating clinically relevant transcripts of hearing loss genes (DiStefano et al., 2018) can be used as a reference for other genes.
-
Optional : Use the “Position Converter” tool in Mutalyzer to convert variant nomenclature and select the correct nomenclature with clinically relevant transcript as the reference.
Example : The NM_001193495.1(ADAR):c.-615_-614del variant can be converted to NM_001111.4(ADAR):c.271_272del, NM_015840.3(ADAR):c.271_272del, NM_001025107.2(ADAR):c.-615_-614del, and so on by using the Mutalyzer “Position Converter” tool. NM_001111.4 is translated into the clinically relevant p150 isoform of ADAR causing dyschromatosis symmetrica hereditaria (Zhang et al., 2013). Therefore, NM_001111.4(ADAR):c.271_272del is the preferred choice for further interpretation.
Evaluate population data
-
5.
Query variant population frequency. A simple way is to search gnomAD (https://gnomad.broadinstitute.org/). Go to the website, select data set version v2.1.1, and search by gene, region, or variant. However, we recommend using VarSome, VarCards, Mastermind, or wANNOVAR to query multiple databases for a more comprehensive result.
Large population databases, such as gnomAD and Bravo, have been widely used to obtain population allele frequency information. These databases have also been integrated into many online tools, such as those listed in this step, so that multiple databases can be queried simultaneously.
Example : Input “NM_004004.6(GJB2):c.109G>A,” “GJB2 c.109G>A,” “GJB2 Val37Ile,” “GJB2 V37I,” or “chr13-20763612-C-T” in the VarSome search box to query information about the variant of NM_004004.6(GJB2):c.109G>A, p.(Val37Ile). On the variant page, click “Frequencies” on the right panel to view the population frequencies of the variant in gnomAD, Kaviar, and Bravo. Click on “View this on gnomAD Browser” to access detailed variant information in gnomAD.
-
6.
Apply one of the following criteria to the variant to be classified based on its population allele frequency according to the ACMG/AMP and ClinGen recommendations. Use ClinGen allele frequency thresholds specified by VCEPs whenever available. If specifications are not available, use the following four general ACMG/AMP criteria related to population data evaluation (Richards et al., 2015):
-
BA1 (allele frequency >5%): The updated statement proposed by the SVI is: “Allele frequency is >0.05 in any general continental population dataset of at least 2,000 observed alleles and found in a gene without a gene- or variant-specific BA1 modification” (Ghosh et al., 2018). It is important to note that some variants in continental populations with a frequency of >0.05 have also been found to be pathogenic. SVI provided an exception list for non-benign alleles with a frequency of >0.05 (Ghosh et al., 2018). Apply BA1 if the population allele frequency is >0.05 and the variant is not on the exception list.
The BA1 exception list and nomination form can be found on the SVI webpage (https://www.clinicalgenome.org/working-groups/sequence-variant-interpretation/).
Example: The NM_004004.6(GJB2):c.109G>A variant has an allele frequency of 0.08345 in the East Asian population, but it is classified as pathogenic (Shen et al., 2019). This variant was on the BA1 exception list (Ghosh et al., 2018).
BS1 (allele frequency higher than expected): An online alleleFrequencyApp (Whiffin et al., 2017) can be used to calculate the maximum credible population allele frequency by adjusting parameters that describe the genetic architecture of the disorder. It can also calculate the maximum tolerated allele count in a specific reference population (such as in gnomAD) based on the size of the population and at a user-specified confidence level. Apply BS1 if the population allele frequency is greater than the maximum credible population allele frequency without conflicting evidence.
BS2 (variant observed in healthy adult individuals): The evidence can be accessed from gnomAD, the literature, or in-house data. gnomAD shows the age distribution of heterozygous or homozygous variant carriers. Use caution if a low number of individuals is observed in gnomAD when the disease is adult-onset or has variable penetrance/expressivity. Apply BS2 if it is reasonable to believe that individuals with the variant in large population databases are truly unaffected.
PM2 (absent/rare in population databases): The threshold proposed for dominant disorders is “within pathogenic range” but not “absent.” The gnomAD population should be viewed as “the general population” rather than as “unaffected controls.” Although individuals with severe pediatric diseases have been excluded from the gnomAD dataset, particular disorders with adult onset or incomplete penetrance cannot be ruled out.
-
Query literature/database and in-house evidence
-
7.
Search the literature to obtain cases with the variant to be interpreted. Variant databases such as ClinVar, HGMD, LOVD, and ClinGen Evidence Repository can be used to query previously reported cases with the variant. “Variants in Papers” tracks from the UCSC genome browser can be used to obtain variants extracted from full-text publications gathered by Mastermind and Automatic Variant evidence DAtabase (AVADA) (Birgmeier et al., 2020). Identify references associated with the cases and obtain the full-text articles.
ClinVar, HGMD, and LOVD variants have been linked to UCSC genome browser tracks for query. Protocols have been published for using ClinVar and HGMD (see Current Protocols articles Harrison et al., 2016; Stenson et al., 2012). Certain variants in ClinVar are assigned review status with 3 or 4 stars, for which the interpretation can be adopted directly. Literature review is mandatory for other variants.
-
8.
Perform a literature search using search engines if no publications have been found by searching databases. PubMed and Google Scholar are commonly used. Other useful tools, including variant2literature (Lin et al., 2019), LitVar, Mastermind Genomic Search Engine, and Variant Information Search Tool (VIST) (Seva et al., 2019) can extract information on a variant from text or images, thus allowing the search to be more efficient.
Consult the ClinGen General Sequence Variant Curation Process SOP (https://clinicalgenome.org/docs/variant-curation-sop/) for detailed guidance regarding literature searches and curation.
-
9.
Retrieve case information from public databases, publications, and in-house databases. Case information includes phenotype descriptions of affected individuals, number of affected probands, pedigree structures of affected probands, affected status and genotypes of proband family members, and ethnic origins.
Double-check the clinical information in publications from the same authors or institutions to avoid recounting the same probands or families.
Example : The NM_004523.4(KIF11):c.139C>T variant has been reported in three publications (Hazan et al., 2012; Jones et al., 2014; Robitaille et al., 2014), all of them described the same Turkish proband with microcephaly, lymphedema, and chorioretinal dysplasia (Hazan et al., 2012). In this situation, duplicated counting of the proband should be avoided.
-
10.
Apply PP4 to the variant to be classified if the phenotype or family history is specific to the condition. This criterion can be used when the patient’s phenotype is consistent with a highly specific genetic etiology. The usage of PP4 should not be restricted to diseases with single genetic etiology. The criterion is applicable if a disorder has a limited number of genetic etiologies and all those genes have been tested. This strength has also been proposed to upgrade to a moderate or strong level in some VCEPs.
Example : PP4 can be applied to the PAH variant identified in a phenylketonuria patient with plasma Phe > 120 μM and normal BH4 cofactor metabolism, according to ClinGen PAH expert panel specifications (Zastrow et al., 2018).
-
11.
Evaluate case-control data and previously reported rare cases.
-
Apply PS4 to the variant to be classified based on case-control statistics on variants that are relatively common. Count cases and controls based on genotypes and stratified by ethnic origins. Calculate odds ratios (OR), p values, 95% confidence intervals (CI), and z-statistics using MedCalc or other statistical tools. Apply PS4 if OR > 5.0 and CI does not include 1.0.
