

Protein shells of viruses (capsids) have evolved to undergo specific changes to ensure the timely delivery of genetic material to host cells. The 2-layer capsid of reovirus provides a model system to study the interactions between capsid proteins and the changes they undergo during entry. We tested a virus in which the core proteins were derived from a different strain than the outer capsid. In comparison to the parental T3D strain, we found that this mismatched virus was less stable and completed conformational changes required for entry prematurely. Capsid stability was restored by introduction of specific changes to the outer capsid, indicating that an optimal fit between inner and outer shells maintains capsid function. Separate from this property, mismatch between these protein layers also impacted the capacity of the virus to initiate infection and produce progeny. This study reveals new insights into the roles of capsid proteins and their multiple functions during viral replication.
KEYWORDS: capsid, double-stranded RNA virus, reovirus
ABSTRACT
The capsids of mammalian reovirus contain two concentric protein shells, the core and the outer capsid. The outer capsid is composed of μ1-σ3 heterohexamers which surround the core. The core is composed of λ1 decamers held in place by σ2. After entry into the endosome, σ3 is proteolytically degraded and μ1 is cleaved and exposed to form infectious subvirion particles (ISVPs). ISVPs undergo further conformational changes to form ISVP*s, resulting in the release of μ1 peptides, which facilitate the penetration of the endosomal membrane to release transcriptionally active core particles into the cytoplasm. Previous work identified regions or specific residues within reovirus outer capsid proteins that impact the efficiency of cell entry. We examined the functions of the core proteins λ1 and σ2. We generated a reovirus T3D reassortant that carries strain T1L-derived σ2 and λ1 proteins (T3D/T1L L3S2). This virus displays lower ISVP stability and therefore converts to ISVP*s more readily. To identify the molecular basis for lability of T3D/T1L L3S2, we screened for hyperstable mutants of T3D/T1L L3S2 and identified three point mutations in μ1 that stabilize ISVPs. Two of these mutations are located in the C-terminal ϕ region of μ1, which has not previously been implicated in controlling ISVP stability. Independent of compromised ISVP stability, we also found that T3D/T1L L3S2 launches replication more efficiently and produces higher yields in infected cells than T3D. In addition to identifying a new role for the core proteins in disassembly events, these data highlight the possibility that core proteins may influence multiple stages of infection.
IMPORTANCE Protein shells of viruses (capsids) have evolved to undergo specific changes to ensure the timely delivery of genetic material to host cells. The 2-layer capsid of reovirus provides a model system to study the interactions between capsid proteins and the changes they undergo during entry. We tested a virus in which the core proteins were derived from a different strain than the outer capsid. In comparison to the parental T3D strain, we found that this mismatched virus was less stable and completed conformational changes required for entry prematurely. Capsid stability was restored by introduction of specific changes to the outer capsid, indicating that an optimal fit between inner and outer shells maintains capsid function. Separate from this property, mismatch between these protein layers also impacted the capacity of the virus to initiate infection and produce progeny. This study reveals new insights into the roles of capsid proteins and their multiple functions during viral replication.
INTRODUCTION
In order to successfully launch replication, a virus must protect, transport, and deliver its genome into the host cell. The viral capsid is a complex mechanical container with the primary function of assembling around the viral genome in one host cell, exiting that host cell, and releasing the genome in another host cell. Therefore, the capsid has two demands that appear to be in direct conflict with one another. It must be stable enough to protect and transport the viral genome, but it also must be dynamic, poised to react to the right environment at the right time to perform functions during entry and to allow replication of the genome. Viruses have evolved capsid proteins that are capable of these dynamic mechanical interactions in a number of diverse and fascinating ways. While some capsids are made up of only a single type of capsid protein, others have more complex combinations of proteins. One family of viruses, Reoviridae, have double- or even triple-layered capsids.
Members of the family Reoviridae are made up of nonenveloped virions with capsids that are composed of 1 to 3 concentric protein shells that surround 9 to 12 double-stranded-RNA (dsRNA) genome segments (1–3). While the outer layers of the multilayered capsids are proteolytically processed and undergo conformational changes during entry, the innermost capsid (or core), which contains the genome, remains intact throughout the remainder of replication (4). The core is a complex molecular machine that is capable of producing fully capped and functional RNA transcripts for translation by the host machinery (5, 6). The capsid proteins must, therefore, be capable of multiple functions in addition to their protective and structural roles. The outer capsid proteins must be poised to undergo the conformational changes required to release the cores and allow them to be transcriptionally active. While studies of mammalian reovirus have provided many insights into how outer capsid proteins regulate and mediate entry events that lead to these conformational changes, the role of the core proteins in cell entry events remains unclear.
The capsids of mammalian reovirus are composed of two concentric protein shells, the core and the outer capsid (4). The outer capsid is primarily made up of μ1-σ3 heterohexamers (7) (Fig. 1). These surround the core, which is composed of λ1 decamers held in place by σ2 (8). Turrets, made up of λ2 pentamers, protrude from the core at the fivefold axis of symmetry. The σ1 attachment protein forms trimers that are anchored in the λ2 turret (8, 9). After entry, σ3 is proteolytically degraded within the endosome and μ1 is cleaved into μ1δ and ϕ fragments (10, 11). These μ1 fragments remain particle associated at this stage, and the particle is referred to as an infectious subvirion particle (ISVP) (12–14). Further conformational changes within the endosome result in cleavage of μ1δ to form μ1N and δ (15, 16). These particles are no longer infectious and are referred to as ISVP*. Release of μ1 peptides (μ1N and ϕ) results in penetration of the endosomal membrane and the deposition of the transcriptionally active cores into the cytoplasm (13, 17, 18).
FIG 1.
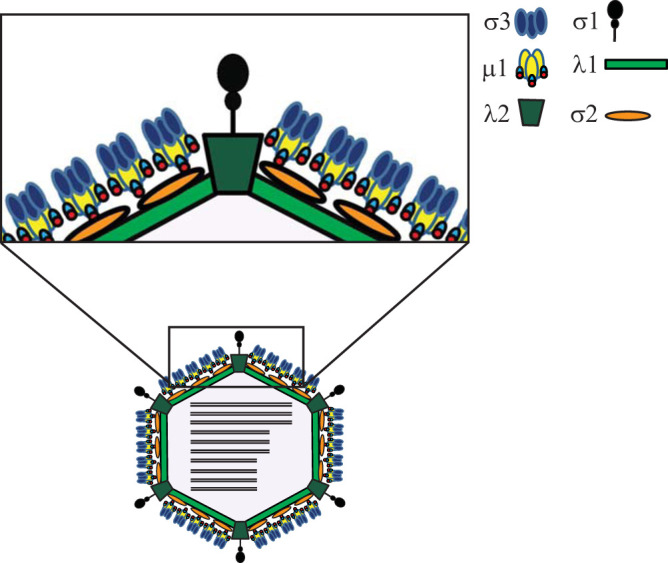
Schematic representation of reovirus capsid proteins.
