

Epstein-Barr virus (EBV) has significant oncogenic potential that is linked to its latent infection of B lymphocytes, during which virus replication is not supported. The establishment of latent infection, which is lifelong and can precede tumor development by years, requires the concerted actions of nearly a dozen EBV proteins and numerous small non-protein-coding RNAs. Elucidating how these EBV products contribute to latency is crucial for understanding EBV’s role in specific malignancies and, ultimately, for clinical intervention. Historically, EBV genes that contribute to virus replication have been excluded from consideration of a role in latency, primarily because of the general incompatibility between virus production and cell survival. However, here, we provide evidence that the genetic locus containing one such gene, BHLF1, indeed contributes to key aspects of EBV latency, including its ability to promote the continuous growth of B lymphocytes, thus providing significant new insight into EBV biology and oncogenic potential.
KEYWORDS: Epstein-Barr virus, human herpesviruses, latency, noncoding gene
ABSTRACT
The Epstein-Barr virus (EBV) BHLF1 gene encodes an abundant linear and several circular RNAs believed to perform noncoding functions during virus replication, although an open reading frame (ORF) is retained among an unknown percentage of EBV isolates. Evidence suggests that BHLF1 is also transcribed during latent infection, which prompted us to investigate the contribution of this locus to latency. Analysis of transcripts transiting BHLF1 revealed that its transcription is widespread among B-cell lines supporting the latency I or III program of EBV protein expression and is more complex than originally presumed. EBV-negative Burkitt lymphoma cell lines infected with either wild-type or two different BHLF1 mutant EBVs were initially indistinguishable in supporting latency III. However, cells infected with BHLF1− virus ultimately transitioned to the more restrictive latency I program, whereas cells infected with wild-type virus either sustained latency III or transitioned more slowly to latency I. Upon infection of primary B cells, which require latency III for growth in vitro, both BHLF1− viruses exhibited variably reduced immortalization potential relative to the wild-type virus. Finally, in transfection experiments, efficient protein expression from an intact BHLF1 ORF required the EBV posttranscriptional regulator protein SM, whose expression is limited to the replicative cycle. Thus, one way in which BHLF1 may contribute to latency is through a mechanism, possibly mediated or regulated by a long noncoding RNA, that supports latency III critical for the establishment of EBV latency and lifelong persistence within its host, whereas any retained protein-dependent function of BHLF1 may be restricted to the replication cycle.
IMPORTANCE Epstein-Barr virus (EBV) has significant oncogenic potential that is linked to its latent infection of B lymphocytes, during which virus replication is not supported. The establishment of latent infection, which is lifelong and can precede tumor development by years, requires the concerted actions of nearly a dozen EBV proteins and numerous small non-protein-coding RNAs. Elucidating how these EBV products contribute to latency is crucial for understanding EBV’s role in specific malignancies and, ultimately, for clinical intervention. Historically, EBV genes that contribute to virus replication have been excluded from consideration of a role in latency, primarily because of the general incompatibility between virus production and cell survival. However, here, we provide evidence that the genetic locus containing one such gene, BHLF1, indeed contributes to key aspects of EBV latency, including its ability to promote the continuous growth of B lymphocytes, thus providing significant new insight into EBV biology and oncogenic potential.
INTRODUCTION
Epstein-Barr virus (EBV) is a potentially oncogenic herpesvirus able to persist for the life of its human host upon the establishment of a latent infection within B lymphocytes. The process through which this occurs is mediated through the concerted actions of a subset of EBV genes that are believed to direct a germinal center-like reaction, ultimately enabling infected cells to specifically enter the memory B-cell pool that serves as the primary reservoir of EBV and from which virus replication can be periodically reactivated (1). Because EBV-positive tumors predominantly support latent infection, and virus replication (i.e., lytic infection) is generally incompatible with cell survival, elucidation of the contributions of EBV to its associated malignancies has primarily focused on the latency-associated genes. These genes are typically expressed exclusively during latency, although some are also expressed upon the activation of the EBV replicative cycle (2–5). Conversely, the expression of EBV lytic-cycle genes within predominantly latently infected cell lines and tumors historically has been attributed to the sporadic and often abortive reactivation of the virus replication cycle in a subpopulation of cells; consequently, their potential contribution to EBV latency and its associated oncogenic potential has only rarely been considered.
An exception to the common presumption that EBV lytic-cycle genes do not contribute to latency came from the realization that a subset of these genes are expressed for a limited period immediately following infection and are required for the efficient immortalization of primary B cells by EBV in vitro, a hallmark property of latent EBV infection. These genes include those encoding the viral BCL-2-related prosurvival proteins BHRF1 (vBCL-2) and BALF1 (6) as well as BZLF1 (also known as Zta) (7, 8). Interestingly, BZLF1, an AP-1-related transcription factor responsible for the initiation of the EBV lytic cycle upon the reactivation of its expression from latent infection, performs a different function upon de novo infection, that of promoting cellular proliferation (8). In what appears to be consistent with this, SCID mice injected with B cells immortalized by BZLF1− EBV and humanized mice infected with BZLF1− virus are less prone to lymphoproliferative disease and lymphoma development, respectively, than upon receiving BZLF1+ cells or virus (9, 10). The EBV BCRF1 gene, which encodes an interleukin-10 (IL-10) homolog (vIL-10) (11), is also expressed early upon infection (12–14), and although there is conflicting evidence for a direct role of this protein in B-cell immortalization in vitro (12, 15, 16), it almost certainly contributes to latency in vivo through downregulation of the early immune response to newly infected B cells (13, 14), as does a second early-expressed lytic-cycle immunomodulatory protein, BNLF2a (13). Because the expression of these lytic-cycle proteins is short-lived, their contributions are believed to be restricted to this prelatency period, i.e., an establishment phase of latency prior to the exclusive expression of the classically defined latency genes in the majority of infected B cells (17).
Whether additional lytic-cycle genes have dual or even distinct roles during latency and virus replication is unclear. One such candidate is BHLF1, an early lytic-cycle gene for which there is mounting evidence of expression during latency as well. BHLF1 abuts oriLytLeft, one of two origins of DNA replication present within the EBV genome that are active only during the lytic cycle (a distinct origin of DNA replication, oriP, functions during latency). BHLF1 encodes a 2.5-kb unspliced, polyadenylated RNA that is highly expressed upon the induction of the lytic cycle within latently infected B-cell lines (18–22), and early DNA sequencing revealed a long open reading frame (ORF) that is within the transcribed region of the gene (18, 23). Interestingly, an apparent paralog of BHLF1 exists, LF3, which is adjacent to the second lytic-cycle origin of DNA replication (oriLytRight) (18, 24) (Fig. 1). BHLF1 transcripts are also detectable within latently infected B-cell lines and tumors by a variety of techniques (25–29), although this is not inconsistent with sporadic reactivation of the virus replication cycle in a subpopulation of cells. Several observations, however, have provided more direct evidence of the latency-associated expression of BHLF1. The first was the detection of BHLF1 transcripts upon infection of primary B cells in the presence of cycloheximide (30), which, along with a recent RNA sequencing (RNA-seq)-based analysis of EBV transcription through the first 2 weeks postinfection (p.i.), supports the transcription of BHLF1, at least during the prelatency phase (31). The second was the identification of putative latency-specific transcription initiation sites shortly upstream of the BHLF1 start site that is used upon the induction of the lytic cycle (32).
FIG 1.

Organization of the gene locus for BHLF1 and that of its paralog LF3. The EBV genome (top) is shown in its linear configuration (not to scale) bounded by its terminal repeats (TR); major internal direct repeat elements (IR1 to IR4) and the origin of EBV DNA replication utilized during latency (oriP) are shown for reference, as are the common EBNA promoters Cp and Wp and the EBNA1-only promoter Qp. The BHLF1 and LF3 genes overlap the two highly homologous origins of EBV DNA replication, oriLytLeft and oriLytRight, respectively, active during productive (lytic) infection. In some EBV genomes, a long ORF (colored arrows) is present within the transcribed regions of BHLF1 and LF3 that are composed primarily of related direct repeats (vertical lines) of 125 bp (IR2) and 102 bp (IR4), respectively; copy numbers of IR2 and IR4 repeats may vary and are based here on the complete composite EBV genome derived from the B95.8 and Raji isolates of EBV (GenBank accession number NC_007605.1). The ∼1-kbp duplicated-sequence domains DSL and DSR that encompass oriLytLeft and oriLytRight, respectively, are underscored by the green bar. Solid horizontal arrows depict previously characterized transcripts that are highly expressed from the BHLF1 and LF3 P1 promoters upon the induction of the EBV replicative cycle and that are unspliced and polyadenylated at sites indicated by short vertical arrows. Transcription start sites upstream of P1 that implicate latency-specific promoters have been mapped by a nuclease protection assay (P2) or were localized by RT-PCR (P3′, P3, and P4) (32). The structures of these transcripts have not been defined and thus are represented here as dashed arrows 3′ coterminal with the P1 transcripts from either locus. The ΔBHLF1 and ΔB-S deletions within mutant rEBVs used in this study are depicted below the expanded BHLF1 locus.
BHLF1 is remarkable in that 61% of the RNA-coding portion and 78% of the ORF are comprised of ∼12.3 copies of a 125-bp direct repeat that make up the internal repeat 2 (IR2) domain, alternatively known as the NotI repeats because each repeat contains a single NotI restriction site (18, 33, 34). Furthermore, its ORF has an unusually high GC content of 82% (79% within the RNA-encoding portion of the gene), which would contribute to a high percentage of Pro (21%) and Gly (15%) in the polypeptide that it is predicted to encode (as well as 14% Arg and 16% Ala) (18). In contrast, the average GC content of the EBV genome is 57% (23). These properties of the gene early on raised the possibility that BHLF1’s primary function may not be as a protein-coding gene. Direct evidence supporting a noncoding function was ultimately revealed by sequence analyses of the genomes of the Akata and Mutu EBV isolates, which revealed BHLF1 ORFs containing a premature termination codon and lacking a methionine initiation codon, respectively, relative to the ORF of the prototypical strain of EBV, B95.8 (35). Furthermore, while BHLF1 transcripts in latently infected B-cell lines are readily detected by RNA-seq (28, 31, 36, 37), analysis of the EBV transcriptome and proteome in parallel failed to detect BHLF1-encoded polypeptides in the same cell population, even upon the induction of the lytic cycle (38). Collectively, these observations are highly indicative of a non-protein-coding role for BHLF1, which may function primarily instead via its transcript as a long noncoding RNA (lncRNA). This appears to be true in the context of lytic infection, during which BHLF1 transcripts contribute to RNA-DNA duplexes at their coding locus to promote DNA replication mediated by the adjacent oriLytLeft (39). Recently, additional noncoding roles of BHLF1 during productive infection have been implicated by the detection of its RNA in virus-induced nodular structures on the periphery of nuclear viral replication compartments (40) and the discovery of circular RNAs (circRNAs) expressed from this locus upon the activation of the lytic cycle (41, 42). There are no clear indications, however, of how BHLF1 may contribute directly to EBV latency and long-term persistence.
To address the potential latency function of BHLF1, we generated recombinant EBVs (rEBVs) in which either the BHLF1 coding and 5′ regulatory regions or the DNA corresponding to the ORF alone had been deleted and monitored viral latency gene expression upon the infection of EBV-negative Burkitt lymphoma (BL) cell lines. Following infection of BL2 cells, both BHLF1− rEBVs were initially indistinguishable from wild-type (WT) rEBV in supporting the latency III (Lat III) program, in which the full complement of EBV latency-associated proteins are expressed. Ultimately, however, these BL cells infected with BHLF1− rEBV transitioned to Lat I, defined by the exclusive expression of the latency genome maintenance protein, EBNA1, whereas cells infected with WT rEBV sustained Lat III. A second cell line, BL30, likewise supported Lat III initially but in all infections transitioned to Lat I; the transition to Lat I, however, was noticeably delayed in BHLF1− relative to WT rEBV infections.
The complete inability to sustain Lat III in BL2 cells could not be attributed to an effect of the BHLF1 deletions on adjacent latency-associated genes encoding EBNA2, BHRF1, or the BHRF1-derived microRNAs (miRNAs). Furthermore, because our rEBVs were derived from the Akata isolate of EBV (whose genome lacks an intact BHLF1 ORF), this contribution of BHLF1 to latency is likely through a noncoding mechanism. This interpretation was strengthened by our finding that the transient expression of the BHLF1 protein from an intact ORF required the coexpression of the EBV posttranscriptional regulator protein SM, the expression of which is exclusive to the lytic cycle. Thus, even among isolates that retain a translatable ORF, protein function may be limited to lytic infection. Finally, BHLF1− rEBVs overall were less efficient in their immortalization of primary B cells, which, unlike EBV-negative BL cells, require the Lat III program of EBV for growth in vitro. Thus, one way in which BHLF1 may contribute to EBV latency is through a noncoding mechanism that favors the Lat III program of EBV, which is critical for the establishment of EBV latency and lifelong persistence within its host.
RESULTS
BHLF1 RNA is widely expressed within latently infected B-cell lines.
To obtain a clearer picture of the range of BHLF1 expression within the different latency programs maintained within EBV-infected B cells, we performed reverse transcription-quantitative PCR (RT-qPCR) on total RNA from B-cell lines that maintain either Lat I or Lat III. Because of the high degree of homology between BHLF1 and its paralog LF3, to ensure the specific detection of BHLF1 RNA, we amplified a region unique to BHLF1 that is immediately upstream of the IR2 repeats (Fig. 1) (18). Furthermore, to gauge to what degree BHLF1 RNA expression in these cell lines might be associated with spontaneous entry into the virus replication cycle, we determined in parallel the level of the early-lytic-cycle mRNA encoding the EBV protein SM. SM mRNA is abundant within EBV-positive B-cell lines that have been induced to replicate EBV (43). SM mRNA or the protein that it encodes has not been found to be expressed during latency; thus, the detection of its expression is a good indication that at least a subpopulation of otherwise latently infected cells have entered the lytic cycle even if abortively so.
BHLF1 transcripts were readily detected in all latently infected cell lines examined (Fig. 2A) regardless of whether the cells maintained Lat I (Kem I, Mutu I, and Akata clone 21 [A.21]) or Lat III (Ak-LCL, Kem III, and MH-LCL). As an additional indication of specificity for BHLF1, RNA was not detected in the BL line Sal, in which the entire BHLF1 locus is deleted from its endogenous EBV genomes (44), but was present within MH-LCL cells, which carry the B95.8 EBV genome from which LF3 has been deleted (24, 45). As expected, the induction of the EBV replicative cycle in A.21 BL cells resulted in substantial increases in BHLF1 and SM RNA expression (Fig. 2A). While SM RNA was also detected in all cell lines, the relative level of this lytic-cycle transcript did not always correlate positively with BHLF1 RNA. For example, and somewhat unexpectedly, Kem III cells exhibited the highest level of SM RNA (even higher than that in induced A.21 cells), yet BHLF1 RNA levels within Kem III cells were in line with those in cells that expressed relatively low levels of SM RNA, e.g., Kem I and MH-LCL. Conversely, MH-LCL cells expressed one of the higher levels of the BHLF1 transcript (exceeded only by induced A.21 cells) yet expressed one of the lower relative levels of SM RNA. In summary of the data presented in Fig. 2A, while in general, the BHLF1 transcript levels differed only modestly among latently infected B-cell lines, it was apparent that there was not always a direct correlation with SM expression. We concluded, therefore, that the regulation of BHLF1 expression is likely more complex and that the presence of its RNA in a population of otherwise latently infected cells is not ostensibly due to a subpopulation of cells that have entered the EBV replication cycle.
FIG 2.

