

Abstract
Metastasis, a process that requires tumor cell dissemination followed by tumor growth, is the primary cause of death in cancer patients. An essential step of tumor cell dissemination is intravasation, a process by which tumor cells cross the blood vessel endothelium and disseminate to distant sites. Studying this process is of utmost importance given that intravasation in the primary tumor, as well as the secondary and tertiary metastases, is the key step in the systemic spread of tumor cells, and that this process continues even after removal of the primary tumor. High-resolution intravital imaging of the tumor microenvironment of breast carcinoma has revealed that tumor cell intravasation exclusively occurs at doorways, termed “Tumor MicroEnvironment of Metastasis” (TMEM), composed of three different cell types: a Tie2high/VEGFhigh perivascular macrophage, a Mena overexpressing tumor cell, and an endothelial cell, all in direct contact. In this review article, we discuss the interactions between these cell types, the subsequent signaling events which lead to tumor cell intravasation, and the role of invadopodia in supporting tumor cell invasion and dissemination. We end our review by discussing how the knowledge acquired from the use of intravital imaging is now leading to new clinical trials targeting tumor cell dissemination and preventing metastatic progression.
Keywords: Metastasis, Intravasation, Dissemination, Tumor Microenvironment, MenaINV, Invadopodia, TMEM Doorways, Intravital Imaging
Introduction
Despite significant advances in understanding tumor progression, metastatic disease remains the major cause of death in cancer patients. The traditional view of metastatic progression, framed by the Paget’s 126-year-old “seed-and-soil” hypothesis (1), is that circulating tumor cells (CTCs, the “seed”) detach from the primary tumor, enter into the circulation, and disseminate from the primary tumor to secondary sites (the “soil”) that are favorable for colonization: supporting extravasation, survival, and growth of secondary tumors. However, the recent development of new scientific methods and technologies, including intravital imaging, has challenged the implicit view that metastasis is a uni-directional process. For instance, it has been demonstrated that CTCs can disseminate in multiple directions, seeding not only to secondary sites, but also re-seeding primary site and seeding onward from the secondary to tertiary sites: a new metastatic model defined as “metastasis from metastases” (2–9).
However, it is becoming clear that tumor cells do not act alone in the process of dissemination, seeding, and metastasis, but instead interact with the extracellular matrix and the surrounding normal cells that constitute the tumor microenvironment (TME) (10–13). The TME includes several components. One such component is the set of ECM proteins (such as collagen, laminin, fibronectin, glycoproteins, and proteoglycans) that function as physical scaffolds for tissues and as sources of biochemical and biomechanical cues (14–17). Another component consists of secreted soluble factors and micro-vesicles that mediate bi-directional communication between tumor and host cells. These factors not only facilitate the growth and survival of the tumor cells, but also serve as chemoattractants by enhancing the recruitment and migration of other cells into the TME (18–20). Yet another component consists of host cells that are present in the tumor such as fibroblasts, myofibroblasts, endothelial cells, pericytes, adipocytes, macrophages, myeloid cells, etc. (21–28). All these cells work in concert to maintain tissue homeostasis. However, as the disease progresses, the tumor can recruit a variety of stromal cells, such as endothelial cells, pericytes, adipocytes, fibroblasts, and mesenchymal stromal cells, as well as immune cells, such as monocytes, NK, leukocytes, from the surrounding microenvironment, and from the bone marrow, to promote tumor progression and accelerate metastasis.
The interactions between tumor cells and the TME, and their role in promoting tumor progression and metastasis, have been described extensively (29,30). In this regard, the contribution of tumor-associated macrophages (a major component of the TME) in regulating immune response, angiogenesis, and tumor growth has been well recognized (31–37). However, less focus has been placed on the role of macrophages in tumor cell intravasation and dissemination, two crucial steps in the metastatic cascade (38). Part of the reason for this is the lack of tools which can allow the direct in vivo observation of, at single cell resolution, the interaction between macrophages and tumor cells that leads to tumor cell intravasation at the primary site, or extravasation at secondary sites. Recently, high-resolution intravital imaging of the murine breast TME has revealed dynamic heterotypic interactions (between tumor cells, macrophages, and other stromal cells) that mediate tumor cell intravasation and dissemination (39–42). These heterotypic interactions include: (i) the paracrine interaction between macrophages and tumor cells that is responsible for migration of tumor cells on collagen fibers towards the blood vasculature, a process known as tumor cell “streaming” migration (43–47); and (ii) the direct physical interaction between macrophages, tumor cells and endothelial cells, which is responsible for the assembly of the “Tumor MicroEnvironment of Metastasis” (TMEM) doorway, a stable portal into the vasculature that allows tumor cells to intravasate and disseminate to secondary, and possibly tertiary, sites (9,39,42,48–53). In this review, we will focus on describing the signaling and molecular mechanisms of TMEM doorways leading to tumor cell intravasation, and the role of invadopoda in orchestrating TMEM doorway assembly and tumor cell dissemination. Finally, this review will conclude with a brief description of a new therapeutic strategy aimed at preventing metastasis by directly targeting intravasation and dissemination.
MenaINV promotes Tumor Cell “Streaming” Migration with Macrophages toward Blood Vessels
To intravasate, tumor cells must detach from the primary tumor, migrate, cross the endothelium, and enter the circulation (54–56). A key event in promoting the migration of tumor cells and invasion of the vasculature is the epithelial-mesenchymal transition (EMT) program during which tumor cells down-regulate the epithelial cadherin proteins (E- or P-cadherin) that are required for maintenance of the integrity of the epithelium, and up-regulate mesenchymal cadherin proteins (N-cadherin) (57–60). These events lead to a subsequent disassembly of cell-cell adhesion junctions and the acquisition of a migratory and invasive phenotype through the reorganization of the actin cytoskeleton (59,61).
