

Abstract
Background
Trichloroethylene (TCE) was negative for developmental toxicity after inhalation and oral gavage exposure of pregnant rats but fetal cardiac defects were reported following drinking water exposure throughout gestation. Because of the deficiencies in this latter study, we performed another drinking water study to evaluate whether TCE causes heart defects.
Methods
Groups of 25 mated Sprague Dawley rats consumed water containing 0, 0.25, 1.5, 500, or 1,000 ppm TCE from gestational day 1–21. TCE concentrations were measured at daily formulation, when placed into water bottles each day and when water bottles were removed from cages. Four additional mated rats per group were used for plasma measurements. At termination, fetal hearts were carefully dissected fresh and examined.
Results
All TCE concentrations were >90% of target when initially placed in water bottles and when bottles were placed on cages. All dams survived with no clinical signs. Rats in the two higher dose groups consumed less water/day than other groups but showed no changes in maternal or fetal weights. The only fetal cardiac observation was small (<1 mm) membranous ventricular septal defect occurring in all treated and water control groups; incidences were within the range of published findings for naive animals. TCE was not detected in maternal blood, but systemic exposure was confirmed by detecting its primary oxidative metabolite, trichloroacetic acid, although only at levels above the quantitation limit in the two higher dose groups.
Conclusions
Ingesting TCE in drinking water ≤1,000 ppm throughout gestation does not cause cardiac defects in rat offspring.
Keywords: TCE, toxicokinetics, trichloroacetic acid, ventricular septal defects
1. INTRODUCTION
Trichloroethylene (TCE) is a chlorinated hydrocarbon that has been in use for more than a century and a half. Historically, TCE has been used as an extractant for fats, oils, and spices and to decaffeinate coffee (Linak, Leder, & Yoshida, 1992). By the mid‐1970's, the U.S. Food and Drug Administration (FDA) banned its use as a food additive (U.S. Food and Drug Administration, 1977). TCE has been used medically as an anesthetic for dental, medical, and obstetrical procedures (Nowill, Stephen, & Searles, 1953; Prior, 1972), to disinfect minor wounds and surgical instruments (Sadove, Wyant, & Gittelson, 1953), as well as in the treatment of trigeminal neuralgia (Defalque, 1961). TCE was used widely as a dry‐cleaning agent until it was replaced by the more fabric‐friendly perchloroethylene in the 1950's (Reich & Cormany, 1979). Currently, the vast majority of TCE is used as a chemical intermediate in the closed‐system manufacture of refrigerants (U.S. Department of Health and Human Services, 2014), although it is still used as an industrial metal‐degreasing agent (U.S. Department of Health and Human Services, 2014). It also continues to have minor use as a solvent in the production of paints, lacquers, and varnishes (Mertens, 1993).
The extensive past uses and its environmental persistence in soil and groundwater (Wu & Schaum, 2000) have made TCE a commonly detected environmental pollutant, albeit at low concentrations (U.S. Department of Health and Human Services, 2014). The 50th and 95th percentile ambient air concentrations at hundreds of locations across the United States between 1998 and 2008 have been reported to range from 0.025 to 0.030 and 0.091 to 0.250 ppb, respectively (U.S. Department of Health and Human Services, 2014). TCE is detected in 4.5–18% of monitored U.S. ground‐ or surface‐water drinking water supplies with median and 95th percentile concentrations ranging from 1.10 to 2.50 and 11.0 to 29.0 ppb, respectively, for the years 1998–2005 (U.S. Department of Health and Human Services, 2014). A median occupational exposure of 43 ppm TCE has been described for degreasing operations from the 1940's to the 1980's, but interpretation of these measurements is challenging due to the generally small numbers and short‐term nature of the air samples (Bakke, Stewart, & Waters, 2007). In recent decades, however, the advent of closed‐loop machines has substantially reduced worker exposures in TCE degreasing operations (von Grote, Hürlimann, Scheringer, & Hungerbühler, 2003). The ACGIH occupational threshold limit value is 10 ppm with a short‐term exposure limit of 25 ppm (American Conference of Governmental Industrial Hygienists [ACGIH], 2018). TCE has been thoroughly investigated for its potential to cause adverse health effects by authoritative bodies over the past four decades (European Commission‐Joint Research Centre, 2004; International Agency for Research on Cancer, 1979, 1995, 2014; National Research Council [NRC], 2006; U.S. Department of Health and Human Services, 2014; U.S. Environmental Protection Agency [EPA], 1985, 2001, 2011). Of particular concern is a series of studies reporting TCE as adversely impacting cardiac development in animals and humans at very low to very high TCE exposures (Dawson, Johnson, Goldberg, & Ulreich, 1990, 1993; Forand, Lewis‐Michl, & Gomez, 2012; Goldberg, Lebowitz, Graver, & Hicks, 1990; Johnson, Dawson, & Goldberg, 1998; Johnson, Goldberg, Mays, & Dawson, 2003). These findings have been questioned due to limitations in experimental design and interpretation of the animal data (Hardin, Kelman, & Brent, 2005; Watson, Jacobson, Williams, Howard, & DeSesso, 2006) and of several human studies reporting positive associations of TCE exposures and cardiac malformations (Bukowski, 2014; Hardin et al., 2005; Watson et al., 2006). The key drinking water study (Johnson et al., 2003) has been the subject of two published errata (Johnson, Goldberg, Mays, & Dawson, 2005, 2014) and one letter to the editor (Johnson, Goldberg, Mays, & Dawson, 2004) to explain apparent inconsistencies in their data and to clarify some of their methods. In large, well‐conducted guideline‐ (or near guideline‐) and good laboratory practices (GLP)‐compliant studies, TCE did not cause adverse developmental effects by the inhalation (Carney, Thorsrud, Dugard, & Zablotny, 2006) or oral gavage (Fisher et al., 2001) routes. The primary oxidative metabolite of TCE, trichloroacetic acid (TCA) also did not adversely affect cardiac development in rats following 300 mg/kg/day dosing on gestational day (GD) 6–15 (Fisher et al., 2001). The top inhalation concentration of 600 ppm, 6 hr/day has been estimated as equivalent to a systemic dose of 141–240 mg/kg/day (Koizumi, 1989; Stott, Quast, & Watanabe, 1982), and the oral gavage study was conducted at a single daily dose of 500 mg/kg/day (Fisher et al., 2001). The highest TCE dosage administered in the drinking water study reported as causing increased cardiac malformations, 1,100 ppm (the water solubility limit of TCE; O'Neil, Heckelman, & Roman, 2006), was estimated as a daily systemic dose of 129 mg/kg/day. However, increased cardiac lesions were also described at the very much lower daily drinking water dose of 0.048 mg/kg/day (0.25 ppm) (Johnson et al., 2003). Such a dose–response pattern is highly unusual given the 4,400‐fold differences in the range of responding doses. The doses delivered in this study are questionable as there were no analytical analyses reported to confirm concentrations of TCE in the water. This imposes considerable limits on the interpretation of Johnson et al. (2003), especially at the lower nominal drinking water concentrations.
These divergent observations have been the subject of numerous subsequent assessments that either support (Chiu et al., 2013; Makris et al., 2016) or assail (Hardin et al., 2005; Watson et al., 2006) the drinking water findings, or question the use of this study as the basis for risk assessment (Wikoff, Urban, Harvey, & Haws, 2018).
In its toxicological review of TCE, the EPA derived a reference concentration of 0.4 ppb (2 μg/m3) and reference dose of 0.0005 mg/kg‐day based, in part, on the Johnson et al. (2003) cardiac malformation data for its Integrated Risk Information System (IRIS) (U.S. Environmental Protection Agency, 2011). As a consequence, there has been a great deal of public concern regarding the potential for induction of cardiac malformations from TCE exposure in indoor air. Inhalation exposure limits of 2 μg/m3 have been issued by EPA Region 9 and some states for short‐term exposures to TCE to protect women in their first trimester of pregnancy, resulting in mitigation measures that have included evacuation of residents or workers from buildings (U.S. Environmental Protection Agency, 2014). TCE is also listed as a high priority chemical for evaluating the human health risk to workers and the general population under the amended Toxic Substance Control Act (TSCA) known as the Frank R. Lautenberg Chemical Safety for the 21st Century Act. EPA is required by Congress to complete this assessment by the end of 2019. Hence, there is an important need to resolve the reproducibility of the Johnson et al. (2003) study.
The purpose of the experiments described herein is to test the hypothesis that exposing mated rats to drinking water containing TCE at concentrations similar to those of Johnson et al. (2003; ranging from 0.250 to 1,000 ppm) throughout gestation (GD 1–21) causes cardiac malformations in offspring. The experimental design adhered as closely as possible to published guidance for performing developmental toxicity studies and followed GLPs. However, the present investigation was focused primarily on the internal anatomy of the heart, to the exclusion of other structures, and used procedures for detailed inspection of the septa and valves that exceeded the attention given to those structures during typical cardiac examinations. Because the volatility of TCE raises concern for substantial loss of the test material during preparation of drinking water formulations (especially at the low concentrations) and to address one of the limitations in the Johnson studies (Johnson et al., 2003), particular attention was given to this aspect of the experimental design. To ensure that the water concentrations delivered to test animals were acceptably close to the nominal concentrations, meticulous methods were employed to avoid loss by volatilization of TCE content from the drinking water. In addition, TCE was measured at the time of the daily preparation of test solutions, as well as at the time of filling of water bottles and 24 hr later at the time of replacement of the water bottles. Systemic exposure was determined in a separate toxicokinetic arm that measured TCA (a stable metabolite of TCE) in the plasma of dams and fetuses at various times during gestation.
2. MATERIALS AND METHODS
2.1. Test materials
TCE, 99.98% pure was procured from Spectrum Chemical Manufacturing Corporation (New Brunswick, NJ). TCE was kept at 18–24°C in a temperature‐controlled room and protected from light. Solubility, stability, and concentrations of TCE in dosing solutions were analyzed by high performance liquid chromatography (HPLC) with an ultraviolet (UV) detector. The TCE dosing formulations were prepared daily, in a closed system, under amber light, without sonication, and stored and transported in the same closed system. All formulation batches were prepared at volumes large enough to minimize headspace. A known volume of prechilled (2–8°C) water was placed in an 11.5 L amber glass bottle. The bottle was purged with nitrogen and sealed with a foil liner and silicone septum fitted with a fabricated siphon valve system built at the test facility (Figure 1). The required volume of TCE was injected via a glass syringe and metal cannula (~24″) through the silicone septum cap affixed to the bottle; the length of the cannula ensured that TCE was injected into the water, near the bottom of the amber glass bottle to minimize contact with ambient air/nitrogen. The 500 and 1,000 ppm concentrations were prepared on the day prior to dosing and stirred slowly at room temperature for at least 24 hr to ensure dissolution of TCE in water. The 0.25 and 1.5 ppm concentrations were prepared via dilution of higher concentrations on the day of dose administration.