Widespread expression of BHLF1 RNA in latently infected B-cell lines. RT-qPCR was used with three different primer sets to determine the relative levels of BHLF1 transcripts within B-cell lines that maintain Lat I or III or in Akata BL-derived Lat I lines (A.21 and BX1) that had been treated by cross-linking of surface IgG to induce the EBV replication cycle. (A, left) BHLF1 RNAs were amplified with a primer set specific for the unique-sequence domain (Unique BHLF1) immediately upstream of the IR2 repeats within the known BHLF1 mRNA and not present within RNAs encoded by LF3. (Right) Levels of SM transcripts were determined in parallel to provide an indication of the degree of EBV lytic-cycle gene transcription supported in each cell line. Levels of expression shown for both BHLF1 and SM transcripts are relative to those of the respective RNAs in Ak-LCL cells (Lat III), which were arbitrarily set at 1.0. (B) In addition to the primer set used to obtain the BHLF1 results in panel A, primer sets expected to detect the putative latency-specific transcripts initiating from P2 or P3′ (P2/P3′) or P3′ were used. Because these primers target the duplicated sequence elements present in the highly homologous BHLF1 and LF3 loci, RNA from MH-LCL cells was used as a reference, since the genome of the B95.8 isolate of EBV within this LCL lacks the LF3 locus, a consequence of an 11.5-kbp deletion. Conversely, the BHLF1 locus is absent from the EBV genomes within the Sal BL line, also due to a naturally occurring deletion; consequently, amplification of RNA from Sal cells with the P2/P3′ and/or P3′ primer sets would be indicative of LF3 transcription. The relative positions of the primer sets employed for reverse transcription and qPCR are shown as shaded bars in relation to their position within the various BHLF1 transcripts (see diagram). Kem I, Mutu I, A.21, and BX1 maintain Lat I, and Ak-LCL, Kem III, and MH-LCL maintain Lat III. Sal cells support Wp-restricted latency (Lat III but minus the expression of EBNA2, LMP1, and LMP2). BL2 and A.2 are EBV-negative BL cell lines.
Contributing to this complexity may be the expression of more recently identified transcripts originating shortly upstream of the BHLF1 promoter (P1) that directs the expression of the originally defined 2.5-kb BHLF1 mRNA. These are at least two leftward transcripts originating from the putative promoters P2 and P3′, which were implicated by apparent transcription start sites that were either mapped by RNase protection assays (P2) or localized by RT-PCR (P3′) (Fig. 1) (32). Importantly, these RNAs, whose structures have yet to be defined, were originally detected within B-cell lines maintaining Lat III, and their abundance, unlike that of P1-originating transcripts, was not notably increased upon chemical induction of the EBV replicative cycle, i.e., consistent with their expression during Lat III (32). To further explore the expression of these transcripts, we performed RT-qPCR with primer sets that would detect P3′-derived transcripts alone or P2 and P3′ together (assuming that P3′ transcripts overlap the primer-annealing sites within the body of the P2 RNAs). Because the amplified portions of the P2 and P3′ transcripts are either completely (P2) or partially (P3′) within the duplicated regions of the EBV genome that overlap the BHLF1 and LF3 genes (Fig. 1), to help distinguish between transcripts originating from these highly homologous loci, we again included in our analysis RNA isolated from the cell lines Sal and MH-LCL. The deletion in the Sal EBV genomes has removed the complete BHLF1 locus, including P2 and P3′ (44), and is thus BHLF1− LF3+; MH-LCL was generated by infection in vitro with the B95.8 isolate of EBV, the genome of which lacks 11.5 kbp of DNA due to a deletion that spans the LF3 locus and is thus BHLF1+ LF3− (24, 45). (Note that P3′ lies outside the duplicated domain in BHLF1, whereas LF3 P3 lies within its respective duplicated region; i.e., BHLF1 P3′ and LF3 P3 would be distinct promoters [32].)
As shown in Fig. 2B, transcripts consistent with initiation from P3′ or P2 and P3′ were detected in all cell lines tested but were more abundant in those that maintained Lat III (MH-LCL and Kem III) than in those that maintained Lat I (Kem I and BX1). Although we cannot completely exclude the possibility that some of these transcripts originated from LF3, we detected little or no transcripts within Sal BL cells (BHLF1− LF3+). While the previous report noted a lack of inducible expression of these transcripts upon the activation of the EBV lytic cycle within B-cell lines that maintain Lat III (32), we noted an 8- to 14-fold increase in their expression upon the induction of the lytic cycle in BX1 BL cells, which normally maintain Lat I (Fig. 2B, compare BX1 to induced BX1 results for the respective transcripts). This observation may be comparable to the induction of the three latency-associated latent membrane protein (LMP) genes upon the activation of the lytic cycle in Lat I BL lines, which do not express LMP1, -2A, or -2B during latent infection (5). The 60-fold induction in BX1 cells of transcripts amplified with a primer set specific for the unique region of BHLF1 (immediately upstream of the IR2 domain) would represent the previously characterized lytic-cycle BHLF1 transcript, in addition to P2 and/or P3′ transcripts, assuming that these extend through this BHLF1-unique domain that was targeted for amplification. Interestingly, in the experiments represented in Fig. 2B, we detected relatively few transcripts in Kem III cells with the primer set specific for the unique-region domain; in contrast, transcripts could be readily amplified from the same Kem III RNA with the P3 and P2/P3′ primer sets, possibly indicating that Lat III-specific transcripts expressed from P2 and P3′ are not entirely colinear with those from lytic-cycle-specific P1. In summary, our results indicated that the BHLF1 locus is transcribed to various degrees in all latently infected B-cell lines examined but that the expression of transcripts putatively originating from the promoters P2 and/or P3′ may be Lat III specific (their expression in Lat I-maintaining B cell lines had not been examined previously [32]). Moreover, the structures of these RNAs are likely to be more complex than the originally defined linear BHLF1 transcript.
BHLF1 protein expression is enhanced by SM.
In determining a role for BHLF1 in EBV latency, we considered a potential contribution by the protein that it has been reported to encode (46, 47). As a BHLF1-specific antibody was not available, we cloned the BHLF1 ORF from the genome of the prototypic EBV strain, B95.8, with a FLAG epitope-encoding tag at its 5′ end into a eukaryotic expression vector. Upon the transfection of EBV-negative BL cells with this vector, however, we repeatedly detected little or no FLAG-BHLF1 by immunoblotting, even though in our experience, the simian virus 40 (SV40) promoter in this vector (pSG5; Stratagene) is very active in EBV-negative BL cell lines. Because the EBV SM protein enhances the expression of a number of EBV replicative-cycle mRNAs through several posttranscriptional mechanisms (48, 49), we tested whether SM might be required for BHLF1 protein expression. As shown in Fig. 3, notable expression of FLAG-BHLF1 was achieved only by cotransfection with an SM expression vector and in a dose-dependent manner, suggesting that the efficient expression of the BHLF1 protein is SM dependent and thus would be limited to the EBV replication cycle.
FIG 3.
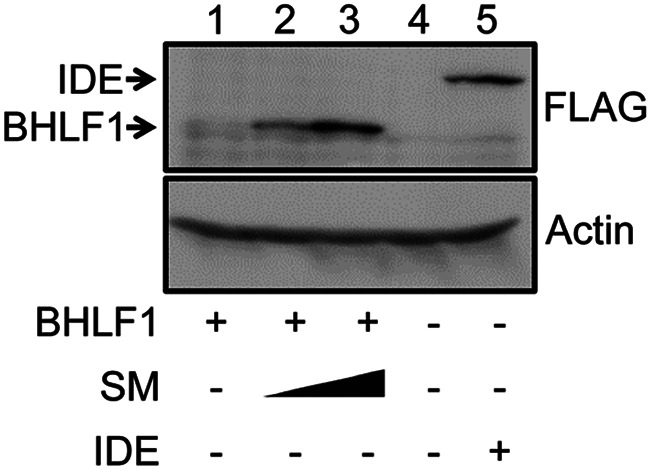
BHLF1 protein expression is enhanced by SM. EBV-negative Louckes BL cells were cotransfected with an expression vector encoding FLAG-tagged BHLF1 (origin, B95.8 EBV DNA) and 5 or 10 μg of the expression vector for the EBV SM protein or the empty pcDNA3 expression vector. +, −, and the closed triangle indicate the presence, absence, and increasing amounts of the indicated expression vector, respectively. All transfection mixtures contained an equal amount of plasmid DNA, adjusted with the empty expression vector (20 μg total plasmid DNA [lanes 1 to 3] and 10 μg total plasmid DNA [lanes 4 and 5]). Protein expression was detected by immunoblotting for the FLAG epitope (BHLF1 and insulin-degrading enzyme [IDE], a positive control for the detection of FLAG) or α-actin (gel-loading control). Results are representative of data from four experiments, each of which revealed a dependence on SM for efficient BHLF1 protein expression.
To test this in the context of virus infection, we originally sought to engineer an rEBV that would encode FLAG-BHLF1. During the generation of this rEBV on an Akata EBV genetic background, we discovered a single-base deletion (relative to the B95.8 EBV genome) 57 nucleotides after the start of the ORF, shifting the translational reading frame and resulting in a termination codon after an additional 16 nucleotides. Concurrent with our unpublished findings, Lin and colleagues reported the whole-genome nucleotide sequences for the Akata and Mutu EBV isolates, revealing an identical single-base change in the Akata BHLF1 ORF and the absence of a likely ORF in the BHLF1 locus as well within the Mutu EBV genome (35). Taken together, these observations and the data in Fig. 3 suggested that during latency, BHLF1 transcripts may function as lncRNAs and are able to function efficiently as mRNAs for BHLF1 protein expression only upon the activation of the EBV replication cycle (and SM expression) but only from the genomes of EBV isolates for which the BHLF1 ORF has been conserved.
BHLF1 supports Lat III in established B-cell lines.
To determine the potential contribution of BHLF1 to EBV latency, we infected the EBV-negative BL cell line BL2 with either WT rEBV or our previously described mutant rEBV (ΔB-S) in which the entire BHLF1 ORF and 5′ promoter region (including P2 and P3′) had been deleted (50). This 3,264-bp deletion also removes oriLytLeft and extends to the right boundary of the largest reported naturally occurring deletion found in EBV genomes within a subset of BLs and the cell lines derived from them that maintain Wp-restricted latency (44) (Fig. 1). Six independently derived cell lines infected with either WT or ΔB-S rEBV were analyzed for latency-associated gene expression upon outgrowth in the presence of G418, the resistance to which is encoded within these bacterial artificial chromosome (BAC)-derived rEBVs. As shown in Fig. 4A (left), we observed the establishment of Lat III in all WT and ΔB-S rEBV infections, as indicated by the detection of EBNA1, EBNA2, the three EBNA3 proteins (EBNA3A, -3B, and -3C), and LMP1 through at least 30 days postinfection (p.i.). However, by ∼2 months p.i., lines infected with ΔB-S rEBV appeared to have all transitioned to a Lat I program, as suggested by the detection of EBNA1 only (Fig. 4A, right).
FIG 4.

The BHLF1 locus is required to sustain Lat III. (A) Immunoblot detection of EBV latency-associated proteins expressed in BL2 cells 30 and 68 days after infection with either WT or ΔB-S rEBV revealed a shift from Lat III to a Lat I-specific pattern of EBV protein expression (EBNA1 only) in cells infected with BHLF1− rEBV. All BL2 cell lines (cells lines 1 to 6) resulted from independent infections by WT or mutant rEBV. The detection of α-actin or β-tubulin served as a loading control. (B) Analysis of EBNA1 promoter usage by endpoint RT-PCR at 28, 41, and 68 days postinfection (dpi) revealed a shift from Cp/Wp- to Qp-driven EBNA1 mRNA expression (Lat III to Lat I) in BL2 cells infected with ΔB-S rEBV, which corresponded to the loss of Lat III-specific protein expression as seen in panel A. Amplifications of EBNA1 cDNAs generated from the RNAs of Kem I (Lat I), Kem III (Lat III), and BL2 (EBV-negative) cells served as controls. (C) A similar decrease in EBNA2 and EBNA3C mRNA expression was detected in BL2 cells infected with ΔB-S but not with WT rEBV.
To determine whether this was likely to be a bona fide Lat III-to-Lat I transition, we assessed whether EBNA1 promoter usage had indeed shifted from Cp/Wp (Lat III) to Qp (Lat I) in ΔB-S rEBV infections. As shown in Fig. 4B, RT-PCR analysis of the EBNA1 mRNA structure revealed a gradual transition from Cp/Wp- to Qp-driven expression of EBNA1 mRNAs in ΔB-S infections, whereas Cp/Wp usage was sustained in WT rEBV infections (the primers used do not distinguish between a Cp and a Wp origin of these transcripts). Consistent with the apparent silencing of Cp/Wp in ΔB-S infections, the levels of EBNA2 and EBNA3C mRNAs (which originate only from Cp or Wp) also decreased (Fig. 4C), although these Cp/Wp-specific transcripts remained detectable when their encoded proteins were not, most likely due to a greater sensitivity of RT-PCR than of immunoblotting for the assessment of EBNA gene expression. We also confirmed that the BHLF1 P2/P3′ locus was indeed transcribed in BL2 cells infected with WT rEBV, and the inability to amplify these transcripts upon infection with ΔB-S rEBV supports the conclusion that these transcripts in WT rEBV infections originated from the BHLF1 locus and not from the highly homologous LF3 locus (Fig. 5). Based on these data, we concluded that the deletion in the ΔB-S rEBV genome precluded the long-term maintenance of Lat III, ultimately resulting in an apparent shift to the Lat I transcriptional program.
FIG 5.
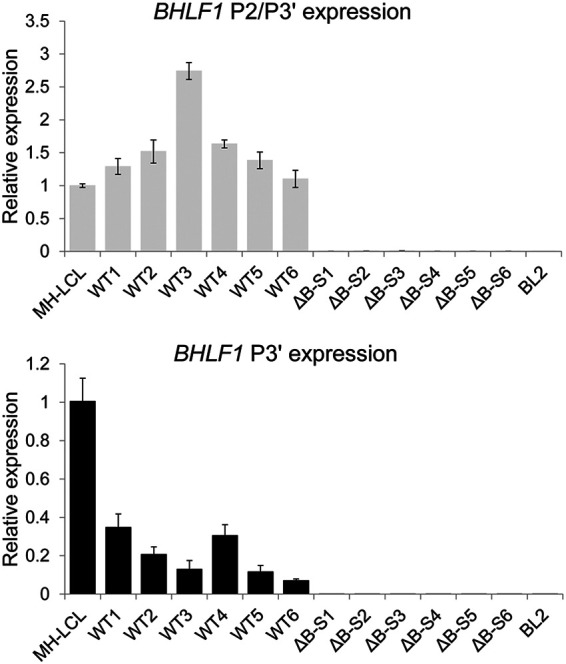
BHLF1 RNA levels within six independently derived BL2 cell lines approximately 1 month after infection with either WT or ΔB-S rEBV were determined by RT-qPCR in triplicate. Error bars indicate the standard deviations. Values are relative to the level of RNA in MH-LCL (LF3−) cells determined using primer sets specific for transcripts initiating from P2 and/or P3′ (P3′-initiating transcripts may overlap those from P2) or at P3′ alone (see diagram in Fig. 2B). RNA from BL2 cells infected with ΔB-S rEBV (BHLF1− LF3+) was included to help exclude the possibility that products amplified with the P2/P3′ and P3′ primer sets had originated from the highly homologous LF3 locus. Comparable results were obtained in separate experiments when amplification was done using a primer set specific for the unique-sequence domain of BHLF1 (data not shown).