During EMT, Mammalian enabled (Mena), a member of the Enabled (Ena)/Vasodilator-Stimulated Phosphoprotein (VASP) family which is involved in actin dynamics, cell adhesion, and motility (62–65), is alternatively spliced giving rise to different isoforms of Mena (66–69). Mena contains two highly conserved regions, an N-terminal EnaA/ASP Homology-1 (EVH-1) domain, and a C-terminal Ena/VASP Homology-2 (EVH-2) domain (67). The EVH-1 domain mediates protein-protein interactions important for Ena/VASP localization and regulation (62,63), while the EVH-2 domain mediates interactions with the G- and F-actin (70,71). As tumor cells undergo EMT, Mena11a, an isoform containing an additional 21 amino acids in the EVH-2 domain and which confers an epithelial phenotype, is down-regulated (Mena11alow) resulting in a loss of epithelial cohesion (72–74). Next, the direct contact between Mena11alow tumor cells and macrophages in the microenvironment of the primary tumor induces in the tumor cells an increase in overall Mena expression (panMena), including the isoforms Menaclassic and MenaINV (MenaINVhigh) which contains an additional 19 amino acids inserted in the EVH-1 domain (66,69). The Mena expression pattern Mena11alow and panMenahigh, also known as MenaCalc, has been shown to be prognostic for the development of metastatic diseases in some subsets of breast cancer patients (75,76). Overexpression of the MenaINVhigh isoform confers to tumor cells a 50 fold increased sensitivity to gradients of Epidermal Growth Factor (EGF) and Hepatocyte Growth Factor (HGF) (77) secreted by the surrounding cells. This is accomplished by sequestering Protein Tyrosine Phosphatase 1B (PTB1B) away from the receptor tyrosine kinases (77–79) and results in a highly migratory and invasive tumor cell phenotype (73,80). The enhanced sensitivity to chemotactic growth factors (by 25-50 fold) enables MenaINVhigh tumor cells to increase cell protrusion and migrate along with macrophages on collagen fibers (blue line in the Fig 1A, left panel) toward an HGF-secreting endothelial cell generated gradient, a process known as multicellular “streaming” migration (Fig. 1A and 1B, left panels) (42–47). The multicellular streaming migration consists of a co-migration of MenaINVhigh tumor cells and macrophages, without cell-cell contacts, on collagen fibers toward the blood vasculature. At the molecular level, this co-migration is coordinated by an EGF - Colony Stimulating Factor 1 (CSF1) paracrine loop (81,82). In this loop, tumor cells secrete CSF1, which attracts CSF1 receptor (CSF1R)-expressing macrophages toward them and stimulates the macrophages to secrete EGF (82). In turn, the EGF increases the migration of EGF receptor (EGFR)-expressing tumor cells, which respond by migrating towards the macrophages and secreting more CSF1 (77,81–85). These events lead to a chemotactic axis in which the EGF/CSF1 signals are amplified from one cell to the next and keep macrophages and MenaINVhigh tumor cells in close proximity. However, while EGF/CSF1 signals between macrophages and MenaINVhigh tumor cells mediates pairing and streaming, HGF/C-Met signaling attracts tumor cells towards blood vessels (86) and TMEM associated blood vessels in particular (39,42). MenaINV expression drastically increases the sensitivity of the c-Met receptor in tumor cells to its HGF ligand, leading to a directional migration of tumor cells along with macrophages towards blood vessels (86) where they intravasate at TMEM doorways (42,51,52). The essential role of the paracrine EGF/CSF1 loop and the HGF/C-Met signaling were confirmed by the fact that a genetic or pharmacological ablation of either EGF or CSF1, or pharmacological inhibition of the c-Met receptor significantly impairs migration of both cell types and blocks tumor cell streaming and metastasis in in vivo models (47,81,82,84,86). Furthermore, macrophage depletion by genetic ablation in transgenic mice (Csf1op/Csf1op) (39,81), or by systemic treatment with clodronate liposomes (39,43), leads to a significant decrease of streaming motility.
Figure 1. The structure and function of TMEM Doorway.
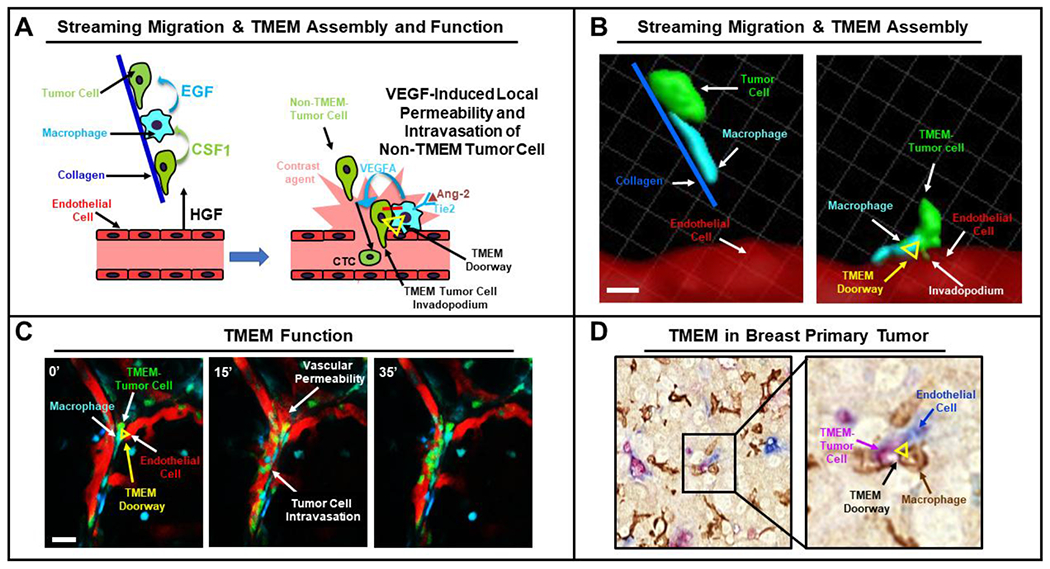
A. Left. Streaming migration. Mena expressing tumor cells (green) and macrophages (cyan) pair utilizing a CSF1/ EGF paracrine loop and migrate uni-directionally along collagen 1 fibers (blue line) towards endothelial cells that attract tumor cells by secreting HGF. Right. TMEM Assembly & Function. A Tie2high/VEGFhigh perivascular macrophage interacts via Jagged with the Notch receptor on the tumor cell (red line) thereby leading to invadopodium formation that degrades the basement membrane surrounding the endothelial cells. Secretion of VEGF by the macrophage destabilizes the vascular junction, induces transient vascular permeability (shown as extravasation of contrast agent from the vasculature, pink) and enables the trans-endothelial migration of tumor cells from the neighboring stream. B. Left. Three-dimensional reconstruction of time-lapse intravital imaging data showing a tumor cell (green) and a macrophage (cyan) streaming along collagen 1 fibers (blue line) toward the blood vessel (red). Right. A later time point showing the three cell types stably bound together forming a TMEM doorway (vertices of yellow triangle indicate positions of the three cell types). The TMEM-tumor cell’s invadopodium is inserted between two endothelial cells of the blood vessel to create a site of vascular weakness and intravasation. Scale bar, 50 μm. C. Stills from a time lapse movie showing TMEM activity and tumor cell intravasation. At time t=0’ a TMEM doorway can be observed (yellow triangle). At time t=15’, leakage of the red blood serum marker (155 kDa dextran-TMR) and intravasation of green tumor cells can be observed. Finally, at t=35’ leakage from the vasculature ceases, indicating that the vascular permeability is transient and regulated. Scale bar, 50 μm. D. Immunostaining of a formaline fixed, paraffin embedded section of murine primary tumor for TMEM doorways. The black box, in the left panel, indicates one TMEM doorway, which is magnified in the right panel to show the three constituent cells of TMEM. A Mena over-expressing tumor cell (pink), a macrophage (brown) and a blood vessel endothelial cell (Blue) in direct contact (yellow triangle) form the TMEM doorway.
MenaINV is important for Invadopodium Formation.