Previous work in identifying interactions and proteins involved in reovirus disassembly and cell entry have focused on the outer capsid proteins. Here, we concentrated on the structural functions of the core proteins λ1 and σ2. We characterized a reassortant virus containing the λ1-encoding gene segment L3 and the σ2-encoding gene segment S2 (T3D/T1L L3S2). We found that this new virus displays a higher ISVP-to-ISVP* conversion efficiency or lower ISVP stability. We identified three point mutations in μ1 that increase the stability of T3D/T1L L3S2. Two of these mutations are located in the ϕ region of μ1, which has not previously been implicated in maintaining ISVP stability or controlling ISVP-to-ISVP* conversion. Additionally, this reassortant virus has increased growth resulting from higher transcription levels and, subsequently, higher protein production. This role does not appear to be related to the enhanced ISVP-to-ISVP* conversion of this virus. These results provide insights into the structural functions of the core proteins and how their interactions may influence disassembly and early replication steps during infection.
RESULTS
ISVP-to-ISVP* conversion efficiency of T3D is altered by the T1L core proteins λ1 and σ2.
Single-gene reassortants between prototype reovirus strains T1L and T3D, which contain mismatches between outer capsid proteins, display altered capsid stability and efficiency of disassembly intermediate formation (19, 20). To determine whether the core proteins λ1 and σ2 play a role in ISVP-to-ISVP* conversion efficiency, we characterized the properties of T3D/T1L L3S2. This virus contains the λ1-encoding gene segment L3 and the σ2-encoding gene segment S2 from T1L in an otherwise T3D genetic background. The resulting virus therefore contains major core proteins that do not match the outer capsid. The λ1 protein from T3D and T1L are 99.3% identical (9 amino acid differences out of 1,275), and the σ2 proteins are 98.8% identical (5 amino acid differences out of 418) (21–23). While σ2 interacts with the outer capsid μ1 trimer, λ1 is not known to interact with any outer capsid proteins (24).
To test overall virion stability, we incubated T3D and T3D/T1L L3S2 over a series of elevated temperatures and tested the protease stability of the major viral capsid proteins. Such an approach has been used previously to test stability of virions and viral entry intermediates (19). Based on the similarity in their protease sensitivity profiles, we think that virions of T3D/T1L L3S2 do not display significant changes in stability in comparison to the parent strain, T3D (Fig. 2A). To determine if the mismatches in this reassortant virus alter stability of ISVPs or efficiency of ISVP-to-ISVP* conversion, we generated ISVPs of T3D and T3D/T1L L3S2 and incubated them over a gradient of increasing temperatures. ISVP* conversion was determined as a measure of trypsin sensitivity of the μ1δ fragment (25). In comparison to the parent strain T3D, T3D/T1L L3 ISVPs have significantly reduced stability or enhanced ISVP-to-ISVP* conversion in vitro (Fig. 2B).
FIG 2.

T3D/T1L L3S2 exhibits increased efficiency of ISVP-to-ISVP* conversion in vitro. (A) T3D and T3D/T1L L3S2 virions (2 × 1012 particles/ml) were divided into aliquots of equal volume and incubated either at 4°C or over a range of temperatures (65 to 85°C) for 5 min. The reaction mixtures were chilled on ice and digested with 0.10 mg/ml trypsin for 30 min. Following addition of loading dye, the samples were subjected to SDS-PAGE analysis. The positions of major capsid proteins are shown. μ1 runs as μ1C (15). (B) ISVPs (2 × 1011 particles/ml) of T3D or T3D/T1L L3S2 were divided into aliquots of equivalent volume and incubated either at 4°C or over a range of temperatures (22 to 42°C) for 20 min. The reactions were chilled on ice and digested with 0.10 mg/ml trypsin for 30 min. Following addition of loading dye, the samples were subjected to SDS-PAGE analysis. The gels shown are representative of at least 3 independent experiments. The positions of major capsid proteins are shown. μ1 runs as μ1C. (C) ISVPs generated from P2 stocks of the indicated virus strain were divided into aliquots of equivalent volume and incubated at either 4°C or 40°C for 20 min. Reactions were then diluted in PBS and subjected to plaque assay. The data are plotted as mean loss of infectivity for three independent samples in comparison to samples incubated at 4°C. Error bars indicate SD. ***, P < 0.001 in comparison to T3D, as determined by Student's t test.
Conversion to ISVP* results in loss of outer capsid proteins that are essential for entry (26, 27). As a consequence, ISVPs that are heated to temperatures that result in ISVP* conversion have significantly reduced titers. Measuring thermal stability of ISVP infectivity, therefore, is an alternate method to evaluate the efficiency of ISVP* formation. T3D/T1L L3S2 and T3D ISVPs were heated to 40°C, and loss of infectivity was measured by plaque assay. Consistent with previous work (20), at 40°C, most of the T3D ISVPs did not convert to ISVP* and the change in titer was minimal (Fig. 2C). However, heating T3D/T1L L3S2 ISVPs to 40°C resulted in ISVP* conversion and a significant loss in infectivity (Fig. 2C). These data indicate that the reovirus core proteins play a role in stability of the ISVP, a function previously not attributed to the major core proteins λ1 and σ2.
Enhanced ISVP-to-ISVP* conversion is not due to interactions with RNA.
In addition to its structural functions, λ1 plays a variety of other roles during infection. One of those functions involves interaction with RNA (28, 29). If λ1-RNA interactions affect stability of the particles, it may explain why viruses with different core proteins and, consequently, the potential for different RNA interaction properties may have altered stability. To rule out this possibility, we tested the ISVP-to-ISVP* conversion efficiency of genome-containing particles in comparison to genome-deficient particles of T3D. This comparison would allow us to uncover the contribution of viral genomic RNA to ISVP stability. ISVP-to-ISVP* conversion was again determined as a measure of trypsin sensitivity of μ1δ. The stability of ISVPs of genome-containing particles and that of ISVPs of genome-deficient particles were not significantly different, which suggests that λ1-RNA interactions do not affect ISVP-to-ISVP* conversion. Thus, the ISVP-to-ISVP* phenotype of T3D/T1L L3S2 is likely not related to differences in RNA interactions (Fig. 3).
FIG 3.

Increased ISVP-to-ISVP* conversion efficiency in T3D/T1L L3S2 is not due to altered interactions with viral RNA. ISVPs (2 × 1011 particles/ml) derived from genome-containing or genome-deficient particles of strain T3D were divided into aliquots of equivalent volume and incubated either at 4°C or over a range of temperatures (22 to 40°C) for 20 min. The reaction mixtures were chilled on ice and digested with 0.10 mg/ml trypsin for 30 min. Following addition of loading dye, the samples were subjected to SDS-PAGE analysis. The positions of major capsid proteins are shown. μ1 runs as μ1C.
Isolation of heat-resistant mutants in T3D/T1L L3S2.