While the results presented in Fig. 4 implicated a role for the BHLF1 locus in the maintenance of Lat III, the deletion in ΔB-S rEBV also removed DNA encoding the EBV miRNA miR-BHRF1-1, one of three miRNAs that are encoded within the BHRF1 locus upstream of BHLF1 in the opposite transcriptional orientation and that are expressed during Lat III (51, 52). Although the phenotypes associated with a targeted mutation of the three BHRF1 miRNAs (either together or individually) in the context of Lat III in primary B lymphocytes did not appear to be consistent with an inability to sustain Lat III gene expression (53–55), we nonetheless sought to exclude the possibility that the loss of miR-BHRF1-1 expression contributed to the inability of BL2 cells to sustain Lat III. We therefore generated a second mutant rEBV, ΔBHLF1, in which only the DNA corresponding to the BHLF1 ORF (as in B95.8-like isolates) was deleted (Fig. 1). The BL2 infection experiments described above were then repeated with WT and ΔBHLF1 rEBVs, and the results are shown in Fig. 6. In all cases, BL2 lines infected with WT rEBV sustained Lat III, as observed previously (data not shown), whereas those infected with ΔBHLF1 ultimately transitioned to Lat I, as had the lines infected with ΔB-S rEBV. This was evident at both the protein and mRNA levels (Fig. 6A and B, respectively), although overall, in multiple experiments, it seemed that the cells infected with ΔBHLF1 took slightly longer to complete the transition to Lat I than what was observed for the ΔB-S rEBV infections. Finally, we also considered whether the transition to Lat I may have occurred due to a silencing of the EBV genome as a consequence of the integration of the BHLF1− genomes in EBV-negative BL cells (56). However, we were readily able to rescue episomal copies of the EBV genome into Escherichia coli from Hirt extracts of these infected BL2 lines, and the amount of rescued episomes corresponded well to the total EBV DNA copy number (data not shown), arguing against the integration of a substantial fraction of viral genomes.
FIG 6.
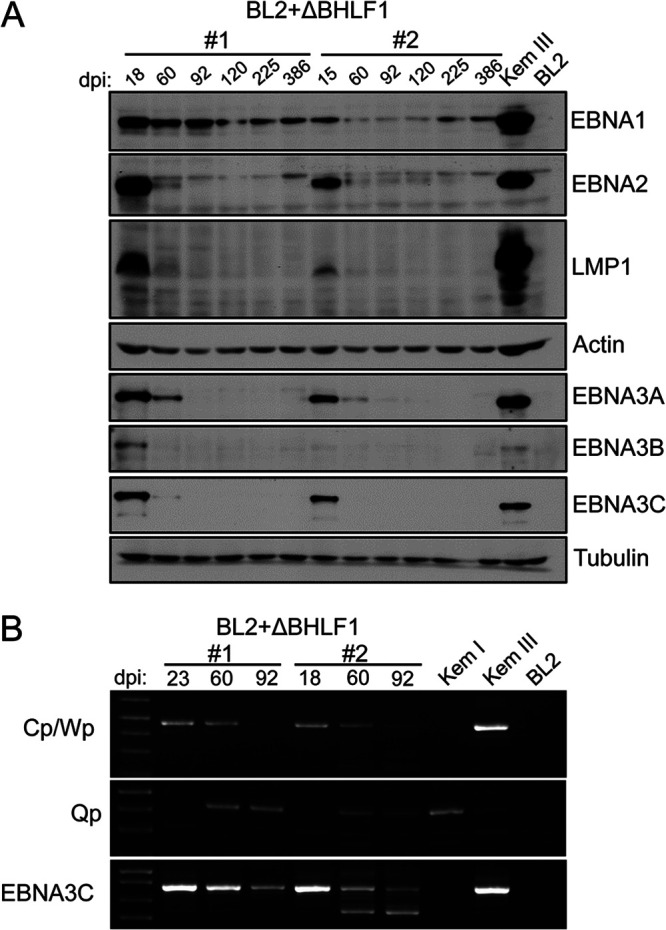
rEBV lacking the BHLF1 ORF alone is unable to sustain Lat III. EBV latency gene expression was assessed in BL2 cells infected with ΔBHLF1 rEBV at 18 to 386 days p.i., as described in the legend for Fig. 4. (A) Detection of EBNA and LMP1 expression by immunoblotting. (B) Detection by endpoint RT-PCR of EBNA1 mRNAs from Cp/Wp or Qp and of EBNA3C mRNA at 18/23, 60, and 92 dpi. The smaller EBNA3C cDNA bands detected at 60 and 92 days p.i. in ΔBHLF1 rEBV-infected cell line 2 were determined by DNA sequence analysis to be splicing variants of the EBNA3C mRNA. BL2 cells infected in parallel with WT rEBV failed to transition to Lat I (data not shown), as also observed independently in Fig. 4.
We next repeated our analysis of both BHLF1− rEBVs within the context of a second EBV-negative BL line, BL30. While every infection of BL2 cells with WT rEBV resulted in sustained Lat III, this was less pronounced upon infection of BL30 cells. As revealed by RT-PCR analysis of Cp/Wp usage in BL30 cells at 1 and 3 months p.i., two of the six infections with WT rEBV (infections 2 and 6) appeared to transition to Lat I by 3 months p.i. (Fig. 7A, top). While we observed notable Cp/Wp usage at 3 months p.i. in the remaining four infections with WT rEBV, all WT rEBV infections ultimately transitioned to Lat I (data not shown). In contrast, upon infection with either BHLF1− rEBV, the transition to Lat I was more pronounced by 3 months p.i. (Fig. 7A, compare ΔBHLF1 and ΔB-S to the WT). The generally delayed conversion to Lat I in BL30 cells infected with WT relative to BHLF1− rEBV was more pronounced upon the reduction of PCR cycles, as shown by the results for three representative BL30 lines for each infection (Fig. 7B). (Accurate analysis of Cp/Wp usage by RT-qPCR is difficult due to the presence of submolar concentrations of cDNA products representing alternative splicing events and with more than one copy of the W1-W2 exon repeat unit of the EBNA mRNAs spanning the large internal repeat [IR1] domain.) Analysis by RT-qPCR of Qp usage in each infection revealed an increase in Lat I EBNA1 expression over 3 months p.i. (Fig. 7C), consistent with reduced Cp/Wp usage over the same time frame. For reasons that are not clear, the levels of Qp-specific transcripts in the ΔB-S infections were approximately 10-fold higher than those in the WT and ΔBHLF1 infections and generally were equal to or higher than the level of Qp-derived EBNA1 transcripts in the Lat I positive-control line, Kem I. In summary, while BL30 cells infected with WT rEBV ultimately supported the transition to Lat I, unlike BL2 cells, the deletion of the BHLF1 locus appeared to accelerate this process. We concluded, therefore, that BHLF1 supports Lat III, but the degree to which it does this is cell specific.
FIG 7.

BHLF1 contributes to but is not essential for the transition to Lat I in BL30 BL cells. EBV-negative BL30 cells were infected with either WT, ΔBHLF1, or ΔB-S rEBV, and cell lines resulting from six independent infections with each virus were assessed by RT-PCR for Cp/Wp (Lat III) and Qp (Lat I) usage at 1 and 3 months p.i. (mpi). The results indicated that BL30 cells infected with WT rEBV were able to support Lat I, but their apparent transition to Lat I was generally delayed relative to cells infected with either BHLF1− virus. (A) Detection by endpoint RT-PCR (35 cycles) of Cp/Wp usage for EBNA1 mRNA expression in six cell lines 1 and 3 months after infection with either WT, ΔBHLF1, or ΔB-S rEBV. GAPDH mRNA was amplified in parallel as a control for RNA integrity. Kem I (Lat I), Kem III (Lat III), and uninfected BL30 cells served as controls. (B) Cell lines 3, 4, and 5 from each infection were reassessed by endpoint RT-PCR at 30 cycles to highlight the generally delayed transition (loss of Cp/Wp usage) to Lat I in BL30 cells infected with WT rEBV, as in WT lines 4 and 5. The smaller, submolar amplification product seen in panels A and B represents an alternatively spliced transcript, which complicates assessment by RT-qPCR. (C) Quantification by RT-qPCR of Qp usage for EBNA1 mRNA expression indicative of Lat I. Data were analyzed by the ΔΔCT method, with expression values normalized to GAPDH mRNA values; the Qp values for Kem I cells (Lat I) were set at 1. Note that the expression scale is different in the bottom panel due to the larger amounts of Qp-derived transcripts in BL30 cells infected with ΔB-S rEBV.
Deletion of the BHLF1 locus has a minimal influence on BHRF1 mRNA and miRNA expression.
The deletions introduced to generate ΔB-S and ΔBHLF1 rEBVs were within ∼550 bp of the 3′ end of the EBNA2 mRNA to the left and within ∼1,800 and ∼530 bp of the BHRF1 3′ coding exon and ORF to the right. Also, as noted above, ΔB-S completely removes the DNA encoding the EBV miRNA miR-BHRF1-1 within the BHRF1 locus, whereas the ΔBHLF1 deletion is approximately 1,200 bp upstream of the miR-BHRF1-1 locus; the remaining BHRF1 miRNAs are derived from the 3′ end of the BHRF1 gene. We did not observe gross differences in EBNA2 expression from either deletion for at least 2 to 4 weeks p.i. (Fig. 4 and 6), arguing against a negative influence on EBNA2 expression being directly responsible for the transition to Lat I, e.g., due to the activation of Qp by default in the absence of sufficient EBNA2 to sustain transcription from Cp. We also did not observe any effect of these deletions during this time frame on the expression of the EBNA3s and EBNA1, whose primary transcripts from Cp/Wp transverse the BHLF1 locus, the deletion of which might have interfered with pre-mRNA processing, altering the expression of their mRNAs. To determine whether the phenotype common to both deletions might have been due to an influence on the expression of the adjacent BHRF1 locus, we measured the levels of the latency-associated BHRF1 mRNAs that encode EBV vBCL-2, an antiapoptotic homolog of BCL-2 (57–59). We also assessed the expression of the EBV miRNAs miR-BHRF1-1, miR-BHRF1-2, and miR-BHRF1-3, which are generated from transcripts transiting the BHRF1 locus and which are normally expressed during Lat III (51, 52).
BHRF1 mRNA expression during latency is driven by the EBNA promoter Wp and possibly Cp, and consequently, the 5′ leaders of these BHRF1 mRNAs share an exonic structure with the leaders of the EBNA mRNAs (57–59). These contain multiple copies of the two-exon repeat W1-W2, each derived from a copy of the IR1/BamHI-W restriction fragment; the last W2 exon is typically spliced to the first of three short unique-sequence exons (Y1, Y2, and then Y3) encoded within the adjacent BamHI-Y restriction fragment, although we have observed that the Y3 exon is rarely included within BHRF1 mRNAs (50). The Y2 or Y3 exon is ultimately spliced to the single long 3′ exon that contains the entire BHRF1 ORF within the BamHI-H fragment (57–59). A separate promoter ∼600 bp upstream of the BHRF1 ORF (and removed by ΔB-S but not ΔBHLF1) is used for BHRF1 transcription during lytic infection (57).
To measure BHRF1 mRNA levels by RT-qPCR, we employed a common reverse primer specific for the 3′ coding exon and forward (5′) primers specific for either the W2 or Y2 exon. The results for each of six independently derived BL2 lines infected with either WT, ΔB-S, or ΔBHLF1 rEBV are shown in Fig. 8. In WT rEBV infections, when employing a W2-specific forward primer, the level of BHRF1 mRNA in all BL2 lines was equivalent (on average, <2-fold higher) to BHRF1 mRNA levels in the reference cell line Ak-LCL. When using the Y2-specific primer, the levels observed for BHRF1 mRNAs in the same WT rEBV-infected BL2 lines ranged from one-half to equivalent to the levels observed for Ak-LCL. In the BL2 lines infected with ΔB-S rEBV, the levels of BHRF1 mRNA were approximately 2-fold higher than those in their WT rEBV-infected counterparts when assessed with a W2-specific primer and 3-fold higher than those for the WT when using the Y2 primer. In ΔBHLF1 rEBV infections, the levels of BHRF1 mRNAs were only marginally higher than those in the WT control infections, regardless of the forward primer used. Thus, although the increased levels of BHRF1 mRNA associated with either deletion were minimal, we noted that the larger deletion (ΔB-S) was associated with a greater increase in expression. This was not unexpected, as previous work demonstrated that large deletions that remove all of the EBNA2 and most or all of the BHLF1 loci in the EBV genomes within the so-called Wp-restricted BL lines are associated with increased BHRF1 expression (59, 60), as shown here for the BHRF1 mRNAs amplified with the W2-specific primer from RNA isolated from the Wp-restricted BL line Sal. Given the prosurvival function of the BHRF1 protein and that neither of the BHLF1 deletions resulted in a decrease in BHRF1 mRNA expression over several weeks p.i., we considered it unlikely that the reduced ability to support Lat III was a direct consequence of any change in BHRF1 expression.
FIG 8.

Deletion of the BHLF1 locus does not negatively impact the expression of BHRF1 mRNA. The expression of latency-associated BHRF1 mRNA was analyzed by RT-qPCR 1 month after infection with either WT versus ΔB-S rEBV (A) or WT versus ΔBHLF1 rEBV (B). Each of the 6 WT lines in panel A is distinct from the 6 WT lines in panel B; i.e., a total of 12 independent BL2 lines infected with WT rEBV were analyzed. Each bar represents the mean relative level of expression determined, in triplicate, for BHRF1 mRNA in the tested line relative to the respective BHRF1 mRNA level (set at 1.0) in the Lat III reference line Ak-LCL. The forward (5′) PCR primers used were specific for either the W2 or Y2 exon present within EBNA and BHRF1 mRNAs expressed from either Cp or Wp. The Wp-restricted BL line Sal was included as an additional reference, as it contains a deletion in its endogenous EBV genomes that removed the DNA encoding the Y2 exon, rightward through EBNA2 and BHLF1, to the same relative 3′ coordinate as the deletion within ΔB-S rEBV (consequently, Y2-exon-specific detection of BHRF1 mRNA in Sal was negative). Error bars indicate the standard errors of the means.
We next addressed whether our BHLF1 deletions had perturbed the expression of miR-BHRF1-1, miR-BHRF1-2, or miR-BHRF1-3. As shown in Fig. 9, we observed variable but generally only minor differences in the expression of any of the BHRF1 miRNAs between infections with the WT and infections with ΔB-S and ΔBHLF1 rEBVs. The exception was the expected absence of miR-BHRF1-1 in BL2 cells infected with the ΔB-S virus and in the reference line Sal (a Wp-restricted BL line), in which the introduced deletion in ΔB-S and the naturally occurring deletion in the Sal EBV genome extend through the coding region for miR-BHRF1-1. Thus, we concluded that the observed inability to sustain Lat III in BL2 cells was a direct consequence of the loss of the BHLF1 locus and a specific function that it performs.
FIG 9.

Deletion of the BHLF1 locus does not impact the expression of BHRF1 miRNAs. The expression of miR-BHRF1-1, miR-BHRF1-2, and miR-BHRF1-3 at 1 month p.i. was assessed by RT-qPCR for the same BL2 cells as the ones analyzed for BHRF1 mRNA expression in Fig. 8. BL2 cells were infected with WT or ΔB-S rEBV (A) or WT or ΔBHLF1 rEBV (B). Each bar represents the mean relative level of expression determined, in triplicate, for each of the three BHRF1 miRNAs relative to the respective BHRF1 miRNA level (set at 1.0) in the Lat III reference line Ak-LCL. miR-BHRF1-1 is absent in BL2 cells infected with ΔB-S rEBV and the BL line Sal due to introduced or naturally occurring deletions in the EBV genomes within these lines, respectively. Error bars indicate the standard errors of the means.
BHLF1 contributes to EBV immortalization and growth of primary B lymphocytes.