Once tumor cells reach blood vessels by streaming migration with macrophages, they must enter the blood space to travel to distant organs. This process, called “transendothelial migration”, requires tumor cells to penetrate the basement membrane and the endothelial barrier surrounding blood vessels. Invadopodia are F-actin-rich specialized protrusions that degrade extracellular matrix and are required for transendothelial migration of tumor cells through TMEM doorways during tumor cell intravasation (87,88). MenaINVhigh expression is critical for the formation of mature invadopodia during transendothelial migration (50,88,89). Mechanistically, invadopodium formation requires the recruitment of cofilin, neural Wiskott-Aldrich syndrome protein (N-WASP), and the Actin-related protein 2/3 (ARP2/3) complex (90–93). Various extracellular stimuli including EGF, cofilin, and N-WASP induce the rapid actin polymerization required for membrane protrusion by activating the actin nucleation activity of the Arp2/3 complex (88,94,95). Both cofilin and N-WASP have been shown to initially activate Arp2/3 in the invadopodium precursor core (88,96). Finally, invadopodium maturation is associated with an actin polymerization causing protrusion elongation, matrix metalloproteinase (MMP) recruitment, and ECM degradation (88,97). The MenaINVhigh expression stimulates invadopodial assembly and function in tumor cells by enhancing the phosphorylation of cortactin at tyrosine 421, a well-known regulatory step in tumor cell invasion (98). Specifically, MenaINVhigh expression inhibits dephosphorylation of cortactin by displacing PTP1B from the invadopodia (98). In preventing the localization of PTP1B to invadopodia, MenaINVhigh sensitizes tumor cells to a range of extracellular stimuli such the growth factors EGF and HGF (77) as well as ECM signals that promote invadopodial maturation (88). The role of Mena in tumor cell intravasation was demonstrated by genetic ablation of the Mena gene in a mouse model of spontaneous breast carcinoma which resulted in dramatically reduced CTCs and overall metastatic burden (99).
Invadopodia are important for TMEM Doorway Function.
Intravital microscopy studies have demonstrated that invadopodia are also important for transendothelial migration and vascular invasion (42,50,88,89), showing that TMEM-tumor cells use invadopodia to keep the doorways open, resulting in tumor cell intravasation of non-TMEM tumor cells (42,50,89). Juxtacrine interactions between macrophages and tumor cells in and around TMEM doorways result in Notch-Jagged signaling (red line in Fig. 1A, right panel) (89) and MenaINV-dependent invadopodium formation in the TMEM-tumor cell (Fig. 1B, right panel) (73,87,89). Indeed, pharmacological inhibition of the Notch pathway, or suppression of direct cell-cell contact, significantly reduced MenaINV expression in tumor cells (50,89).
TMEM Doorways in the Primary Tumor Are Sites of Tumor Cell Intravasation and Dissemination to Secondary Sites.
Direct imaging of intravasation sites in breast tumors has demonstrated that tumor cell intravasation does not occur in the stroma surrounding the tumors, nor randomly along the tumor associated vasculature, but is instead restricted to within the tumor nests (100) and occurs exclusively at TMEM doorways (42,50,53). Given the location of these doorways, and to distinguish them from TMEM doorways located in secondary sites, we have in this review termed TMEM in the primary tumor as P-TMEM doorways, and those at secondary sites (such as the lung and lymph nodes) as S-TMEM doorways.
The P-TMEM perivascular macrophages are Tie2high (42), originate from a committed bone marrow-derived monocyte lineage, and are capable of transdifferentiating into M2-like, noninflammatory macrophages (101–103). Macrophage depletion strategies have recently provided insight in the kinetics of macrophage recruitment and homing into the perivascular niche, and at TMEM in particular (104). Specifically, it was shown that peripheral monocytes expressing the chemotactic receptor CCR2 are recruited in the tumor microenvironment through increased intratumoral expression of CCL2 (104,105). Once CCR2+ monocytes infiltrate the tumor parenchyma, they transdifferentiate into macrophages, migrate towards hypoxic areas away from blood vessels, and start expressing the chemokine receptor CXCR4 (104), possibly under the control of TGF-beta (104,106). This newly-generated CXCR4+ macrophage subpopulation becomes responsive to CXCL12 chemotactic gradients (generated by the blood vascular endothelium and perivascular fibroblasts (104,107,108) and migrates towards the perivascular niche where it subsequently assembles P-TMEM doorways with a Mena expressing tumor cell and an endothelial cell (104).
It has recently been demonstrated that the opening of P-TMEM doorways depends on the release of VEGF from the Tie2high perivascular macrophage, which in turn causes a local disruption of the underlying endothelial-specific junction proteins Zonula Occludens-1 (ZO-1) and Vascular-Endothelial Cadherin (VE-CAD) (42). This disruption results in a transient opening of the endothelial wall during which MenaINVhigh migrating tumor cells are able to intravasate, enter into the blood circulation, and disseminate to secondary sites (Fig. 1A, right panel) (42). During transendothelial migration of tumor cells, the P-TMEM-associated tumor cell and the P-TMEM-associated macrophage neither migrate nor intravasate, but rather remain affixed to the endothelium and define a site of vascular weakness exploited by other dissemination competent tumor cells (42,104). The opening of a P-TMEM doorway can be directly visualized (in real time, using multiphoton microscopy in live tissue) as the localized transient leakage into the interstitial space of an intravascularly injected high-molecular weight fluorescently labeled dextran dye that is otherwise incapable of perfusing from normal or even angiogenic vessels (Fig. 1C, middle panel) (42,53,109,110). The critical role of Tie2high/VEGFhigh perivascular macrophages in the activity of P-TMEM doorways was demonstrated by the fact that conditional ablation of either macrophages themselves in PyMT transgenic mice, or VEGF specifically within the monocyte/macrophage lineage in FVB mice, dramatically inhibits P-TMEM function, and consequently, tumor cell intravasation and CTCs (42,81).
Importantly, the P-TMEM doorways have also been identified in primary tumors resected from human breast cancer patients using a triple immunostain identifying Mena-overexpressing tumor cells, macrophages, and endothelial cells, all in direct contact (Fig. 1D). Scoring of P-TMEM counts within these paraffin-embedded samples has been shown to be prognostic for the development of distant metastasis in patients (49,51,52,111) and clinical validation of P-TMEM doorways as a prognostic maker is discussed in the “P-TMEM Doorways in Human Breast Carcinoma: A Prognostic Marker for Risk of Distant Recurrence” section below.
TMEM Doorways at Secondary Sites Are Sources of Tumor Cell Re-Intravasation and Re-Dissemination to Tertiary Sites.
The dynamic interaction between macrophages, tumor cells, and endothelial cells has also been reported in the microenvironment of the metastatic (secondary) site, defined here as S-TMEM doorways. TMEM doorways have been identified in samples of metastatic sites (the lung) taken from mouse models of breast cancer as well as human patients (112). In addition, TMEM doorways have been identified in association with angiogenic blood vessels within metastatic lymph node tumor cell nests (113).
High-resolution intravital imaging of lung metastasis in live mice has revealed that S-TMEM doorways are indeed active. The activity of S-TMEM doorways has been demonstrated by the visualization, in live tissue, of the transient permeability and leakage of vascular contents into the interstitium, identical to that observed in P-TMEM (112). The presence of functional S-TMEM doorways in lung metastases indicates that dissemination of tumor cells may also occur from secondary sites, allowing tumor cells to re-intravasate, enter the blood circulation, seed back to the primary tumor, and/or re-disseminate to tertiary sites.
