

Abstract
The circulation of the lung is unique both in volume and function. For example, it is the only organ with two circulations: the pulmonary circulation, the main function of which is gas exchange, and the bronchial circulation, a systemic vascular supply that provides oxygenated blood to the walls of the conducting airways, pulmonary arteries and veins. The pulmonary circulation accommodates the entire cardiac output, maintaining high blood flow at low intravascular arterial pressure. As compared with the systemic circulation, pulmonary arteries have thinner walls with much less vascular smooth muscle and a relative lack of basal tone. Factors controlling pulmonary blood flow include vascular structure, gravity, mechanical effects of breathing, and the influence of neural and humoral factors. Pulmonary vascular tone is also altered by hypoxia, which causes pulmonary vasoconstriction. If the hypoxic stimulus persists for a prolonged period, contraction is accompanied by remodeling of the vasculature, resulting in pulmonary hypertension. In addition, genetic and environmental factors can also confer susceptibility to development of pulmonary hypertension. Under normal conditions, the endothelium forms a tight barrier, actively regulating interstitial fluid homeostasis. Infection and inflammation compromise normal barrier homeostasis, resulting in increased permeability and edema formation. This article focuses on reviewing the basics of the lung circulation (pulmonary and bronchial), normal development and transition at birth and vasoregulation. Mechanisms contributing to pathological conditions in the pulmonary circulation, in particular when barrier function is disrupted and during development of pulmonary hypertension, will also be discussed.
Introduction
The pulmonary vasculature is unique, both in volume and function. During fetal life, the pulmonary circulation is a low flow, high-resistance circuit. With transition to postnatal life, the pulmonary vasculature dilates, accommodating the entire cardiac output (CO), with high blood flow maintained at low intravascular pulmonary arterial pressure (PPA). As compared with the systemic circulation, pulmonary arteries have thinner walls with much less vascular smooth muscle and a relative lack of basal tone, likely a function of high production of endogenous vasodilators and low production of vasoconstrictors, resulting in a normal pulmonary vascular resistance (PVR) that is approximately one-tenth that of the systemic circulation. In the adult lung, factors controlling pulmonary blood flow include vascular structure, gravity, mechanical effects of breathing, and the influence of neural and humoral factors.
While autoregulation is a well-recognized feature of most systemic vascular beds, this phenomenon is absent in the adult pulmonary circulation. The pulmonary circulation also differs functionally from the systemic in that the pulmonary arteries carry mixed venous blood. From the pulmonary arteries, deoxygenated blood is channeled through the alveolar/capillary units where a large component of gas exchange occurs, and returned to the left heart by the pulmonary veins for distribution to the systemic circulation. The pulmonary vascular pressor response to hypoxia is also unique, as systemic arteries dilate as oxygen concentrations decrease. The acute pressor response occurs immediately and is rapidly reversible with return to normoxic conditions. If the hypoxic stimulus is maintained over a prolonged period, as can occur with various chronic lung diseases, contraction is accompanied by remodeling of the vasculature, resulting in elevated PVR and pulmonary hypertension, a potentially devastating disease. In addition to hypoxia, other genetic and environmental factors can also confer susceptibility to development of pulmonary hypertension even in the absence of a hypoxic stimulus.
In addition to gas exchange, the pulmonary vasculature also serves to filter the blood, removing microemboli and participates in the metabolic regulation of a variety of vasoactive hormones. The endothelium forms a tight barrier, actively regulating paracellular extravasation of proteins, solutes and fluids to control interstitial fluid homeostasis. Infection and inflammation compromise normal barrier homeostasis, resulting in increased permeability, fluid, protein and cellular extravasation, edema formation, and ultimately acute respiratory distress syndrome (ARDS).
The lung also has a systemic vascular supply, the bronchial circulation, which provides oxygenated blood from the systemic circulation to the walls of the conducting airways, pulmonary arteries and veins. The historical concepts (233) and anatomy (635) of the pulmonary circulation, comparative physiology (686), effects of exercise (445) and high altitude (233), the mechanics (172) and distribution of blood flow (217, 220), and mechanisms matching ventilation and perfusion (161,172,220) have been covered extensively in previous reviews. In this article, we will focus on reviewing the basics of the lung circulation (pulmonary and bronchial), normal development and transition at birth, and vasoregulation. We will also discuss the mechanisms contributing to pathological conditions in the pulmonary circulation, in particular when barrier function is disrupted and during development of pulmonary hypertension.
Anatomy—A Brief Overview
Any discussion of the pulmonary circulation requires at least a rudimentary understanding of the anatomy of the vasculature. A thorough review detailing the structure of the pulmonary vessels, including wall composition, has been published recently (635); herein we provide a brief overview to orient the reader.
In general, normal embryogenesis gives rise to a pulmonary circulation with an arterial network that closely parallels the airways, a capillary network at the level of the alveoli, and an irregular venous system that drains to the left heart. In the postnatal lung, blood exits the right ventricle into the main pulmonary artery, the diameter of which is similar to that of the aorta (<3 cm) (440). During gestation and immediately following birth, the wall thickness of the pulmonary artery is nearly identical to that of the aorta, but, postnatally, the elastic tissue gradually diminishes resulting in a pulmonary artery wall that is much thinner than the aorta [reviewed in (202)].
The main pulmonary artery divides into the right and left main branches, with the caliber of each branch approximately half that of the main pulmonary artery (440). The right and left branches further divide to supply each lobe before entering the lung. Within the lung, each lobar artery subdivides into rather irregular branches corresponding to the bronchial tree. The close proximity of the pulmonary arteries and airways under-scores the relationship between ventilation and perfusion that defines the normal function of the lung. The large pulmonary arteries (>2000 μm o.d.) are classified as elastic, with the media comprised primarily of elastic fibers and some smooth muscle (523,635). As vascular diameter decreases, these elastic arteries gradually give rise to vessels with increased smooth muscle content (523). In general, arteries between 2000 and 150 μm o.d. can be considered muscular arteries, but are still more thin-walled than systemic arteries of the same diameter (155, 523, 635). The pulmonary arterial wall also comprises collagen and advential fibroblasts, a longitudinal elastic lamina, which allows for expansion during inspiration and a thin intima composed of endothelial cells (ECs).
The small arterioles have a nonuniform smooth muscle cell (SMC) layer, giving way to the small nonmuscular preacinar arterioles, which are located proximal to terminal bronchi (264, 418, 523, 524). At the alveoli, the terminal arterioles break into a network of pulmonary capillaries within the alveolar walls. Although it had long been surmised that blood passed from the arterial to venous circulation via small vessels not visible to the eye or with traditional light microscopy [reviewed in (687)], the use of electron microscopy in the early 1950s allowed the first visualization of the pulmonary capillaries (375). The capillaries have a very thin wall consisting of a single layer of ECs (210,687) and, at an estimated 126 m2 in the adult human (210), contain most of the surface area of the pulmonary vasculature. It should be noted that pulmonary capillaries are not uniform in thickness (267, 635, 678). For example, the gas-exchange portion of the intra-acinar alveolar capillaries wall can thin to essentially just a bit of cytoplasm between the two sections of plasma membrane, resulting in a thickness of 20 to 30 nm (635,678) with the cell contents and nucleus shifting to the “thick” side of the septum.
Gas exchange between the alveolar gas and blood takes place largely within the pulmonary capillary bed after which the blood flows into venules, which are almost indistinguishable in gross structure from arterioles (635). It should be noted, however, that oxygen uptake has been proposed to also occur across the walls of larger arteries (118, 297, 586), a notion recently confirmed by elegant in vivo imaging techniques (611). While each small arteriole supplies a specific unit of respiratory tissue, the venules drain several portions of the lung. Venules do not follow the bronchiole tree and unite to form the pulmonary veins, which conduct the oxygenated blood into the left ventricle.
Anatomically, vessels can be categorized as extrapulmonary or intrapulmonary. Extrapulmonary vessels are outside the lung, in the mediastinum, and the pressure surrounding these vessels is related to pleural pressures. The intrapulmonary vessels, including the capillaries, can be further subdivided anatomically and functionally into extra-alveolar or alveolar. Alveolar vessels are comprised of capillaries that are adjacent to alveoli, while extra-alveolar vessels are larger and more proximal. These two types of vessels also have different perivascular pressures due to the differences in the tissues that surround them, and are exposed to alveolar pressure (PALV) or pressure exerted by lung tissue connections. Both of these forces increase with lung inflation; however, they act in opposite directions, with PALV directed inward and tissue pressure directed outward. Thus, the alveolar vessels are surrounded by PALV, due to localization within the septal wall, whereas the extra-alveolar vessels are surrounded by septa and are typically contained in a sheath of connective tissue. These vessels include a subset of the pulmonary capillaries, called the corner vessels, which are located in the alveolar parenchyma at the alveolar corners. Detailed morphologic measurements of the alveolar vessel density, radius, path length from artery to vein, and other measurements have been made and are discussed in detail elsewhere (635).
Fetal and Neonatal Pulmonary Circulation
Vascular morphogenesis
In humans, during fetal development the pulmonary circulation begins to form during the embryonic stage (0–7 weeks) from the truncus arteriosus (332), which originates from the ventricles and subsequently divides into the ascending aorta and pulmonary trunk. The arteries and veins continue to develop during the pseudoglandular stage (7–17 weeks), following the branching patterns of the airways (248, 261). By the 17th week of gestation, development of the pulmonary circulation down to the preacinar pulmonary arteries is complete (202,261,262) (Fig. 1).
Figure 1.

Timeline of vascular development. Adapted, with permission, from Hislop and Pierce (265).
During organogenesis, vascularization can occur via vasculogenesis or angiogenesis, or a combination of the two. Immunohistochemical analysis of fetal lung specimens to characterize the temporal and spatial expression of endothelial markers suggested that intrapulmonary arteries in humans arise primarily via vasculogenesis, or the development of new blood vessels from endothelial progenitor cells (248). In these studies, endothelial tubes containing blood cells could be identified in the mesenchyme surrounding the airways and the foregut (248). In mice, however, a combination of vascular casting and electron microscopy was used to determine that angiogenesis, or sprouting of new vessels from existing ones, was a more prominent process (137). Examination of lung sections from mice expressing the bacterial lacZ gene under control of an endothelial promoter also revealed a major role for angiogenesis (475), although in this case, instead of deriving from more proximal vessels, sprouts were proposed to arise from the capillary network and remodel (i.e., gain smooth muscle) to form the more proximal vessels. Possible explanations for the apparent contradictions in reported results between studies include differences in the techniques (i.e., casting versus immunohistochemical visualization of structures) and terminology employed. Nonetheless, the exact contribution of vasculogenesis and angiogenesis to proximal vessel development remains unresolved and a topic of ongoing investigation.
As the pulmonary arteries and veins develop, the primitive vessels acquire a smooth muscle layer and extracellular matrix proteins are deposited to form a basement membrane that both separates different layers within the vascular wall and maintains the wall structure. In humans, the SMCs surrounding the arteries appear to originate from three sites (248). First, between 38 to 98 days gestation, pulmonary arterial SMCs (PASMCs) derive from the bronchial smooth muscle in adjacent airways. During this time period, PASMCs also appear to derive from the mesenchyme surrounding the arteries. Later in gestation (98–140 days gestation), PASMCs coexpressed smooth muscle specific α-actin and endothelial markers, suggesting endothelial to SMC transdifferentiation. In contrast, venous SMCs derive from the mesenchyme (247).
The pulmonary capillaries appear to arise separately from the arteries and veins (137), becoming connected to the more proximal vasculature during the pseudoglandular stage (136, 248). As the fetus enters the canalicular stage, the capillaries increase in number, a process thought to involve a combination of angiogenesis, vasculogenesis and intussesception, or formation of pillars within existing capillaries to increase the surface area (84,136,137,475).
At the time of birth, the branching pattern of the pulmonary circulation is similar to that observed in the adult (Fig. 2) (265), although the size of the vessels increases with lung growth. Since the majority of alveolarization of the human lung continues after birth, the postnatal capillary bed continues to expand via angiogenesis and intussesception, with the surface area for gas exchange increasing approximately 20-fold from birth to adulthood (83).
Figure 2.
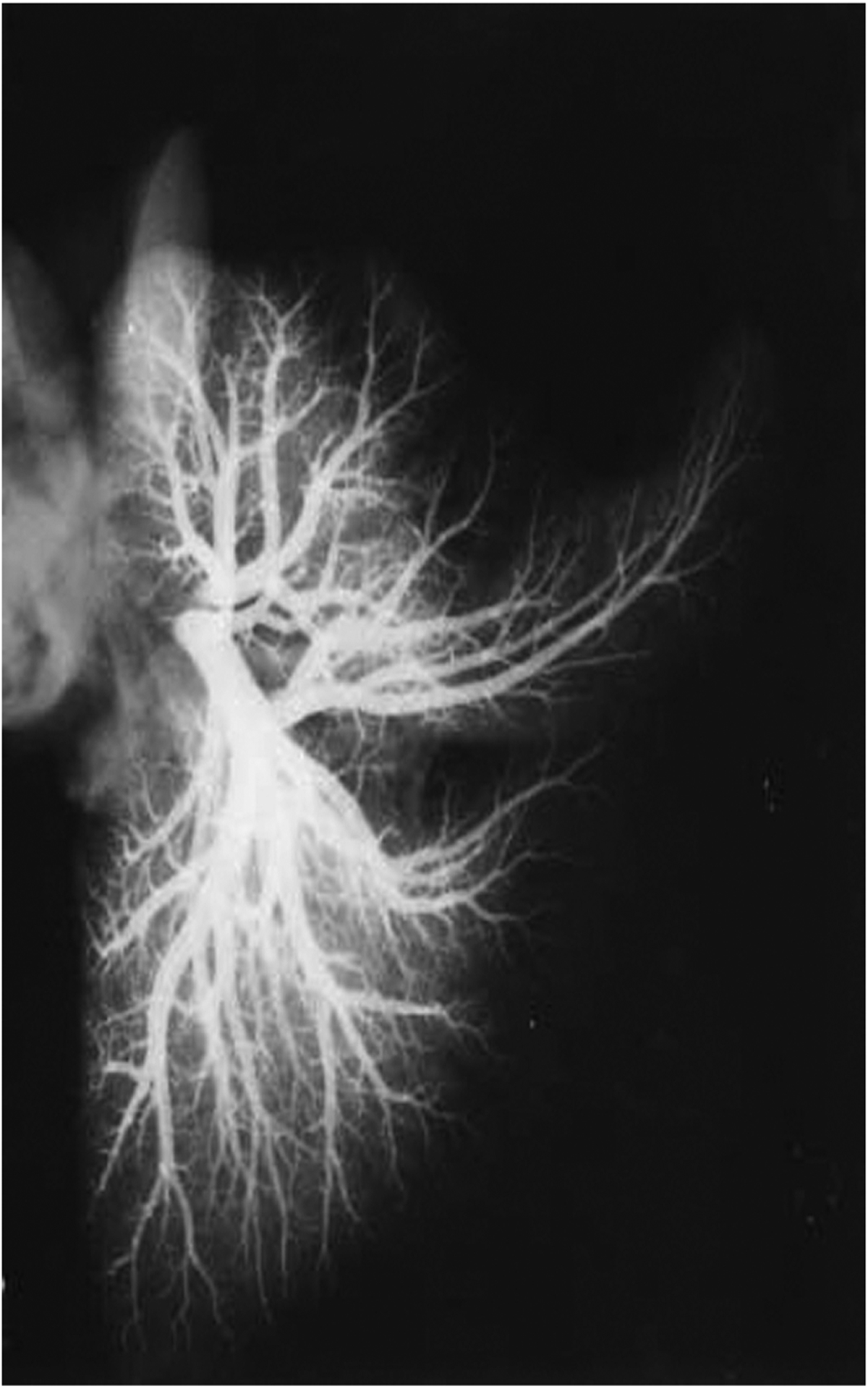
Arteriogram of a human lung obtained postmortem from a fetus (39 weeks gestation) showing distal vascularization. The arteries were injected with barium sulphate (mag × 0.75). Reprinted, with permission, from (265).
Factors regulating vascular development
Growth factors
While histological studies provided early details regarding vascular morphogenesis, more recent studies utilizing transgenic animals have yielded important clues as to the molecular aspects of vascular development. It is now recognized that a number of factors contribute to the development of the pulmonary vasculature, including transcription factors, growth factors and the extracellular matrix. One of the most widely studied angiogenic factors, vascular endothelial growth factor (VEGF), plays a central role in vascular development through receptor/ligand interactions with both VEGFR1 (Flt-1) and VEGFR2 (KDR/Flk-1) receptors. Mice with global heterozygosity for the Vegf allele exhibit defective vascularization and embryonic lethality (89). Similarly, mice with global knock-out of VEGFR2 are embryonic lethal with poor vascular formation (562) while VEGFR1-deficient mice die in utero due to lack of structural organization of vessel walls (178). Thus, both VEGF receptor subtypes are required for vascular development, with VEGFR2 and VEGFR1 controlling angiogenesis and vascular organization, respectively.
Early in development, expression of both VEGF and VEGFR2 are observed in the rat lung, with VEGF localized to epithelium (52,209), and VEGFR2 found primarily in the mesenchyme (52, 209). Moreover, transgenic mice with conditional inactivation of the Vegf gene in lung epithelium exhibited almost complete absence of pulmonary capillaries (707). These genetic studies coupled with observations on the pattern of VEGF/VEGF receptor localization suggested an important interaction between tissues in regulating vascular development.
In addition to VEGF, the glycoprotein growth factors, angiopoietins (Ang 1, 2, 3, and 4), also play a major role in vascular development. These proteins signal through their major receptor, Tie-2, to modulate survival and migration of ECs, vascular remodeling and vascular integrity (127, 373). Indeed, loss of either Ang 1 (604) or Tie-2 receptors (545) is embryonic lethal due to substantial microvascular defects leading to hemorrhage and edema. The immature and improperly organized vessels in these mice suggest that the normal role of Ang 1/Tie-2 is to facilitate branching, sprouting and remodeling. In mice, Ang 1 is produced by lung mesenchyme and smooth muscle, whereas Tie-2 expression is restricted to endothelium (545, 553). The expression of Ang 1 and Tie-2 in mouse lung is measureable early on (E9.5) and increases throughout gestation (116, 236), although the spatial expression was not determined in these studies.
The Wnt family of proteins bind to receptors of the Frizzled family on the cell surface, primarily signaling through a canonical pathway involving β-catenin (352). Wnt proteins are clearly involved in vascular development (115). With respect to the lung, the isoform Wnt7b has been shown to influence maturation of vessels. Mice with null alleles for the Wnt7b gene died perinatally from respiratory failure, with lungs that were poorly inflated and hemorrhagic (576). While the endothelium appeared to develop normally, the large pulmonary vessels were dilated, exhibited defective SMC layers and herniation of the endothelial lining. These observations suggest that Wnt7b may regulate the development and/or recruitment of the mural support cells during maturation of the lung vessels.
The transforming growth factor-β (TGF-β) superfamily consists of over 40 secreted cytokines that signal through serine/threonine kinase receptors. TGF-β family members, including TGF-β1 and bone morphogenetic proteins (BMPs), as well as their receptors, are expressed in the developing lungs (204, 289, 422, 491, 613, 734), but their roles in lung vascular development have not been completely defined. TGF-β signaling is important in the development of many tissues, including the vasculature (142), with complete loss of TGF-β1 resulting in 50% prenatal lethality associated with inadequate capillary tube formation and impaired endothelial differentiation. Loss of TGF-β type-I receptors also results in embryonic lethality due to vascular defects (344). Mice deficient in Smad1 or Smad5, the main signaling intermediaries downstream of the BMP receptors, die midgestation due to vascular abnormalities including enlarged blood vessels with insufficient surrounding vascular SMCs (712).
Other potential factors that are known to be involved in vascular morphogenesis include notch, platelet-derived growth factor, angiostatic proteins, ephrins, insulin-like growth factors, and endothelial monocyte activating polypeptides [reviewed in (202, 485, 592)]. While there is ample evidence to suggest that these factors could contribute to lung vascular development, confirmation of their exact roles has yet to be reported.
Transcription factors
The regulation of many of the aforementioned pathways is coordinated by transcription factors. For example, Forkhead box (Fox) proteins are a family of transcription factors that play diverse roles in development due to their control of regulation of cell cycle progression, cell survival, expression of differentiated genes, and cell metabolism (88). Mice with complete loss of the Foxf1 transcription factor died midgestation (396), whereas heterozygous mice developed further but showed a variety of foregut developmental defects, including malformed lungs, impaired lung vascular development and perinatal lethality (310, 395). In the lungs of these mice, distal lung capillary density was reduced, and tight junctions between endothelial and distal lung epithelial cells were disrupted, leading to hemorrhage (310). Foxf1 regulates a variety of genes and is expressed in the mesenchyme during lung branching morphogenesis [reviewed in (392)]. Although the exact Foxf1 targets that modulate lung vascular formation have not been identified, lungs from Foxf1−/− mice exhibited reduced VEGF expression (310), suggesting a potential mechanism by which Foxf1 may regulate pulmonary vascular development.
Hundreds of genes are controlled by activation of hypoxia-inducible factors (HIFs), oxygen-sensitive transcription factors. HIFs are highly conserved transcription factors that are tightly regulated by oxygen availability. The first family member identified (560), HIF-1, is found in all nucleated cells and exists as a heterodimer, consisting of HIF-1α and HIF-1β subunits. HIF-1β is ubiquitously expressed, whereas HIF-1α is found at very low levels under normoxic conditions. When oxygen levels are normal, HIF-1α protein is ubiquitinated and subjected to proteasomal degradation. This process is interrupted during hypoxia [reviewed in (505)], allowing for rapid accumulation of HIF-1α protein and activation of the transcriptional complex. Thus, HIF-1α confers sensitivity and specificity for hypoxic induction of HIF-1 transcriptional activity. Subsequently, HIF-2α was identified (156). Like HIF-1α, HIF-2α is regulated in a similar manner by oxygen and dimerizes with HIF-1β, but HIF-2α exhibits a much more restricted pattern of expression. During fetal development, the lung of the normal embryo is hypoxic; thus, it is not surprising that HIFs would be important contributors to lung vascular development. HIF mRNA and protein levels are quite high in the fetal lung (23, 232, 234), with the entire HIF system in place as early as 8 weeks of gestation in the human (232). Analysis of the spatial pattern of expression in human lungs during initial development of the pulmonary vasculature revealed high levels of HIF-1α protein localized predominantly in branching epithelium (232). HIF-2α protein was also present in epithelium, as well as in mesenchymal structures (232). The different patterns of expression suggest the paralogs may serve distinct and specific functions in epithelial and vascular morphogenesis, a possibility supported by studies in HIF-1α and HIF-2α loss-of-function models. In mice, global deletion of HIF-1α or HIF-1β results in severe cardiovascular malformations and embryonic lethality at day 10 (117, 295, 331, 399). Loss of HIF-2α, on the other hand, leads to death in approximately 50% of embryos, with the viable offspring exhibiting impaired lung development, reduced production of surfactant, postnatal respiratory distress, and neonatal lethality (80, 117). Interestingly, animals heterozygous for either HIF-1α or HIF-2α develop normally and survive to adulthood, with outwardly normal lung vasculature and function under normoxic conditions. In late development, fetal lung levels of HIF-1α and HIF-2α protein are normally high, with a rapid decrease in HIF-1α protein levels upon delivery (23,234), and in utero depletion of HIF-1α using antisense oligonucleotides reduced lung branching morphogenesis and vascularization (645). Similarly, a decline in HIF-1α and HIF-2α protein levels in the lungs of mechanically ventilated preterm animals is associated with vascular and alveolar hypoplasia, neonatal respiratory distress, and bronchopulmonary dysplasia (23,234). Taken together, these studies point to a central role for HIFs in pulmonary vascular morphogenesis.
Blood flow in the fetal lung
During gestation, the placenta serves as the primary gas-exchange organ for the fetus; thus, the fetal lung receives modest blood flow. Rather than traversing the pulmonary circulation, oxygenated blood crosses from the right atrium to the left atrium through the foramen ovale. The majority of blood pumped out by the right ventricle returns to the aorta via the ductus arteriosus, a wide muscular vessel connecting the pulmonary arterial trunk to the descending aorta (325, 519). In the human, blood flow to the lung increases from 10% to 15% of the combined CO at 20 week gestation (421, 519) to 25% at 30 weeks (519). Between 30 and 38 week gestation, fetal lung blood flow decreases to 21% of CO (519). Slightly lower values have been reported in fetal lamb (538), with the lung receiving <10% of the CO. As the fetal lamb matures from midgestation to near term, PPA and aortic blood pressure increase in parallel from 30 to 50 mm Hg, while pulmonary blood flow increases approximately 40-fold and PVR decreases from 6 mm Hg/mL*min to 0.3 mm Hg/mL*min (539). Similar increases in pressure were measured in the right and left ventricles of the human fetus (305). In both the lamb and human, it is likely that the early increase in blood flow at midgestation is due to expansion of the pulmonary vascular bed since reactivity of the pulmonary circulation during this period is largely unaffected by increasing maternal oxygenation (518). Nearer term, both the pulmonary arteries and veins actively respond to vasoactive factors (86,93,128,351,735), suggesting the development of vasoreactivity during this period and that at this stage, high PVR is maintained by a combination of the thick SMC layer surrounding the vessels and significant release of vasoconstrictor substances. Interestingly, evidence in lambs also suggests that the fetal pulmonary circulation possesses a myogenic response that contributes to high PVR by opposing vasodilatory stimuli (5,47,603).
Postnatal vasodilation
In utero, the oxygen tension in pulmonary arterial blood is approximately 18 mmHg and oxygen saturation 50% (202). In this low oxygen fetal environment, the high levels of endogenous vasoconstrictors, high myogenic tone and low basal release of vasodilators such as nitric oxide (NO) and prostacyclin (PGI2) contribute to the high PVR. Toward the end of gestation, increasing oxygen tension from 24 to 46 mmHg increases pulmonary blood flow by 10-fold in lambs (432) and maternal hyperoxygenation reduces PVR in the human fetus (518, 519) and lamb (329), indicating that oxygen concentration is a major factor regulating vasomotor tone in the mature fetus. Similar results have been reported in primates (46).
After birth, the lung becomes the organ of gas exchange. As breathing commences, pulmonary blood flow increases, reaching total CO in the human infant (519). During the transition from fetal to neonatal life PPA gradually decreases, and within hours approaches 50% of systemic pressure (158,540). As the systemic vascular resistance and pressure become greater than PPA, the foramen ovale closes. The ductus arteriosus begins to close within the first few hours after birth (436), closing completely by 2 days after birth (280). In the normal infant, PPA reaches adult levels within the first 2 weeks of life (158,540). During maturation, the myogenic response is also lost.
The rapid postnatal reduction in PPA can be attributed to a reduction in PVR, achieved primarily via marked vasodilation. Initiated by ventilation, oxygenation, and shear stress due to increasing blood flow, a variety of vasorelaxant mediators are produced, including NO and PGI2, in conjunction with falling levels of vasoconstrictor agents such as endothelin-1 (ET-1) (202, 257, 701). The initial, immediate changes in PVR that occur with the onset of breathing are followed by postnatal vascular reorganization and remodeling that occur rapidly after birth and continue over the first few months of life. The factors controlling the changes in vasomotor tone that accompany the normal transition of the pulmonary circulation at birth, including mechanical factors and vasoactive agents, have recently been reviewed in detail (202).
The Adult Pulmonary Circulation
To effectively distribute CO to the vascular beds of various organs that possess unique requirements with respect to blood flow and, in humans, are separated by vertical distances up to 180 cm, the systemic circulation requires a high-pressure gradient. Thus, the vascular resistance of the systemic vessels is precisely controlled to maintain adequate pressure. While baroreceptors monitor pressure and utilize neural pathways to regulate intravascular pressure during changes in posture, physical activity, temperature, and blood volume, it is well recognized that the systemic vasculature also exhibits substantial autoregulation. This is in stark contrast to the adult pulmonary circulation, which is required to accommodate the total CO and, thus, cannot actively adjust its total blood flow. Instead, the adult pulmonary circulation is a low-resistance, highly compliant vascular bed whose properties are greatly influenced by external forces such as PALV, CO, and posture.
As noted earlier in this review, the pulmonary circulation can be broadly divided into extra-alveolar, alveolar, arterial and venular segments. In addition to serving as a conduit for blood and, in the case of the microvasculature, gas exchange, each segment of the pulmonary vasculature is unique in its responses to hemodynamic stress, and is subject to a specific set of luminal and perivascular pressures due to the anatomic differences between the segments. These differences in turn produce differential responses to mechanical events such as lung inflation. Given the existing regional inhomogeneities in structure, mechanical forces, ventilation, and gas exchange, the design of the cardiorespiratory apparatus provides for local differences in blood flow and compensatory mechanisms for rearrangement that match blood flow to alveolar ventilation insofar as is possible.
General aspects of pulmonary hemodynamics
As might be expected, the pattern of blood flow in the pulmonary circulation varies with vessel caliber. In earlier studies, flow in the pulmonary artery was noted to be borderline turbulent with flow velocities approximating 18 cm/s (174). Newer methods of blood flow imaging, using magnetic resonance imaging, demonstrated similar average flow velocities (22 cm/s) at rest in supine healthy volunteers, but complex blood flow patterns in these large vessels (26). Average and peak flow velocities increased with exercise, although the magnitude of these changes may be position dependent. Exercise is also associated with increased flow and capillary recruitment, but the effect of exercise on hemodynamic parameters of the pulmonary circulation also depends on exercise intensity and degree of hypoxia [as reviewed in (445)].
Effects of posture on hemodynamics
Starting in the 1960s, heterogeneity of lung perfusion has been investigated in a variety of species and using multiple methods. It is now widely recognized that pulmonary flow distribution is affected by numerous factors including local/regional changes in flow and other nongravitational factors, such as genetic determination of pulmonary vascular branching and changes in alveolar capillary perfusion that are independent of alveolar and arterial pressures (the mechanisms of which are yet unclear), as discussed in detail by Glenny et al. (220). Briefly, in the upright lung, the net result of forces that increase flow (gravity) and decrease flow (regional areas of shunting, areas of high PALV) is an overall increase in blood flow in the bases compared to the apex, a finding that has been shown using radiotracers as well as multiple newer methods, including single photon emission tomography and positron emission tomography (278). Regardless of the vertical effects of gravity in the prone versus supine position, the presence of isogravimetric gradients suggest that factors unrelated to gravity likely also play a role in perfusion heterogeneity (15, 217, 244, 306). While earlier studies focused on changes in perfusion between standing and supine subjects (688), there has been renewed interest in the effects of the different types of horizontal posture (supine vs. prone) on ventilation and perfusion due to the clinical benefit of prone positioning reported in patients with ARDS (275); however, results of the published studies have been somewhat contradictory. Multiple animal studies have shown changes in distribution of flow between supine and prone position in mechanically ventilated animals, even in the absence of additional parenchymal injury (12, 438). In contrast, use of PET imaging in healthy, nonsmoking adults on no ventilatory support revealed flow gradients favoring dependent regions in both supine and prone positions, but did not detect appreciable changes in flow between positions (444). Similar findings were noted in healthy nonsmokers not on mechanical ventilation, where regions of interest examined on MRI images also showed similar gradients in both positions (602). Interestingly, SPECT imaging (with microaggregate albumin as tracer) on healthy adults during application of continuous positive airway pressure (CPAP) showed more uniform perfusion in the prone position but only when CPAP was not used, suggesting that the presence of positive ventilation further complicates existing postural perfusion gradients (439). The finding of more uniform perfusion in the prone position in ventilated and nonventilated healthy humans was also corroborated by other studies (256, 463). In addition to the aforementioned reports, several other publications have also shown opposing results. These studies and potential reasons underlying the conflicting data has been discussed by Hughes et al. (277).
The studies described above were performed in healthy adults; however, the possibility exists that the effects of prone position on lung perfusion may differ in the diseased lung. Positional effects are of most interest in ARDS, where positive pressure and changes in CO may exert more significant changes in perfusion compared to healthy lungs (245,254). For example, in studies where CT scans were performed in patients with ARDS, prone position had salutary effects on alveolar recruitment and improved stress and strain, which may lead to more homogeneous perfusion (205, 490). Positional studies in animal models of lung injury also showed benefits to prone positioning (144, 341, 530). In dogs undergoing oleic acid injury, more uniform distribution of perfusion was noted in the prone position before injury and redistribution of perfusion away from dependent regions in prone but not supine position following injury (693). These results are consistent with studies in multiple animal models of injury where, species differences between animals studied and between quadrupeds and humans notwithstanding, beneficial alterations in perfusion in the prone position have been reported (661, 686). Taken as a whole, it would appear that whether or not vertical gradients of flow exist and are changed in the prone position independent of redistribution of lung parenchyma and changes in oxygenation in healthy humans, in humans with diseased lungs, aeration, and compliance improve with prone positioning (238,490,530). Thus, clinical improvement observed with prone position in pathologic states such as ARDS may reflect increased lung perfusion distribution due to changes in tissue distribution (495) and/or mechanics.
Effects of respiration on blood flow
Owing to low intravascular pressure, surrounding pressures exert a substantial effect on pulmonary vessel caliber. As noted earlier, alveolar vessels are highly influenced by PALV since changes in PALV produce similar (but not equivalent) changes in alveolar vessel transmural pressures (the difference between the pressure inside and outside the vessel). Thus, these vessels undergo compression at higher PALV. Blood flow control in alveolar vessels can be independent of changes in upstream pressures, and may be regulated by local factors (672). In addition to the effects of PALV, the connection of these vessels to lung parenchyma subjects them to substantial tissue forces.
Extra-alveolar vessels are, by definition, not affected by changes in PALV; however, changes in lung inflation also influence transmural pressures in these vessels. For instance, with inflation, extra-alveolar vessels dilate. This interdependence between lung volume and extra-alveolar vessel diameter is related to the effects of inflation on the perivascular interstitium surrounding these vessels (48), which is composed of loose parenchymal tissue, collagenous fibers and lymph vessels. Inflation deforms the surrounding tissue to produce changes in transmural pressures that distend extra-alveolar vessels independent of luminal vascular pressures (340). This effect is minimal at functional residual capacity, but increases with increasing lung volumes. Though individual responses of alveolar and extra-alveolar vessels to changes in lung volume are different, the net effect of lung inflation on PVR is predictable (Fig. 3). Resistance is lowest at normal breathing and increases with both increased and decreased lung volume due to the differential effects of lung volume on the diameters of alveolar and extra-alveolar vessels.
Figure 3.
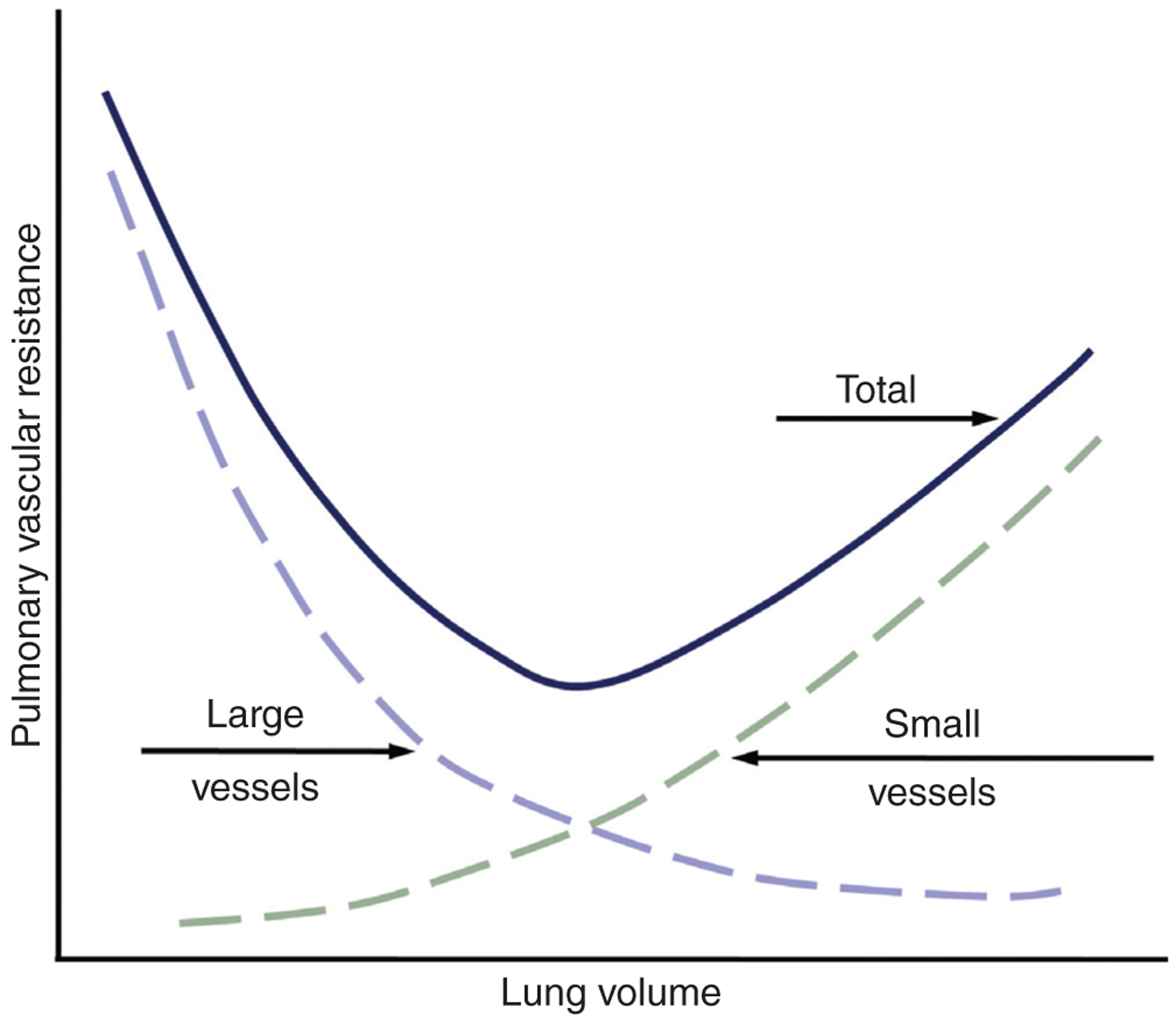
Graph illustrating the relationship between PVR and volume. Adapted, with permission, from (581).
Since perfusion of alveolar vessels is dependent on PPA, pulmonary venous pressure (PV) and PALV, the lung is described as being divided to three major functional zones based on these pressures (Fig. 4). In Zone 1, PALV exceeds PPA and, as a consequence, alveolar vessels are collapsed. In this situation, the V/Q ratio approaches infinity. The behavior of the lung vasculature under varying venous and alveolar pressures has been described using the principles of starling resistors (described below). However, alternate models have also been proposed to explain zonal blood flow (172); for instance, Fung et al. (191) have modeled the lung vasculature using a sheet-flow framework, where blood flow is envisioned to occur in between two alveolar sheets that are subject to elastic deformation due to changes in alveolar pressures.
Figure 4.

Original three-zone model proposed by West et al., (688) to illustrate the regional heterogeneity in blood flow. Within each of the three zones, the behavior of the blood vessels is different, based on the relative magnitudes of pulmonary arterial, alveolar and venous pressures (Pa, PA, and Pv, respectively). In zone 1, PA is greater than Pa, occluding collapsible vessels and preventing flow. In zone 2, Pa is greater than PA, which exceeds Pv, such that blood flow is dictated by the Pa-PA pressure gradient. In zone 3, both Pa and Pv exceed PA, and vessels are held open, allowing blood flow based on the Pa-Pv pressure gradient. Reproduced, with permission, from West et al. (688).
In the upright lung, gravitational forces cause increases in PPA such that when PPA exceeds PALV, flow is determined by the pressure gradient between PPA and PALV. This condition is described as Zone 2, and is characterized by PALV exceeding PV, but remaining below PPA. Thus, in Zone 2, blood flow is determined by the difference between PPA and PALV, rather than the arterial-venous pressure difference (as seen in Zone 3). This hemodynamic situation, in which flow is independent of downstream pressure, has been likened to a vascular waterfall or Starling resistor. Since flow is independent of the difference between arterial and venous pressure in this zone, lower downstream pressures (i.e., increasing the height of the waterfall) have no effect on flow. This phenomenon was illustrated by Permutt et al. (494), who showed that, when flow is held constant, PPA falls linearly with PALV at higher alveolar pressures, but at lower PALV, PPA becomes invariant to changes in PALV. This behavior would not be expected if PPA and PALV were the only factors that influenced flow. Cessation of flow at an arterial-alveolar gradient >0 suggests an additional source of resistance (371, 494). One possibility is that when PALV drops below the critical closing pressure of a segment of the pulmonary vasculature, critical closure, or vessel collapse, occurs. Thus, critical closure is defined as a state of zero flow despite a positive driving pressure (PPA − PALV).