Lat III is critical for the initial stage of EBV infection of B lymphocytes that leads to lifelong EBV persistence within B cells of its human host and is also required for sustained growth (immortalization) of primary B cells upon EBV infection in vitro, a hallmark property of EBV linked to its oncogenic potential. Given the defect or inefficiency in maintaining Lat III that was exhibited by our two BHLF1− viruses in BL2 and BL30 cells, which do not require EBV for sustained growth in vitro, and the relatively late manifestation of this effect in BL2 cells, we asked whether this would translate to an inability of BHLF1− EBV to immortalize primary B cells. Purified B cells from four healthy adult donors were therefore infected with WT, ΔB-S, or ΔBHLF1 rEBV, and B-cell growth transformation/immortalization was scored at 6 weeks p.i. (due to a limiting number of B cells, only those from donors 1 and 4 were infected with both BHLF1− rEBVs). As illustrated in Fig. 10, in a total of five independent experiments (B cells from donor 1 were assessed twice), 100% transformation was achieved by WT rEBV infection of B cells from all four donors at multiplicities of infection (MOIs) of ∼0.8 × 10−2 to 1.3 × 10−2. In contrast, for donor 1 B cells, ΔB-S rEBV was unable to immortalize B cells over the range of MOIs tested, whereas we observed inefficient transformation by ΔBHLF1 rEBV, i.e., >10-fold lower than that by WT rEBV. Similarly, ΔBHLF1 rEBV was clearly deficient relative to WT rEBV in the transformation of B cells from donors 3 and 4. For reasons that are unclear, transformations of donor 3 and 4 B cells by ΔBHLF1 and ΔB-S rEBVs, respectively, occurred only in wells at the midrange of MOIs tested. Finally, unlike for donors 1, 3, and 4, we did not observe a difference between WT and ΔBHLF1 rEBVs in growth transformation of B cells from donor 2, although we noted that B cells from this donor appeared to be slightly (∼2 to 3 times) more sensitive to transformation by WT rEBV than B cells from donors 1, 3, and 4.
FIG 10.
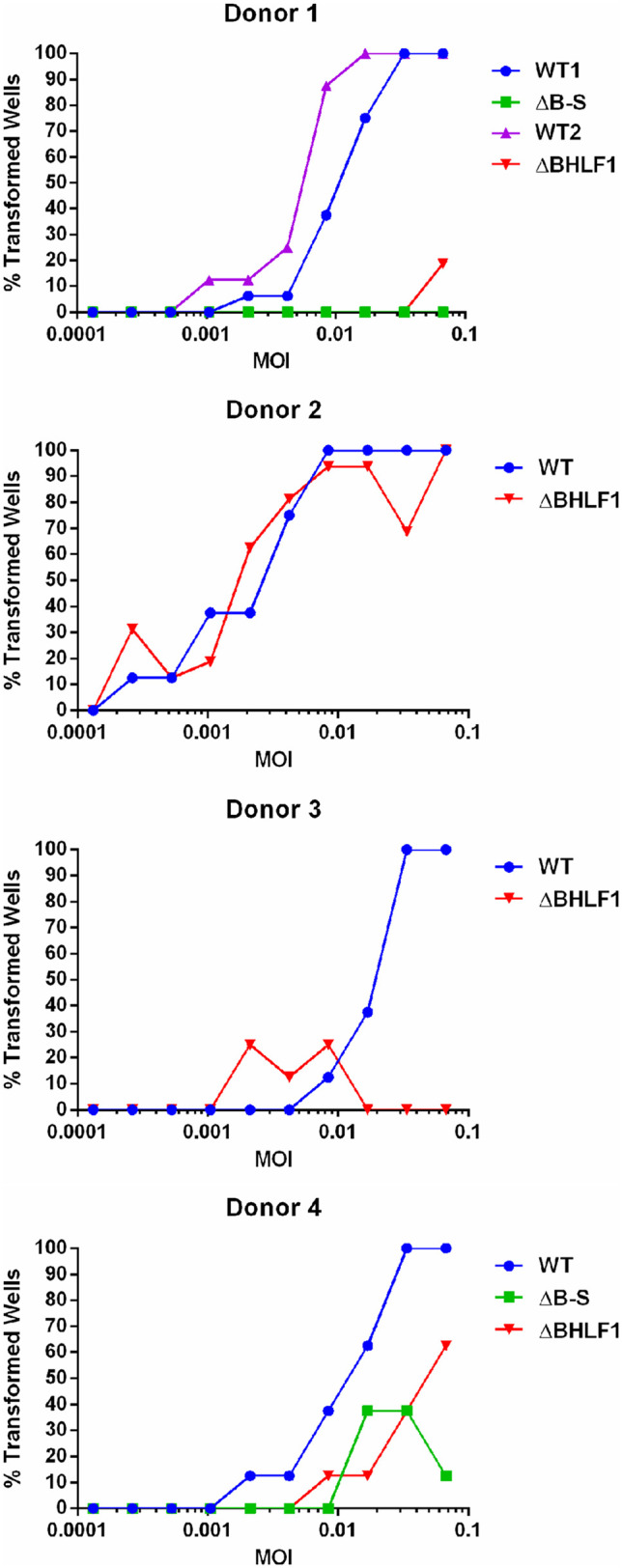
BHLF1 contributes to EBV-mediated B-cell immortalization. Primary B lymphocytes from four adult donors were infected in vitro with either WT, ΔB-S (donors 1 and 4 only), or ΔBHLF1 rEBV at the indicated multiplicity of infection (MOI). Immortalization was scored at 6 weeks p.i. and is presented as a percentage of wells out of eight for each MOI that exhibited outgrowth of cells. The paired WT1 and ΔB-S infections and WT2 and ΔBHLF1 infections of donor 1 B cells were done at different times (cells were frozen for later infection with WT and ΔBHLF1 rEBVs). Unlike for B cells from donors 1, 3, and 4, donor 2 B cells were equivalently susceptible to immortalization by WT and ΔBHLF1 rEBVs; however, donor 2 cells were ∼3 to 4 times more sensitive to immortalization than those of the other three donors.
Given the variable requirement for BHLF1 in our immortalization assays, we expanded B cells from donor 3 that had scored positive for immortalization following infection with either WT or ΔBHLF1 rEBV and performed a comparative analysis of their growth properties. Interestingly, lymphoblastoid cell lines (LCLs) could not be as easily established from ΔBHLF1 rEBV-infected B cells as from those infected with WT rEBV. Of 11 ΔBHLF1 lines that were established, 2 were found by PCR to contain BHLF1 DNA, presumably due to the outgrowth of B cells containing the donor’s endogenous EBV (note that this did not explain the transformation by ΔBHLF1 rEBV observed only in infections at midrange MOIs). An analysis of EBV EBNA and LMP expression in the nine ΔBHLF1 lines by immunoblotting (as in Fig. 4 and 6) did not reveal any gross differences relative to LCLs transformed with WT rEBV (data not shown). To determine whether ΔBHLF1 LCLs had lower growth potentials, several lines each of WT and ΔBHLF1 rEBV-infected B cells were seeded at a moderately low density (105 cells per ml), and the viable-cell concentration was monitored daily. As shown by the representative growth curves in Fig. 11 (top), LCLs infected with WT rEBV exhibited virtually identical growth rates, with each line going through ∼4.5 doublings to reach a maximum density of ∼1.2 × 106 cells per ml. In contrast, LCLs infected with ΔBHLF1 rEBV either did not expand at all (Fig. 11, bottom, cell lines 1 and 2) or did so more slowly than their WT rEBV-infected counterparts, going through ∼3 doublings to reach a slightly lower maximum density of ∼1 × 106 cells per ml (Fig. 11, bottom, cell lines 5 and 6). Consistent with this apparent growth deficiency, cultures of ΔBHLF1 LCLs routinely exhibited lower cell viabilities than those infected with WT rEBV.
FIG 11.
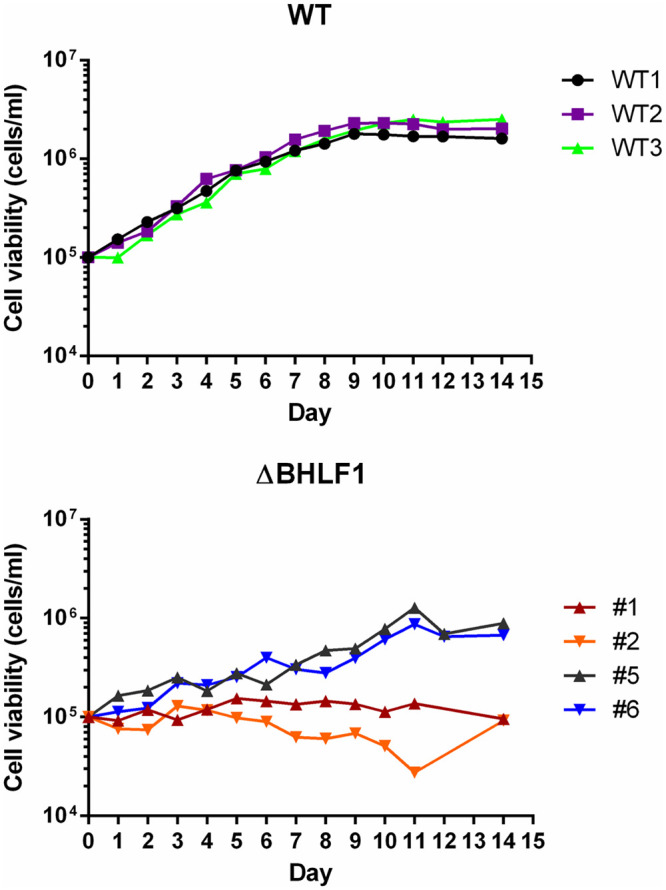
B lymphocytes immortalized with ΔBHLF1 rEBV exhibit reduced growth properties. Shown are representative growth curves of three B LCLs immortalized by WT rEBV (top) and four immortalized by infection with ΔBHLF1 rEBV (bottom). All LCLs were derived from B-cell donor 3 (Fig. 10). Cells were seeded in triplicate at 1 × 105 cells per ml, and the mean viable-cell number per milliliter of culture medium was determined daily afterwards.
These experiments were subsequently repeated with early-passage LCLs derived from the B cells from donor 4, which had also been infected with ΔB-S rEBV (LCLs from donors 1 and 2 were unavailable). Interestingly, while we initially observed little difference in the growth curves of donor 4 LCLs infected with either WT, ΔBHLF1, or ΔB-S virus (4 lines each), each of the LCLs infected with BHLF1− virus subsequently went through crisis and could no longer be propagated, preventing us from performing further analysis on long-term LCLs as we had for the BHLF1− LCLs derived from donor 3 (Fig. 11). Collectively, the results presented in Fig. 10 and 11, and our difficulty in expanding and maintaining LCLs infected with either BHLF1− virus, support our conclusion that the BHLF1 locus contributes to B-cell growth.
DISCUSSION
Originally assigned to the early class of EBV genes (20), BHLF1 has long been believed to contribute to EBV biology exclusively within the context of the virus replication cycle. Here, we provide evidence of a contribution of the BHLF1 locus to the latent phase of EBV infection, which is intimately linked to the oncogenic potential of this herpesvirus. Specifically, EBV-negative BL cells (BL2) that upon infection stably support a Lat III program of EBV protein expression were unable to do so after infection with BHLF1− virus, instead transitioning to the more restrictive Lat I program. In a BL line that naturally transitioned from Lat III to Lat I (BL30), the loss of the BHLF1 locus appeared to accelerate the transition to Lat I. Seemingly consistent with this, upon infection of primary B cells, which, unlike BL cells, require the Lat III program for sustained growth in vitro, BHLF1− rEBVs exhibited decreased growth-transforming potential relative to WT rEBV. We found no evidence that a disruption of the BHLF1 locus itself significantly influenced the expression of adjacent genes that encode proteins or miRNAs during Lat III, thus supporting our conclusion that the defects exhibited by our BHLF1− rEBVs are likely to result directly from the loss of a latency-related function(s) of the BHLF1 locus.
While this is the first direct evidence for a role of the BHLF1 locus in latency, from a historical perspective, it is likely relevant that the naturally occurring deletion (∼6.8 to 8.5 kbp) that targets BHLF1 within the EBV genomes present in the subset of BL tumors and cell lines that maintain so-called Wp-restricted latency (44) was long ago associated with a lack of growth-transforming potential of EBV carried by the BL cell line P3HR-1 (alternatively, P3J-HR-1 or HR-1) (19, 61–64). This deletion variably extends rightward to either well within or completely across the BHLF1 locus, and to the left of BHLF1, it removes the entire EBNA2 ORF and a variable portion of the DNA encoding the C-terminal domain of EBNA-LP (44). Repair or complementation of the deletion within the P3HR-1 EBV genome rescues the transforming potential of the virus, and while this restoration has been determined to require EBNA2 and, to a lesser extent, EBNA-LP, a requirement for BHLF1 was not assessed (65–68). Later work, seeking to introduce a selectable marker into the EBV genome by taking advantage of efficient recombination able to repair the deletion in the P3HR-1 genome, succeeded in inserting a 1.8-kbp hygromycin resistance gene into BHLF1 at a site corresponding to the 44th codon of the ORF and in a transcriptional orientation opposite that of BHLF1 (69). While the resulting rEBVs in which BHLF1 had been disrupted in this manner were able to transform primary B lymphocytes, the efficiency of transformation was less than 100% (range, 31% to 100%; mean, 65%) (69). These results appear to be consistent with our findings here, although for several reasons, it is not possible to conclude this with certainty. Most notably, only a single inoculum of an unknown MOI was used for the transformation assays in the previous study, preventing accurate comparisons to our results in Fig. 10. Also, transformation results were not provided for an equivalent inoculum (MOI) of an appropriate WT rEBV control, and it was not clear if the primary B cells used in the four experiments reported were from a single or multiple donors. And finally, it is not known if the insertion of the transgene in the opposite transcriptional orientation would have actually inhibited a noncoding function of BHLF1. Given our current findings, we consider it likely that the complete or partial removal of the BHLF1 locus contributes to the loss of EBV transforming potential associated with this naturally occurring deletion.
There is increasing evidence that BHLF1 functions via a noncoding mechanism(s), possibly through lncRNAs that it encodes. The originally characterized BHLF1 transcript is a 2.5-kb unspliced, polyadenylated RNA whose expression is highly induced upon the activation of the EBV replication cycle (18, 19). Northern blot analyses in early studies revealed little or no detectable presence of the transcript within latently infected B-cell lines prior to the induction of the lytic cycle, suggesting that the transcription of BHLF1 is limited to productive infection. Assessment of EBV gene expression upon infection of primary B cells in the presence of a protein synthesis inhibitor, however, indicated the transcription of BHLF1 in at least the prelatency period (30). Consistent with this, a recent RNA-seq analysis identified BHLF1 as a member of the first cluster of EBV genes to be transcribed upon infection of primary B cells (31). RNA-seq analyses of the EBV transcriptome within established BL cell lines that maintain Lat I (28) and B LCLs that support Lat III (36) have suggested that BHLF1 transcripts are also present during established latent infections, although one could argue that these represent RNAs from highly transcribed BHLF1 loci in a minor population of cells that have spontaneously entered the lytic cycle.