Together, the findings in lung and lymph nodes indicate that TMEM doorways are a universal mechanism for tumor cell dissemination that is not restricted solely to the primary tumor microenvironment, but additionally implicated in tumor cell re-dissemination from metastatic sites. The clinical observation that CTCs can be found in the blood of patients years or decades after the removal of their primary tumor (114,115) may thus be a result of S-TMEM doorways allowing metastatic tumor cells to continually disseminate into the blood circulation, supporting a new model of metastasis where tumor cells disseminate not only from primary to secondary sites, but, but rather multi-directionally.
P-TMEM Doorways in Human Breast Carcinoma: A Prognostic Invadopodium Associated Marker of Risk of Recurrence
As previously mentioned, P-TMEM doorways have been identified not only in mouse, but also in human breast carcinoma, and many studies have been conducted to assess the prognostic ability of TMEM for risk of recurrence (49,51,52,111). In a first-retrospective study, a case-control analysis was performed on 30 patients who developed distant metastasis and 30 matched patients who did not (49). P-TMEM density was not correlated with tumor size or grade, lymph node metastasis, lymphovascular invasion, nor hormone receptor status. However, P-TMEM density was significantly greater in the group of patients who developed distant metastases compared to the patients who did not In a second-retrospective study, a case-control analysis was performed in a cohort of 250 matched pairs (51). Case patients were women who subsequently developed metastasis and were matched (1:1) with control subjects who did not developed distant recurrence. TMEM density within the highest tertile was associated with increased risk of distant metastasis in patients with ER+ / HER2– breast cancer (51,111).
The P-TMEM score (also known as MetaSite Score) has now been clinically validated in ER+/HER2– breast cancer, making TMEM the first ever biomarker specifically for metastasis, and it gives complimentary information regarding distant recurrence in patients with low OncotypeDx score (111). Clinically, patients who have high P-TMEM score would presumably need to be monitored very closely, even after resection of the primary tumor, as they have a high risk of tumor cell dissemination to the secondary site that could subsequently re-disseminate via S-TMEM doorways to tertiary sites. These patients would thus be candidates for a combination treatment containing chemo- or other targeted therapies (to suppress the growth of tumor cells) and drugs designed to “seal” the TMEM doorways and prevent dissemination. A discussion of this therapeutic strategy to block tumor cell dissemination is presented below in the “Sealing the Doorways to Tumor Cells: Therapeutic Strategy” section.
Sealing the Doorways to Tumor Cells: Therapeutic Strategy
The identification of TMEM doorways both in the primary tumor and at secondary sites clearly indicates that TMEM is a common mechanism of tumor cell dissemination from all metastatic sites. Therefore, a pharmacological inhibition capable of “sealing” the TMEM doorways and blocking dissemination is a promising therapeutic strategy to eliminate CTCs and prevent metastatic progression.
As previously discussed, TMEM-dependent vascular permeability is mediated by VEGF released from the TMEM-bound Tie2high/VEGFhigh macrophages. We have previously shown that macrophage specific depletion of VEGFA reduces transient vascular permeability and circulating tumor cells (42). However, while anti-angiogenic treatments targeting VEGF (such as bevacizumab and other VEGFA pathway inhibitors) have shown efficacy in decreasing tumor angiogenesis and disease burden in both the preclinical and clinical settings (116,117), it has been demonstrated that cancer patients develop therapeutic resistance (118). One of the mechanisms of tumor resistance (or recurrence after anti-angiogenic therapy) has been attributed to tumor-infiltrating myeloid cells in response to cell death and hypoxia after vascular regression (118). Therefore, Tie2 has become an attractive pharmacological target for the suppression of tumor dissemination. In fact, a small molecule inhibitor of Tie2 signaling, rebastinib, was recently shown to be useful for this purpose (53,119). Rebastinib, a selective inhibitor of the Tie2 receptor tyrosine kinase that has pico-molar affinity, inhibits kinase activity by an allosteric “switch pocket control” mechanism and prevents Tie2high macrophages from inducing TMEM-dependent vascular permeability and cancer cell dissemination (53,119). A pre-clinical study demonstrated that rebastinib blocks TMEM function in both mouse models of spontaneous breast carcinoma and pancreatic neuroendocrine carcinoma, resulting in reduced vascular permeability and tumor cell intravasation, and decreased CTCs in the blood (119). In addition, the combination of rebastinib with paclitaxel significantly increased the overall survival of paclitaxel-treated mice when compared to mice treated with chemotherapy alone, even after resection of the primary tumor. The increased survival after primary tumor resection demonstrates that rebastinib is capable of blocking tumor cell re-dissemination from metastatic sites and shows that rebastinib has clinical utility, even after surgical removal of the primary tumor. Furthermore, the observation that rebastinib specifically inhibits TMEM-dependent intravasation and dissemination has been confirmed using breast cancer patient-derived xenografts (53).
Based on these pre-clinical studies, rebastinib has been approved for several clinical trials, including a phase 1b trial in women with metastatic breast cancer (NCT02824575) where the safety profile of rebastanib in combination with anti-tubulin therapy (paclitaxel or eribulin) is being evaluated along with its efficacy on preventing tumor cell dissemination (determined by measuring the levels of CTCs). Two other multi-center phase 1b/2 clinical trials are testing the safety profiles of rebastinib in combination with carboplatin (NCT03717415) and paclitaxel (NCT03601897) in patients with locally advanced or metastatic solid tumors; specifically, breast cancer, ovarian cancer, mesothelioma, and endometrial cancer.
It is becoming increasingly clear that classic chemotherapy treatments that aim to reduce tumor growth need to be combined with treatments aimed at blocking dissemination from all metastatic sites to effectively block metastatic progression (120–122). Such a therapeutic combination is believed to be capable of efficiently eradicating minimal residual disease and prevent metastatic relapse. Indeed, early results of the clinical trials are revealing promising results with dramatic reductions in CTCs observed in many patients (123).
Conclusion
Despite the progress that has been made in cancer therapeutics, metastatic disease remains the main cause of cancer-related deaths. Metastasis, defined as the growth of secondary tumors at distant sites, is the endpoint of a dynamic and complex series of events that begins with the growth of a primary tumor, is followed by intravasation and dissemination of motile tumor cells, and ends with the growth of a secondary tumor. Most studies of metastasis to date have focused on the steps that occur after extravasation at the secondary site (i.e. growth of the secondary tumor). As such, current therapeutic strategies have been designed to target only the growth of primary and/or secondary tumors. However, tumor cell dissemination and growth are not always linked (124). For instance, clinical studies have found CTCs in the blood of patients years or decades after the removal of their primary tumor (125–128) indicating that the systematic dissemination of tumor cells from secondary metastatic sites regularly continues, even when the primary tumor has been removed. Nevertheless, intravasation and dissemination have been less studied, in part due to a lack of methods that allow the direct observation of these processes in vivo in real time.
Recently, advances in technology, including high-resolution multiphoton intravital imaging of breast tumors, have provided unparalleled insight into the mechanisms of cell migration and the interactions of tumor cells with their surrounding microenvironment during intravasation. Intravital imaging of the breast tumor microenvironment has led to the discovery of: 1) the paracrine interaction between tumor cells and macrophages; 2) the role macrophages play in assisting the migration of tumor cells towards blood vessels; 3) how the direct interaction between macrophages, tumor cells, and endothelial cells leads to the assembly of TMEM doorways; and 4) the involvement of invadopodia in TMEM doorway function as an essential step in intravasation.