Although aggregated population data in gnomAD can be used as a control, individually determined genotype data from laboratories are more powerful. Statistical analyses on ethnicity-stratified subpopulations should be used to avoid different ethnic compositions of cases and controls as a confounding factor (Shen et al., 2019).
Example : The GJB2 p.(Val37Ile) variant was observed with the highest frequency in the East Asian population. Shen et al. (2019) compared frequencies of individuals with this variant between cases and controls. The homozygote of the variant is statistically enriched in cases with OR = 20 (95% CI 17–24, Z = 31, p < 0.0001). It was still significant when ethnicity-stratified subpopulation analysis was performed (OR = 12, 95% CI = 9.1–15, Z = 19, p < 0.0001). Therefore, the PS4 criterion can be applied.
-
Apply a modified strength of PS4 to the variant when it has been previously identified in unrelated affected individual(s) and is absent from large population databases (meeting PM2). Case-control analysis cannot be performed for rare variants associated with rare disorders due to small sample sizes that do not provide sufficient power to reach statistical significance. Based on the Bayesian approach to the ACMG/AMG recommendations, an exponential increase in thresholds as the strength level increases has been suggested (Tavtigian et al., 2018). The threshold for “PS4_Supporting,” “PS4_Moderate,” “PS4,” and “PS4_Very Strong” has been specified as 1, 2, 4, and 16 previously reported cases or scores, respectively (Lee et al., 2018; Mester et al., 2018).
The modified strength of PS4 is most applicable to autosomal dominant or X-linked diseases. For autosomal recessive diseases, use adjusted PM3 instead.
-
-
12.
Determine the segregation of a variant with disease phenotypes in pedigrees with affected patients. Apply PP1 with an appropriate strength level if a variant was found to co-segregate with multiple affected family members. Apply BS4 if there is a lack of segregation of a variant with affected members. The ClinGen Gene Curation Working Group and Hearing Loss Working Group have proposed recommendations for segregation evaluation and calculating a simplified estimated logarithm of the odds (LOD) score (Oza et al., 2018; Strande et al., 2017). Although the Hearing Loss Working Group has referenced an SVI general recommendation, the formal SVI recommendation is still under development. Based on the recommendations from the Hearing Loss Working Group, the strength level for “PP1,” “PP1_Moderate,” and “PP1_Strong” may be 0.6, 1.2, and 1.5 of LOD scores, respectively (Oza et al., 2018).
Segregation counting: Generally, the number of affected segregations will be the number of affected individuals minus one, but there may be exceptions. For dominant or X-linked disorders, segregations from affected individuals (genotype+, phenotype+) and obligate carriers (regardless of phenotype) are counted. For recessive disorders, segregations from affected individuals and unaffected individuals (typically siblings who are at risk to inherit the two variants identified in the proband) are counted.
Calculate LOD scores: Use one of the following equations to calculate LOD scores for dominant/X-linked or recessive segregations. The LOD scores calculated in different families with the same variant can be added to assign a final score. The final LOD score is then used to determine the applicable level of strength of the evidence.
Equation for dominant/X-linked segregations:
Equation for recessive segregations:
It should be noted that the limit for counting segregations is different for gene curation and variant curation. For gene curation, families with <4 affected segregations for dominant/X-linked disorders, or <3 affected individuals for recessive disorders, cannot be included in a segregation analysis (https://clinicalgenome.org/working-groups/gene-curation/). For variant curation, two affected segregations for dominant/X-linked disorders, or one affected segregation for recessive disorders, are sufficient to apply PP1 according to the ClinGen Hearing Loss Working Group recommendations (Oza et al., 2018).
BS4 may be applied for an affected individual without the specific variant (genotype−, phenotype+), but not for unaffected individuals with the variant because of age-related penetrance and variable expression.
A simplified method for calculating N for segregation analysis has been proposed (Jarvik & Browning, 2016). The authors recommended that the N threshold should be smaller if the evidence of segregation comes from one family, instead of two or more families, simply because of the concern that evidence from one family can be generated by the physical linkage between the observed variant and an unobserved causal variant.
Example : The NM_001009944.3(PKD1):c.7288C>T, p.(Arg2430*) variant was detected in a family with polycystic kidney disease (Fig. 2). Although the genotypes of II-1 and II-3 are unknown, they are counted as obligate carriers because they connect two affected members with a positive genotype. There are five segregations in this family, as illustrated by colored lines. III-2 harbors the variant but is unaffected, and therefore it should not be considered as a segregation. The estimated LOD score for five segregations is 1.5, and thus “PP1_Strong” may be applied. III-2 does not count as a non-segregation, because the penetrance of the condition may be age-dependent and III-2 is still young.
Figure 2. Pedigree of a family with polycystic kidney disease.

Black solid circles/squares represent affected members; positive signs indicate members who harbor an NM_001009944.3(PKD1):c.7288C>T heterozygous variant; negative signs indicate members who were genotyped and do not harbor the variant; colored lines indicate five segregations in this family.
-
13.
Apply PS2 or PM6 to the variant to be classified based on de novo occurrence evaluation. The SVI has specified these criteria using a point-based system. The strength of these criteria is adjustable based on the following evidence: confirmed or assumed parental status, phenotypic consistency, and the number of de novo observations. The detailed SVI recommendation for de novo criteria (PS2 & PM6) can be found on the SVI webpage (https://www.clinicalgenome.org/working-groups/sequence-variant-interpretation/).
Example: A patient with early-onset seizures has a de novo NM_003042.4(SLC6A1):c.1070C>A variant. Parental identities were confirmed. The variant is awarded one point, because it is confirmed de novo and the patient’s phenotype is consistent with the disease associated with SLC6A1. However, having seizures is not a highly specific phenotype. The variant has not been reported in the literature or patient databases. Therefore, PS2_Moderate is applied according to the SVI recommendation for de novo criteria.
-
14.
Evaluate the cis/trans phase of the variants when multiple variants in the same gene are identified.
For recessive disorders, if a variant is detected in trans with a pathogenic or likely pathogenic variant in an affected individual, PM3 may be applied. The strength may be adjusted depending on the number of cases with trans phase and the allele frequency of the variant. If a variant is detected in cis with a pathogenic or likely pathogenic variant, apply BP2. The SVI specified the PM3 criterion using a comprehensive point-based system based on allele frequency, phasing, variant classification, and zygosity. The detailed SVI recommendation for in trans criteria (PM3) can be found on the SVI webpage (https://clinicalgenome.org/working-groups/sequence-variant-interpretation/).
-
For dominant disorders, apply BP2 to the variant to be classified if it is observed in cis with a pathogenic or likely pathogenic variant, or in trans with a pathogenic or likely pathogenic variant with complete penetrance.
Example: The NM_00277.3(PAH):c.1197A>T, p.(Val399=) variant was confirmed to be in trans with the pathogenic variant p.(Arg408Trp) (award 1.0 point) in a proband, and with the likely pathogenic variant p.(Ala434Asp) in another proband (award 1.0 point) (Gu et al., 2014). In another study, the variant was reported as homozygous in five probands (award 1.0 point, as the maximum points allowed for homozygous occurrence is 1.0). The variant was detected with another pathogenic variant Tyr356* in 9 probands but the phase was unknown (4.5 points) (Liu et al., 2017). The population allele frequency is <0.0001 in gnomAD. Therefore, PM3_Very Strong is applied as the combined points (7.5 points) reach the Very Strong threshold.