While we found BHLF1 transcripts not to be remarkably abundant in latently infected cell lines, their levels also did not always correlate with that of the mRNA encoding the early-lytic-cycle-specific protein SM (Fig. 2A). Interestingly, in one report, mentioned above (36), that analyzed EBV transcriptome data from ENCODE RNA-seq results generated from EBV-immortalized LCLs, spliced versions of BHLF1 transcripts were identified in these Lat III-maintaining B cells. In these transcripts, a novel splice acceptor site within the body of the previously characterized BHLF1 mRNA is spliced to at least one of eight donor sites ∼1.3 to 97.3 kbp upstream. While the complete structures of these novel BHLF1 transcripts could not be deciphered from RNA-seq data, this is potentially significant, as all known latency-associated EBV genes (except those encoding the small ncRNAs EBER1 and EBER2) encode spliced mRNAs, with all but one (LMP1) containing multiple large introns of 3 to upwards of 40 kb. In contrast, only a subset of lytic-cycle mRNAs is spliced, and most of these contain 2 or 3 exons separated by short introns of a few hundred to several hundred bases in length. It should be noted, however, that the number of reads specific for these spliced BHLF1 RNAs suggests that they are quite low in abundance (36), and indeed, we have had difficulty in amplifying them by RT-PCR. It now appears, however, that they most likely belong to a family of circular RNAs that result from the back-splicing of novel and cryptic splice sites within BHLF1 and are primarily if not exclusively expressed during the lytic cycle of EBV infection (41, 42).
Several observations collectively provide more definitive evidence of the latency-specific transcription of BHLF1. Our early analysis of the transcription of the EBV genome in a latently infected LCL revealed a level of transcription across the BHLF1 locus that was at least equivalent to that of the adjacent, latency-specific, EBNA2-encoding exon (70), yet by Northern blotting, we did not detect polyadenylated BHLF1 transcripts in the cytoplasmic fraction (as would be expected for the highly abundant 2.5-kb BHLF1 mRNA if a small percentage of cells had spontaneously entered the lytic cycle). This suggested that the transcription of BHLF1 can occur during latency but that a posttranscriptional “block” may prevent its expression as an mRNA in the cytoplasm. We find this intriguing in light of our current finding (Fig. 3) that the efficient expression of protein from an intact BHLF1 ORF may require the EBV protein SM, a broadly acting posttranscriptional regulator of EBV gene expression known to affect mRNA stability, processing, export, and translation (48, 49).
More recently, analysis of RNA encoded by DNA encompassing oriLytLeft and oriLytRight, which abut BHLF1 and its paralog LF3, respectively, implicated the existence of novel BHLF1 and LF3 transcription start sites within B-cell lines that maintain Lat III (Fig. 1) (32). Notably, these BHLF1 transcription start sites are 360 bp (P2) and ∼1 kbp (P3′) upstream of those used for the 2.5-kb BHLF1 transcript expressed from P1 during the lytic cycle (Fig. 1). Furthermore, the expression of these novel transcripts did not increase upon the chemical induction of the lytic cycle in B-cell lines maintaining Lat III, suggesting that these RNAs originate from latency-specific promoters (P2 and P3′) (32). Our RT-qPCR-based detection of transcripts originating upstream of the lytic-cycle-specific BHLF1 promoter P1 (Fig. 2B) is consistent with the existence of such transcripts in the total-RNA fraction of latently infected B-cell lines. We also detected the expression of these transcripts during Lat I although less so than in B cells maintaining Lat III, consistent with the previous finding that P2/P3′-specific transcripts are less abundant or undetectable in biopsy specimens of BL tumors (32), which typically maintain Lat I. Furthermore, although we noted an increase in the expression of these transcripts upon the induction of the lytic cycle in BL cells that maintain Lat I, the level of induction was modest compared to that seen for the SM gene. The structures of these putative latency-specific BHLF1 RNAs have yet to be defined, although they appear to be polyadenylated (32). More recent work has indicated that within BL cells, albeit those maintaining Lat I, BHLF1 transcripts are predominantly nuclear (71), which may explain why these transcripts were not observed by Northern blotting in some early studies that assessed RNAs from the cytoplasmic fraction of cells.
The strongest evidence of latency-associated BHLF1 transcription can be gleaned from a recent analysis of the cellular transcriptome within individual cells of an EBV-immortalized LCL (72). Our examination of these single-cell RNA-seq data for the detection of EBV transcripts revealed the presence of BHLF1 RNA in each of 10 cells whose RNA was profiled (Fig. 12). While some of these BHLF1 transcripts are likely to be lytic-cycle specific (suggested by the codetection in some cells of known lytic-cycle mRNAs, e.g., the 3′-coterminal BaRF1, BMRF1, and BMRF2 transcripts [43]), the detection of BHLF1 transcripts in all cells supports our contention that there is latency-specific transcription of this locus. In contrast, the mRNAs of EBNA2 and LMP1, well-established latency-associated genes that encode the most abundant EBV latency-associated mRNAs during Lat III (70), were detected in only a minority of the cells and at numbers far below the number of reads of the BHLF1 transcripts. In this study, cDNA synthesis was primed with an oligo(dT)-containing oligonucleotide, and BHLF1-specific reads matched the unique-sequence domain immediately upstream of the known BHLF1 polyadenylation site. Thus, at least a subset of BHLF1 transcripts expressed during latency appear to be polyadenylated and 3′ coterminal with the previously characterized 2.5-kb BHLF1 mRNA.
FIG 12.

The BHLF1 locus is uniformly transcribed during Lat III. Single-cell RNA-seq analysis of the EBV transcriptome in 10 cells of the B LCL GM12878, immortalized by the B95.8 isolate of EBV, indicated that BHLF1 transcripts were present in each cell. This was in contrast to the heterogeneous detection of transcripts encoded by known latency-associated genes, e.g., EBNA1, LMP2A/2B, EBNA2, and LMP1 (note that transcripts from the latter two genes are not evident in this figure due to the scale used for the vertical axis). Other peaks represent various lytic-cycle transcripts. The results shown were obtained by analysis, as described previously (81), of publicly available data sets from https://www.encodeproject.org/experiments/ENCSR673UIY/ (72).
Given the apparent transcription of BHLF1 during the phases of EBV latency within B cells that are assessable in vitro (Lat I and III) and that the Akata EBV genome used to generate our WT rEBV lacks an intact BHLF1 ORF (35), we consider it likely that BHLF1’s contribution to EBV latency is noncoding in nature and possibly dependent on an lncRNA acting either in trans or in cis. With respect to the latter, BHLF1 RNAs have been shown to form RNA-DNA duplexes or R-loops at their site of synthesis, which appears to contribute to the function of the adjacent oriLytLeft (39). Currently, it is unclear how such hybrids might contribute to latency. One could envision, for example, a contribution to the regulation of histone modifications and effects on local chromatin structure that, in turn, could positively influence EBV transcription over long distances (e.g., from Cp/Wp), possibly by influencing the looping of the EBV genome. Alternatively, an effect may not be specifically dependent on the lncRNA but simply the maintenance of active transcription through this locus; i.e., features of the lncRNA itself may be largely irrelevant. We also considered that, as antisense to the EBNA and BHRF1 primary transcripts originating upstream, BHLF1 lncRNAs might regulate the rate of transcription or mRNA processing through duplex formation with either DNA or RNA. However, we did not observe a notable increase or decrease in any of the EBNA mRNAs or proteins (i.e., prior to the apparent transition to Lat I) as a consequence of deleting BHLF1. While we noted a small increase in the levels of BHRF1 mRNAs (Fig. 8), it is difficult to rationalize how a small increase in the levels of mRNAs encoding the prosurvival BHRF1/vBCL-2 protein would negatively impact the maintenance of Lat III and the transforming efficiency of BHLF1− virus.
Many lncRNAs act in trans and do so through a variety of mechanisms that involve an interaction with regulatory proteins, often existing in a multiprotein-RNA complex (reviewed in reference 73). Because RNA-binding proteins frequently recognize RNA structure rather than solely a specific nucleotide sequence motif, we find it potentially noteworthy that in silico prediction of the secondary structure within BHLF1 transcripts reveals that these RNAs are highly structured (our unpublished observation). This is particularly evident within the IR2 domain, which is comprised of ∼12.3 copies of the 125-nucleotide NotI repeat and accounts for ∼60% of the length of the unspliced 2.5-kb BHLF1 transcript originating from P1 (Fig. 1). Although the actual secondary structure of these RNAs is not known, given the largely repetitive nature of BHLF1 transcripts spanning IR2, an attractive hypothesis is that repeating stem-loop structures within the repeat domain serve as protein-binding sites that, collectively, could act as a sink for an RNA-binding protein(s) to sequester them or otherwise block their normal activity or possibly serve as a scaffold for the assembly of a functional protein complex. Along these lines, in addition to its presumed role in the function of oriLytLeft (39), Park and Miller (40) recently identified the 2.5-kb BHLF1 lncRNA as a component of novel virus-induced nodules on replication compartments (VINORCs) and that they propose may function to facilitate the selective processing and export of viral mRNAs. However, the absence of BHLF1-containing VINORCs prior to the activation of the lytic cycle (40) argues against such a role of this lncRNA during latency.
Our attempts to rescue the defect of BHLF1− rEBV in BL2 cells by the expression of BHLF1 in trans have been unsuccessful. Alternatively, any lncRNA may be acting in cis, and/or the BHLF1 DNA locus itself may be the critical factor contributing to its apparent influence on Lat III and B-cell growth. It is also possible that latency-specific lncRNAs expressed from this locus differ in structure and, thus, function from those of the characterized 2.5-kb lytic-cycle RNA encoded by BHLF1, which at best would appear to be expressed at a very low level during latency. Of particular note in this respect are the transcripts initiating from the putative promoters P2 and P3′ (Fig. 1) that we found to be more abundant in cells maintaining Lat III than in those maintaining Lat I (Fig. 2B), which correlates with our observed positive influence of BHLF1 on Lat III. In addition to these RNAs, an apparent family of transcripts antisense to BHLF1 between P1 and P2 that also appears to be latency associated was previously reported (32); it is not clear whether these transcripts may represent long unspliced versions of BHRF1 mRNAs reported to originate from the same region, i.e., within oriLytLeft (58). Moreover, recent mappings of mature 5′ and 3′ termini of EBV transcripts identified clusters of previously unknown transcription initiation and polyadenylation sites located ∼200 to 500 and ∼500 to 800 bp downstream, respectively, of the major transcription start site for the 2.5-kb BHLF1 transcript, although these sites also appear to be used exclusively upon the activation of the lytic cycle within latently infected cells (74, 75). Given the extensive complexity of transcription within and through the BHLF1 locus, an important objective moving forward will be the elucidation of the structures of all BHLF1 transcripts, as this knowledge will be important to determine the ranges of RNA expression and subcellular location during the different latency programs and, ultimately, the mechanism of action of the transcripts and whether they indeed contribute to the functions regulated by the BHLF1 locus.
Regardless of whether BHLF1 acts through an lncRNA, our observations suggest that it contributes to the maintenance of Lat III in EBV-negative BL cells, and such a function is not inconsistent with the diminished growth-transforming properties of BHLF1− rEBV. It is notable that the apparent inability to sustain Lat III in BL2 cells infected with BHLF1− viruses was consistently observed at between 1 and 2 months p.i. This delay in a measurable phenotype is not inconsistent with selection against cells that support Lat III in the absence of BHLF1, resulting in the eventual outgrowth of cells able to transition to Lat I by default. Mechanistically speaking, therefore, BHLF1’s role may be an indirect or supportive one rather than one as a direct regulator of the Lat III program. This interpretation is also not inconsistent with the less-than-complete requirement for BHLF1 for the immortalization of B cells (which, to some extent, appeared to be donor dependent) and the generally poorer growth properties of LCLs that resulted from infection with BHLF1− rEBV. Thus, upon infection, the net activity or level of a BHLF1 target that must be optimally regulated to promote Lat III may dictate the degree to which BHLF1 is required. In other words, BHLF1 may have evolved to fine-tune a specific molecular process to ensure the efficient establishment of latency rather than to directly regulate latency gene expression itself. Furthermore, the apparent absolute requirement for BHLF1 to sustain Lat III in BL2 cells but not in BL30 cells, an EBV-negative BL line that naturally favored the transition to Lat I, perhaps should not be surprising given the qualitative differences in mutational loads between endemic (EBV-positive) and sporadic (EBV-negative) BL and among EBV-negative BLs themselves (76). For example, due to mutations distinct from those in BL2 cells, BL30 cells may have a greater propensity to epigenetically silence the EBV genome, overriding any positive effect of BHLF1 on Lat III as revealed in BL2 cells.
In conclusion, we have shown that the deletion of the BHLF1 locus of the EBV genome results in a diminished ability of the virus to immortalize B cells in vitro, a hallmark property of EBV latency and one intimately associated with successful colonization by EBV of its human host as well as its significant oncogenic potential. While the mechanism through which BHLF1 functions during latency is currently unclear, our results suggest that the diminished growth-transforming potential of BHLF1− EBV may be due to an inability to fully support the Lat III program of EBV infection that is critical during the establishment of a B-cell reservoir of EBV in vivo and that is required for the continued growth of primary B cells in vitro. Our results are consistent with increasing evidence over the past decade and earlier of the latency-associated expression of BHLF1, which has heretofore been thought to contribute exclusively to the productive phase of EBV infection. Additionally, the lack of conservation of the ORF within BHLF1 among some EBV isolates has raised the likelihood that BHLF1 functions not through a protein that it encodes but also through either an lncRNA that acts in trans or cis or an inherent property of the DNA within the locus itself that may be regulated by its active transcription. Our results also suggest that in those isolates that have retained the BHLF1 ORF, the protein that it encodes may require the presence of the EBV posttranscriptional regulator SM for efficient expression, thus limiting a protein-coding role to productive infection. And finally, our evidence supporting a latency-related function of BHLF1 raises the question of what the contribution may be of its paralog LF3, for which latency-associated expression and a noncoding function have also been implicated (28, 35).
MATERIALS AND METHODS
Cell lines.
Akata clone 21 (A.21), Kem I, Mutu I, and BX1 are BL-derived cell lines that support the EBV Lat I program. A.21 was originally cloned from the parental Akata BL line (77). BX1 (a gift of L. Hutt-Fletcher) was generated by infection of an EBV-negative clone of Akata cells with an rEBV (BLX/Rc-TK) in which an expression cassette encoding G418 resistance and green fluorescent protein (GFP) had been inserted into the BXLF1 (thymidine kinase) ORF within the genome of the Akata isolate of EBV (78). The B-cell lines used in this study that support the EBV Lat III program were the BL-derived cell lines Kem III and Raji and the LCLs Ak-LCL and MH-LCL generated by the immortalization of primary human B lymphocytes in vitro with either rEBV derived from Ak-GFP-BAC (see below) or the B95.8 isolate of EBV, respectively. Note that while the Kem III line was originally believed to be of BL origin, it has recently come to our attention that some Kem III lines may instead be spontaneous B LCLs that arose during the primary culture of Kem BL tumor cells. Sal is a BL cell line that maintains a Wp-restricted program of EBV latency gene expression and contains a deletion in its endogenous EBV genomes that has removed the adjacent EBNA2 and BHLF1 loci and the C-terminal coding region of EBNA-LP upstream of EBNA2 (44). BL2, BL30, and Louckes are EBV-negative BL cell lines, as is the A.2 clone of the Akata BL line. All B-cell lines indicated above were maintained in RPMI 1640 medium (HyClone) supplemented with 2 mM l-glutamine (HyClone) and 10% fetal bovine serum (FBS; HyClone). Primary B lymphocytes infected in vitro and the LCLs that resulted from these infections (see below) were maintained in RPMI 1640 medium supplemented with 2 mM l-glutamine, 15% FBS, and 50 μg gentamicin sulfate per ml (Lonza). The human embryonic kidney cell line HEK293 was maintained in Dulbecco’s modified Eagle medium (DMEM; Lonza) supplemented with 10% FBS, except as noted below for the production of rEBV. All cell lines were maintained at 37°C in a humidified 5% CO2 atmosphere. For the induction of the EBV replication cycle, A.21 or BX1 cells were plated in 6-well plates at a density of 3 × 106 cells per well, and surface IgG was cross-linked with the goat F(ab′)2 fragment to human IgG (Cappel; MP Biomedicals) added to a concentration of 100 μg per ml; cells were harvested for subsequent analysis 48 h after the addition of F(ab′)2.