Interestingly, the identification of functional TMEM doorways, not only in primary tumors but also in secondary metastatic sites, demonstrates that TMEM doorways are not restricted solely to the primary tumor microenvironment but are additionally implicated in tumor cell re-dissemination from metastatic sites. This indicates that TMEM doorways are a common mechanism of tumor cell dissemination, present at all tumor sites and during all stages of breast cancer progression, supporting the concept that it is “never too late” to block dissemination and to treat metastatic patients. Currently, TMEM score, the first biomarker of metastasis, is clinically validated as a biomarker for prognostication of metastatic risk in breast cancer patients. As we are rapidly entering the era of precision medicine for cancer treatment where biomarkers are used to determine the first-line treatment for cancer patients, it is crucial to continue to develop additional reliable metastasis specific biomarkers that can be used to determine optimal treatments for these patients.
Acknowledgments
This work was supported by grants from the NCI (CA150344, CA100324 and CA216248), the Gruss-Lipper Biophotonics Center and its Integrated Imaging Program, Montefiore’s Ruth L. Kirschstein T32 Training Grant of Surgeons for the Study of the Tumor Microenvironment (CA200561), METAvivor Early Career Investigator Award and an IRACDA fellowship where the content is solely the responsibility of the authors and does not necessarily represent the official views supporting K12GM102779.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Paget S The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev 1989;8:98–101 [PubMed] [Google Scholar]
- 2.Hoover HC Jr., Ketcham AS. Metastasis of metastases. Am J Surg 1975;130:405–11 [DOI] [PubMed] [Google Scholar]
- 3.Norton L, Massague J. Is cancer a disease of self-seeding? Nat Med 2006;12:875–8 [DOI] [PubMed] [Google Scholar]
- 4.Kim MY, Oskarsson T, Acharyya S, Nguyen DX, Zhang XH, Norton L, et al. Tumor self-seeding by circulating cancer cells. Cell 2009;139:1315–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aguirre-Ghiso JA. On the theory of tumor self-seeding: implications for metastasis progression in humans. Breast Cancer Res 2010;12:304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Comen E, Norton L, Massague J. Clinical implications of cancer self-seeding. Nat Rev Clin Oncol 2011;8:369–77 [DOI] [PubMed] [Google Scholar]
- 7.Turajlic S, Swanton C. Metastasis as an evolutionary process. Science 2016;352:169–75 [DOI] [PubMed] [Google Scholar]
- 8.de Groot AE, Roy S, Brown JS, Pienta KJ, Amend SR. Revisiting Seed and Soil: Examining the Primary Tumor and Cancer Cell Foraging in Metastasis. Mol Cancer Res 2017;15:361–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karagiannis GS, Goswami S, Jones JG, Oktay MH, Condeelis JS. Signatures of breast cancer metastasis at a glance. J Cell Sci 2016;129:1751–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DeClerck YA, Pienta KJ, Woodhouse EC, Singer DS, Mohla S. The Tumor Microenvironment at a Turning Point Knowledge Gained Over the Last Decade, and Challenges and Opportunities Ahead: A White Paper from the NCI TME Network. Cancer Res 2017;77:1051–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Borriello L, DeClerck YA. [Tumor microenvironment and therapeutic resistance process]. Med Sci (Paris) 2014;30:445–51 [DOI] [PubMed] [Google Scholar]
- 12.Borriello L, Seeger RC, Asgharzadeh S, DeClerck YA. More than the genes, the tumor microenvironment in neuroblastoma. Cancer Lett 2016;380:304–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Swartz MA, Iida N, Roberts EW, Sangaletti S, Wong MH, Yull FE, et al. Tumor microenvironment complexity: emerging roles in cancer therapy. Cancer Res 2012;72:2473–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Varani J Interaction of tumor cells with the extracellular matrix. Revis Biol Celular 1987;12:1–113 [PubMed] [Google Scholar]
- 15.Gould VE, Koukoulis GK, Virtanen I. Extracellular matrix proteins and their receptors in the normal, hyperplastic and neoplastic breast. Cell Differ Dev 1990;32:409–16 [DOI] [PubMed] [Google Scholar]
- 16.Oudin MJ, Jonas O, Kosciuk T, Broye LC, Guido BC, Wyckoff J, et al. Tumor Cell-Driven Extracellular Matrix Remodeling Drives Haptotaxis during Metastatic Progression. Cancer Discov 2016;6:516–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alowami S, Troup S, Al-Haddad S, Kirkpatrick I, Watson PH. Mammographic density is related to stroma and stromal proteoglycan expression. Breast Cancer Res 2003;5:R129–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thery C Exosomes: secreted vesicles and intercellular communications. F1000 Biol Rep 2011;3:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sung BH, Ketova T, Hoshino D, Zijlstra A, Weaver AM. Directional cell movement through tissues is controlled by exosome secretion. Nat Commun 2015;6:7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Becker A, Thakur BK, Weiss JM, Kim HS, Peinado H, Lyden D. Extracellular Vesicles in Cancer: Cell-to-Cell Mediators of Metastasis. Cancer Cell 2016;30:836–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leek RD, Harris AL. Tumor-associated macrophages in breast cancer. Journal of Mammary Gland Biology and Neoplasia 2002;7:177–89 [DOI] [PubMed] [Google Scholar]
- 22.Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 2012;21:309–22 [DOI] [PubMed] [Google Scholar]
- 23.Bussard KM, Mutkus L, Stumpf K, Gomez-Manzano C, Marini FC. Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Res 2016;18:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kalluri R The biology and function of fibroblasts in cancer. Nat Rev Cancer 2016;16:582–98 [DOI] [PubMed] [Google Scholar]
- 25.Borriello L, Nakata R, Sheard MA, Fernandez GE, Sposto R, Malvar J, et al. Cancer-Associated Fibroblasts Share Characteristics and Protumorigenic Activity with Mesenchymal Stromal Cells. Cancer Res 2017;77:5142–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hida K, Maishi N, Annan DA, Hida Y. Contribution of Tumor Endothelial Cells in Cancer Progression. Int J Mol Sci 2018;19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pietras K, Ostman A. Hallmarks of cancer: interactions with the tumor stroma. Exp Cell Res 2010;316:1324–31 [DOI] [PubMed] [Google Scholar]
- 28.Sanchez LR, Borriello L, Entenberg D, Condeelis JS, Oktay MH, Karagiannis GS. The emerging roles of macrophages in cancer metastasis and response to chemotherapy. J Leukoc Biol 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer 2009;9:239–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roussos ET, Condeelis JS, Patsialou A. Chemotaxis in cancer. Nat Rev Cancer 2011;11:573–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crowther M, Brown NJ, Bishop ET, Lewis CE. Microenvironmental influence on macrophage regulation of angiogenesis in wounds and malignant tumors. J Leukoc Biol 2001;70:478–90 [PubMed] [Google Scholar]
- 32.Lin EY, Pollard JW. Macrophages: modulators of breast cancer progression. Novartis Found Symp 2004;256:158–68; discussion 68–72, 259–69 [PubMed] [Google Scholar]