-
15.
Apply BP5 to the variant to be classified if an alternate molecular basis that can explain the patient’s phenotype has been identified. Generally, this criterion is more applicable to dominant disorders with an obvious alternate mechanism. For recessive disorders, incidental carriers can harbor pathogenic variants unrelated to the disease phenotype.
Use caution for multigenic disorders and disorders in which multiple variants can contribute to more severe phenotypes.
Example : In a Chinese family with hypertrophic cardiomyopathy (HCM), Zhang et al. (2017) reported that family members harboring the heterozygous NM_000256.3(MYBPC3):c.3624del variant presented less-severe phenotypes than those with this variant plus the heterozygous NM_201596.3(CACNB2):c.1598C>T variant. Because −15%-50% of familial HCM patients have been observed to harbor two heterozygous variants and this has been speculated to result in a more severe HCM phenotype (Emrahi, Tabrizi, Gharehsouran, Ardebili, & Estiar, 2016; Zhao et al., 2017), BP5 should not be applied in this case.
-
16.
Evaluate experimental evidence in functional studies. Apply PS3 if well-controlled reproducible functional assays prove a damaging effect. Apply BS3 if validated functional assays show no effect of the variant on the gene or gene product. The evidence strength may be adjusted based on the validity of the assay and the effective size of the impact. Generally, in vivo studies provide stronger evidence than in vitro studies, and assays that rescue phenotypes provide stronger evidence than assays manipulating them. Current recommendations of VCEPs for functional assays were comparatively analyzed (Kanavy et al., 2019). A structured approach to evaluate functional assays for variant interpretation has been proposed by the SVI (Brnich et al., 2019) and should be consulted. The approach includes an assessment of the clinical validity of functional assays and a four-step decision tree for determining the strength of criteria to be applied.
PS3 should only be used to evaluate functional data that demonstrate effect caused by the variant to be classified.
Perform variant-type-specific analysis
-
17.
Null variants : PVS1 is applicable if a variant is predicted to lead to a null protein product where loss-of-function (LoF) is the known disease mechanism. These include nonsense mutations, frameshifts, ±1 or 2 canonical splice sites, initiation codons, and large deletions. PVS1 is the only criterion with a very strong strength level in the ACMG/AMP recommendations (Richards et al., 2015). Recommendations for applying PVS1 (Abou Tayoun et al., 2018) have been published by the SVI, which provides a framework for evaluating whether LoF is the cause of disease and whether a variant causes LoF.
For large copy-number loss or gain variant, the ACMG/ClinGen technical standards for copy-number variant (CNV) interpretation (Riggs et al., 2019) should be consulted.
-
Determine if LoF is the disease mechanism. For recessive disorders, LoF is generally the disease mechanism (thus, proceed to step 17b). For dominant disorders, use the following two methods: (i) Access the ClinGen Dosage Sensitivity Map (https://www.ncbi.nlm.nih.gov/projects/dbvar/clingen/) to check the dosage-sensitivity of a curated gene. If the gene has been established to cause disease due to haploinsufficiency (ClinGen HI score: 3), proceed to step 17b. (ii) For unestablished genes, the gnomAD pLI score (Lek et al., 2016) and DECIPHER HI index (Huang, Lee, Marcotte, & Hurles, 2010) can be used as HI predictors. Genes with high pLI scores (pLI ≥ 0.9) and high HI index ranks (0%–10%) are more likely to exhibit HI and more intolerance to LoF variants. However, these scores are just a predictor and will not always be accurate. Generally, they are not applicable to late-onset diseases occurring after reproductive age.
Example: The ClinGen Haploinsufficiency Score for ZEB2 is 3. This score means there is sufficient evidence suggesting that dosage sensitivity is associated with the clinical phenotype.
Example : LoF variants in BRCA1 have been curated as the mechanism of disease, although the gnomAD pLI is 0. This is an example where pLI is an inaccurate predictor, because BRCA1 is associated with breast/ovarian cancer, which has age-related penetrance, and patients usually develop BRCA1-related cancer after reproductive age.
-
Evaluate whether a variant causes LoF. The SVI suggested using a PVS1 decision tree to determine the LoF effect of a variant in the context of gene structure and molecular etiology, such as alternative splicing or nonsense-mediated mRNA decay (NMD) (Abou Tayoun et al., 2018). An online automatic PVS1 interpretation (AutoPVS1) tool (Xiang et al., 2019) has been developed to facilitate determination of the PVS1 strength level for null variants. The strength level should be adjusted based on the clinical validity classification of the gene (refer to Table 1 in Abou Tayoun et al., 2018).
Example : NM_000441.1(SLC26A4):c.1920G>A, p.(Trp640*) is a nonsense variant and predicted to undergo NMD because it is located in exon 17/21 far from the NMD-resistant region. Exon 17 is present in the biologically relevant transcript of SLC26A4. SLC26A4 is definitively associated with autosomal recessive Pendred syndrome (https://search.clinicalgenome.org/kb/genes/HGNC:8818). Therefore, PVS1 can be applied.
-
-
18.
Missense variants : Evaluate pathogenic/benign variant spectrum.
-
Apply PP2 if missense variants are a common cause of the disease. Use the gnomAD browser to check whether a gene is intolerant to missense variants. The SVI suggests a gene with a missense gene constraint z-score > 3.09 is more likely to be intolerant of missense changes. The z-score is calculated by comparing the expected versus the observed number of missense variants. The higher the z-score, the higher the constraint or intolerance to variation.
Example : COL1A1 has a missense gene constraint z- score of 3.53 in gnomAD (https://gnomad.broadinstitute.org/gene/ENSG00000108821?dataset=gnomad_r2_1). Therefore, missense variants identified in COL1A1 meet PP2.
-
Apply BP1 if a missense variant is identified in a gene where the disease is primarily caused by truncations. Use Simple ClinVar (Perez-Palma et al., 2019) to estimate the primary types of variants causing a disease. BP1 was proposed to apply if all or nearly all (e.g., >90%) of the disease-causing variants are truncations (Amendola et al., 2016).
Example : There are 180 pathogenic/likely pathogenic variants in ASPM when choosing the corresponding “clinical significance filter” in the Simple ClinVar. Of these, only three are missense variants (date of access: March 8, 2020). Therefore, missense variants in ASPM meet BP1.
-
-
19.
Missense variant : Evaluate variants affecting the same amino acid residue. Apply PS1 if the same missense change at an amino acid residue has been previously determined as pathogenic or likely pathogenic. Apply PM5 if a different missense change at the same amino acid residue has been previously determined as pathogenic or likely pathogenic. This information can be obtained from variant databases (e.g., ClinVar, HGMD) or from a literature search. Notably, variant2literature (Lin et al., 2019) has a “Position offset” option, which allows users to search neighboring variants of the query. VarSome can automatically link this information to PS1/PM5 in the “ACMG Classification” tab.