Due to the nonrigid nature of blood vessels, critical closure occurs in both the systemic and pulmonary circulation when-ever the outflow pressure in a vessel is less than the pressure at which the vessel collapses (critical pressure) (172). In addition to Zone 2 hemodynamics, this concept also explains why venous return (VR) in the vena cava fails to continue increasing when right atrial pressure falls below a certain thresh-old (381). In the sheet-flow model, Fung et al. (192) have described this phenomenon as a “sluicing gate.” The exact critical closing pressure of a vessel depends on both factors intrinsic to the vessel itself as well as perivascular pressures. Since the latter changes in the lung based on factors such as lung volume and local edema, the exact critical closing pressure for any particular segment of the vascular tree is hard to determine. Based on isolated, perfused lung experiments, both the extra-alveolar (79, 371, 494) and alveolar (230) vessels have been implicated as sites of critical closure, and the exact site likely depending on lung volume. Moreover, each site of critical closure within Zone 2 may be determined by local factors controlling interstitial pressures and, thus, the measured critical closing pressure likely represents an average. Lastly, changes in resistance and flow in larger, upstream vessels may contribute to the lack of decrease in arterial pressures at low alveolar pressures, unrelated to whether or not critical closure occurs. In sum, Zone 2 flow is determined not only by PALV but also by Starling resistor behavior both at the level of the alveolus and possibly other segments of the vasculature, producing variable capillary opening and closing that depends on PALV and interstitial pressures.
In Zone 3, both PPA and PV are higher than PALV and, thus, in theory flow is completely independent of PALV. Additionally, regional heterogeneity in perfusion exists even in isogravimetric planes, implying that additional factors related to vascular structure and/or the capillary endothelium may play a role in blood flow in the lung microvasculature (220). For example, the most dependent part of the upright lung would be expected to have high flow due to gravitational effects; however, this area has paradoxically decreased flow relative to Zone 3. This area, known as Zone 4, is thought to experience decreased flow due to increased PVR in the extra-alveolar vessels as a consequence of mechanical forces, such that the effective precapillary vascular pressures are diminished. This point can be further illustrated in experiments using fluorescent microspheres to measure blood flow distribution in baboons studied in the upright, supine, prone, and head-down postures (218). In all positions, vertical gradients of perfusion were observed, although multiple stepwise linear regression analysis revealed that both gravity and geometry of the vascular tree influenced regional blood flow. Further complexity is added by heterogeneity in blood flow even between neighboring groups of alveoli, as shown by Baumgartner et al. (39). Consideration of these factors has led to the proposal of a refined model of lung zones (217, 219) whereby all three zones can exist within the same horizontal (isogravimetric) plane, but the numbers of different zones within each plane shifts with increasing hydrostatic pressure down the lung, from predominantly zones 1 and 2 at the apex to all zone 3 conditions in the dependent lung regions.
Pressure-flow relationships
The pulmonary vasculature exhibits high distensibility and low resistance. Propagation of flow in the pulmonary circulation is dependent on a myriad of factors including blood viscosity, vascular compliance, and transmural pressures. Additionally, nonlinear flow patterns originate as a result of vascular branching that occurs with each subsequent generation of pulmonary artery bifurcations. Though both the systemic and the pulmonary circulation feature a system of branching vessels downstream of a ventricular pump, they have different patterns of reflection, as evidenced by differences in pressure and flow curves between these two circuits (643). Like the systemic circuit, flow is pulsatile throughout the pulmonary circuit, remaining pulsatile, though with signal attenuation, to approximately the midcapillary region. In a mathematical model of wave propagation, the drop in pressure pulse appears to be greatest at the entry to the microcirculation (694). The venular segment of the pulmonary circulation exhibits flow patterns that are more dependent on left atrial pressures than changes in PPA (516). Together, the pulmonary arterial, capillary, and venular segments serve to maintain synchrony between right ventricular output and left ventricular filling. Though often described in relation to resistance (see below), it should be noted that the pressure-flow relationships have been modeled as a function of vessel distensibility rather than resistance (357), especially when describing pressure-flow dependence on perfusate viscosity.
The pulmonary arterial tree maintains flow by adjusting resistance and capacitance (in opposite directions) at any given PPA. According to this concept of the time constant (resistance times capacitance), the pulmonary arterial tree tends to be self-adjusting. Consequently, agents that decrease the caliber of the resistance vessels (small arteries and arterioles) in the lungs simultaneously stiffen the larger pulmonary arteries (capacitance vessels). The total PVR of the pulmonary circulation is dispersed across the various types of vessels, with each type (large arteries, smaller arteries, microcirculation, and veins) contributing to a portion of the total PVR. Given that PVR is dependent on various mechanical and hemodynamic factors, it is not surprising that the portion contributed by each subsegment of the vasculature, as well as total PVR, changes with conditions such as hypoxia, volume loading, and positive pressure ventilation (74).
Pressure and flow in the pulmonary arteries
PPA is pulsatile and decreases slightly with inspiration. By comparison, PPA is significantly smaller in magnitude than aortic pressure, displaying a rapid rise to a rounded peak during systole, a brisk small, and gradual decrease in pressure during diastole. End diastolic pressure in the pulmonary artery is approximately 7 to 12 mmHg; PPA rises to approximately 20 to 30 mmHg during systole. PVR is calculated by measuring the drop in pressures across the pulmonary circulation and dividing by pulmonary blood flow. Pulmonary capillary wedge pressure (PCWP), measured with a Swan-Ganz catheter introduced into the right-sided vessels and advanced via the pulmonary artery into the pulmonary circulation, is often used as a measure of PV to make this calculation. In the normal human lung, PVR is approximately 50 to 100 dynes*s/cm5 (174, 445). Calculation of PVR is based on several assumptions including homogeneity of the intraluminal fluid, rigid vessels, and laminar flow; however, blood is a nonhomogeneous fluid, the pulmonary vessels are not rigid and flow is often turbulent or nonlaminar. Furthermore, increases in PPA decrease PVR by way of recruitment, a process that essentially alters the circuit by adding additional sources of capacitance and resistance. Lung volumes affect PVR in a parabolic fashion. At volumes less than FRC, extra-alveolar and alveolar vessel resistance increase and decrease, respectively, contributing to a net increase in PVR and consequently, reduced flow. At volumes higher than FRC, the opposite occurs; alveolar vessel resistance increases due to increasing alveolar pressures, and extra-alveolar vessel resistance decreases, with the same net effect of increasing PVR. In summary, in addition to the limitations associated with interpreting isolated PVR measurements, interpreting changes in measured PVR in the lung is confounded by several factors, including: (i) functional zone of the vessels surrounding the catheter; (ii) lung volume; (iii) position; and (iv) changes in PPA. For example, if the vessels surrounding the site of PV measurement (i.e., the vessel in which the Swan-Ganz catheter resides) do not display Zone 3 behavior, the measured PV may actually reflect PALV instead of true PV, thus artificially lowering calculated PVR. Moreover, there is a preexisting gradient of decreasing vascular resistance from top to bottom in the vertical lung and since resistance and arterial pressures are correlated, rises in PPA are sufficient to cause large decreases in PVR due to recruitment alone.
Pressure and flow in the microcirculation
Obtaining precise values of pressures in the microcirculation has proven difficult due to technical challenges. Studies utilizing isogravimetric, isolated perfused lobes have suggested that the majority of PVR occurs at the precapillary (i.e., arterial) level (481). However, micropuncture measurements performed in subpleural microvessels suggest a significant contribution to total PVR in this segment, with marked drops in pressure occurring just proximal (i.e., <50 μm diameter vessels) to the pulmonary capillaries (54). Exact measurements of pressure gradients across the microcirculation are also confounded by the effects of inflation, the choice of experimental preparation (i.e., isolated and perfused lung), differences between subpleural and deeper vessels, and lack of anatomic precision regarding the location of the vessels studied in a given experimental preparation. PVR measurements are further complicated by capillary recruitment, where the number of vessels with blood flow is subject to change based on ventilatory factors and subject position. In sum, precapillary resistance plays a large role in total PVR, but venous and microcirculatory sources of resistance exist as well.
Pulmonary capillary wedge pressure
As discussed above, PCWP, an estimate of left atrial pressure, can be recorded by introducing a Swan-Ganz catheter through the great veins and advancing the catheter through the right ventricle, pulmonary artery and, ultimately, “wedging” the catheter, with a balloon, in a small precapillary vessel. Assuming Zone 3 conditions, there is an open stream of blood between the tip of the catheter and the left atrium and thus, the pressure recording is said to reflect left atrial pressures. This method of measuring right sided pulmonary vascular pressures and left atrial pressures remains in clinical use, though the utility of PCWP to meaningfully alter clinical care is not clear (517). Multiple efforts to ensure proper position of the catheter are often taken, including analysis of the characteristic wedge tracing, measurement of oxygen saturation from the distal port of the catheter (which, if positioned properly, should reflected pulmonary venous, oxygenated blood) and comparison of the PCWP to pulmonary arterial mean and diastolic pressures. However, despite these efforts, the catheter may still provide incorrect information due to the dynamic nature of zones in the lung. The catheter may be wedged in a Zone 1 or Zone 2 area and thus may not accurately reflect PV. Other considerations such as the presence of parenchymal disease, such as fibrosis or emphysema, and presence of embolic disease may further confound the reliability of these measurements. Finally, two additional factors must be taken into consideration when analyzing pulmonary vascular pressures: transmural pressures and the effects of mechanical ventilation.
Intravascular pressure measurements solely provide information regarding luminal pressures. Since transmural pressure (and not luminal pressure) governs filling and distension, interpreting measurements of intravascular pressure relies largely on the assumption that perivascular pressures are constant between measurements, which may not be the case. While pleural pressure can be measured using a variety of techniques including esophageal balloons, extrapolation of pressures on the surface of the pulmonary vessels from pleural pressure is confounded by several factors including known vertical gradients in pleural pressure in the upright lung and variable transmission of pleural pressure to the various segments of the pulmonary vasculature.
Mechanical ventilation, especially with high positive end expiratory pressure (PEEP) in the setting of hypoxic respiratory failure, has multiple effects on both left sided CO as well as VR. The exact mechanisms behind decreased VR with application of PEEP in both healthy subjects and ill patients have been critically reviewed elsewhere (164, 381). PEEP affects both the mean systemic pressure within the venous system as well as right atrial pressure, and also changes the nature of flow limitation in the vena cava. In a manner similar to Zone 2 of the lung, application of PEEP alters the critical closing pressure of the vena cava, thus preventing further drops in right atrial pressure from affecting VR and producing the “flat” portion of the VR curve (Fig. 5). Transmission of elevations of airway pressure to other thoracic structures (such as the pericardium or the great veins) is variable and depends on a host of anatomic and physiologic factors, such as lung compliance, heart size, and volume status. In general, mechanical ventilation decreases VR and left ventricular afterload in patients with heart failure; however, it should be noted that precise calculations of the effects of mechanical ventilation and/or PEEP on transmural pressures in various segments of the pulmonary circulation are technically challenging and likely vary based on a number of patient-specific factors.
Figure 5.

Effect of PEEP and no, or zero, end expiratory pressure on VR and CO. (A) Effect of PEEP on CO and VR if PEEP has no effects on venous return flow limitation. Points A and B represent effects of PEEP without changes in mean systemic pressure (Pms). Point C represents effect of PEEP with Pms compensation. (B) Effect of PEEP on CO and venous return if increased VR flow limitation (FL2) occurs with higher PEEP. Reprinted, with permission, from Luecke and Pelosi (381).
Regulation of Pulmonary Vascular Tone
Although a low resistance, low-tone circulation under normal conditions, several factors influence vascular SMC tone under a variety of physiologic and pathologic conditions, including neural and circulating factors and oxygen concentration. These potent regulators of both pulmonary vasomotor responses and vascular caliber exert a substantial influence on pulmonary blood flow. If the changes in vasomotor tone induced by these factors are not uniform, significant redistribution of blood flow can occur. While the following discussion focuses on major factors involved in vasomotor regulation, it should be noted that in some cases, factors elicit differential responses depending on the level of initial tone, as discussed in (172).
Methods used to measure vascular tone
Various technical approaches have been utilized to examine vasomotor responses in the lung. Previous reviews have explained in detail some of the caveats of the methods used for detecting changes in vasomotor tone (172). Briefly, measurements in the lungs of intact animals are complicated by potential changes in heart rate, CO, and regional redistribution of pressure/flow. Interpretation of data from intact animals must also consider potential contributions from changes in neural and systemic inputs. In an attempt to remove these confounding factors, many investigators have turned to isolated, perfused lung/lobe preparations, where nervous influences are removed and pulmonary blood flow and left atrial pressure can be precisely regulated. Even more reduced experimental conditions, consisting of isolated pulmonary artery/vein preparations, are used to eliminate potentially confounding effects of extravascular influences, such as circulating mediators, and allow evaluation of influences localized to the vessel wall, such as the endothelium. In these preparations, vasomotor tone has been measured in vascular rings or strips mounted on wire myographs for isometric force measurement or in cannulated vessel segments where vascular diameter can be directly measured under conditions of constant transmural pressure.
Vasodilators
A number of humoral factors participate in active regulation of pulmonary vascular tone (36,129,172). Among the factors that induce pulmonary vasodilation, NO, adenosine, atrial natriuretic factor (ANP) and the eicosanoid, PGI2, are the most studied. Some of these factors are endogenous, derived from the vascular endothelium, while others are produced by circulating cells or in other vascular beds. In this section, we will discuss these factors, and their role in regulating tone under normal and pathologic conditions.
Nitric oxide
NO is produced primarily by the endothelium and is perhaps the most widely studied of the pulmonary vasodilators. NO is generated by oxidation of L-arginine to L-citrulline in a reaction that requires molecular oxygen and is catalyzed by the enzyme NO synthase (NOS) (473). Three NOS isoforms have been identified, with endothelial (eNOS) and neuronal (nNOS) isoforms being Ca2+ dependent and constitutively expressed. In contrast, the inducible form (iNOS) is Ca2+ independent and is expressed in response to cytokines and other stimuli (471, 472). In ECs, constitutive NO synthesis occurs via the activity of eNOS, whereas NO levels can be enhanced by activation of iNOS. Given its short half-life, NO is not suited for action as a circulating factor and instead, once released by ECs, quickly diffuses to the underlying smooth muscle and promotes relaxation via activating soluble guanylate cyclase, leading to generation of cGMP and cGMP-dependent decreases in intracellular calcium concentration ([Ca2+]i) and/or myofilament Ca2+ sensitivity that can limit or reverse ongoing contraction (90).
Numerous studies have utilized NOS antagonists to investigate the role of NO in maintaining low normal pulmonary vascular tone. In the neonate, studies clearly demonstrate a critical role for NO as a modulator of PVR (143, 228, 455). In the adult lung, however, contradictory results have been reported. While NO antagonists increased normoxic pulmonary vasomotor tone in humans (64), and rats (35,92,139), suggesting that NO was exerting a vasodilatory influence, in other studies these interventions had minimal effect on baseline pulmonary vascular tone (35, 157, 163, 249). These data suggest that the contribution of NO to maintenance of low pulmonary vasomotor tone during normoxia depends on the presence or absence of contractile influences and/or relaxing influences other than NO, which in turn may depend on species, preparation, experimental conditions, and other factors.
As a highly reactive gas with short biological half-life, NO was initially thought of as functioning primarily as an autocrine/paracrine signaling molecule, at most diffusing the short distance from the endothelium where it was produced to the underlying smooth muscle. What has only been appreciated more recently is the potential for NO to be stored as nitrite. With a half-life of approximately 50 min (498), nitrite is relatively stable compared to NO. Originally, nitrite was viewed simply as a byproduct of NO metabolism, useful mostly as a biomarker for NO production. However, it is now recognized that nitrite can readily be reduced to NO, thus forming an in vivo physiological reservoir from which NO can be recycled independent of NOS (119, 738). Experiments where exogenous nitrite was administered showed that nitrite had the ability to dilate the systemic vasculature (193,288,324,345) and the pulmonary circulation under conditions where tone was increased (281, 739). Whether NO derived from nitrite contributes to the maintenance of low pulmonary vasomotor tone is unclear at this point, but is a likely possibility, especially under conditions where NO bioavailability is compromised.
Bradykinin
Bradykinin, a peptide product of the renin-angiotensin system, dilates isolated pulmonary vascular rings in many species. In the intact pulmonary vascular bed, however, whether bradykinin is a vasodilator may depend in part on the species tested and/or the level of preexisting tone. Under basal conditions, bradykinin minimally constricted the pulmonary vascular bed of the rabbit (250), but reduced PPA in isolated perfused dog lung (350). Vasodilation became evident, however, when the vasculature was precontracted (183,359).
Bradykinin exerts its dilatory influence by stimulating endothelial NO, and in some cases PGI2, release (21, 287, 474). Indeed, in vitro, removal of the endothelium results in a loss of dilation in response to bradykinin, with vasoconstriction observed in some cases due to activation of receptors on the smooth muscle.
Adenosine
Adenosine, a metabolite of AMP or S-adenosylhomocysteine, dilates most vascular beds. In the lung of various species, including humans, adenosine causes relaxation of pulmonary arteries, via binding to its cell surface receptors and transducing signals through G-protein coupled adenylyl cyclase (189,229,251,330,415,453,454,486,525,535). In some cases, adenosine has also been reported to elicit pulmonary vasoconstriction (56, 77, 103, 535). The exact reason for the discrepancy between studies is unclear, but may depend on species, concentration, or initial vasomotor tone. Consistent with this possibility, adenosine contracted isolated pulmonary vessel preparations at resting tone (364,695), but induced relaxation when vessels were precontracted (153,364,403).
Adenosine infusion increases pulmonary blood flow in healthy humans (255), suggesting that under normal conditions, adenosine exerts a vasodilatory action. Although infusion of adenosine receptor inhibitors had no effect on the high PVR in near-term fetal lamb (329), the effects of adenosine receptor inhibitors in the adult human lung have not been tested and whether endogenous adenosine contributes to maintenance of low basal tone is unknown.
Atrial natriuretic peptide
ANP is produced from cardiomyocytes primarily in response to stretch of the right heart, such that with an increase in PPA, circulating ANP levels are elevated (24). ANP is produced and stored in cardiomyocytes as an inactive precursor, pro-ANP, and upon secretion is cleaved to the active 28-amino acid peptide (α-ANP) (708). Given the source, the pulmonary circulation is the first vascular bed to see ANP; not surprisingly, ANP plasma levels are 30% greater in the pulmonary than systemic circulation. Additionally, pulmonary arteries exhibit greater sensitivity to the vasodilatory effects of ANP than arteries from other vascular beds. Vasodilation is mediated by ANP-A and ANP-B receptors, which are coupled to guanylate cyclase (461). These receptors have been localized in the lung (4, 573, 639) and pulmonary vessels exhibit a high density of ANP-binding sites (16). Exogenous application of ANP causes relaxation of precontracted isolated pulmonary vessels (298,356) and vasodilation in the perfused pulmonary vascular bed under basal conditions (108, 302) that was enhanced when the lung was precontracted (108). In COPD patients, ANP reduces pulmonary vascular pressure and resistance (8), suggesting that in vivo ANP vasodilates the pulmonary circulation, especially under conditions of increased tone.
Eicosanoids
Under basal conditions, when tone is low, infusion of arachidonic acid constricts the perfused pulmonary vascular bed (559, 692) but causes relaxation when vascular tone is elevated (211). Arachidonic acid is the precursor of several vasoactive mediators, including the cyclooxygenase product, PGI2. Released from endothelium, PGI2 stimulates adenylate cyclase and increases production of cAMP in the underlying smooth muscle, vasodilating both the pulmonary and systemic circulations (282). The role of PGI2 in maintaining low vasomotor tone is incompletely understood. Several investigators found no effect of the cyclooxygenase inhibitors, indomethacin or meclofenamate, on baseline PPA and/or PVR in either intact animals or isolated, perfused lungs (94, 487, 496, 522, 680). In contrast, other investigators observed increases in PPA with administration of these inhibitors (307, 522, 618, 659), suggesting basal endogenous production of a vasodilating prostanoid, likely PGI2. The reason for these differences is unclear, but may be related to concentrations of drugs used or the duration of exposure to drug before measurements were made, since most of the measurements showing no effect were made shortly (<30 min) after drug administration. Along these lines, long-term administration of cyclooxygenase inhibitors (3 weeks) was reported to increase PPA (417). Overall, these data suggest that, at least under certain conditions, vasodilating cyclooxygenase products may contribute to low pulmonary vasomotor tone.
In some cases, endothelium-dependent relaxations induced by acetylcholine and other agents were not completely abolished with inhibition of NO and PGI2, indicating the presence of additional endothelium-dependent vasodilation pathways (67, 102, 162, 328). Since these vasodilatory effects occurred in association with smooth muscle hyperpolarization, it was postulated that release of an endothelium-derived hyperpolarizing factor (EDHF) was responsible. The exact identity of EDHF is still unclear, but one of the most likely possibilities is a cytochrome P-450 product of arachidonic acid metabolism, perhaps an epoxyeicosatrienoic (EET) acid (68, 175, 177, 206). In both the systemic and pulmonary circulations, cytochrome P-450 metabolites can elicit either dilation or contraction, although in the systemic circulation, EETs mediate relaxation while 20-HETE is a constrictor (176). Remarkably, the opposite appears true in the pulmonary vascular bed, with infusion of 11,12-EET increasing PPA in isolated perfused mouse lungs (320) and 20-HETE causing dilation in human PAs (59). Although EDHF appeared to play a role in vasomotor responses of isolated pulmonary arteries or lungs (198, 732), whether it contributes to maintenance of low basal tone is unclear.
Vasoconstrictors
Pulmonary vasoconstriction is caused by a variety of factors, including serotonin, ET-1, angiotensin II (ANG II), and prostaglandins, some of which are derived from the vascular endothelium. Circulating cells are also an important source of vasoconstrictors that can act on the pulmonary circulation, including serotonin, a primary product of activated platelets.
Endothelin-1
Discovered in 1988, ET-1 is a peptide secreted by primarily ECs (709). Of the three isoforms that have been identified (ET-1, −2, and −3), ET-1 is the most widely expressed, and thus, most studied. ET-1 is a 21-amino acid peptide that causes profound pulmonary vasoconstriction in every species tested to date [reviewed in (606)], and is perhaps the most potent endogenous vasoconstrictor in the lung. ET-1 binds to two membrane bound receptors (ETA and ETB) (629), both of which are expressed in PASMCs (124,188), and mediate contraction, proliferation and migration. In contrast, ECs express only ETB receptors, which are thought to act as a “sink” for circulating ET-1. While ET-1 can elicit NO production and transient dilation when EC receptors are activated, the majority of ET-1 is secreted basolaterally; thus, in vivo PASMCs are likely to be the main target of ET-1 with the main action of ET-1 being vasoconstriction. At concentrations as low as 10−10 M, ET-1 constricted isolated pulmonary arteries through activation of ETA or ETB receptors on PASMC, while infusion of ET-1 caused long-lasting increases in vascular resistance in isolated perfused lungs [reviewed in (570, 606)]. ET-1 secretion from the pulmonary endothelium has been shown to increase in response to shear stress and hypoxia [reviewed in (563, 570, 606)]. Binding of PASMC ET receptors initiates a complicated signaling pathway (570, 629), involving inhibition of K+ channel expression and activity, increased [Ca2+]i and activation of NHE1 and Rho kinase (ROCK) (570,642,691).
Acute infusion of an ETA receptor antagonist causes pulmonary vasodilation in fetal sheep, suggesting that basal ET-1 activity contributes to high in utero PVR (293, 700). Similar results were observed in intact adult pigs (268, 683), but not dogs (624), perhaps reflecting a species-dependent difference in either endogenous ET-1 synthesis or participation in regulating basal tone.
Arachidonic acid metabolites
Arachidonic acid, which is readily taken up in the pulmonary circulation, is metabolized into a number of vasoactive eicosanoids, including PGE2, PGF2α, thromboxane, and leukotrienes, all of which diffuse to the smooth muscle and cause contraction. Thromboxane, or thromboxane mimetics, constrict isolated perfused lungs (166, 169, 515) as well as isolated pulmonary arteries (114, 166, 299, 360, 443). Synthesized via the lipoxygenase pathway, leukotrienes C4, D4, and E4 caused pulmonary vasoconstriction in a variety of species, including humans [reviewed in (36, 658)]. Arachidonic acid can also be metabolized by cytochrome P-450, which produces midchain hydroxyeicosatetraenoic (HETE) and cis-EET acids in pulmonary arterial ECs and PASMCs (731, 737), the latter of which can be further metabolized by soluble epoxide hydrolase, forming dihydroxy derivatives. In the precontracted pulmonary vasculature, HETE and EET can cause vasodilation (59, 296, 599, 722) but primarily mediate vasoconstriction under unstimulated conditions (190,372,706,736). Since administration of arachidonic acid constricts the normal perfused pulmonary vascular bed (559, 692), it is likely that under normal conditions of low tone, the balance of arachidonic acid metabolism is shifted toward a vasoconstrictor effect.
Serotonin (5-HT)
Pulmonary neuroendocrine cells secrete various vasoactive substances, including 5-HT (304, 346). A 5-HT transporter protein [serotonin transporter (SERT)] in plasma membranes allows circulating 5-HT to be taken up by cells, including pulmonary ECs and platelets, where it is stored and subsequently released during aggregation. At baseline, storage in platelets and the presence of reuptake mechanisms ensure low circulating 5-HT levels, but plasma levels can be elevated by selective 5-HT reuptake inhibitors (SSRIs). Application of exogenous 5-HT constricts the pulmonary vasculature, although its potency varies greatly among species [reviewed in (36,606)]. When tone is low, contraction to 5-HT is mediated primarily by 5-HT2A receptors; with elevated tone, a portion of 5-HT-induced contraction can be mediated by 5-HT1 receptors (388). In the intact animal, infusion of the 5-HT2A receptor antagonist, ketanserin, reduced PVR in intact cats (75), fetal sheep (135) and in patients with stable angina (270), but had no effect on PVR in isolated perfused dog lung (34). Similar contradictory results were obtained with application of SSRIs. Infusion of the SSRI, sertraline, increased PVR in fetal sheep (135), whereas the SSRI, dexfenfluramine, had had no effect on PVR in porcine lungs and had limited effect in human lungs, with vasoconstriction only noted at high concentrations (258). Whether these situations reflect differences between intact animals and isolated perfused lung preparations, or conditions where tone and/or endogenous 5-HT levels were elevated, is unclear.
Angiotensin II
Circulating angiotensin I, a product of the renin-angiotensin system, is converted into the vasoactive peptide, angiotensin II (ANG II), in the lung by means of endothelium-bound angiotensin converting enzyme. In point of fact, the lung endothelium is the primary site of metabolism of angiotensin I, with approximately 60% to 80% of plasma angiotensin I converted to ANG II in a single pass through the pulmonary circulation. Exogenous ANG II causes pulmonary vasoconstriction in most species, both in isolated vessel and intact lung preparations (37,70,87,160,185,222,404,408,462,506,534, 614, 682). Some studies suggest that basal activation of the renin-angiotensin system and production of ANG II exerts a slight vasoconstrictor effect on the pulmonary circulation (222, 462), although other studies failed to show an effect of ANG II blockade at baseline (246,276,323,335,506).
Summary
In addition to the vasodilators and vasoconstrictors described in the preceding sections, a number of other factors have been shown to exert regulatory influences on tone in the pulmonary vasculature, including histamine, platelet-activating factor, vasopressin, vasoactive intestinal peptide, and substance P. The actions of these agents on the pulmonary circulation have been reviewed in detail elsewhere (36, 129) and interested readers are encouraged to consult these previous reviews for further information regarding pulmonary vasoregulation by these factors.
The preponderance of evidence suggests that although most of these factors can produce changes in vasomotor tone, with the exception of NO and PGI2, none appear to exert a major regulatory influence on basal pulmonary vascular tone in the normal lung. However, under pathological conditions modulation of tone by a variety of factors may become more pronounced. For example, bradykinin induces a moderate fall in PPA in patients with hypoxic pulmonary hypertension (62). Similarly, adenosine acts as a pulmonary vasodilator in pigs following infarction (589), in patients with pulmonary hypertension (252, 431) and in cardiac surgical patients (189). Moreover, vasoconstrictors can modulate tone under pathological conditions, such as in pulmonary hypertension induced by anorexic agents, where serotonin uptake is impaired, and excessive production of ET-1 is a common feature of pulmonary hypertension.
Nervous control
The pulmonary circulation is richly innervated by the autonomic nervous system, with both sympathetic and parasympathetic connections (36, 173, 337, 529). In most organs, arterioles exhibit the highest level of sympathetic innervation (561); in the lung the opposite is true, with sympathetic noradrenergic innervation density highest in the extrapulmonary vessels (96, 171, 243). The degree to which sympathetic innervation penetrates the vascular tree varies with species [reviewed in (36)]. Rat and mouse intrapulmonary arteries do not appear to be innervated (96,171); however, in most animals, including humans, sympathetic axons extend down to the level of small arterioles (96, 171, 243). Intrapulmonary veins are also innervated (96,309,338), although to a lesser extent than the arterial network.
Given evidence indicating that pulmonary arteries are abundantly innervated, it is perhaps surprising that there appears to be minimal nervous control of basal vascular caliber in the pulmonary circulation, with adrenergic antagonists causing a slight, if any, increase in PVR in anesthetized and conscious dogs and sheep (312,398,442). While these results suggest that there may be a mild degree of basal sympathetically driven dilatory influence on tone under normal physiological conditions, nervous stimulation under experimental conditions clearly modulates tone. In general, increased sympathetic activity leads to release of catecholamines (e.g., dopamine, norepinephrine, epinephrine, and neuropeptide Y) that cause vasoconstriction and an increase in PVR (82,338). Indeed, in the intact lung, sympathetic nerve stimulation caused an increase in PVR that could be inhibited by chemical denervation or adrenergic blockade (283,308,309). Similarly, stimulation of sympathetic nerves constricted resistance-sized pulmonary arteries, which was reversed by an α-adrenergic blocker (574). This point is further illustrated in numerous studies where infusion of α-adrenergic agonists caused pulmonary vasoconstriction in intact animals with controlled pulmonary perfusion (32, 499, 536, 580). Interestingly, infusion of β-adrenergic agonists dilated the pulmonary vasculature (500,580), suggesting β-adrenergics mediate vasodilation in response to circulating catecholamines (65,285) and locally released norepinephrine (284). Taken together, control of the pulmonary circulation by the sympathetic nervous system may vary with stimulus and/or ultimately may be a coordinated response including both α- and β-adrenergic components.
Pulmonary arteries contain fewer cholinergic than adrenergic nerve fibers, although the distribution is similar, with highest density at the extrapulmonary arteries (152, 243). Parasympathetic stimulation causes release of acetylcholine and vasoactive intestinal polypeptide (11,96,237,407), which mediate vascular dilation and act to decrease PVR. However, cholinergic blockade had no effect on basal PVR (442). The lung also contains nonadrenergic, noncholinergic (NANC) nerves that can release vasoactive intestinal peptide, calcitonin-gene related peptide, ATP, substance P and NO to mediate vasodilation (9,227,241,362,363,404,557). The presence of nNOS has been demonstrated in nerve fibers in and around the walls of pulmonary arteries (168,241), consistent with NO being the primary neurotransmitter responsible for NANC-induced pulmonary vasodilation (227,241,362,363). However, NOS blockade did not fully inhibit NANC-mediated dilation (241), suggesting that the other factors may also participate in the response. While the existence and activity of NANC nerves have been demonstrated in vitro [reviewed in (36)], regulation of tone in vivo by these nerves has not been demonstrated.
Thus, the majority of evidence suggests little in the way of contribution from the nervous system in control of resting vasomotor tone in the lung; however, stimulation of adrenergic nerves may modulate PVR and blood flow during exercise and cold exposure and may increase in regulatory contribution during pathological states, particularly during pulmonary edema and embolism.
Hypoxia
One of the unique aspects of the pulmonary circulation is the hypoxic pressor response. Unlike the systemic circulation, where hypoxia causes vasodilation to increase blood flow and oxygen delivery to tissues, reductions in alveolar and arterial oxygen content cause profound constriction in the pulmonary vasculature. This phenomenon was suggested as early as 1852 (51), when the first measurements of PPA were reported and showed that stopping ventilation increased PPA. Later studies by von Euler and Lillestrand in 1946 (652) confirmed this early finding, and provided the first detailed characterization of hypoxic pulmonary vasoconstriction (HPV).
While systemic hypoxic vasodilation plays an obvious role in working to meet the metabolic demands of the tissue, the exact reason why the pulmonary circulation contracts in response to hypoxia remains a matter of some debate. HPV is most commonly believed to be either a residual feature from fetal development, where the hypoxic environment maintains a closed pulmonary circulation until birth, or a mechanism to match ventilation and perfusion during periods of localized hypoxia, by diverting blood flow from poorly oxygenated portions of the lung. What is not in debate is the response itself. Numerous studies have explored the effects of hypoxia on the pulmonary circulation in a variety of species [reviewed in (606)]. Uniformly, these studies have described an increase in PVR that occurs within 1 to 2 min after a reduction in alveolar oxygen tension that reaches a maximal response in 5 min. This immediate contraction is maintained for the duration of the hypoxic exposure, but varies in intensity by level of hypoxia, sex, and species (606). Upon reoxygenation, HPV rapidly reverses, with PPA typically returning to normal levels within minutes.
Although the large pulmonary arteries are capable of responding to hypoxia, it is now generally accepted that the main site of increased resistance during HPV is in the small, muscular arterioles. Postcapillary hypoxic venoconstriction may also occur, although the magnitude of the contribution of these vessels to the increase in total PVR is uncertain.
The exact mechanisms by which the pulmonary vasculature senses a drop in oxygen tension and transduces this signal to vasoconstriction are still an area of active investigation. Studies using reduced preparations, including isolated vessels and single cells, clearly established that hypoxia had a direct effect on PASMCs, although it is also evident that the maximal contractile response requires a coordinated response between ECs and SMCs(1, 606). For example, it is well documented that ECs provide a priming or modulating influence through the release of vasoactive agents [reviewed in (1, 606)]. In addition, ECs communicate with SMCs via myoendothelial gap junctions, which allow transmission of electrical current and small signaling molecules between cells and facilitate HPV (326, 433). Indeed, myoendothelial gap junction signaling contributes to HPV via Rho kinase-dependent increases in Ca2+ sensitization of the contractile apparatus (326). More recently, studies revealed a fundamental role for the endothelium as part of the initiating sensor of HPV, with capillary ECs exhibiting depolarization in response to alveolar hypoxia that is propagated upstream via endothelial connexin 40 gap junctions, eventually leading to production of EET acids (669). Thus, the in vivo response to hypoxia likely results from a coordinated response including sensing by, and communication between, both the ECs and SMCs.
Considerable effort has gone into delineating the SMC response to hypoxia, sometimes with confusing and/or contradictory results [reviewed in (606)]. For example, substantial evidence suggests that reactive oxygen species (ROS) appear to be key mediators of HPV; however, the source, direction of change during hypoxia and downstream targets have varied across laboratories [reviewed in (606)]. Initial reports indicated that ROS production decreased during hypoxia, leading to inhibition of redox-sensitive K+ channels, depolarization and activation of voltage-gated Ca2+ channels (19, 20, 419) (redox hypothesis; Fig. 6A). Later studies, however, suggested that ROS formation in fact increased during hypoxia (347, 361, 520, 665, 674, 675, 677). While perhaps counterintuitive, given the fact that oxygen is a substrate for ROS production, it was suggested that despite a fall in oxygen concentrations, ROS production from mitochondrial complex III was enhanced (ROS hypothesis; Fig. 6B), leading to increased [Ca2+]i due to both Ca2+ release and influx. Interestingly, Ca2+ release has also been shown to inhibit K+ channels during hypoxia (502), suggesting potential links between these two proposed hypotheses.
Figure 6.
Proposed mechanisms by which hypoxia caused pulmonary vasoconstriction. Reproduced, with permission, from Sylvester et al. (606).
The exact reasons underlying the differences in results remains to be fully explained, although the recent development of ratiometric ROS sensors that could be targeted to specific compartments within cells has provided potential insights. Using these probes, it was found that there were substantial differences in ROS production during hypoxia that varied by cellular compartment; ROS levels increased in the cytosol and mitochondrial intermembrane space, but decreased in the mitochondrial matrix (676). Thus, significant differences in results could be due to the compartment within the cell in which measurements were obtained. Overall, it would seem that the majority of data now supports that, in PASMCs, hypoxia leads to production of mitochondrial ROS (98, 674), localized areas in which ROS are reduced (19, 676), calcium release from the sarcoplasmic reticulum (458,663,705), opening of calcium permeable nonselective cation channels (NSCCs) (458,664,679), inhibition of voltage-gated potassium channels (19, 503, 728, 729), membrane depolarization (503, 729), and subsequent activation of Ca2+ influx through voltage-gated Ca2+ channels (409,664).
Although the exact mechanisms by which hypoxia activates Ca2+ signaling are still being investigated, recent data demonstrated a role for sphingolipid signaling that might link ROS with Ca2+ responses and HPV (610). Oxidant stress activates neutral sphingomyelinase, releasing ceramide, which meditates both the recruitment of the NSCC, TRPC6, to the plasma membrane and activation of TRPC6 via PLC. The sphingolipid product, sphingolipid-1-phosphate (S1P), concomitantly increases Rho kinase activation, combining with Ca2+ influx to cause PASMC contraction.
Increased production of vasoconstricting (i.e., ET-1), and/or reduced production of vasodilating (i.e., NO), factors by the neighboring ECs augment PASMC responses to hypoxia (606), indicating that while the main contractile mechanics underpinning HPV reside in PASMCs, the endothelium certainly plays a modulatory role. Other factors that modulate HPV include prostaglandins, angiotensin II, serotonin and leukotrienes, although all of these have been ruled out as mediators of the response [reviewed in (606)]. The autonomic nervous system has also been ruled out as a mediator, as the response can be observed in isolated, perfused lungs that lack nervous input (123, 367, 368) and most experiments in intact animals suggest that sympathetic activity has no effect on HPV [reviewed in (606)]. Indeed, in some cases, blockade of β-adrenergic receptors enhanced HPV (32, 76, 367, 499), suggesting that while sympathetic activity does not mediate HPV, it could modulate the response under certain conditions. In contrast, parasympathetic nerves do not appear to modulate HPV (349,351).
The fact that the pulmonary circulation contracts almost immediately upon initiation of hypoxia would also appear to rule out a role for transcriptional regulation in the acute HPV response. However, HPV is enhanced in individuals with Chuvash polycythemia, a rare genetic condition where germline hypomorphic mutations in the gene encoding von-Hippel Lindau protein—a major component of the HIF degradation pathway—result in augmented HIF expression under nonhypoxic conditions (585), suggesting a potential role for HIFs in regulating acute pulmonary vasomotor responses to hypoxia. Similarly, studies of humans with HIF-2α gain-of-function mutations also exhibit enhanced HPV (179). Although it is unlikely that acute changes in gene transcription underlie the immediate pulmonary vascular response to hypoxia, these findings support an intriguing possibility that HIF may modulate the basal expression of targets believed to be directly involved in the contractile response (i.e., K+ or Ca2+ channels) or circulating factors known to be required for enhancement or priming of PASMCs (i.e., ET-1).
Pulmonary Barrier Function and Edema
The pulmonary endothelium is a contiguous structure, one of the main functions of which is to protect the alveolus from flooding and injury due to flux of electrolytes and other small molecules, proteins, and fluid from the vascular compartment. The thinness of the pulmonary endothelium and minimal distance between the capillary wall and alveolar epithelium are ideal for gas exchange, but also predispose the lungs to edema formation. In recent years, the barrier protective function of the pulmonary vasculature has been investigated intensely [reviewed in (53, 60, 327, 476)]. Recent studies from several laboratories suggest that the pulmonary microvascular endothelium is much less permeable than that of capillaries in other organs and less permeable than the pulmonary macrovasculature (134, 477, 480, 601). Phenotypic variation between the ECs that comprise the intima of these vascular segments likely contributes to the differences in their responses to various barrier-disrupting stimuli. Understanding the cellular mechanisms that control endothelial barrier function has been the focus of many recent studies of the pulmonary circulation, especially given that increases in microvascular and macrovascular endothelial permeability have been identified as key processes in the development of diseases like ARDS.