Figure 1.
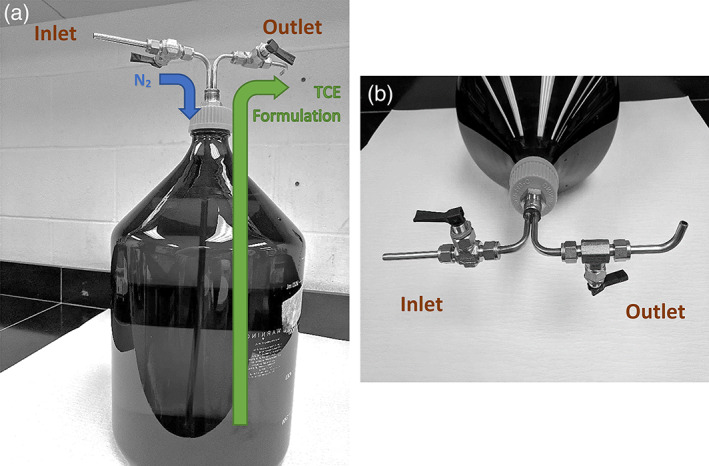
An 11.5 L amber glass bottle used for batch formulation preparation is shown fitted with a fabricated siphon valve to ensure that formulations are prepared in a closed system (a). The bottle was purged with nitrogen; it was then capped and sealed with a foil liner and silicone septum. Note the two cannulas, each with its own siphon valve. Each siphon valve had an inlet and an outlet outfitted with a spigot (b), whereby the inlet valve pumped nitrogen into the bottle while the TCE‐containing water formulation was removed via the outlet valve. TCE, trichloroethylene
The TCE formulations were stored at room temperature (18–24°C) until dispensation and use. During transfer into drinking water bottles, nitrogen displaced the dosing formulations that were removed via the outlet value. Purging headspace with nitrogen reduced volatilization of TCE and ensured that residual water formulations did not come in contact with ambient air. Drinking water bottles were filled by allowing the water to flow along the inner wall to reduce splashing, bubbling, and volatilization of TCE.
To ensure the animals received a known dose, the TCE formulated water was analyzed directly immediately after preparation using HPLC/UV and authentic standards (methodological details are provided in the Supporting Information section). The concentrations of TCE were measured in the drinking water formulations and vehicle control at the time of preparation and again after placement in the water bottles (immediately following transfer from the 11.5 L formulation bottles). Measurements were made from the 1st, 2nd, 3rd, 7th, 12th, 15th, 22nd, and last batch of drinking water formulations. Two 10 mL samples were collected from the middle stratum and transferred into amber glass auto‐sampler vials with rubber stoppers, and crimped tops for concentration analysis. The loss of TCE from the water bottles in the animal cages over the 24‐hr exposure was also determined for the 1st, 22nd, and last batch of dosing formulations. Two 10 mL samples were collected from three randomly selected water bottles at the end of the 24‐hr exposure period. Loss of TCE was calculated by subtracting the TCE concentration in the water bottle at the end of the 24‐hr exposure period from the measurement of TCE concentration made at the time of preparation.
All‐trans retinoic acid (RA), 100% pure, was purchased from Sigma Aldrich (St. Louis, MO); it was kept at −20°C and protected from light. RA was used as a positive control for heart malformations. The RA dosing formulations in soybean oil were prepared approximately weekly, and an adequate amount of each formulation was dispensed into daily aliquots, which were stored frozen at −20°C, purged with nitrogen and protected from light until use. The dosing formulation for each day was thawed and stirred for at least 30 min. before administration.
2.2. Animals
Virgin female Crl:CD(SD) rats were received from Charles River Laboratories, Inc. (Raleigh, NC) on July 17, 2018. The animals were approximately 12 weeks old at receipt. During the acclimation period the rats were weighed, identified with a microchip, and examined for general appearance and behavior. Females were paired on a 1:1 basis with resident males. Vaginal lavages were performed daily during the mating period until evidence of mating was observed. Positive evidence of mating was considered by the presence of either a vaginal copulatory plug or sperm in a vaginal lavage. Animals were assigned to groups by a stratified randomization scheme designed to achieve similar group mean body weights. Animals at extremes of body weight ranges were not assigned to groups. Mated females weighed between 221 and 281 g on GD 0.
During cohabitation, the animals were paired for mating in the home cage of the male. Following the breeding period, the females were individually housed. Throughout the study animals were housed in solid‐bottom cages containing corn cob bedding (Bed‐ O'Cobs®) and were kept on a 12‐hr light/dark cycle in an environmentally controlled room. The ventilation rate was set at a minimum of 10 room air changes per hour. Animals were maintained in accordance with the Guide for the Care and Use of Laboratory Animals (NRC, 2011). The animal facilities were accredited by the Association for Assessment and Accreditation of Laboratory Care International (AAALAC International). Animals were fed PMI Nutrition International, LLC Certified Rodent LabDiet® 5002 ad libitum. Municipal water was reverse osmosis‐purified (with TCE added for the appropriate animal groups during the treatment period) and available ad libitum via amber glass water bottles with metal sipper tubes. The water bottles were changed daily during the treatment period.
2.3. Study design
One hundred and forty‐five mated female Crl:CD(SD) rats were randomly assigned to five groups of 25 rats per group and were given drinking water alone or with TCE at concentrations of 0.25, 1.5, 500, or 1,000 ppm from GD 1–21, which covered the range of doses (0.25; 1.5; 1,100 ppm) where cardiac malformation were reported by Johnson et al. (2003). Four additional mated rats per group were included for the measurement of plasma TCA only. The RA positive control group was included to verify that the study animals were susceptible to a known cardiac teratogen. The RA group consisted of an additional 25 mated rats that were dosed once daily by oral gavage with 15 mg/kg/day RA on GD 6–15. Each animal was observed twice daily for moribundity and mortality during the study. Body weights, food, and water consumption were measured daily. On GD 21, all rats were euthanized by carbon dioxide asphyxiation. Immediately thereafter, a complete gross necropsy was performed that included evaluation of the thoracic, abdominal, and pelvic cavities. The ovaries were inspected for corpora lutea. The gravid uterus was weighed and examined for the number and distribution of implantation sites, live and dead fetuses, and early and late resorptions. The placenta was also examined. Each viable fetus was examined externally in detail, sexed (confirmed by internal examination), weighed, and euthanized by a subcutaneous injection of sodium pentobarbital in the scapular region. All fetuses were examined for visceral cardiac anomalies by dissection in the fresh (nonfixed) state. Assessors were blinded with regard to the treatment group of the specimens being necropsied and examined. The thoracic cavity was opened, and the external and internal anatomy of the heart examined using a technique described by Stuckhardt and Poppe (1984). This examination was limited to a thorough examination of the heart and great and major blood vessels using a dissecting microscope. Any observed ventricular septal defects (VSDs) were characterized by size (<1 mm, 1–2 mm, or > 2 mm) and location (muscular or membranous). Observations were verified by a senior teratologist and were photographed along with representative controls. All carcasses were discarded following completion of the internal examination according to standard operating procedures.
2.4. Determination of TCA plasma levels
Preliminary experiments showed that TCE concentrations in maternal blood were below the level of quantitation (50 ng/mL; Supporting Information). This was likely due to extensive oral first‐pass hepatic metabolism of TCE aided by the drinking water (“trickle‐in”) versus bolus gavage mode of administration (Liu, Bartlett, White, Muralidhara, & Bruckner, 2009). We note that TCE concentrations similarly were not detectable in the blood of pregnant rats administered 350 ppm TCE in drinking water, while TCE and TCA were quantitated in pregnant rats orally gavaged with 2.3 mg/kg/day TCE or exposed for 4 hr to 600 ppm TCE (Fisher, Whittaker, Taylor, Clewell, & Andersen, 1989). As a result, plasma levels of TCA, which is a nonvolatile oxidative metabolite of TCE, were measured in this study to gauge TCE exposure.
Briefly, maternal blood (0.5 mL) was collected from the jugular veins of animals that were assigned to the exposure assessment phase of the study between 0830 and 0930 hr on GD 8 and 12 and just prior to euthanasia on GD 21. Immediately after maternal blood collection and euthanasia on GD 21, fetal blood was collected via cardiac puncture under isoflurane inhalation and pooled by litter. Once collected, the blood samples were transferred into chilled tubes containing lithium heparin, capped, and maintained on wet ice, protected from light. The plasma was separated by centrifugation and stored at −70°C, protected from light, until assayed. The levels of TCA were determined as follows. Under yellow light, an aliquot of 250 μL of the internal standard trifluoroacetic acid (TFA) solution (1 μg/mL in a solution of 2.5% formic acid in acetonitrile) was added to each 50 μL plasma sample in a 96‐well plate on wet ice. The mixture was vortexed, centrifuged, and a 200 μL aliquot was then transferred to another 96‐well plate and evaporated under N2. The residue was reconstituted with 275 μL 80:20 water/methanol. The quantity of TCA was determined by a validated UHPLC–MS/MS method in the negative electrospray ionization (ESI‐) mode, using trifluoroacetic acid as an internal standard. The lower limit of quantification was 150 ng/mL.
2.5. Statistical analyses
Analyses were conducted using two‐tailed tests (except as noted otherwise) for minimum significance levels of 1 and 5%, comparing each test substance‐treated group to the vehicle control group by sex. The positive control group was compared separately to the drinking water control group. Where applicable, the litter was used as the experimental unit. Maternal body weights and body weight changes (absolute and net), food and water consumption, gravid uterine weights, numbers of corpora lutea, implantation sites, and viable fetuses, and fetal body weights (separated by sex and combined) were subjected to a parametric one‐way analysis of variance (ANOVA) to determine intergroup differences (Snedecor & Cochran, 1980). If the results of the ANOVA were significant (p < .05), the Dunnett's test (Dunnett, 1964) was used to compare all the treatment groups to the control group. For the positive control group, the data were compared to the drinking water control group using a two‐sample t test (Sokal & Rohlf, 1981). Mean litter proportions of prenatal data (viable and nonviable fetuses), early and late resorptions, total resorptions, pre‐ and post‐implantation loss, and fetal sex distribution), total fetal cardiac malformations and developmental variations, and each particular visceral cardiac malformation or variation were analyzed by the Kruskal–Wallis nonparametric ANOVA test (Kruskal & Wallis, 1952) to determine intergroup differences. If the nonparametric ANOVA showed statistical significance (p < .05), the Dunn's test (Dunn, 1964) was used to compare the test substance‐treated groups and the positive control group to the drinking water control group.