The other variant affecting the amino acid residue should be classified as pathogenic (or likely pathogenic) without depending on PS1 or PM5. Do not apply the same variant being assessed because it is not yet classified as pathogenic. Analysis of previously reported cases should use PS4 with an appropriate level of strength (see Step 11 above).
Example:When querying the COMP p.(Arg718Trp) variant using VarSome, PM5 is automatically checked in the “ACMG Classification” tab. The other missense variant at this codon, p.(Arg718Pro), is listed in the PM5 tab. Because p.(Arg718Pro) is classified as likely pathogenic, PM5 can be applied.
-
20.
In-frame insertions or deletions : Evaluate repetitive region for in-frame insertion or deletion. Apply PM4 if in-frame indels or a stop loss is present in the nonrepeat region of a gene, and BP3 if an in-frame indel is in the repetitive region of a gene. In-frame indels and surrounding sequences can be accessed through the “Region Browser” tab in VarSome. The RepeatMasker track from the UCSC genome browser can be used to evaluate repetitive elements. The protocol for using RepeatMasker has been published (see Current Protocols article Tarailo-Graovac & Chen, 2009).
Example : NM_001453.3(FOXC1):c.1136_1141dup, p.(Gly379_Gly380dup) is an in-frame insertion variant in FOXC1, but is in a repetitive region (chr6:1611803-1611852) (GRCh37/hg19) marked by RepeatMasker. Thus, it meets the criterion for BP3.
Perform computational predictions
-
21.
Missense variants : Apply PP3 to the variant to be classified if the REVEL score of a missense variant is >0.75. The SVI recommended a meta-predictor REVEL that combines 13 individual tools with high sensitivity and specificity compared to any single tool or to other Ensembl methods (Ioannidis et al., 2016). Apply BP4 if the REVEL score is low. Note, however, that the threshold for BP4 has not yet been proposed by the SVI. Some VCEPs have specified thresholds for BP4. A REVEL score ≤0.15 was recommended by the ClinGen Hearing Loss Working Group as the threshold for BP4, given that 95% of pathogenic variants at this range are excluded (Oza et al., 2018). VarSome and VarCards (J. Li et al., 2018) can be used to retrieve REVEL and other predictors’ scores.
The predictions are not always accurate for all variants. Professional judgment should be used when considering the prediction evidence.
VarCards contains a “damaging score,” which shows the proportion of in silico predictors predicted to be deleterious. Another meta-predictor included in VarCards, ClinPred (Alirezaie et al., 2018), can also be considered for use.
It should be noted that missense variants may alter splicing. Evaluation of splicing may occasionally be necessary.
Example : The NM_000095.2(COMP):c.2153G>C variant has a REVEL score of 0.9419, and thus meets PP3. The NM_001009944.2(PKD1):c.10529C>T variant has a REVEL score of 0.103, and only 1/23 algorithms in VarCards predict a damaging impact, so BP4 can be applied.
-
22.
Silent, intronic, or missense variants : Apply PP3 to the variant to be classified if a noncanonical splice site variant is predicted to impact splicing. Apply BP7 if a silent variant is not highly conserved and is not predicted to impact splicing. Human Splicing Finder, MaxEntScan, NNSPLICE, and SpliceAI can be useful for predicting splicing effects. The database dbscSNV (Jian, Boerwinkle, & Liu, 2014) includes all potential human SNVs within splicing consensus regions (−3 to +8 at the 5’ splice site and −12 to +2 at the 3’ splice site) and two Ensembl prediction scores for predicting their potential for alternative splicing. If the adaptive boosting and random forest scores of a variant from dbscSNV are <0.6, then BP7 may be applied. These scores can be accessed through VarSome. Conservation scores of the nucleotide across species can be easily accessed through VarSome, VarCards, or most genome browsers. If abnormal splicing/protein-level function is shown in functional studies, an appropriate strength level of PS3 should be used instead (Brnich et al., 2019).
PP3 should not be applied to canonical +/−1 or 2 splice variants to prevent double-counting the same predictive evidence used to assign PVS1.
Example : LRG_1191t1(JAG1):c.2917-8C>A is predicted to activate an intronic cryptic acceptor site by Human Splicing Finder. Further, cDNA sequencing indicates that the variant destroyed the intron 23/exon 24 splice acceptor site and activated a new stronger acceptor site in intron 23 (position c.2917-7/c.2917-6). Consequently, the intron 23 sequence from positions c.2917-6 to c.2917-1 (TTTTAG) was inserted into the mutant transcript (r.2916_2917ins2917-6_2917-1; p.Gly973Phefs*2) (Chen, Liu, Chen, Zhang, & Xu, 2019). In this situation, an appropriate strength level of PS3 can be applied instead of PP3.
Example : The NM_022124.6(CDH23):c.4842G>C, p.(Leu1614=) variant is not predicted to affect splicing, and the position is not conserved (GERP++ rejected substitutions = 3.06, which is <6.8). Thus, the BP7 criterion is met.
-
23.
In-frame insertions or deletion s : Apply PP3 or BP4 to an in-frame insertion or deletion if the variant is predicted to be detrimental or benign. For example, the ClinGen lysosomal storage disorders expert panel has proposed using PROVEAN (Choi & Chan, 2015) and MutationTaster (Schwarz et al., 2014) to determine the potential functional effect and usage of PP3/BP4 criteria.
Example : BP4 may be applied to the NM_000218.2(KCNQ1):c.160_168dup variant, as it is predicted as “neutral” and “polymorphism” by PROVEAN and MutationTaster, respectively.
-
24.Missense variants or in-frame indels : Apply PM1 if a variant occurs within a mutational hot spot and/or critical and well-established functional domain. The SVI proposed using a regional missense constraint o/e score (Samocha et al., 2017) to obtain the critical domain by comparing observed versus expected missense variants for specific regions of a gene. The regional missense constraint score can be accessed by selecting the ExAC dataset in the gnomAD browser. It is shown only for genes that exhibit regional missense constraints. This score can also be accessed via DECIPHER. Currently, the threshold of the regional missense constraint score to be used for PM1 has not been set by SVI. Several online tools are available to help determine functional domains of a particular gene.
- The constraint coding regions (CCRs) Browser (Havrilla et al., 2019) can be used to search CCRs in the human genome. Higher CCR percentiles indicate more significant predicted constraints.
- subRVIS (Gussow et al., 2016) can be used to plot functional domains or exons with a subRVIS score, which indicates how tolerant or intolerant a region is to functional variation.
- PER viewer (Perez-Palma et al., 2020) can be used to plot pathogenic enriched regions (PERs) across genes and gene families.
- MISCAST (Iqbal et al., 2020) can interpret the effect of missense variants on protein 3D structure in 1330 genes and thus provide structure-based insights into their biological impact.
PM1 only applies to missense variants and in-frame indels. The ACGS recommended an upgrade to be strong for particular amino acid residues that are critical for protein structure or function (Ellard et al., 2020). For example, invariant cysteines in the EGF-like calcium-binding domain of FBN1, cysteine substitutions that result in an uneven number of cysteine residues within an EGF-like repeat of NOTCH3, glycine substitutions in the Gly-X-Y repeat of collagen genes, and cysteine or histidine substitutions in C2H4 zinc fingers such as GLI3.
Classify variant
-
25.