Vascular phenotypes: Microvascular and microvascular endothelium
Since the pulmonary vascular endothelium plays a key role in the pathogenesis of various lung diseases, recent investigations into the molecular biology of populations of lung ECs led to the classification of vessels based on their endothelial phenotype, as either macrovascular or microvascular. Like older classification systems, this system is also based on vessel diameter size; however, it includes additional functional and morphologic characteristics that differentiate these two cell types. While differences between alveolar and extra-alveolar vessels have been studied in the context of mechanical forces such as inspiration or changes in vascular pressure, the differences between the vascular endothelial phenotypes have been studied largely in the context of their gene and protein expression, cytoskeletal function and responses to barrier disrupting stimuli.
Differences between the various segments of the pulmonary endothelium begin in utero, where it has been shown that the microvascular ECs develop from a pool of progenitor cells distinct from arterial and venular ECs (208). All ECs in the lung share some features that are common to ECs throughout the body, such as size, fine control of cell adhesion and a basic array of cell surface markers such as von Willebrand factor and PECAM-1 (208). However, it is logical that differences in local milleu dictate the genotypic and phenotypic variation seen within the pulmonary endothelium. For instance, macrovascular ECs are exposed to more pulsatile flow and are not subjected to nearby alveolar forces, unlike microvascular ECs. Consequently, these two cell types exhibit unique morphologic characteristics, distinct genomic and proteomic profiles and differential responses to a variety of injurious stimuli (635). In general, microvascular ECs form a tighter barrier and are more restrictive to fluid efflux through paracellular routes, and when barrier disruptive stimuli such as pathogens, alterations in intracellular Ca2+ or oxidative stress are applied, macrovascular and microvascular cells respond differently to these injuries (13,109). For example, in experiments utilizing various Ca2+ channel agonists in isolated, perfused lung preparations, increased permeability in microvascular endothelium can be induced with agonism of the Ca2+ channel, TRPV4, but not depletion of Ca2+ stores with thapsigargin, whereas macrovascular endothelial permeability can be increased by store depletion (104). In vitro, store depletion and activation of store-activated Ca2+ channels appears to be sufficient to induce paracellular leak in macrovascular, but not microvascular, ECs, suggesting differential expression of these channels as well as fundamental differences in barrier disrupting signal transduction (376). There are additional differences in virtually all domains of cell function, from cytoskeletal dynamics to proliferation and responses to flow (600). For instance, compared to macrovascular ECs, microvascular ECs express binding motifs for interactions with Griffonia simplicifolia lectin, but lack Weibel-Palade bodies (600). Microvascular ECs also express more nucleosome assembly protein-1, which has been implicated in their ability to proliferate faster than macrovascular ECs (111). In vitro, application of cyclic stretch produces more changes in cell-cell adhesions in macrovascular endothelium (7). Indeed, more global analysis of gene expression using microarrays has demonstrated distinct gene expression profiles between micro- and macrovascular ECs suggesting large differences in cell function and signaling between these two adjacent EC types (107). In vivo, there appears to be a distinct transition zone, occurring around vessels with outer diameters of approximately 25 μm, when microvascular ECs appear and macrovascular ECs disappear (635). The exact reasons underlying the observed functional differences between pulmonary micro- and macrovascular ECs are still being investigated, but may lie in differential expression of various ion channels, kinases and signaling molecules that participate in barrier regulation, as will be discussed later in this review.
Methods of measuring endothelial permeability
To compare across studies, a fundamental understanding of the multiple methods that have been developed to assess lung endothelial permeability in vitro and in vivo is helpful [reviewed in (727)]. In vivo measures of endothelial permeability measure leak of fluid into the interstitial space due to either increase in intravascular pressures or endothelial permeability (478). An important consideration is that the net efflux of fluid from the vascular space is a function of Starling forces, as well as properties of the luminal fluid, permeability state of the endothelium, flooding in the alveolus and lymphatic flow (401), and occurs in both the alveolar and extra-alveolar vessels [reviewed in (478)]. Methods of quantifying endothelial leak in vivo in intact animals include gravimetric methods (e.g., wet/dry ratios), light and electron microscopy, and visual quantification of dye extravasation from the vascular compartment (i.e., Evans Blue Dye). These methods are not without drawbacks, however, including arti-factual increases in perceived permeability due to changes in lung blood volume and dye binding to native proteins.
Isolated, perfused lung models offer the advantage of maintaining the integrity of the vasculature and its relationship to ventilated alveoli, while allowing for more precise measurements of filtration across the endothelium by manipulating arterial and venous pressures. First described in 1967 (194), measuring the endothelial filtration coefficient (Kf) using an isolated, perfused lung preparation remains the gold-standard tool for assessing pulmonary vascular permeability (479) particularly via the paracellular pathway (478). Assuming that further recruitment is not taking place (479), rises in Kf directly correlate to increased conductance of fluid across the endothelium. Accordingly, this method has been used in both wild-type and transgenic mice and rats in multiple models of lung injury. However, as explained in detail in a recent review (55), Kf measurements are prone to errors due to changes in pulmonary volume when the atrial pressure is artificially increased during the measurement process. Additional considerations include injury to the ECs due to the measurement procedure itself, and whether or not concomitant alveolar edema is present. Much work has been invested developing techniques to minimize these sources of error as much as possible, and detailed descriptions of specific recommendations for experimental procedures have been published (55,478). Once an appropriate isolated, perfused lung preparation is in place, in addition to Kf, further measures of permeability such as the reflection coefficient and permeability surface area product can be obtained as well (478). Techniques to measure flux occurring via vesicular and transcellular pathways have also been developed, and are discussed elsewhere (319,532,727).
The choice of a specific method to measure endothelial permeability may be dictated, to some degree, by the type of model in which lung injury is incited. Physiologically functional models where the cardiopulmonary circuit is left intact (e.g., survival ischemia-reperfusion models, endotoxin (lipopolysaccharide; LPS) or cecal-ligation puncture models of lung injury), while closest to native physiology, do not allow for enough manipulation of hemodynamic parameters to reliably make Kf measurements unless the animal is sacrificed afterward for use in the isolated, perfused lung preparation. Instead, for these models, a general dye-based method such as extravasation of Evans Blue Dye is often used (670). It should be noted, however, that limitations in this method related to binding of dye to albumin and other proteins (122) and changes in vascular tone and recruitment between animals (727) have been noted and may complicate interpretation of results.
Injury models in intact animals are particularly useful for determination and assessment of in vivo endothelial barrier function, but do not allow for detailed mechanistic exploration of the precise cellular mechanisms involved in injury. To combat this shortcoming, in vitro models of EC barrier permeability have been developed. The use of cultured EC monolayers allows for multiple modes of manipulation of pathways and evaluation of cell dynamics, albeit in a less physiologic setting, due to the absence of structures such as a proper glycocalyx, basement membrane, and surrounding extracellular matrix as well as the lack of tonic shear from luminal flow and changes in the endothelial proteome that occur due to in vitro handling (641). Methods for assessing permeability in cultured ECs include transwell assays, which determine permeability by measuring migration of a large weight molecule from an upper chamber to a lower chamber across a confluent layer of ECs (579) and electrical cell impedance sensing (ECIS), which utilizes the electrical properties of an EC monolayer, with the cells acting as capacitors, and pores that open when permeability increases acting as resistors, to measure total impedance (269). In the case of the latter method, increases in permeability will thus decrease measured ECIS resistance. One of the most exciting recent developments in the field is the newer “lung on a chip” in vitro model (279), which recapitulates many of the physiologic processes that occur in the native lung (Fig. 7). In this preparation, epithelial cells and ECs are appositionally seeded, separated by a porous synthetic membrane that has been coated with matrix components. The epithelial side is then exposed to air while the endothelial side is exposed to perfusate. Additional side chambers allow for simulation of cyclic stretch that occurs with normal tidal breathing. Thus, several of the limitations inherent in traditional static culture models such as lack of endothelial shear stress, alveolar stretch, alveolar-matrix, and matrix-endothelial interactions are overcome in this powerful new technique, providing the potential to combine more physiologically relevant parameters with the ability to manipulate individual cell populations in a coculture setting.
Figure 7.

The lung-on-a-chip microdevice. (A) Compartmentalized channels form an alveolar-capillary barrier on a thin, porous, flexible membrane. Physiological breathing movements are mimicked by applying a vacuum to the side chambers, causing mechanical stretching of the membrane that forms the alveolar-capillary barrier. (B) Cartoon illustrating the mechanical effects of inspiration in the living lung, where contraction of the diaphragm reduced pleural pressure (Pip), resulting in distension of alveoli and physical stretching of the alveolar-capillary interface. (C) The three-layer system forming parallel microchannels with a porous membrane. Scale bar, 200 μm. (D) After the three layers are bonded together, the membrane layers in the side channels are removed, producing two side chambers to which vacuum is applied to cause mechanical stretching. Scale bar, 200 μm. (E) Actual images of a lung-on-a-chip device. Reproduced, with permission, from (279).
Mechanisms regulating endothelial permeability
Numerous investigators have invested considerable effort to understanding the complex mechanistic underpinnings of barrier homeostasis and dysregulation. Our understanding of the molecular mechanisms of endothelial permeability is complicated by the numerous agents that have been used to experimentally induce injury. Confounding matters further is the variable behavior of signaling pathways in vivo compared to in vitro cell culture systems, due to critical modifications in endothelial structure and/or environment (i.e., loss of glycocalyx, lack of interactions with underlying smooth muscle and matrix proteins, absence of flow, etc). As a consequence, agonism of several endothelial barrier disrupting pathways have produced differential effects on lung EC permeability in vivo compared to in vitro (641). Recent excellent reviews have discussed some of the controversies, and the signaling cascades underlying maintenance and loss of endothelial barrier function and various mechanisms by which fluid flux occurs in detail (146,327,413,646,651,727). Thus, in this section, we will highlight major pathways that have been implicated, focusing on some of the more recent findings.
Signals leading to initiation of barrier disruption
Multiple upstream mediators of intracellular events leading to cellular contraction, loss of cell-cell interactions and increased transcellular and paracellular flux have been identified [reviewed in (727)]. In particular, VEGF/VEGFR binding has been associated with a myriad of second messaging systems (727), including: (i) increased [Ca2+]i; (ii) generation of inositol triphosphate (IP3) and diacylglycerol, which activate Ca2+ release from intracellular stores and protein kinase C (PKC), respectively; and (iii) activation of Src and MAP family kinases, all of which have been implicated in endothelial permeability (as discussed later in this section). Human studies examining VEGF levels in patients with ARDS showed higher VEGF levels in plasma and lower levels in bronchoalveolar lavage fluid (626). When plasma from patients with ARDS was perfused atop confluent microvascular ECs, paracellular leak occurred that was attenuated with neutralization of plasma VEGF (626). Although animal models have shown injurious effects of VEGF on the lung vasculature, there is also evidence suggesting that VEGF may be protective (437). It is likely that VEGF plays a dual role in the pulmonary vasculature, either pathologic or protective depending on the stage and type of injury.
Clinically, sepsis is one of the major causes of ARDS. Bacterial toxins activate inflammatory cascades upon recognition by the family of toll-like receptors. As might be expected in this case, ligation of toll-like receptor 4 by LPS plays a key role in initiation of endothelial hyperpermeability in endotoxin-mediated barrier injury (130). In addition, other upstream receptors have been shown to be involved in barrier regulation; thrombin increased EC permeability in EC monolayers (315) and increased macrovascular (but not microvascular) EC permeability in isolated, perfused rat lungs (636).
In addition to direct effects on ECs via surface bound receptor signaling, LPS promotes barrier disruption via initiating derangements of extracellular endothelial components, including the glycocalyx (552,715). For example, degradation of the glycocalyx allows for exposure of previously hidden binding sites for inflammatory cells and other proinflammatory molecules on the EC surface (715). Moreover, the glycocalyx is connected to the cytoskeleton and focal adhesions, allowing for potential direct cross-talk between alterations in the extracellular environment and intracellular components known to modify cell shape. During inflammatory states, neutrophils traversing the lung exit the vasculature and cross the endothelial barrier at the capillary level (657). LPS-mediated loss of the glycocalyx resulted in neutrophil attachment to ECs, the first step in endothelial transmigration. It is well recognized that neutrophilic infiltration of the interstitium contributes to the pathogenesis of ARDS by generating local stimuli that are injurious to the endothelium (699), such as ROS (generated by neutrophils via NADPH oxidase pathways) and release of inflammatory factors stored in neutrophil granules, including various proteases (such as matrix metalloproteinase and elastase) which may injure the endothelium through indiscriminate proteolysis of nearby proteins. Proteases likely represent one in a series of neutrophil-dependent injurious stimuli that are released in ARDS; indeed, inhibition of neutrophil elastase alone has not been shown to be of clinical benefit in ARDS (294).
Finally, changes in blood flow during ischemia may also contribute to barrier dysregulation. It is now appreciated that the presence of pulsatile flow in the luminal side of ECs exerts tonic effects, and that changing flow across the endothelial layer initiates multiple secondary signaling pathways that affect the function of junctional proteins such as α-catenin and VE-cadherin, destabilizing cell-cell adhesions and promoting paracellular fluid flux (240).
Transduction of the barrier disruption signal
Regardless of the method by which the signal for barrier disruption is generated, it is now recognized that intracellular signaling converges on several key pathways. In particular, production of ROS (from NADPH oxidase or other sources), is a common event, whether generated in ECs or surrounding cells (i.e., neutrophils). ROS serve important signaling functions at baseline levels, with activation of many more pathways when ROS levels are increased (97). Indeed, recent evidence has implicated proteins that participate in endogenous ROS generation and regulation, such as NADPH oxidase and peroxiredoxin (170, 200) as playing critical roles in initiation of the intracellular signaling in acute lung injury and ARDS.
Another early signaling event activated by numerous barrier disrupting agents is increased [Ca2+]i. The increase in [Ca2+]i may occur as a direct consequence of receptor activation, or secondary to ROS (66). Both influx from extracellular sources, through TRP channels such as TRPC1, TRPC4, and TRPV4 (13, 631, 634), store operated channels such as Orai1 (200) and release from internal stores (via IP3) lead to activation of various Ca2+-dependent systems. For instance, increases in [Ca2+]i can directly activate components of the contractile apparatus [primarily through myosin light chain kinase (MLCK)]. [Ca2+]i may also play an important role in linking membrane events to the cell-cell junction complex that controls paracellular permeability, with a rise in [Ca2+]i ultimately inducing hyperpermeability via disruption of the junctional complex and/or via effects on cytoskeletal elements (13,200,634).
While much of the work examining the role of [Ca2+]i in EC barrier function has been performed in vitro, in vivo Ca2+ dynamics in ECs include mechanisms such as wave propagation of rises of [Ca2+]i from cell to cell in an oscillatory fashion (717) and spread of Ca2+ though gap junctions (483) that are not seen in vitro. The extent to which these mechanisms may also play a role in regulating endothelial permeability, under both basal and stimulated conditions, is still being investigated.
Elevations in [Ca2+]i can also activate certain isoforms of PKC, which can lead to disassembly of junctional components as well as other barrier disrupting effects through activation of MLCK (631). In addition, Ca2+-dependent activation of PKC may serve as a starting point for activation of several downstream second messenger systems, including MAP kinases, focal adhesion kinase, and small GTPases (727), several of which have been implicated as part of a complex system of second messengers that effect hyperpermeability in ECs. For example, Rho-GTPase members of the Ras family of proteins associate with the actin cytoskeleton in ECs and are involved in thrombin-induced changes in endothelial permeability. Within this family, RhoA diminishes barrier function through its effects on myosin light chains (146) while Rac1 improves barrier function by stabilizing adherens junctions (61, 147, 327). Along these lines, silencing or pharmacological inhibition of the downstream effector molecule, ROCK1, prevented F-actin organization induced by in vivo agonism of the thrombin receptor, but had no effect on hyperpermeability (45). Rather, ROCK2 activation was established as the main mediator of pulmonary edema, via regulation of basal tension and cell-cell adhesion. Interestingly, dual ROCK1/ROCK2 inhibition was additive. These results challenge the notion that formation of actin stress fibers is sufficient for reducing barrier function and suggest that F-fiber formation only increases permeability when cell junctions are weakened. The MAP kinase pathway has also been implicated in exerting control over actin and adherens junction organization (44,321,327).
Other kinase signaling cascades, including members of the Src family kinases (SFKs), have been shown to be activated as part of the global increase in kinase/phosphatase ratio that occurs following an increase in ROS. As such, various extracellular mediators known to increase ROS, including TNFα and thrombin, activate SFK members (273). SFK have been subcellularly localized near cell junctions, and interact with virtually all of the major components of the junctional/contractile apparatus (272). In particular, SFKs have been shown to directly phosphorylate adherens junction targets, including VE-cadherin, and facilitate adherens junction disassembly (207). SFKs can also interact with components of transcellular flux pathways such as caveolin-1 (272). While SFKs are known to be expressed in lung ECs (273) and phosphorylate targets critical to barrier function in various types of ECs in vitro (727), the role of specific SFK members in regulating macro- and microvascular EC function and their participation in mechanisms mediating barrier dysfunction and lung injury are largely not understood.
Finally, NO is another signaling molecule that has been studied intensely as a regulator of endothelial barrier function. The effect of NO on the microvasculature is complex and not fully understood, with lines of evidence supporting both barrier protective and barrier disruptive behavior (149,726). For instance, cGMP may offer protection from oxidative stress in microvascular ECs (598) and ROS-induced endothelial barrier dysfunction in vitro (427), yet NO-induced elevations in cGMP levels have been implicated in permeability increases following high tidal volume ventilator injury in mice (551). The reasons for these discordant results is unclear; however, emerging evidence suggests that subcompartmentalization of messengers like cGMP, cAMP, kinases and eNOS (541) may allow variable effects on barrier function based on subcellular localization. Lipid raft domains may serve as platforms for aggregation of several culprit second messenger proteins such as various protein kinases, tyrosine kinases, receptors such as VEGR and TRPs and other Ca2+ channels, allowing for rapid, localized signal transduction to induce rises in intracellular Ca2+. Interactions between signaling pathways may also complicate the issue, as the cGMP-dependent injury observed with high tidal volume was coupled with increased hydrolysis of cAMP (551), which offers barrier protective effects when localized near the plasma membrane (548,646).
For most of the pathways outlined earlier, the effect of various second messaging systems on barrier function likely varies as a function of injury type (e.g., ROS vs. shear stress-induced endothelial injury) as well as the endothelial phenotype under study (macrovascular vs. microvascular). Moreover, other experiment-dependent characteristics, such as species used, the presence of flow and in vivo versus in vitro preparations may also have an impact.
Targets of barrier disruption signaling
As touched upon in the preceding section, activation of the multiple kinase families, intracellular Ca2+ and other messenger systems ultimately converge on several key effector proteins that control endothelial barrier function. These include: (i) the contractile apparatus (Table 1), which is comprised of actin filaments, phosphorylated myosin light chains, and various associated proteins which control actin fiber polymerization, rearrangement, degradation and actin-myosin binding (146); (ii) members of the adherens junctions, encompassing VE-cadherin, catenins, and other adaptor molecules that connect the junctions to the contractile apparatus; (iii) components of focal adhesions such as integrins, which tether the cell to the underlying matrix; and (iv) pathways of transcellular fluid transport, controlled primarily via interactions between second messenger kinases and caveolin-1 (303).
Table 1.
Proteins Involved in Regulating Endothelial Cell Barrier Function [Adapted from Dudek and Garcia (146)]
| Protein | Function |
|---|---|
| Spectrin | Cross-links F actin at periphery and stimulates myosin II ATPase |
| α-Actinin | Links actin cytoskeleton to focal adhesions; displacement from actin stress fibers disrupts the microfilaments |
| Fimbrin | Links actin cytoskeleton to vimentin network at cell adhesion sites |
| Cortactin | Phosphorylation by p60src reduces actin bundling activity |
| Cofilin | Rho pathway inhibits depolymerization activity during stress fiber formation |
| hsp27 | Phosphorylation by p38/MAPKAP induces stress fiber formation |
| VASP | Stimulates actin nucleation and polymerization at focal adhesions |
| Arp2/3 | Produces branching actin network at cell periphery through interaction with Wiskott-Aldrich syndrome protein and cortactin |
| Profilin | Overexpression inhibits actin stress fiber formation |
| Gelsolin | Inhibition of activity decreases stress fiber-dependent contraction |
| MLCK | Activation produces stress fibers, cellular contraction, EC permeability |
| Filamin | Phosphorylation by CaMKII permits reorganization of cortical actin |
| Caldesmon | Facilitates actomyosin interaction |
| Vinculin | Binds actin and catenin at junctional sites |
Abbreviations: hsp27, heat shock protein 27; VASP, vasodilator-stimulated phosphoprotein; MLCK, myosin light chain kinase; CAMKII, Ca2+/calmodulin-dependent protein kinase II; MAPKAP, MAP kinase-activated protein kinase 2.
Of all of these systems, the cytoskeleton plays a critical role in regulating barrier strength. Complete reviews of the role of the cytoskeleton in regulating permeability have been published (146, 651). Adherens junctions, in particular, are directly linked to cytoskeletal elements, like actin, such that actin/myosin contraction “tugs” on the adherens junction and VE-cadherin pairs from adjacent cells to dissociate them from each other. Thus, the major regulation of junction integrity occurs either at the level of expression of adherens junction proteins (i.e., assembly or disassembly of VE-Cadherin on the cell membrane) or at the level of contractile apparatus attachment (146). Actin filament formation and degradation is controlled by a family of proteins (i.e., cofilin) and these proteins then serve as targets for kinase systems as a means of regulating paracellular barrier function. Other pathways that are independent of actin and myosin light chain interaction have been identified as well (646), including pathways that utilize nonactin components of the cytoskeleton, such as intermediate filaments and microtubules, although these mechanisms are less well understood (146,632). Lastly, while paracellular leak is thought to be the primary mechanism driving increases in endothelial permeability, the transcellular pathway has been shown to be important in maintaining basal barrier homeostasis as well contributing (albeit to a smaller extent) to lung injury. Transcellular permeability involves caveolae-mediated transport of molecules like albumin and, like paracellular mechanisms, is tightly regulated by multiple families of kinases, including Src kinases (272).
It is clear that under pathological conditions, numerous stimuli can act at a variety of points to modify EC behavior and promote EC barrier dysregulation. In addition to investigation of signaling pathways that worsen barrier function, there has been recent interest in upstream proteins that exert barrier protective effects, such as S1P and angiopoietins. Interactions between S1P and one of its receptors, S1P1R, are thought to strengthen the endothelial barrier through a variety of mechanisms, including stabilization of the adherens junction and rearrangement of the actin cytoskeleton in a protective configuration [reviewed in (452,640,668)]. Consistent with these suppositions, S1P administration attenuated LPS-induced increases in bronchoalveolar lavage fluid protein and Evans blue dye extravasation in a murine model of endotoxin-induced lung injury (493) and attenuated high tidal volume induced increases in bronchoalveolar lavage fluid protein in a canine model of lung injury (608). Interestingly, prolonged applications of S1P promoted barrier dysfunction, suggesting that S1P-S1P1R binding can have temporally different effects, perhaps due to receptor internalization following ligation (564). Unlike S1P1R, ligation of S1P2R has opposing effects, increasing vascular permeability in response to exogenous ROS (542). The exact effects of the other members of the S1P receptor family on vascular permeability remain incompletely understood.
Ultimately, understanding the pathways involved in the pathogenesis of edema in patients with acute lung injury and ARDS is required to develop therapeutics to reduce mortality. Whether pharmacological targeting to disrupt pathways that have already been identified to be involved in reducing barrier integrity, or to enhance pathways that may improve barrier function, can be translated to decrease mortality associated with ARDS remains to be determined.
Pulmonary Hypertension
Pulmonary hypertension (PH) is a complex, progressive, and ultimately fatal condition arising from a variety of etiologies. Defined by the hemodynamic criteria of mean PPA ≥ 25 mmHg at rest, PH has been classified into five major categories: (i) pulmonary arterial hypertension (PAH), including idiopathic, heritable and drug/toxin-induced PH; (ii) PH due to left heart disease; (iii) PH due to interstitial lung diseases and/or hypoxia; (iv) chronic thromboembolic PH; and (v) PH with unclear and/or multifactorial origin, including hematologic and systemic disorders (27,197). Due to the complicated nature of the disease, the exact pathogenesis of PH is poorly understood; however, genetic mutations have been identified as an underlying cause in some cases of idiopathic and familial PAH, while global alveolar hypoxia is thought to be a causal factor in PH due to high altitude exposure, sleep-disordered breathing, and chronic lung diseases. In all cases, however, owing to increased afterload, right ventricular hypertrophy becomes a serious complication with the potential for ensuing right ventricular failure.
While the exact causes of PH remain under investigation, and are likely to vary with the underlying pathogenic or genetic cause, it is widely recognized that pathophysiology results from sustained active vasoconstriction and pulmonary vascular remodeling. Dysfunction of the pulmonary endothelium, which is a rich source of both vasodilators and vasoconstrictors, is a likely contributing factor. In the normal lung, the balance of vasodilators to vasoconstrictors favors low PVR. However, with EC dysfunction, release of NO and PGI2 is impaired while release of ET-1 and thromboxane may be augmented. A shift in the balance between endogenous vasodilators and vasoconstrictors combined with remodeling of pulmonary blood vessels results in a net vasoconstriction and increased vascular resistance.
The remodeling of the pulmonary vasculature in PH is characterized to varying degrees by thickening of the intimal and/or medial layer of muscular vessels and the appearance of cells expressing smooth muscle specific markers in precapillary arterioles (distal muscularization), resulting from excessive proliferation and migration of PASMCs (567, 596, 650). In some forms of severe PAH, increased medial muscularity is coupled with the development of occlusive lesions, involving PASMCs, ECs and possibly cells of nonvascular origin (597, 637). Reductions in arteriolar caliber, whether from vasoconstriction or occlusion, clearly exert the greatest influence on PVR; however, decreased compliance (i.e., increased stiffness) in the elastic proximal pulmonary arteries may also increase right ventricular afterload (199,394,596,647).
Depending on the underlying cause, the relative contributions of vasoreactivity and vascular remodeling to elevated PPA vary. Initially, all forms of PH were thought to arise from “fixed” constriction due to inward remodeling and narrowing of the vascular lumen. Over the last 15 years, however, accumulating evidence suggests a large portion of the “fixed” component was actually due to incomplete relaxation and that remodeling resulting in luminal encroachment is less prevalent (286, 410, 595), restricted primarily to severe forms of PAH. This is particularly true in the case of hypoxia-induced PH, where medial remodeling was shown to occur in an outward manner, and likely contributes to elevated PVR via hyperreactivity to constricting agents. Several recent reviews have provided detailed descriptions of the evidence for remodeling in various forms of PH, as well as in depth coverage of the potential cellular mechanisms (508, 549, 567, 595, 597). In the following section, we will briefly review the current knowledge and highlight recent advances in identifying some of the factors involved in both the contractile and remodeling process.
Pulmonary hypertension in humans
In humans, vasoconstriction is relatively easy to identify in PH with administration of vasodilators, including Ca2+ channel blockers, inhaled NO, prostacyclin or ROCK inhibitors (597, 627). In contrast, most evidence for remodeling comes from postmortem or postoperative tissue specimens. For example, examination of lung tissue from PAH patients revealed early abnormalities that included medial hypertrophy, adventitial thickening, and extension of muscle down the vascular tree, with vaso-occlusive lesions developing at later stages (596, 637). Similarly, COPD patients with PH (31, 697, 703) and South Americans living at altitude (22, 253, 446, 447, 492) also exhibited arteriolar muscularization, increased intimal and/or medial thickness and reduced lumen area.
While development of PH is common in individuals that are born at sea level and subsequently move to high altitude, several populations have been identified that have evolutionarily adapted to living at altitude, including highlander populations in the South American Andes, Tibet and the East African Plateau. All reside at high altitude without developing substantial PH, yet genetic diversity appears to underlie the adaptation achieved by these populations, as can be inferred from the differences in both phenotype and genotype. For example, Tibetans and Andeans differ substantially in resting ventilation, hypoxic ventilatory response, oxygen saturation, and hemoglobin concentration [reviewed in (40,41)]. In contrast to both the Tibetans and Andeans, Ethiopians have high arterial oxygen saturations at altitude (43). While data is not available from the African population, the native high-altitude-adapted population of Tibet does not have increased pulmonary vascular smooth muscle (235,242), while healthy Andeans exhibit some remodeling (22).
Given that Tibetans have resided at high altitude longer than any other population, the difference in vascular response may be due to variations in genetic adaptation. Indeed, a loss-of-function mutation in the Hif2a gene was identified in Tibetans and found to correlate with reduced PPA (42,58,583,644,716). Conversely, genetic mutations leading to HIF-2α gain of functionality are associated with development of PH in humans (179, 195) and in mice (619). Finally, individuals with Chuvash polycythemia, a rare congenital genetic disorder resulting in hypomorphic alleles for the gene encoding von Hippel-Lindau protein, a main component in the HIF degradation pathway, exhibit elevated HIF levels, increased pulmonary vascular sensitivity to hypoxia and susceptibility to developing PH at sea level (85,585).
Genetics also play a role in some cases of PAH. Initial discovery of a gene locus at chromosome 2q31–32 (435,459) led to the subsequent identification of bone morphogenetic protein receptor II (BMPR2) as the gene of interest (138, 290). BMPR2 is a serine/threonine kinase receptor that binds members of the TGF-β superfamily, including BMPs. BMP ligands bind type-II receptors (BMPR2 and Act2A and 2B), which recruit type-I receptors (ALK2, 3, and 6) to form a heterodimeric complex whereby type-II receptors induce type-1 receptor phosphorylation, leading to initiation of intracellular signaling cascades that ultimately lead to activation of nuclear transcription factors to upregulate or suppress target genes (577).
Mutations in the gene encoding BMPR2 occur in ~70% of patients with heritable PAH (10,112,113,138,385,628) and ~25% of idiopathic PAH patients (154). Although the number of unique BMPR2 mutations identified in patients approaches 300, all result in loss of protein function, encompassing insufficient protein production, dysfunctional protein trafficking and localization, or abnormal protein signaling (126). Interestingly, the penetrance of the BMPR2 mutation is low with an estimated lifetime risk of 20% (343), suggesting a “second hit” may be required for disease manifestation. Factors influencing development of PAH in those with BMPR2 mutations include female gender (343). Additional genes that may be associated with susceptibility include ALK-1, SMAD9, ENG, BMPR1B (ALK6), CAV1, KCNK3, and EIF1AK4 [reviewed in (25,384)].
Pulmonary hypertension in animal models
To better understand functional and structural changes in the hypertensive lung and to begin to identify underlying causes, several animal models of PH have been utilized (685). Unfortunately, no single model perfectly replicates human PAH. Nevertheless, these models provide opportunities for studying the development, progression and mechanistic underpinnings of PH and evaluating potential therapeutic treatments.
Hypoxia
In 1915, altitude was identified as a primary cause of lower chest edema, cardiac dilation and enlargement, and eventual heart failure in cattle living in the Colorado mountains (>8000 ft) (221). This recognition provided the first animal model of hypoxic PH, which has been used to explore the pulmonary and cardiac changes induced by exposure to chronic hypoxia. In later studies, extensive vascular remodeling was reported, characterized by collagen deposition, adventitial and medial thickening, and arteriolar muscularization (527, 594, 596). As with humans, there is variability in the development of hypoxic PH in cattle, which may have a genetic basis (575, 681, 698). Recently, SNP analysis of four candidate genes in cattle with severe hypoxic PH failed to uncover obvious SNPs associated with high-altitude susceptibility, although SNPs in additional candidate genes were identified and will require further investigation (456). Moreover, genetic comparison between altitude-susceptible and -resistant cattle revealed alterations in several gene networks whose biological function involve signaling pathways known to contribute to human PH, including BMP, TGF-β, VEGF, ERK/MAPK, NF-kB, and HIF signaling (456). The extent to which each of the identified pathways confers susceptibility to hypoxia-induced PH in these animals remains to be determined, but it should be noted that all of the pathways identified have been implicated in humans and other species.
Rodents exposed to CH develop predictable and reproducible vasoconstriction and vascular remodeling reminiscent of humans with hypoxia-associated PH. Rats exposed to several weeks of simulated high-altitude exhibit substantial increases in PPA and right ventricular mass, accompanied by increased vascular wall thickness, due to PASMC hypertrophy and hyperplasia, and distal muscularization (513,596). While imaging studies initially appeared to suggest that remodeling in these models reduced luminal diameter and resulted in rarefaction, or vascular pruning (263, 512), later observations showing that remodeling in this model appears to occur in an outward manner (271) and that administration of ROCK inhibitors acutely normalized PPA (448), suggest that the imaging results may have reflected incomplete reversal of vasoconstriction or variable perfusion (50,406,595). Mice also develop PH upon exposure to CH, although PASMC proliferation is minimal (38, 470, 596) and remodeling is most often observed as muscularization of distal vessels and thickening and functional stiffening of proximal, conduit arteries (593,594,596). Across species, the phenotype observed with chronic hypoxia exposure is consistent, with lack of occlusive lesions and reversal of PPA with return to normoxic conditions.
Other models in which hypoxia plays a contributing factor have been developed, including the Fawn-hooded rat (FHR) and the Sugen/hypoxia (Su/Hx) model. FHRs spontaneously develop PH with age, with mild CH accelerating the process (450, 544). In this model, extension of muscle into peripheral vessels and medial hypertrophy of proximal arteries was observed, as was reduced vessel filling in imaging studies (544), but whether this reflected development of vaso-occlusive lesions, which have not been reported, or severe vasoconstriction (595) is unclear. In light of findings that acute administration of ROCK inhibitors resulted in near normalization of PPA, it would appear that the latter is more likely. In the Su/Hx model, a single dose of the VEGF receptor inhibitor, SU5416, is followed by exposure to CH (3,621). VEGF receptor inhibition initially causes EC apoptosis; subsequent hypoxic exposure may stimulate proliferation of a subset of ECs that are apoptosis-resistant, causing occlusive lesions reminiscent of PAH in humans and PASMC proliferation. PH induced in the Su/Hx model is irreversible even after return to normoxia, and ROCK inhibitors only partially reversed PPA (633), suggesting a significant portion of the increased PVR may be due to remodeling and occlusion of flow in small vessels. Interestingly, a detailed temporal characterization of the Su/Hx model revealed that while vaso-occlusive lesions are present early (2-week posthypoxia) in the course of the Su/Hx model (621,633) concentric neointimal, or plexiform, lesions did not form until later (i.e., 8-to 13-week posthypoxia) time points (3). These results would suggest that the plexiform lesions observed in PAH patients, while perhaps contributing to progression/severity of the rise in PPA in late disease, may in fact be a consequence of EC damage during PH rather than the inciting cause. The Su/Hx model has been adapted to mice, which exhibited increased arterial muscularization and collagen deposition in the media and adventitia, vaso-occlusive lesions and ultimately right heart dysfunction; however, in contrast to rats, mice required weekly injections of SU5416 and PH resolved upon return to normoxia (110).
Monocrotaline
Following ingestion of seeds from Crotalaria spectabilis, monocrotaline (MCT) is metabolized in the liver into MCT pyrrole, a toxic substance that causes inflammation and endothelial injury. In rats, a single injection of MCT results in the development of PH within 2 to 3 weeks (317). The MCT model is widely used due to the ease of inducing severe PH without requiring specialized equipment or housing as is needed with hypoxia models. Histological analysis has shown that while smooth muscle content increases in arteries of all sizes, occlusive lesions do not develop (416). However, when MCT-induced wall injury/EC dysfunction is combined with pneumonectomy, intimal changes occur, including development of distal vascular occlusive lesions (467, 620). Despite substantial remodeling, ROCK inhibitors acutely normalize PPA in MCT-treated rats (448), suggesting that sustained contraction is a major component of PH in this model. Unlike human disease, the fact that >30 agents effectively reverse MCT-induced PH (596) raises questions as to the relevance of the MCT model.
Genetically engineered murine models
With advances in knowledge regarding some of the underlying causes of human PH, attempts have been made to recapitulate the disease using genetically engineered mice. For example, it is now clear that 5-HT was involved in the development of PAH associated with use of the diet medications, aminorex or fenfluramines, which functioned to increase 5-HT availability. Under normal conditions, the vast majority (>99%) of 5-HT is stored in platelets and rapid metabolism of 5-HT results in near negligible plasma levels (390). With elevations in plasma 5-HT, activation of cell surface receptors can mediate pulmonary vasoconstriction (386,389,429), or 5-HT can be taken up into SMCs. Receptor engagement and/or internalization and receptor-independent signaling results in generation of ROS, activation of MAPKs and ROCK, and induction of genes involved in regulating cell growth (390). In PASMCs, 5-HT influx and efflux is controlled by membrane SERTs (390), the expression of which are upregulated in PASMCs from PAH patients (390), and increased 5-HT transport into PASMCs enhances, and SERT inhibition prevents, proliferation (150, 387). To mimic the enhanced SERT expression observed in PAH patients, mice overexpressing the gene for human SERT were generated, with SERT overexpression inducing medial thickening and distal muscularization in normoxic female, but not male, mice (390, 690), mimicking the disproportionate female predominance seen in human PAH. On the other hand, male mice deficient in tryptophan hydroxylase 1, the enzyme responsible for peripheral 5-HT synthesis, were resistant to hypoxia-induced PH (428). While increasing 5-HT internalization via SERT over-expression is sufficient to induce ROCK-dependent remodeling in mice (397), coordination between SERT and 5-HT receptor binding may also occur. For example, inhibiting 5-HT1B receptors by silencing or pharmacological blockade reduced PASMC proliferation and CH-induced remodeling (390,430), responses mediated by SERT (150,387).
Another model of PAH generated based on data from patients is a murine model with loss-of-function mutations in BMPR2. Transgenic mice with global heterozygous BMPR2 mutations exhibit variable PH morphology, with some, but not all, developing increased wall thickness compared to wild type mice (49). Subjecting these mice to hypoxia for 3 weeks had no effect on right ventricular pressure (182,588), whereas extending hypoxic exposure to 5 weeks resulted in mild disease presentation (182), suggesting that the mutation alone may not be sufficient to generate PH. When BMPR2 loss-of-function was restricted to smooth muscle, mice developed PH with increased medial thickness and muscularization of distal pulmonary arteries, but no intimal lesions (684). Clearly, these models present complex morphologies and the results are consistent with the fact that only ~20% of individuals with heterozygous BMPR2 mutations develop PAH, suggesting a second genetic or environmental “hit” may be required (457).
Factors involved in the pathogenesis of pulmonary hypertension
The animal models described above have been utilized extensively to identify mechanisms responsible for development of PH and test potential treatments. Given the multiple classifications of human PH, none of the preclinical models perfectly captures the pulmonary-specific characteristics of severe human disease; however, these models have provided valuable insight into the cellular modifications and signaling pathways contributing to disease pathogenesis in PH, with the majority of work centered on PASMCs and ECs (Fig. 8). Detailed reviews regarding the cellular mechanisms involved in the pathogenesis of vasoconstriction and vascular remodeling in PH have been published (549,567,593,596). In this section, we will briefly review the most well-established candidates.
Figure 8.

Illustration of putative mechanisms involved in the pathogenesis of pulmonary hypertension. Abbreviations: 5-HT, 5-hydroxytryptamin; K- and Ca-channels, potassium and calcium channels; AEC, alveolar epithelial cells; BMP, bone morphogenetic protein; cGMP, cyclic guanosine monophosphate; ECM, extracellular matrix; EGF, epidermal growth factor; EPC, endothelial progenitor cells; HIF, hypoxia inducible factor; MMPs, matrix metalloproteinases; NADPH, nicotinamide adenine dinucleotide phosphate; NO, nitric oxide; PDE, phosphodiesterase; PDGF, platelet-derived growth factor; PGI2, prostaglandin I2; Rho-Ki, Rho kinases; ROS, reactive oxygen species; sGC, soluble guanylate cyclase; TGF, transforming growth factor-β; TK, tyrosine kinase; TKi, tyrosine kinase inhibitor; TRPC, transient receptor potential cation channels; VEGF, vascular endothelial growth factor. Reproduced, with permission, from Schermuly et al. (549).