3. RESULTS
3.1. Analysis of dosing formulations
Analyses of the drinking water formulations for TCE content at the end of the preparation stage demonstrated that all samples contained at least 90% of the target concentrations, indicating minimal loss of TCE to volatilization during preparation (Table S1). The concentrations of TCE in the drinking water bottles immediately after transfer from dosing formulation containers and before placement in the animal cages to begin the exposure period were also all greater than 90% of nominal (Table S2). Comparison of the values in Tables S1 and S2 demonstrates negligible losses of TCE from the dosing formulations during this transfer step. The TCE drinking water concentrations within each dose group were generally consistent throughout the study. As animals received fresh batches of drinking water formulations on a daily basis, the TCE concentrations in the drinking water bottles were also measured at the end of the 24‐hr exposure period. The loss of TCE over the 24‐hr period ranged from 30.2 to 48.6% (Table S3), which is similar to the losses reported in other TCE rat drinking water studies that evaluated this parameter (Fisher et al., 1989; Johnson et al., 2003).
3.2. Trichloroacetic acid concentrations
TCE blood levels were not measured in this study because a preliminary study did not detect TCE in the blood of pregnant rats following exposure to 1,000 ppm TCE in drinking water on GD 12 and 13 (Below the Level of Quantitation, <50 ng/mL) (methodological details are provided in the Supporting Information section; data not shown). As a consequence, plasma TCA was measured as a surrogate of TCE exposure. Maternal and fetal plasma samples were collected from the four dams per group assigned to toxicokinetic analyses. All of the dams selected for toxicokinetic analyses survived to study termination and all were pregnant. The concentrations of TCA in maternal plasma on GDs 8, 12, and 21 and in fetal plasma concentrations at GD 21 are tabulated in Table 1.
Table 1.
TCA concentrations (μg/mL) in maternal plasma during gestation and in fetal plasma at study termination
| TCE exposure | Maternal plasma | Fetal plasma | ||
|---|---|---|---|---|
| GD 8 | GD 12 | GD 21 | GD 21 | |
| 0 ppm | NDa | ND | ND | ND |
| 0.25 ppm | ND | ND | ND | ND |
| 1.5 ppm | ND | ND | ND | ND |
| 500 ppm | 1.71 ± 0.46 | 1.81 ± 0.88 | 1.11 ± 0.24 | 1.17 ± 0.27 |
| 1,000 ppm | 1.70 ± 0.59 | 2.24 ± 0.62 | 1.165 ± 0.37 | 1.24 ± 0.43 |
Abbreviation: GD, gestational day; TCE, trichloroethylene.
ND, not detected, < limit of quantitation (150 ng/mL).
The plasma concentration of TCA did not increase during gestation and the concentrations were comparable at both of the higher exposure levels. In addition, the ratios of maternal to fetal plasma concentrations at study termination (GD 21) were 0.95 and 0.94, respectively. These values indicate that TCA was not preferentially transported to or stored in the fetus.
3.3. Observations in maternal animals
When mated rats received drinking water containing 0, 0.25, 1.5, 500, or 1,000 ppm TCE from GD 1–21, their calculated mean daily intakes1 of TCE were 0, 0.04, 0.21, 58.03, and 113.45 mg TCE/kg/day, respectively. No deaths or clinical signs of toxicity were reported, although rats in the 500 and 1,000 ppm TCE groups drank significantly less water than rats in the other groups (Figure 2). However, because the maternal body weights throughout gestation (Figure 3), body weight gains (Figure 4), and food consumption (data presented in Table S4) were similar among all groups, these findings were considered to be nonadverse.
Figure 2.
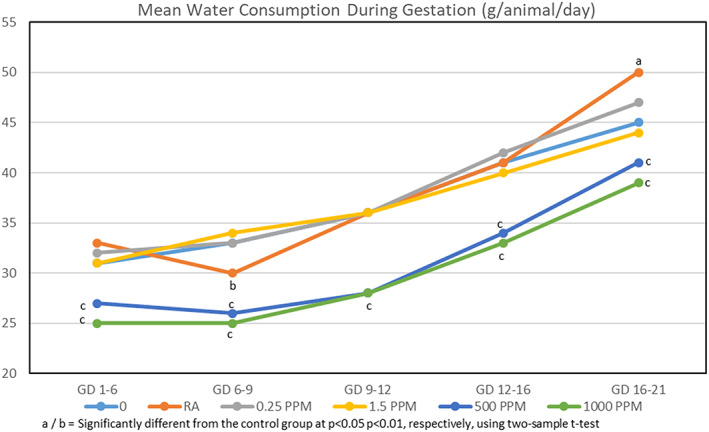
Mean water consumption during gestation. Values based on measured concentrations of TCE in dosing formulations prior to transfer to water bottles and the amount of water consumed; it does not account for the loss of TCE from the drinking water when given to the animals in water bottles over the 24‐hr exposure period. TCE, trichloroethylene
Figure 3.
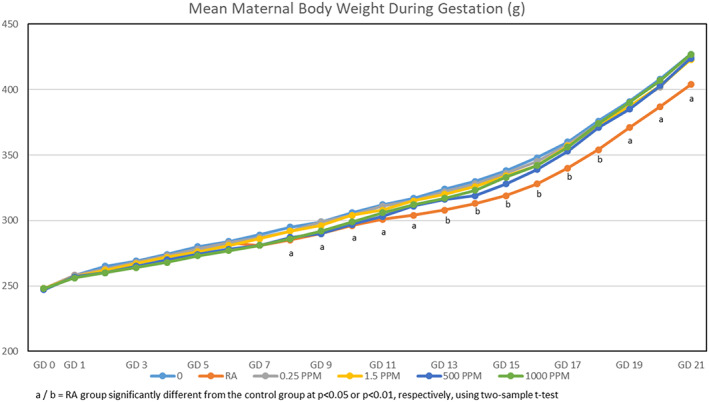
Mean body weights during gestation
Figure 4.
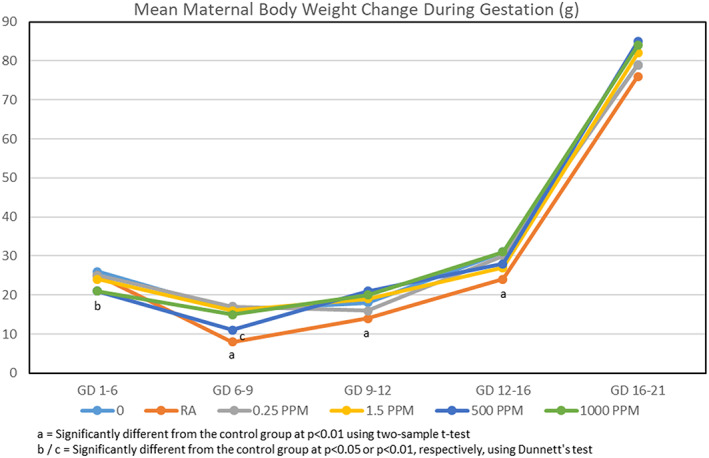
Mean body weight gains during gestation
Maternal necropsies revealed no significant macroscopic findings in TCE‐exposed rats compared to water controls. The mean number of corpora lutea, implantation sites, and early or late resorptions did not differ significantly among the TCE and control groups (Table 2). Rates of both pre‐ and post‐implantation loss were similar among TCE and control groups.
Table 2.
Summary of ovarian and uterine data
| TCE (ppm) | No. pregnant females | Mean no. corpora lutea | Mean no. implantation sites | Pre‐implantation loss (%) | Resorptions | Dead fetuses | Post implantation loss (%) | Mean no. viable fetuses | Mean fetal weights (g) | |
|---|---|---|---|---|---|---|---|---|---|---|
| Early | Late | |||||||||
| 0 | 24 | 15.0 ± 2.7 | 13.7 ± 4.4 | 1.3 ± 2.2 | 0.8 ± 1.2 | 0.0 ± 0.2 | 0 | 0.9 ± 1.4 | 12.8 ± 4.3 |
6.0 ± 0.3 combined 6.1 ± 0.3 ♂ 5.9± 0.3 ♀ |
| 0.25 | 23 | 15.6 ± 2.6 | 13.2 ± 3.4 | 2.4 ± 4.7 | 1.2 ± 2.5 | 0.0 ± 0.0 | 0 | 1.2 ± 2.5 | 12.0 ± 4.1 |
6.2 ± 0.3 combined 6.3 ± 0.4 ♂ 6.0± 0.3 ♀ |
| 1.5 | 24 | 15.1 ± 2.1 | 13.8 ± 3.2 | 1.3 ± 1.8 | 0.5 ± 0.9 | 0.0 ± 0.0 | 0 | 0.5 ± 0.9 | 13.4 ± 3.1 |
5.9 ± 0.4 combined 6.1 ± 0.5 ♂ 5.8± 0.4 ♀ |
| 500 | 24 | 15.4 ± 1.9 | 14.4 ± 1.8 | 1.0 ± 1.2 | 0.7 ± 1.1 | 0.0 ± 0.0 | 0 | 0.7 ± 1.1 | 13.8 ± 2.2 |
6.0 ± 0.3 combined 6.2 ± 0.4 ♂ 5.9± 0.3 ♀ |
| 1,000 | 24 | 16.3 ± 2.2 | 14.9 ± 2.5 | 1.3 ± 2.2 | 0.6 ± 0.7 | 0.0 ± 0.2 | 0 | 0.7 ± 0.8 | 14.3 ± 2.4 |
5.9 ± 0.3 combined 6.1 ± 0.3 ♂ 5.8± 0.3 ♀ |
Note: Data are presented as mean ± SD, where appropriate.
Abbreviation: TCE, trichloroethylene.
3.4. Fetal evaluations
Neither the mean number of fetuses per litter nor the mean fetal weights (combined and by separate sex) differed among the groups. External examination of fetuses found no TCE‐related malformations. A total of two fetuses with malformations were identified. One was a control fetus and the other was a fetus from a high dose litter. Both presented with microphthalmia. Given their presence in a control fetus, these cases of microphthalmia were considered to be incidental findings and not related to treatment.
Skeletal examinations were not performed, and visceral examination was limited to detailed inspection of the heart and the great and major vessels. All cardiac defects identified in the TCE‐exposed fetuses were VSDs (Table 3) and their numbers were not significantly different from the negative controls (p ≤ .01). In the control group, there were eight fetuses with defective hearts, seven of which presented with VSDs. The eighth malformed control fetus presented with situs inversus. Greater than 40% of the fetuses in the RA positive control group presented with VSDs, which clearly demonstrated the susceptibility of the developing rat heart to the malformation of interest in this study.
Table 3.