Immunoblot analysis.
For the detection of proteins by immunoblotting, cells were harvested by centrifugation, washed once with phosphate-buffered saline (PBS), and then lysed at 106 cells per 80 μl 2× SDS-PAGE buffer containing 5% β-mercaptoethanol. Samples were then sonicated and boiled for 5 min, and proteins were resolved by SDS-PAGE, after which they were subjected to semidry transfer onto a polyvinylidene difluoride (PVDF) membrane. Cellular and EBV proteins were detected by standard immunoblotting techniques using primary antibodies to EBNA1 (rabbit antiserum; gift of J. Herring); EBNA2 (monoclonal antibody [mAb] PE2); LMP1 (mAb S12); EBNA3A, -3B, and -3C (sheep antiserum to each; Exalpha Biologicals, Inc.); actin (mAb JLA20; Calbiochem); β-tubulin (clone H-234; Santa Cruz Biotechnology); and FLAG (mAb M2; Sigma-Aldrich). The following secondary antibodies were used: donkey anti-rabbit for EBNA1 and β-tubulin (GE Healthcare UK Limited); anti-mouse for LMP1, actin, and FLAG (GE Healthcare UK Limited); and rabbit anti-sheep (Chemicon) for EBNA3A, -3B, and -3C. Immunoblots were developed using either the Pierce ECL Western blotting substrate (Thermo Fisher Scientific) or the Immobilon Western chemiluminescent horseradish peroxidase (HRP) substrate (Millipore Sigma).
Expression of the BHLF1 protein.
For the transient expression of the BHLF1 protein, a BHLF1 ORF derived from the B95.8 EBV genome was cloned with a FLAG epitope-encoding sequence at its 5′ terminus into the expression vector pSG5 (Stratagene). Coexpression of the EBV SM protein was achieved from the vector pcDNA3-SM (gift of S. Swaminathan). A pSG5-derived vector encoding FLAG-tagged insulin-degrading enzyme (pSG5-IDE) was used as a positive control for transfection and detection of FLAG by immunoblotting. Louckes cells (5 × 106 cells per transfection) were transfected by nucleofection with plasmid DNA, as indicated (Fig. 3), using an Amaxa Nucleofector with solution V and program G-16 according to the manufacturer’s instructions (Lonza). Transfected cells were plated in 6-well plates and after 48 h of incubation were harvested for immunoblot analysis.
Isolation and analysis of RNA.
Reverse transcription-PCR (RT-PCR) was used to assess the expression levels of EBV RNAs. For the analysis of mRNA and lncRNA in Fig. 2A, Fig. 4B, Fig. 6B, and Fig. 7, total cellular RNA was extracted using RNA-Bee (Tel-Test) according to the manufacturer’s instructions, followed by digestion with either RQ1-DNase (Promega) or Turbo DNase (Thermo Fisher Scientific) to remove residual DNA. The cDNA template for PCR was generated from 2 μg total RNA in 19-μl reaction mixtures containing 200 U SuperScript III reverse transcriptase (Invitrogen) according to the manufacturer’s instructions, using either 0.1 μM gene-specific primer (GSP) or 2.5 μM random decamers (the latter were used for the reverse transcription of EBNA1, EBNA2, EBNA3C, and GAPDH [glyceraldehyde-3-phosphate dehydrogenase] mRNAs); GAPDH mRNA served as an expression reference. A complete description of oligonucleotide primers and probes is provided in Table 1, with the exception of GAPDH primers and the TaqMan probe, which were purchased as a kit (Applied Biosystems). Corresponding negative-control reaction mixtures did not contain reverse transcriptase (−RT). For endpoint RT-PCR (Fig. 4 , 6, and 7), 2 μl of the cDNA synthesis or −RT control reaction mixture was amplified in a 25-μl reaction mixture containing 0.5 μM each PCR primer, 0.25 mM deoxynucleoside triphosphates (dNTPs), 1× PCR buffer without Mg, 1.5 mM MgCl2, and 4 U Platinum Taq DNA polymerase (Invitrogen) using the following cycling conditions: 95°C for 3 min; 30 to 35 cycles of 95°C for 15 s, the appropriate annealing temperature (55°C or 60°C) for 30 s, and 72°C for 1 min; and a final extension step at 72°C for 10 min. For quantitative (real-time) analysis of RNA levels by RT-qPCR (Fig. 2A and Fig. 7), cDNA synthesis was primed as described above with GSPs for BHLF1, BHRF1, and SM RNAs (Table 1) or random decamers for GAPDH. Quantitative PCR was performed using 2 μl of the cDNA template in a 20-μl volume containing 1× TaqMan universal master mix II (Applied Biosystems), 900 nM each primer, and 250 nM 6-carboxyfluorescein (FAM)-labeled TaqMan probe. A GAPDH TaqMan probe (Applied Biosystems) was included as an internal control. Parameters for qPCR were 10 min at 95°C, followed by 40 cycles of 15 s at 95°C and 60 s at 60°C. Relative gene expression compared to a reference LCL was determined using the comparative threshold cycle (CT) method ().
TABLE 1.
RT-PCR primers and TaqMan probes for detection of mRNA and lncRNAa
| Primer or probe | Annealing site | Sequence (5′–3′) | EBV genome coordinatesb | Purpose(s) (figure[s]) |
|---|---|---|---|---|
| RT-PCR primers | ||||
| BHLF1-GSP | BHLF1-unique region | TCTGGGGGTCGCTGCAT | 39927–39943 | Synthesis of BHLF1 cDNA (Fig. 2) |
| BHLF1-Fwd | BHLF1-unique region | GTACGCCTGGATTGCCG | 39992–40008 | Amplification of BHLF1 cDNA (Fig. 2) |
| BHLF1-Rev | BHLF1-unique region | AGGTCGGACTAGCGGATG | 40130–40113 | Amplification of BHLF1 cDNA (Fig. 2) |
| P2-BHLF1-GSP | BHLF1 downstream of the P2 promoter | TTAAGGTTTGCTCAGGAGTGG | 40557–40577 | Synthesis of P2/P3′-BHLF1 cDNA (Fig. 2 and 5) |
| P2-BHLF1-Fwd | BHLF1 downstream of the P2 promoter | GCTTAGGATACCTCCAGGATAATG | 40856–40833 | Amplification of P2/P3′-BHLF1 cDNA (Fig. 2 and 5) |
| P2-BHLF1-Rev | BHLF1 downstream of the P2 promoter | CCATAGGGTTGAACCAGGAG | 40745–40764 | Amplification of P2/P3′-BHLF1 cDNA (Fig. 2 and 5) |
| P3′-BHLF1-GSP | BHLF1 downstream of the P3′ promoter | TAGAACCTAGAGGAAGGGAACC | 41043–41064 | Synthesis of P3′-BHLF1 cDNA (Fig. 2 and 5) |
| P3′-BHLF1-Fwd | BHLF1 downstream of the P3′ promoter | GAGCCGTCCTTATTCTTGCT | 41468–41449 | Amplification of P3′-BHLF1 cDNA (Fig. 2 and 5) |
| P3′-BHLF1-Rev | BHLF1 downstream of the P3′ promoter | GCCTCACCATGACACACTAA | 41333–41352 | Amplification of P3′-BHLF1 cDNA (Fig. 2 and 5) |
| SM-GSP | SM | ACCGCCAGCATCGACTGT | 71146–71163 | Synthesis of SM cDNA (Fig. 2) |
| SM-Fwd | SM | GGGCAAGGTGACAAATGTAATC | 71211–71232 | Amplification of SM cDNA (Fig. 2) |
| SM-Rev | SM | AAGAACGCAGCCAGAGG | 71302–71286 | Amplification of SM cDNA (Fig. 2) |
| Random decamers | NA | NA | NA | Synthesis of EBNA1, EBNA2, EBNA3C (Fig. 4 and 6), and GAPDH (Fig. 7) cDNAs |
| BHRF1-GSP | BHRF1 exon HF | TTCTCTTGCTGCTAGCT | 42192–42176 | Synthesis of BHRF1 cDNA (Fig. 8) |
| W2-Fwd | EBNA/BHRF1 exon W2 | TGGTAAGCGGTTCACCTTCAG | 14810–14830 | Amplification of Cp/Wp-derived BHRF1 cDNA (Fig. 8) |
| Y2-Fwd | EBNA/BHRF1 exon Y2 | GAGGATGAAGACTAAGTCACAGGCTTA | 35680–35706 | Amplification of Cp/Wp-derived EBNA1, EBNA2, EBNA3C, and BHRF1 cDNAs (Fig. 4, 6, and 8) |
| Qp-Fwd | EBNA1 5′ exon Q | AAGGCGCGGGATAGC | 50137–50151 | Amplification of Qp-derived EBNA1 cDNAs (Fig. 4, 6, and 7) |
| Qp-Rev | EBNA1 exon U | TCTACTGGCGGTCTATGATGC | 55322–55302 | Amplification of Qp-derived EBNA1 cDNA (Fig. 7) |
| EBNA1-Rev/GSP | EBNA1 3′ exon K | CTCTATGTCTTGGCCCT | 95863–95847 | Amplification of Cp/Wp- and Qp-derived EBNA1 cDNA (Fig. 4 and 6); synthesis of EBNA1 cDNA (Fig. 7) |
| EBNA1-Rev′ | EBNA1 3′ exon K | GTACCTGGCCCCTCGTCA | 95684–95667 | Amplification of Cp/Wp-derived EBNA1 cDNA (Fig. 7) |
| EBNA2-Rev | EBNA2 3′ exon YH | GAGAGTGACGGGTTTCCAAGb | 36301–36282b | Amplification of EBNA2 cDNA (Fig. 4) |
| GAGAGTGACGGGTTTCCAACc | 36205–36186c | |||
| EBNA3C-Rev | EBNA3C exon BERF4 | GGAGATGTTAGAAGCCAATGTC | 86683–86662 | Amplification of EBNA3C cDNA (Fig. 4 and 6) |
| BHRF1-Rev | BHRF1 exon HF | TCCCGTATACACAGGGCTAACAGT | 42134–42111 | Amplification of BHRF1 cDNA (Fig. 8) |
| TaqMan probes | ||||
| Unique-BHLF1 | BHLF1-unique domain | CTTGCCTGGTCCTGGAGCTCATC | 40079–40101 | Detection of BHLF1 cDNA (Fig. 2) |
| P2/P3′-BHLF1 | Between P1 and P2 | TACCTACCTCTAGGCTCCACCCAC | 40820–40797 | Detection of P2/P3′-BHLF1 cDNA (Fig. 2 and 5) |
| P3′-BHLF1 | Between P2 and P3′ | CCGGGACGTGGTGCTTCCTAAA | 41400–41379 | Detection of P3′-BHLF1 cDNA (Fig. 2 and 5) |
| SM | SM | ACCGTGGTTTGACATGAGTCTGGTT | 71271–71247 | Detection of SM cDNA (Fig. 2) |
| BHRF1 | BHRF1 HF exon | AATAGGCCATCTTGCTCTACAAGATCTGGCA | 42097–42067 | Detection of BHRF1 cDNA (Fig. 8) |
Primers and probes used for the assessment of GAPDH mRNA and EBV miRNAs were obtained from and used in conjunction with commercially obtained RT-qPCR kits (see Materials and Methods) and are not listed here. GSP, gene-specific primer used for the synthesis of cDNA by reverse transcription; Fwd, forward/5′ PCR primer; Rev, reverse/3′ PCR primer; NA, not applicable.
Based on the B95.8-Raji composite EBV genome sequence (GenBank accession number NC_007605.1).
Based on the Akata EBV genome sequence (GenBank accession number KC_207813).
A slightly different protocol was employed for the quantification of BHLF1 RNA levels reported in Fig. 2B and Fig. 5. Briefly, total cellular RNA was extracted using the QIAshredder and RNeasy Plus minikit (Qiagen) according to the manufacturer’s instructions, digested with Turbo DNase, and then purified according to the RNA cleanup protocol of the RNeasy minikit (Qiagen). cDNA was synthesized from 1 μg RNA in a 20-μl reaction mixture containing the appropriate GSP (100 nM) and 200 U SuperScript III according to the manufacturer’s instructions. As described above, −RT controls were run in parallel. Either the GSPs for the P3′, P2/P3′, and unique BHLF1 primer sets (Table 1) were individually multiplexed with a GAPDH GSP (instead of random decamers) in the same cDNA synthesis reaction (Fig. 2B) or cDNA synthesis was primed with each BHLF1 and GAPDH GSP in a separate reaction (Fig. 5). Singleplex BHLF1 (P3′, P2/P3′, or unique primer sets) and GAPDH qPCRs were performed in triplicate in 20-μl reaction mixtures containing 2.0 μl of cDNA synthesis (undiluted or diluted 8-fold) or −RT control reaction mixture, 1× TaqMan universal master mix II, BHLF1 forward and reverse PCR primers (0.5 μM each) or 1× GAPDH TaqMan assay reagent (Thermo Fisher Scientific), and the appropriate TaqMan probe (250 nM). The qPCR cycling parameters were 10 min at 95°C, followed by 45 cycles of 15 s at 95°C and 60 s at 57°C or 60°C. GAPDH reaction mixtures were amplified in the same 96-well plate as BHLF1 reaction mixtures to ensure equivalent amplification conditions. Relative gene expression was determined using the comparative CT method ().
For the analysis of EBV miRNA levels by RT-qPCR (Fig. 8), RNA was isolated from cells using the Ambion mirVana miRNA isolation kit (Life Technologies) as directed by the manufacturer. RT-qPCR employing 10 ng of RNA was performed using the TaqMan microRNA reverse transcription kit and miRNA assays for the EBV miRNAs miR-BHRF1-1, -2, and -3 (Applied Biosystems) according to the manufacturer’s instructions. Amplification of U6 snRNA was used as an internal control, and sample values were normalized to the value for an EBV-positive reference cell line. The parameters for qPCR were the same as the ones described above, with 40 cycles and 60°C for annealing/extension. All RT-qPCRs (mRNA, lncRNA, and miRNA) were done with a StepOnePlus real-time PCR system (Applied Biosystems).
Generation of BHLF1− rEBV.