Ion channels
In chronic hypoxia models, the earliest study examining functional changes in PASMCs revealed depolarization of the membrane potential (605), a finding confirmed in other species [reviewed in (569)]. Reduced K+ channel expression and activity were subsequently identified as contributing factors to depolarization in hypoxic PH (81, 419, 569, 584, 662). Experimental manipulations that augmented PASMC K+ channel expression/activity reduced remodeling and reduced right ventricular systolic pressure in hypoxic animals (419, 504). The initial reports of downregulated K+ channels and membrane depolarization were hypothesized to drive activation of voltage-gated Ca2+ channels and Ca2+ influx, leading to elevated [Ca2+]i. Increased [Ca2+]i results in enhanced PASMC contraction (572), growth (223,342,426) and migration (348) and has been documented in cells from hypoxic animals (572). However, inhibitors of voltage-gated Ca2+ channels have little effect on PASMCs from hypoxic animals (355,571,666), but can be activated by agonists (259,382) and contribute to stimulated proliferation (533). Thus, voltage-gated Ca2+ channels may participate in the remodeling process in PH, particularly in the presence of excessive growth factors.
It is now well established that sustained Ca2+ influx is mediated primarily by upregulation of NSCCs (565, 569), which are composed of TRPC proteins (355, 666). Unlike voltage-gated Ca2+ channels, NSCCs are not activated by depolarization, but can be modulated by phosphorylation, receptor activation or store depletion (260, 464, 665). Increased abundance of TRPC proteins was observed in PASMCs derived from animals subjected to chronic hypoxia (355,666). Decreasing the activity of NSCCs, either pharmacologically or by RNA silencing, reduced [Ca2+]i (355,666).
Consistent with results obtained in the chronic hypoxia model, PASMCs from humans with PAH are also depolarized (725) and exhibit reduced K+ channel expression and activity (73, 730). Similar reductions in K+ channel activity were reported in the FHR model (72). Augmenting PASMC K+ channel expression/activity increased apoptosis in PASMCs from PAH patients (73). Thus, modulating K+ channel activity modifies PASMC growth; likely by conferring resistance to apoptosis (81,526).
Increased [Ca2+]i also has been documented in PASMCs from MCT-treated rats (365) and PAH patients (223, 587). This is consistent with findings demonstrating increased abundance of TRPC proteins in PASMCs derived from PAH patients (339,723). Moreover, a gain-of-function single nucleotide polymorphism in TRPC6 was recently identified in patients with PAH (724).
Na+/H+ exchange
The Na+/H+ exchanger (NHE) is a major contributor to maintenance of PASMC pH homeostasis (391, 511). Increased NHE activity was associated with growth factor-induced proliferation (509) suggesting a potential role for NHE in PH. Indeed, exposure to CH increased expression of NHE isoform 1 (NHE1) in PASMCs and NHE activity causes an alkaline shift in pH (531,566), while pharmacological inhibition (510) or genetic deletion of NHE1 decreased hypoxia-induced PH, vascular remodeling and PASMC proliferation and migration (720, 721). These data provided firm evidence that NHE1 plays an important role in the pathogenesis of PH, although the exact mechanism by which this occurs is unclear. Recent studies have provided tantalizing clues, suggesting that loss of NHE1 increased p27, a cyclin-dependent kinase inhibitor, and decreased activation of ROCK and E2F1, a nuclear transcription factor that controls proliferation (720, 721). These results suggest NHE1 represses a growth inhibitor pathway while stimulating proliferation.
Rho kinase
Even under basal conditions, ROCK appears to be activated and provides a minimal amount of basal tone in the lung, as demonstrated by reduced PPA when ROCK inhibitors are infused (92). With exposure to hypoxia, the ROCK system is upregulated (78, 300), and contributes to elevated PVR. Indeed, the ROCK signaling pathway plays a central role in mediating vasoconstriction in all PH models [reviewed in (466, 671)] and is implicated in the remodeling process (167, 465). ROCK activation is necessary for PASMC migration and proliferation (212,366,465), and while acute administration of ROCK inhibitors markedly reduces right ventricular systolic pressure in most preclinical models (448, 449, 647), prolonged inhibition of ROCK reduces vascular remodeling in CH mice (160, 286). At least part of the mechanism by which ROCK modulates PASMC function occurs via cytoskeletal rearrangement in PASMCs and ECs, with RhoB contributing to hypoxia-induced migration and proliferation in both cell types (702). ROCK also converges with other mechanisms involved in remodeling, as ROCK activation is a consequence of several growth factors (546), opens Ca2+ channels (382,665), and stimulates the activity of NHE1 (642).
BMP signaling
BMPs signal through activation of type-I and type-II receptors, activating SMAD-dependent and -independent pathways [reviewed in (577)]. Interactions with type-I receptors activate SMAD1/5/8, which subsequently bind SMAD4, leading to activation of transcriptional responses (377); whereas SMAD-independent signaling involves MAP kinases, phosphatidylinositol 3-kinase/AKT and PKC (713). In PH, the ligands BMP2, −4, and −7 and the receptors, BMPR1 and BMPR2 have been most studied. BMP2 and BMP7 appear to play protective roles in the pulmonary circulation (434); indeed, application of BMP2 exerted an antiproliferative effect on PASMCs, and loss of BMP2 resulted in greater susceptibility to hypoxia-induced PH (17). BMP4 also exerts a growth inhibiting effect in PASMCs from proximal vessels (434), but in PASMCs from distal vessels BMP4 increased [Ca2+]i via TRPC upregulation (378) and induced proliferation (714). Moreover, mice with partial deficiency for BMP4 were protected against development of hypoxia-induced PH (181,182). In addition to BMP2, −4 and −7, the BMP family contains a variety of endogenous antagonists and binding proteins which may regulate BMP receptor binding and activity in vivo (377,577).
In PAH patients, BMPR2 mutations lead to loss of BMP signaling, preventing the antiproliferative effects of BMP2 (126). Expression of both BMPR1 and BMPR2 is decreased in non-PAH forms of the disease (145) and in CH models (612), suggesting abnormal BMP signaling may be common in many forms of PH. On ECs, activation of BMPR2 by BMP2 or BMP4 was linked to NO production (201), while loss of BMPR2 results in apoptosis (625). Since EC apoptosis and/or dysfunctional production of vasodilators are likely early events in the pathogenesis of PAH, these results may explain some of the protective effects of BMPs in vivo.
Endothelin-1
A role for ET-1 in mediating PH has been suspected nearly since the discovery of the protein. Indeed, early on it was recognized that circulating ET-1 levels were increased in all animal models and human forms of PH (563, 570). In addition to vasoconstriction, ET-1 exerts promigration and proliferation actions on vascular SMCs (563). Moreover, ET receptor inhibitors prevented and reversed PH and vascular remodeling in several animal models [reviewed in (125)], and reduced proliferation in PASMCs from PAH patients (339), paving the way for use of ET receptor inhibitors in clinical practice. ET receptor inhibitors improve exercise capacity and survival (125), but whether this is due to reduced vasoconstriction or remodeling, or other effects of the drugs, is unclear.
Transcription factors
Two major transcription factors have been implicated in the development of PH: HIFs and nuclear factor of activated T-cells (NFATs). Increases in [Ca2+]i activate several downstream signal transduction pathways and transcription factors are activated that could be involved in PASMC proliferation [reviewed in (336)], in particular, NFAT. In turn, NFAT reduces K+ channel expression and increases proliferation (73), linking alterations in [Ca2+]i to dysregulated K+ channel expression/activity and PASMC growth. Consistent with this possibility, genetic deletion or pharmacological inhibition of NFAT prevented and/or reversed hypoxia-induced remodeling and PH (57,73).
The discovery of HIF-1 (560) provided an attractive candidate for mediating hypoxia-induced PH. As noted earlier, HIF-1β is constitutively expressed whereas HIF-1α is typically not detectable under normoxic conditions. Initial studies in mice heterozygous for a null HIF-1α allele (Hif1a+/−) revealed a role for HIF-1 in the development of hypoxia-induced PH (568, 719), with Hif1a+/− mice exhibiting impaired development of PH and reduced vascular remodeling in response to CH (719). PASMCs from Hif1a+/− mice also exhibited reduced hypoxia-induced proliferation (565). Consistent with the results obtained in Hif1a+/− mice, mice with homozygous, conditional deletion of HIF-1α in smooth muscle during exposure to chronic hypoxia exhibited attenuated development of hypoxia-induced pulmonary vascular remodeling and PH (29). Similar attenuation in CH-induced PH and vascular remodeling was observed in Hif2a+/− mice (80).
HIFs are likely to be involved in numerous mechanisms that mediate PH. In addition to regulating both Ca2+ and pH homeostasis (566, 666), HIFs also regulate other factors involved in the pathogenesis of PH, including ET-1 and VEGF [reviewed in (505)]. While hypoxia is an obvious stimulant for HIF activation, HIFs also can be upregulated under nonhypoxic conditions by a number of factors (505), including ET-1 (497). Moreover, HIF-1α protein is detectable in PASMCs from FHRs (72) and lungs of PAH patients (18, 165), which is thought to be linked to mitochondrial dysfunction. Upregulation of HIF under nonhypoxic conditions could also be due to ROS generated from NADPH oxidase and activation of mTOR, both of which have been observed in cells from PAH patients (225) and lead to induction of prosurvival pathways (226,231,379).
Dysregulated cell metabolism
In recent years, the role of metabolic reprogramming has emerged as a potential mechanism underlying cellular responses during PH. PASMCs from Fawn-hooded (18), chronically hypoxic and Su/Hx rats (105) and PAH patients (704) exhibit a phenotypic switch such that instead of mitochondrial metabolism, these cells preferentially use aerobic glycolysis. This type of bioenergetic shift confers a survival/growth advantage since glucose/glycolytic intermediates are substrates utilized by the pentose phosphate shunt to generate reduced nicotinamide adenine dinucleotide phosphate and nucleotides. The mechanisms underlying the switch to aerobic glycolysis may be related to overactivation of glucose-6-phosphate dehydrogenase (105) and HIF-dependent upregulation of glycolytic (710) and pro-proliferative (105,106) genes. Additionally, hypoxia-induced upregulation of glucose-6-phosphate dehydrogenase activity was correlated with reduced expression of contractile proteins in PASMCs (106), suggesting that metabolic derangements may underlie the phenotypic switch from a “contractile” to “synthetic” SMC phenotype. Activation of HIF-1 was also associated with reductions in mitochondrial number in PAH ECs (165), while mitochondrial fragmentation was observed in PAH PASMCs (72). In PASMCs from PAH patients and hypoxia- and MCT-treated rats, HIF activation was associated with mitochondrial fission mediated by dynamin related protein-1 (400), which was reduced when dynamin-related protein-1 was inhibited with the peptide Mdiv-1. Mdiv-1 also attenuated PH and proliferation (400). Consistent with the metabolic derangements observed in cultured cells and animal models, in vivo imaging studies using 18-fluorodeoxyglucose in PAH patients verified increased glucose uptake in the lungs (383,704).
The immune system
It is now recognized that in addition to, or because of, genetic influences and shifts in cell metabolism, inflammation is a contributing factor in the development of PH. While certain autoimmune diseases (i.e., systemic sclerosis and systemic lupus erythematous) and infections (i.e., HIV and schistosomiasis) are known causes of PAH, examination of patient specimens has revealed perivascular inflammation in almost all forms of PH [(638) and reviewed in (151,507,514)]. Infiltrating immune cells, including T-cells, B-cells, macrophages, dendritic cells, and mast cells have been observed in the vascular lesions of PAH patients and substantial evidence has now accumulated demonstrating a critical role for macrophages (184, 630, 648) and mast cells (30, 121, 266) in promoting vascular dysfunction in animal models of PH. High levels of procontractile and pro-proliferative cytokines are observed in both patients and animal models of PH [reviewed in (151, 507, 514)], providing a link between inflammation and the vascular hallmarks of PH. In addition, circulating anti-endothelial (141, 615), anti-fibroblast (616) and anti-nuclear (528) autoantibodies have also been reported in PAH patients, although the extent to which these autoantibodies initiate or facilitate progression of disease is still under investigation.
Surprisingly, athymic rats given an injection of SU5416, which were hypothesized to exhibit less inflammation and thus diminished PH, instead developed severe PH in the absence of hypoxic stimulus and worse inflammation (622). Subsequent studies demonstrated that loss of regulatory T-cells, which limit perivascular inflammation and vascular injury, accounted for the exaggerated phenotype in the athymic rats (617). Similarly, hypoxia was not required to induce severe PH in rats given SU5416 following sensitization with ovalbumin (425), suggesting that an allergic inflammatory phenotype can facilitate development of PH.
Therapeutics for pulmonary hypertension
Work from preclinical models allowed the identification of individual modifiers of PH and pulmonary vascular remodeling and has provided most of the rationale for the development of therapies aimed at human disease, such as PGI2 and endothelin receptor blockade. While these studies lead to the introduction of several clinically useful therapies for PH, all current therapies are aimed at reducing vasoconstriction or slowing the progression of vascular remodeling. There are currently no therapeutic options available as a means to deremodel the pulmonary vasculature after the disease is already established. While PGI2 and ET-1 receptor blockers have been used clinically for over a decade (180), recent drugs added to the armamentarium include the phosphodiesterase type-V inhibitor, sildenafil, and the HMG-CoA reductase inhibitor, simvastatin (314). Both drugs were originally marketed for treatment of other disorders (erectile dysfunction and dyslipidemia, respectively), but based on their pharmacological actions were hypothesized to provide benefit by targeting abnormal signaling pathways in PAH. In the case of sildenafil, increasing cGMP could provide vasodilatory and anti-proliferative actions, while statins have multiple effects on endothelial function and may inhibit ROCK. In vitro experiments revealed that sildenafil restored BMP signaling in PAH PASMCs (711), whereas simvastatin increased BMPR2 expression in ECs (274), suggesting the possibility that these drugs might rescue BMP function in vivo. Sildenafil also reduced hypoxia-induced increases in [Ca2+]i and TRPC expression in PASMCs (667), attenuated PASMC proliferation (689,711) and attenuated PH and vascular remodeling in preclinical models (239,711). Sildenafil is now routinely used clinically as an add-on therapy in the PH population. Promising trials using the next generation long-acting PDE5-specific inhibitor, tadalafil (196, 469), and an activator of sGC, riociguat (69,213,214,537,582), and subsequent recent approval of these drugs for clinical use demonstrate that the cGMP pathway remains a prime target in PAH.
Simvastatin also prevented PH in preclinical models (215, 314, 733), but had variable success in reversal protocols (216, 411, 623). Unfortunately, clinical trials in the PH population have failed to find sustained improvement in PH (316, 696). Among the potential therapies suggested by data from animal models, restoration of K+ channel activity/expression provides an attractive target for therapeutics aimed at reducing remodeling in PAH and may be the closest to actual clinical use. Animal studies using dehydroepiandrosterone, a steroid hormone that, among other actions, opens K+ channels (71) or dichloroacetate, a metabolic modulator that also increases the expression of K+ channels (412, 419), showed promising outcomes in CH and MCT-treated animals. In the Su/Hx model, dehydroepiandrosterone attenuated PH and preserved RV contractile function via reduced RV oxidative stress, capillary rarefaction, apoptosis, autophagy and fibrosis (14, 105, 521). While it is unclear whether the beneficial effects of either drug were specifically due to actions on K+ channels, the use of dichloroacetate to treat PAH is currently in Phase 1 clinical trials in Canada and a small pilot study found dehydroepiandrosterone reduced mean PPA and PVR and improved 6-min walk distances in COPD patients with PH (148).
In contrast to K+ channels, little has been pursued with respect to other ion channels, exchangers, and PH therapeutics. Selective inhibitors for NSCCs have not been tested in PH, and given the widespread distribution of these channels; side-effects are likely to be significant. A similar problem arises with therapies aimed at targeting NHE1. Selective inhibitors of NHE1 have been developed, originally for treatment of myocardial infarction, but are associated with major side effects, including cerebrovascular events (414, 441). Given the wide distribution of TRPCs and NHE1 throughout the body, it is likely that inhibitors of these targets may only be useful in the treatment of PAH if cell-specific targeting could be achieved.
There was considerable excitement for the potential for administration of ROCK inhibitors, given that these agents reduced PPA and PVR in PH models and PAH patients (187, 291). Unfortunately, substantial systemic hypotension accompanies intravenous delivery, limiting their use. Inhaled formulations of ROCK inhibitors can circumvent the systemic side effects (448), and were effective in acutely reducing PVR in PAH patients (186). Chronic ROCK inhibition reduced PH and remodeling in animal models (2, 160, 286) suggesting the possibility that in addition to acute vasodilator effects, prolonged use could slow and/or reverse remodeling in the patient population. Interestingly, sildenafil and statins can inhibit ROCK (216, 239), although whether the clinical dosing of these drugs inhibits ROCK activation in patients is unclear.
Studies targeting HIF activity using the pharmacologic inhibitors, digoxin or acriflavine, demonstrated reduced hypoxia-induced PH and vascular remodeling in rodents (6). Moreover, when treatment was initiated after PH was established, digoxin could slow the progression of PH (6), providing evidence supporting a therapeutic potential for drugs that block or reduce HIF in the treatment of PH. Finally, given the known role of BMPR2 dysfunction in many PAH cases, identifying means of increasing BMPR2 expression/activity in these patients remains an active area of investigation. A recent screen of clinical compounds identified low concentrations of FK506 as activators of BMPR2 signaling (591). FK506, a calcineurin inhibitor, displaces FK-binding protein-12 from type-I BMP receptors, enhancing BMP signaling. Currently in phase II trials, preliminary reports of compassionate use of FK506 in 3 advanced PAH patients revealed improvements in symptoms and clinical outcomes (590). While these initial reports are highly encouraging, whether this approach will be successful in a large population or in patients with loss-of-function BMPR2 mutations remains to be seen. Other studies in preclinical models targeting the BMPR signaling axis demonstrated that administration of low concentrations of BMP9 increased BMPR2 expression and selectively enhanced BMPR signaling in the endothelium (369). These early studies suggest that therapeutics aimed at modulating BMP signaling may be a promising strategy for treating PAH.
Pediatric pulmonary hypertension
As discussed in earlier sections, soon after birth, PPA normally falls to levels comparable to adult values. Similar to adults, in children (term babies at sea level >3 months old), PH is diagnosed when the mean PPA exceeds 25 mmHg in the presence of an equal distribution of blood flow to all segments of both lungs (292). While PH in children is relatively rare, persistent pulmonary hypertension of the newborn (PPHN) is more common, with an incidence of approximately 2 per 1000 live births (660). PPHN results from failure of the perinatal circulatory to transition after birth, and is characterized primarily by sustained elevation of PVR; resulting in decreased perfusion of the pulmonary circulation and continued right-to-left shunting of blood through the foramen ovale and ductus arteriosus. Common causes of PPHN include: (i) abnormal constriction of the pulmonary vasculature secondary to meconium aspiration, respiratory distress syndrome, and sepsis; (ii) structural abnormality of the vasculature due to failed remodeling after birth or congenital malformations; and (iii) bronchopulmonary dysplasia (501).
Hypoxia is a common consequence of PPHN. The majority of work addressing the effects of chronic hypoxia on the pulmonary circulation has been performed in adult animals, even though increased PPA, remodeling and contraction also occur in the pediatric population. It is widely recognized that regulation of PPA is influenced by postnatal age and it is unclear which mechanisms identified in the adult translate to the neonate. A recent review provided an in depth dissection of the role of hypoxia in PPHN, and the mediators and cellular mechanisms involved (203).
Summary
Regardless of inciting cause, PH is clearly a complex disorder in humans. Preclinical models have aided in understanding the remodeling that occurs, identifying cellular mechanisms involved, and investigating potential therapies. While progress has been made, much is yet to be learned regarding the mechanisms underlying the phenotypic changes in PASMCs and ECs. Unfortunately, none of the currently available treatments for PAH are curative and are often accompanied by negative side-effects or inconvenient routes of administration. Recent data from preclinical models has pointed to several promising potential therapies (224,508), including dichloroacetate, ROCK, NFAT, and HIF inhibitors. Clinical trials are needed to assess whether targeting any of these pathways will be beneficial in the patient population, and further investigation to identify additional pathways involved in the pathogenesis of PH will be required to develop new pharmacological agents aimed at targeting and reversing PH.
Bronchial Circulation
Although most agree that by the 1600s it was recognized that the lung had a second circulation, attributing the discovery of the bronchial circulation to a particular individual has been controversial, ranging from Galen to Domenici de Marchettis, to Leonardo da Vinci (99, 120, 424). In 1721, Frederich Ruysch was the first to definitively describe the bronchial circulation in detail (Fig. 9) and identify the bronchial artery originating from the aorta [reviewed in (424)]. Regardless of who initially described this circulation, it is now understood that the bronchial vasculature plays an important role in health and disease. The bronchial arteries supply oxygenated blood from the systemic circulation to the conducting airways, regional lymph nodes, nerves, walls of the pulmonary arteries and veins (vasa vasorum), and the visceral pleura, although additional functions of the bronchial circulation and its regulation in humans continue to be explored.
Figure 9.

Drawing by Fredrick Ruysch detailing the bronchial circulation. Reproduced, with permission, from Mitzner (424).
Development
The anatomy of the bronchial circulation varies substantially across species (172), with mice lacking a bronchial circulation altogether (423). Development of the human bronchial circulation occurs in two stages, with initial formation of arteries from the dorsal aorta in the fourth week of gestation. In the second trimester, these “primitive” arteries are subsequently replaced by bronchial arteries primarily arising from the superior thoracic aorta, (120, 353), although bronchial arteries either originating from, or communicating with, coronary arteries have been reported (322,374). Examination of human autopsy specimens revealed that in many, but not all, cases two bronchial arteries supply each lung (313, 353). Branching of the intrapulmonary bronchial arteries along the length of the bronchial tree down to the terminal bronchioles results in a vast network of capillaries, which form extensive anastomoses with other bronchial arteries as well as the pulmonary vasculature, creating a peribronchial plexus (120, 554). Arterioles arising from this plexus penetrate the muscular layer, forming a submucosal microvascular network (653). Venous drainage of the proximal conducting airway (first two divisions) follows the same pattern as the arterial supply, with blood being returned to the right heart via the azygos and hemiazygos veins.
“Bronchial” circulation may in fact be a bit of a misnomer, as in addition to providing systemic blood supply to the airways, these vessels also deliver systemic arterial blood to the walls of the pulmonary vessels, regional lymph nodes, nerves, and the visceral pleura (172, 405). Moreover, the distal bronchial microvasculature forms anastomoses with the pulmonary circulation. Due to these multiple connections with the pulmonary circulation, the deoxygenated bronchial venous blood drains via pulmonary veins to the left heart, producing an anatomic right to left shunt often referred to as the “bronchopulmonary anastomotic” flow or the “pulmonary collateral” circulation. While bronchopulmonary anastomoses can occur throughout the vasculature (101, 554, 656), it is now thought that drainage of bronchial venous blood is primarily a postpulmonary capillary event (482).
Determination of bronchial blood flow
The bronchial arteries are small in diameter (<1.5 mm at origin); consequently, the bronchial circulation is a high-resistance, low-capacitance system (95). The vast majority of the lung blood flow passes through the pulmonary circulation, with only a small percent carried by the bronchial circulation. Until recent years, measuring the exact blood flow carried by the bronchial circulation was a difficult task, given its low flow, small size vessels, and variable anatomic nature. Researchers relied on indicator-dilution techniques or radiolabeled microsphere tracers, both of which carried significant drawbacks (172), including the fact that these techniques measure total airway perfusion, not just that supplied by the bronchial arteries. With the use of invasive techniques, such as mounting a doppler ultrasound probe on the bronchial artery, approximations of bronchial artery blood flow were possible, with bronchial blood flow reported to be 13 to 50 mL/min in sheep (63, 100, 370). Despite the difficulties inherent in the microsphere technique, values obtained in piglets (42.1 ± 10.4 mL/min) were similar (550) to those measured using flow probes. At these levels of flow, under normal conditions bronchial flow accounts for approximately 1% to 5% of CO (370,393,550).
A number of factors can influence bronchial blood flow. Several studies have shown that flow in the bronchial arteries is increased during alveolar hypoxia (133, 673), consistent with the response observed in most systemic vascular beds. The mechanical effects of respiration also modulate flow in the bronchial circulation, with reductions in flow and increased draining via the pulmonary veins observed during lung inflation (28, 131). It is likely that these inspiration-induced alterations in bronchial flow are a consequence of changing lung volume and transpulmonary pressure compressing and/or stretching the systemic-to-pulmonary anastomotic bronchial blood vessels.
Various vasoactive factors have also been shown to alter the pattern of blood flow in the bronchial arteries. As might be expected from results in other systemic circulations, NO (100, 420) and PGI2 (132) have each been shown to increase the bronchial blood flow. Intravenous administration of acetylcholine also vasodilates the bronchial circulation (543, 558), as does histamine (334, 358, 370). Interestingly, ET-1 also dilated bronchial arteries when administered intravenously (402, 607), perhaps due to activation of endothelial ETB receptors.
Function of the bronchial circulation in the normal lung
A primary function of the bronchial circulation is the delivery of oxygen and nutrients to the airway wall. This becomes obvious when bronchial flow is disrupted, as can occur during lung transplant or when interventions are required for pulmonary atresia, ventricular septal defect, and major aortopulmonary collaterals. In such cases, necrosis of the airway mucosa was a frequent complication (451, 484, 555, 556). The bronchial circulation may also play an important role in acute airway responses and airway defense. Recruitment of inflammatory cells that participate in immune function to the airway mucosa also occurs via the bronchial circulation (354). Another key function of the bronchial circulation is airway clearance. Bronchial blood flow contributes to washout of bronchoconstrictors, proinflammatory mediators, and particulate matter from the airways (318, 654, 655). In instances where particulate matter causes pulmonary infarcts, the bronchial circulation also provides collateral blood flow to the lungs.
The bronchial circulation in disease
The bronchial circulation plays a substantial role in a variety of pathologic conditions, including hemoptysis, lung transplant and supplemental lung perfusion. The bronchial arteries possess a remarkable capacity for remodeling, exhibiting both enlargement and angiogenesis depending on the underlying cause. For example, infection is a common cause of bronchial arterial enlargement and increased blood flow, contributing to hemoptysis (468). Indeed, the bronchial circulation is the source of bronchial bleeding in up to 90% of patients (718). With recent development of sophisticated imaging techniques the anatomy of the bronchial arteries can be visualized (91) to accurately identify the enlargement of the bronchial arteries and the potential source of the bleed.
Remodeling of the bronchial circulation also occurs in conditions where pulmonary gas exchange is compromised, as in the case of pulmonary atresias or ventral septal defects (460). While the vast majority of gas exchange occurs in the pulmonary vasculature, the bronchial circulation can contribute to gas exchange through its anastomotic connections. In the absence of a normal pulmonary circulation, the bronchial arteries increase in number and caliber to provide the blood supply to the entire lungs (159, 333, 460, 489, 609, 649). Upon restoration of the pulmonary circulation, the bronchial blood flow returns to normal and the expanded vasculature partially regresses (159). While the exact cellular mechanisms responsible for this proliferation of vessels is still under investigation, it is clear that certain growth factors enhance the angiogenic capacity of the vessels, including VEGF (547) and stromal cell-derived factor-1 (301).
During transplantation, the pulmonary arterial, but not bronchial, circulation is surgically restored. Given that the bronchial circulation supplies approximately 50% of the airways wall perfusion (33) with the remainder coming from the pulmonary arteries, it is increasingly recognized that the bronchial circulation plays an important role in lung transplant and that loss of flow to the distal airways may result in further ischemia and hypoxic injury. In animal models, transplant with bronchial artery revascularization improved airway oxygenation compared to traditional transplant (311). It has been postulated that the loss of the bronchial circulation in lung transplant patients causes airway hypoxia, despite adequate pulmonary artery blood flow (140). In animal models, the bronchial artery circulation can be slowly restored following transplantation through the process of angiogenesis (488, 578); however, both imaging and autopsy specimens exhibit a lack of identifiable bronchial arteries and/or significant microvascular loss around airways within human transplanted lungs (140, 380). These results suggest that airway ischemia could contribute to airway fibrosis and poor outcomes with transplantation.
Conclusion
Over the past century, incredible progress has been made in understanding the structure and function of the lung circulations, spurred on by the development of advanced imaging techniques, the advent of the molecular biology revolution and availability of genetically altered animals. The adaptation of these techniques, and others, to the study of the pulmonary and bronchial vasculatures has provided a wealth of information, and while much has been learned, it is clear that there is still a great deal more to be understood. As investigators delve deeper into the cellular mechanisms that provide the basis of genetic and phenotypic changes in the lung circulations, and develop better models of pulmonary vascular disease, including lung injury and PH, undoubtedly new insights into both the physiology and factors contributing to pathology of the pulmonary circulation will be uncovered. Translation of these findings will potentially lead to better outcomes in disease states.
Acknowledgments
Work on this review was supported by NIH grants HL126514, HL124930, HL073859, and HL007534.