Summary of ventricular septal defects
| Fetal parameter | TCE concentration | Positive control | ||||
|---|---|---|---|---|---|---|
| 0 ppm | 0.25 ppm | 1.5 ppm | 500 ppm | 1,000 ppm | RA 15 mg/kg/day | |
| Available fetuses (litters) | 308 (24) | 275 (22)a | 321 (24) | 330 (24) | 342 (24) | 269 (25) |
| Affected fetuses (litters) | 7 (5) | 4 (4) | 5 (3) | 13 (8) | 12 (6) | 112 (23) |
| Mean litter proportion (% per litter) | 2.4 | 1.4 | 1.5 | 3.8 | 3.7 | 42.2** |
| No. fetuses (size of opening) | 6 (<1 mm) 1 (1‐2 mm) | All (<1 mm) | All (<1 mm) | All (<1 mm) | All (<1 mm) | 103 (<1 mm); 8 (1–2 mm); 1 (>2 mm) |
| Location of opening | Mem | Mem | Mem | Mem | Mem | Mem (111 fetuses) Mus/Mem (1 fetus) |
Abbreviations: Mem, membranous portion of ventricular septum; Mus/Mem, muscular and membranous portions of ventricular septum; RA retinoic acid; TCE, trichloroethylene.
One female had a very early total litter resorption and therefore had no available fetuses.
**Significantly different from vehicle control group at p ≤.01.
Fetuses in the RA group (positive control for offspring effects) demonstrated the expected array of congenital defects including craniofacial and digital malformations observed at external evaluation and cardiac and great and major vessel malformations reported during the visceral examination.
4. DISCUSSION
The impetus for the present study was the data reported by Johnson et al. (2003) in which TCE exposure to 9–13 litters per drinking water dose group resulted in statistically increased congenital heart defects in fetuses at exposure concentrations of 0.25 and 1,100 ppm, but not in the 1.5 ppm group. The objective of this hypothesis‐driven study was to evaluate the occurrence of cardiac defects in offspring in guideline‐acceptable numbers of pregnant dams consuming TCE formulated drinking water from the day following copulation throughout the entire gestational period. No deaths or treatment‐related clinical signs were reported in any of the TCE or water‐control groups. The rats in the two higher dose groups (500 and 1,000 ppm TCE) experienced reduced mean daily water consumption; however, this was ascribed to poor palatability because there were no changes in terminal mean body weights of the dams, mean body weight gains, or mean fetal body weights when compared to controls. Thus, there was no overt maternal toxicity at concentrations that approached the aqueous solubility limit of TCE.
The dominant finding during the visceral examination of the heart was the occurrence of VSDs at similar litter proportions across all TCE‐exposed and control groups. To appreciate the scale and location of this defect, it is important to briefly review the anatomy and embryology of the ventricular septum, which is similar between rats and humans. The anatomy of VSDs has been described thoroughly (e.g., Penny & Vick, 2011). The right and left ventricles of the heart are separated by the ventricular septum, which is composed of two parts (Gardner, Gray, & O'Rahilly, 1969; Warwick & Williams, 1973) and is depicted in Figure 5. The larger portion is the muscular ventricular septum which is made up of cardiac muscle that originates in the bulboventricular ridge in the floor of the right ventricle and extends posteromedially toward the endocardial cushion tissue in the floor of the atria (Harvey & Rosenthal, 1999). The muscular portion ultimately accounts for >90% of the ventricular septum.
Figure 5.
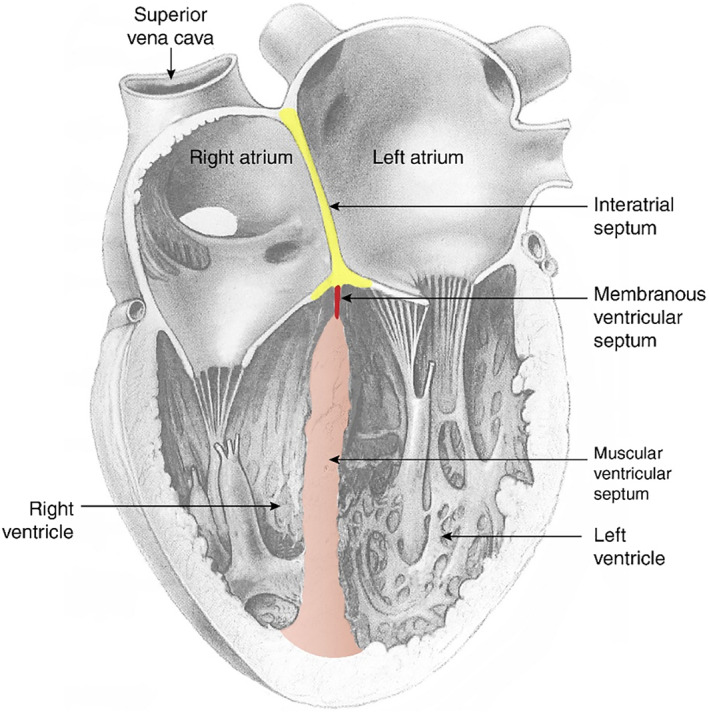
This diagram illustrates a coronal section through a postnatal heart. The muscular ventricular septum, shown in section, forms the medial walls of the right and left ventricles. Note that the muscular septum (pink) contributes more than 90% of the vertical height of the ventricular septum. The membranous portion of the ventricular septum (red) is the small thin‐walled structure near the floor of the atria. In the present study, the reported VSDs in the TCE exposed groups were all located in the membranous ventricular septum and were smaller than 1 mm in diameter. The right and left atria are separated by the interatrial septum (yellow), which is attached to the floor of the atria. TCE, trichloroethylene; VSDs, ventricular septal defects
In the embryo, the right and left ventricles are connected by a space (interventricular foramen) that allows blood from the low‐functioning embryonic pulmonary circulation to be shunted from the right ventricle to the left ventricle to join the systemic circulation. The interventricular foramen is gradually diminished by continued growth of the muscular septum. Final closure occurs when (a) the inferior edge of the conotruncal ridge (precursor to the aorticopulmonary septum, which separates the aorta from the pulmonary artery) is anchored at the tip of the muscular septum and (b) contributions from the adjacent endocardia cushion tissue reinforce the junction (Carlson, 2014). Typically, these processes are complete by GD 15–15.5 in rats, although the membranous portion of the interventricular septum may fully close perinatally (DeSesso, 2012). This small final structure (membranous ventricular septum) is made up of thin‐walled tissue that fills in the gap between the tip of the muscular septum and the floor of the atria. The membranous ventricular septum is quite small compared to the muscular potion of the septum. It is found at the apex of the septum in a location where there is relatively low pressure on the septum. The foramina identified in the control and TCE‐exposed rat fetuses were all smaller than 1 mm. Such small foramina would transmit little (if any) blood and would close spontaneously rather quickly after birth. No increases in septal defects in the membranous region or the muscular portion of the septum were observed in the TCE‐treatment groups.
The natural history of VSDs in rat fetuses has been studied by following pups throughout lactation or later. Solomon et al. (1997) examined untreated Sprague Dawley rat offspring for cardiac defects perinatally, at the end of lactation and as adults. Examination included careful dissection of the heart followed by light or scanning electron microscopic examination the ventricular septum. VSDs were apparent in 2.0% of near‐term fetuses and measured approximately 1 mm in diameter (in close agreement with the 7 of 308, 2.4%, VSDs of late‐term control fetuses in Table 3). Pups examined at weaning had no VSDs nor were VSDs found among rats allowed to survive for 5 or 10 months. The authors concluded that small VSDs close spontaneously during neonatal life. In another part of the study, positive control animals were administered 50 mg/kg of trypan blue subcutaneously on GD 7 to 9 or 400 mg/kg b.i.d. of trimethadione orally on GD 9 and 10. VSDs occurred in 93% of fetuses in the trimethadione group and in 9% of the fetuses in the trypan blue group, showing that cardiac teratogens cause a greater incidence of VSDs. However, no positive control pups were examined postnatally, not allowing for further evaluation of whether small chemically induced VSDs were likewise capable of rapid postnatal closure.
The hypothesis concerning postnatal closure of small VSDs has been examined by Fleeman, Cappon, and Hurtt (2004), who administered 400 (200 b.i.d.) or 600 (300 b.i.d.) mg/kg of trimethadione orally to mated rats on GD 9 and 10. A total of 7.4% of the fetuses from the 400 mg/kg trimethadione group presented with small VSDs, but no VSDs were found in pups at weaning. In the 600 mg/kg group, 39.6% of fetuses at term presented with VSDs of 2 sizes; at weaning 6.4% of pups had VSDs, none of which were small. The authors concluded that closure of VSDs depends on the severity of lesion seen at term. The foramina identified in the present study are as small as, or smaller than, the experimentally created foramina that have been found to close spontaneously during the lactation period in rats. Our results support the conclusions of Fleeman et al. (2004) that there are two populations of VSDs: small VSDs which close spontaneously and should be considered normal developmental delay; and larger VSDs that are not expected to close and are considered severe malformations.
The incidence of VSDs in all groups was far above the rate of 0.26% in the Charles River Ashland Historical Control Database for fetal observations. The mean litter proportion of VSDs in the control group was more than ninefold higher than the maximum mean value for this parameter in the historical controls. The increased reporting of VSDs is the result of the design of this study, which placed a singular focus on the heart and great vessels during the visceral examination. This resulted in a heightened sensitivity of the observers to interior heart anatomy and additional time in exploring that region of the heart compared to a typical fetal necropsy. Consequently, the laboratory's historical control database was not a useful comparator for VSDs. Rather, VSD data from previously published journal articles of studies that had similar meticulous examinations of interior heart anatomy were used (Table 4; Haring, 1965, 1966; Inomata & Yasuda, 1971). It is important to note that VSDs also are the most common congenital heart defect, accounting for ~39% of reported congenital heart defects, in human infants (Hoffman, 1995). It is also important to note that the natural history of these defects has been studied for more than 50 years and that 85–90% of the observed defects close spontaneously during the first year of life (Bloomfield, 1964; Hoffman & Kaplan, 2002; Hoffman & Rudolph, 1965; Turner, Hunter, & Wylie, 1999). These authors point out that virtually all small VSDs close and that the larger VSDs often diminish in size during the same period.
Table 4.
Fetal incidence of ventricular septal defects (VSDs) in control Sprague Dawley rats after enhanced cardiac evaluations
| Study | Ventricular septal defects (%) |
|---|---|
| Haring (1965)a | 3.2 |
| Haring (1966)a | 2.8 |
| Inomata and Yasuda (1971)b | 5.2 |
| Inomata and Yasuda (1971)b | 3.6 |
| Solomon et al. (1997) | 2.0 |
| Current studyc | 2.4 |
Hearts were embedded in paraffin, serially sectioned and examined by light microscopy.
Examined by a combination of Wilson freehand razor sections plus microdissection of the cardiac outflow tract.
Examined by fresh dissection.