Because ISVP particles that have converted to ISVP* are less infectious, it is possible to select for and isolate variants with mutations that render ISVPs more stable and thus still retain infectivity after exposure to heat (27, 30, 31). Mapping such mutations could reveal key interactions between viral structural proteins that contribute to maintaining ISVP stability. In order to better understand the basis for the lower stability of this reassortant, we sought to identify such mutations in T3D/T1L L3S2 (Fig. 4A). ISVPs of T3D/T1L L3S2 were heated to 40°C (a temperature at which they display significantly lower infectivity than that of similarly treated wild-type ISVPs derived from T3D) and subjected to plaque assay. Resulting plaques were isolated as potential heat-resistant mutants. To confirm the heat resistance of these isolates, ISVPs of each isolate were again incubated at 40°C, and loss of infectivity in comparison to ISVPs incubated at 4°C was determined by plaque assay. Of the 20 isolates tested, 7 were determined to have little to no loss in infectivity. These isolates were considered heat resistant (HR) (Fig. 4B). For each of these 7 isolates, genome segments encoding λ1, σ2, and μ1 (L3, S2, and M2, respectively) were sequenced. We reasoned that these genome segments may bear mutations, because T3D/T1L L3S2 differs from T3D in the properties of λ1 and σ2 and because μ1 was previously implicated in controlling stability of ISVPs (27, 32–34). None of the isolates contained mutations in L3 or S2. Four isolates were identified with mutations in μ1. HR16 had a mutation at amino acid 459 (lysine to glutamic acid) which is located in the δ fragment of μ1. HR2, HR15, and HR17 had mutations in the ϕ fragment of μ1. The remaining HR mutants identified did not contain mutations in the gene segments sequenced and were not examined further. The mutation in HR15 was at amino acid 607 (proline to glutamine), and HR2 and HR17 had the same mutation at amino acid 615 (alanine to threonine) (Fig. 4C to E). Notably, the K459E mutation was previously identified as a stabilizing mutation (30). However, mutations in μ1ϕ that contribute to ISVP stability or ISVP-to-ISVP* conversion efficiency had not been previously identified.
FIG 4.

Selection of viruses with mutations that confer stability to T3D/T1L L3S2 ISVPs. (A) Diagram depicting the process for selecting for mutants with reduced ISVP-ISVP* conversion efficiency of T3D/T1L L3S2. ISVPs of T3D/T1L L3S2 were incubated at 40°C for 20 min. Reaction mixtures were then diluted in PBS and subjected to plaque assay. Viruses from resulting plaques were isolated and propagated to generate P0 stocks. Heat resistance of these putative heat-resistant (HR) plaque isolates was confirmed by measuring the thermal stability of ISVPs incubated at 4°C or 40°C using a plaque assay. Mutants that were confirmed as heat resistant were sequenced. (B) ISVPs generated from P0 stocks were incubated at either 4°C or 40°C for 20 min. Reaction mixtures were then diluted in PBS and subjected to plaque assay. Note that HR1 to -10 and HR11 to -20 are from two separate isolation experiments, collected and tested at different times. ND, not detectable. (C and D) Top (left) and side (right) views of the μ1 trimer (C) and monomer (D). Positions of mutations identified in HR viruses are shown in green. μ1 cleavage fragments are colored as in panel E, with one μ1 monomer shown with darker colors.
Mutations in μ1 stabilize T3D/T1L L3S2 ISVPs.
Because we did not sequence the entire genome of HR viruses, it remains possible that mutations in genome segments other than L3, S2, and M2 influence the thermal stability of ISVPs generated from the second-site revertants. To evaluate the stabilizing effect of the identified mutations on T3D/T1L L3S2 ISVPs, each mutation was introduced individually into a T3D/T1L L3S2 background. Each new mutant virus was tested for ISVP-to-ISVP* conversion efficiency. ISVPs of each virus were generated and incubated over a gradient of temperatures. ISVP* conversion was determined as a measure of trypsin sensitivity of the μ1δ fragment (19). In comparison to T3D/T1L L3S2, each of the ISVPs with μ1 mutations had increased stability, suggesting that these amino acid residues in μ1 play important roles in ISVP-to-ISVP* conversion (Fig. 5A). To verify these results, the infectivities of ISVPs of T3D/T1L L3S2 and each of the μ1 mutants at 4°C and 40°C were compared by plaque assay. Consistent with the results seen in Fig. 2C, T3D/T1L L3S2 ISVPs experienced a loss of infectivity at 40°C. In contrast, introduction of μ1 changes identified in heat-resistant mutants into T3D/T1L L3S2 resulted in ISVP particles that displayed greater stability (Fig. 5B). These data indicate that mutations in μ1 are sufficient to restore wild-type-like ISVP-to-ISVP* conversion efficiency and thermal stability to T3D/T1L L3S2.
FIG 5.

Mutations in μ1 restore stability. (A) ISVPs (2 × 1011 particles/ml) of T3D/T1L L3S2 with the indicated M2 mutations were divided into aliquots of equivalent volume and incubated at either 4°C or over a range of temperatures (22 to 42°C) for 20 min. The reaction mixtures were chilled on ice and digested with 0.10 mg/ml trypsin for 30 min. Following addition of loading dye, the samples were subjected to SDS-PAGE analysis. The gels shown are representative of at least 3 independent experiments. The positions of major capsid proteins are shown. μ1 runs as μ1C. (B) ISVPs generated from P2 stocks of the indicated virus strain were divided into aliquots of equivalent volume and incubated at either 4°C or 40°C for 20 min. Reaction mixtures were then diluted in PBS and subjected to plaque assay. The data are plotted as mean loss of infectivity for three independent samples in comparison to samples incubated at 4°C. Error bars indicate SD. **, P < 0.01, and ***, P < 0.001, in comparison to T3D/T1L L3S2, as determined by Student's t test.
Mutations in μ1 also affect ISVP-to-ISVP* conversion in wild-type T3D.
The μ1 mutations that restored thermal stability of ISVPs and normal ISVP-to-ISVP* conversion efficiency of T3D/T1L L3S2 are not in a position to contact proteins that make up the core (7, 24). Thus, it seems unlikely that these mutations stabilize the capsid by directly altering core-outer capsid interactions. One possibility is that the changes in μ1 simply stabilize the capsid by strengthening interactions between μ1 monomers or between μ1 trimers. If so, the μ1 mutations would be expected to further stabilize ISVPs of T3D, which contains different L3 and S2 alleles. To test this idea, we also generated viruses containing one of each of the three μ1 changes found in the HR viruses in a wild-type T3D background. As before, ISVPs of each virus were generated and incubated over a gradient of temperatures. ISVP* conversion was again determined as a measure of trypsin sensitivity of the μ1δ fragment. In comparison to wild-type T3D, each mutant underwent ISVP-to-ISVP* conversion much less efficiently (Fig. 6A). To verify these results, each virus was tested for loss of infectivity. ISVPs of T3D along with each mutant were heated to 49°C, and loss in infectivity was measured by plaque assay (Fig. 6B). The 49°C temperature was determined empirically as the lowest temperature at which T3D ISVPs exhibit a loss in infectivity (data not shown). Consistent with the data in Fig. 6A, at this temperature each of the μ1 mutants displayed no loss of infectivity compared to wild-type T3D. These data suggest that the mutations identified in μ1 are generally stabilizing mutations and not directly related to the effects of the mismatched core proteins.