References
- 1.Aaronson PI, Robertson TP, Knock GA, Becker S, Lewis TH, Snetkov V, Ward JP. Hypoxic pulmonary vasoconstriction: mechanisms and controversies. J Physiol 570: 53–58, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abe K, Shimokawa H, Morikawa K, Uwatoku T, Oi K, Matsumoto Y, Hattori T, Nakashima Y, Kaibuchi K, Sueishi K, Takeshit A. Long-term treatment with a Rho-kinase inhibitor improves monocrotaline-induced fatal pulmonary hypertension in rats. Circ Res 94: 385–393, 2004. [DOI] [PubMed] [Google Scholar]
- 3.Abe K, Toba M, Alzoubi A, Ito M, Fagan KA, Cool CD, Voelkel NF, McMurtry IF, Oka M. Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation 121: 2747–2754, 2010. [DOI] [PubMed] [Google Scholar]
- 4.Abe T, Arakawa Y, Rajasekaran AK, Yu TH, Wada O. Interaction of atrial natriuretic peptide with its receptors in bovine lung membranes. J Biol Chem 270: 7672–7678, 1995. [DOI] [PubMed] [Google Scholar]
- 5.Abman SH, Accurso FJ. Acute effects of partial compression of ductus arteriosus on fetal pulmonary circulation. Am J Physiol 257: H626–H634, 1989. [DOI] [PubMed] [Google Scholar]
- 6.Abud EM, Maylor J, Undem C, Punjabi A, Zaiman AL, Myers AC, Sylvester JT, Semenza GL, Shimoda LA. Digoxin inhibits development of hypoxic pulmonary hypertension in mice. Proc Natl Acad Sci U S A 109: 1239–1244, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Adkison JB, Miller GT, Weber DS, Miyahara T, Ballard ST, Frost JR, Parker JC. Differential responses of pulmonary endothelial phenotypes to cyclical stretch. Microvasc Res 71: 175–184, 2006. [DOI] [PubMed] [Google Scholar]
- 8.Adnot S, Andrivet P, Chabrier PE, Piquet J, Plas P, Braquet P, Roudot-Thoraval F, Brun-Buisson C. Atrial natriuretic factor in chronic obstructive lung disease with pulmonary hypertension. Physiological correlates and response to peptide infusion. J Clin Invest 83: 986–993, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adnot S, Cigarini I, Herigault R, Harf A. Effects of substance P and calcitonin gene-related peptide on the pulmonary circulation. J Appl Physiol 70: 1707–1712, 1991. [DOI] [PubMed] [Google Scholar]
- 10.Aldred MA, Vijayakrishnan J, James V, Soubrier F, Gomez-Sanchez MA, Martensson G, Galie N, Manes A, Corris P, Simonneau G, Humbert M, Morrell NW, Trembath RC. BMPR2 gene rearrangements account for a significant proportion of mutations in familial and idiopathic pulmonary arterial hypertension. Hum Mutat 27: 212–213, 2006. [DOI] [PubMed] [Google Scholar]
- 11.Allen KM, Wharton J, Polak JM, Haworth SG. A study of nerves containing peptides in the pulmonary vasculature of healthy infants and children and of those with pulmonary hypertension. Brit Heart J 62: 353–360, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Altemeier WA, McKinney S, Krueger M, Glenny RW. Effect of posture on regional gas exchange in pigs. J Appl Physiol 97: 2104–2111, 2004. [DOI] [PubMed] [Google Scholar]
- 13.Alvarez DF, King JA, Weber D, Addison E, Liedtke W, Townsley MI. Transient receptor potential vanilloid 4-mediated disruption of the alveolar septal barrier: A novel mechanism of acute lung injury. Circ Res 99: 988, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alzoubi A, Toba M, Abe K, O’Neill KD, Rocic P, Fagan KA, McMurtry IF, Oka M. Dehydroepiandrosterone restores right ventricular structure and function in rats with severe pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol 304: H1708–H1718, 2013. [DOI] [PubMed] [Google Scholar]
- 15.Amis TC, Jones HA, Hughes JM. Effect of posture on inter-regional distribution of pulmonary perfusion and VA/Q ratios in man. Resp Physiol 56: 169–182, 1984. [DOI] [PubMed] [Google Scholar]
- 16.Anand-Srivastava MB, Trachte GJ. Atrial natriuretic factor receptors and signal transduction mechanisms. Pharmacol Rev 45: 455–497, 1993. [PubMed] [Google Scholar]
- 17.Anderson L, Lowery JW, Frank DB, Novitskaya T, Jones M, Mortlock DP, Chandler RL, de Caestecker MP. Bmp2 and Bmp4 exert opposing effects in hypoxic pulmonary hypertension. Am J Physiol Regul Integr Comp Physiol 298: R833–R842, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Archer SL, Gomberg-Maitland M, Maitland ML, Rich S, Garcia JG, Weir EK. Mitochondrial metabolism, redox signaling, and fusion: a mitochondria-ROS-HIF-1α-Kv1.5 O2-sensing pathway at the intersection of pulmonary hypertension and cancer. Am J Physiol Heart Circ Physiol 294: H570–H578, 2008. [DOI] [PubMed] [Google Scholar]
- 19.Archer SL, Huang J, Henry T, Peterson D, Weir EK. A redox-based O2 sensor in rat pulmonary vasculature. Circ Res 73: 1100–1112, 1993. [DOI] [PubMed] [Google Scholar]
- 20.Archer SL, Reeve HL, Michelakis E, Puttagunta L, Waite R, Nelson DP, Dinauer MC, Weir EK. O2 sensing is preserved in mice lacking the gp91 phox subunit of NADPH oxidase. Proc Nat Acad Sci 96: 7944–7949, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Archer SL, Tolins JP, Raij L, Weir EK. Hypoxic pulmonary vasoconstriction is enhanced by inhibition of the synthesis of an endothelium derived relaxing factor. Biochem Biophys Res Commun 164: 1198–1205, 1989. [DOI] [PubMed] [Google Scholar]
- 22.Arias-Stella J, Saldana M. The terminal portion of the pulmonary arterial tree in people native to high altitudes. Circulation 28: 915–925, 1963. [DOI] [PubMed] [Google Scholar]
- 23.Asikainen TM, Ahmad A, Schneider BK, White CW. Effect of preterm birth on hypoxia-inducible factors and vascular endothelial growth factor in primate lungs. Pediatric Pulm 40: 538–546, 2005. [DOI] [PubMed] [Google Scholar]
- 24.Atlas S, Maack T. Atrial natriuretic factor. Compr Physiol (Suppl 25): 1577–1673, 2011. [Google Scholar]
- 25.Austin ED, Loyd JE. The genetics of pulmonary arterial hypertension. Circ Res 115: 189–202, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bächler P, Pinochet N, Sotelo J, Crelier G, Irarrazaval P, Tejos C, Uribe SC. Assessment of normal flow patterns in the pulmonary circulation by using 4D magnetic resonance velocity mapping. Magn Reson Imaging 31: 178–188. [DOI] [PubMed] [Google Scholar]
- 27.Badesch DB, Champion HC, Sanchez MA, Hoeper MM, Loyd JE, Manes A, McGoon M, Naeije R, Olschewski H, Oudiz RJ, Torbicki A. Diagnosis and assessment of pulmonary arterial hypertension. J Am Col Cardiol 54: S55–S66, 2009. [DOI] [PubMed] [Google Scholar]
- 28.Baile EM. The anatomy and physiology of the bronchial circulation. J Aerosol Med 9: 1–6, 1996. [DOI] [PubMed] [Google Scholar]
- 29.Ball MK, Waypa GB, Mungai PT, Nielsen JM, Czech L, Dudley VJ, Beussink L, Dettman RW, Berkelhamer SK, Steinhorn RH, Shah SJ, Schumacker PT. Regulation of hypoxia-induced pulmonary hypertension by vascular smooth muscle HIF-1a. Am J Respir Crit Care Med 189: 314–324, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Banasova A, Maxova H, Hampl V, Vizek M, Povysilova V, Novotna J, Vajnerova O, Hnilickova O, Herget J. Prevention of mast cell degranulation by disodium cromoglycate attenuates the development of hypoxic pulmonary hypertension in rats exposed to chronic hypoxia. Respiration 76: 102–107, 2008. [DOI] [PubMed] [Google Scholar]
- 31.Barberà JA, Riverola A, Roca J, Ramirez J, Wagner PD, Ros D, Wiggs BR, Rodriguez-Roisin R. Pulmonary vascular abnormalities and ventilation-perfusion relationships in mild chronic obstructive pulmonary disease. Am J Resp Crit Care Med 149: 423–429, 1994. [DOI] [PubMed] [Google Scholar]
- 32.Barer GR. The effect of catecholamine blocking and depleting agents on the changes in the pulmonary circulation caused by hypoxia and hypercapnia. J Physiol 186: 97P–99P, 1966. [PubMed] [Google Scholar]
- 33.Barman SA, Ardell JL, Parker JC, Perry ML, Taylor AE. Pulmonary and systemic blood flow contributions to upper airways in canine lung. Am J Physiol 255: H1130–H1135, 1988. [DOI] [PubMed] [Google Scholar]
- 34.Barman SA, Pauly JR, Isales CM. Canine pulmonary vasoreactivity to serotonin: Role of protein kinase C and tyrosine kinase. Am J Physiol 272: H740–H747, 1997. [DOI] [PubMed] [Google Scholar]
- 35.Barnard JW, Wilson PS, Moore TM, Thompson WJ, Taylor AE. Effect of nitric oxide and cyclooxygenase products on vascular resistance in dog and rat lungs. J Appl Physiol 74: 2940–2948, 1993. [DOI] [PubMed] [Google Scholar]
- 36.Barnes PJ, Liu SF. Regulation of pulmonary vascular tone. Pharmacol Rev 47: 87–131, 1995. [PubMed] [Google Scholar]
- 37.Baudouin SV, Messent M, Evans TW. Effect of Intralipid on hypoxic and angiotensin-II induced pulmonary vasoconstriction in the isolated rat lung. Crit Care Med 22: 1964–1968, 1994. [PubMed] [Google Scholar]
- 38.Bauer NR, Moore TM, McMurtry IF. Rodent models of PAH: Are we there yet? Am J Physiol Lung Cell Mol Physiol 293: L580–L582, 2007. [DOI] [PubMed] [Google Scholar]
- 39.Baumgartner WA Jr., Jaryszak EM, Peterson AJ, Presson RG Jr., Wagner WW Jr. Heterogeneous capillary recruitment among adjoining alveoli. J Appl Physiol (1985) 95: 469–476, 2003. [DOI] [PubMed] [Google Scholar]
- 40.Beall CM. Tibetan and Andean contrasts in adaptation to high-altitude hypoxia. Adv Exp Med Biol 475: 63–74, 2000. [DOI] [PubMed] [Google Scholar]
- 41.Beall CM. Tibetan and Andean patterns of adaptation to high-altitude hypoxia. Human Biol 72: 201–228, 2000. [PubMed] [Google Scholar]
- 42.Beall CM, Cavalleri GL, Deng L, Elston RC, Gao Y, Knight J, Li C, Li JC, Liang Y, McCormack M, Montgomery HE, Pan H, Robbins PA, Shianna KV, Tam SC, Tsering N, Veeramah KR, Wang W, Wangdui P, Weale ME, Xu Y, Xu Z, Yang L, Zaman MJ, Zeng C, Zhang L, Zhang X, Zhaxi P, Zheng YT. Natural selection on EPAS1 (HIF2a) associated with low hemoglobin concentration in Tibetan highlanders. Proc Nat Acad Sci 107: 11459–11464, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Beall CM, Decker MJ, Brittenham GM, Kushner I, Gebremedhin A, Strohl KP. An Ethiopian pattern of human adaptation to high-altitude hypoxia. Proc Nat Acad Sci U S A 99: 17215–17218, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Becker PM, Verin AD, Booth MA, Liu F, Birukova A, Garcia JG. Differential regulation of diverse physiological responses to VEGF in pulmonary endothelial cells. Am J Physiol Lung Cell Mol Physiol 281: L1500–L1511, 2001. [DOI] [PubMed] [Google Scholar]
- 45.Beckers CM, Knezevic N, Valent ET, Tauseef M, Krishnan R, Rajendran K, Hardin CC, Aman J, van Bezu J, Sweetnam P, van Hinsbergh VW, Mehta D, van Nieuw Amerongen GP. ROCK2 primes the endothelium for vascular hyperpermeability responses by raising baseline junctional tension. Vascul Pharmacol 70: 45–54, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Behrman RE, Lees MH, Peterson EN, De Lannoy CW, Seeds AE. Distribution of the circulation in the normal and asphyxiated fetal primate. Am J Obstet Gyn 108: 956–969, 1970. [DOI] [PubMed] [Google Scholar]
- 47.Belik J The myogenic response of arterial vessels is increased in fetal pulmonary hypertension. Ped Res 37: 196–201, 1995. [DOI] [PubMed] [Google Scholar]
- 48.Benjamin JJ, Murtagh PS, Proctor DF, Menkes HA, Permutt S. Pulmonary vascular interdependence in excised dog lobes. J Appl Physiol 37: 887–894, 1974. [DOI] [PubMed] [Google Scholar]
- 49.Beppu H, Ichinose F, Kawai N, Jones RC, Yu PB, Zapol WM, Miyazono K, Li E, Bloch KD. BMPR-II heterozygous mice have mild pulmonary hypertension and an impaired pulmonary vascular remodeling response to prolonged hypoxia. Am J Physiol Lung Cell Mol Physiol 287:L1241–1247, 2004. [DOI] [PubMed] [Google Scholar]
- 50.Berg JT. Chronic hypoxia-induced pulmonary hypertension does/does not lead to loss of pulmonary vasculature. J Appl Physiol 103: 1455, 2007. [DOI] [PubMed] [Google Scholar]
- 51.Beutner A Ueber die strom-und druckkrafte des blutes in der arteria pulmonalis. Z Rationelle Med 2: 97–138, 1852. [Google Scholar]
- 52.Bhatt AJ, Amin SB, Chess PR, Watkins RH, Maniscalco WM. Expression of vascular endothelial growth factor and Flk-1 in developing and glucocorticoid-treated mouse lung. Ped Res 47: 606–613, 2000. [DOI] [PubMed] [Google Scholar]
- 53.Bhattacharya J, Matthay MA. Regulation and repair of the alveolar-capillary barrier in acute lung injury. Annu Rev Physiol 75: 593–615, 2013. [DOI] [PubMed] [Google Scholar]
- 54.Bhattacharya J, Nanjo S, Staub NC. Micropuncture measurement of lung microvascular pressure during 5-HT infusion. J Appl Physiol 52: 634–637, 1982. [DOI] [PubMed] [Google Scholar]
- 55.Bhattacharya JC. Interpreting the lung microvascular filtration coefficient. Am J Physiol Lung Cell Mol Physiol 293: L9–L10, 2007. [DOI] [PubMed] [Google Scholar]
- 56.Biaggioni I, King LS, Enayat N, Robertson D, Newman JH. Adenosine produces pulmonary vasoconstriction in sheep. Evidence for thromboxane A2/prostaglandin endoperoxide-receptor activation. Circ Res 65: 1516–1525, 1989. [DOI] [PubMed] [Google Scholar]
- 57.Bierer R, Nitta CH, Friedman J, Codianni S, de Frutos S, Dominguez Bautista JA, Howard TA, Resta TC, Bosc LV. NFATc3 is required for chronic hypoxia-induced pulmonary hypertension in adult and neonatal mice. Am J Physiol Lung Cell Mol Physiol 301: L872–L880, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bigham A, Bauchet M, Pinto D, Mao X, Akey JM, Mei R, Scherer SW, Julian CG, Wilson MJ, Lopez Herraez D, Brutsaert T, Parra EJ, Moore LG, Shriver MD. Identifying signatures of natural selection in Tibetan and Andean populations using dense genome scan data. PLoS genetics 6: e1001116, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Birks EK, Bousamra M, Presberg K, Marsh JA, Effros RM, Jacobs ER. Human pulmonary arteries dilate to 20-HETE, an endogenous eicosanoid of lung tissue. The Am J Physiol 272: L823–L829, 1997. [DOI] [PubMed] [Google Scholar]
- 60.Birukov KG, Zebda N, Birukova AA. Barrier enhancing signals in pulmonary edema. Compr Physiol 3: 429–484, 2013. [DOI] [PubMed] [Google Scholar]
- 61.Birukov KGC. Small GTPases in mechanosensitive regulation of endothelial barrier. Microvasc Res 77: 46–52, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bishop JM, Harris P, Segel N. The circulatory effects of bradykinin in normal subjects and patients with chronic bronchitis. Br J Pharmac Chemotherapy 25: 456–469, 1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bishop R, McLeod D, McIlveen S, Blake R, Gunther R, Davis J, Talken L, Cottee D, Quail A, Parsons G, White S. Effects of graded exercise on bronchial blood flow and airway dimensions in sheep. Pulm Pharmacol Thera 20: 178–189, 2007. [DOI] [PubMed] [Google Scholar]
- 64.Blitzer ML, Loh E, Roddy MA, Stamler JS, Creager MA. Endothelium-derived nitric oxide regulates systemic and pulmonary vascular resistance during acute hypoxia in humans. J Am Coll Cardiol 28: 591–596, 1996. [DOI] [PubMed] [Google Scholar]
- 65.Boe J, Simonsson BG. Adrenergic receptors and sympathetic agents in isolated human pulmonary arteries. Eur J Respir Dis 61: 195–202, 1980. [PubMed] [Google Scholar]
- 66.Bogeski I, Kappl R, Kummerow C, Gulaboski R, Hoth M, Niemeyer BA. Redox regulation of calcium ion channels: chemical and physiological aspects. Cell Calcium 50: 407–423, 2011. [DOI] [PubMed] [Google Scholar]
- 67.Bolton TB, Clapp LH. Endothelial-dependent relaxant actions of car-bachol and substance P in arterial smooth muscle. Br J Pharmacol 87: 713–723, 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bolz SS, Fisslthaler B, Pieperhoff S, De Wit C, Fleming I, Busse R, Pohl U. Antisense oligonucleotides against cytochrome P450 2C8 attenuate EDHF-mediated Ca2+ changes and dilation in isolated resistance arteries. Faseb J 14: 255–260, 2000. [DOI] [PubMed] [Google Scholar]
- 69.Bonderman D, Ghio S, Felix SB, Ghofrani HA, Michelakis E, Mitrovic V, Oudiz RJ, Boateng F, Scalise AV, Roessig L, Semigran MJ, Left Ventricular Systolic Dysfunction Associated With Pulmonary Hypertension Riociguat Trial Study Group. Riociguat for patients with pulmonary hypertension caused by systolic left ventricular dysfunction: a phase IIb double-blind, randomized, placebo-controlled, dose-ranging hemodynamic study. Circulation 128: 502–511, 2013. [DOI] [PubMed] [Google Scholar]
- 70.Bonnet S, Belus A, Hyvelin JM, Roux E, Marthan R, Savineau JP. Effect of chronic hypoxia on agonist-induced tone and calcium signaling in rat pulmonary artery. Am J Physiol Lung Cell Mol Physiol 281: L193–L201, 2001. [DOI] [PubMed] [Google Scholar]
- 71.Bonnet S, Dumas-de-La-Roque E, Begueret H, Marthan R, Fayon M, Dos Santos P, Savineau JP, Baulieu EE. Dehydroepiandrosterone (DHEA) prevents and reverses chronic hypoxic pulmonary hypertension. Proc Natl Acad Sci U S A 100: 9488–9493, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bonnet S, Michelakis ED, Porter CJ, Andrade-Navarro MA, Thebaud B, Bonnet S, Haromy A, Harry G, Moudgil R, McMurtry MS, Weir EK, Archer SL. An abnormal mitochondrial-hypoxia inducible factor-1a-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: Similarities to human pulmonary arterial hypertension. Circulation 113: 2630–2641, 2006. [DOI] [PubMed] [Google Scholar]
- 73.Bonnet S, Rochefort G, Sutendra G, Archer SL, Haromy A, Webster L, Hashimoto K, Bonnet SN, Michelakis ED. The nuclear factor of activated T cells in pulmonary arterial hypertension can be therapeutically targeted. Proc Natl Acad Sci U S A 104: 11418–11423, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bouwmeester JC, Belenkie I, Shrive NG, Tyberg JV. Partitioning pulmonary vascular resistance using the reservoir-wave model. J Appl Physiol 115: 1838–1845, 2013. [DOI] [PubMed] [Google Scholar]
- 75.Breuer J, Meschig R, Breuer HW, Arnold G. Effects of serotonin on the cardiopulmonary circulatory system with and without 5-HT2-receptor blockade by ketanserin. J Cardiovasc Pharmacol 7(Suppl 7): S64–S66, 1985. [DOI] [PubMed] [Google Scholar]
- 76.Brimioulle S, Vachiery JL, Brichant JF, Delcroix M, Lejeune P, Naeije R. Sympathetic modulation of hypoxic pulmonary vasoconstriction in intact dogs. Cardiovasc Res 34: 384–392, 1997. [DOI] [PubMed] [Google Scholar]
- 77.Broadley KJ, Maddock HL. P1-purinoceptor-mediated vasodilatation and vasoconstriction in hypoxia. J Auton Pharmacol 16: 363–366, 1996. [DOI] [PubMed] [Google Scholar]
- 78.Broughton BR, Walker BR, Resta TC. Chronic hypoxia induces Rho kinase-dependent myogenic tone in small pulmonary arteries. Am J Physiol Lung Cell Mol Physiol 294: L797–L806, 2008. [DOI] [PubMed] [Google Scholar]
- 79.Brower R, Wise RA, Hassapoyannes C, Bromberger-Barnea B, Permutt S. Effect of lung inflation on lung blood volume and pulmonary venous flow. J Appl Physiol 58: 954–963, 1985. [DOI] [PubMed] [Google Scholar]
- 80.Brusselmans K, Compernolle V, Tjwa M, Wiesener MS, Maxwell PH, Collen D, Carmeliet P. Heterozygous deficiency of hypoxia-inducible factor-2α protects mice against pulmonary hypertension and right ventricular dysfunction during prolonged hypoxia. J Clin Invest 111: 1519–1527, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Burg ED, Remillard CV, Yuan JX. Potassium channels in the regulation of pulmonary artery smooth muscle cell proliferation and apoptosis: Pharmacotherapeutic implications. Br J Pharmacol 153(Suppl 1): S99–S111, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Burnstock G Purinergic cotransmission. F1000 Biol Rep 1: 46, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Burri PH. Structural aspects of postnatal lung development - alveolar formation and growth. Biol Neonate 89: 313–322, 2006. [DOI] [PubMed] [Google Scholar]
- 84.Burri PH, Hlushchuk R, Djonov V. Intussusceptive angiogenesis: Its emergence, its characteristics, and its significance. Dev Dynamics 231: 474–488, 2004. [DOI] [PubMed] [Google Scholar]
- 85.Bushuev VI, Miasnikova GY, Sergueeva AI, Polyakova LA, Okhotin D, Gaskin PR, Debebe Z, Nekhai S, Castro OL, Prchal JT, Gordeuk VR. Endothelin-1, vascular endothelial growth factor and systolic pulmonary artery pressure in patients with Chuvash polycythemia. Haematologica 91: 744–749, 2006. [PubMed] [Google Scholar]
- 86.Campbell AG, Dawes GS, Fishman AP, Hyman AI, Perks AM. The release of a bradykinin-like pulmonary vasodilator substance in foetal and new-born lambs. J Physiol 195: 83–96, 1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cargill RI, Lipworth BJ. Acute effects of hypoxaemia and angiotensin II in the human pulmonary vascular bed. Pulm Pharmacol 7: 305–310, 1994. [DOI] [PubMed] [Google Scholar]
- 88.Carlsson P, Mahlapuu M. Forkhead transcription factors: Key players in development and metabolism. Developmental biology 250: 1–23, 2002. [DOI] [PubMed] [Google Scholar]
- 89.Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C, Declercq C, Pawling J, Moons L, Collen D, Risau W, Nagy A. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature 380: 435–439, 1996. [DOI] [PubMed] [Google Scholar]
- 90.Carvajal JA, Germain AM, Huidobro-Toro JP, Weiner CP. Molecular mechanism of cGMP-mediated smooth muscle relaxation. J Cell Physiol 184: 409–420, 2000. [DOI] [PubMed] [Google Scholar]
- 91.Carvalho P, Anderson DK, Charan NB. Bronchial arterial imaging using helical computed tomography. Pulm Pharmacol Ther 20: 104–108, 2007. [DOI] [PubMed] [Google Scholar]
- 92.Casey DB, Badejo AM, Dhaliwal JS, Sikora JL, Fokin A, Golwala NH, Greco AJ, Murthy SN, Nossaman BD, Hyman AL, Kadowitz PJ. Analysis of responses to the Rho-kinase inhibitor Y-27632 in the pulmonary and systemic vascular bed of the rat. Am J Physiol Heart Circ Physiol 299: H184–H192, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cassin S The role of eicosanoids and endothelium-dependent factors in regulation of the fetal pulmonary circulation. J Lipid Med 6: 477–485, 1993. [PubMed] [Google Scholar]
- 94.Catravas JD, Buccafusco JJ, El Kashef H. Effects of acetylcholine in the pulmonary circulation of rabbits. J Pharmacol Exp Ther 231: 236–241, 1984. [PubMed] [Google Scholar]
- 95.Cauldwell EW, Siekert RG, Lininger RE, Anson BJ. The bronchial arteries; an anatomic study of 150 human cadavers. Surg Gynecol Obstet 86: 395–412, 1948. [PubMed] [Google Scholar]
- 96.Cech S Adrenergic innervation of blood vessels in the lung of some mammals. Acta Anatomica 74: 169–182, 1969. [PubMed] [Google Scholar]
- 97.Chabot F, Mitchell JA, Gutteridge JM, Evans TW. Reactive oxygen species in acute lung injury. Eur Resp J 11: 745, 1998. [PubMed] [Google Scholar]
- 98.Chandel NS, Schumacker PT. Cellular oxygen sensing by mitochondria: Old questions, new insight. J Appl Physiol 88: 1880–1889, 2000. [DOI] [PubMed] [Google Scholar]
- 99.Charan NB, Carvalho P, Agostoni PG. On the purported discovery of the bronchial circulation by Leonardo da Vinci: A rebuttal. J Appl Physiol 76: 1836–1838, 1994. [DOI] [PubMed] [Google Scholar]
- 100.Charan NB, Johnson SR, Lakshminarayan S, Thompson WH, Carvalho P. Nitric oxide and beta-adrenergic agonist-induced bronchial arterial vasodilation. J Appl Physiol 82: 686–692, 1997. [DOI] [PubMed] [Google Scholar]
- 101.Charan NB, Turk GM, Dhand R. Gross and subgross anatomy of bronchial circulation in sheep. J Appl Physiol 57: 658–664, 1984. [DOI] [PubMed] [Google Scholar]
- 102.Chen G, Yamamoto Y, Miwa K, Suzuki H. Hyperpolarization of arterial smooth muscle induced by endothelial humoral substances. Am J Physiol: Heart Circ Physiol 260: H1888–H1892, 1991. [DOI] [PubMed] [Google Scholar]
- 103.Cheng DY, DeWitt BJ, Suzuki F, Neely CF, Kadowitz PJ. Adenosine A1 and A2 receptors mediate tone-dependent responses in feline pulmonary vascular bed. Am J Physiol: Heart Circ Physiol 270: H200–H207, 1996. [DOI] [PubMed] [Google Scholar]
- 104.Chetham PM, Babal P, Bridges JP, Moore TM, Stevens T. Segmental regulation of pulmonary vascular permeability by store-operated Ca2+ entry. Am J Physiol 276: L41–L50, 1999. [DOI] [PubMed] [Google Scholar]
- 105.Chettimada S, Gupte R, Rawat D, Gebb SA, McMurtry IF, Gupte SA. Hypoxia-induced glucose-6-phosphate dehydrogenase overexpression and -activation in pulmonary artery smooth muscle cells: Implication in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 308: L287–L300, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chettimada S, Rawat DK, Dey N, Kobelja R, Simms Z, Wolin MS, Lincoln TM, Gupte SA. Glc-6-PD and PKG contribute to hypoxia-induced decrease in smooth muscle cell contractile phenotype proteins in pulmonary artery. Am J Physiol Lung Cell Mol Physiol 303: L64–L74, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chi JT, Chang HY, Haraldsen G, Jahnsen FL, Troyanskaya OG, Chang DS, Wang Z, Rockson SG, van de Rijn M, Botstein D, Brown PO. Endothelial cell diversity revealed by global expression profiling. Proc Natl Acad Sci U S A 100: 10623–10628, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cigarini I, Adnot S, Chabrier PE, Viossat I, Braquet P, Gaujour B. Pulmonary vasodilator responses to atrial natriuretic factor and sodium nitroprusside. J Appl Physiol 67: 2269–2275, 1989. [DOI] [PubMed] [Google Scholar]
- 109.Cioffi DL, Lowe K, Alvarez DF, Barry C, Stevens T. TRPing on the lung endothelium: calcium channels that regulate barrier function. Antioxidants Redox Signal 11: 765, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ciuclan L, Bonneau O, Hussey M, Duggan N, Holmes AM, Good R, Stringer R, Jones P, Morrell NW, Jarai G, Walker C, Westwick J, Thomas M. A novel murine model of severe pulmonary arterial hypertension. Am J Resp Crit Care Med 184: 1171–1182, 2012. [DOI] [PubMed] [Google Scholar]
- 111.Clark J, Alvarez DF, Alexeyev M, King JA, Huang L, Yoder MC, Stevens T. Regulatory role for nucleosome assembly protein-1 in the proliferative and vasculogenic phenotype of pulmonary endothelium. Am J Physiol Lung Cell Mol Physiol 294: L431–L439, 2008. [DOI] [PubMed] [Google Scholar]
- 112.Cogan JD, Pauciulo MW, Batchman AP, Prince MA, Robbins IM, Hedges LK, Stanton KC, Wheeler LA, Phillips JA III, Loyd JE, Nichols WC. High frequency of BMPR2 exonic deletions/duplications in familial pulmonary arterial hypertension. Am J Resp Crit Care Med 174: 590–598, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cogan JD, Vnencak-Jones CL, Phillips JA III, Lane KB, Wheeler LA, Robbins IM, Garrison G, Hedges LK, Loyd JE. Gross BMPR2 gene rearrangements constitute a new cause for primary pulmonary hypertension. Genet Med 7: 169–174, 2005. [DOI] [PubMed] [Google Scholar]
- 114.Cogolludo A, Moreno L, Bosca L, Tamargo J, Perez-Vizcaino F. Thromboxane A2-induced inhibition of voltage-gated K+ channels and pulmonary vasoconstriction: Role of protein kinase Czeta. Circ Res 93: 656–663, 2003. [DOI] [PubMed] [Google Scholar]
- 115.Cohen ED, Tian Y, Morrisey EE. Wnt signaling: An essential regulator of cardiovascular differentiation, morphogenesis and progenitor self-renewal. Development 135: 789–798, 2008. [DOI] [PubMed] [Google Scholar]
- 116.Colen KL, Crisera CA, Rose MI, Connelly PR, Longaker MT, Gittes GK. Vascular development in the mouse embryonic pancreas and lung. J Pediat Surg 34: 781–785, 1999. [DOI] [PubMed] [Google Scholar]
- 117.Compernolle V, Brusselmans K, Franco D, Moorman A, Dewerchin M, Collen D, Carmeliet P. Cardia bifida, defective heart development and abnormal neural crest migration in embryos lacking hypoxia-inducible factor-1α. Cardiovas Res 60: 569–579, 2003. [DOI] [PubMed] [Google Scholar]
- 118.Conhaim RL, Staub NC. Reflection spectrophotometric measurement of O2 uptake in pulmonary arterioles of cats. J Appl Physiol 48: 848–856, 1980. [DOI] [PubMed] [Google Scholar]
- 119.Cosby K, Partovi KS, Crawford JH, Patel RP, Reiter CD, Martyr S, Yang BK, Waclawiw MA, Zalos G, Xu X, Huang KT, Shields H, Kim-Shapiro DB, Schechter AN, Cannon RO III, Gladwin MT. Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat Med 9: 1498–1505, 2003. [DOI] [PubMed] [Google Scholar]
- 120.Cudkowicz L, Armstrong JB. Observations on the normal anatomy of the bronchial arteries. Thorax 6: 343–358, 1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Dahal BK, Kosanovic D, Kaulen C, Cornitescu T, Savai R, Hoffmann J, Reiss I, Ghofrani HA, Weissmann N, Kuebler WM, Seeger W, Grimminger F, Schermuly RT. Involvement of mast cells in monocrotaline-induced pulmonary hypertension in rats. Respir Res 12: 60, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Dallal MM, Chang SW. Evans blue dye in the assessment of permeability-surface area product in perfused rat lungs. J Appl Physiol 77: 1030–1035, 1994. [DOI] [PubMed] [Google Scholar]
- 123.Daly ID, Michel CC, Ramsay DJ, Waaler BA. Conditions governing the pulmonary vascular response to ventilation hypoxia and hypoxaemia in the dog. J Physiol 196: 351–379, 1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Davenport AP, O’Reilly G, Kuc RE. Endothelin ETA and ETB mRNA and receptors expressed by smooth muscle in the human vasculature: Majority of the ETA sub-type. Br J Pharmacol 114: 1110–1116, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Davie NJ, Schermuly RT, Weissmann N, Grimminger F, Ghofrani HA. The science of endothelin-1 and endothelin receptor antagonists in the management of pulmonary arterial hypertension: current understanding and future studies. Eur J Clin Invest 39(Suppl 2): 38–49, 2009. [DOI] [PubMed] [Google Scholar]
- 126.Davies RJ, Morrell NW. Molecular mechanisms of pulmonary arterial hypertension: Role of mutations in the bone morphogenetic protein type II receptor. Chest 134: 1271–1277, 2008. [DOI] [PubMed] [Google Scholar]
- 127.Davis S, Yancopoulos GD. The angiopoietins: Yin and Yang in angiogenesis. Curr Top Microbiol Immunol 237: 173–185, 1999. [DOI] [PubMed] [Google Scholar]
- 128.Dawes GS, Mott JC. The vascular tone of the foetal lung. J Physiol 164: 465–477, 1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Dawson CA. Role of pulmonary vasomotion in physiology of the lung. Physiol Rev 64: 544–616, 1984. [DOI] [PubMed] [Google Scholar]
- 130.DebRoy A, Vogel SM, Soni D, Sundivakkam PC, Malik AB, Tiruppathi C. Cooperative signaling via transcription factors NF-κB and AP1/c-Fos mediates endothelial cell STIM1 expression and hyperpermeability in response to endotoxin. J Biol Chem 289(35): 24188–24201, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Deffebach ME. Lung mechanical effects on the bronchial circulation. Eur Resp J Supplement 12: 586s–590s, 1990. [PubMed] [Google Scholar]
- 132.Deffebach ME, Agostoni P, Kirk W, Lakshminarayan S. Pulmonary artery infusion of prostacyclin increases lobar bronchial blood flow. Resp Physiol 77: 147–156, 1989. [DOI] [PubMed] [Google Scholar]
- 133.Deffebach ME, Charan NB, Lakshminarayan S, Butler J. The bronchial circulation. Small, but a vital attribute of the lung. Am Rev Resp Dis 135: 463–481, 1987. [DOI] [PubMed] [Google Scholar]
- 134.Del Vecchio PJ, Siflinger-Birnboim A, Belloni PN, Holleran LA, Lum H, Malik AB. Culture and characterization of pulmonary microvascular endothelial cells. In Vitro Cell Dev Biol 28A: 711–715, 1992. [DOI] [PubMed] [Google Scholar]
- 135.Delaney C, Gien J, Grover TR, Roe G, Abman SH. Pulmonary vascular effects of serotonin and selective serotonin reuptake inhibitors in the late-gestation ovine fetus. Am J Physiol Lung Cell Mol Physiol 301: L937–L944, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.deMello DE, Reid LM. Embryonic and early fetal development of human lung vasculature and its functional implications. Pediat Dev Path 3: 439–449, 2000. [DOI] [PubMed] [Google Scholar]
- 137.deMello DE, Sawyer D, Galvin N, Reid LM. Early fetal development of lung vasculature. Am J Resp Cell Mol Bio 16: 568–581, 1997. [DOI] [PubMed] [Google Scholar]
- 138.Deng Z, Morse JH, Slager SL, Cuervo N, Moore KJ, Venetos G, Kalachikov S, Cayanis E, Fischer SG, Barst RJ, Hodge SE, Knowles JA. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet 67: 737–744, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Dhaliwal JS, Casey DB, Greco AJ, Badejo AM, Gallen TB, Murthy SN, Nossaman BD, Hyman AL, Kadowitz PJ. Rho kinase and Ca2+ entry mediate increased pulmonary and systemic vascular resistance in L-NAME-treated rats. Am J Physiol Lung Cell Mol Physiol 293: L1306–L1313, 2007. [DOI] [PubMed] [Google Scholar]
- 140.Dhillon GS, Zamora MR, Roos JE, Sheahan D, Sista RR, Van der Starre P, Weill D, Nicolls MR. Lung transplant airway hypoxia: A diathesis to fibrosis? Am J Resp Crit Care Med 182: 230–236, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Dib H, Tamby MC, Bussone G, Regent A, Berezne A, Lafine C, Broussard C, Simonneau G, Guillevin L, Witko-Sarsat V, Humbert M, Mouthon L. Targets of anti-endothelial cell antibodies in pulmonary hypertension and scleroderma. Eur Respir J 39: 1405–1414, 2012. [DOI] [PubMed] [Google Scholar]
- 142.Dickson MC, Martin JS, Cousins FM, Kulkarni AB, Karlsson S, Akhurst RJ. Defective haematopoiesis and vasculogenesis in transforming growth factor-b 1 knock out mice. Development 121: 1845–1854, 1995. [DOI] [PubMed] [Google Scholar]
- 143.Domkowski PW, Cockerham JT, Kot PA, Myers JL, Wallace RB, Hopkins RA. The role of N omega-nitro-L-arginine in modulation of pulmonary vascular tone in the maturing newborn pig. J Thorac Cardiovasc Surg 110: 1486–1492, 1995. [DOI] [PubMed] [Google Scholar]
- 144.Du HL, Yamada Y, Orii R, Suzuki S, Sawamura S, Suwa K, Hanaoka K. Beneficial effects of the prone position on the incidence of barotrauma in oleic acid-induced lung injury under continuous positive pressure ventilation. Acta Anaesthesiol Scand 41: 701–707, 1997. [DOI] [PubMed] [Google Scholar]
- 145.Du L, Sullivan CC, Chu D, Cho AJ, Kido M, Wolf PL, Yuan JX, Deutsch R, Jamieson SW, Thistlethwaite PA. Signaling molecules in nonfamilial pulmonary hypertension. N Engl J Med 348: 500–509, 2003. [DOI] [PubMed] [Google Scholar]
- 146.Dudek SM, Garcia JG. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol 91: 1487–1500, 2001. [DOI] [PubMed] [Google Scholar]
- 147.Duluc L, Wojciak-Stothard B. Rho GTPases in the regulation of pulmonary vascular barrier function. Cell Tissue Res 355: 675–685, 2014. [DOI] [PubMed] [Google Scholar]
- 148.Dumas de La Roque E, Savineau JP, Metivier AC, Billes MA, Kraemer JP, Doutreleau S, Jougon J, Marthan R, Moore N, Fayon M, Baulieu EE, Dromer C. Dehydroepiandrosterone (DHEA) improves pulmonary hypertension in chronic obstructive pulmonary disease (COPD): A pilot study. Annales d’endocrinologie 73: 20–25, 2012. [DOI] [PubMed] [Google Scholar]
- 149.Duran WN, Breslin JW, Sanchez FACP. The NO cascade, eNOS location, and microvascular permeability. Cardiovas Res 87: 254–261, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Eddahibi S, Fabre V, Boni C, Martres MP, Raffestin B, Hamon M, Adnot S. Induction of serotonin transporter by hypoxia in pulmonary vascular smooth muscle cells. Relationship with the mitogenic action of serotonin. Circ Res 84:329–336, 1999. [DOI] [PubMed] [Google Scholar]
- 151.El Chami H, Hassoun PM. Inflammatory mechanisms in the pathogenesis of pulmonary arterial hypertension. Compr Physiol 1: 1929–1941, 2011. [DOI] [PubMed] [Google Scholar]
- 152.El-Bermani AW, Bloomquist EI, Montvilo JA. Distribution of pulmonary cholinergic nerves in the rabbit. Thorax 37: 703–710, 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.El-Kashef H, Elmazar MM, Al-Shabanah OA, Al-Bekairi AM. Effect of adenosine on pulmonary circulation of rabbits. Gen Pharmacol 32: 307–313, 1999. [DOI] [PubMed] [Google Scholar]
- 154.Elliott CG. Genetics of pulmonary arterial hypertension: Current and future implications. Semin Respir Crit Care Med 26: 365–371, 2005. [DOI] [PubMed] [Google Scholar]
- 155.Elliott FM, Reid L. Some new facts about the pulmonary artery and its branching pattern. Clin Radiol 16: 193–198, 1965. [DOI] [PubMed] [Google Scholar]
- 156.Ema M, Taya S, Yokotani N, Sogawa K, Matsuda Y, Fujii-Kuriyama Y. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1α regulates the VEGF expression and is potentially involved in lung and vascular development. Proc Nat Acad Sci U S A 94: 4273–4278, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Emery CJ, Teng GQ, Liu X, Barer GR. Vasoreactions to acute hypoxia, whole lungs and isolated vessels compared: modulation by NO. Resp Physiol Neurobiol 134: 115–129, 2003. [DOI] [PubMed] [Google Scholar]
- 158.Emmanouilides GC, Moss AJ, Duffie ER Jr., Adams FH. Pulmonary arterial pressure changes in human newborn infants from birth to 3 days of age. J Pediat 65: 327–333, 1964. [DOI] [PubMed] [Google Scholar]
- 159.Fadel E, Wijtenburg E, Michel R, Mazoit JX, Bernatchez R, Decante B, Sage E, Mazmanian M, Herve P. Regression of the systemic vasculature to the lung after removal of pulmonary artery obstruction. Am J Resp Crit Care Med 173: 345–349, 2006. [DOI] [PubMed] [Google Scholar]
- 160.Fagan KA, Oka M, Bauer NR, Gebb SA, Ivy DD, Morris KG, McMurtry IF. Attenuation of acute hypoxic pulmonary vasoconstriction and hypoxic pulmonary hypertension in mice by inhibition of Rho-kinase. Am J Physiol Lung Cell Mol Physiol 287: L656–L664, 2004. [DOI] [PubMed] [Google Scholar]
- 161.Farhi L Ventilation-perfusion relationships. Compr Physiol (Suppl 13): 199–215, 2011. [Google Scholar]
- 162.Feletou M, Vanhoutte PM. Endothelium-dependent hyperpolarization of canine coronary smooth muscle. Br J Pharmacol 93: 515–524, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Feng CJ, Cheng DY, Kaye AD, Kadowitz PJ, Nossaman BD. Influence of N omega-nitro-L-arginine methyl ester, LY83583, glybenclamide and L158809 on pulmonary circulation. Eur J Pharmacol 263: 133–140, 1994. [DOI] [PubMed] [Google Scholar]