Combine all applied criteria to reach a classification according to the scoring rules in Table 5 of the ACMG/AMP recommendations (Richards et al., 2015).
Some VCEPs have revised the original ACMG/AMP rules, so they should be consulted for guidance. Expert judgment should be applied when considering the evidence and assigning a classification.
Support Protocol
REEVALUATING THE CLINICAL SIGNIFICANCE OF SEQUENCE VARIANTS
The growing use of NGS technologies not only increases the diagnostic yield in certain genetic disorders, but also increases the report of variants of uncertain significance (VUS) (Turner, Rao, Morgan, Vnencak-Jones, & Wiesner, 2019). With information regarding variant pathogenicity accumulated over time, these variants have a high rate of reclassification (Bennett et al., 2019; Mersch et al., 2018; SoRelle et al., 2019), thus requiring reevaluation on a routine basis. Recent studies of variant reevaluation of patients with inherited cancer and cardiac disease have shown that variant reclassifications have the potential to improve clinical management and reproductive choices (Slavin et al., 2019; So et al., 2019; Turner et al., 2019; Wong et al., 2019). The ACMG has published a statement on variant-level reevaluation (Deignan et al., 2019). They proposed that clinical laboratories should develop protocols or policies regarding variant reevaluation, external request, and counseling, as well as establish an up-to-date in-house database for reported variants (Deignan et al., 2019). Variant reevaluation involves consideration of new information and reassessment of the whole body of evidence. This protocol describes the initiation and procedures for variant reevaluation.
Materials
- Initiate variant reevaluation. The initiation typically comes from periodic reevaluation specified by the laboratory. Laboratories can set an internal timeframe for periodic reevaluation. In ClinGen, the VCEPs are expected to reevaluate all likely pathogenic and VUS classifications at least every 2 years. Likely benign classifications are reevaluated when new population datasets become available (Rivera-Munoz et al., 2018). However, reevaluation should be triggered as soon as possible in certain circumstances, such as:
- Upon external request.
- Upon observation of a previously unclassified variant in a new patient or the literature.
- Upon updating of population database or guidelines.
- Prior to clinical decision making.
- Reclassify variants (following the Basic Protocol) by considering all updated databases, literature, and in-house findings. Frequently updated information includes:
- New findings from pedigree analysis or additional phenotypic information/family history that has developed over time.
- Updated population database or variant databases.
- Cases with the same variant reported in the literature.
-
Improved computational algorithms for variant pathogenicity prediction.Variant-level functional study.
Release an updated report if variant reclassification leads to a clinically significant change (e.g., a change from pathogenic/likely pathogenic to VUS/benign/likely benign or vice versa).
Update variant databases, including in-house database and public databases (e.g., ClinVar).
COMMENTARY
Background Information
Understanding the gene-disease relationship is essential for accurately interpreting variants. Prior to following this protocol, laboratories should first check the gene-disease relationship, which can be queried through the ClinGen webpage (https://www.clinicalgenome.org/) or curated using the published framework (Strande et al., 2017). Useful gene-disease databases are also listed in this protocol. ClinGen has developed a framework for evaluating gene-disease associations (Strande et al., 2017) using a semiquantitative measurement for the gene-disease relationship defined as: “definitive,” “strong,” “moderate,” “limited,” “no reported evidence,” or “conflicting evidence.” As indicated in the ACMG/AMP recommendations, variants found in genes with uncertain significance should always be classified as VUS (Richards et al., 2015).
Laboratories should also check whether variant-related genes/disorders have been specified by the ClinGen VCEPs. If the VCEPs have specifications, the sequence variant interpretation process should follow those specifications with the highest priority. Otherwise, a general sequence variant curation SOP proposed by the SVI should be consulted. The VCEPs and curation status can be found in the ClinGen Clinical Domain Working Groups website (https://clinicalgenome.org/working-groups/clinical-domain/). The SVI general recommendations and VCEPs specified variant interpretation guidelines are located at the SVI webpage (https://www.clinicalgenome.org/working-groups/sequence-variant-interpretation/).
It should be noted that the SVI proposed to remove PP5/BP6 criteria from the ACMG/AMP framework (Biesecker et al., 2018). Thus, these criteria are not involved in this protocol.
The SVI has recommended using the original criteria code followed by an underscore and a new modified level of strength for modifications of ACMG/AMP criteria code (refer to “Guidance on how to rename criteria codes when strength of evidence is modified” on the SVI webpage). For example, co-segregation with disease in multiple affected family members is a criterion for pathogenicity at supporting strength level (PP1); however, this criterion can be upgraded to moderate (PP1_Moderate) or strong (PP1_Strong) with increasing segregations.
Critical Parameters
Published literature, population and variant databases, in-house data, recommendations for guidelines, and online tools are constantly being updated. It is important to keep all resources up to date. In addition, collaboration with other laboratories will increase case information and enhance the ability to make accurate interpretations. Contributing variant and associated clinical information to ClinVar will help the community improve the variant interpretation process.
Acknowledgments
We thank Zinan Zhou, Yong Gao, Qifei Li, Zheng Yan, Xiaoting Ma, and Jingyi Li for their advice and comments, and Linda Johnson and Abigail E. Lorenzo for editing of the language. This work is supported by the National Natural Science Foundation of China (no. 81401219 to JZ), the Shanghai Municipal Commission of Science and Technology Program (no. 19ZR1462300 to JZ), the Municipal Human Resources Development Program for Outstanding Young Talents in Medical and Health Sciences in Shanghai (no. 2018YQ39 to JZ), the Shanghai Jiao Tong University Program (no. YG2017MS39 to JZ), and the U.S. National Institutes of Health/National Institute on Deafness and Other Communication Disorder (nos. R01DC015052 and R03DC013866 to JS).