FIG 6.

Mutations in μ1 hyperstabilize T3D. (A) ISVPs (2 × 1011 particles/ml) of T3D and T3D with the indicated M2 mutations were divided into aliquots of equivalent volume and incubated at either 4°C or over a range of temperatures (32 to 46°C) for 20 min. The reaction mixtures were chilled on ice and digested with 0.10 mg/ml trypsin for 30 min. Following addition of loading dye, the samples were subjected to SDS-PAGE analysis. The gels shown are representative of at least 3 independent experiments. The positions of major capsid proteins are shown. μ1 runs as μ1C. (B) ISVPs generated from purified virions were divided into aliquots of equivalent volume and incubated at either 4°C or 49°C for 20 min. Reaction mixtures were then diluted in PBS and subjected to plaque assay. The data are plotted as mean loss of infectivity for three independent samples in comparison to samples incubated at 4°C. Error bars indicate SD. ***, P < 0.001, in comparison to T3D, as determined by Student’s t test.
Mismatches between the core and outer capsid proteins affect viral replication and transcription.
To determine if the differences in the ISVP-to-ISVP* conversion efficiency of T3D and T3D/T1L L3S2 are relevant during a viral infection, we next examined viral growth following infection of cells at an MOI of 0.1 PFU/cell. Virus titer at 24 h following infection was determined by plaque assay, and viral yield was calculated as an increase in titer from 0 h postinfection (which measured virus adsorbed to cells at the start of infection). We observed that infection with T3D/T1L L3S2 resulted in an increased yield in comparison to T3D (Fig. 7A). To determine if this growth phenotype correlates with increased ISVP-to-ISVP* conversion efficiency, we tested the impact of introducing a representative μ1 mutation, A615T, in these viruses. While growth of T3D was significantly higher than growth of T3D M2 A615T, growth of T3D/T1L L3S2 was not significantly different from that of T3D/T1L L3S2 M2A615T (Fig. 7A). As the M2 A615T mutation in T3D/T1L L3S2 restores the stability of its ISVPs, these data indicate that the difference in replication efficiency of T3D and T3D/T1L L3S2 is not a consequence of the capacity of the reassortant virus to more easily convert to ISVP*. These data also suggest that T1L-derived L3 and S2 genome segments can influence the properties of T3D in multiple independent ways.
FIG 7.

T3D/T1L L3S2 affects viral replication. (A) L cell monolayers were infected with T3D or T3D/T1L L3S2 or with the indicated mutant viruses at an MOI of 0.1 PFU/cell. At 0 and 24 h postinfection, the infected cells were lysed and the viral yield was quantified by plaque assay. Error bars indicate SD. *, P < 0.05, and ***, P < 0.001, in comparison to T3D, as determined by Student's t test. Data are representative of at least 3 different experiments. (B) L cell monolayers were infected with the indicated viruses at an MOI of 10 PFU/cell. At 10 h postinfection, the cells were lysed and protein production was determined by immunoblotting. Protein quantification of 3 replicates normalized to PSTAIR is shown. Error bars indicate SD. ***, P < 0.001, in comparison to T3D, as determined by Student's t test. (C) L cell monolayers were infected with the indicated viruses at an MOI of 10 PFU/cell. The cells were lysed at 6 h postinfection, and total RNA was isolated. cDNA was generated using primers for T3D S1 and GAPDH. mRNA production was measured by RT-qPCR. Data are fold change compared to mock-infected samples and normalized to GAPDH. Error bars indicate SD. ***, P < 0.001, in comparison to T3D, as determined by Student's t test.
The higher replication potential of T3D/T1L L3S2 could be related to the capacity of this virus to produce more viral gene products with faster kinetics or to a greater extent. To test this idea, we next examined viral gene expression early during infection using immunoblots. Cells were infected with ISVPs at equal multiplicities of infection (MOI) and harvested at 0 or 10 h postinfection. Expression of the μ1 protein was assessed as a representative. While expression of μ1 was visible at 10 h postinfection in all samples, T3D/T1L L3S2 displayed significantly higher protein expression than T3D (Fig. 7B). To test the impact of ISVP-to-ISVP* conversion phenotypes on protein expression, we also examined T3D M2 A615T and T3D/T1L L3S2 M2 A615T. While both viruses displayed lower protein levels than T3D/T1L L3S2, the mutant in the T3D background had significantly lower μ1 expression than the mutant in the T3D/T1L L3S2 background, suggesting that the ISVP-to-ISVP* conversion phenotype alone is not responsible for the increase in protein expression. To determine whether the greater level of viral protein expression is a consequence of a higher level of viral mRNA, we measured transcription of viral S1 mRNA early during infection using reverse transcription-quantitative PCR (RT-qPCR). Cells were infected with ISVPs at equal MOI and harvested at 6 h postinfection. At this time point, T3D/T1L L3S2 showed significantly more reovirus S1 transcripts than T3D, indicating that the increase in protein production is likely due to enhanced transcription by the reassortant virus (Fig. 7C). It is worth noting that while there is a significant increase in protein between the T3D and T3D/T1L L3S2 viruses with the M2 A615T mutation, this does not appear to correlate with increased transcription in these viruses. More studies are needed to clarify why this may be the case. Nonetheless, these data indicate that swapping core proteins between T1L and T3D in a reassortant virus also influences postentry viral replication events.
DISCUSSION
To test the role of the inner core proteins on disassembly and early entry events we generated a reassortant virus with major core proteins from T1L and all other proteins (including the outer capsid) from T3D (T3D/T1L L3S2). T3D/T1L L3S2 undergoes ISVP-to-ISVP* conversion much more efficiently than the parental strain. We identified mutations in μ1 that stabilize ISVPs. T3D/T1L L3S2 also demonstrated increased growth kinetics compared to the parental T3D strain. Surprisingly, the more efficient growth of T3D/T1L L3S2 was not related to its capacity to undergo more efficient ISVP-to-ISVP* transition. When T3D and T3D/T1L L3S2 with the M2 A615T mutation, both with lower levels of ISVP-to-ISVP* conversion efficiency, are compared to one another, it is clear that the L3S2 reassortant virus still has increased growth compared to its wild-type (WT) T3D counterpart, suggesting that the ability of the virus to form ISVP*s is not related to the increased growth seen here. Instead, increased growth of T3D/T1L L3S2 relates to more rapid protein and mRNA production early in infection. These data suggest that alterations in properties of core proteins can impact the function of the outer capsid proteins in cell entry. Additionally, properties of core proteins can also influence the enzymatic functions of the capsid that are required to establish efficient infection of host cells.