- 33.Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res 2006;66:605–12 [DOI] [PubMed] [Google Scholar]
- 34.Laoui D, Movahedi K, Van Overmeire E, Van den Bossche J, Schouppe E, Mommer C, et al. Tumor-associated macrophages in breast cancer: distinct subsets, distinct functions. Int J Dev Biol 2011;55:861–7 [DOI] [PubMed] [Google Scholar]
- 35.Ruffell B, Affara NI, Coussens LM. Differential macrophage programming in the tumor microenvironment. Trends Immunol 2012;33:119–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lewis CE, Harney AS, Pollard JW. The Multifaceted Role of Perivascular Macrophages in Tumors. Cancer Cell 2016;30:18–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rivera Sanchez L, Borriello L, Entenberg D, Condeelis JS, Oktay MH, Karagiannis GS. The Emerging Roles of Macrophages in Cancer Metastasis and Response to Chemotherapyt. Journal of Leukocyte Biology 2018;In Press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell 2006;124:263–6 [DOI] [PubMed] [Google Scholar]
- 39.Wyckoff JB, Wang Y, Lin EY, Li JF, Goswami S, Stanley ER, et al. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res 2007;67:2649–56 [DOI] [PubMed] [Google Scholar]
- 40.Wyckoff J, Gligorijevic B, Entenberg D, Segall J, Condeelis J. High-resolution multiphoton imaging of tumors in vivo. Cold Spring Harb Protoc 2011;2011:1167–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dovas A, Patsialou A, Harney A, Condeelis J, Cox D. Imaging interactions between macrophages and tumour cells that are involved in metastasis in vivo and in vitro. Journal of microscopy 2013;251:261–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harney AS, Arwert EN, Entenberg D, Wang Y, Guo P, Qian BZ, et al. Real-Time Imaging Reveals Local, Transient Vascular Permeability, and Tumor Cell Intravasation Stimulated by TIE2hi Macrophage-Derived VEGFA. Cancer Discovery 2015;5:932–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roussos ET, Balsamo M, Alford SK, Wyckoff JB, Gligorijevic B, Wang Y, et al. Mena invasive (MenaINV) promotes multicellular streaming motility and transendothelial migration in a mouse model of breast cancer. J Cell Sci 2011;124:2120–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sharma VP, Beaty BT, Patsialou A, Liu H, Clarke M, Cox D, et al. Reconstitution of in vivo macrophage-tumor cell pairing and streaming motility on one-dimensional micro-patterned substrates. Intravital 2012;1:77–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bravo-Cordero JJ, Hodgson L, Condeelis J. Directed cell invasion and migration during metastasis. Curr Opin Cell Biol 2012;24:277–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Patsialou A, Bravo-Cordero JJ, Wang Y, Entenberg D, Liu H, Clarke M, et al. Intravital multiphoton imaging reveals multicellular streaming as a crucial component of in vivo cell migration in human breast tumors. Intravital 2013;2:e25294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leung E, Xue A, Wang Y, Rougerie P, Sharma VP, Eddy R, et al. Blood vessel endothelium-directed tumor cell streaming in breast tumors requires the HGF/C-Met signaling pathway. Oncogene 2017;36:2680–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wyckoff JB, Jones JG, Condeelis JS, Segall JE. A critical step in metastasis: in vivo analysis of intravasation at the primary tumor. Cancer Res 2000;60:2504–11 [PubMed] [Google Scholar]
- 49.Robinson BD, Sica GL, Liu YF, Rohan TE, Gertler FB, Condeelis JS, et al. Tumor microenvironment of metastasis in human breast carcinoma: a potential prognostic marker linked to hematogenous dissemination. Clinical Cancer Research 2009;15:2433–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pignatelli J, Goswami S, Jones JG, Rohan TE, Pieri E, Chen X, et al. Invasive breast carcinoma cells from patients exhibit MenaINV- and macrophage-dependent transendothelial migration. Sci Signal 2014;7:ra112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rohan TE, Xue X, Lin HM, D’Alfonso TM, Ginter PS, Oktay MH, et al. Tumor microenvironment of metastasis and risk of distant metastasis of breast cancer. J Natl Cancer Inst 2014;106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Oktay MH, Jones JG. TMEM: a novel breast cancer dissemination marker for the assessment of metastatic risk. Biomark Med 2015;9:81–4 [DOI] [PubMed] [Google Scholar]
- 53.Karagiannis GS, Pastoriza JM, Wang Y, Harney AS, Entenberg D, Pignatelli J, et al. Neoadjuvant chemotherapy induces breast cancer metastasis through a TMEM-mediated mechanism. Science Translational Medicine 2017;9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gupta GP, Massague J. Cancer metastasis: Building a framework. Cell 2006;127:679–95 [DOI] [PubMed] [Google Scholar]
- 55.Nguyen DX, Bos PD, Massague J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer 2009;9:274–84 [DOI] [PubMed] [Google Scholar]
- 56.Lambert AW, Pattabiraman DR, Weinberg RA. Emerging Biological Principles of Metastasis. Cell 2017;168:670–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kalluri R EMT: when epithelial cells decide to become mesenchymal-like cells. J Clin Invest 2009;119:1417–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest 2009;119:1420–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chaffer CL, San Juan BP, Lim E, Weinberg RA. EMT, cell plasticity and metastasis. Cancer Metastasis Rev 2016;35:645–54 [DOI] [PubMed] [Google Scholar]
- 60.Aiello NM, Kang Y. Context-dependent EMT programs in cancer metastasis. J Exp Med 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yilmaz M, Christofori G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev 2009;28:15–33 [DOI] [PubMed] [Google Scholar]
- 62.Gertler FB, Niebuhr K, Reinhard M, Wehland J, Soriano P. Mena, a relative of VASP and Drosophila Enabled, is implicated in the control of microfilament dynamics. Cell 1996;87:227–39 [DOI] [PubMed] [Google Scholar]
- 63.Krause M, Dent EW, Bear JE, Loureiro JJ, Gertler FB. Ena/VASP proteins: regulators of the actin cytoskeleton and cell migration. Annu Rev Cell Dev Biol 2003;19:541–64 [DOI] [PubMed] [Google Scholar]