- 164.Fessler HE. Heart-lung interactions: Applications in the critically ill. Eur Respir J 10: 226–237, 1997. [DOI] [PubMed] [Google Scholar]
- 165.Fijalkowska I, Xu W, Comhair SA, Janocha AJ, Mavrakis LA, Krishnamachary B, Zhen L, Mao T, Richter A, Erzurum SC, Tuder RM. Hypoxia inducible-factor1α regulates the metabolic shift of pulmonary hypertensive endothelial cells. Am J Pathol 176: 1130–1138, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Fike CD, Kaplowitz MR, Pfister SL. Arachidonic acid metabolites and an early stage of pulmonary hypertension in chronically hypoxic newborn pigs. Am J Physiol Lung Cell Mol Physiol 284: L316–L323, 2003. [DOI] [PubMed] [Google Scholar]
- 167.Firth AL, Choi I-W, Park WS. Animal models of pulmonary hypertension: Rho kinase inhibition. Prog Biophys Mol Biol 109: 67–75, 2012. [DOI] [PubMed] [Google Scholar]
- 168.Fischer A, Mundel P, Mayer B, Preissler U, Philippin B, Kummer W. Nitric oxide synthase in guinea pig lower airway innervation. Neurosci Let 149: 157–160, 1993. [DOI] [PubMed] [Google Scholar]
- 169.Fischer LG, Honemann CW, Patrie JT, Durieux ME, Rich GF. Ropivacaine attenuates pulmonary vasoconstriction induced by thromboxane A2 analogue in the isolated perfused rat lung. Reg Anesth Pain Med 25: 187–194, 2000. [DOI] [PubMed] [Google Scholar]
- 170.Fisher AB. The serpentine path to a novel mechanism-based inhibitor of acute inflammatory lung injury. J Appl Physiol 116:1521–1530, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Fisher AW. The intrinsic innervation of the pulmonary vessels. Acta Anatomica 60: 481–496, 1965. [DOI] [PubMed] [Google Scholar]
- 172.Fishman A Pulmonary Circulation. Comp Physiol 93–165, 2011. [Google Scholar]
- 173.Fishman AP. Autonomic vasomotor tone in the pulmonary circulation. Anesthesiology 45: 1–2, 1976. [DOI] [PubMed] [Google Scholar]
- 174.Fishman AP. The pulmonary circulation. JAMA 239: 1299–1301, 1978. [PubMed] [Google Scholar]
- 175.Fisslthaler B, Fleming I, Busse R. EDHF: A cytochrome P450 metabolite in coronary arteries. Semin Perinatol 24: 15–19, 2000. [DOI] [PubMed] [Google Scholar]
- 176.Fleming I DiscrEET regulators of homeostasis: Epoxyeicosatrienoic acids, cytochrome P450 epoxygenases and vascular inflammation. Trends Pharmacol Sci 28: 448–452, 2007. [DOI] [PubMed] [Google Scholar]
- 177.Fleming I, Busse R. Endothelium-derived epoxyeicosatrienoic acids and vascular function. Hyperten 47: 629–633, 2006. [DOI] [PubMed] [Google Scholar]
- 178.Fong GH, Rossant J, Gertsenstein M, Breitman ML. Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nat 376: 66–70, 1995. [DOI] [PubMed] [Google Scholar]
- 179.Formenti F, Beer PA, Croft QP, Dorrington KL, Gale DP, Lappin TR, Lucas GS, Maher ER, Maxwell PH, McMullin MF, O’Connor DF, Percy MJ, Pugh CW, Ratcliffe PJ, Smith TG, Talbot NP, Robbins PA. Cardiopulmonary function in two human disorders of the hypoxia-inducible factor (HIF) pathway: von Hippel-Lindau disease and HIF-2α gain-of-function mutation. FASEB J 25: 2001–2011, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Fraidenburg D, Yuan J. Current and future therapeutic targets for pulmonary arterial hypertension. High Alt Med Biol 14: 134–143, 2013. [DOI] [PubMed] [Google Scholar]
- 181.Frank DB, Abtahi A, Yamaguchi DJ, Manning S, Shyr Y, Pozzi A, Baldwin HS, Johnson JE, de Caestecker MP. Bone morphogenetic protein 4 promotes pulmonary vascular remodeling in hypoxic pulmonary hypertension. Circ Res 97: 496–504, 2005. [DOI] [PubMed] [Google Scholar]
- 182.Frank DB, Lowery J, Anderson L, Brink M, Reese J, de Caestecker M. Increased susceptibility to hypoxic pulmonary hypertension in Bmpr2 mutant mice is associated with endothelial dysfunction in the pulmonary vasculature. Am J Physiol Lung Cell Mol Physiol 294:L98–109, 2008. [DOI] [PubMed] [Google Scholar]
- 183.Frantz E, Soifer SJ, Clyman RI, Heymann MA. Bradykinin produces pulmonary vasodilation in fetal lambs: Role of prostaglandin production. J Appl Physiol 67: 1512–1517, 1989. [DOI] [PubMed] [Google Scholar]
- 184.Frid MG, Brunetti JA, Burke DL, Carpenter TC, Davie NJ, Reeves JT, Roedersheimer MT, van Rooijen N, Stenmark KR. Hypoxia-induced pulmonary vascular remodeling requires recruitment of circulating mesenchymal precursors of a monocyte/macrophage lineage. Am J Path 168: 659–669, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Fujimoto K, Sakai A, Yoshikawa S, Shinozaki S, Matsuzawa Y, Kubo K, Kobayashi T, Ueda G, Sekiguchi M, Voelkel NF. Effect of cyclic guanosine monophosphate on hypoxic and angiotensin-II-induced pulmonary vasoconstriction. Lung 168: 333–343, 1990. [DOI] [PubMed] [Google Scholar]
- 186.Fujita H, Fukumoto Y, Saji K, Sugimura K, Demachi J, Nawata J, Shimokawa H. Acute vasodilator effects of inhaled fasudil, a specific Rho-kinase inhibitor, in patients with pulmonary arterial hypertension. Heart Vessels 25: 144–149, 2010. [DOI] [PubMed] [Google Scholar]
- 187.Fukumoto Y, Matoba T, Ito A, Tanaka H, Kishi T, Hayashidani S, Abe K, Takeshita A, Shimokawa H. Acute vasodilator effects of a Rho-kinase inhibitor, fasudil, in patients with severe pulmonary hypertension. Heart 91: 391–392, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Fukuroda T, Kobayashi M, Ozaki S, Yano M, Miyauchi T, Onizuka M, Sugishita Y, Goto K, Nishikibe M. Endothelin receptor subtypes in human versus rabbit pulmonary arteries. J Appl Physiol 76: 1976–1982, 1994. [DOI] [PubMed] [Google Scholar]
- 189.Fullerton DA, Kirson LE, Jones SD, McIntyre RC Jr. Adenosine is a selective pulmonary vasodilator in cardiac surgical patients. Chest 109: 41–46, 1996. [DOI] [PubMed] [Google Scholar]
- 190.Fuloria M, Eckman DM, Leach DA, Aschner JL. 20-hydroxyeicosatetraenoic acid is a vasoconstrictor in the newborn piglet pulmonary microcirculation. Am J Physiol Lung Cell Mol Physiol 287: L360–L365, 2004. [DOI] [PubMed] [Google Scholar]
- 191.Fung YC, Sobin SS. Pulmonary alveolar blood flow. Circ Res 30: 470–490, 1972. [DOI] [PubMed] [Google Scholar]
- 192.Fung YC, Zhuang FY. An analysis of the sluicing gate in pulmonary blood flow. J Biomech Eng 108: 175–182, 1986. [DOI] [PubMed] [Google Scholar]
- 193.Furchgott RF, Bhadrakom S. Reactions of strips of rabbit aorta to epinephrine, isopropylarterenol, sodium nitrite and other drugs. J Pharm Exp Ther 108: 129–143, 1953. [PubMed] [Google Scholar]
- 194.Gaar KA Jr., Taylor AE, Owens LJ, Guyton AC. Pulmonary capillary pressure and filtration coefficient in the isolated perfused lung. Am J Physiol 213: 910–914, 1967. [DOI] [PubMed] [Google Scholar]
- 195.Gale DP, Harten SK, Reid CD, Tuddenham EG, Maxwell PH. Auto-somal dominant erythrocytosis and pulmonary arterial hypertension associated with an activating HIF2 α mutation. Blood 112: 919–921, 2008. [DOI] [PubMed] [Google Scholar]
- 196.Galie N, Brundage BH, Ghofrani HA, Oudiz RJ, Simonneau G, Safdar Z, Shapiro S, White RJ, Chan M, Beardsworth A, Frumkin L, Barst RJ, Pulmonary Arterial H, Response to Tadalafil Study G. Tadalafil therapy for pulmonary arterial hypertension. Circulation 119: 2894–2903, 2009. [DOI] [PubMed] [Google Scholar]
- 197.Galie N, Hoeper MM, Humbert M, Torbicki A, Vachiery JL, Barbera JA, Beghetti M, Corris P, Gaine S, Gibbs JS, Gomez-Sanchez MA, Jondeau G, Klepetko W, Opitz C, Peacock A, Rubin L, Zellweger M, Simonneau G. Guidelines for the diagnosis and treatment of pulmonary hypertension: the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J 30: 2493–2537, 2009. [DOI] [PubMed] [Google Scholar]
- 198.Gambone LM, Murray PA, Flavahan NA. Synergistic interaction between endothelium-derived NO and prostacyclin in pulmonary artery: Potential role for K+ATP channels. Br J Pharmacol 121: 271–279, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Gan CT-J, Lankhaar J-W, Westerhof N, Marcus JT, Becker A, Twisk JWR, Boonstra A, Postmus PE, Vonk-Noordegraaf A. Noninvasively assessed pulmonary artery stiffness predicts mortality in pulmonary arterial hypertension. Chest 132:1906–1912, 2007. [DOI] [PubMed] [Google Scholar]
- 200.Gandhirajan RK, Meng S, Chandramoorthy HC, Mallilankaraman K, Mancarella S, Gao H, Razmpour R, Yang XF, Houser SR, Chen J, Koch WJ, Wang H, Soboloff J, Gill DL, Madesh M. Blockade of NOX2 and STIM1 signaling limits lipopolysaccharide-induced vascular inflammation. J Clin Invest 123: 887, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Gangopahyay A, Oran M, Bauer EM, Wertz JW, Comhair SA, Erzurum SC, Bauer PM. Bone morphogenetic protein receptor II is a novel mediator of endothelial nitric-oxide synthase activation. J Biol Chem 286: 33134–33140, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Gao Y, Raj JU. Regulation of the pulmonary circulation in the fetus and newborn. Physiol Rev 90: 1291–1335, 2010. [DOI] [PubMed] [Google Scholar]
- 203.Gao Y, Raj JU. Hypoxic pulmonary hypertension of the newborn. Compr Physiol 1: 61–79, 2011. [DOI] [PubMed] [Google Scholar]
- 204.Gatherer D, Ten Dijke P, Baird DT, Akhurst RJ. Expression of TGF-beta isoforms during first trimester human embryogenesis. Development 110: 445–460, 1990. [DOI] [PubMed] [Google Scholar]
- 205.Gattinoni L, Pesenti A, Carlesso E. Body position changes redistribute lung computed-tomographic density in patients with acute respiratory failure: Impact and clinical fallout through the following 20 years. Inten Care Med 39: 1909–1915, 2013. [DOI] [PubMed] [Google Scholar]
- 206.Gauthier KM, Edwards EM, Falck JR, Reddy DS, Campbell WB. 14,15-Epoxyeicosatrienoic acid represents a transferable endothelium-dependent relaxing factor in bovine coronary arteries. Hypertension 45: 666–671, 2005. [DOI] [PubMed] [Google Scholar]
- 207.Gavard J, Gutkind JS. VEGF controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of VE-cadherin. Nat Cell Biol 8: 1223–1234, 2006. [DOI] [PubMed] [Google Scholar]
- 208.Gebb S, Stevens T. On lung endothelial cell heterogeneity. Microvasc Res 68: 1–12, 2004. [DOI] [PubMed] [Google Scholar]
- 209.Gebb SA, Shannon JM. Tissue interactions mediate early events in pulmonary vasculogenesis. Dev Dynam 217: 159–169, 2000. [DOI] [PubMed] [Google Scholar]
- 210.Gehr P, Bachofen M, Weibel ER. The normal human lung: Ultrastructure and morphometric estimation of diffusion capacity. Resp Physiol 32: 121–140, 1978. [DOI] [PubMed] [Google Scholar]
- 211.Gerber JG, Voelkel N, Nies AS, McMurtry IF, Reeves JT. Moderation of hypoxic vasoconstriction by infused arachidonic acid: Role of PGI2. J Appl Physiol 49: 107–112, 1980. [DOI] [PubMed] [Google Scholar]
- 212.Gerthoffer WT. Mechanisms of vascular smooth muscle cell migration. Circ Res 100: 607–621, 2007. [DOI] [PubMed] [Google Scholar]
- 213.Ghofrani HA, D’Armini AM, Grimminger F, Hoeper MM, Jansa P, Kim NH, Mayer E, Simonneau G, Wilkins MR, Fritsch A, Neuser D, Weimann G, Wang C, Group C-S. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. N Eng J Med 369: 319–329, 2013. [DOI] [PubMed] [Google Scholar]
- 214.Ghofrani HA, Galie N, Grimminger F, Grunig E, Humbert M, Jing ZC, Keogh AM, Langleben D, Kilama MO, Fritsch A, Neuser D, Rubin LJ, Group P-S. Riociguat for the treatment of pulmonary arterial hypertension. N Eng J Med 369: 330–340, 2013. [Google Scholar]
- 215.Girgis RE, Li D, Zhan X, Garcia JG, Tuder RM, Hassoun PM, Johns RA. Attenuation of chronic hypoxic pulmonary hypertension by simvastatin. Am J Physiol Heart Circ Physiol 285: H938–H945, 2003. [DOI] [PubMed] [Google Scholar]
- 216.Girgis RE, Mozammel S, Champion HC, Li D, Peng X, Shimoda L, Tuder RM, Johns RA, Hassoun PM. Regression of chronic hypoxic pulmonary hypertension by simvastatin. Am J Physiol Lung Cell Mol Physiol 292: L1105–L1110, 2007. [DOI] [PubMed] [Google Scholar]
- 217.Glenny R, Robertson HT. Distribution of perfusion. Compr Physiol 1: 245–262, 2011. [DOI] [PubMed] [Google Scholar]
- 218.Glenny RW, Bernard S, Robertson HT, Hlastala MP. Gravity is an important but secondary determinant of regional pulmonary blood flow in upright primates. J Appl Physiol 86: 623–632, 1999. [DOI] [PubMed] [Google Scholar]
- 219.Glenny RW, Robertson HT. A computer simulation of pulmonary perfusion in three dimensions. J Appl Physiol 79: 357–369, 1995. [DOI] [PubMed] [Google Scholar]
- 220.Glenny RW, Robertson HT. Determinants of pulmonary blood flow distribution. Compr Physiol 1: 39–59, 2011. [DOI] [PubMed] [Google Scholar]
- 221.Glover GH, Newsom IE. Brisket disease: dropsy of high altitudes. Colo Agric Exp Station 204: 3–24, 1915. [Google Scholar]
- 222.Goll HM, Nyhan DP, Geller HS, Murray PA. Pulmonary vascular responses to angiotensin II and captopril in conscious dogs. J Appl Physiol 61: 1552–1559, 1986. [DOI] [PubMed] [Google Scholar]
- 223.Golovina VA, Platoshyn O, Bailey CL, Wang J, Limsuwan A, Sweeney M, Rubin LJ, Yuan JX. Upregulated TRP and enhanced capacitative Ca2+ entry in human pulmonary artery myocytes during proliferation. Am J Physiol Heart Circ Physiol 280: H746–H755, 2001. [DOI] [PubMed] [Google Scholar]
- 224.Gomberg-Maitland M, Bull TM, Saggar R, Barst RJ, Elgazayerly A, Fleming TR, Grimminger F, Rainisio M, Stewart DJ, Stockbridge N, Ventura C, Ghofrani AH, Rubin LJ. New trial designs and potential therapies for pulmonary artery hypertension. J Am Col Card 62: D82–D91, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Goncharov DA, Kudryashova TV, Ziai H, Ihida-Stansbury K, DeLisser H, Krymskaya VP, Tuder RM, Kawut SM, Goncharova EA. Mammalian target of rapamycin complex 2 (mTORC2) coordinates pulmonary artery smooth muscle cell metabolism, proliferation, and survival in pulmonary arterial hypertension. Circulation 129: 864–874, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Goncharova EA. mTOR and vascular remodeling in lung diseases: Current challenges and therapeutic prospects. FASEB J 27: 1796–1807, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Gonzales-Luis G, Fletcher AJ, Moreno L, Perez-Vizcaino F, Blanco CE, Villamor E. Nitric oxide-mediated nonadrenergic noncholinergic relaxation of piglet pulmonary arteries decreases with postnatal age. J Physiol Pharmacol 58: 45–56, 2007. [PubMed] [Google Scholar]
- 228.Gordon JB, Tod ML. Effects of N omega-nitro-L-arginine on total and segmental vascular resistances in developing lamb lungs. J Appl Physiol 75: 76–85, 1993. [DOI] [PubMed] [Google Scholar]
- 229.Gottlieb JE, Peake MD, Sylvester JT. Adenosine and hypoxic pulmonary vasodilation. Am J Physiol Heart Circ Physiol 247: H541–H547, 1984. [DOI] [PubMed] [Google Scholar]
- 230.Graham R, Skoog C, Oppenheimer L, Rabson J, Goldberg HS. Critical closure in the canine pulmonary vasculature. Circ Res 50: 566–572, 1982. [DOI] [PubMed] [Google Scholar]
- 231.Green DE, Murphy TC, Kang BY, Kleinhenz JM, Szyndralewiez C, Page P, Sutliff RL, Hart CM. The Nox4 inhibitor GKT137831 attenuates hypoxia-induced pulmonary vascular cell proliferation. Am J Respir Cell Mol Biol 47: 718–726, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Groenman F, Rutter M, Caniggia I, Tibboel D, Post M. Hypoxia-inducible factors in the first trimester human lung. J Histochem Cytochem 55: 355–363, 2007. [DOI] [PubMed] [Google Scholar]
- 233.Grover RF, Wagner WW, McMurtry IF, Reeves JT. Pulmonary Circulation. Compr Physiol (Suppl 8): 103–136, 2011. [Google Scholar]
- 234.Grover TR, Asikainen TM, Kinsella JP, Abman SH, White CW. Hypoxia-inducible factors HIF-1α and HIF-2α are decreased in an experimental model of severe respiratory distress syndrome in preterm lambs. Am J Physiol Lung Cell Mol Physiol 292: L1345–L1351, 2007. [DOI] [PubMed] [Google Scholar]
- 235.Groves BM, Droma T, Sutton JR, McCullough RG, McCullough RE, Zhuang J, Rapmund G, Sun S, Janes C, Moore LG. Minimal hypoxic pulmonary hypertension in normal Tibetans at 3,658 m. J Appl Physiol 74: 312–318, 1993. [DOI] [PubMed] [Google Scholar]
- 236.Grzenda A, Shannon J, Fisher J, Arkovitz MS. Timing and expression of the angiopoietin-1-Tie-2 pathway in murine lung development and congenital diaphragmatic hernia. Dis Mod Mech 6: 106–114, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Guembe L, Villaro AC. Histochemical demonstration of neuronal nitric oxide synthase during development of mouse respiratory tract. Am J Respir Cell Mol Biol 20: 342–351, 1999. [DOI] [PubMed] [Google Scholar]
- 238.Guerin C, Badet M, Rosselli S, Heyer L, Sab JM, Langevin B, Philit F, Fournier G, Robert D. Effects of prone position on alveolar recruitment and oxygenation in acute lung injury. Intensive Care Med 25: 1222–1230, 1999. [DOI] [PubMed] [Google Scholar]
- 239.Guilluy C, Sauzeau V, Rolli-Derkinderen M, Guerin P, Sagan C, Pacaud P, Loirand G. Inhibition of RhoA/Rho kinase pathway is involved in the beneficial effect of sildenafil on pulmonary hypertension. Br J Pharmacol 146: 1010–1018, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Gulino-Debrac DCP. Mechanotransduction at the basis of endothelial barrier function. Tissue Barriers 1: e24180, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Gumusel B, Orhan D, Tolunay O, Uma S. The role of nitric oxide in mediating nonadrenergic, noncholinergic relaxation in rat pulmonary artery. Nitric Oxide 5: 296–301, 2001. [DOI] [PubMed] [Google Scholar]
- 242.Gupta ML, Rao KS, Anand IS, Banerjee AK, Boparai MS. Lack of smooth muscle in the small pulmonary arteries of the native Ladakhi. Is the Himalayan highlander adapted? Am Rev Resp Dis 145:1201–1204, 1992. [DOI] [PubMed] [Google Scholar]
- 243.Haberberger R, Schemann M, Sann H, Kummer W. Innervation pattern of guinea pig pulmonary vasculature depends on vascular diameter. J Appl Physiol 82: 426–434, 1997. [DOI] [PubMed] [Google Scholar]
- 244.Hakim TS, Dean GW, Lisbona R. Effect of body posture on spatial distribution of pulmonary blood flow. J Appl Physiol 64: 1160–1170, 1988. [DOI] [PubMed] [Google Scholar]
- 245.Hakim TS, Lisbona R, Dean GW. Effect of cardiac output on gravity-dependent and nondependent inequality in pulmonary blood flow. J Appl Physiol 66: 1570–1578, 1989. [DOI] [PubMed] [Google Scholar]
- 246.Hales CA, Rouse ET, Kazemi H. Failure of saralasin acetate, a competitive inhibitor of angiotensin II, to diminish alveolar hypoxic vasoconstriction in the dog. Cardiovasc Res 11: 541–546, 1977. [DOI] [PubMed] [Google Scholar]
- 247.Hall SM, Hislop AA, Haworth SG. Origin, differentiation, and maturation of human pulmonary veins. Am J Resp Cell Mol Biol 26: 333–340, 2002. [DOI] [PubMed] [Google Scholar]
- 248.Hall SM, Hislop AA, Pierce CM, Haworth SG. Prenatal origins of human intrapulmonary arteries: Formation and smooth muscle maturation. Am J Resp Cell Mol Biol 23: 194–203, 2000. [DOI] [PubMed] [Google Scholar]
- 249.Hasunuma K, Yamaguchi T, Rodman DM, O’Brien RF, McMurtry IF. Effects of inhibitors of EDRF and EDHF on vasoreactivity of perfused rat lungs. Am J Physiol Lung Cell Mol Physiol 260: L97–L104, 1991. [DOI] [PubMed] [Google Scholar]
- 250.Hauge A, Lunde PK, Waaler BA. The effect of bradykinin, kallidin and eledoisin upon the pulmonary vascular bed of an isolated blood-perfused rabbit lung preparation. Acta Physiol Scan 66: 269–277, 1966. [DOI] [PubMed] [Google Scholar]
- 251.Haynes J Jr., Obiako B, Thompson WJ, Downey J. Adenosine-induced vasodilation: Receptor characterization in pulmonary circulation. Am J Physiol Heart Circ Physiol 268: H1862–H1868, 1995. [DOI] [PubMed] [Google Scholar]
- 252.Haywood GA, Sneddon JF, Bashir Y, Jennison SH, Gray HH, McKenna WJ. Adenosine infusion for the reversal of pulmonary vasoconstriction in biventricular failure. A good test but a poor therapy. Circulation 86: 896–902, 1992. [DOI] [PubMed] [Google Scholar]
- 253.Heath D, Smith P, Rios Dalenz J, Williams D, Harris P. Small pulmonary arteries in some natives of La Paz, Bolivia. Thorax 36: 599–604, 1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Hedenstierna G, White FC, Wagner PDC. Spatial distribution of pulmonary blood flow in the dog with PEEP ventilation. J Appl Physiol 47:938–946, 1979. [DOI] [PubMed] [Google Scholar]
- 255.Heinonen I, Savolainen AM, Han C, Kemppainen J, Oikonen V, Luotolahti M, Duncker DJ, Merkus D, Knuuti J, Kalliokoski KK. Pulmonary blood flow and its distribution in highly trained endurance athletes and healthy control subjects. J Appl Physiol 114: 329–334, 2013. [DOI] [PubMed] [Google Scholar]
- 256.Henderson AC, Sa RC, Theilmann RJ, Buxton RB, Prisk GK, Hopkins SRCP. The gravitational distribution of ventilation-perfusion ratio is more uniform in prone than supine posture in the normal human lung. J Appl Physiol 115: 313–324, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Heymann MA. Control of the pulmonary circulation in the fetus and during the transitional period to air breathing. Eur J Ob Gyn Repro Biol 84: 127–132, 1999. [DOI] [PubMed] [Google Scholar]
- 258.Higenbottam T, Marriott H, Cremona G, Laude E, Bee D. The acute effects of dexfenfluramine on human and porcine pulmonary vascular tone and resistance. Chest 116: 921–930, 1999. [DOI] [PubMed] [Google Scholar]
- 259.Hirenallur SD, Haworth ST, Leming JT, Chang J, Hernandez G, Gordon JB, Rusch NJ. Upregulation of vascular calcium channels in neonatal piglets with hypoxia-induced pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 295: L915–L924, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Hisatsune C, Kuroda Y, Nakamura K, Inoue T, Nakamura T, Michikawa T, Mizutani A, Mikoshiba K. Regulation of TRPC6 channel activity by tyrosine phosphorylation. J Biol Chem 279: 18887, 2004. [DOI] [PubMed] [Google Scholar]
- 261.Hislop A, Reid L. Intra-pulmonary arterial development during fetal life-branching pattern and structure. J Anat 113: 35–48, 1972. [PMC free article] [PubMed] [Google Scholar]
- 262.Hislop A, Reid L. Fetal and childhood development of the intrapulmonary veins in man–branching pattern and structure. Thorax 28: 313–319, 1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Hislop A, Reid L. New findings in pulmonary arteries of rats with hypoxia-induced pulmonary hypertension. Br J Exp Pathol 57: 542–554, 1976. [PMC free article] [PubMed] [Google Scholar]
- 264.Hislop A, Reid L. Normal structure and dimensions of the pulmonary arteries in the rat. J Anat 125: 71–83, 1978. [PMC free article] [PubMed] [Google Scholar]
- 265.Hislop AA, Pierce CM. Growth of the vascular tree. Paediat Resp Rev 1: 321–327, 2000. [DOI] [PubMed] [Google Scholar]
- 266.Hoffmann J, Yin J, Kukucka M, Yin N, Saarikko I, Sterner-Kock A, Fujii H, Leong-Poi H, Kuppe H, Schermuly RT, Kuebler WM. Mast cells promote lung vascular remodelling in pulmonary hypertension. Eur Respir J 37: 1400–1410, 2011. [DOI] [PubMed] [Google Scholar]
- 267.Hogan J, Smith P, Heath D, Harris P. The thickness of the alveolar capillary wall in the human lung at high and low altitude. Br J Dis Chest 80: 13–18, 1986. [DOI] [PubMed] [Google Scholar]
- 268.Holm P Endothelin in the pulmonary circulation with special reference to hypoxic pulmonary vasoconstriction. Scand Cardiovas J Supplement 46: 1–40, 1997. [PubMed] [Google Scholar]
- 269.Hong J, Kandasamy K, Marimuthu M, Choi CS, Kim S. Electrical cell-substrate impedance sensing as a non-invasive tool for cancer cell study. Analyst 136: 237–245, 2011. [DOI] [PubMed] [Google Scholar]
- 270.Hood S, Birnie D, Nasser A, Hillis WS. Effect of ketanserin on central haemodynamics and coronary circulation. J Cardiovasc Pharmacol 32: 983–987, 1998. [DOI] [PubMed] [Google Scholar]
- 271.Howell K, Preston RJ, McLoughlin P. Chronic hypoxia causes angiogenesis in addition to remodelling in the adult rat pulmonary circulation. J Physiol 547: 133–145, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Hu G, Minshall RD. Regulation of transendothelial permeability by Src kinase. Microvasc Res 77: 21–25, 2009. [DOI] [PubMed] [Google Scholar]
- 273.Hu G, Place AT, Minshall RD. Regulation of endothelial permeability by Src kinase signaling: Vascular leakage versus transcellular transport of drugs and macromolecules. Chem Biol Interact 171: 177–189, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Hu H, Sung A, Zhao G, Shi L, Qiu D, Nishimura T, Kao PN. Simvastatin enhances bone morphogenetic protein receptor type II expression. Biochem Biophys Res Commun 339: 59–64, 2006. [DOI] [PubMed] [Google Scholar]
- 275.Hu SL, He HL, Pan C, Liu AR, Liu SQ, Liu L, Huang YZ, Guo FM, Yang Y, Qiu HBC. The effect of prone positioning on mortality in patients with acute respiratory distress syndrome: a meta-analysis of randomized controlled trials. Crit Care 18: R109, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Hubloue I, Rondelet B, Kerbaul F, Biarent D, Milani GM, Staroukine M, Bergmann P, Naeije R, Leeman M. Endogenous angiotensin II in the regulation of hypoxic pulmonary vasoconstriction in anaesthetized dogs. Crit Care 8: R163–R171, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Hughes M, West JB. Last word on Point:Counterpoint: Gravity is/is not the major factor determining the distribution of blood flow in the human lung. J Appl Physiol 104: 1539, 2008. [DOI] [PubMed] [Google Scholar]
- 278.Hughes M, West JB. Point: Gravity is the major factor determining the distribution of blood flow in the human lung. J Appl Physiol 104: 1531–1533, 2008. [DOI] [PubMed] [Google Scholar]
- 279.Huh D, Matthews BD, Mammoto A, Montoya-Zavala M, Hsin HY, Ingber DEC. Reconstituting organ-level lung functions on a chip. Science 328: 1662–1668, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Huhta JC, Cohen M, Gutgesell HP. Patency of the ductus arteriosus in normal neonates: Two-dimensional echocardiography versus Doppler assessment. J Am Col Cardiol 4: 561–564, 1984. [DOI] [PubMed] [Google Scholar]
- 281.Hunter CJ, Dejam A, Blood AB, Shields H, Kim-Shapiro DB, Machado RF, Tarekegn S, Mulla N, Hopper AO, Schechter AN, Power GG, Gladwin MT. Inhaled nebulized nitrite is a hypoxia-sensitive NO-dependent selective pulmonary vasodilator. Nat Med 10: 1122–1127, 2004. [DOI] [PubMed] [Google Scholar]
- 282.Hyman AL, Kadowitz PJ. Pulmonary vasodilator activity of prostacyclin (PGI2) in the cat. Circ Res 45: 404–409, 1979. [DOI] [PubMed] [Google Scholar]
- 283.Hyman AL, Lippton HL, Kadowitz PJ. Autonomic regulation of the pulmonary circulation. J Cardiovasc Pharmacol 7(Suppl 3): S80–S95, 1985. [DOI] [PubMed] [Google Scholar]
- 284.Hyman AL, Lippton HL, Kadowitz PJ. Analysis of pulmonary vascular responses in cats to sympathetic nerve stimulation under elevated tone conditions. Evidence that neuronally released norepinephrine acts on alpha 1-, alpha 2-, and beta 2-adrenoceptors. Circ Res 67: 862–870, 1990. [DOI] [PubMed] [Google Scholar]
- 285.Hyman AL, Nandiwada P, Knight DS, Kadowitz PJ. Pulmonary vasodilator responses to catecholamines and sympathetic nerve stimulation in the cat. Evidence that vascular beta-2 adrenoreceptors are innervated. Circ Res 48: 407–415, 1981. [DOI] [PubMed] [Google Scholar]
- 286.Hyvelin J-M, Howell K, Nichol A, Costello CM, Preston RJ, McLoughlin P. Inhibition of Rho-kinase attenuates hypoxia-induced angiogenesis in the pulmonary circulation. Circ Res 97: 185–191, 2005. [DOI] [PubMed] [Google Scholar]
- 287.Ignarro LJ, Byrns RE, Buga GM, Wood KS. Mechanisms of endothelium-dependent vascular smooth muscle relaxation elicited by bradykinin and VIP. Am J Physiol 253: H1074–H1082, 1987. [DOI] [PubMed] [Google Scholar]
- 288.Ignarro LJ, Lippton H, Edwards JC, Baricos WH, Hyman AL, Kadowitz PJ, Gruetter CA. Mechanism of vascular smooth muscle relaxation by organic nitrates, nitrites, nitroprusside and nitric oxide: Evidence for the involvement of S-nitrosothiols as active intermediates. J Pharmacol Exp Ther 218: 739–749, 1981. [PubMed] [Google Scholar]
- 289.Ikeda T, Takahashi H, Suzuki A, Ueno N, Yokose S, Yamaguchi A, Yoshiki S. Cloning of rat type I receptor cDNA for bone morphogenetic protein-2 and bone morphogenetic protein-4, and the localization compared with that of the ligands. Dev Dynam 206: 318–329, 1996. [DOI] [PubMed] [Google Scholar]
- 290.International PPHC, Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA III, Loyd JE, Nichols WC, Trembath RC. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat Gen 26: 81–84, 2000. [DOI] [PubMed] [Google Scholar]
- 291.Ishikura K, Yamada N, Ito M, Ota S, Nakamura M, Isaka N, Nakano T. Beneficial acute effects of rho-kinase inhibitor in patients with pulmonary arterial hypertension. Circ J 70: 174–178, 2006. [DOI] [PubMed] [Google Scholar]
- 292.Ivy DD, Abman SH, Barst RJ, Berger RM, Bonnet D, Fleming TR, Haworth SG, Raj JU, Rosenzweig EB, Schulze Neick I, Steinhorn RH, Beghetti M. Pediatric pulmonary hypertension. J Am Col Cardiol 62: D117–D126, 2013. [DOI] [PubMed] [Google Scholar]
- 293.Ivy DD, Kinsella JP, Abman SH. Endothelin blockade augments pulmonary vasodilation in the ovine fetus. J Appl Physiol 81: 2481–2487, 1996. [DOI] [PubMed] [Google Scholar]
- 294.Iwata K, Doi A, Ohji G, Oka H, Oba Y, Takimoto K, Igarashi W, Gremillion DH, Shimada T. Effect of neutrophil elastase inhibitor (sivelestat sodium) in the treatment of acute lung injury (ALI) and acute respiratory distress syndrome (ARDS): A systematic review and meta-analysis. Intern Med 49: 2423–2432, 2010. [DOI] [PubMed] [Google Scholar]
- 295.Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, Gassmann M, Gearhart JD, Lawler AM, Yu AY, Semenza GL. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1α. Genes Dev 12: 149–162, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Jacobs ER, Zeldin DC. The lung HETEs (and EETs) up. Am J Physiol Heart Circ Physiol 280: H1–H10, 2001. [DOI] [PubMed] [Google Scholar]
- 297.Jameson AG. Diffusion of gases from alveolus to precapillary arteries. Science 139: 826–828, 1963. [DOI] [PubMed] [Google Scholar]
- 298.Jansen TL, Morice AH, Brown MJ. A comparison of the vasodilator responses to atrial peptides in the pulmonary and renal arteries of the pig in vitro. Br J Pharmacol 91: 687–691, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Janssen LJ, Premji M, Netherton S, Coruzzi J, Lu-Chao H, Cox PG. Vasoconstrictor actions of isoprostanes via tyrosine kinase and Rho kinase in human and canine pulmonary vascular smooth muscles. Br J Pharmacol 132: 127–134, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Jernigan NL, Walker BR, Resta TC. Reactive oxygen species mediate RhoA/Rho kinase-induced Ca2+ sensitization in pulmonary vascular smooth muscle following chronic hypoxia. Am J Physiol Lung Cell Mol Physiol 295: L515–L529, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Jiang X, Malkovskiy AV, Tian W, Sung YK, Sun W, Hsu JL, Manickam S, Wagh D, Joubert LM, Semenza GL, Rajadas J, Nicolls MR. Promotion of airway anastomotic microvascular regeneration and alleviation of airway ischemia by deferoxamine nanoparticles. Biomaterials 35: 803–813, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Jin HK, Yang RH, Thornton RM, Chen YF, Jackson R, Oparil S. Atrial natriuretic peptide lowers pulmonary arterial pressure in hypoxia-adapted rats. J Appl Physiol 65: 1729–1735, 1988. [DOI] [PubMed] [Google Scholar]
- 303.Jin Y, Lee SJ, Minshall RD, Choi AM. Caveolin-1: A critical regulator of lung injury. Am J Physiol Lung Cell Mol Physiol 300: L151, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Johnson DE, Georgieff MK. Pulmonary neuroendocrine cells. Their secretory products and their potential roles in health and chronic lung disease in infancy. Am Rev Respir Dis 140: 1807–1812, 1989. [DOI] [PubMed] [Google Scholar]
- 305.Johnson P, Maxwell DJ, Tynan MJ, Allan LD. Intracardiac pressures in the human fetus. Heart 84: 59–63, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Jones AT, Hansell DM, Evans TWC. Pulmonary perfusion in supine and prone positions: An electron-beam computed tomography study. J Appl Physiol 90: 1342–1348, 2001. [DOI] [PubMed] [Google Scholar]
- 307.Kadowitz PJ, Chapnick BM, Joiner PD, Hyman AL. Influence of inhibitors of prostaglandin synthesis on the canine pulmonary vascular bed. Am J Physiol 229: 941–946, 1975. [DOI] [PubMed] [Google Scholar]
- 308.Kadowitz PJ, Hyman AL. Effect of sympathetic nerve stimulation on pulmonary vascular resistance in the dog. Circ Res 32: 221–227, 1973. [DOI] [PubMed] [Google Scholar]
- 309.Kadowitz PJ, Knight DS, Hibbs RG, Ellison JP, Joiner PD, Brody MJ, Hyman AL. Influence of 5-and 6-hydroxydopamine on adrenergic transmission and nerve terminal morphology in the canine pulmonary vascular bed. Circ Res 39: 191–199, 1976. [DOI] [PubMed] [Google Scholar]
- 310.Kalinichenko VV, Lim L, Stolz DB, Shin B, Rausa FM, Clark J, Whitsett JA, Watkins SC, Costa RH. Defects in pulmonary vasculature and perinatal lung hemorrhage in mice heterozygous null for the Forkhead Box f1 transcription factor. Dev Biol 235: 489–506, 2001. [DOI] [PubMed] [Google Scholar]
- 311.Kamler M, Nowak K, Bock M, Herold U, Motsch J, Hagl S, Gebhard MM, Jakob H. Bronchial artery revascularization restores peribronchial tissue oxygenation after lung transplantation. J Heart Lung Trans 23: 763–766, 2004. [DOI] [PubMed] [Google Scholar]
- 312.Kane DW, Tesauro T, Koizumi T, Gupta R, Newman JH. Exercise-induced pulmonary vasoconstriction during combined blockade of nitric oxide synthase and beta adrenergic receptors. J Clin Invest 93: 677–683, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Kasai T, Chiba S. Macroscopic anatomy of the bronchial arteries. Anatomischer Anzeiger 145: 166–181, 1979. [PubMed] [Google Scholar]
- 314.Katsiki N, Wierzbicki AS, Mikhailidis DP. Pulmonary arterial hypertension and statins: An update. Cur Opin Cardiol 26: 322–326, 2011. [DOI] [PubMed] [Google Scholar]
- 315.Kawkitinarong K, Linz-McGillem L, Birukov KG, Garcia JG. Differential regulation of human lung epithelial and endothelial barrier function by thrombin. Am J Respir Cell Mol Biol 31: 517–527, 2004. [DOI] [PubMed] [Google Scholar]
- 316.Kawut SM, Bagiella E, Lederer DJ, Shimbo D, Horn EM, Roberts KE, Hill NS, Barr RG, Rosenzweig EB, Post W, Tracy RP, Palevsky HI, Hassoun PM, Girgis RE, Group A-SS. Randomized clinical trial of aspirin and simvastatin for pulmonary arterial hypertension: ASA-STAT. Circulation 123: 2985–2993, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Kay JM, Harris P, Heath D. Pulmonary hypertension produced in rats by ingestion of Crotalaria spectabilis seeds. Thorax 22: 176–179, 1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Kelly L, Kolbe J, Mitzner W, Spannhake EW, Bromberger-Barnea B, Menkes H. Bronchial blood flow affects recovery from constriction in dog lung periphery. J Appl Physiol 60: 1954–1959, 1986. [DOI] [PubMed] [Google Scholar]
- 319.Kern DF, Malik AB. Microvascular albumin permeability in isolated perfused lung: Effects of EDTA. J Appl Physiol 58: 372–375, 1985. [DOI] [PubMed] [Google Scholar]
- 320.Keseru B, Barbosa-Sicard E, Popp R, Fisslthaler B, Dietrich A, Guder-mann T, Hammock BD, Falck JR, Weissmann N, Busse R, Fleming I. Epoxyeicosatrienoic acids and the soluble epoxide hydrolase are determinants of pulmonary artery pressure and the acute hypoxic pulmonary vasoconstrictor response. FASEB J 22: 4306–4315, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Kevil CG, Payne DK, Mire E, Alexander JS. Vascular permeability factor/vascular endothelial cell growth factor-mediated permeability occurs through disorganization of endothelial junctional proteins. J Biol Chem 273: 15099–15103, 1998. [DOI] [PubMed] [Google Scholar]
- 322.Khalpey Z, Gilfeather MS, Camp PC Jr., Jaklitsch MT. Controlled antegrade single lung reperfusion during double lung transplant. In Cardiovas Thor Surg 9: 932–933, 2009. [DOI] [PubMed] [Google Scholar]
- 323.Kiely DG, Cargill RI, Lipworth BJ. Angiotensin II receptor blockade and effects on pulmonary hemodynamics and hypoxic pulmonary vasoconstriction in humans. Chest 110: 698–703, 1996. [DOI] [PubMed] [Google Scholar]
- 324.Kimura H, Mittal CK, Murad F. Activation of guanylate cyclase from rat liver and other tissues by sodium azide. J Biol Chem 250: 8016–8022, 1975. [PubMed] [Google Scholar]
- 325.Kiserud T Physiology of the fetal circulation. Sem Fetal Neonat Med 10: 493–503, 2005. [DOI] [PubMed] [Google Scholar]
- 326.Kizub IV, Strielkov IV, Shaifta Y, Becker S, Prieto-Lloret J, Snetkov VA, Soloviev AI, Aaronson PI, Ward JP. Gap junctions support the sustained phase of hypoxic pulmonary vasoconstriction by facilitating calcium sensitization. Cardiovas Res 99: 404–411, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Komarova Y, Malik AB. Regulation of endothelial permeability via paracellular and transcellular transport pathways. Ann Rev Physiol 72: 463–493, 2010. [DOI] [PubMed] [Google Scholar]
- 328.Komori K, Lorenz RR, Vanhoutte PM. Nitric oxide, ACh, and electrical and mechanical properties of canine arterial smooth muscle. Am J Physiol 255: H207–H212, 1988. [DOI] [PubMed] [Google Scholar]
- 329.Konduri GG, Mital S, Gervasio CT, Rotta AT, Forman K. Purine nucleotides contribute to pulmonary vasodilation caused by birth-related stimuli in the ovine fetus. Am J Physiol 272: H2377–H2384, 1997. [DOI] [PubMed] [Google Scholar]