The reovirus core is a T=1 icosahedron, and it is surrounded by the outer capsid (T=13). The outer capsid is made up of 200 heterohexamers of μ1 and σ3 proteins that cover the core (7, 24). It is penetrated by λ2 pentameric turrets at each fivefold axis of symmetry. Trimers of the σ1 attachment protein are situated inside these λ2 turrets (8, 24). The core is made up of 120 copies of λ1 arranged in asymmetric pairs of pentamers to form decamers. Twelve such decamers make up the core. The σ2 protein clamps onto λ1 at 3 different sites within an asymmetric unit, resulting in 150 copies of σ2 stabilizing the core shell (8). At each fivefold axis of symmetry, channels penetrate the shell via the λ2 pentameric turrets. It is at each of these channels that the λ3 polymerase interacts with λ1 and is thought to interact with the polymerase cofactor μ2 (5, 35, 36). There are no known contacts between λ1 and the outer capsid. A majority of contacts between the core and the outer capsid occur between μ1 and σ2. These interactions involve the bottom surface of μ1 and the top surface of σ2 (24). The “hub and spoke” structure formed by the C-terminal 33 residues of μ1, which is thought to be important for stabilizing the μ1 lattice, also makes contact with λ2 and/or σ2 (24). This interaction is thought to be stabilized in part by μ1 residues 51 to 62, which form flexible loops (24). It is possible that the polymorphic differences between σ2 proteins of T1L and T3D influence interaction with μ1 sufficiently that disassembly is altered. λ1 can also impact interaction of the core with μ1, thereby changing disassembly. However, because μ1 does not contact λ1, this effect would occur indirectly if the structure or conformation of λ2 or σ2, two proteins that do interact with μ1, is altered due to differences in λ1 residues. The relative contribution of differences in properties of σ2 and λ1 and the potential subtle differences in structure remain the focus of our ongoing work.
Differences in entry efficiency between different serotypes and laboratory strains of reovirus along with genetic approaches have been used to study how conformational changes and cleavage events required for entry are regulated. Panels of reassortant viruses have linked differences in ISVP-to-ISVP* conversion to μ1 (27). The autocatalytic cleavage of μ1 and efficiency of ISVP* formation have both been linked to distinct portions of the δ fragment of μ1 (32). Multiple other studies have linked efficiency of entry-related disassembly events to the δ fragment of μ1 (7, 30, 32, 34, 37). The μ1N fragment is released from the particle during ISVP* conversion, and the released fragment is thought to function in a positive feedback loop to further drive ISVP* conversion (38). The ISVP*-promoting activity of μ1N is most efficient in the presence of membranes, likely because membrane-associated μ1N recruits ISVP-like particles (39, 40). Cleavage of ϕ, which is required for its release from particles during ISVP* conversion, is also required for efficient interaction of ISVPs with membranes (40). However, precisely how ϕ functions in this step is not known. Here, we identified two mutations in the ϕ fragment of μ1 that influence ISVP-to-ISVP* conversion. These mutations are the first indication that the ϕ fragment may also be involved in ISVP-to-ISVP* conversion efficiency. Because our ISVP-to-ISVP* reactions described here were performed in the absence of membranes, the mutations are unlikely to influence ISVP-to-ISVP* conversion by affecting particle-membrane interaction. Thus, the precise mechanism by which the identified ϕ residues influence ISVP* remains unclear. While the majority of μ1-σ3 interactions occur in the jelly roll domains (residues 306 to 514) of μ1, both of the mutations identified are in a region proposed to form the cradle for the base of σ3 (7). This region has not been shown to interact with any core proteins or with neighboring μ1 monomers (7). Therefore, it is unlikely that these mutations stabilize ISVPs by strengthening inter- or intra-μ1 trimer interactions or by interacting with core proteins. The identified mutations are in a region of μ1 that may unfold in order to accommodate μ1N release, which is a necessary step of ISVP-to-ISVP* conversion (7, 24). Thus, one likely explanation is that ϕ properties affect the release of μ1N.
The L3 gene segment-encoded λ1 protein is thought of first as a structural protein that makes up the inner core of reovirus. However, as is the case with most viruses, proteins are capable of playing multiple roles during infection. In addition to its structural roles, λ1 has RNA helicase activity and phosphohydrolase activity and is known to interact with RNA (29). The role of each of these functions during infection is currently unknown. The polymerase λ3 interacts with λ1 on the inside of the shell at each fivefold axis (8, 41). While the position of the transcription cofactor protein μ2 within the capsid is not known, it is possible that it also interacts with λ1. Thus, the interaction of λ1 with these encapsidated enzymes could alter viral transcription efficiency. Reovirus serotypes T1L and T3D replicate with different efficiency in some cell lines, with T1L replicating to a higher extent and with faster kinetics (42). Reassortant analyses have partially linked this phenotype to the T1L-derived λ1-encoding L3 gene segment (42). Our studies indicate that the enhanced infection efficiency of T3D/T1L L3S2 is not related to its greater propensity for ISVP-to-ISVP* transition. Instead, we propose that enhanced efficiency of infection is a result of differences in the activity of λ1 itself or its impact on the activity of the transcriptional machinery composed of λ3 and μ2. While the role of σ2 as a structural protein is well established, additional roles in replication have not been confidently identified. σ2 has weak interactions with dsRNA, and reassortant studies have linked σ2 with increased induction and sensitivity to interferon in some cell types (43, 44). Additional studies with monoreassortants bearing only S2 and L3 gene segments in the T3D background are needed to precisely ascertain the basis for the enhanced replicative efficiency of T3D/T1L L3S2.
Recent studies from our laboratory have revealed that reassortant viruses display phenotypes that are unexpected and extend beyond the known function of the protein. First, we found that, even though the primary function of the M2-encoded protein μ1 is in membrane penetration, an M2 reassortant virus displays greater attachment to host cells (45). Second, we found that although the S1-encoded σ1 protein is the attachment factor, an S1 reassortant impacts the stability of the μ1 layer, with which it has no interactions (20). The study presented here reveals that core proteins, previously thought to have only a structural role in packaging the genomic material, influence cell entry events regulated by the outer capsid. Until the advent of reverse genetics, reassortant analyses were used as the main strategy to assign function to proteins of segmented viruses (46, 47). This approach has also been useful to determine the genetic basis of viral disease. While this approach has been invaluable, we think our work suggests that the structure-function explanation of some phenotypes reported for reovirus and possibly other members of the family Reoviridae may be more complicated than previously appreciated.
MATERIALS AND METHODS
Cells and viruses.
Spinner-adapted murine L929 (L) cells were grown at 37°C in Joklik's minimal essential medium (Lonza) supplemented with 5% fetal bovine serum (FBS) (Life Technologies), 2 mM l-glutamine (Invitrogen), 100 U/ml penicillin (Invitrogen), 100 μg/ml streptomycin (Invitrogen), and 25 ng/ml amphotericin B (Sigma-Aldrich). All virus strains used in this study were derived from reovirus type 3 Dearing (T3D) and reovirus type 1 Lang (T1L) and were generated by plasmid-based reverse genetics (46, 47). Mutations within the T3D M2 gene were generated by QuikChange (Agilent Technologies) site-directed mutagenesis. Primer sequences are available upon request.