In the current study, our findings fail to find evidence of a TCE‐related increase in the occurrence of large (and likely persistent) VSDs or any other heart malformation. With respect to the widespread finding of VSDs, it is important to note that an effort was made to specify the location of each VSD and to measure its size. In all cases in fetuses from the TCE and water control groups, the VSDs were small (<1 mm in size) and were located in the membranous ventricular septum. These are the types of foramina that are expected to close spontaneously in humans and were shown to close in the studies of rats by Solomon et al. (1997) and Fleeman et al. (2004).
There are some important differences between our study and that of Johnson et al. (2003). Some of the data reported in the Johnson et al. (2003) paper had been previously reported 10 years earlier by Dawson et al. (1993), which reported cardiac defects but did not report the number of dams per group and presented limited maternal data. Control data were not tabulated. These shortcomings greatly impede the ability to independently evaluate their data (Hardin et al., 2005; Watson et al., 2006).
During the approximate 10 year interval between publication of the Dawson et al. (1993) and the Johnson et al. (2003) studies, it appears that data from additional control and TCE‐treated (0.0025 and 0.25 ppm) animals were collected. The control data presented in Johnson et al. (2003) were apparently derived from a pooling of control data collected from multiple experiments performed within the overall time period (Johnson et al., 2005). All of their data across these studies were then collated and presented in the Johnson et al. (2003) paper as a single study. The Johnson group evaluated the fetal hearts in a nonstandard fashion that involved removal of the atria from the heart, flushing of the heart with fixatives, and manipulation of the heart to evaluate valvular function. Over the course of the decade between publication of the Dawson et al. (1993) and the Johnson et al. (2003) studies, their method changed with respect to the order of surgical procedures (in some studies the heart was examined in situ, in others the heart was removed from the chest before examination). The ingredients of the fluid used to flush the hearts were altered as well.
The Johnson et al. (2003) paper reported several cardiac malformations not seen in any of the more robust studies (Table 5). These included atrial septal defects and various atrioventricular valve aberrations. Each of these anomalies could have been caused by the surgical methods used by the Johnson team. Hearts were placed in (or the mediastinum was flooded with) fixative prior to surgical removal of the atria (and detachment of the inferior border of the interatrial septum) and subsequent investigator manipulation of the delicate cardiac valve tissue (which would have become friable from the effects of the fixation fluid). Thus, it is possible that atrial and valvular damage could have resulted from iatrogenic causes rather than by any effect of TCE. Interestingly, however, when one compares the litter proportions of VSDs in the TCE‐treated groups in Johnson et al. (2003) study to those in the current study, they are nearly identical. Johnson et al. (2003) found VSDs in 1.7% of fetuses at 1.5 ppm (compared to 1.5% in the present study) and 3.8% of fetuses at 1,100 ppm (compared 3.7% at 1,000 ppm in the present study). These data show remarkable consistency across the studies and suggest that, when compared to the Charles River Laboratories historical control database (0.26%), the apparent increased VSD incidence could be due to the enhanced examinations of internal heart anatomy employed in these studies. An important difference between these two studies, however, is that Johnson et al. (2003) reported statistically increased cardiac malformations (including VSDs plus atrial septal defects, valvular malformations, transposition of the great vessels, and enlarged coronary sinus) when compared against pooled controls evaluated over an extended time period with varying evaluation techniques. In contrast, the present study identified only VSDs, which were not increased when measured against an appropriately sized and contemporaneous control group.
Table 5.
Heart abnormalities reported by Johnson et al. (2003)
| TCE concentration (ppm) | No. fetuses examined | Ventricular septal defects | Atrial septal defects | Malformed valves | Other cardiac findings | Total fetuses with abnormal heartsa |
|---|---|---|---|---|---|---|
| 0 | 606 | 4 | 7 | 1b | 3c | 13 |
| 0.25 | 110 | 0 | 1 | 3d | 1e | 5 |
| 1.5 | 181 | 3 | 4 | 0 | 2f | 9 |
| 1,100 | 105 | 5 | 7 | 2g | 0 | 11 |
Abbreviation: TCE, trichloroethylene.
Some fetuses had multiple abnormalities.
Atretic mitral valve leaflet.
Two fetuses with D‐loop in right chest; one with an endocardial cushion defect.
One fetus with small tricuspid valve; one with abnormal tricuspid valve; one with dysplastic aortic valve.
One fetus with a large coronary sinus.
One fetus with transposition of great vessels and one with absent right pulmonary artery.
One fetus with unicuspid aortic valve and one with a fenestrated aortic valve leaflet.
Importantly, two other robustly designed studies in rats, in addition to the present study using drinking water treatment similar to Johnson et al. (2003), have also failed to confirm the results of the Johnson group (Table 6). Carney et al. (2006) used TCE inhalation concentrations as high as 600 ppm (limit dose) for 6 hr/day from GD 6–20. Fisher et al. (2001) administered TCE at 500 mg/kg/day or its primary oxidative metabolite TCA and minor metabolite dichloroacetic acid at 300 mg/kg/day from GD 6–15 (Dr. Johnson was part of the pathology examination team for this study, which used examination techniques similar to that of Johnson et al., 2003). In contrast to Fisher et al. (2001) who did not detect an increase in cardiac malformations induced by 300 mg/kg/day TCA gavage, Johnson et al. (1998) reported that TCA (2,730 ppm in drinking water; 291 mg/kg/day) induced cardiac malformations in rats similar to those reported in the TCE drinking water studies from their laboratory. In the current study, TCA also was detected in maternal plasma on GD 8, 12, and 21 only in the 500 and 1,000 ppm drinking water dose groups, but not at the lower concentrations. TCA was also detected in fetal plasma on GD 21 at concentrations equivalent to those in maternal plasma. These data indicate that the absence of TCE‐related cardiac effects in the present study was likely not attributable to an absence of substantial maternal metabolism to TCA, or to a failure of maternal TCA to be distributed to the fetus. Further confidence in the maternal GD 21 TCA concentrations of 1.1–1.2 μg/mL measured in the present study is supported by their reasonable agreement with the maternal GD 21 TCA blood concentration of 2.8 μg/mL reported for SD rats exposed to 350 ppm TCE in drinking water for 3 weeks of pregnancy (Fisher et al., 1989).
Table 6.
Comparison of various studies investigating TCE‐induced cardiac defects in Sprague Dawley rats
| Study | Route | TCE exposure | Duration | Number of females/group | Maternal toxicity | Exam method | Abnormal heartsa |
|---|---|---|---|---|---|---|---|
| Fisher et al. (2001) | Oral gavage | 500 mg/kg/day | GD 6–15 | 20 | None observed | Johnson | 4.5% (13/290) |
| 0 (soybean oil) | 25 | 6.5% (24/367) | |||||
| Johnson et al. (2003) | Drinking water | 0 ppm | GD 1–21 | 55 | Not assessed | Johnson | 2.2% (13/606) |
| 0.0025 ppm (0.0005 mg/kg/day) | 12 | 0 | |||||
| 0.25 ppm (0.05 mg/kg/day) | 9 | 4.5% (5/110) | |||||
| 1.5 ppm (0.22 mg/kg/day) | 13 | 5.0% (9/181) | |||||
| 1,100 ppm (129 mg/kg/day) | 9 | 10.5% (7/308) | |||||
| Present study | Drinking water | 0 ppm | GD 1–21 | 24 | None observed | Enhanced Stuckhardt and Poppe | 2.3% (7/308) |
| 0.25 ppm (0.04 mg/kg/day) | 23 | 1.5% (4/275) | |||||
| 1.5 ppm (0.21 mg/kg/day) | 24 | 1.6% (5/321) | |||||
| 500 ppm (58 mg/kg/day) | 24 | 3.9% (13/330) | |||||
| 1,000 ppm (113.5 mg/kg/day) | 24 | 3.5% (12/342) | |||||
| Carney et al. (2006) | Inhalation | 0 ppm | GD 6–20 (6 hr/day; 7 day/week) | 22 | None observed | Stuckhardt and Poppe | 0 |
| 50 ppm | 22 | None observed | 0 | ||||
| 150 ppm | 22 | None observed | 0 | ||||
| 600 ppm | 22 | 22% decrease in body weight gain GD 6–9 | 0 |
Abbreviations: GD, gestational day; TCE, trichloroethylene.
Values presented are percent overall incidence (incidence data).
A comparison of TCE toxicokinetics following drinking water, inhalation or gavage administration indicates that parent TCE is a dosimetrically implausible source of potential TCE cardiac malformations (Table 7). The inability of a preliminary investigation in this study to detect TCE in maternal blood in rats treated at the near limit of aqueous solubility (1,000 ppm) in drinking water is consistent with other findings that TCE was not detected in maternal blood of SD rats on GD 21 following 3 weeks of 350 ppm TCE drinking water exposures (Fisher et al., 1989). The analytical TCE level of detection (LOD) of 30 ng/mL in this earlier study was similar to that of 50 ng/mL in the present study. In contrast to an absence of TCE detection during drinking water exposures, the toxicokinetic data in Table 7 indicate that TCE would clearly be present in maternal blood in the earlier 500 mg/kg/day gavage (Fisher et al., 2001) and 600 ppm 6 hr/day inhalation (Carney et al., 2006) developmental toxicity studies. Peak TCE concentrations of 0.26, 33, and 24 μg/mL were observed following gavage doses of 2.3 mg/kg to SD rats on GD 21, of 591 mg/kg to adult male SD rats, and a 4 hr 600 ppm inhalation exposure to SD rats on GD 21, respectively (Fisher et al., 1989; Larson & Bull, 1992). The inability to detect TCE following drinking water dosing is likely due to enhanced first‐pass hepatic oxidative metabolism of TCE administered by “trickle‐in” drinking water administration compared to gavage bolus administration (Liu et al., 2009). Taken together, these kinetic data cast substantial doubt on the speculation in Johnson et al. (2003) that a drinking water concentration of 0.25 ppm, which is four orders of magnitude smaller than the highest exposure used in the present study, could result in maternal TCE plasma concentrations sufficient to directly disrupt embryonic heart development.
Table 7.
Comparison of peak plasma concentration of TCE and TCA after various exposures
| Peak (AUC) concentration (μg/mL or mg·hr/L) | ||||||
|---|---|---|---|---|---|---|
| TCE | TCA | |||||
| Exposure Route | Drinking watera | Drinking waterb | Inhalationb | Gavageb | Gavagec | Gavagec |
| TCE/TCA | 0.25–1,000 ppm | 350 ppm | 600 ppm, 4 hr | 2.3 mg/kg | 591 mg/kg | 98 mg/kg |
| TCE blood | ND | ND | 24 (1,864) | 0.26 (0.72) | 33 | — |
| TCA plasma | 1.1–1.2 | 2.7 (585) | 13 (5,201) | 0.70 (106) | 25 | 201 |
Abbreviations: AUC, area under the curve; TCA, trichloroacetic acid; TCE, trichloroethylene.