- 330.Konduri GG, Woodard LL, Mukhopadhyay A, Deshmukh DR. Adenosine is a pulmonary vasodilator in newborn lambs. Am Rev Respir Dis 146: 670–676, 1992. [DOI] [PubMed] [Google Scholar]
- 331.Kotch LE, Iyer NV, Laughner E, Semenza GL. Defective vascularization of HIF-1a-null embryos is not associated with VEGF deficiency but with mesenchymal cell death. Dev Biol 209: 254–267, 1999. [DOI] [PubMed] [Google Scholar]
- 332.Kotecha S Lung growth for beginners. Paediat Resp Rev 1: 308–313, 2000. [DOI] [PubMed] [Google Scholar]
- 333.Kowalski TF, Guidotti S, Deffebach M, Kubilis P, Bishop M. Bronchial circulation in pulmonary artery occlusion and reperfusion. J Appl Physiol 68: 125–129, 1990. [DOI] [PubMed] [Google Scholar]
- 334.Kramer GC, Lindsey DC, Wu CH, Mertens S, Russell LA, Cross CE. Airway blood flow distribution and lung edema after histamine infusion in awake sheep. J Appl Physiol 65: 1847–1854, 1988. [DOI] [PubMed] [Google Scholar]
- 335.Krebs MO, Boemke W, Simon S, Wenz M, Kaczmarczyk G. Acute hypoxic pulmonary vasoconstriction in conscious dogs decreases renin and is unaffected by losartan. J Appl Physiol 86: 1914–1919, 1999. [DOI] [PubMed] [Google Scholar]
- 336.Kuhr FK, Smith KA, Song MY, Levitan I, Yuan JX-J. New mechanisms of pulmonary arterial hypertension: role of Ca2+ signaling. Am J Physiol Heart Circ Physiol 302: H1546–H1562, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Kummer W Pulmonary vascular innervation and its role in responses to hypoxia: Size matters! Proc Am Thorac Soc 8: 471–476, 2011. [DOI] [PubMed] [Google Scholar]
- 338.Kummer W, Fischer A, Kurkowski R, Heym C. The sensory and sympathetic innervation of guinea-pig lung and trachea as studied by retrograde neuronal tracing and double-labelling immunohistochemistry. Neuroscience 49: 715–737, 1992. [DOI] [PubMed] [Google Scholar]
- 339.Kunichika N, Landsberg JW, Yu Y, Kunichika H, Thistlethwaite PA, Rubin LJ, Yuan JX. Bosentan inhibits transient receptor potential channel expression in pulmonary vascular myocytes. Am J Respir Crit Care Med 170: 1101–1107, 2004. [DOI] [PubMed] [Google Scholar]
- 340.Lai-Fook SJ. A continuum mechanics analysis of pulmonary vascular interdependence in isolated dog lobes. J Appl Physiol 46: 419–429, 1979. [DOI] [PubMed] [Google Scholar]
- 341.Lamm WJ, Graham MM, Albert RK. Mechanism by which the prone position improves oxygenation in acute lung injury. Am J Respir Crit Care Med 150: 184–193, 1994. [DOI] [PubMed] [Google Scholar]
- 342.Landsberg JW, Yuan JX. Calcium and TRP channels in pulmonary vascular smooth muscle cell proliferation. News Physiol Sci 19: 44–50, 2004. [DOI] [PubMed] [Google Scholar]
- 343.Larkin EK, Newman JH, Austin ED, Hemnes AR, Wheeler L, Robbins IM, West JD, Phillips JA III, Hamid R, Loyd JE. Longitudinal analysis casts doubt on the presence of genetic anticipation in heritable pulmonary arterial hypertension. Am J Respir Crit Care Med 186: 892–896, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Larsson J, Goumans MJ, Sjostrand LJ, van Rooijen MA, Ward D, Leveen P, Xu X, ten Dijke P, Mummery CL, Karlsson S. Abnormal angiogenesis but intact hematopoietic potential in TGF-beta type I receptor-deficient mice. EMBO J 20: 1663–1673, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Lauer T, Preik M, Rassaf T, Strauer BE, Deussen A, Feelisch M, Kelm M. Plasma nitrite rather than nitrate reflects regional endothelial nitric oxide synthase activity but lacks intrinsic vasodilator action. Proc Nat Acad Sci U S A 98: 12814–12819, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Lauweryns JM, de Bock V, Guelinckx P, Decramer M. Effects of unilateral hypoxia on neuroepithelial bodies in rabbit lungs. J Appl Physiol 55: 1665–1668, 1983. [DOI] [PubMed] [Google Scholar]
- 347.Leach RM, Hill HM, Snetkov VA, Robertson TP, Ward JP. Divergent roles of glycolysis and the mitochondrial electron transport chain in hypoxic pulmonary vasoconstriction of the rat: identity of the hypoxic sensor. J Appl Physiol 536: 211–224, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Leggett K, Maylor J, Undem C, Lai N, Lu W, Schweitzer KS, King LS, Myers AC, Sylvester JT, Sidhaye VK, Shimoda LA. Hypoxia-induced migration in pulmonary arterial smooth muscle cells requires calcium-dependent upregulation of aquaporin 1. Am J Physiol Lung Cell Mol Physiol 303: L343–L353, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Lejeune P, Vachiery JL, Leeman M, Brimioulle S, Hallemans R, Melot C, Naeije R. Absence of parasympathetic control of pulmonary vascular pressure-flow plots in hyperoxic and hypoxic dogs. Respir Physiol 78: 123–133, 1989. [DOI] [PubMed] [Google Scholar]
- 350.Levine BW, Talamo RC, Kazemi H. Action and metabolism of bradykinin in dog lung. J Appl Physiol 34: 821–826, 1973. [DOI] [PubMed] [Google Scholar]
- 351.Lewis AB, Heymann MA, Rudolph AM. Gestational changes in pulmonary vascular responses in fetal lambs in utero. Circ Res 39: 536–541, 1976. [DOI] [PubMed] [Google Scholar]
- 352.Li VS, Ng SS, Boersema PJ, Low TY, Karthaus WR, Gerlach JP, Mohammed S, Heck AJ, Maurice MM, Mahmoudi T, Clevers H. Wnt signaling through inhibition of beta-catenin degradation in an intact Axin1 complex. Cell 149: 1245–1256, 2012. [DOI] [PubMed] [Google Scholar]
- 353.Liebow AA. Patterns of origin and distribution of the major bronchial arteries in man. Am J Anat 117: 19–32, 1965. [DOI] [PubMed] [Google Scholar]
- 354.Lim LH, Bochner BS, Wagner EM. Leukocyte recruitment in the airways: An intravital microscopic study of rat tracheal microcirculation. Am J Physiol Lung Cell Mol Physiol 282: L959–L967, 2002. [DOI] [PubMed] [Google Scholar]
- 355.Lin MJ, Leung GP, Zhang WM, Yang XR, Yip KP, Tse CM, Sham JS. Chronic hypoxia-induced upregulation of store-operated and receptor-operated Ca2+ channels in pulmonary arterial smooth muscle cells: A novel mechanism of hypoxic pulmonary hypertension. Circ Res 95: 496–505, 2004. [DOI] [PubMed] [Google Scholar]
- 356.Lindberg F, Andersson KE. Vasodilator responses to alpha-humanatrial natriuretic peptide in isolated omental and pulmonary arteries from rabbit and man. Acta Physiol Scand 134: 391–397, 1988. [DOI] [PubMed] [Google Scholar]
- 357.Linehan JH, Haworth ST, Nelin LD, Krenz GS, Dawson CA. A simple distensible vessel model for interpreting pulmonary vascular pressure-flow curves. J Appl Physiol 73: 987–994, 1992. [DOI] [PubMed] [Google Scholar]
- 358.Link DP, Parsons GH, Lantz BM, Gunther RA, Green JF, Cross CE. Measurement of bronchial blood flow in the sheep by video dilution technique. Thorax 40: 143–149, 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Lippton HL, Nandiwada PA, Hyman AL, Kadowitz PJ. Influence of cyclooxygenase blockade on responses to isoproterenol, bradykinin and nitroglycerin in the feline pulmonary vascular bed. Prostaglandins 28: 253–270, 1984. [DOI] [PubMed] [Google Scholar]
- 360.Liu F, Wu JY, Beasley D, Orr JA. TxA2-induced pulmonary artery contraction requires extracellular calcium. Respir Physiol 109: 155–166, 1997. [DOI] [PubMed] [Google Scholar]
- 361.Liu JQ, Sham JS, Shimoda LA, Kuppusamy P, Sylvester JT. Hypoxic constriction and reactive oxygen species in porcine distal pulmonary arteries. Am J Physiol Lung Cell Mol Physiol 285: L322–L333, 2003. [DOI] [PubMed] [Google Scholar]
- 362.Liu SF, Crawley DE, Evans TW, Barnes PJ. Endothelium-dependent nonadrenergic, noncholinergic neural relaxation in guinea pig pulmonary artery. J Pharmacol Exp Ther 260: 541–548, 1992. [PubMed] [Google Scholar]
- 363.Liu SF, Crawley DE, Rohde JA, Evans TW, Barnes PJ. Role of nitric oxide and guanosine 3’,5’-cyclic monophosphate in mediating nonadrenergic, noncholinergic relaxation in guinea-pig pulmonary arteries. Br J Pharmacol 107: 861–866, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Liu SF, McCormack DG, Evans TW, Barnes PJ. Evidence for two P2-purinoceptor subtypes in human small pulmonary arteries. Br J Pharmacol 98: 1014–1020, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Liu XR, Zhang MF, Yang N, Liu Q, Wang RX, Cao YN, Yang XR, Sham JS, Lin MJ. Enhanced store-operated Ca2+ entry and TRPC channel expression in pulmonary arteries of monocrotaline-induced pulmonary hypertensive rats. Am J Physiol Cell Physiol 302: C77–C87, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Liu Y, Suzuki YJ, Day RM, Fanburg BL. Rho kinase-induced nuclear translocation of ERK1/ERK2 in smooth muscle cell mitogenesis caused by serotonin. Circ Res 95: 579–586, 2004. [DOI] [PubMed] [Google Scholar]
- 367.Lloyd TC. Effect of alveolar hypoxia on pulmonary vascular resistance. J Appl Physiol 19: 1086–1094, 1964. [DOI] [PubMed] [Google Scholar]
- 368.Lloyd TC Jr. Role of nerve pathways in the hypoxic vasoconstriction of lung. J Appl Physiol 21: 1351–1355, 1966. [DOI] [PubMed] [Google Scholar]
- 369.Long L, Ormiston ML, Yang X, Southwood M, Graf S, Machado RD, Mueller M, Kinzel B, Yung LM, Wilkinson JM, Moore SD, Drake KM, Aldred MA, Yu PB, Upton PD, Morrell NW. Selective enhancement of endothelial BMPR-II with BMP9 reverses pulmonary arterial hypertension. Nature Med 21: 777–785, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Long WM, Sprung CL, el Fawal H, Yerger LD, Eyre P, Abraham WM, Wanner A. Effects of histamine on bronchial artery blood flow and bronchomotor tone. J Appl Physiol 59: 254–261, 1985. [DOI] [PubMed] [Google Scholar]
- 371.Lopez-Muniz R, Stephens NL, Bromberger-Barnea B, Permutt S, Riley RLC. Critical closure of pulmonary vessels analyzed in terms of Starling resistor model. J Appl Physiol 24: 625–635, 1968. [DOI] [PubMed] [Google Scholar]
- 372.Losapio JL, Sprague RS, Lonigro AJ, Stephenson AH. 5,6-EET-induced contraction of intralobar pulmonary arteries depends on the activation of Rho-kinase. J Appl Physiol 99: 1391–1396, 2005. [DOI] [PubMed] [Google Scholar]
- 373.Loughna S, Sato TN. Angiopoietin and Tie signaling pathways in vascular development. Matrix Biol 20: 319–325, 2001. [DOI] [PubMed] [Google Scholar]
- 374.Loukas M, Hanna M, Chen J, Tubbs RS, Anderson RH. Extracardiac coronary arterial anastomoses. Clin Anat 24: 137–142, 2011. [DOI] [PubMed] [Google Scholar]
- 375.Low FN. Electron microscopy of the rat lung. Anat Rec 113: 437–449, 1952. [DOI] [PubMed] [Google Scholar]
- 376.Lowe K, Alvarez D, King J, Stevens T. Phenotypic heterogeneity in lung capillary and extra-alveolar endothelial cells. Increased extra-alveolar endothelial permeability is sufficient to decrease compliance. J Surg Res 143: 70–77, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Lowery JW, de Caestecker MP. BMP signaling in vascular development and disease. Cytokine Growth Factor Rev 21: 287–298, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Lu W, Ran P, Zhang D, Lai N, Zhong N, Wang J. Bone morphogenetic protein 4 enhances canonical transient receptor potential expression, store-operated Ca2+ entry, and basal [Ca2+]i in rat distal pulmonary arterial smooth muscle cells. Am J Physiol Cell Physiol 299: C1370–C1378, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Lu X, Bijli KM, Ramirez A, Murphy TC, Kleinhenz J, Hart CM. Hypoxia downregulates PPARgamma via an ERK1/2-NF-kappaB-Nox4-dependent mechanism in human pulmonary artery smooth muscle cells. Free Radic Biol Med 63: 151–160, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Luckraz H, Goddard M, McNeil K, Atkinson C, Charman SC, Stewart S, Wallwork J. Microvascular changes in small airways predispose to obliterative bronchiolitis after lung transplantation. J Heart Lung Trans 23: 527–531, 2004. [DOI] [PubMed] [Google Scholar]
- 381.Luecke T, Pelosi PCP. Clinical review: Positive end-expiratory pressure and cardiac output. Crit Care 9: 607–621, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Luke T, Maylor J, Undem C, Sylvester JT, Shimoda LA. Kinase dependent activation of voltage-gated Ca2+ channels by ET-1 in pulmonary arterial myocytes during chronic hypoxia. Am J Physiol Lung Cell Mol Physiol 302: L1128–L1139, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Lundgrin EL, Park MM, Sharp J, Tang WH, Thomas JD, Asosingh K, Comhair SA, DiFilippo FP, Neumann DR, Davis L, Graham BB, Tuder RM, Dostanic I, Erzurum SC. Fasting 2-deoxy-2-[18F]fluoro-D-glucose positron emission tomography to detect metabolic changes in pulmonary arterial hypertension hearts over 1 year. Ann Am Thorac Soc 10: 1–9, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Ma L, Chung WK. The genetic basis of pulmonary arterial hypertension. Human Gen 133: 471–479, 2014. [DOI] [PubMed] [Google Scholar]
- 385.Machado RD, Pauciulo MW, Thomson JR, Lane KB, Morgan NV, Wheeler L, Phillips JA III, Newman J, Williams D, Galie N, Manes A, McNeil K, Yacoub M, Mikhail G, Rogers P, Corris P, Humbert M, Donnai D, Martensson G, Tranebjaerg L, Loyd JE, Trembath RC, Nichols WC. BMPR2 haploinsufficiency as the inherited molecular mechanism for primary pulmonary hypertension. Am J Human Gen 68: 92–102, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.MacIntyre PD, Bhargava B, Hogg KJ, Gemmill JD, Hillis WS. Effect of subcutaneous sumatriptan, a selective 5HT1 agonist, on the systemic, pulmonary, and coronary circulation. Circulation 87: 401–405, 1993. [DOI] [PubMed] [Google Scholar]
- 387.MacLean MR, Alexander D, Stirrat A, Gallagher M, Douglas SA, Ohlstein EH, Morecroft I, Polland K. Contractile responses to human urotensin-II in rat and human pulmonary arteries: Effect of endothelial factors and chronic hypoxia in the rat. Br J Pharmacol 130: 201–204, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.MacLean MR, Clayton RA, Hillis SW, McIntyre PD, Peacock AJ, Templeton AG. 5-HT1-receptor-mediated vasoconstriction in bovine isolated pulmonary arteries: influences of vascular endothelium and tone. Pulm Pharmacol 7: 65–72, 1994. [DOI] [PubMed] [Google Scholar]
- 389.MacLean MR, Clayton RA, Templeton AG, Morecroft I. Evidence for 5-HT1-like receptor-mediated vasoconstriction in human pulmonary artery. Br J Pharmacol 119: 277–282, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Maclean MR, Dempsie Y. The serotonin hypothesis of pulmonary hypertension revisited. Adv Exp Med Biol 661: 309–322, 2010. [DOI] [PubMed] [Google Scholar]
- 391.Madden JA, Ray DE, Keller PA, Kleinman JG. Ion exchange activity in pulmonary artery smooth muscle cells: the response to hypoxia. Am J Physiol Lung Cell Mol Physiol 280: L264–L271, 2001. [DOI] [PubMed] [Google Scholar]
- 392.Maeda Y, Dave V, Whitsett JA. Transcriptional control of lung morphogenesis. Physiol Rev 87: 219–244, 2007. [DOI] [PubMed] [Google Scholar]
- 393.Magno MG, Fishman AP. Origin, distribution, and blood flow of bronchial circulation in anesthetized sheep. J Appl Physiol 53: 272–279, 1982. [DOI] [PubMed] [Google Scholar]
- 394.Mahapatra S, Nishimura RA, Sorajja P, Cha S, McGoon MD. Relationship of pulmonary arterial capacitance and mortality in idiopathic pulmonary arterial hypertension. J Am Col Cardiol 21:799–803, 2006. [DOI] [PubMed] [Google Scholar]
- 395.Mahlapuu M, Enerback S, Carlsson P. Haploinsufficiency of the forkhead gene Foxf1, a target for sonic hedgehog signaling, causes lung and foregut malformations. Development 128: 2397–2406, 2001. [DOI] [PubMed] [Google Scholar]
- 396.Mahlapuu M, Ormestad M, Enerback S, Carlsson P. The forkhead transcription factor Foxf1 is required for differentiation of extra-embryonic and lateral plate mesoderm. Development 128: 155–166, 2001. [DOI] [PubMed] [Google Scholar]
- 397.Mair KM, MacLean MR, Morecroft I, Dempsie Y, Palmer TM. Novel interactions between the 5-HT transporter, 5-HT1B receptors and Rho kinase in vivo and in pulmonary fibroblasts. Br J Pharmacol 155:606–616, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Malik AB, Mewmark JM. Adrenergic mechanisms and the pulmonary vascular response to respiratory acidosis. Respiration 33: 179–187, 1976. [DOI] [PubMed] [Google Scholar]
- 399.Maltepe E, Schmidt JV, Baunoch D, Bradfield CA, Simon MC. Abnormal angiogenesis and responses to glucose and oxygen deprivation in mice lacking the protein ARNT. Nat 386: 403–407, 1997. [DOI] [PubMed] [Google Scholar]
- 400.Marsboom G, Toth PT, Ryan JJ, Hong Z, Wu X, Fang YH, Thenappan T, Piao L, Zhang HJ, Pogoriler J, Chen Y, Morrow E, Weir EK, Rehman J, Archer SL. Dynamin-related protein 1-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ Res 110: 1484–1497, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.Martin GS, Brigham KL. Fluid flux and clearance in acute lung injury. Compr Physiol 2: 2471–2480, 2012. [DOI] [PubMed] [Google Scholar]
- 402.Matran R, Alving K, Hemsen A, Lundberg JM. Endothelin-1 increases airway mucosa blood flow in the pig. Agents Actions 31: 237–241, 1990. [DOI] [PubMed] [Google Scholar]
- 403.McCormack DG, Clarke B, Barnes PJ. Characterization of adenosine receptors in human pulmonary arteries. Am J Physiol 256: H41–H46, 1989. [DOI] [PubMed] [Google Scholar]
- 404.McCormack DG, Rees RG, Crawley D, Barnes PJ, Evans TW. Sensory neuropeptides and hypoxic pulmonary vasoconstriction in the rat. Thorax 48: 554–557, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405.McCullagh A, Rosenthal M, Wanner A, Hurtado A, Padley S, Bush A. The bronchial circulation–worth a closer look: a review of the relationship between the bronchial vasculature and airway inflammation. Pediat Pulm 45: 1–13, 2010. [DOI] [PubMed] [Google Scholar]
- 406.Mcloughlin P, Mcmurtry I. Counterpoint: Chronic hypoxia-induced pulmonary hypertension does not lead to loss of pulmonary vasculature. J Appl Physiol 103: 1451–1453; discussion 1453–1454, 2007. [DOI] [PubMed] [Google Scholar]
- 407.McMahon TJ, Hood JS, Kadowitz PJ. Pulmonary vasodilator response to vagal stimulation is blocked by N omega-nitro-L-arginine methyl ester in the cat. Circ Res 70: 364–369, 1992. [DOI] [PubMed] [Google Scholar]
- 408.McMurtry IF. Angiotensin is not required for hypoxic constriction in salt solution-perfused rat lungs. J Appl Physiol 56: 375–380, 1984. [DOI] [PubMed] [Google Scholar]
- 409.McMurtry IF, Davidson AB, Reeves JT, Grover RF. Inhibition of hypoxic pulmonary vasoconstriction by calcium antagonists in isolated rat lungs. Circ Res 38: 99–104, 1976. [DOI] [PubMed] [Google Scholar]
- 410.McMurtry IV, Abe K, Ota H, Fagan KA, Oka M. Rho kinase-mediated vasoconstriction in pulmonary hypertension. Adv Exp Med Biol 661: 299–308, 2010. [DOI] [PubMed] [Google Scholar]
- 411.McMurtry MS, Bonnet S, Michelakis ED, Bonnet S, Haromy A, Archer SL. Statin therapy, alone or with rapamycin, does not reverse monocrotaline pulmonary arterial hypertension: the rapamycin-atorvastatin-simvastatin study. Am J Physiol Lung Cell Mol Physiol 293: L933–L940, 2007. [DOI] [PubMed] [Google Scholar]
- 412.McMurtry MS, Bonnet S, Wu X, Dyck JR, Haromy A, Hashimoto K, Michelakis ED. Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ Res 95: 830–840, 2004. [DOI] [PubMed] [Google Scholar]
- 413.Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev 86: 279, 2006. [DOI] [PubMed] [Google Scholar]
- 414.Mentzer RM Jr., Bartels C, Bolli R, Boyce S, Buckberg GD, Chaitman B, Haverich A, Knight J, Menasche P, Myers ML, Nicolau J, Simoons M, Thulin L, Weisel RD. Sodium-hydrogen exchange inhibition by cariporide to reduce the risk of ischemic cardiac events in patients undergoing coronary artery bypass grafting: results of the EXPEDITION study. Ann Thorac Surg 85: 1261–1270, 2008. [DOI] [PubMed] [Google Scholar]
- 415.Mentzer RM Jr., Rubio R, Berne RM. Release of adenosine by hypoxic canine lung tissue and its possible role in pulmonary circulation. Am J Physiol 229: 1625–1631, 1975. [DOI] [PubMed] [Google Scholar]
- 416.Meyrick B, Gamble W, Reid L. Development of Crotalaria pulmonary hypertension: Hemodynamic and structural study. Am J Physiol 239: H692–H702, 1980. [DOI] [PubMed] [Google Scholar]
- 417.Meyrick B, Niedermeyer ME, Ogletree ML, Brigham KL. Pulmonary hypertension and increased vasoreactivity caused by repeated indomethacin in sheep. J Appl Physiol (1985) 59: 443–452, 1985. [DOI] [PubMed] [Google Scholar]
- 418.Michel RP. Arteries and veins of the normal dog lung: Qualitative and quantitative structural differences. Am J Anat 164: 227–241, 1982. [DOI] [PubMed] [Google Scholar]
- 419.Michelakis ED, McMurtry MS, Wu XC, Dyck JR, Moudgil R, Hopkins TA, Lopaschuk GD, Puttagunta L, Waite R, Archer SL. Dichloroacetate, a metabolic modulator, prevents and reverses chronic hypoxic pulmonary hypertension in rats: Role of increased expression and activity of voltage-gated potassium channels. Circulation 105: 244–250, 2002. [DOI] [PubMed] [Google Scholar]
- 420.Middelveld RJ, Alving K. Responses of the bronchial and pulmonary circulations to short-term nitric oxide inhalation before and after endo toxaemia in the pig. Acta Physiol Scand 176: 71–78, 2002. [DOI] [PubMed] [Google Scholar]
- 421.Mielke G, Benda N. Cardiac output and central distribution of blood flow in the human fetus. Circulation 103: 1662–1668, 2001. [DOI] [PubMed] [Google Scholar]
- 422.Millan FA, Denhez F, Kondaiah P, Akhurst RJ. Embryonic gene expression patterns of TGF beta 1, beta 2 and beta 3 suggest different developmental functions in vivo. Development 111: 131–143, 1991. [DOI] [PubMed] [Google Scholar]
- 423.Mitzner W, Lee W, Georgakopoulos D, Wagner E. Angiogenesis in the mouse lung. Am J Path 157: 93–101, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Mitzner W, Wagner E. On the purported discovery of the bronchial circulation by Leonardo da Vinci. J Appl Physiol 73: 1196–1201, 1992. [DOI] [PubMed] [Google Scholar]
- 425.Mizuno S, Farkas L, Al Husseini A, Farkas D, Gomez-Arroyo J, Kraskauskas D, Nicolls MR, Cool CD, Bogaard HJ, Voelkel NF. Severe pulmonary arterial hypertension induced by SU5416 and ovalbumin immunization. Am J Resp Cell Mol Biol 47: 679–687, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426.Mogami H, Kojima I. Stimulation of calcium entry is prerequisite for DNA synthesis induced by platelet-derived growth factor in vascular smooth muscle cells. Biochem Biophys Res Commun 196: 650–658, 1993. [DOI] [PubMed] [Google Scholar]
- 427.Moldobaeva A, Welsh-Servinsky LE, Shimoda LA, Stephens RS, Verin AD, Tuder RM, Pearse DB. Role of protein kinase G in barrier-protective effects of cGMP in human pulmonary artery endothelial cells. Am J Physiol Lung Cell Mol Physiol 290: L919–L930, 2006. [DOI] [PubMed] [Google Scholar]
- 428.Morecroft I, Dempsie Y, Bader M, Walther DJ, Kotnik K, Loughlin L, Nilsen M, MacLean MR. Effect of tryptophan hydroxylase 1 deficiency on the development of hypoxia-induced pulmonary hypertension. Hypertension 49: 232–236, 2007. [DOI] [PubMed] [Google Scholar]
- 429.Morecroft I, Heeley RP, Prentice HM, Kirk A, MacLean MR. 5-hydroxytryptamine receptors mediating contraction in human small muscular pulmonary arteries: Importance of the 5-HT1B receptor. Br J Pharmacol 128: 730–734, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430.Morecroft I, Loughlin L, Nilsen M, Colston J, Dempsie Y, Sheward J, Harmar A, MacLean MR. Functional interactions between 5-hydroxytryptamine receptors and the serotonin transporter in pulmonary arteries. J Pharmacol Exp Ther 313: 539–548, 2005. [DOI] [PubMed] [Google Scholar]
- 431.Morgan JM, McCormack DG, Griffiths MJ, Morgan CJ, Barnes PJ, Evans TW. Adenosine as a vasodilator in primary pulmonary hypertension. Circulation 84: 1145–1149, 1991. [DOI] [PubMed] [Google Scholar]
- 432.Morin FC III, Egan EA. Pulmonary hemodynamics in fetal lambs during development at normal and increased oxygen tension. J Appl Physiol 73: 213–218, 1992. [DOI] [PubMed] [Google Scholar]
- 433.Morio Y, Carter EP, Oka M, McMurtry IF. EDHF-mediated vasodilation involves different mechanisms in normotensive and hypertensive rat lungs. Am J Physiol Heart Circ Physiol 284: H1762–H1770, 2003. [DOI] [PubMed] [Google Scholar]
- 434.Morrell NW, Yang X, Upton PD, Jourdan KB, Morgan N, Sheares KK, Trembath RC. Altered growth responses of pulmonary artery smooth muscle cells from patients with primary pulmonary hypertension to transforming growth factor-β1 and bone morphogenetic proteins. Circulation 104: 790–795, 2001. [DOI] [PubMed] [Google Scholar]
- 435.Morse JH, Jones AC, Barst RJ, Hodge SE, Wilhelmsen KC, Nygaard TG. Mapping of familial primary pulmonary hypertension locus (PPH1) to chromosome 2q31-q32. Circulation 95: 2603–2606, 1997. [DOI] [PubMed] [Google Scholar]
- 436.Moss AJ, Emmanouilides G, Duffie ER Jr. Closure of the ductus arteriosus in the newborn infant. Pediatrics 32: 25–30, 1963. [PubMed] [Google Scholar]
- 437.Mura M, dos Santos CC, Stewart D, Liu M. Vascular endothelial growth factor and related molecules in acute lung injury. J Appl Physiol (1985) 97: 1605–1617, 2004. [DOI] [PubMed] [Google Scholar]
- 438.Mure M, Domino KB, Lindahl SG, Hlastala MP, Altemeier WA, Glenny RW. Regional ventilation-perfusion distribution is more uniform in the prone position. J Appl Physiol 88: 1076–1083, 2000. [DOI] [PubMed] [Google Scholar]
- 439.Mure M, Nyren S, Jacobsson H, Larsson SA, Lindahl SG. High continuous positive airway pressure level induces ventilation/perfusion mismatch in the prone position. Crit Care Med 29: 959–964, 2001. [DOI] [PubMed] [Google Scholar]
- 440.Murillo H, Cutalo MJ, Jones RP, Lane MJ, Fleischmann D, Restrepo CS. Pulmonary circulation imaging: Embryology and normal anatomy. Sem Ultrasound, CT, and MR 33: 473–484, 2012. [DOI] [PubMed] [Google Scholar]
- 441.Murphy E, Allen DG. Why did the NHE inhibitor clinical trials fail? J Mol Cell Cardiol 46: 137–141, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442.Murray PA, Lodato RF, Michael JR. Neural antagonists modulate pulmonary vascular pressure-flow plots in conscious dogs. J Appl Physiol (1985) 60: 1900–1907, 1986. [DOI] [PubMed] [Google Scholar]
- 443.Murtha YM, Allen BM, Orr JA. The role of protein kinase C in thromboxane A2-induced pulmonary artery vasoconstriction. J Biomed Sci 6: 293–295, 1999. [DOI] [PubMed] [Google Scholar]
- 444.Musch G, Layfield JDH, Harris RS, Melo MFV, Winkler T, Callahan RJ, Fischman AJ, Venegas JGC. Topographical distribution of pulmonary perfusion and ventilation, assessed by PET in supine and prone humans. J Appl Physiol 93: 1841–1851, 2002. [DOI] [PubMed] [Google Scholar]
- 445.Naeije R, Chesler NC. Pulmonary circulation at exercise. Compr Physiol 2: 711–741, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446.Naeye RL. Hypoxemia and pulmonary hypertension. A study of the pulmonary vasculature. Arch Pathol 71: 447–452, 1961. [PubMed] [Google Scholar]
- 447.Naeye RL. Children at high altitude: Pulmonary and renal abnormalities. Circ Res 16: 33–38, 1965. [DOI] [PubMed] [Google Scholar]
- 448.Nagaoka T, Fagan KA, Gebb SA, Morris KG, Suzuki T, Shimokawa H, McMurtry IF, Oka M. Inhaled Rho kinase inhibitors are potent and selective vasodilators in rat pulmonary hypertension. Am J Resp Crit Care Med 171: 494–499, 2005. [DOI] [PubMed] [Google Scholar]
- 449.Nagaoka T, Morio Y, Casanova N, Bauer N, Gebb S, McMurtry I, Oka M. Rho/Rho kinase signaling mediates increased basal pulmonary vascular tone in chronically hypoxic rats. Am J Physiol Lung Cell Mol Physiol 287: L665–L672, 2004. [DOI] [PubMed] [Google Scholar]
- 450.Nagaoka T, Muramatsu M, Sato K, McMurtry I, Oka M, Fukuchi Y. ild hypoxia causes severe pulmonary hypertension in fawn-hooded but not in Tester Moriyama rats. Resp Physiol 127: 53–60, 2001. [DOI] [PubMed] [Google Scholar]
- 451.Nakanishi R Revascularization of trachea in lung and tracheal transplantation. Clin Transplant 21: 668–674, 2007. [DOI] [PubMed] [Google Scholar]
- 452.Natarajan V, Dudek SM, Jacobson JR, Moreno-Vinasco L, Huang LS, Abassi T, Mathew B, Zhao Y, Wang L, Bittman R, Weichselbaum R, Berdyshev E, Garcia JG. Sphingosine-1-phosphate, FTY720, and sphingosine-1-phosphate receptors in the pathobiology of acute lung injury. Am J Respir Cell Mol Biol 49: 6–17, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 453.Neely CF, Kadowitz PJ, Lippton H, Neiman M, Hyman AL. Adenosine does not mediate the pulmonary vasodilator response of adenosine 5’-triphosphate in the feline pulmonary vascular bed. J Pharmacol Exp Ther 250: 170–176, 1989. [PubMed] [Google Scholar]
- 454.Neely CF, Matot I, Batra VK, Bo X, Burnstock G. P2X purinoceptors in the feline pulmonary vascular bed: distribution and selective in vivo pharmacological probes. Am J Physiol 270: L889–L897, 1996. [DOI] [PubMed] [Google Scholar]
- 455.Nelin LD, Dawson CA. The effect of N omega-nitro-L-arginine methylester on hypoxic vasoconstriction in the neonatal pig lung. Pediatr Res 34: 349–353, 1993. [DOI] [PubMed] [Google Scholar]
- 456.Newman JH, Holt TN, Hedges LK, Womack B, Memon SS, Willers ED, Wheeler L, Phillips JA III, Hamid R. High-altitude pulmonary hypertension in cattle (brisket disease): Candidate genes and gene expression profiling of peripheral blood mononuclear cells. Pulm Circ 1: 462–469, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457.Newman JH, Phillips JA, Loyd JE. Narrative review: The enigma of pulmonary arterial hypertension: New insights from genetic studies. Ann Intern Med 148: 278–283, 2008. [DOI] [PubMed] [Google Scholar]
- 458.Ng LC, Wilson SM, Hume JR. Mobilization of sarcoplasmic reticulum stores by hypoxia leads to consequent activation of capacitative Ca2+ entry in isolated canine pulmonary arterial smooth muscle cells. J Physiol 563: 409–419, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459.Nichols WC, Koller DL, Slovis B, Foroud T, Terry VH, Arnold ND, Siemieniak DR, Wheeler L, Phillips JA III, Newman JH, Conneally PM, Ginsburg D, Loyd JE. Localization of the gene for familial primary pulmonary hypertension to chromosome 2q31–32. Nat Gen 15: 277–280, 1997. [DOI] [PubMed] [Google Scholar]
- 460.Norgaard MA, Alphonso N, Cochrane AD, Menahem S, Brizard CP, d’Udekem Y. Major aorto-pulmonary collateral arteries of patients with pulmonary atresia and ventricular septal defect are dilated bronchial arteries. Eur J Cardio Surg 29: 653–658, 2006. [DOI] [PubMed] [Google Scholar]
- 461.Nussenzveig DR, Lewicki JA, Maack T. Cellular mechanisms of the clearance function of type C receptors of atrial natriuretic factor. J Biol Chem 265: 20952–20958, 1990. [PubMed] [Google Scholar]
- 462.Nyhan DP, Chen BB, Fehr DM, Rock P, Murray PA. Anesthesia alters pulmonary vasoregulation by angiotensin II and captopril. J Appl Physiol 72: 636–642, 1992. [DOI] [PubMed] [Google Scholar]
- 463.Nyrén S, Radell P, Lindahl SGE, Mure M, Petersson J, Larsson SA, Jacobsson H, Sánchez-Crespo AC. Lung ventilation and perfusion in prone and supine postures with reference to anesthetized and mechanically ventilated healthy volunteers. Anesthesiology 112: 682–687, 2010. [DOI] [PubMed] [Google Scholar]
- 464.Odell AF, Scott JL, Van Helden DF. EGF induces tyrosine phosphorylation, membrane insertion and activation of transient receptor potential channel 4. J Biol Chem 280: 37974–37987, 2005. [DOI] [PubMed] [Google Scholar]
- 465.Oka M, Fagan KA, Jones PL, McMurtry IF. Therapeutic potential of RhoA/Rho kinase inhibitors in pulmonary hypertension. Br J Pharmacol 155: 444–454, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 466.Oka M, Homma N, Taraseviciene-Stewart L, Morris KG, Kraskauskas D, Burns N, Voelkel NF, McMurtry IF. Rho kinase-mediated vasoconstriction is important in severe occlusive pulmonary arterial hypertension in rats. Circ Res 100: 923–929, 2007. [DOI] [PubMed] [Google Scholar]
- 467.Okada K, Tanaka Y, Bernstein M, Zhang W, Patterson GA, Botney MD. Pulmonary hemodynamics modify the rat pulmonary artery response to injury. A neointimal model of pulmonary hypertension. Am J Path 151: 1019–1025, 1997. [PMC free article] [PubMed] [Google Scholar]
- 468.Osaki S, Nakanishi Y, Wataya H, Takayama K, Inoue K, Takaki Y, Murayama S, Hara N. Prognosis of bronchial artery embolization in the management of hemoptysis. Respiration 67: 412–416, 2000. [DOI] [PubMed] [Google Scholar]
- 469.Oudiz RJ, Brundage BH, Galie N, Ghofrani HA, Simonneau G, Botros FT, Chan M, Beardsworth A, Barst RJ, Group PS. Tadalafil for the treatment of pulmonary arterial hypertension: A double-blind 52-week uncontrolled extension study. J Am Col Cardiol 60: 768–774, 2012. [DOI] [PubMed] [Google Scholar]
- 470.Paddenberg R, Stieger P, Von Lilien A-L, Faulhammer P, Goldenberg A, Tillmanns HH, Kummer W, Braun-Dullaeus RC. Rapamycin attenuates hypoxia-induced pulmonary vascular remodeling and right ventricular hypertrophy in mice. Respir Res 8: 15, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471.Palmer LA, Johns RA. Hypoxia upregulates inducible (Type II) nitric oxide synthase in an HIF-1 dependent manner in rat pulmonary microvascular but not aortic smooth muscle cells. Chest 114: 33S–34S, 1998. [DOI] [PubMed] [Google Scholar]
- 472.Palmer LA, Semenza GL, Stoler MH, Johns RA. Hypoxia induces type II NOS gene expression in pulmonary artery endothelial cells via HIF-1. Am J Physiol Lung Cell Mol Physiol 274: L212–L219, 1998. [DOI] [PubMed] [Google Scholar]
- 473.Palmer RM, Ashton DS, Moncada S. Vascular endothelial cells synthesize nitric oxide from L-arginine. Nature 333: 664–666, 1988. [DOI] [PubMed] [Google Scholar]
- 474.Palmer RM, Ferrige AG, Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 327: 524–526, 1987. [DOI] [PubMed] [Google Scholar]
- 475.Parera MC, van Dooren M, van Kempen M, de Krijger R, Grosveld F, Tibboel D, Rottier R. Distal angiogenesis: A new concept for lung vascular morphogenesis. Am J Physiol Lung Cell Mol Physiol 288: L141–L149, 2005. [DOI] [PubMed] [Google Scholar]
- 476.Parker JC. Acute lung injury and pulmonary vascular permeability: Use of transgenic models. Compr Physiol 1: 835–882. [DOI] [PubMed] [Google Scholar]
- 477.Parker JC, Stevens T, Randall J, Weber DS, King JA. Hydraulic conductance of pulmonary microvascular and macrovascular endothelial cell monolayers. Am J Physiol Lung Cell Mol Physiol 291: L30–L37, 2006. [DOI] [PubMed] [Google Scholar]
- 478.Parker JC, Townsley MI. Evaluation of lung injury in rats and mice. Am J Physiol Lung Cell Mol Physiol 286: L231–L246, 2004. [DOI] [PubMed] [Google Scholar]
- 479.Parker JC, Townsley MIC. Physiological determinants of the pulmonary filtration coefficient. Am J Physiol Lung Cell Mol Physiol 295: L235–L237, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480.Parker JC, Yoshikawa S. Vascular segmental permeabilities at high peak inflation pressure in isolated rat lungs. Am J Physiol Lung Cell Mol Physiol 283: L1203–L1209, 2002. [DOI] [PubMed] [Google Scholar]
- 481.Parker RE, Granger DN, Taylor AE. Estimates of isogravimetric capillary pressures during alveolar hypoxia. Am J Physiol 241: H732–H739, 1981. [DOI] [PubMed] [Google Scholar]
- 482.Parsons GH, Bommer WJ, Siefkin AD, Brock J, Lantz BM. Drainage routes of bronchial blood flow in anaesthetized sheep. Pulm Pharmacol Ther 20: 109–111, 2007. [DOI] [PubMed] [Google Scholar]
- 483.Parthasarathi K, Ichimura H, Monma E, Lindert J, Quadri S, Issekutz A, Bhattacharya JCP. Connexin 43 mediates spread of Ca2+-dependent proinflammatory responses in lung capillaries. J Clin Invest 116: 2193–2200, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 484.Patterson GA, Todd TR, Cooper JD, Pearson FG, Winton TL, Maurer J. Airway complications after double lung transplantation. Toronto Lung Transplant Group. J Thoracic Cardiovasc Surg 99: 14–20; discussion 20–11, 1990. [PubMed] [Google Scholar]
- 485.Pauling MH, Vu TH. Mechanisms and regulation of lung vascular development. Cur Topics Dev Biol 64: 73–99, 2004. [DOI] [PubMed] [Google Scholar]
- 486.Pearl RG. Adenosine produces pulmonary vasodilation in the perfused rabbit lung via an adenosine A2 receptor. Anesth Analg 79: 46–51, 1994. [DOI] [PubMed] [Google Scholar]
- 487.Pearl RG, Prielipp RC. Leukotriene synthesis inhibition and receptor blockade do not inhibit hypoxic pulmonary vasoconstriction in sheep. Anesth Analg 72: 169–176, 1991. [DOI] [PubMed] [Google Scholar]
- 488.Pearson FG, Goldberg M, Stone RM, Colapinto RF. Bronchial arterial circulation restored after reimplantation of canine lung. Can J Surg 13: 243–250, 1970. [PubMed] [Google Scholar]
- 489.Pelage JP. Bronchial artery embolization: Anatomy and technique. Tech Vasc Intervent Radiol 10: 274–275, 2007. [DOI] [PubMed] [Google Scholar]
- 490.Pelosi P, Tubiolo D, Mascheroni D, Vicardi P, Crotti S, Valenza F, Gattinoni L. Effects of the prone position on respiratory mechanics and gas exchange during acute lung injury. Am J Resp Crit Care Med 157: 387–393, 1998. [DOI] [PubMed] [Google Scholar]
- 491.Pelton RW, Saxena B, Jones M, Moses HL, Gold LI. Immunohistochemical localization of TGF β1, TGF β2, and TGF β3 in the mouse embryo: Expression patterns suggest multiple roles during embryonic development. J Cell Biol 115: 1091–1105, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 492.Penaloza D, Arias-Stella J, Sime F, Recavarren S, Marticorena E. the heart and pulmonary circulation in children at high altitudes: Physiological, anatomical, and clinical observations. Pediatrics 34: 568–582, 1964. [PubMed] [Google Scholar]