Virus propagation and purification.
All wild-type and mutant viruses used in this study were propagated and purified as previously described (32, 48). Briefly, plaques isolated from plasmid-based reverse genetics were propagated successively in T-25, T-75, and T-175 flasks to generate P0, P1, and P2 virus stocks, respectively. To generate purified virus, L cells infected with P2 reovirus stocks were lysed by sonication. Virus particles were extracted from the lysates using Vertrel-XF specialty fluid (Dupont) (49). The extracted particles were layered onto 1.2- to 1.4-g/cm3 CsCl step gradients. The gradients were then centrifuged at 187,000 × g for 4 h at 4°C. Bands corresponding to purified virus particles and top component “empty” particles (∼1.36 g/cm3) (50) were isolated and dialyzed into virus storage buffer (10 mM Tris-HCl [pH 7.4], 15 mM MgCl2, and 150 mM NaCl). Following dialysis, the particle concentration was determined by measuring the optical density at 260 nm (OD260) of the purified virus stocks (1 unit at OD260 is equal to 2.1 × 1012 particles/ml).
Generation of ISVPs.
Purified virions of the virus strains indicated on the figures (2 × 1012 particles/ml or 4 × 1012 particles/ml) were digested with 200 μg/ml TLCK (Nα-p-tosyl-l-lysine chloromethyl ketone)-treated chymotrypsin (Worthington Biochemical) in a total volume of 100 μl for 1 h at 32°C. After 1 h, the reaction mixtures were incubated for 20 min on ice and quenched by the addition of 1 mM phenylmethylsulfonyl fluoride (Sigma-Aldrich). The generation of ISVPs was confirmed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Coomassie brilliant blue staining.
Analysis of ISVP-to-ISVP* conversion.
ISVPs (2 × 1012 particles/ml) of the indicated viral strains were divided into aliquots of equivalent volumes and heated at the indicated temperatures for 20 min. The reaction mixtures were cooled on ice and then digested with 0.10 mg/ml trypsin (Sigma-Aldrich) for 30 min on ice. Following addition of the SDS-PAGE loading dye, the samples were subjected to SDS-PAGE analysis. For analysis by quantitative infectivity assay, P2 stocks or purified virus stocks of the indicated viruses were diluted 1:10 in virion storage buffer (10 mM Tris-HCl [pH 7.4], 15 mM MgCl2, and 150 mM NaCl). A 200-μg/ml concentration of TLCK-treated chymotrypsin (Worthington Biochemical) was added to each sample. Samples were heated to 37°C for 30 min. The reaction was quenched by the addition of 1 mM phenylmethylsulfonyl fluoride (Sigma-Aldrich) and cooled on ice for 10 min. The reaction mixtures were divided into equivalent volumes and incubated at 4°C or 40°C for 20 min. These mixtures were used to initiate infection of L929 cells, and infectivity was determined by plaque assay. The change in infectivity at a given temperature (T) was calculated using the following formula: log10(PFU/ml)T − log10(PFU/ml)4°C.
Analysis of virion stability.
Virions (2 × 1012 particles/ml) of the indicated viral strains were divided into aliquots of equivalent volumes and heated at the indicated temperatures for 20 min. The reaction mixtures were cooled on ice and then digested with 0.10 mg/ml trypsin (Sigma-Aldrich) for 30 min on ice. Following addition of SDS loading dye, the samples were subjected to analysis by SDS-PAGE.
Isolation and verification of HR mutants.
ISVPs of purified T3D/T1L L3S2 were generated and subsequently heated to 40°C for 20 min. Resulting reactions were diluted in phosphate-buffered saline (PBS) supplemented with 2 mM MgCl2 and subjected to plaque assay. Heat-resistant mutants were selected by plaque purification and propagated in L cells to obtain P0 viral stocks. P0 stocks were diluted 1:10 in virion storage buffer (10 mM Tris-HCl [pH 7.4], 15 mM MgCl2, and 150 mM NaCl). A 200-μg/ml concentration of TLCK-treated chymotrypsin (Worthington Biochemical, Lakewood, NJ) was added to each sample. Samples were heated to 37°C for 30 min. The reaction was quenched by the addition of 1 mM phenylmethylsulfonyl fluoride (Sigma-Aldrich) and cooled on ice for 10 min. The reaction mixtures were divided in equivalent volumes and incubated at 4°C or 40°C for 20 min. These mixtures were used to initiate infection of L929 cells, and infectivity was determined by plaque assay. The change in infectivity at a given temperature (T) was calculated using the following formula: log10(PFU/ml)T − log10(PFU/ml)4°C.
Plaque titration.
Plaque assays were conducted in spinner-adapted L929 cells plated in 6-well plates (Greiner Bio-One). Cells were adsorbed with dilutions of virus in phosphate-buffered saline (PBS). Cells were overlaid with a molten mixture composed of 1× medium 199 and 1% Bacto agar supplemented with 10 μg/ml chymotrypsin. Five days following infection, the monolayers were fixed by addition of 4% formaldehyde solution in PBS and incubated overnight. The agar overlay was peeled off, and the monolayers were stained with 1% crystal violet stain in 5% ethanol for 5 h at room temperature. The monolayers were washed with water. Virus titer was quantified by manual counting of plaques.
Analysis of protein levels by immunoblotting.
The samples were whole-cell lysates of infected cells prepared using radioimmunoprecipitation assay (RIPA) lysis buffer (50 mM NaCl, 1 mM EDTA at pH 8, 50 mM Tris at pH 7.5, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS) supplemented with protease inhibitor cocktail (Roche) and 500 μM phenylmethylsulfonyl fluoride (PMSF), and they were resolved on 10% SDS-PAGE gels and transferred to nitrocellulose membranes. For immunoblotting using polyclonal rabbit antireovirus serum, the membranes were blocked with 5% milk in Tris-buffered saline (TBS) at room temperature for 1 h. Following blocking, rabbit antireovirus serum (1:1,000) or anti-PSTAIR was incubated with the membrane in appropriate blocking buffer at room temperature for 1 h. The membranes were washed with TBS supplemented with 0.1% Tween 20 (TBS-T) twice for 15 min and then incubated with Alexa Fluor-conjugated anti-rabbit IgG or anti-mouse IgG in blocking buffer. Following three washes, membranes were scanned using an Odyssey infrared imager (Li-Cor), and intensities of bands were quantified using Image Studio Lite software (Li-Cor).
Analysis of mRNA levels by RT-qPCR.