LITERATURE CITED:
- Abou Tayoun AN, Pesaran T, DiStefano MT, Oza A, Rehm HL, Biesecker LG, … ClinGen Sequence Variant Interpretation Working, G. (2018). Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum Mutat, 39(11), 1517–1524. doi: 10.1002/humu.23626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adzhubei I, Jordan DM, & Sunyaev SR (2013). Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet, Chapter 7, Unit7 20. doi: 10.1002/0471142905.hg0720s76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alirezaie N, Kernohan KD, Hartley T, Majewski J, & Hocking TD (2018). ClinPred: Prediction Tool to Identify Disease-Relevant Nonsynonymous Single-Nucleotide Variants. Am J Hum Genet, 103(4), 474–483. doi: 10.1016/j.ajhg.2018.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amendola LM, Jarvik GP, Leo MC, McLaughlin HM, Akkari Y, Amaral MD, … Rehm HL (2016). Performance of ACMG-AMP Variant-Interpretation Guidelines among Nine Laboratories in the Clinical Sequencing Exploratory Research Consortium. Am J Hum Genet, 98(6), 1067–1076. doi: 10.1016/j.ajhg.2016.03.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balmana J, Digiovanni L, Gaddam P, Walsh MF, Joseph V, Stadler ZK, … Domchek SM (2016). Conflicting Interpretation of Genetic Variants and Cancer Risk by Commercial Laboratories as Assessed by the Prospective Registry of Multiplex Testing. J Clin Oncol, 34(34), 4071–4078. doi: 10.1200/JCO.2016.68.4316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett JS, Bernhardt M, McBride KL, Reshmi SC, Zmuda E, Kertesz NJ, … Kamp AN (2019). Reclassification of Variants of Uncertain Significance in Children with Inherited Arrhythmia Syndromes is Predicted by Clinical Factors. Pediatr Cardiol, 40(8), 1679–1687. doi: 10.1007/s00246-019-02203-2 [DOI] [PubMed] [Google Scholar]
- Biesecker LG, & Harrison SM (2018). The ACMG/AMP reputable source criteria for the interpretation of sequence variants. Genet Med. doi: 10.1038/gim.2018.42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birgmeier J, Deisseroth CA, Hayward LE, Galhardo LMT, Tierno AP, Jagadeesh KA, … Bejerano G. (2020). AVADA: toward automated pathogenic variant evidence retrieval directly from the full-text literature. Genet Med, 22(2), 362–370. doi: 10.1038/s41436-019-0643-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brnich SE, Abou Tayoun AN, Couch FJ, Cutting GR, Greenblatt MS, Heinen CD, … Clinical Genome Resource Sequence Variant Interpretation Working, G. (2019). Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework. Genome Med, 12(1), 3. doi: 10.1186/s13073-019-0690-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Liu X, Chen S, Zhang J, & Xu C (2019). Targeted Sequencing and RNA Assay Reveal a Noncanonical JAG1 Splicing Variant Causing Alagille Syndrome. Front Genet, 10, 1363. doi: 10.3389/fgene.2019.01363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y, & Chan AP (2015). PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics, 31(16), 2745–2747. doi: 10.1093/bioinformatics/btv195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deignan JL, Chung WK, Kearney HM, Monaghan KG, Rehder CW, Chao EC, & Committee ALQA (2019). Points to consider in the reevaluation and reanalysis of genomic test results: a statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med. doi: 10.1038/s41436-019-0478-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Dunnen JT, Dalgleish R, Maglott DR, Hart RK, Greenblatt MS, McGowan-Jordan J, … Taschner PE (2016). HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum Mutat, 37(6), 564–569. doi: 10.1002/humu.22981 [DOI] [PubMed] [Google Scholar]
- Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, & Beroud C (2009). Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res, 37(9), e67. doi: 10.1093/nar/gkp215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiStefano MT, Hemphill SE, Cushman BJ, Bowser MJ, Hynes E, Grant AR, … Abou Tayoun AN (2018). Curating Clinically Relevant Transcripts for the Interpretation of Sequence Variants. J Mol Diagn, 20(6), 789–801. doi: 10.1016/j.jmoldx.2018.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellard S, Baple EL, Callaway A, Berry I, Forrester N, Turnbull C, … McMullan DJ (2020). ACGS Best Practice Guidelines for Variant Classification in Rare Disease 2020. Retrieved from https://www.acgs.uk.com/media/11631/uk-practice-guidelines-for-variant-classification-v4-01-2020.pdf
- Emrahi L, Tabrizi MT, Gharehsouran J, Ardebili SM, & Estiar MA (2016). Spectrum of MYBPC3 Gene Mutations in Patients with Hypertrophic Cardiomyopathy, Reporting Two Novel Mutations from North-West of Iran. Clin Lab, 62(5), 757–764. doi: 10.7754/clin.lab.2014.141134 [DOI] [PubMed] [Google Scholar]
- Fokkema IF, Taschner PE, Schaafsma GC, Celli J, Laros JF, & den Dunnen JT (2011). LOVD v.2.0: the next generation in gene variant databases. Hum Mutat, 32(5), 557–563. doi: 10.1002/humu.21438 [DOI] [PubMed] [Google Scholar]
- Freeman PJ, Hart RK, Gretton LJ, Brookes AJ, & Dalgleish R (2018). VariantValidator: Accurate validation, mapping, and formatting of sequence variation descriptions. Hum Mutat, 39(1), 61–68. doi: 10.1002/humu.23348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelb BD, Cave H, Dillon MW, Gripp KW, Lee JA, Mason-Suares H, … Vincent LM (2018). ClinGen’s RASopathy Expert Panel consensus methods for variant interpretation. Genet Med. doi: 10.1038/gim.2018.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh R, Harrison SM, Rehm HL, Plon SE, Biesecker LG, & ClinGen Sequence Variant Interpretation Working, G. (2018). Updated recommendation for the benign stand-alone ACMG/AMP criterion. Hum Mutat, 39(11), 1525–1530. doi: 10.1002/humu.23642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y, Lu K, Yang G, Cen Z, Yu L, Lin L, … Huang J (2014). Mutation spectrum of six genes in Chinese phenylketonuria patients obtained through next-generation sequencing. PLoS One, 9(4), e94100. doi: 10.1371/journal.pone.0094100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gussow AB, Petrovski S, Wang Q, Allen AS, & Goldstein DB (2016). The intolerance to functional genetic variation of protein domains predicts the localization of pathogenic mutations within genes. Genome Biol, 17, 9. doi: 10.1186/s13059-016-0869-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison SM, Biesecker LG, & Rehm HL (2019). Overview of Specifications to the ACMG/AMP Variant Interpretation Guidelines. Current Protocols in Human Genetics, 103(1). doi: 10.1002/cphg.93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison SM, Dolinsky JS, Knight Johnson AE, Pesaran T, Azzariti DR, Bale S, … Rehm HL (2017). Clinical laboratories collaborate to resolve differences in variant interpretations submitted to ClinVar. Genet Med. doi: 10.1038/gim.2017.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison SM, Riggs ER, Maglott DR, Lee JM, Azzariti DR, Niehaus A, … Rehm HL (2016). Using ClinVar as a Resource to Support Variant Interpretation. Curr Protoc Hum Genet, 89, 8 16 11–18 16 23. doi: 10.1002/0471142905.hg0816s89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havrilla JM, Pedersen BS, Layer RM, & Quinlan AR (2019). A map of constrained coding regions in the human genome. Nat Genet, 51(1), 88–95. doi: 10.1038/s41588-018-0294-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazan F, Ostergaard P, Ozturk T, Kantekin E, Atlihan F, Jeffery S, & Ozkinay F (2012). A novel KIF11 mutation in a Turkish patient with microcephaly, lymphedema, and chorioretinal dysplasia from a consanguineous family. Am J Med Genet A, 158A(7), 1686–1689. doi: 10.1002/ajmg.a.35371 [DOI] [PubMed] [Google Scholar]