- 64.Barzik M, Kotova TI, Higgs HN, Hazelwood L, Hanein D, Gertler FB, et al. Ena/VASP proteins enhance actin polymerization in the presence of barbed end capping proteins. J Biol Chem 2005;280:28653–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Applewhite DA, Barzik M, Kojima S, Svitkina TM, Gertler FB, Borisy GG. Ena/VASP proteins have an anti-capping independent function in filopodia formation. Mol Biol Cell 2007;18:2579–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Goswami S, Philippar U, Sun D, Patsialou A, Avraham J, Wang W, et al. Identification of invasion specific splice variants of the cytoskeletal protein Mena present in mammary tumor cells during invasion in vivo. Clin Exp Metastasis 2009;26:153–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gertler F, Condeelis J. Metastasis: tumor cells becoming MENAcing. Trends Cell Biol 2011;21:81–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shapiro IM, Cheng AW, Flytzanis NC, Balsamo M, Condeelis JS, Oktay MH, et al. An EMT-driven alternative splicing program occurs in human breast cancer and modulates cellular phenotype. PLoS Genet 2011;7:e1002218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Di Modugno F, Iapicca P, Boudreau A, Mottolese M, Terrenato I, Perracchio L, et al. Splicing program of human MENA produces a previously undescribed isoform associated with invasive, mesenchymal-like breast tumors. Proc Natl Acad Sci U S A 2012;109:19280–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kuhnel K, Jarchau T, Wolf E, Schlichting I, Walter U, Wittinghofer A, et al. The VASP tetramerization domain is a right-handed coiled coil based on a 15-residue repeat. Proc Natl Acad Sci U S A 2004;101:17027–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Loureiro JJ, Rubinson DA, Bear JE, Baltus GA, Kwiatkowski AV, Gertler FB. Critical roles of phosphorylation and actin binding motifs, but not the central proline-rich region, for Ena/vasodilator-stimulated phosphoprotein (VASP) function during cell migration. Molecular Biology of the Cell 2002;13:2533–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pino MS, Balsamo M, Di Modugno F, Mottolese M, Alessio M, Melucci E, et al. Human Mena+11a isoform serves as a marker of epithelial phenotype and sensitivity to epidermal growth factor receptor inhibition in human pancreatic cancer cell lines. Clin Cancer Res 2008;14:4943–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Roussos ET, Goswami S, Balsamo M, Wang Y, Stobezki R, Adler E, et al. Mena invasive (Mena(INV)) and Mena11a isoforms play distinct roles in breast cancer cell cohesion and association with TMEM. Clin Exp Metastasis 2011;28:515–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Balsamo M, Mondal C, Carmona G, McClain LM, Riquelme DN, Tadros J, et al. The alternatively-included 11a sequence modifies the effects of Mena on actin cytoskeletal organization and cell behavior. Sci Rep 2016;6:35298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Agarwal S, Gertler FB, Balsamo M, Condeelis JS, Camp RL, Xue X, et al. Quantitative assessment of invasive mena isoforms (Menacalc) as an independent prognostic marker in breast cancer. Breast Cancer Res 2012;14:R124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Forse CL, Agarwal S, Pinnaduwage D, Gertler F, Condeelis JS, Lin J, et al. Menacalc, a quantitative method of metastasis assessment, as a prognostic marker for axillary node-negative breast cancer. BMC Cancer 2015;15:483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chan AY, Raft S, Bailly M, Wyckoff JB, Segall JE, Condeelis JS. EGF stimulates an increase in actin nucleation and filament number at the leading edge of the lamellipod in mammary adenocarcinoma cells. J Cell Sci 1998; 111 ( Pt 2):199–211 [DOI] [PubMed] [Google Scholar]
- 78.Philippar U, Roussos ET, Oser M, Yamaguchi H, Kim HD, Giampieri S, et al. A Mena invasion isoform potentiates EGF-induced carcinoma cell invasion and metastasis. Dev Cell 2008;15:813–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hughes SK, Oudin MJ, Tadros J, Neil J, Del Rosario A, Joughin BA, et al. PTP1B-dependent regulation of receptor tyrosine kinase signaling by the actin-binding protein Mena. Mol Biol Cell 2015;26:3867–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Oudin MJ, Hughes SK, Rohani N, Moufarrej MN, Jones JG, Condeelis JS, et al. Characterization of the expression of the pro-metastatic Mena(INV) isoform during breast tumor progression. Clin Exp Metastasis 2016;33:249–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wyckoff J, Wang W, Lin EY, Wang Y, Pixley F, Stanley ER, et al. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res 2004;64:7022–9 [DOI] [PubMed] [Google Scholar]
- 82.Goswami S, Sahai E, Wyckoff JB, Cammer M, Cox D, Pixley FJ, et al. Macrophages promote the invasion of breast carcinoma cells via a colony-stimulating factor-1/epidermal growth factor paracrine loop. Cancer Res 2005;65:5278–83 [DOI] [PubMed] [Google Scholar]
- 83.Patsialou A, Wyckoff J, Wang Y, Goswami S, Stanley ER, Condeelis JS. Invasion of human breast cancer cells in vivo requires both paracrine and autocrine loops involving the colony-stimulating factor-1 receptor. Cancer Res 2009;69:9498–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Patsialou A, Wang Y, Pignatelli J, Chen X, Entenberg D, Oktay M, et al. Autocrine CSF1R signaling mediates switching between invasion and proliferation downstream of TGFbeta in claudin-low breast tumor cells. Oncogene 2015;34:2721–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mader CC, Oser M, Magalhaes MA, Bravo-Cordero JJ, Condeelis J, Koleske AJ, et al. An EGFR-Src-Arg-cortactin pathway mediates functional maturation of invadopodia and breast cancer cell invasion. Cancer Res 2011;71:1730–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Leung E, Xue A, Wang Y, Rougerie P, Sharma VP, Eddy R, et al. Blood vessel endothelium-directed tumor cell streaming in breast tumors requires the HGF/C-Met signaling pathway. Oncogene 2016;36:2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Roh-Johnson M, Bravo-Cordero JJ, Patsialou A, Sharma VP, Guo P, Liu H, et al. Macrophage contact induces RhoA GTPase signaling to trigger tumor cell intravasation. Oncogene 2014;33:4203–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Eddy RJ, Weidmann MD, Sharma VP, Condeelis JS. Tumor Cell Invadopodia: Invasive Protrusions that Orchestrate Metastasis. Trends Cell Biol 2017;27:595–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pignatelli J, Bravo-Cordero JJ, Roh-Johnson M, Gandhi SJ, Wang Y, Chen X, et al. Macrophage-dependent tumor cell transendothelial migration is mediated by Notch1/MenaINV-initiated invadopodium formation. Sci Rep 2016;6:37874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Carlier MF, Nioche P, Broutin-L’Hermite I, Boujemaa R, Le Clainche C, Egile C, et al. GRB2 links signaling to actin assembly by enhancing interaction of neural Wiskott-Aldrich syndrome protein (N-WASp) with actin-related protein (ARP2/3) complex. J Biol Chem 2000;275:21946–52 [DOI] [PubMed] [Google Scholar]
- 91.Gligorijevic B, Wyckoff J, Yamaguchi H, Wang Y, Roussos ET, Condeelis J. N-WASP-mediated invadopodium formation is involved in intravasation and lung metastasis of mammary tumors. J Cell Sci 2012;125:724–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rohatgi R, Ma L, Miki H, Lopez M, Kirchhausen T, Takenawa T, et al. The interaction between N-WASP and the Arp2/3 complex links Cdc42-dependent signals to actin assembly. Cell 1999;97:221–31 [DOI] [PubMed] [Google Scholar]