RNA was extracted from infected cells, at various times after infection, using a total RNA minikit (Bio-Rad). For RT-qPCR, 0.5 to 2 μg of RNA was reverse transcribed with a high-capacity cDNA RT kit (Applied Biosystems), using random hexamers included with the kit for amplification of cellular and viral genes. Undiluted cDNA was subjected to PCR using SYBR Select Master Mix (Applied Biosystems) and primers specific for T3D S1 and GAPDH (glyceraldehyde-3-phosphate dehydrogenase). Fold increases in gene expression with respect to control samples (indicated in the figure legends) were measured using the ΔΔCT method (51). ΔΔCT values and relative levels of gene expression were determined as follows: fold increase in viral gene expression = 2−ΔΔCT.
Statistical analyses.
The reported values are the means from three independent biological replicates. The error bars in the figures indicate standard deviations (SD). P values were calculated using Student's t test (two tailed; unequal variance assumed).
Modeling.
Molecular graphics were created and analysis were performed with the UCSF Chimera package (52).
REFERENCES
- 1.Sabin AB. 1959. Reoviruses. A new group of respiratory and enteric viruses formerly classified as ECHO type 10 is described. Science 130:1387–1389. doi: 10.1126/science.130.3386.1387. [DOI] [PubMed] [Google Scholar]
- 2.Gomatos PJ, Tamm I. 1963. The secondary structure of reovirus RNA. Proc Natl Acad Sci U S A 49:707–714. doi: 10.1073/pnas.49.5.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bellamy AR, Shapiro L, August JT, Joklik WK. 1967. Studies on reovirus RNA. I. Characterization of reovirus genome RNA. J Mol Biol 29:1–17. doi: 10.1016/0022-2836(67)90177-5. [DOI] [PubMed] [Google Scholar]
- 4.Dermody TS, Parker JSL, Sherry B. 2013. Orthoreoviruses, p 1304–1346. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 5.Gillies S, Bullivant S, Bellamy AR. 1971. Viral RNA polymerases: electron microscopy of reovirus reaction cores. Science 174:694–696. doi: 10.1126/science.174.4010.694. [DOI] [PubMed] [Google Scholar]
- 6.Skehel JJ, Joklik WK. 1969. Studies on the in vitro transcription of reovirus RNA catalyzed by reovirus cores. Virology 39:822–831. doi: 10.1016/0042-6822(69)90019-1. [DOI] [PubMed] [Google Scholar]
- 7.Liemann S, Chandran K, Baker TS, Nibert ML, Harrison SC. 2002. Structure of the reovirus membrane-penetration protein, μ1, in a complex with its protector protein, σ3. Cell 108:283–295. doi: 10.1016/S0092-8674(02)00612-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reinisch KM, Nibert ML, Harrison SC. 2000. Structure of the reovirus core at 3.6 Å resolution. Nature 404:960–967. doi: 10.1038/35010041. [DOI] [PubMed] [Google Scholar]
- 9.Mao ZX, Joklik WK. 1991. Isolation and enzymatic characterization of protein lambda 2, the reovirus guanylyltransferase. Virology 185:377–386. doi: 10.1016/0042-6822(91)90785-a. [DOI] [PubMed] [Google Scholar]
- 10.Baer GS, Dermody TS. 1997. Mutations in reovirus outer-capsid protein sigma3 selected during persistent infections of L cells confer resistance to protease inhibitor E64. J Virol 71:4921–4928. doi: 10.1128/JVI.71.7.4921-4928.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Silverstein SC, Astell C, Levin DH, Schonberg M, Acs G. 1972. The mechanisms of reovirus uncoating and gene activation in vivo. Virology 47:797–806. doi: 10.1016/0042-6822(72)90571-5. [DOI] [PubMed] [Google Scholar]
- 12.Dryden KA, Wang G, Yeager M, Nibert ML, Coombs KM, Furlong DB, Fields BN, Baker TS. 1993. Early steps in reovirus infection are associated with dramatic changes in supramolecular structure and protein conformation: analysis of virions and subviral particles by cryoelectron microscopy and image reconstruction. J Cell Biol 122:1023–1041. doi: 10.1083/jcb.122.5.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nibert ML, Fields BN. 1992. A carboxy-terminal fragment of protein mu 1/mu 1C is present in infectious subvirion particles of mammalian reoviruses and is proposed to have a role in penetration. J Virol 66:6408–6418. doi: 10.1128/JVI.66.11.6408-6418.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Borsa J, Sargent MD, Lievaart PA, Copps TP. 1981. Reovirus: evidence for a second step in the intracellular uncoating and transcriptase activation process. Virology 111:191–200. doi: 10.1016/0042-6822(81)90664-4. [DOI] [PubMed] [Google Scholar]
- 15.Nibert ML, Odegard AL, Agosto MA, Chandran K, Schiff LA. 2005. Putative autocleavage of reovirus mu1 protein in concert with outer-capsid disassembly and activation for membrane permeabilization. J Mol Biol 345:461–474. doi: 10.1016/j.jmb.2004.10.026. [DOI] [PubMed] [Google Scholar]
- 16.Odegard AL, Chandran K, Zhang X, Parker JSL, Baker TS, Nibert ML. 2004. Putative autocleavage of outer capsid protein micro1, allowing release of myristoylated peptide micro1N during particle uncoating, is critical for cell entry by reovirus. J Virol 78:8732–8745. doi: 10.1128/JVI.78.16.8732-8745.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang L, Chandran K, Nibert ML, Harrison SC. 2006. Reovirus mu1 structural rearrangements that mediate membrane penetration. J Virol 80:12367–12376. doi: 10.1128/JVI.01343-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chandran K, Farsetta DL, Nibert ML. 2002. Strategy for nonenveloped virus entry: a hydrophobic conformer of the reovirus membrane penetration protein μ1 mediates membrane disruption. J Virol 76:9920–9933. doi: 10.1128/jvi.76.19.9920-9933.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Snyder AJ, Wang J-Y, Danthi P. 2018. Components of the reovirus capsid differentially contribute to stability. J Virol 93:e01894-18. doi: 10.1128/JVI.01894-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thete D, Danthi P. 2018. Protein mismatches caused by reassortment influence functions of the reovirus capsid. J Virol 92:e00858-18. doi: 10.1128/JVI.00858-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harrison SJ, Farsetta DL, Kim J, Noble S, Broering TJ, Nibert ML. 1999. Mammalian reovirus L3 gene sequences and evidence for a distinct amino-terminal region of the lambda1 protein. Virology 258:54–64. doi: 10.1006/viro.1999.9707. [DOI] [PubMed] [Google Scholar]