Both WT and mutant rEBVs were generated from the Akata EBV genome present within the BAC Ak-GFP-BAC (clone 12-15) (79). Two BHLF1− rEBVs were employed in these studies, ΔB-S and ΔBHLF1, which contained deletions removing different amounts of the BHLF1 locus (Fig. 1). The generation of ΔB-S rEBV by BAC recombineering has been described in detail previously (50). Briefly, a 3.3-kbp deletion (ΔB-S) was introduced within Ak-GFP-BAC that corresponded to nucleotide coordinates 38287 to 41550 of the composite WT EBV genome derived from the B95.8 and Raji isolates (GenBank accession number NC_007605.1). This deletion extends rightward from the stop codon of the BHLF1 ORF to the right boundary of the naturally occurring 8.5-kbp deletion present in the endogenous EBV genomes within the Sal BL line and thus removes the entire BHLF1 ORF (positions 38287 to 40269) and approximately 1.3 kbp of DNA upstream of it that spans the transcription start sites for the BHLF1 promoters P1 (position 40520), P2 (position 40879), and P3′ (position ∼41514) as well as oriLytLeft (positions 40301 to 41293) (Fig. 1). The final deletion step in E. coli was mediated by flippase recombinase (Flp), and thus, a single 34-bp Flp recognition target (FRT) element is present at the site of the deletion. For the generation of ΔBHLF1 rEBV, a deletion (ΔBHLF1) was introduced within Ak-GFP-BAC that corresponded to the BHLF1 ORF in the B95.8 EBV genome (positions 38287 to 40269). The introduction of ΔBHLF1 was accomplished with the galactokinase (galK) positive/negative selection method of recombineering in E. coli strain SW105 (80). Briefly, a galK expression cassette flanked by 50 bp of DNA homologous to the regions immediately upstream and downstream of the BHLF1 ORF was generated by PCR; this DNA fragment was introduced by electroporation into SW105 cells carrying Ak-GFP-BAC for the replacement of the BHLF1 target with galK by homologous recombination. The DNA was isolated from multiple GalK-positive clones (able to grow on minimal medium agar plates with galactose as the sole carbon source), and correct recombination within the BHLF1 locus was confirmed by PCR amplification across the galK-EBV DNA junctions, followed by sequence analysis of amplified DNA. Next, to remove the galK cassette, SW105 cells carrying Ak-GFP-BAC DNA in which the ΔBHLF1 deletion had been appropriately introduced were electroporated with a double-stranded DNA (dsDNA) oligonucleotide representing the fused 50-bp homology arms of EBV that had been used to introduce the galK cassette. GalK-negative clones, which would carry ΔBHLF1-Ak-GFP-BAC, were identified by growth on minimal medium agar containing a glycerol carbon source and 2-deoxy-galactose, which selects against bacteria still containing and expressing galK. From these clones, DNA was isolated and subjected to amplification and DNA sequence analysis to verify the appropriate removal of the galK cassette, resulting in ΔBHLF1. Unlike for ΔB-S, the final deletion step was not mediated by Flp, thus resulting in a seamless deletion. The Ak-GFP-BAC DNAs with ΔB-S and ΔBHLF1 deletions were transferred into E. coli DH10B, from which BAC DNA was purified with the NucleoBond BAC100 kit (Clontech) and subjected to restriction analysis and Southern blot hybridization to ensure against unintended recombination, deletions, and rearrangements.
Virus production and infection of BL2 cells.
For the production of rEBV, 4 × 105 HEK293 cells were seeded into each well of a 6-well plate and transfected with 2 μg Ak-GFP-BAC (WT) or its ΔB-S or ΔBHLF1 derivative using TransIT-293 transfection reagent (Mirus), and stable transfectants were selected with G418 (Geneticin) (500 μg/ml). Individual G418-resistant colonies that were also GFP positive were selected, expanded, and assessed for rEBV production. To induce EBV replication, cells were seeded at 6.3 × 106 cells per 150-mm plate 24 h prior to transient transfection as described above with 15.6 μg each of the expression vectors for the EBV BZLF1 and BALF4 (gB) proteins; at 24 h posttransfection, sodium butyrate and 12-O-tetradecanoylphorbol-13-acetate (TPA) were added to the culture medium at final concentrations of 4 mM and 20 ng/ml, respectively. After 3 h, the cell monolayers were rinsed and then incubated in fresh RPMI growth medium (instead of DMEM) for 4 days, after which the culture medium was clarified by centrifugation and passed through a 0.45-μm filter. To select clones that produced the largest amount of rEBV, virus titers were determined by the conversion of Raji BL cells to GFP expression. Briefly, 5 × 105 Raji BL cells in 1 ml were added to each well of a six-well plate and mixed with 1 ml of increasing dilutions of the filtered rEBV-containing HEK293 culture supernatant. Plates were then centrifuged at 200 × g for 1 h at 4°C and then incubated at 37°C for 24 h, at which time 2 ml of fresh RPMI 1640 culture medium was added to each well. At 3 days p.i., the cells were analyzed for GFP expression by flow cytometry using a BD FACSCalibur instrument (BD Biosciences). Flow cytometry data were analyzed using FlowJo software, and the viral titer was expressed as green Raji units (GRU) per milliliter. For large-scale production of virus, HEK293 clones producing the highest titer of WT or BHLF1− mutant rEBVs were expanded in 150-mm plates and processed as described above to induce virus replication. Filtered culture supernatants were then concentrated approximately 25- to 45-fold by tangential-flow filtration using a MidiKros hollow-fiber filter module with a pore rating of 500 kDa (Spectrum Labs) and a Bio-Rad model EP-1 Econo pump at a rate of 10 ml/min. Aliquots of the concentrated virus were frozen and stored at −70°C prior to determining the virus titer as described above. The virus used in individual experiments was never exposed to more than one freeze-thaw cycle. To infect BL2 and BL30 cells, 5 × 105 cells were infected at an MOI of no greater than 1 in 6-well plates as described above. At 24 h p.i., most of the culture medium was removed and replaced with 3 ml of fresh RPMI 1640 growth medium and 1 ml of conditioned medium taken from cultures of uninfected BL2 or BL30 cells. At 5 to 7 days p.i., infected cells were placed under G418 selection (Geneticin) (500 μg/ml) and subsequently expanded for further analysis.
Isolation, infection, and analysis of primary B lymphocytes.
Human CD19+ B lymphocytes were isolated from the whole blood of anonymous adult donors. Following the isolation of peripheral blood mononuclear cells (PBMCs) by gradient centrifugation on lymphocyte separation medium (LSM; MP Biomedicals), B cells were isolated from the PBMC fraction by positive selection using human CD19 MicroBeads (Miltenyi Biotec) according to the manufacturer’s instructions. Alternatively, purified CD19+ B cells were purchased from Stemcell Technologies. For infection, primary B lymphocytes were plated in 96-well plates at 5 × 104 cells per well, WT or mutant rEBV was added at a starting MOI of 0.068 and 2-fold serial dilutions thereof, and the plates were then centrifuged at 1,000 rpm for 1 h at 13°C prior to incubation at 37°C. Growth medium was replaced weekly, and at 6 weeks p.i., the wells of infected cells were scored for transformation, indicated by yellowing of the growth medium, cell clumping, and GFP expression. The cells from transformation-positive wells were then expanded to establish cell lines for further analysis. To assess growth properties, cells were seeded in triplicate in 6-well plates (5 ml per well) at 1 × 105 cells per ml in LCL growth medium containing 15% FBS. The viable-cell concentration was then determined daily by trypan blue dye exclusion using a Countess II FL automated cell counter (Life Technologies).
ACKNOWLEDGMENTS
We thank Teru Kanda and Kenzo Takada for their kind gift of Ak-GFP-BAC; Sankar Swaminathan for the SM expression vector; Janet Herring for EBNA1 antiserum; Lindsey Hutt-Fletcher and Cliona Rooney for the BX1 and MH-LCL cell lines, respectively; Rebekah Templin for excellent technical support; and the Flow Cytometry Core Facility of the Penn State Hershey Cancer Institute for their excellent service.
This work was supported by U.S. Public Health Service grant AI110328 to J.T.S. and in part by Commonwealth Universal Research Enhancement (CURE) funds. L.N.L.-G. received support from National Cancer Institute training grant T32 CA060395 and is a Lymphoma Research Foundation grantee.
We declare no conflict of interest.
This work is dedicated to the memory of Ingrid K. Ruf and Richard J. Courtney.
REFERENCES
- 1.Thorley-Lawson DA. 2015. EBV persistence—introducing the virus. Curr Top Microbiol Immunol 390:151–209. doi: 10.1007/978-3-319-22822-8_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Torii T, Konishi K, Sample J, Takada K. 1998. The truncated form of the Epstein-Barr virus LMP-1 is dispensable or complimentable by the full-length form in virus infection and replication. Virology 251:273–278. doi: 10.1006/viro.1998.9411. [DOI] [PubMed] [Google Scholar]
- 3.Nonkwelo C, Skinner J, Bell A, Rickinson A, Sample J. 1996. Transcription start sites downstream of the Epstein-Barr virus (EBV) Fp promoter in early-passage Burkitt lymphoma cells define a fourth promoter for expression of the EBV EBNA-1 protein. J Virol 70:623–627. doi: 10.1128/JVI.70.1.623-627.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schaefer BC, Strominger JL, Speck SH. 1995. The Epstein-Barr virus BamHI F promoter is an early lytic promoter: lack of correlation with EBNA 1 gene transcription in group 1 Burkitt’s lymphoma cell lines. J Virol 69:5039–5047. doi: 10.1128/JVI.69.8.5039-5047.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rowe M, Lear AL, Croom-Carter D, Davies AH, Rickinson AB. 1992. Three pathways of Epstein-Barr virus gene activation from EBNA1-positive latency in B lymphocytes. J Virol 66:122–131. doi: 10.1128/JVI.66.1.122-131.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Altmann M, Hammerschmidt W. 2005. Epstein-Barr virus provides a new paradigm: a requirement for the immediate inhibition of apoptosis. PLoS Biol 3:e404. doi: 10.1371/journal.pbio.0030404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wen W, Iwakiri D, Yamamoto K, Maruo S, Kanda T, Takada K. 2007. Epstein-Barr virus BZLF1 gene, a switch from latency to lytic infection, is expressed as an immediate-early gene after primary infection of B lymphocytes. J Virol 81:1037–1042. doi: 10.1128/JVI.01416-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kalla M, Schmeinck A, Bergbauer M, Pich D, Hammerschmidt W. 2010. AP-1 homolog BZLF1 of Epstein-Barr virus has two essential functions dependent on the epigenetic state of the viral genome. Proc Natl Acad Sci U S A 107:850–855. doi: 10.1073/pnas.0911948107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hong GK, Gulley ML, Feng WH, Delecluse HJ, Holley-Guthrie E, Kenney SC. 2005. Epstein-Barr virus lytic infection contributes to lymphoproliferative disease in a SCID mouse model. J Virol 79:13993–14003. doi: 10.1128/JVI.79.22.13993-14003.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma SD, Hegde S, Young KH, Sullivan R, Rajesh D, Zhou Y, Jankowska-Gan E, Burlingham WJ, Sun X, Gulley ML, Tang W, Gumperz JE, Kenney SC. 2011. A new model of Epstein-Barr virus infection reveals an important role for early lytic viral protein expression in the development of lymphomas. J Virol 85:165–177. doi: 10.1128/JVI.01512-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hsu DH, de Waal Malefyt R, Fiorentino DF, Dang MN, Vieira P, de Vries J, Spits H, Mosmann TR, Moore KW. 1990. Expression of interleukin-10 activity by Epstein-Barr virus protein BCRF1. Science 250:830–832. doi: 10.1126/science.2173142. [DOI] [PubMed] [Google Scholar]
- 12.Miyazaki I, Cheung RK, Dosch HM. 1993. Viral interleukin 10 is critical for the induction of B cell growth transformation by Epstein-Barr virus. J Exp Med 178:439–447. doi: 10.1084/jem.178.2.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jochum S, Moosmann A, Lang S, Hammerschmidt W, Zeidler R. 2012. The EBV immunoevasins vIL-10 and BNLF2a protect newly infected B cells from immune recognition and elimination. PLoS Pathog 8:e1002704. doi: 10.1371/journal.ppat.1002704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zeidler R, Eissner G, Meissner P, Uebel S, Tampe R, Lazis S, Hammerschmidt W. 1997. Downregulation of TAP1 in B lymphocytes by cellular and Epstein-Barr virus-encoded interleukin-10. Blood 90:2390–2397. doi: 10.1182/blood.V90.6.2390.2390_2390_2397. [DOI] [PubMed] [Google Scholar]
- 15.Swaminathan S, Hesselton R, Sullivan J, Kieff E. 1993. Epstein-Barr virus recombinants with specifically mutated BCRF1 genes. J Virol 67:7406–7413. doi: 10.1128/JVI.67.12.7406-7413.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stuart AD, Stewart JP, Arrand JR, Mackett M. 1995. The Epstein-Barr virus encoded cytokine viral interleukin-10 enhances transformation of human B lymphocytes. Oncogene 11:1711–1719. [PubMed] [Google Scholar]
- 17.Kalla M, Hammerschmidt W. 2012. Human B cells on their route to latent infection—early but transient expression of lytic genes of Epstein-Barr virus. Eur J Cell Biol 91:65–69. doi: 10.1016/j.ejcb.2011.01.014. [DOI] [PubMed] [Google Scholar]
- 18.Laux G, Freese UK, Bornkamm GW. 1985. Structure and evolution of two related transcription units of Epstein-Barr virus carrying small tandem repeats. J Virol 56:987–995. doi: 10.1128/JVI.56.3.987-995.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jeang KT, Hayward SD. 1983. Organization of the Epstein-Barr virus DNA molecule. III. Location of the P3HR-1 deletion junction and characterization of the NotI repeat units that form part of the template for an abundant 12-O-tetradecanoylphorbol-13-acetate-induced mRNA transcript. J Virol 48:135–148. doi: 10.1128/JVI.48.1.135-148.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hummel M, Kieff E. 1982. Epstein-Barr virus RNA. VIII. Viral RNA in permissively infected B95-8 cells. J Virol 43:262–272. doi: 10.1128/JVI.43.1.262-272.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shin S, Tanaka A, Nonoyama M. 1983. Transcription of the Epstein-Barr virus genome in productively infected cells. Virology 124:13–20. doi: 10.1016/0042-6822(83)90286-6. [DOI] [PubMed] [Google Scholar]
- 22.Freese UK, Laux G, Hudewentz J, Schwarz E, Bornkamm GW. 1983. Two distant clusters of partially homologous small repeats of Epstein-Barr virus are transcribed upon induction of an abortive or lytic cycle of the virus. J Virol 48:731–743. doi: 10.1128/JVI.48.3.731-743.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baer R, Bankier AT, Biggin MD, Deininger PL, Farrell PJ, Gibson TJ, Hatfull G, Hudson GS, Satchwell SC, Seguin C, Tuffnell PS, Barrell BG. 1984. DNA sequence and expression of the B95-8 Epstein-Barr virus genome. Nature 310:207–211. doi: 10.1038/310207a0. [DOI] [PubMed] [Google Scholar]
- 24.Parker BD, Bankier A, Satchwell S, Barrell B, Farrell PJ. 1990. Sequence and transcription of Raji Epstein-Barr virus DNA spanning the B95-8 deletion region. Virology 179:339–346. doi: 10.1016/0042-6822(90)90302-8. [DOI] [PubMed] [Google Scholar]
- 25.Ryon JJ, Hayward SD, MacMahon EM, Mann RB, Ling Y, Charache P, Phelan JA, Miller G, Ambinder RF. 1993. In situ detection of lytic Epstein-Barr virus infection: expression of the NotI early gene and viral interleukin-10 late gene in clinical specimens. J Infect Dis 168:345–351. doi: 10.1093/infdis/168.2.345. [DOI] [PubMed] [Google Scholar]