- 93.Yamaguchi H, Lorenz M, Kempiak S, Sarmiento C, Coniglio S, Symons M, et al. Molecular mechanisms of invadopodium formation: the role of the N-WASP-Arp2/3 complex pathway and cofilin. J Cell Biol 2005;168:441–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bravo-Cordero JJ, Magalhaes MA, Eddy RJ, Hodgson L, Condeelis J. Functions of cofilin in cell locomotion and invasion. Nat Rev Mol Cell Biol 2013;14:405–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Beaty BT, Condeelis J. Digging a little deeper: the stages of invadopodium formation and maturation. Eur J Cell Biol 2014;93:438–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Helgeson LA, Nolen BJ. Mechanism of synergistic activation of Arp2/3 complex by cortactin and N-WASP. Elife 2013;2:e00884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Murphy DA, Courtneidge SA. The ‘ins’ and ‘outs’ of podosomes and invadopodia: characteristics, formation and function. Nat Rev Mol Cell Biol 2011;12:413–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Weidmann MD, Surve CR, Eddy RJ, Chen X, Gertler FB, Sharma VP, et al. MenaINV dysregulates cortactin phosphorylation to promote invadopodium maturation. Sci Rep 2016;6:36142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Roussos ET, Wang Y, Wyckoff JB, Sellers RS, Wang W, Li J, et al. Mena deficiency delays tumor progression and decreases metastasis in polyoma middle-T transgenic mouse mammary tumors. Breast Cancer Res 2010;12:R101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Deryugina EI, Kiosses WB. Intratumoral Cancer Cell Intravasation Can Occur Independent of Invasion into the Adjacent Stroma. Cell Rep 2017;19:601–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Venneri MA, De Palma M, Ponzoni M, Pucci F, Scielzo C, Zonari E, et al. Identification of proangiogenic TIE2-expressing monocytes (TEMs) in human peripheral blood and cancer. Blood 2007;109:5276–85 [DOI] [PubMed] [Google Scholar]
- 102.Pucci F, Venneri MA, Biziato D, Nonis A, Moi D, Sica A, et al. A distinguishing gene signature shared by tumor-infiltrating Tie2-expressing monocytes, blood “resident” monocytes, and embryonic macrophages suggests common functions and developmental relationships. Blood 2009;114:901–14 [DOI] [PubMed] [Google Scholar]
- 103.Forget MA, Voorhees JL, Cole SL, Dakhlallah D, Patterson IL, Gross AC, et al. Macrophage colony-stimulating factor augments Tie2-expressing monocyte differentiation, angiogenic function, and recruitment in a mouse model of breast cancer. PLoS One 2014;9:e98623. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 104.Arwert EN, Harney AS, Entenberg D, Wang Y, Sahai E, Pollard JW, et al. A Unidirectional Transition from Migratory to Perivascular Macrophage Is Required for Tumor Cell Intravasation. Cell Rep 2018;23:1239–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011;475:222–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chen S, Tuttle DL, Oshier JT, Knot HJ, Streit WJ, Goodenow MM, et al. Transforming growth factor-beta1 increases CXCR4 expression, stromal-derived factor-1alpha-stimulated signalling and human immunodeficiency virus-1 entry in human monocyte-derived macrophages. Immunology 2005;114:565–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005;121:335–48 [DOI] [PubMed] [Google Scholar]
- 108.Boimel PJ, Smirnova T, Zhou ZN, Wyckoff J, Park H, Coniglio SJ, et al. Contribution of CXCL12 secretion to invasion of breast cancer cells. Breast Cancer Res 2012;14:R23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Harney AS, Wang Y, Condeelis JS, Entenberg D. Extended Time-lapse Intravital Imaging of Real-time Multicellular Dynamics in the Tumor Microenvironment. J Vis Exp 2016:e54042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Karagiannis GS, Pastoriza JM, Borriello L, Jafari R, Coste A, Condeelis JS, et al. Assessing Tumor Microenvironment of Metastasis Doorway-Mediated Vascular Permeability Associated with Cancer Cell Dissemination using Intravital Imaging and Fixed Tissue Analysis. J Vis Exp 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sparano JA, Gray R, Oktay MH, Entenberg D, Rohan T, Xue X, et al. A metastasis biomarker (MetaSite Breast Score) is associated with distant recurrence in hormone receptor-positive, HER2-negative early-stage breast cancer. Nature PJ Breast Cancer 2017;3:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Entenberg D, Voiculescu S, Guo P, Borriello L, Wang Y, Karagiannis GS, et al. A permanent window for the murine lung enables high-resolution imaging of cancer metastasis. Nature Methods 2018;15:73–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ginter PS, Karagiannis GS, Entenberg D, Lin Y, Condeelis J, Jones JG, et al. Tumor Microenvironment of Metastasis (TMEM) Doorways Are Restricted to the Blood Vessel Endothelium in Both Primary Breast Cancers and Their Lymph Node Metastases. Cancers (Basel) 2019;11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Muller V, Stahmann N, Riethdorf S, Rau T, Zabel T, Goetz A, et al. Circulating tumor cells in breast cancer: correlation to bone marrow micrometastases, heterogeneous response to systemic therapy and low proliferative activity. Clin Cancer Res 2005;11:3678–85 [DOI] [PubMed] [Google Scholar]
- 115.Meng S, Tripathy D, Frenkel EP, Shete S, Naftalis EZ, Huth JF, et al. Circulating tumor cells in patients with breast cancer dormancy. Clin Cancer Res 2004;10:8152–62 [DOI] [PubMed] [Google Scholar]
- 116.Ferrara N VEGF and Intraocular Neovascularization: From Discovery to Therapy. Transl Vis Sci Technol 2016;5:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jayson GC, Kerbel R, Ellis LM, Harris AL. Antiangiogenic therapy in oncology: current status and future directions. Lancet 2016;388:518–29 [DOI] [PubMed] [Google Scholar]
- 118.Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer 2008;8:592–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Harney AS, Karagiannis GS, Pignatelli J, Smith BD, Kadioglu E, Wise SC, et al. The Selective Tie2 Inhibitor Rebastinib Blocks Recruitment and Function of Tie2(Hi) Macrophages in Breast Cancer and Pancreatic Neuroendocrine Tumors. Mol Cancer Ther 2017;16:2486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sleeman J, Steeg PS. Cancer metastasis as a therapeutic target. Eur J Cancer 2010;46:1177–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nat Med 2006;12:895–904 [DOI] [PubMed] [Google Scholar]
- 122.Fernandes M, Rosel D, Brabek J. Solid cancer: the new tumour spread endpoint opens novel opportunities. Br J Cancer 2019;121:513–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Anampa Mesias JDS, Oktay MH, Xue X, Condeelis J, Sparano JA. Phase Ib study of rebastinib plus antitubulin therapy with paclitaxel or eribulin in patients with metastatic breast cancer (MBC). 2017 [Google Scholar]
- 124.Harper KL, Sosa MS, Entenberg D, Hosseini H, Cheung JF, Nobre R, et al. Mechanism of early dissemination and metastasis in Her2(+) mammary cancer. Nature 2016;540:589–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Pantel K, Brakenhoff RH. Dissecting the metastatic cascade. Nat Rev Cancer 2004;4:448–56 [DOI] [PubMed] [Google Scholar]
- 126.Pukazhendhi G, Gluck S. Circulating tumor cells in breast cancer. J Carcinog 2014;13:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Pierga JY, Bidard FC, Mathiot C, Brain E, Delaloge S, Giachetti S, et al. Circulating tumor cell detection predicts early metastatic relapse after neoadjuvant chemotherapy in large operable and locally advanced breast cancer in a phase II randomized trial. Clin Cancer Res 2008;14:7004–10 [DOI] [PubMed] [Google Scholar]
- 128.Bidard FC, Proudhon C, Pierga JY. Circulating tumor cells in breast cancer. Mol Oncol 2016;10:418–30 [DOI] [PMC free article] [PubMed] [Google Scholar]