Current study, ND = Below LOD (50 ng/mL); TCA detected only 500 and 1,000 ppm, not at 0.25 and 1.5 ppm.
Fisher et al. (1989); TCE and TCA measured in pregnant GD21 SD rats following dosing at 5 days/week for 3 weeks.
Larson and Bull (1992); TCE and TCA measured after single dose to adult male rats; TCE C max estimated from graphical data.
Examination of the toxicokinetics of TCA in pregnant and nonpregnant rats dosed with TCE or TCA across varying routes and modes of administration provide additional evidence informing the interpretation of the Johnson et al. (2003) drinking water study compared to multiple studies that did not detect TCE‐ or TCA‐induced cardiac malformations following oral bolus (TCE, TCA; Fisher et al., 2001), inhalation (TCE; Carney et al., 2006), and drinking water (TCE; current study) exposures (Tables 6 and 7). Compared to plasma TCA concentrations of 1.1–1.2 μg/mL detected only in rats administered 500 or 1,000 ppm TCE in drinking water, a peak plasma concentration of 13 μg/mL TCA was observed after a 4 hr 600 ppm TCE inhalation exposure to GD 21 rats, and 25 μg/mL in adult male rats receiving a 591 mg/kg TCE gavage dose close to the 500 mg/kg/day dose used in the Fisher et al. (2001) TCE developmental toxicity study. A peak TCA concentration of 0.70 μg/mL was detected in GD 21 rats gavaged with 2.3 mg/kg TCE, a dose approximately 200‐fold lower than the 500 mg/kg/day gavage dosing used Fisher et al. (2001). A TCA C max of 201 μg/mL was observed in adult male rats gavaged with 98 mg/kg TCA, indicating that peak plasma TCA concentrations were likely very high following the 300 mg/kg TCA gavage treatment regimen used in the Fisher et al. (2001) developmental toxicity study. These findings indicate that the reason that the high‐quality oral bolus and inhalation studies did not cause cardiac malformations is not due to lower systemic peak concentrations of TCA compared to those in either the present negative drinking water study or to the Johnson et al. (2003).
The toxicokinetic AUC data in Table 7 further support a conclusion that the gavage and inhalation modes of administration delivered substantively higher TCA doses compared to the drinking water route. Fisher et al. (1989) used physiologically based pharmacokinetic modeling to estimate maternal blood TCA area under the curve values of 106, 585, and 5,201 mg‐hr/L reported, respectively, for pregnant SD rats receiving 2.3 mg/kg TCE by oral bolus, 350 ppm drinking water or a 4 hr 600 ppm inhalation exposure. These data indicate that a 4 hr 600 ppm inhalation exposure (compared to 6 hr 600 ppm reported in in Carney et al., 2006) resulted in a total systemic dose of TCA that was approximately ninefold greater than that delivered by a drinking water dose (350 ppm). As noted above, gavage TCA dosing substantially increased peak plasma TCA concentrations relative to that achieved following all modes of TCE administration, indicating the 300 mg/kg/day gavage TCA developmental toxicity study of Fisher et al. (2001) resulted in the highest systemic TCA dose challenge.
In their assessment of the potential impact of TCE on heart development, Makris et al. (2016) cite in vitro chick embryo data (Boyer, Finch, & Runyan, 2000) in which explanted embryonic heart tissue from the presumptive atrioventricular canal of stage 16 chick embryos was cultured in media containing TCE at concentrations of 50–250 ppm. Under these in vitro study conditions TCE inhibited the process of epithelial–mesenchymal transformation, which occurs during formation of the fibrous cardiac skeleton. Importantly, Boyer et al. (2000) state that concentrations below 50 ppm were not tested due to data indicating the insensitivity of the assay to concentrations lower than 50 ppm. Thus, these cardiac cells were unresponsive when bathed in fluid containing TCE concentrations nominally 1,000‐fold greater than the plasma of rats treated with 1,000 ppm in drinking water, assuming a conservatively estimated LOD of 0.05 ppm for TCE. Makris et al. (2016) also cite Mishima, Hoffman, Hill, and Krug (2006) who observed responses similar to those of Boyer et al. (2000) in chick embryo culture at 80 ppm TCE, but not at 10 ppm. Despite the incongruity of these in vitro chick embryo exposures (>50 ppm) to those conservatively assumed as present in blood of rats exposed to TCE by drinking water, Makris et al. (2016) used these studies to speculate that a key event in the TCE adverse outcome pathway (AOP) for cardiac malformations, inferred as relevant to rats, is TCE inhibition of endothelial–mesenchymal transition, which was not observed at TCE exposure concentrations <50 ppm.
The extremely large differential between the in vitro test concentrations producing the response and actual in vivo kinetic data indicate this proposed AOP is not dosimetrically plausible for TCE. Importantly, scientists from the computational toxicology program at EPA have published multiple papers indicating that margins of exposure of this size relative to concentrations eliciting in vitro results indicate a low priority health concern (Rotroff et al., 2010; Thomas et al., 2013; Wetmore et al., 2012).
The fact that TCE and TCA developmental studies conducted by inhalation and oral gavage failed to show an increase in fetal heart defects, even at TCE and TCA plasma levels that greatly exceed the levels achievable in drinking water studies, provides additional confirmatory evidence of an absence of adverse developmental health risks associated with drinking water exposures to TCE.
It is also important to emphasize the consistency in findings among the three robustly performed studies of the potential effect of TCE on heart development (Carney et al., 2006; Fisher et al., 2001; the present study). The first two of these studies were nearly contemporaneous with the Johnson study. The in‐life portion of the Fisher et al. (2001) study, which produced a higher dose of TCE than the Johnson study, occurred in 1997, 2 years after the experimental period reported by the Johnson team in their first erratum (Johnson et al., 2005). The Carney et al. (2006) inhalation study, which also delivered a higher dose of TCE than Johnson and also found no cardiac defects in offspring, was performed approximately 4 years after the Johnson exposures. It has been speculated (Makris et al., 2016) that genetic drift could explain why the findings of a single laboratory (Dawson et al., 1993; Johnson et al., 2003) have not been reproduced by subsequent studies. This speculation seems highly remote given the timing outlined above and because the heart and its development are highly conserved across vertebrate species as demonstrated by the consistent incidence of VSDs in the control Sprague Dawley rats from multiple breeders located on multiple continents over several decades (Table 4). Consequently, it is unlikely that disparity in the incidence of cardiac anomalies observed by Johnson et al. (2003) compared to the other studies is explained by genetic drift and more likely that it can be explained by differences in the dissection methodologies. Additionally, the pharmacokinetic data in the current study match fairly well with those of Fisher et al. (1989), indicating no difference in the handling of TCE and its metabolites by Sprague Dawley rats despite the 30 years difference in study executions.
5. CONCLUSIONS
Under the conditions of this GLP drinking water study conducted in pregnant Sprague Dawley rats, the NOAEL for maternal systemic and reproductive toxicity was 1,000 ppm (the highest dose tested). The NOAEL for fetal cardiac anomalies was also 1,000 ppm (the highest dose tested). The present study not only tested exposures of TCE that covered the range of doses employed in the drinking water study reported previously (Johnson et al., 2003), but also ensured that the deficiencies of the Johnson study were corrected. These included (a) following a robust design with balanced numbers of animals in each group with an adequate number of pregnant dams in each treatment group per guideline requirements; (b) performing the study during a single, defined time period; (c) obtaining animals from a single source at the same time; (d) identifying and ensuring the chemicals were from the same lots; (e) following the standard method for evaluating fetal hearts; (f) recording maternal clinical data, as well as ovarian and uterine parameters; (g) including a positive control group; and (h) obtaining contemporaneous steady state blood level data, in addition to measuring TCE concentrations in water formulations throughout the gestation treatment period.
In a wider perspective, it is important to recognize that the results of the present study, when added to the previously published results of Fisher et al. (2001) and Carney et al. (2006), conclusively demonstrate the absence of TCE‐induced cardiac defects in three high‐quality and robust studies, conducted in different laboratories, using three distinct routes/modes of administration. Further, the totality of the data indicates that the Carney et al. (2006) inhalation study is the most reliable and appropriate study for addressing essentially all environmental exposures associated with TCE (i.e., occupational, general population [air]), as well as special concerns such as vapor intrusion or secondary inhalation from TCE contaminated water (e.g., bathing, showers).
CONFLICT OF INTEREST
Dr. Pottenger is retired from the Olin Corporation, a member company of HSIA and a producer of TCE. Dr. Bevan is the Director, Scientific Programs at HSIA and was formerly a consultant to Westlake Chemical Company, a member of HSIA. Dr. Budinsky is an employee of The Dow Chemical Company, a member company of HSIA. The employer of Drs. Bus and DeSesso, Exponent, Inc., has contracts with HSIA. Dr. York is a consultant to HSIA. Drs. Coder and Sen and Ms. Lucarell are employees of Charles River Laboratories Ashland, the contract research organization that performed the research.
Supporting information
Appendix S1: Supplemental Information
Table S1: Concentration of TCE in Dosing Formulations (End of Preparation Stage)
Table S2: Concentration of TCE in Dosing Formulations (Water Bottles at Start of Exposure Period)
Table S3: Loss of TCE from Water Bottles During the 24‐hour Exposure Period
Table S4: Food Consumption during Various Periods of Gestation (g/animal/day_
ACKNOWLEDGMENTS
The authors acknowledge the Halogenated Solvents Industry Alliance and American Chemistry Council for funding and the staff of Charles River Laboratories Ashland for their technical contribution to this research. In addition, the authors thank Ms. Betty Dowd for preparation of the illustration of the heart.
DeSesso JM, Coder PS, York RG, et al. Trichloroethylene in drinking water throughout gestation did not produce congenital heart defects in Sprague Dawley rats. Birth Defects Research. 2019;111:1217–1233. 10.1002/bdr2.1531
This article was published online on 13 June 2019. An error was subsequently identified. This notice is included in the online and print versions to indicate that both have been corrected 22 June 2019.
Funding information Halogenated Solvents Industry Alliance (HSIA); American Chemistry Council
Data Availability: The data that support the findings of this study are available for download at the EPA docket for TSCA evaluation at https://www.regulations.gov/document?D=EPA-HQ-OPPT-2016-0737-0120. The report therein is Coder, P. S. (2019) Final Report. An Oral (Drinking Water) Study of the Effects of Trichloroethylene (TCE) on Fetal Heart Development in Sprague Dawley Rats. Charles River Laboratories, Laboratory Project ID 00459506.
Endnote
Values based on measured concentrations of TCE in dosing formulations prior to transfer to water bottles and the amount of water consumed; it does not account for the loss of TCE from the drinking water when given to the animals in water bottles over the 24‐hr exposure period.