- 26.Xue SA, Lu QL, Poulsom R, Karran L, Jones MD, Griffin BE. 2000. Expression of two related viral early genes in Epstein-Barr virus-associated tumors. J Virol 74:2793–2803. doi: 10.1128/jvi.74.6.2793-2803.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xue SA, Labrecque LG, Lu QL, Ong SK, Lampert IA, Kazembe P, Molyneux E, Broadhead RL, Borgstein E, Griffin BE. 2002. Promiscuous expression of Epstein-Barr virus genes in Burkitt’s lymphoma from the central African country Malawi. Int J Cancer 99:635–643. doi: 10.1002/ijc.10372. [DOI] [PubMed] [Google Scholar]
- 28.Lin Z, Xu G, Deng N, Taylor C, Zhu D, Flemington EK. 2010. Quantitative and qualitative RNA-Seq-based evaluation of Epstein-Barr virus transcription in type I latency Burkitt’s lymphoma cells. J Virol 84:13053–13058. doi: 10.1128/JVI.01521-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Labrecque LG, Xue SA, Kazembe P, Phillips J, Lampert I, Wedderburn N, Griffin BE. 1999. Expression of Epstein-Barr virus lytically related genes in African Burkitt’s lymphoma: correlation with patient response to therapy. Int J Cancer 81:6–11. doi:. [DOI] [PubMed] [Google Scholar]
- 30.Yamamoto M, Tabata T, Smith M, Tanaka A, Nonoyama M. 1989. Cycloheximide-resistant gene of Epstein-Barr virus in freshly infected B lymphocytes. Virology 170:307–310. doi: 10.1016/0042-6822(89)90385-1. [DOI] [PubMed] [Google Scholar]
- 31.Mrozek-Gorska P, Buschle A, Pich D, Schwarzmayr T, Fechtner R, Scialdone A, Hammerschmidt W. 2019. Epstein-Barr virus reprograms human B lymphocytes immediately in the prelatent phase of infection. Proc Natl Acad Sci U S A 116:16046–16055. doi: 10.1073/pnas.1901314116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xue SA, Griffin BE. 2007. Complexities associated with expression of Epstein-Barr virus (EBV) lytic origins of DNA replication. Nucleic Acids Res 35:3391–3406. doi: 10.1093/nar/gkm170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dambaugh TR, Kieff E. 1982. Identification and nucleotide sequences of two similar tandem direct repeats in Epstein-Barr virus DNA. J Virol 44:823–833. doi: 10.1128/JVI.44.3.823-833.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jones MD, Griffin BE. 1983. Clustered repeat sequences in the genome of Epstein Barr virus. Nucleic Acids Res 11:3919–3937. doi: 10.1093/nar/11.12.3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin Z, Wang X, Strong MJ, Concha M, Baddoo M, Xu G, Baribault C, Fewell C, Hulme W, Hedges D, Taylor CM, Flemington EK. 2013. Whole-genome sequencing of the Akata and Mutu Epstein-Barr virus strains. J Virol 87:1172–1182. doi: 10.1128/JVI.02517-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arvey A, Tempera I, Tsai K, Chen HS, Tikhmyanova N, Klichinsky M, Leslie C, Lieberman PM. 2012. An atlas of the Epstein-Barr virus transcriptome and epigenome reveals host-virus regulatory interactions. Cell Host Microbe 12:233–245. doi: 10.1016/j.chom.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang C, Li D, Zhang L, Jiang S, Liang J, Narita Y, Hou I, Zhong Q, Zheng Z, Xiao H, Gewurz BE, Teng M, Zhao B. 2019. RNA sequencing analyses of gene expression during Epstein-Barr virus infection of primary B lymphocytes. J Virol 93:e00226-19. doi: 10.1128/JVI.00226-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dresang LR, Teuton JR, Feng H, Jacobs JM, Camp DG II, Purvine SO, Gritsenko MA, Li Z, Smith RD, Sugden B, Moore PS, Chang Y. 2011. Coupled transcriptome and proteome analysis of human lymphotropic tumor viruses: insights on the detection and discovery of viral genes. BMC Genomics 12:625. doi: 10.1186/1471-2164-12-625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rennekamp AJ, Lieberman PM. 2011. Initiation of Epstein-Barr virus lytic replication requires transcription and the formation of a stable RNA-DNA hybrid molecule at OriLyt. J Virol 85:2837–2850. doi: 10.1128/JVI.02175-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Park R, Miller G. 2018. Epstein-Barr virus-induced nodules on viral replication compartments contain RNA processing proteins and a viral long noncoding RNA. J Virol 92:e01254-18. doi: 10.1128/JVI.01254-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Toptan T, Abere B, Nalesnik MA, Swerdlow SH, Ranganathan S, Lee N, Shair KH, Moore PS, Chang Y. 2018. Circular DNA tumor viruses make circular RNAs. Proc Natl Acad Sci U S A 115:E8737–E8745. doi: 10.1073/pnas.1811728115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ungerleider N, Concha M, Lin Z, Roberts C, Wang X, Cao S, Baddoo M, Moss WN, Yu Y, Seddon M, Lehman T, Tibbetts S, Renne R, Dong Y, Flemington EK. 2018. The Epstein Barr virus circRNAome. PLoS Pathog 14:e1007206. doi: 10.1371/journal.ppat.1007206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sample J, Lancz G, Nonoyama M. 1986. Mapping of genes in BamHI fragment M of Epstein-Barr virus DNA that may determine the fate of viral infection. J Virol 57:145–154. doi: 10.1128/JVI.57.1.145-154.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kelly G, Bell A, Rickinson A. 2002. Epstein-Barr virus-associated Burkitt lymphomagenesis selects for downregulation of the nuclear antigen EBNA2. Nat Med 8:1098–1104. doi: 10.1038/nm758. [DOI] [PubMed] [Google Scholar]
- 45.Raab-Traub N, Dambaugh T, Kieff E. 1980. DNA of Epstein-Barr virus VIII: B95-8, the previous prototype, is an unusual deletion derivative. Cell 22:257–267. doi: 10.1016/0092-8674(80)90173-7. [DOI] [PubMed] [Google Scholar]
- 46.Lieberman PM, Hardwick JM, Hayward SD. 1989. Responsiveness of the Epstein-Barr virus NotI repeat promoter to the Z transactivator is mediated in a cell-type-specific manner by two independent signal regions. J Virol 63:3040–3050. doi: 10.1128/JVI.63.7.3040-3050.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nuebling CM, Mueller-Lantzsch N. 1989. Identification and characterization of an Epstein-Barr virus early antigen that is encoded by the NotI repeats. J Virol 63:4609–4615. doi: 10.1128/JVI.63.11.4609-4615.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Swaminathan S. 2005. Post-transcriptional gene regulation by gamma herpesviruses. J Cell Biochem 95:698–711. doi: 10.1002/jcb.20465. [DOI] [PubMed] [Google Scholar]
- 49.Mure F, Panthu B, Zanella-Cleon I, Delolme F, Manet E, Ohlmann T, Gruffat H. 2018. Epstein-Barr virus protein EB2 stimulates translation initiation of mRNAs through direct interactions with both poly(A)-binding protein and eukaryotic initiation factor 4G. J Virol 92:e01917-17. doi: 10.1128/JVI.01917-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hughes DJ, Dickerson CA, Shaner MS, Sample CE, Sample JT. 2011. trans-repression of protein expression dependent on the Epstein-Barr virus promoter Wp during latency. J Virol 85:11435–11447. doi: 10.1128/JVI.05158-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cai X, Schafer A, Lu S, Bilello JP, Desrosiers RC, Edwards R, Raab-Traub N, Cullen BR. 2006. Epstein-Barr virus microRNAs are evolutionarily conserved and differentially expressed. PLoS Pathog 2:e23. doi: 10.1371/journal.ppat.0020023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pfeffer S, Zavolan M, Grasser FA, Chien M, Russo JJ, Ju J, John B, Enright AJ, Marks D, Sander C, Tuschl T. 2004. Identification of virus-encoded microRNAs. Science 304:734–736. doi: 10.1126/science.1096781. [DOI] [PubMed] [Google Scholar]
- 53.Seto E, Moosmann A, Gromminger S, Walz N, Grundhoff A, Hammerschmidt W. 2010. Micro RNAs of Epstein-Barr virus promote cell cycle progression and prevent apoptosis of primary human B cells. PLoS Pathog 6:e1001063. doi: 10.1371/journal.ppat.1001063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Feederle R, Haar J, Bernhardt K, Linnstaedt SD, Bannert H, Lips H, Cullen BR, Delecluse HJ. 2011. The members of an Epstein-Barr virus microRNA cluster cooperate to transform B lymphocytes. J Virol 85:9801–9810. doi: 10.1128/JVI.05100-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Feederle R, Linnstaedt SD, Bannert H, Lips H, Bencun M, Cullen BR, Delecluse HJ. 2011. A viral microRNA cluster strongly potentiates the transforming properties of a human herpesvirus. PLoS Pathog 7:e1001294. doi: 10.1371/journal.ppat.1001294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hurley EA, Agger S, McNeil JA, Lawrence JB, Calendar A, Lenoir G, Thorley-Lawson DA. 1991. When Epstein-Barr virus persistently infects B-cell lines, it frequently integrates. J Virol 65:1245–1254. doi: 10.1128/JVI.65.3.1245-1254.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pearson GR, Luka J, Petti L, Sample J, Birkenbach M, Braun D, Kieff E. 1987. Identification of an Epstein-Barr virus early gene encoding a second component of the restricted early antigen complex. Virology 160:151–161. doi: 10.1016/0042-6822(87)90055-9. [DOI] [PubMed] [Google Scholar]
- 58.Austin PJ, Flemington E, Yandava CN, Strominger JL, Speck SH. 1988. Complex transcription of the Epstein-Barr virus BamHI fragment H rightward open reading frame 1 (BHRF1) in latently and lytically infected B lymphocytes. Proc Natl Acad Sci U S A 85:3678–3682. doi: 10.1073/pnas.85.11.3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kelly GL, Long HM, Stylianou J, Thomas WA, Leese A, Bell AI, Bornkamm GW, Mautner J, Rickinson AB, Rowe M. 2009. An Epstein-Barr virus anti-apoptotic protein constitutively expressed in transformed cells and implicated in Burkitt lymphomagenesis: the Wp/BHRF1 link. PLoS Pathog 5:e1000341. doi: 10.1371/journal.ppat.1000341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kelly GL, Stylianou J, Rasaiyaah J, Wei W, Thomas W, Croom-Carter D, Kohler C, Spang R, Woodman C, Kellam P, Rickinson AB, Bell AI. 2013. Different patterns of Epstein-Barr virus latency in endemic Burkitt lymphoma (BL) lead to distinct variants within the BL-associated gene expression signature. J Virol 87:2882–2894. doi: 10.1128/JVI.03003-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rabson M, Gradoville L, Heston L, Miller G. 1982. Non-immortalizing P3J-HR-1 Epstein-Barr virus: a deletion mutant of its transforming parent, Jijoye. J Virol 44:834–844. doi: 10.1128/JVI.44.3.834-844.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Miller G, Robinson J, Heston L, Lipman M. 1974. Differences between laboratory strains of Epstein-Barr virus based on immortalization, abortive infection, and interference. Proc Natl Acad Sci U S A 71:4006–4010. doi: 10.1073/pnas.71.10.4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Menezes J, Leibold W, Klein G. 1975. Biological differences between Epstein-Barr virus (EBV) strains with regard to lymphocyte transforming ability, superinfection and antigen induction. Exp Cell Res 92:478–484. doi: 10.1016/0014-4827(75)90404-8. [DOI] [PubMed] [Google Scholar]
- 64.Heller M, Dambaugh T, Kieff E. 1981. Epstein-Barr virus DNA. IX. Variation among viral DNAs from producer and nonproducer infected cells. J Virol 38:632–648. doi: 10.1128/JVI.38.2.632-648.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hammerschmidt W, Sugden B. 1989. Genetic analysis of immortalizing functions of Epstein-Barr virus in human B lymphocytes. Nature 340:393–397. doi: 10.1038/340393a0. [DOI] [PubMed] [Google Scholar]
- 66.Cohen JI, Wang F, Mannick J, Kieff E. 1989. Epstein-Barr virus nuclear protein 2 is a key determinant of lymphocyte transformation. Proc Natl Acad Sci U S A 86:9558–9562. doi: 10.1073/pnas.86.23.9558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Skare J, Farley J, Strominger JL, Fresen KO, Cho MS, zur Hausen H. 1985. Transformation by Epstein-Barr virus requires DNA sequences in the region of BamHI fragments Y and H. J Virol 55:286–297. doi: 10.1128/JVI.55.2.286-297.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mannick JB, Cohen JI, Birkenbach M, Marchini A, Kieff E. 1991. The Epstein-Barr virus nuclear protein encoded by the leader of the EBNA RNAs is important in B-lymphocyte transformation. J Virol 65:6826–6837. doi: 10.1128/JVI.65.12.6826-6837.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Marchini A, Cohen JI, Wang F, Kieff E. 1992. A selectable marker allows investigation of a nontransforming Epstein-Barr virus mutant. J Virol 66:3214–3219. doi: 10.1128/JVI.66.5.3214-3219.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sample J, Kieff E. 1990. Transcription of the Epstein-Barr virus genome during latency in growth-transformed lymphocytes. J Virol 64:1667–1674. doi: 10.1128/JVI.64.4.1667-1674.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Concha M, Wang X, Cao S, Baddoo M, Fewell C, Lin Z, Hulme W, Hedges D, McBride J, Flemington EK. 2012. Identification of new viral genes and transcript isoforms during Epstein-Barr virus reactivation using RNA-Seq. J Virol 86:1458–1467. doi: 10.1128/JVI.06537-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Marinov GK, Williams BA, McCue K, Schroth GP, Gertz J, Myers RM, Wold BJ. 2014. From single-cell to cell-pool transcriptomes: stochasticity in gene expression and RNA splicing. Genome Res 24:496–510. doi: 10.1101/gr.161034.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kopp F, Mendell JT. 2018. Functional classification and experimental dissection of long noncoding RNAs. Cell 172:393–407. doi: 10.1016/j.cell.2018.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Djavadian R, Hayes M, Johannsen E. 2018. CAGE-seq analysis of Epstein-Barr virus lytic gene transcription: 3 kinetic classes from 2 mechanisms. PLoS Pathog 14:e1007114. doi: 10.1371/journal.ppat.1007114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Majerciak V, Yang W, Zheng J, Zhu J, Zheng Z-M. 2019. A genome-wide Epstein-Barr virus polyadenylation map and its antisense RNA to EBNA. J Virol 93:e01593-18. doi: 10.1128/JVI.01593-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Grande BM, Gerhard DS, Jiang A, Griner NB, Abramson JS, Alexander TB, Allen H, Ayers LW, Bethony JM, Bhatia K, Bowen J, Casper C, Choi JK, Culibrk L, Davidsen TM, Dyer MA, Gastier-Foster JM, Gesuwan P, Greiner TC, Gross TG, Hanf B, Harris NL, He Y, Irvin JD, Jaffe ES, Jones SJM, Kerchan P, Knoetze N, Leal FE, Lichtenberg TM, Ma Y, Martin JP, Martin M-R, Mbulaiteye SM, Mullighan CG, Mungall AJ, Namirembe C, Novik K, Noy A, Ogwang MD, Omoding A, Orem J, Reynolds SJ, Rushton CK, Sandlund JT, Schmitz R, Taylor C, Wilson WH, Wright GW, Zhao EY, et al. 2019. Genome-wide discovery of somatic coding and noncoding mutations in pediatric endemic and sporadic Burkitt lymphoma. Blood 133:1313–1324. doi: 10.1182/blood-2018-09-871418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ruf IK, Rhyne PW, Yang H, Borza CM, Hutt-Fletcher LM, Cleveland JL, Sample JT. 1999. Epstein-Barr virus regulates c-MYC, apoptosis, and tumorigenicity in Burkitt lymphoma. Mol Cell Biol 19:1651–1660. doi: 10.1128/mcb.19.3.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Molesworth SJ, Lake CM, Borza CM, Turk SM, Hutt-Fletcher LM. 2000. Epstein-Barr virus gH is essential for penetration of B cells but also plays a role in attachment of virus to epithelial cells. J Virol 74:6324–6332. doi: 10.1128/jvi.74.14.6324-6332.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kanda T, Yajima M, Ahsan N, Tanaka M, Takada K. 2004. Production of high-titer Epstein-Barr virus recombinants derived from Akata cells by using a bacterial artificial chromosome system. J Virol 78:7004–7015. doi: 10.1128/JVI.78.13.7004-7015.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG. 2005. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res 33:e36. doi: 10.1093/nar/gni035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Phan AT, Fernandez SG, Somberg JJ, Keck KM, Miranda JL. 2016. Epstein-Barr virus latency type and spontaneous reactivation predict lytic induction levels. Biochem Biophys Res Commun 474:71–75. doi: 10.1016/j.bbrc.2016.04.070. [DOI] [PMC free article] [PubMed] [Google Scholar]