- 22.Xu W, Coombs KM. 2009. Conserved structure/function of the orthoreovirus major core proteins. Virus Res 144:44–57. doi: 10.1016/j.virusres.2009.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chapell JD, Goral MI, Rodgers SE, dePamphilis CW, Dermody TS. 1994. Sequence diversity within the reovirus S2 gene: reovirus genes reassort in nature, and their termini are predicted to form a panhandle motif. J Virol 68:750–756. doi: 10.1128/JVI.68.2.750-756.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang X, Ji Y, Zhang L, Harrison SC, Marinescu DC, Nibert ML, Baker TS. 2005. Features of reovirus outer capsid protein mu1 revealed by electron cryomicroscopy and image reconstruction of the virion at 7.0 Angstrom resolution. Structure 13:1545–1557. doi: 10.1016/j.str.2005.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Snyder AJ, Danthi P. 2018. Infectious subviral particle to membrane penetration active particle (ISVP-to-ISVP*) conversion assay for mammalian orthoreovirus. Bio Protoc 8:e2700. doi: 10.21769/BioProtoc.2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Snyder AJ, Danthi P. 2015. Lipid membranes facilitate conformational changes required for reovirus cell entry. J Virol 90:2628–2638. doi: 10.1128/JVI.02997-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Middleton JK, Severson TF, Chandran K, Gillian AL, Yin J, Nibert ML. 2002. Thermostability of reovirus disassembly intermediates (ISVPs) correlates with genetic, biochemical, and thermodynamic properties of major surface protein mu1. J Virol 76:1051–1061. doi: 10.1128/jvi.76.3.1051-1061.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lemay G, Danis C. 1994. Reovirus λ1 protein: affinity for double-stranded nucleic acids by a small amino-terminal region of the protein independent from the zinc finger motif. J Gen Virol 75:3261–3266. doi: 10.1099/0022-1317-75-11-3261. [DOI] [PubMed] [Google Scholar]
- 29.Bisaillon M, Bergeron J, Lemay G. 1997. Characterization of the nucleoside triphosphate phosphohydrolase and helicase activities of the reovirus λ1 protein. J Biol Chem 272:18298–18303. doi: 10.1074/jbc.272.29.18298. [DOI] [PubMed] [Google Scholar]
- 30.Middleton JK, Agosto MA, Severson TF, Yin J, Nibert ML. 2007. Thermostabilizing mutations in reovirus outer-capsid protein mu1 selected by heat inactivation of infectious subvirion particles. Virology 361:412–425. doi: 10.1016/j.virol.2006.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Agosto MA, Middleton JK, Freimont EC, Yin J, Nibert ML. 2007. Thermolabilizing pseudoreversions in reovirus outer-capsid protein μ1 rescue the entry defect conferred by a thermostabilizing mutation. J Virol 81:7400–7409. doi: 10.1128/JVI.02720-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sarkar P, Danthi P. 2010. Determinants of strain-specific differences in efficiency of reovirus entry. J Virol 84:12723–12732. doi: 10.1128/JVI.01385-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Drayna D, Fields BN. 1982. Biochemical studies on the mechanism of chemical and physical inactivation of reovirus. J Gen Virol 63(Pt 1):161–170. doi: 10.1099/0022-1317-63-1-161. [DOI] [PubMed] [Google Scholar]
- 34.Wessner DR, Fields BN. 1993. Isolation and genetic characterization of ethanol-resistant reovirus mutants. J Virol 67:2442–2447. doi: 10.1128/JVI.67.5.2442-2447.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Farsetta DL, Chandran K, Nibert ML. 2000. Transcriptional activities of reovirus RNA polymerase in recoated cores. Initiation and elongation are regulated by separate mechanisms. J Biol Chem 275:39693–39701. doi: 10.1074/jbc.M004562200. [DOI] [PubMed] [Google Scholar]
- 36.McDonald SM, Tao YJ, Patton JT. 2009. The ins and outs of four-tunneled Reoviridae RNA-dependent RNA polymerases. Curr Opin Struct Biol 19:775–782. doi: 10.1016/j.sbi.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hooper JW, Fields BN. 1996. Role of the mu 1 protein in reovirus stability and capacity to cause chromium release from host cells. J Virol 70:459–467. doi: 10.1128/JVI.70.1.459-467.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Agosto MA, Myers KS, Ivanovic T, Nibert ML. 2008. A positive-feedback mechanism promotes reovirus particle conversion to the intermediate associated with membrane penetration. Proc Natl Acad Sci U S A 105:10571–10576. doi: 10.1073/pnas.0802039105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Snyder AJ, Danthi P. 2016. Lipids cooperate with the reovirus membrane penetration peptide to facilitate particle uncoating. J Biol Chem 291:26773–26785. doi: 10.1074/jbc.M116.747477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ivanovic T, Agosto MA, Zhang L, Chandran K, Harrison SC, Nibert ML. 2008. Peptides released from reovirus outer capsid form membrane pores that recruit virus particles. EMBO J 27:1289–1298. doi: 10.1038/emboj.2008.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang X, Walker SB, Chipman PR, Nibert ML, Baker TS. 2003. Reovirus polymerase λ3 localized by cryo-electron microscopy of virions at a resolution of 7.6 Å. Nat Struct Biol 10:1011–1018. doi: 10.1038/nsb1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Simon EJ, Howells MA, Stuart JD, Boehme KW. 2017. Serotype-specific killing of large cell carcinoma cells by reovirus. Viruses 9:140. doi: 10.3390/v9060140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dermody TS, Schiff LA, Nibert ML, Coombs KM, Fields BN. 1991. The S2 gene nucleotide sequences of prototype strains of the three reovirus serotypes: characterization of reovirus core protein sigma 2. J Virol 65:5721–5731. doi: 10.1128/JVI.65.11.5721-5731.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sherry B, Torres J, Blum MA. 1998. Reovirus induction of and sensitivity to beta interferon in cardiac myocyte cultures correlate with induction of myocarditis and are determined by viral core proteins. J Virol 72:1314–1323. doi: 10.1128/JVI.72.2.1314-1323.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thete D, Danthi P. 2015. Conformational changes required for reovirus cell entry are sensitive to pH. Virology 483:291–301. doi: 10.1016/j.virol.2015.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boehme KW, Ikizler M, Kobayashi T, Dermody TS. 2011. Reverse genetics for mammalian reovirus. Methods 55:109–113. doi: 10.1016/j.ymeth.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kobayashi T, Antar AAR, Boehme KW, Danthi P, Eby EA, Guglielmi KM, Holm GH, Johnson EM, Maginnis MS, Naik S, Skelton WB, Wetzel JD, Wilson GJ, Chappell JD, Dermody TS. 2007. A plasmid-based reverse genetics system for animal double-stranded RNA viruses. Cell Host Microbe 1:147–157. doi: 10.1016/j.chom.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Furlong DB, Nibert ML, Fields BN. 1988. Sigma 1 protein of mammalian reoviruses extends from the surfaces of viral particles. J Virol 62:246–256. doi: 10.1128/JVI.62.1.246-256.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mendez II, Hermann LL, Hazelton PR, Coombs KM. 2000. A comparative analysis of Freon substitutes in the purification of reovirus and calicivirus. J Virol Methods 90:59–67. doi: 10.1016/s0166-0934(00)00217-2. [DOI] [PubMed] [Google Scholar]
- 50.Smith RE, Zweerink HJ, Joklik WK. 1969. Polypeptide components of virions, top component and cores of reovirus type 3. Virology 39:791–810. doi: 10.1016/0042-6822(69)90017-8. [DOI] [PubMed] [Google Scholar]
- 51.Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 52.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]