DATA AVAILABILITY
The data that support the findings of this study are available for download at the EPA docket for TSCA evaluation at https://www.regulations.gov/document?D=EPA-HQ-OPPT-2016-0737-0120. The report therein is Coder, P. S. (2019) Final Report. An Oral (Drinking Water) Study of the Effects of Trichloroethylene (TCE) on Fetal Heart Development in Sprague Dawley Rats. Charles River Laboratories, Laboratory Project ID 00459506.
REFERENCES
- American Conference of Governmental Industrial Hygienists . (2018). 2018 TLVs® and BEIs® Based on the Documentation of the Threshold Limit Values for Chemical Substances and Physical Agents & Biological Exposure Indices. Cincinnati, OH: ACGIH® Worldwide.
- Bakke, B. , Stewart, P. A. , & Waters, M. A. (2007). Uses of and exposure to trichloroethylene in U.S. industry: A systematic literature review. Journal of Occupational and Environmental Hygiene, 4(5), 375–390. 10.1080/15459620701301763 [DOI] [PubMed] [Google Scholar]
- Bloomfield, D. K. (1964). The natural history of ventricular septal defect in patients surviving infancy. Circulation, 29, 914–955. [DOI] [PubMed] [Google Scholar]
- Boyer, A. S. , Finch, W. T. , & Runyan, R. B. (2000). Trichloroethylene inhibits development of embryonic heart valve precursors in vitro . Toxicological Sciences, 53, 109–117. 10.1093/toxsci/53.1.109 [DOI] [PubMed] [Google Scholar]
- Bukowski, J. (2014). Critical review of the epidemiological literature regarding the association between congenital heart defects and exposure to trichloroethylene. Critical Reviews in Toxicology, 44, 581–589. 10.3109/10408444.2014.910755 [DOI] [PubMed] [Google Scholar]
- Carlson, B. M. (2014). Human embryology and developmental biology (5th ed.). Philadelphia, PA: Elsevier Saunders. [Google Scholar]
- Carney, E. W. , Thorsrud, B. A. , Dugard, P. H. , & Zablotny, C. L. (2006). Developmental toxicity studies in Crl:CD (SD) rats following inhalation exposure to trichloroethylene and perchloroethylene. Birth Defects Research. Part B, Developmental and Reproductive Toxicology, 77, 405–412. 10.1002/bdrb.20091 [DOI] [PubMed] [Google Scholar]
- Chiu, W. A. , Jinot, J. , Scott, C. S. , Makris, S. L. , Cooper, G. S. , Dzubow, R. C. , … Caldwell, J. C. (2013). Human health effects of trichloroethylene: Key findings and scientific issues. Environmental Health Perspectives, 121, 303–311. 10.1289/ehp.1205879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson, B. V. , Johnson, P. D. , Goldberg, S. J. , & Ulreich, J. B. (1990). Cardiac teratogenesis of trichloroethylene and dichloroethylene in a mammalian model. Journal of the American College of Cardiology, 16, 1304–1309. 10.1016/0735-1097(90)90569-B [DOI] [PubMed] [Google Scholar]
- Dawson, B. V. , Johnson, P. D. , Goldberg, S. J. , & Ulreich, J. B. (1993). Cardiac teratogenesis of halogenated hydrocarbon‐contaminated drinking water. Journal of the American College of Cardiology, 21, 1466–1472. 10.1016/0735-1097(93)90325-U [DOI] [PubMed] [Google Scholar]
- Defalque, R. L. (1961). The specific analgesic effect of trichloroethylene upon the trigeminal nerve. Anesthesiology, 22, 379–384. [DOI] [PubMed] [Google Scholar]
- DeSesso, J. M. (2012). Comparative gestational milestones in vertebrate development In Hood R. D. (Ed.), Developmental and reproductive toxicology: A practical approach (3rd ed., pp. 93–138). Informa Healthcare: New York, NY. [Google Scholar]
- Dunn, O. J. (1964). Multiple comparisons using rank sums. Technometrics, 6, 241–252. [Google Scholar]
- Dunnett, C. W. (1964). New tables for multiple comparisons with a control. Biometrics, 20, 482–491. [Google Scholar]
- European Commission ‐ Joint Research Centre, Institute for Health and Consumer Protection, European Chemicals Bureau . (2004). European Union Risk Assessment Report: Trichloroethylene, CAS No. 79–01‐6, EINECS No. 201–167‐4, Volume 31. EUR 21057 EN. Retrieved from https://echa.europa.eu/documents/10162/83f0c99f-f687-4cdf-a64b-514f1e26fdc0
- Fisher, J. W. , Channel, S. R. , Eggers, J. S. , Johnson, P. D. , MacMahon, K. L. , … Graeter, L. J. (2001). Trichloroethylene, trichloroacetic acid, and dichloroacetic acid: Do they affect fetal rat heart development? International Journal of Toxicology, 20, 257–267. [DOI] [PubMed] [Google Scholar]
- Fisher, J. W. , Whittaker, T. A. , Taylor, D. H. , Clewell, H. J., III , & Andersen, M. E. (1989). Physiologically based pharmacokinetic modeling of the pregnant rat: A multiroute exposure model for trichloroethylene and its metabolite, trichloroacetic acid. Toxicology and Applied Pharmacology, 99, 395–414. [DOI] [PubMed] [Google Scholar]
- Fleeman, T. L. , Cappon, G. D. , & Hurtt, M. E. (2004). Postnatal closure of membranous ventricular septal defects in Sprague‐Dawley rat pups after maternal exposure with trimethadione. Birth Defects Research. Part B, Developmental and Reproductive Toxicology, 71, 185–190. 10.1002/bdrb.20011 [DOI] [PubMed] [Google Scholar]
- Forand, S. P. , Lewis‐Michl, E. L. , & Gomez, M. I. (2012). Adverse birth outcomes and maternal exposure to trichloroethylene and tetrachloroethylene through soil vapor intrusion in New York State. Environmental Health Perspectives, 120, 616–621. 10.1289/ehp.1103884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner, E. , Gray, D. J. , & O'Rahilly, R. (1969). Anatomy: A regional study of human structure (3rd ed.). Philadelphia, PA: W. B. Saunders and Company. [Google Scholar]
- Goldberg, S. J. , Lebowitz, M. D. , Graver, E. J. , & Hicks, S. (1990). An association of human congenital cardiac malformations and drinking water contaminants. Journal of the American College of Cardiology, 16, 155–164. 10.1016/0735-1097(90)90473-3 [DOI] [PubMed] [Google Scholar]
- Hardin, B. D. , Kelman, B. J. , & Brent, R. L. (2005). Trichloroethylene and dichloroethylene: A critical review of teratogenicity. Birth Defects Research. Part A, Clinical and Molecular Teratology, 73, 931–955. 10.1002/bdra.20192 [DOI] [PubMed] [Google Scholar]
- Haring, O. M. (1965). Effects of prenatal hypoxia on the cardiovascular system in the rat. Archives of Pathology, 80, 351–356. [PubMed] [Google Scholar]
- Haring, O. M. (1966). Malformation in the rat induced by maternal hypercapnia with hypoxia. Circulation Research, 19, 544–551. [DOI] [PubMed] [Google Scholar]
- Harvey, R. P. , & Rosenthal, N. (1999). Heart development (1st ed.). San Diego, CA: Academic Press. [Google Scholar]
- Hoffman, J. I. (1995). Incidence of congenital disease. I – Postnatal incidence. Pediatric Cardiology, 16, 103–113. [DOI] [PubMed] [Google Scholar]
- Hoffman, J. I. , & Rudolph, A. M. (1965). The natural history of ventricular septal defects in infancy. American Journal of Cardiology, 16, 634–653. [DOI] [PubMed] [Google Scholar]
- Hoffman, J. I. E. , & Kaplan, S. (2002). The incidence of congenital heart disease. Journal of the American College of Cardiology, 39, 1890–1900. 10.1016/S0735-1097(01)01742-9 [DOI] [PubMed] [Google Scholar]
- Inomata, N. , & Yasuda, M. (1971). Prevalence and types of interventricular septal defects in rat fetuses. Experimental Animals, 20, 15–20. [Google Scholar]
- International Agency for Research on Cancer . (1979). Monographs on the evaluation of the carcinogenic risk of chemicals to humans. Some halogenated hydrocarbons (Vol. 20, pp. 491–514). Lyon, France: World Health Organization. [Google Scholar]
- International Agency for Research on Cancer . (1995). Monographs on the evaluation of carcinogenic risks to humans. Dry cleaning, some chlorinated solvents, and other industrial chemicals. Lyon, France: World Health Organization. [PMC free article] [PubMed] [Google Scholar]
- International Agency for Research on Cancer . (2014). Monographs on the evaluation of carcinogenic risks to humans. Trichloroethylene, tetrachloroethylene, and some chlorinated agents (Vol. 106). Lyon, France: World Health Organization. [PMC free article] [PubMed] [Google Scholar]
- Johnson, P. D. , Dawson, B. V. , & Goldberg, S. J. (1998). Cardiac teratogenicity of trichloroethylene metabolites. Journal of the American College of Cardiology, 32, 540–545. 10.1016/S0735-1097(98)00232-0 [DOI] [PubMed] [Google Scholar]
- Johnson, P. D. , Goldberg, S. J. , Mays, M. Z. , & Dawson, B. V. (2003). Threshold of trichloroethylene contamination in maternal drinking waters affecting fetal heart development in the rat. Environmental Health Perspectives, 111, 289–292. 10.1289/ehp.5125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, P. D. , Goldberg, S. J. , Mays, M. Z. , & Dawson, B. V. (2004). Trichloroethylene: Johnson et al.'s response. Environmental Health Perspectives, 112, A608–A609. 10.1289/ehp.112-1247490 [DOI] [Google Scholar]
- Johnson, P. D. , Goldberg, S. J. , Mays, M. Z. , & Dawson, B. V. (2005). Errata. Threshold of trichloroethylene contamination in maternal drinking waters affecting fetal heart development in the rat. Environmental Health Perspectives, 113, A18 10.1289/ehp.113-1253738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, P. D. , Goldberg, S. J. , Mays, M. Z. , & Dawson, B. V. (2014). Erratum: Erratum for Johnson et al. [environ health Perspect 113:A18 (2005)]. Environmental Health Perspectives, 122, A94 https://doi.org/10/1289/ehp.122-A94 [Google Scholar]
- Koizumi, A. (1989). Potential of physiologically based pharmacokinetics to amalgamate kinetic data of trichloroethylene and tetrachloroethylene obtained in rats and man. British Journal of Industrial Medicine, 46, 239–249. 10.1136/oem.46.4.239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruskal, W. H. , & Wallis, W. A. (1952). Use of ranks in one‐criterion variance analysis. Journal of the American Statistical Association, 47, 583–621. [Google Scholar]
- Larson, J. L. , & Bull, R. J. (1992). Species differences in the metabolism of trichloroethylene to the carcinogenic metabolites trichloroacetate and dichloroacetate. Toxicology and Applied Pharmacology, 115, 278–285. 10.1016/0041-008X(92)90332-M [DOI] [PubMed] [Google Scholar]
- Linak, E. , Leder, A. , & Yoshida, Y. (1992). C2 chlorinated solvents In Chemical economics handbook (pp. 632.3000–632.3001). Menlo Park, CA: SRI International. [Google Scholar]
- Liu, Y. , Bartlett, M. G. , White, C. A. , Muralidhara, S. , & Bruckner, J. V. (2009). Presystemic elimination of trichloroethylene in rats following environmentally relevant oral exposures. Drug Metabolism and Disposition, 37, 1994–1998. 10.1124/dmd.109.028100 [DOI] [PubMed] [Google Scholar]
- Makris, S. L. , Scott, C. S. , Fox, J. , Knudsen, T. B. , Hotchkiss, A. K. , Arzuaga, X. , … Narotsky, M. G. (2016). A systematic evaluation of the potential effects of trichloroethylene exposure on cardiac development. Reproductive Toxicology, 65, 321–358. 10.1016/j.reprotox.2016.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertens, J. A. (1993). Chlorocarbons and chlorohydrocarbons In Kirk R. E., Othmer D. F., Grayson M., & Eckroth D. (Eds.), Kirk‐Othmer encyclopedia of chemical technology (Vol. 6, 4th ed., pp. 44–50). New York, NY: John Wiley and Sons. [Google Scholar]
- Mishima, N. , Hoffman, S. , Hill, E. G. , & Krug, E. L. (2006). Chick embryos exposed to trichloroethylene in an ex ovo culture model show selective defects in early endocardial cushion tissue formation. Birth Defects Research. Part A, Clinical and Molecular Teratology, 76, 517–527. 10.1002/bdra.20283 [DOI] [PubMed] [Google Scholar]
- National Research Council . (2006). Assessing the human health risks of trichloroethylene: Key scientific issues. Committee on Human Health Risks of Trichloroethylene, Board on Environmental Studies and Toxicology, Division in Earth and Life Sciences. Washington, DC: The National Academies Press.