- Huang N, Lee I, Marcotte EM, & Hurles ME (2010). Characterising and predicting haploinsufficiency in the human genome. PLoS Genet, 6(10), e1001154. doi: 10.1371/journal.pgen.1001154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, … Sieh W (2016). REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am J Hum Genet, 99(4), 877–885. doi: 10.1016/j.ajhg.2016.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal S, Jespersen JB, Perez-Palma E, May P, Hoksza D, Heyne HO, … Lal (2019). Insights into protein structural, physicochemical, and functional consequences of missense variants in 1,330 disease-associated human genes. bioRxiv, 693259. doi: 10.1101/693259 [DOI] [Google Scholar]
- Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, Darbandi SF, Knowles D, Li YI, … Farh KK (2019). Predicting Splicing from Primary Sequence with Deep Learning. Cell, 176(3), 535–548 e524. doi: 10.1016/j.cell.2018.12.015 [DOI] [PubMed] [Google Scholar]
- Jarvik GP, & Browning BL (2016). Consideration of Cosegregation in the Pathogenicity Classification of Genomic Variants. Am J Hum Genet, 98(6), 1077–1081. doi: 10.1016/j.ajhg.2016.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jian X, Boerwinkle E, & Liu X (2014). In silico prediction of splice-altering single nucleotide variants in the human genome. Nucleic Acids Res, 42(22), 13534–13544. doi: 10.1093/nar/gku1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones GE, Ostergaard P, Moore AT, Connell FC, Williams D, Quarrell O, … Mansour S (2014). Microcephaly with or without chorioretinopathy, lymphoedema, or mental retardation (MCLMR): review of phenotype associated with KIF11 mutations. Eur J Hum Genet, 22(7), 881–887. doi: 10.1038/ejhg.2013.263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanavy DM, McNulty SM, Jairath MK, Brnich SE, Bizon C, Powell BC, & Berg JS (2019). Comparative analysis of functional assay evidence use by ClinGen Variant Curation Expert Panels. Genome Med, 11(1), 77. doi: 10.1186/s13073-019-0683-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly MA, Caleshu C, Morales A, Buchan J, Wolf Z, Harrison SM, … Funke B (2018). Adaptation and validation of the ACMG/AMP variant classification framework for MYH7-associated inherited cardiomyopathies: recommendations by ClinGen’s Inherited Cardiomyopathy Expert Panel. Genet Med, 20(3), 351–359. doi: 10.1038/gim.2017.218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopanos C, Tsiolkas V, Kouris A, Chapple CE, Albarca Aguilera M, Meyer R, & Massouras A (2019). VarSome: the human genomic variant search engine. Bioinformatics, 35(11), 1978–1980. doi: 10.1093/bioinformatics/bty897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Krempely K, Roberts ME, Anderson MJ, Carneiro F, Chao E, … Karam R (2018). Specifications of the ACMG/AMP variant curation guidelines for the analysis of germline CDH1 sequence variants. Hum Mutat, 39(11), 1553–1568. doi: 10.1002/humu.23650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, … Exome Aggregation, C. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature, 536(7616), 285–291. doi: 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Shi L, Zhang K, Zhang Y, Hu S, Zhao T, … Sun, Z. (2018). VarCards: an integrated genetic and clinical database for coding variants in the human genome. Nucleic Acids Res, 46(D1), D1039–D1048. doi: 10.1093/nar/gkx1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, & Wang K (2017). InterVar: Clinical Interpretation of Genetic Variants by the 2015 ACMG-AMP Guidelines. Am J Hum Genet, 100(2), 267–280. doi: 10.1016/j.ajhg.2017.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y-H, Lu Y-C, Chen T-F, Hsu JS, Lee K-H, Cheng Y-W, … Chen C-Y (2019). variant2literature: full text literature search for genetic variants. bioRxiv, 583450. doi: 10.1101/583450 [DOI] [Google Scholar]
- Lionel AC, Costain G, Monfared N, Walker S, Reuter MS, Hosseini SM, … Marshall CR (2018). Improved diagnostic yield compared with targeted gene sequencing panels suggests a role for whole-genome sequencing as a first-tier genetic test. Genet Med, 20(4), 435–443. doi: 10.1038/gim.2017.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Huang Q, Li Q, Zhao D, Li X, Cui L, … Kong X (2017). Spectrum of PAH gene variants among a population of Han Chinese patients with phenylketonuria from northern China. BMC Med Genet, 18(1), 108. doi: 10.1186/s12881-017-0467-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, Feurstein S, Mohan S, Porter CC, Jackson SA, Keel S, … Godley LA (2019). ClinGen Myeloid Malignancy Variant Curation Expert Panel recommendations for germline RUNX1 variants. Blood Adv, 3(20), 2962–2979. doi: 10.1182/bloodadvances.2019000644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mersch J, Brown N, Pirzadeh-Miller S, Mundt E, Cox HC, Brown K, … Ross T (2018). Prevalence of Variant Reclassification Following Hereditary Cancer Genetic Testing. JAMA, 320(12), 1266–1274. doi: 10.1001/jama.2018.13152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mester JL, Ghosh R, Pesaran T, Huether R, Karam R, Hruska KS, … Eng (2018). Gene-specific criteria for PTEN variant curation: Recommendations from the ClinGen PTEN Expert Panel. Hum Mutat, 39(11), 1581–1592. doi: 10.1002/humu.23636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mungall CJ, McMurry JA, Kohler S, Balhoff JP, Borromeo C, Brush M, … Haendel MA (2017). The Monarch Initiative: an integrative data and analytic platform connecting phenotypes to genotypes across species. Nucleic Acids Res, 45(D1), D712–D722. doi: 10.1093/nar/gkw1128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oza AM, DiStefano MT, Hemphill SE, Cushman BJ, Grant AR, Siegert RK, … ClinGen Hearing Loss Clinical Domain Working, G. (2018). Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum Mutat, 39(11), 1593–1613. doi: 10.1002/humu.23630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepin MG, Murray ML, Bailey S, Leistritz-Kessler D, Schwarze U, & Byers PH (2016). The challenge of comprehensive and consistent sequence variant interpretation between clinical laboratories. Genet Med, 18(1), 20–24. doi: 10.1038/gim.2015.31 [DOI] [PubMed] [Google Scholar]
- Perez-Palma E, Gramm M, Nurnberg P, May P, & Lal D (2019). Simple ClinVar: an interactive web server to explore and retrieve gene and disease variants aggregated in ClinVar database. Nucleic Acids Res, 47(W1), W99–W105. doi: 10.1093/nar/gkz411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Palma E, May P, Iqbal S, Niestroj L-M, Du J, Heyne H, … Lal D (2019). Identification of pathogenic variant enriched regions across genes and gene families. bioRxiv, 641043. doi: 10.1101/641043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Palma E, May P, Iqbal S, Niestroj LM, Du J, Heyne HO, … Lal D. (2019). Identification of pathogenic variant enriched regions across genes and gene families. Genome Res, 30(1), 62–71. doi: 10.1101/gr.252601.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese MG, Eeckman FH, Kulp D, & Haussler D (1997). Improved splice site detection in Genie. J Comput Biol, 4(3), 311–323. doi: 10.1089/cmb.1997.4.311 [DOI] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, … Committee, A. L. Q. A. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med, 17(5), 405–424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggs ER, Andersen EF, Cherry AM, Kantarci S, Kearney H, Patel A, … Martin CL (2019). Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). GENETICS in MEDICINE. doi: 10.1038/s41436-019-0686-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera-Munoz EA, Milko LV, Harrison SM, Azzariti DR, Kurtz CL, Lee K, … Berg JS (2018). ClinGen Variant Curation Expert Panel experiences and standardized processes for disease and gene-level specification of the ACMG/AMP guidelines for sequence variant interpretation. Hum Mutat, 39(11), 1614–1622. doi: 10.1002/humu.23645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robitaille JM, Gillett RM, LeBlanc MA, Gaston D, Nightingale M, Mackley MP, … Bedard K (2014). Phenotypic overlap between familial exudative vitreoretinopathy and microcephaly, lymphedema, and chorioretinal dysplasia caused by KIF11 mutations. JAMA Ophthalmol, 132(12), 1393–1399. doi: 10.1001/jamaophthalmol.2014.2814 [DOI] [PubMed] [Google Scholar]
- Samocha KE, Kosmicki JA, Karczewski KJ, O’Donnell-Luria AH, Pierce-Hoffman E, MacArthur DG, … Daly MJ (2017). Regional missense constraint improves variant deleteriousness prediction. bioRxiv, 148353. doi: 10.1101/148353 [DOI] [Google Scholar]
- Schwarz JM, Cooper DN, Schuelke M, & Seelow D (2014). MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods, 11(4), 361–362. doi: 10.1038/nmeth.2890 [DOI] [PubMed] [Google Scholar]
- Seva J, Wiegandt DL, Gotze J, Lamping M, Rieke D, Schafer R, … Leser U (2019). VIST - a Variant-Information Search Tool for precision oncology. BMC Bioinformatics, 20(1), 429. doi: 10.1186/s12859-019-2958-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Oza AM, Del Castillo I, Duzkale H, Matsunaga T, Pandya A, … ClinGen Hearing Loss Working, G. (2019). Consensus interpretation of the p.Met34Thr and p.Val37Ile variants in GJB2 by the ClinGen Hearing Loss Expert Panel. Genet Med. doi: 10.1038/s41436-019-0535-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavin TP, Manjarrez S, Pritchard CC, Gray S, & Weitzel JN (2019). The effects of genomic germline variant reclassification on clinical cancer care. Oncotarget, 10(4), 417–423. doi: 10.18632/oncotarget.26501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- So MK, Jeong TD, Lim W, Moon BI, Paik NS, Kim SC, & Huh J (2019). Reinterpretation of BRCA1 and BRCA2 variants of uncertain significance in patients with hereditary breast/ovarian cancer using the ACMG/AMP 2015 guidelines. Breast Cancer, 26(4), 510–519. doi: 10.1007/s12282-019-00951-w [DOI] [PubMed] [Google Scholar]
- Song C, Duzkale H, & Shen J (2018). Reporting of Clinical Genome Sequencing Results. Curr Protoc Hum Genet, e61. doi: 10.1002/cphg.61 [DOI] [PubMed] [Google Scholar]
- SoRelle JA, Thodeson DM, Arnold S, Gotway G, & Park JY (2019). Clinical utility of reinterpreting previously reported genomic epilepsy test results for pediatric patients. JAMA Pediatr, 173(1), e182302. doi: 10.1001/jamapediatrics.2018.2302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson PD, Ball EV, Mort M, Phillips AD, Shaw K, & Cooper DN (2012). The Human Gene Mutation Database (HGMD) and its exploitation in the fields of personalized genomics and molecular evolution. Curr Protoc Bioinformatics, Chapter 1, Unit1 13. doi: 10.1002/0471250953.bi0113s39 [DOI] [PubMed] [Google Scholar]
- Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S, … Cooper DN (2017). The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet, 136(6), 665–677. doi: 10.1007/s00439-017-1779-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strande NT, Riggs ER, Buchanan AH, Ceyhan-Birsoy O, DiStefano M, Dwight SS, … Berg JS (2017). Evaluating the clinical validity of gene-disease associations: An evidence-based framework developed by the clinical genome resource. Am J Hum Genet, 100(6), 895–906. doi: 10.1016/j.ajhg.2017.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarailo-Graovac M, & Chen N (2009). Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics, Chapter 4, Unit 4 10. doi: 10.1002/0471250953.bi0410s25 [DOI] [PubMed] [Google Scholar]
- Tavtigian SV, Greenblatt MS, Harrison SM, Nussbaum RL, Prabhu SA, Boucher KM, & Biesecker LG (2018). Modeling the ACMG/AMP variant classification guidelines as a Bayesian classification framework. Genet Med. doi: 10.1038/gim.2017.210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner SA, Rao SK, Morgan RH, Vnencak-Jones CL, & Wiesner GL (2019). The impact of variant classification on the clinical management of hereditary cancer syndromes. Genet Med, 21(2), 426–430. doi: 10.1038/s41436-018-0063-z [DOI] [PubMed] [Google Scholar]
- Whiffin N, Minikel E, Walsh R, O’Donnell-Luria AH, Karczewski K, Ing AY, … Ware JS (2017). Using high-resolution variant frequencies to empower clinical genome interpretation. Genet Med, 19(10), 1151–1158. doi: 10.1038/gim.2017.26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildeman M, van Ophuizen E, den Dunnen JT, & Taschner PE (2008). Improving sequence variant descriptions in mutation databases and literature using the Mutalyzer sequence variation nomenclature checker. Hum Mutat, 29(1), 6–13. doi: 10.1002/humu.20654 [DOI] [PubMed] [Google Scholar]
- Wong EK, Bartels K, Hathaway J, Burns C, Yeates L, Semsarian C, … Ingles J (2019). Perceptions of genetic variant reclassification in patients with inherited cardiac disease. Eur J Hum Genet, 27(7), 1134–1142. doi: 10.1038/s41431-019-0377-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang J, Peng J, & Peng Z (2019). AutoPVS1: An automatic classification tool for PVS1 interpretation of null variants. bioRxiv, 720839. doi: 10.1101/720839 [DOI] [PubMed] [Google Scholar]
- Yang H, & Wang K (2015). Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat Protoc, 10(10), 1556–1566. doi: 10.1038/nprot.2015.105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, … Eng CM (2013). Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med, 369(16), 1502–1511. doi: 10.1056/NEJMoa1306555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo G, & Burge CB (2004). Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol, 11(2–3), 377–394. doi: 10.1089/1066527041410418 [DOI] [PubMed] [Google Scholar]
- Zastrow DB, Baudet H, Shen W, Thomas A, Si Y, Weaver MA, … ClinGen Inborn Errors of Metabolism Working, G. (2018). Unique aspects of sequence variant interpretation for inborn errors of metabolism (IEM): The ClinGen IEM Working Group and the Phenylalanine Hydroxylase Gene. Hum Mutat, 39(11), 1569–1580. doi: 10.1002/humu.23649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JY, Chen XD, Zhang Z, Wang HL, Guo L, Liu Y, … Shao FM (2013). The adenosine deaminase acting on RNA 1 p150 isoform is involved in the pathogenesis of dyschromatosis symmetrica hereditaria. Br J Dermatol, 169(3), 637–644. doi: 10.1111/bjd.12401 [DOI] [PubMed] [Google Scholar]
- Zhang X, Xie J, Zhu S, Chen Y, Wang L, & Xu B (2017). Next-generation sequencing identifies pathogenic and modifier mutations in a consanguineous Chinese family with hypertrophic cardiomyopathy. Medicine (Baltimore), 96(24), e7010. doi: 10.1097/MD.0000000000007010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Feng Y, Ding X, Dong S, Zhang H, Ding J, & Xia X (2017). Identification of a novel hypertrophic cardiomyopathy-associated mutation using targeted next-generation sequencing. Int J Mol Med, 40(1), 121–129. doi: 10.3892/ijmm.2017.2986 [DOI] [PMC free article] [PubMed] [Google Scholar]