- National Research Council . (2011). Guide for the Care and Use of Laboratory Animals (8th ed.). Committee for the Update of the Guide for the Care and Use of Laboratory Animals, Institute for Laboratory Animal Research, Division on Earth and Life Sciences. Washington, DC: The National Academies Press.
- Nowill, W. K. , Stephen, C. R. , & Searles, P. W. (1953). Evaluation of trichloroethylene as an anesthetic and analgesic agent. A.M.A Archives of Surgery, 66, 35–47. 10.1001/archsurg.1953.01260030046004 [DOI] [PubMed] [Google Scholar]
- O'Neil, M. J. , Heckelman, P. E. , & Roman, P. B. (2006). The Merck index (14th ed.). Whitehouse Station, NJ: Merck & Co. [Google Scholar]
- Penny, D. J. , & Vick, G. W., III . (2011). Ventricular septal defect. Lancet, 377, 1103–1112. 10.1016/S0140-6736(10)61339-6 [DOI] [PubMed] [Google Scholar]
- Prior, F. N. (1972). Blood levels of trichloroethylene during major surgery. Anaesthesia, 27, 379–389. [DOI] [PubMed] [Google Scholar]
- Reich, D. A. , & Cormany, C. I. (1979). Drycleaning and laundering: Drycleaning In Grayson M. (Ed.), Kirk‐Othmer encyclopedia of chemical technology (Vol. 8, 3rd ed., pp. 50–68). New York, NY: John Wiley and Sons. [Google Scholar]
- Rotroff, D. M. , Wetmore, B. A. , Dix, D. J. , Ferguson, S. S. , Clewell, H. J. , Houck, K. A. , … Sochaski, M. A. (2010). Incorporation human dosimetry and exposure into high‐throughput in vitro toxicity screening. Toxicological Sciences, 117, 348–358. 10.1093/toxsci/kfq220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadove, M. S. , Wyant, G. M. , & Gittelson, L. A. (1953). Trichloroethylene in general practice. Illinois Medical Journal, 103, 95–101. [PubMed] [Google Scholar]
- Snedecor, G. W. , & Cochran, W. G. (1980). One way classifications; analysis of variance In Statistical methods (7th ed., pp. 215–237). Ames, IA: The Iowa State University Press. [Google Scholar]
- Sokal, R. R. , & Rohlf, F. J. (1981). Biometry: The principles and practice of statistics, 2nd Ed. (pp. 222–229). San Francisco, CA: Freeman. [Google Scholar]
- Solomon, H. W. , Wier, P. J. , Fish, C. J. , Hart, T. K. , Johnson, C. M. , Posobiec, L. M. , … Kerns, W. D. (1997). Spontaneous and induced alterations in the cardiac membranous ventricular septum of fetal, weanling, and adult rats. Teratology, 55, 185–194. [DOI] [PubMed] [Google Scholar]
- Stott, W. T. , Quast, J. F. , & Watanabe, P. G. (1982). The pharmacokinetics and macromolecular interactions of trichloroethylene in mice and rats. Toxicology and Applied Pharmacology, 62, 137–151. 10.1016/0041-008X(82)90110-7 [DOI] [PubMed] [Google Scholar]
- Stuckhardt, J. L. , & Poppe, S. M. (1984). Fresh visceral examination of rat and rabbit fetuses used in teratogenicity testing. Teratogenesis Carcinogenesis and Mutagenesis, 4, 181–188. 10.1002/tcm.1770040203 [DOI] [PubMed] [Google Scholar]
- Thomas, R. S. , Philbert, M. A. , Auerbach, S. S. , Wetmore, B. A. , Devito, M. J. , Cote, I. , … Nong, A. (2013). Incorporating new technologies into toxicity testing and risk assessment: Moving from 21st century vision to data‐driven framework. Toxicological Sciences, 136, 4–18. 10.1093/toxsci/kft178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner, S. W. , Hunter, S. , & Wylie, J. P. (1999). The natural history of ventricular septal defects. Archives of Disease in Childhood, 81, 413–416. 10.1136/adc.81.5.413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- U.S. Department of Health and Human Services, Public Health Service, Agency for Toxic Substances and Disease Registry . (2014). Draft toxicological profile for trichloroethylene. Retrieved from https://www.atsdr.cdc.gov/toxprofiles/tp19.pdf [PubMed]
- U.S. Environmental Protection Agency . (1985). Health assessment document for trichloroethylene. Final report. (EPA/600/8–82/006F). Retrieved from U.S. Environmental Protection Agency website: https://nepis.epa.gov
- U.S. Environmental Protection Agency . (2001). Trichloroethylene health risk assessment: Synthesis and characterization. External Review Draft. (EPA/600/P‐01/002A). Retrieved from U.S. Environmental Protection Agency website: https://nepis.epa.gov
- U.S. Environmental Protection Agency . (2011). Toxicological review for trichloroethylene (CAS No. 79–01‐6) in support of summary information on the Integrated Risk Information System (IRIS). (EPA/635/R‐09/011F). Retrieved from U.S. Environmental Protection Agency website: https://cfpub.epa.gov/ncea/iris/iris_documents/documents/toxreviews/0199tr/Chapter7_0199tr.pdf
- U.S. Environmental Protection Agency . (2014). EPA Region 9 response action levels and recommendations to address near‐term inhalation exposures to TCE in air from subsurface vapor intrusion. Memorandum dated July 9, 2014. Retrieved from U.S. Environmental Protection Agency website: https://archive.epa.gov/region9/superfund/web/pdf/r9-tce-interim-action-levels-response-recs-memo-2014.pdf
- U.S. Food and Drug Administration . (1977). Trichloroethylene. Removal from food additive use. 42 Fed. Reg. 187, 49465–49471, September 27, 1977.
- von Grote, J. , Hürlimann, C. , Scheringer, M. , & Hungerbühler, K. (2003). Reduction of occupational exposure to perchloroethylene and trichloroethylene in metal degreasing over the last 30 years: Influences of technology innovation and legislation. Journal of Exposure Analysis and Environmental Epidemiology, 13, 325–340. 10.1038/sj.jea.7500288 [DOI] [PubMed] [Google Scholar]
- Warwick R., & Williams P. L. (Eds.). (1973). Gray's anatomy (35th British ed.). Philadelphia, PA: W. B. Saunders Company. [Google Scholar]
- Watson, R. E. , Jacobson, C. F. , Williams, A. L. , Howard, W. B. , & DeSesso, J. M. (2006). Trichloroethylene‐contaminated drinking water and congenital heart defects: A critical analysis of the literature. Reproductive Toxicology, 21, 117–147. 10.1016/j.reprotox.2005.07.013 [DOI] [PubMed] [Google Scholar]
- Wetmore, B. A. , Wambaugh, J. F. , Ferguson, S. S. , Sochaski, M. A. , Rotroff, D. M. , Freeman, K. , … Thomas, R. S. (2012). Integration of dosimetry, exposure, and high‐throughput screening data in chemical toxicity assessment. Toxicological Sciences, 125, 157‐174. Doi: 10.1093/toxsci/kfr254, 125, 157, 174. [DOI] [PubMed] [Google Scholar]
- Wikoff, D. D. , Urban, J. D. , Harvey, S. , & Haws, L. C. (2018). Role of risk bias in systematic review for chemical risk assessment: A case study in understanding the relationship between congenital heart defects and exposures to trichloroethylene. International Journal of Toxicology, 37, 125–143. 10.1177/1091581818754330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, C. , & Schaum, J. (2000). Exposure assessment of trichloroethylene. Environmental Health Perspectives, 108(Suppl. 2), 359–363. 10.1289/ehp.00108s2359 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supplemental Information
Table S1: Concentration of TCE in Dosing Formulations (End of Preparation Stage)
Table S2: Concentration of TCE in Dosing Formulations (Water Bottles at Start of Exposure Period)
Table S3: Loss of TCE from Water Bottles During the 24‐hour Exposure Period
Table S4: Food Consumption during Various Periods of Gestation (g/animal/day_
Data Availability Statement
The data that support the findings of this study are available for download at the EPA docket for TSCA evaluation at https://www.regulations.gov/document?D=EPA-HQ-OPPT-2016-0737-0120. The report therein is Coder, P. S. (2019) Final Report. An Oral (Drinking Water) Study of the Effects of Trichloroethylene (TCE) on Fetal Heart Development in Sprague Dawley Rats. Charles River Laboratories, Laboratory Project ID 00459506.