- 493.Peng X, Hassoun PM, Sammani S, McVerry BJ, Burne MJ, Rabb H, Pearse D, Tuder RM, Garcia JG. Protective effects of sphingosine 1-phosphate in murine endotoxin-induced inflammatory lung injury. Am J Respir Crit Care Med 169: 1245–1251, 2004. [DOI] [PubMed] [Google Scholar]
- 494.Permutt S, Bromberger-Barnea B, Bane HN. Alveolar pressure, pulmonary venous pressure, and the vascular waterfall. Med Thorac 19: 239–260, 1962. [DOI] [PubMed] [Google Scholar]
- 495.Petersson J, Rohdin M, Sánchez-Crespo A, Nyrén S, Jacobsson H, Larsson SA, Lindahl SGE, Linnarsson D, Neradilek B, Polissar NL, Glenny RW, Mure MC. Posture primarily affects lung tissue distribution with minor effect on blood flow and ventilation. Respir Physiol Neurobiol 156: 293–303, 2007. [DOI] [PubMed] [Google Scholar]
- 496.Pinheiro JM, Malik AB. Mechanisms of endothelin-1-induced pulmonary vasodilatation in neonatal pigs. J Physiol 469: 739–752, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 497.Pisarcik S, Maylor J, Lu W, Yun X, Undem C, Sylvester JT, Semenza GL, Shimoda LA. Activation of hypoxia-inducible factor-1 in pulmonary arterial smooth muscle cells by endothelin-1. Am J Physiol Lung Cell Mol Physiol 304: L549–L561, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498.Pluta RM, Oldfield EH, Bakhtian KD, Fathi AR, Smith RK, Devroom HL, Nahavandi M, Woo S, Figg WD, Lonser RR. Safety and feasibility of long-term intravenous sodium nitrite infusion in healthy volunteers. PloS one 6: e14504, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 499.Porcelli RJ, Bergofsky EH. Adrenergic receptors in pulmonary vasoconstrictor responses to gaseous and humoral agents. J Appl Physiol 34: 483–488, 1973. [DOI] [PubMed] [Google Scholar]
- 500.Porcelli RJ, Bergofsky EH. Effect of pH on pulmonary pressor responses to humoral agents. J Appl Physiol 31: 679–685, 1971. [DOI] [PubMed] [Google Scholar]
- 501.Porta NF, Steinhorn RH. Pulmonary vasodilator therapy in the NICU: Inhaled nitric oxide, sildenafil, and other pulmonary vasodilating agents. Clin Perinat 39: 149–164, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 502.Post JM, Gelband CH, Hume JR. [Ca2+]i inhibition of K+ channels in canine pulmonary artery. Novel mechanism for hypoxia-induced membrane depolarization. Circ Res 77: 131–139, 1995. [DOI] [PubMed] [Google Scholar]
- 503.Post JM, Hume JR, Archer SL, Weir EK. Direct role for potassium channel inhibition in hypoxic pulmonary vasoconstriction. Am J Physiol 262: C882–C890, 1992. [DOI] [PubMed] [Google Scholar]
- 504.Pozeg ZI, Michelakis ED, McMurtry MS, Thebaud B, Wu XC, Dyck JR, Hashimoto K, Wang S, Moudgil R, Harry G, Sultanian R, Koshal A, Archer SL. In vivo gene transfer of the O2-sensitive potassium channel Kv1.5 reduces pulmonary hypertension and restores hypoxic pulmonary vasoconstriction in chronically hypoxic rats. Circulation 107: 2037–2044, 2003. [DOI] [PubMed] [Google Scholar]
- 505.Prabhakar NR, Semenza GL. Adaptive and maladaptive cardiorespiratory responses to continuous and intermittent hypoxia mediated by hypoxia-inducible factors 1 and 2. Physiol Rev 92: 967–1003, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 506.Prewitt RL, Leffler CW. Feline hypoxic pulmonary vasoconstriction is not blocked by the angiotensin I-converting enzyme inhibitor, captopril. J Cardiovasc Pharmacol 3: 293–298, 1981. [DOI] [PubMed] [Google Scholar]
- 507.Price LC, Wort SJ, Perros F, Dorfmuller P, Huertas A, Montani D, Cohen-Kaminsky S, Humbert M. Inflammation in pulmonary arterial hypertension. Chest 141: 210–221, 2012. [DOI] [PubMed] [Google Scholar]
- 508.Pullamsetti SS, Schermuly R, Ghofrani A, Weissmann N, Grimminger F, Seeger W. Novel and emerging therapies for pulmonary hypertension. Am J Resp Criti Care Med 189: 394–400, 2014. [DOI] [PubMed] [Google Scholar]
- 509.Quinn DA, Dahlberg CG, Bonventre JP, Scheid CR, Honeyman T, Joseph PM, Thompson BT, Hales CA. The role of Na+/H+ exchange and growth factors in pulmonary artery smooth muscle cell proliferation. Am J Respir Cell Mol Biol 14: 139–145, 1996. [DOI] [PubMed] [Google Scholar]
- 510.Quinn DA, Du HK, Thompson BT, Hales CA. Amiloride analogs inhibit chronic hypoxic pulmonary hypertension. Am J Respir Crit Care Med 157: 1263–1268, 1998. [DOI] [PubMed] [Google Scholar]
- 511.Quinn DA, Honeyman TW, Joseph PM, Thompson BT, Hales CA, Scheid CR. Contribution of Na+/H+ exchange to pH regulation in pulmonary artery smooth muscle cells. Am J Respir Cell Mol Biol 5: 586–591, 1991. [DOI] [PubMed] [Google Scholar]
- 512.Rabinovitch M, Chesler N, Molthen RC. Point:Counterpoint: Chronic hypoxia-induced pulmonary hypertension does/does not lead to loss of pulmonary vasculature. J Appl Physiol 103: 1449–1451, 2007. [DOI] [PubMed] [Google Scholar]
- 513.Rabinovitch M, Gamble WJ, Nadas AS, Miettinen OS, Reid L. Rat pulmonary circulation after chronic hypoxia: Hemodynamic and structural features. Am J Physiol 236: H818–H827, 1979. [DOI] [PubMed] [Google Scholar]
- 514.Rabinovitch M, Guignabert C, Humbert M, Nicolls MR. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ Res 115: 165–175, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 515.Raj JU, Toga H, Ibe BO, Anderson J. Effects of endothelin, platelet activating factor and thromboxane A2 in ferret lungs. Respir Physiol 88: 129–140, 1992. [DOI] [PubMed] [Google Scholar]
- 516.Rajagopalan B, Friend JA, Stallard T, Lee GD. Blood flow in pulmonary veins: II. The influence of events transmitted from the right and left sides of the heart. Cardiovasc Res 13: 677–683, 1979. [DOI] [PubMed] [Google Scholar]
- 517.Rajaram SS, Desai NK, Kalra A, Gajera M, Cavanaugh SK, Brampton W, Young D, Harvey S, Rowan K. Pulmonary artery catheters for adult patients in intensive care. Cochrane Database Syst Rev 2: Cd003408, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 518.Rasanen J, Wood DC, Debbs RH, Cohen J, Weiner S, Huhta JC. Reactivity of the human fetal pulmonary circulation to maternal hyperoxygenation increases during the second half of pregnancy: A randomized study. Circulation 97: 257–262, 1998. [DOI] [PubMed] [Google Scholar]
- 519.Rasanen J, Wood DC, Weiner S, Ludomirski A, Huhta JC. Role of the pulmonary circulation in the distribution of human fetal cardiac output during the second half of pregnancy. Circulation 94: 1068–1073, 1996. [DOI] [PubMed] [Google Scholar]
- 520.Rathore R, Zheng YM, Niu CF, Liu QH, Korde A, Ho YS, Wang YX. Hypoxia activates NADPH oxidase to increase [ROS]i and [Ca2+]i through the mitochondrial ROS-PKCepsilon signaling axis in pulmonary artery smooth muscle cells. Free Rad Biol Med 45: 1223–1231, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 521.Rawat DK, Alzoubi A, Gupte R, Chettimada S, Watanabe M, Kahn AG, Okada T, McMurtry IF, Gupte SA. Increased reactive oxygen species, metabolic maladaptation, and autophagy contribute to pulmonary arterial hypertension-induced ventricular hypertrophy and diastolic heart failure. Hypertension 64: 1266–1274, 2014. [DOI] [PubMed] [Google Scholar]
- 522.Redding GJ, McMurtry I, Reeves JT. Effects of meclofenamate on pulmonary vascular resistance correlate with postnatal age in young piglets. Pediatr Res 18: 579–583, 1984. [DOI] [PubMed] [Google Scholar]
- 523.Reid L The angiogram and pulmonary artery structure and branching (in the normal and with reference to disease). Proc Royal Soc Med 58: 681–684, 1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 524.Reid LM. Structure and function in pulmonary hypertension. New perceptions. Chest 89: 279–288, 1986. [DOI] [PubMed] [Google Scholar]
- 525.Reid PG, Fraser AG, Watt AH, Henderson AH, Routledge PA. Acute haemodynamic effects of intravenous infusion of adenosine in conscious man. Eur Heart J 11: 1018–1028, 1990. [DOI] [PubMed] [Google Scholar]
- 526.Remillard CV, Yuan JX. Activation of K+ channels: An essential pathway in programmed cell death. Am J Physiol Lung Cell Mol Physiol 286: L49–L67, 2004. [DOI] [PubMed] [Google Scholar]
- 527.Rhodes J Comparative physiology of hypoxic pulmonary hypertension: Historical clues from brisket disease. J Appl Physiol 98: 1092–1100, 2005. [DOI] [PubMed] [Google Scholar]
- 528.Rich S, Kieras K, Hart K, Groves BM, Stobo JD, Brundage BH. Antinuclear antibodies in primary pulmonary hypertension. J Am Col Cardiol 8: 1307–1311, 1986. [DOI] [PubMed] [Google Scholar]
- 529.Richardson JB. Nerve supply to the lungs. Am Rev Resp Dis 119: 785–802, 1979. [DOI] [PubMed] [Google Scholar]
- 530.Richter T, Bellani G, Scott Harris R, Vidal Melo MF, Winkler T, Venegas JG, Musch GCP. Effect of prone position on regional shunt, aeration, and perfusion in experimental acute lung injury. Am J Resp Crit Care Med 172: 480–487, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 531.Rios EJ, Fallon M, Wang J, Shimoda LA. Chronic hypoxia elevates intracellular pH and activates Na+/H+ exchange in pulmonary arterial smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 289(5): L867–874, 2005. [DOI] [PubMed] [Google Scholar]
- 532.Rippe B, Rosengren BI, Carlsson O, Venturoli D. Transendothelial transport: The vesicle controversy. J Vasc Res 39: 375–390, 2002. [DOI] [PubMed] [Google Scholar]
- 533.Rodman DM, Reese K, Harral J, Fouty B, Wu S, West J, Hoedt-Miller M, Tada Y, Li KX, Cool C, Fagan K, Cribbs L. Low-voltage-activated (T-type) calcium channels control proliferation of human pulmonary artery myocytes. Circ Res 96: 864–872, 2005. [DOI] [PubMed] [Google Scholar]
- 534.Rodman DM, Yamaguchi T, O’Brien RF, McMurtry IF. Hypoxic contraction of isolated rat pulmonary artery. J Pharmacol Exp Therap 248: 952–959, 1989. [PubMed] [Google Scholar]
- 535.Roepke JE, Patterson CE, Packer CS, Rhoades RA. Response of perfused lung and isolated pulmonary artery to adenosine. Exp Lung Res 17: 25–37, 1991. [DOI] [PubMed] [Google Scholar]
- 536.Rose JC, Kot PA, Cohn JN, Freis ED, Eckert GE. Comparison of effects of angiotensin and norepinephrine on pulmonary circulation, systemic arteries and veins, and systemic vascular capacity in the dog. Circulation 25: 247–252, 1962. [DOI] [PubMed] [Google Scholar]
- 537.Rubin LJ, Galie N, Grimminger F, Grunig E, Humbert M, Jing ZC, Keogh A, Langleben D, Fritsch A, Menezes F, Davie N, Ghofrani HA. Riociguat for the treatment of pulmonary arterial hypertension: A long-term extension study (PATENT-2). Eur Respir J 45: 1303–1313, 2015. [DOI] [PubMed] [Google Scholar]
- 538.Rudolph AM. Fetal and neonatal pulmonary circulation. Am Rev Resp Dis 115: 11–18, 1977. [DOI] [PubMed] [Google Scholar]
- 539.Rudolph AM. Fetal and neonatal pulmonary circulation. Ann Rev Physiol 41: 383–395, 1979. [DOI] [PubMed] [Google Scholar]
- 540.Rudolph AM. Aortopulmonary transposition in the fetus: Speculation on pathophysiology and therapy. Pediatr Res 61: 375–380, 2007. [DOI] [PubMed] [Google Scholar]
- 541.Sanchez FA, Savalia NB, Duran RG, Lal BK, Boric MP, Duran WNC. Functional significance of differential eNOS translocation. Am J Physiol Heart Circ Physiol 291: H1058–H1064, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 542.Sanchez T, Skoura A, Wu MT, Casserly B, Harrington EO, Hla T. Induction of vascular permeability by the sphingosine-1-phosphate receptor-2 (S1P2R) and its downstream effectors ROCK and PTEN. Arterioscler Thromb Vasc Biol 27: 1312–1318, 2007. [DOI] [PubMed] [Google Scholar]
- 543.Sasaki F, Pare P, Ernest D, Bai T, Verburgt L, March R, Baile E. Endogenous nitric oxide influences acetylcholine-induced bronchovascular dilation in sheep. J Appl Physiol 78: 539–545, 1995. [DOI] [PubMed] [Google Scholar]
- 544.Sato K, Webb S, Tucker A, Rabinovitch M, O’Brien RF, McMurtry IF, Stelzner TJ. Factors influencing the idiopathic development of pulmonary hypertension in the fawn hooded rat. Am Rev Resp Dis 145: 793–797, 1992. [DOI] [PubMed] [Google Scholar]
- 545.Sato TN, Tozawa Y, Deutsch U, Wolburg-Buchholz K, Fujiwara Y, Gendron-Maguire M, Gridley T, Wolburg H, Risau W, Qin Y. Distinct roles of the receptor tyrosine kinases Tie-1 and Tie-2 in blood vessel formation. Nat 376: 70–74, 1995. [DOI] [PubMed] [Google Scholar]
- 546.Satoh K, Fukumoto Y, Shimokawa H. Rho-kinase: Important new therapeutic target in cardiovascular diseases. Am J Physiol Heart Circ Physiol 301: H287–H296, 2011. [DOI] [PubMed] [Google Scholar]
- 547.Saueressig MG, Neto AM, Fortis EA, Westphal D, Edelweiss MI, Meurer L, Matte U. Vascular endothelial growth factor gene therapy induces early re-establishment of canine bronchial circulation. Eur J Cardio Surg 33: 717–722, 2008. [DOI] [PubMed] [Google Scholar]
- 548.Sayner SL. Emerging themes of cAMP regulation of the pulmonary endothelial barrier. Am J Physiol Lung Cell Mol Physiol 300: L667–L678, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 549.Schermuly RT, Ghofrani HA, Wilkins MR, Grimminger F. Mechanisms of disease: Pulmonary arterial hypertension. Nat Rev Cardiol 8: 443–455, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 550.Schlensak C, Doenst T, Preusser S, Wunderlich M, Kleinschmidt M, Beyersdorf F. Cardiopulmonary bypass reduction of bronchial blood flow: A potential mechanism for lung injury in a neonatal pig model. J Thor Cardiovasc Surg 123: 1199–1205, 2002. [DOI] [PubMed] [Google Scholar]
- 551.Schmidt EP, Damarla M, Rentsendorj O, Servinsky LE, Zhu B, Moldobaeva A, Gonzalez A, Hassoun PM, Pearse DBCP. Soluble guanylyl cyclase contributes to ventilator-induced lung injury in mice. Am J Physiol Lung Cell Mol Physiol 295: L1056–L1065, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 552.Schmidt EP, Yang Y, Janssen WJ, Gandjeva A, Perez MJ, Barthel L, Zemans RL, Bowman JC, Koyanagi DE, Yunt ZX, Smith LP, Cheng SS, Overdier KH, Thompson KR, Geraci MW, Douglas IS, Pearse DB, Tuder RM. The pulmonary endothelial glycocalyx regulates neutrophil adhesion and lung injury during experimental sepsis. Nat Med 18: 1217–1223, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 553.Schnurch H, Risau W. Expression of tie-2, a member of a novel family of receptor tyrosine kinases, in the endothelial cell lineage. Development 119: 957–968, 1993. [DOI] [PubMed] [Google Scholar]
- 554.Schraufnagel DE, Pearse DB, Mitzner WA, Wagner EM. Three-dimensional structure of the bronchial microcirculation in sheep. Anatomical Rec 243: 357–366, 1995. [DOI] [PubMed] [Google Scholar]
- 555.Schulze-Neick I, Ho SY, Bush A, Rosenthal M, Franklin RC, Redington AN, Penny DJ. Severe airflow limitation after the unifocalization procedure: clinical and morphological correlates. Circulation 102: III142–III147, 2000. [DOI] [PubMed] [Google Scholar]
- 556.Schulze-Neick I, Penny DJ, Derrick GP, Dhillon R, Rigby ML, Kelleher A, Bush A, Redington AN. Pulmonary vascular-bronchial interactions: Acute reduction in pulmonary blood flow alters lung mechanics. Heart 84: 284–289, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 557.Scott JA, Craig I, McCormack DG. Nonadrenergic noncholinergic relaxation of human pulmonary arteries is partially mediated by nitric oxide. Am J Resp Crit Care Med 154: 629–632, 1996. [DOI] [PubMed] [Google Scholar]
- 558.Scuri M, McCaskill V, Chediak AD, Abraham WM, Wanner A. Effect of inhaled and intravenous acetylcholine on bronchial blood flow in anesthetized sheep. J Appl Physiol 80: 341–344, 1996. [DOI] [PubMed] [Google Scholar]
- 559.Selig WM, Noonan TC, Kern DF, Malik AB. Pulmonary microvascular responses to arachidonic acid in isolated perfused guinea pig lung. J Appl Physiol 60: 1972–1979, 1986. [DOI] [PubMed] [Google Scholar]
- 560.Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol 12: 5447–5454, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 561.Sequeira IM, Haberberger RV, Kummer W. Atrial and ventricular rat coronary arteries are differently supplied by noradrenergic, cholinergic and nitrergic, but not sensory nerve fibres. Ann Anat 187: 345–355, 2005. [DOI] [PubMed] [Google Scholar]
- 562.Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, Schuh AC. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature 376: 62–66, 1995. [DOI] [PubMed] [Google Scholar]
- 563.Shao D, Park JE, Wort SJ. The role of endothelin-1 in the pathogenesis of pulmonary arterial hypertension. Pharmacol Res 63: 504–511, 2011. [DOI] [PubMed] [Google Scholar]
- 564.Shea BS, Brooks SF, Fontaine BA, Chun J, Luster AD, Tager AM. Prolonged exposure to sphingosine 1-phosphate receptor-1 agonists exacerbates vascular leak, fibrosis, and mortality after lung injury. Am J Respir Cell Mol Biol 43: 662–673, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 565.Shimoda LA. 55th Bowditch Lecture: Effects of chronic hypoxia on the pulmonary circulation: Role of HIF-1. J Appl Physiol 113: 1343–1352, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 566.Shimoda LA, Fallon M, Pisarcik S, Wang J, Semenza GL. HIF-1 regulates hypoxic induction of NHE1 expression and alkalinization of intracellular pH in pulmonary arterial myocytes. Am J Physiol Lung Cell Mol Physiol 291: L941–L949, 2006. [DOI] [PubMed] [Google Scholar]
- 567.Shimoda LA, Laurie SS. Vascular remodeling in pulmonary hypertension. J Mol Med 91: 297–309, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 568.Shimoda LA, Manalo DJ, Sham JS, Semenza GL, Sylvester JT. Partial HIF-1α deficiency impairs pulmonary arterial myocyte electrophysio-logical responses to hypoxia. Am J Physiol Lung Cell Mol Physiol 281: L202–L208, 2001. [DOI] [PubMed] [Google Scholar]
- 569.Shimoda LA, Polak J. Theme: Hypoxia hypoxia and ion channel function. Am J Physiol Cell Physiol 300(5): C951–C967, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 570.Shimoda LA, Sham JS, Liu Q, Sylvester JT. Acute and chronic hypoxic pulmonary vasoconstriction: A central role for endothelin-1? Respir Physiol Neurobiol 132: 93–106, 2002. [DOI] [PubMed] [Google Scholar]
- 571.Shimoda LA, Sham JS, Shimoda TH, Sylvester JT. L-type Ca2+ channels, resting [Ca2+]i, and ET-1-induced responses in chronically hypoxic pulmonary myocytes. Am J Physiol Lung Cell Mol Physiol 279: L884–L894, 2000. [DOI] [PubMed] [Google Scholar]
- 572.Shimoda LA, Wang J, Sylvester JT. Ca2+ channels and chronic hypoxia. Microcirculation 13: 657–670, 2006. [DOI] [PubMed] [Google Scholar]
- 573.Shimonaka M, Saheki T, Hagiwara H, Ishido M, Nogi A, Fujita T, Wakita K, Inada Y, Kondo J, Hirose S. Purification of atrial natriuretic peptide receptor from bovine lung. Evidence for a disulfide-linked subunit structure. J Biol Chem 262: 5510–5514, 1987. [PubMed] [Google Scholar]
- 574.Shirai M, Shindo T, Shimouchi A, Ninomiya I. Diameter and flow velocity changes of feline small pulmonary vessels in response to sympathetic nerve stimulation. Pflugers Archiv 429: 267–273, 1994. [DOI] [PubMed] [Google Scholar]
- 575.Shirley KL, Beckman DW, Garrick DJ. Inheritance of pulmonary arterial pressure in Angus cattle and its correlation with growth. J Animal Sci 86: 815–819, 2008. [DOI] [PubMed] [Google Scholar]
- 576.Shu W, Jiang YQ, Lu MM, Morrisey EE. Wnt7b regulates mesenchymal proliferation and vascular development in the lung. Development 129: 4831–4842, 2002. [DOI] [PubMed] [Google Scholar]
- 577.Sieber C, Kopf J, Hiepen C, Knaus P. Recent advances in BMP receptor signaling. Cytokine Growth Factor Rev 20: 343–355, 2009. [DOI] [PubMed] [Google Scholar]
- 578.Siegelman SS, Hagstrom JW, Koerner SK, Veith FJ. Restoration of bronchial artery circulation after canine lung allotransplantation. J Thor Cardiovasc Surg 73: 792–795, 1977. [PubMed] [Google Scholar]
- 579.Siflinger-Birnboim A, Del Vecchio PJ, Cooper JA, Blumenstock FA, Shepard JM, Malik AB. Molecular sieving characteristics of the cultured endothelial monolayer. J Cell Physiol 132: 111–117, 1987. [DOI] [PubMed] [Google Scholar]
- 580.Silove ED, Inoue T, Grover RF. Comparison of hypoxia, pH, and sympathomimetic drugs on bovine pulmonary vasculature. J Appl Physiol 24: 355–365, 1968. [DOI] [PubMed] [Google Scholar]
- 581.Simmons D, Linde L, Miller J, O’Reilly R. Relation between lung volume and pulmonary vascular resistance. Circ res 9: 465–471, 1961. [Google Scholar]
- 582.Simonneau G, D’Armini AM, Ghofrani HA, Grimminger F, Hoeper MM, Jansa P, Kim NH, Wang C, Wilkins MR, Fritsch A, Davie N, Colorado P, Mayer E. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension: A long-term extension study (CHEST-2). Eur Respir J 45: 1293–1302, 2015. [DOI] [PubMed] [Google Scholar]
- 583.Simonson TS, Yang Y, Huff CD, Yun H, Qin G, Witherspoon DJ, Bai Z, Lorenzo FR, Xing J, Jorde LB, Prchal JT, Ge R. Genetic evidence for high-altitude adaptation in Tibet. Science 329: 72–75, 2010. [DOI] [PubMed] [Google Scholar]
- 584.Smirnov SV, Robertson TP, Ward JP, Aaronson PI. Chronic hypoxia is associated with reduced delayed rectifier K+ current in rat pulmonary artery muscle cells. Am J Physiol 266: H365–H370, 1994. [DOI] [PubMed] [Google Scholar]
- 585.Smith TG, Brooks JT, Balanos GM, Lappin TR, Layton DM, Leedham DL, Liu C, Maxwell PH, McMullin MF, McNamara CJ, Percy MJ, Pugh CW, Ratcliffe PJ, Talbot NP, Treacy M, Robbins PA. Mutation of von Hippel-Lindau tumour suppressor and human cardiopulmonary physiology. PLoS Med 3: e290, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 586.Sobol BJ, Bottex G, Emirgil C, Gissen H. Gaseous diffusion from alveoli to pulmonary vessels of considerable size. Circ Res 13: 71–79, 1963. [DOI] [PubMed] [Google Scholar]
- 587.Song MY, Makino A, Yuan JX. STIM2 contributes to enhanced store-operated ca entry in pulmonary artery smooth muscle cells from patients with idiopathic pulmonary arterial hypertension. Pulm Circ 1: 84–94, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 588.Song Y, Jones JE, Beppu H, Keaney JF, Loscalzo J, Zhang Y-Y. Increased susceptibility to pulmonary hypertension in heterozygous BMPR2-mutant mice. Circulation 112: 553–562, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 589.Spalding MB, Ala-Kokko TI, Kiviluoma K, Ruskoaho H, Alahuhta S. The hemodynamic effects of adenosine infusion after experimental right heart infarct in young swine. J Cardiovasc Pharmacol 35: 93–99, 2000. [DOI] [PubMed] [Google Scholar]
- 590.Spiekerkoetter E, Sung YK, Sudheendra D, Bill M, Aldred MA, van de Veerdonk MC, Vonk Noordegraaf A, Long-Boyle J, Dash R, Yang PC, Lawrie A, Swift AJ, Rabinovitch M, Zamanian RT. Low-Dose FK506 (Tacrolimus) in end-stage pulmonary arterial hypertension. Am J Resp Crit Care Med 192: 254–257, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 591.Spiekerkoetter E, Tian X, Cai J, Hopper RK, Sudheendra D, Li CG, El-Bizri N, Sawada H, Haghighat R, Chan R, Haghighat L, de Jesus Perez V, Wang L, Reddy S, Zhao M, Bernstein D, Solow-Cordero DE, Beachy PA, Wandless TJ, Ten Dijke P, Rabinovitch M. FK506 activates BMPR2, rescues endothelial dysfunction, and reverses pulmonary hypertension. J Clin Invest 123: 3600–3613, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 592.Stenmark KR, Abman SH. Lung vascular development: Implications for the pathogenesis of bronchopulmonary dysplasia. Ann Rev Physiol 67: 623–661, 2005. [DOI] [PubMed] [Google Scholar]
- 593.Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling: Cellular and molecular mechanisms. Circ Res 99: 675–691, 2006. [DOI] [PubMed] [Google Scholar]
- 594.Stenmark KR, Fasules J, Hyde DM, Voelkel NF, Henson J, Tucker A, Wilson H, Reeves JT. Severe pulmonary hypertension and arterial adventitial changes in newborn calves at 4,300 m. J Appl Physiol 62: 821–830, 1987. [DOI] [PubMed] [Google Scholar]
- 595.Stenmark KR, McMurtry IF. Vascular remodeling versus vasoconstriction in chronic hypoxic pulmonary hypertension: a time for reappraisal? Circ Res 97: 95–98, 2005. [DOI] [PubMed] [Google Scholar]
- 596.Stenmark KR, Meyrick B, Galie N, Mooi WJ, McMurtry IF. Animal models of pulmonary arterial hypertension: The hope for etiological discovery and pharmacological cure. Am J Physiol Lung Cell Mol Physiol 297: L1013–L1032, 2009. [DOI] [PubMed] [Google Scholar]
- 597.Stenmark KR, Rabinovitch M. Emerging therapies for the treatment of pulmonary hypertension. Pediatr Crit Care Med 11: S85–S90, 2010. [DOI] [PubMed] [Google Scholar]
- 598.Stephens RS, Servinsky LE, Rentsendorj O, Kolb TM, Pfeifer A, Pearse DBCP. Protein kinase G increases antioxidant function in lung microvascular endothelial cells by inhibiting the c-Abl tyrosine kinase. Am J Physiol Cell Physiol 306: C559–C569, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 599.Stephenson AH, Sprague RS, Losapio JL, Lonigro AJ. Differential effects of 5,6-EET on segmental pulmonary vasoactivity in the rabbit. Am J Physiol Heart Circ Physiol 284: H2153–H2161, 2003. [DOI] [PubMed] [Google Scholar]
- 600.Stevens. Functional and molecular heterogeneity of pulmonary endothelial cells. Proc Am Thorac Soc 8: 453–457, 2011. [DOI] [PubMed] [Google Scholar]
- 601.Stevens T, Phan S, Frid MG, Alvarez D, Herzog E, Stenmark KR. Lung vascular cell heterogeneity: Endothelium, smooth muscle, and fibroblasts. Proc Am Thorac Soc 5: 783–791, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 602.Stock KW, Chen Q, Levin D, Hatabu H, Edelman RR. Demonstration of gravity-dependent lung perfusion with contrast-enhanced magnetic resonance imaging. J Magn Reson Imaging 9: 557–561, 1999. [DOI] [PubMed] [Google Scholar]
- 603.Storme L, Rairigh RL, Parker TA, Kinsella JP, Abman SH. In vivo evidence for a myogenic response in the fetal pulmonary circulation. Pediatr Res 45: 425–431, 1999. [DOI] [PubMed] [Google Scholar]
- 604.Suri C, Jones PF, Patan S, Bartunkova S, Maisonpierre PC, Davis S, Sato TN, Yancopoulos GD. Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell 87: 1171–1180, 1996. [DOI] [PubMed] [Google Scholar]
- 605.Suzuki H, Twarog BM. Membrane properties of smooth muscle cells in pulmonary hypertensive rats. Am J Physiol 242: H907–H915, 1982. [DOI] [PubMed] [Google Scholar]
- 606.Sylvester JT, Shimoda LA, Aaronson PI, Ward JP. Hypoxic pulmonary vasoconstriction. Physiol Rev 92: 367–520, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 607.Sylvin H, Weitzberg E, Alving K. Endothelin-induced vascular and bronchial effects in pig airways: Role in acute allergic responses. J Appl Physiol 93: 1608–1615, 2002. [DOI] [PubMed] [Google Scholar]
- 608.Szczepaniak WS, Zhang Y, Hagerty S, Crow MT, Kesari P, Garcia JG, Choi AM, Simon BA, McVerry BJ. Sphingosine 1-phosphate rescues canine LPS-induced acute lung injury and alters systemic inflammatory cytokine production in vivo. Transl Res 152: 213–224, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 609.Tabakin BS, Hanson JS. Congenital absence of the aortic arch associated with patent ductus arteriosus and ventricular septal defect. Am J Cardiol 6: 689–696, 1960. [DOI] [PubMed] [Google Scholar]
- 610.Tabeling C, Yu H, Wang L, Ranke H, Goldenberg NM, Zabini D, Noe E, Krauszman A, Gutbier B, Yin J, Schaefer M, Arenz C, Hocke AC, Suttorp N, Proia RL, Witzenrath M, Kuebler WM. CFTR and sphingolipids mediate hypoxic pulmonary vasoconstriction. Proc Nat Acad Sci U S A 112: E1614–E1623, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 611.Tabuchi A, Styp-Rekowska B, Slutsky AS, Wagner PD, Pries AR, Kuebler WM. Precapillary oxygenation contributes relevantly to gas exchange in the intact lung. Am J Resp Crit Care Med 188: 474–481, 2013. [DOI] [PubMed] [Google Scholar]
- 612.Takahashi H, Goto N, Kojima Y, Tsuda Y, Morio Y, Muramatsu M, Fukuchi Y. Downregulation of type II bone morphogenetic protein receptor in hypoxic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 290: L450–L458, 2006. [DOI] [PubMed] [Google Scholar]
- 613.Takahashi H, Ikeda T. Transcripts for two members of the transforming growth factor-beta superfamily BMP-3 and BMP-7 are expressed in developing rat embryos. Dev Dynam 207: 439–449, 1996. [DOI] [PubMed] [Google Scholar]
- 614.Tamaoki J, Sugimoto F, Tagaya E, Isono K, Chiyotani A, Konno K. Angiotensin II 1 receptor-mediated contraction of pulmonary artery and its modulation by prolylcarboxypeptidase. J Appl Physiol 76: 1439–1444, 1994. [DOI] [PubMed] [Google Scholar]
- 615.Tamby MC, Chanseaud Y, Humbert M, Fermanian J, Guilpain P, Garcia-de-la-Pena-Lefebvre P, Brunet S, Servettaz A, Weill B, Simonneau G, Guillevin L, Boissier MC, Mouthon L. Anti-endothelial cell antibodies in idiopathic and systemic sclerosis associated pulmonary arterial hypertension. Thorax 60: 765–772, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 616.Tamby MC, Humbert M, Guilpain P, Servettaz A, Dupin N, Christner JJ, Simonneau G, Fermanian J, Weill B, Guillevin L, Mouthon L. Antibodies to fibroblasts in idiopathic and scleroderma-associated pulmonary hypertension. Eur Respir J 28: 799–807, 2006. [DOI] [PubMed] [Google Scholar]
- 617.Tamosiuniene R, Tian W, Dhillon G, Wang L, Sung YK, Gera L, Patterson AJ, Agrawal R, Rabinovitch M, Ambler K, Long CS, Voelkel NF, Nicolls MR. Regulatory T cells limit vascular endothelial injury and prevent pulmonary hypertension. Circ Res 109: 867–879, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 618.Tan JZ, Kaley G, Gurtner GH. Nitric oxide and prostaglandins mediate vasodilation to 5,6-EET in rabbit lung. Adv Exp Med Biol 407: 561–566, 1997. [DOI] [PubMed] [Google Scholar]
- 619.Tan Q, Kerestes H, Percy MJ, Pietrofesa R, Chen L, Khurana TS, Christofidou-Solomidou M, Lappin TR, Lee FS. Erythrocytosis and pulmonary hypertension in a mouse model of human HIF2A gain of function mutation. J Biol Chem 288: 17134–17144, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 620.Tanaka Y, Schuster DP, Davis EC, Patterson GA, Botney MD. The role of vascular injury and hemodynamics in rat pulmonary artery remodeling. J Clin Invest 98:434–442, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 621.Taraseviciene-Stewart L, Kasahara Y, Alger L, Hirth P, Mc Mahon G, Waltenberger J, Voelkel NF, Tuder RM. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J 15: 427–438, 2001. [DOI] [PubMed] [Google Scholar]
- 622.Taraseviciene-Stewart L, Nicolls MR, Kraskauskas D, Scerbavicius R, Burns N, Cool C, Wood K, Parr JE, Boackle SA, Voelkel NF. Absence of T cells confers increased pulmonary arterial hypertension and vascular remodeling. Am J Resp Crit Care Med 175: 1280–1289, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 623.Taraseviciene-Stewart L, Scerbavicius R, Choe KH, Cool C, Wood K, Tuder RM, Burns N, Kasper M, Voelkel NF. Simvastatin causes endothelial cell apoptosis and attenuates severe pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 291: L668–L676, 2006. [DOI] [PubMed] [Google Scholar]
- 624.Teerlink JR, Carteaux JP, Sprecher U, Loffler BM, Clozel M, Clozel JP. Role of endogenous endothelin in normal hemodynamic status of anesthetized dogs. Am J Physiol 268: H432–H440, 1995. [DOI] [PubMed] [Google Scholar]
- 625.Teichert-Kuliszewska K, Kutryk MJ, Kuliszewski MA, Karoubi G, Courtman DW, Zucco L, Granton J, Stewart DJ. Bone morphogenetic protein receptor-2 signaling promotes pulmonary arterial endothelial cell survival: Implications for loss-of-function mutations in the pathogenesis of pulmonary hypertension. Circ Res 98: 209–217, 2006. [DOI] [PubMed] [Google Scholar]
- 626.Thickett DR, Armstrong L, Christie SJ, Millar AB. Vascular endothelial growth factor may contribute to increased vascular permeability in acute respiratory distress syndrome. Am J Respir Crit Care Med 164: 1601–1605, 2001. [DOI] [PubMed] [Google Scholar]
- 627.Thomas M Pharmacological targets for pulmonary vascular disease: Vasodilation versus anti-remodelling. Adv Exp Med Biol 661: 475–490, 2010. [DOI] [PubMed] [Google Scholar]
- 628.Thomson JR, Machado RD, Pauciulo MW, Morgan NV, Humbert M, Elliott GC, Ward K, Yacoub M, Mikhail G, Rogers P, Newman J, Wheeler L, Higenbottam T, Gibbs JS, Egan J, Crozier A, Peacock A, Allcock R, Corris P, Loyd JE, Trembath RC, Nichols WC. Sporadic primary pulmonary hypertension is associated with germline mutations of the gene encoding BMPR-II, a receptor member of the TGF-β family. J Med Genet 37: 741–745, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 629.Thorin E, Clozel M. The cardiovascular physiology and pharmacology of endothelin-1. Adv Pharmacol 60: 1–26, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 630.Tian W, Jiang X, Tamosiuniene R, Sung YK, Qian J, Dhillon G, Gera L, Farkas L, Rabinovitch M, Zamanian RT, Inayathullah M, Fridlib M, Rajadas J, Peters-Golden M, Voelkel NF, Nicolls MR. Blocking macrophage leukotriene b4 prevents endothelial injury and reverses pulmonary hypertension. Sci Trans Med 5: 200ra117, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 631.Tiruppathi C, Ahmmed GU, Vogel SM, Malik AB. Ca2+ signaling, TRP channels, and endothelial permeability. Microcirculation 13: 693–708, 2006. [DOI] [PubMed] [Google Scholar]
- 632.Tiruppathi C, Minshall RD, Paria BC, Vogel SM, Malik AB. Role of Ca2+ signaling in the regulation of endothelial permeability. Vascul Pharmacol 39: 173–185, 2002. [DOI] [PubMed] [Google Scholar]
- 633.Toba M, Alzoubi A, O’Neill KD, Gairhe S, Matsumoto Y, Oshima K, Abe K, Oka M, McMurtry IF. Temporal hemodynamic and histological progression in Sugen5416/hypoxia/normoxia-exposed pulmonary arterial hypertensive rats. Am J Physiol Heart Circ Physiol 306: H243–H250, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 634.Townsley MI, King JA, Alvarez DF. Ca2+ channels and pulmonary endothelial permeability: Insights from study of intact lung and chronic pulmonary hypertension. Microcirculation 13: 725–739, 2006. [DOI] [PubMed] [Google Scholar]
- 635.Townsley MI. Structure and composition of pulmonary arteries, capillaries, and veins. Compr Physiol 2: 675–709, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 636.Troyanovsky B, Alvarez DF, King JA, Schaphorst KL. Thrombin enhances the barrier function of rat microvascular endothelium in a PAR-1-dependent manner. Am J Physiol Lung Cell Mol Physiol 294: L266–L275, 2008. [DOI] [PubMed] [Google Scholar]
- 637.Tuder RM. Pathology of pulmonary arterial hypertension. Semin Respir Crit Care Med 30: 376–385, 2009. [DOI] [PubMed] [Google Scholar]
- 638.Tuder RM, Groves B, Badesch DB, Voelkel NF. Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. Am J Path 144: 275–285, 1994. [PMC free article] [PubMed] [Google Scholar]
- 639.Uchida K, Mizuno T, Shimonaka M, Sugiura N, Nara K, Ling N, Hagiwara H, Hirose S. Purification and properties of active atrial-natriuretic-peptide receptor (type C) from bovine lung. Biochem J 263: 671–678, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 640.Uhlig S, Yang Y. Sphingolipids in acute lung injury. Handb Exp Pharmacol 2013(216): 227–246. [DOI] [PubMed] [Google Scholar]
- 641.Uhlig S, Yang Y, Waade J, Wittenberg C, Babendreyer A, Kuebler WM. Differential regulation of lung endothelial permeability in vitro and in situ. Cell Physiol Biochem 34: 1–19, 2014. [DOI] [PubMed] [Google Scholar]
- 642.Undem C, Rios EJ, Maylor J, Shimoda LA. Endothelin-1 augments Na+/H+ exchange activity in murine pulmonary arterial smooth muscle cells via Rho kinase. PloS one 7: e46303, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 643.van den Bos GC, Westerhof N, Randall OS. Pulse wave reflection: can it explain the differences between systemic and pulmonary pressure and flow waves? A study in dogs. Circ Res 51: 479–485, 1982. [DOI] [PubMed] [Google Scholar]
- 644.van Patot MC, Gassmann M. Hypoxia: Adapting to high altitude by mutating EPAS-1, the gene encoding HIF-2α. High Alt Med Biol 12: 157–167, 2011. [DOI] [PubMed] [Google Scholar]
- 645.van Tuyl M, Liu J, Wang J, Kuliszewski M, Tibboel D, Post M. Role of oxygen and vascular development in epithelial branching morphogenesis of the developing mouse lung. Am J Physiol Lung Cell Mol Physiol 288: L167–L178, 2005. [DOI] [PubMed] [Google Scholar]
- 646.Vandenbroucke E, Mehta D, Minshall R, Malik AB. Regulation of endothelial junctional permeability. Ann NY Acad Sci 1123: 134, 2008. [DOI] [PubMed] [Google Scholar]
- 647.Vanderpool RR, Kim AR, Molthen R, Chesler NC. Effects of acute Rho kinase inhibition on chronic hypoxia-induced changes in proximal and distal pulmonary arterial structure and function. J Appl Physiol 110: 188–198, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 648.Vergadi E, Chang MS, Lee C, Liang OD, Liu X, Fernandez-Gonzalez A, Mitsialis SA, Kourembanas S. Early macrophage recruitment and alternative activation are critical for the later development of hypoxia-induced pulmonary hypertension. Circulation 123: 1986–1995, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 649.Viola AR, Abbate EH. Respiratory function and effective collateral flow 8 years after ligation of the left pulmonary artery. Am Rev Resp Dis 108: 1216–1220, 1973. [DOI] [PubMed] [Google Scholar]
- 650.Voelkel NF, Tuder RM. Hypoxia-induced pulmonary vascular remodeling: A model for what human disease? J Clin Invest 106: 733–738, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 651.Vogel SM, Malik AB. Cytoskeletal dynamics and lung fluid balance. Compr Physiol 2: 449–478, 2012. [DOI] [PubMed] [Google Scholar]
- 652.von Euler U, Liljestrand G. Observations on the pulmonary arterial blood pressure in the cat. Acta Phys Scandinav 12: 301–320, 1946. [Google Scholar]
- 653.Wagner EM, Brown RH. Blood flow distribution within the airway wall. J Appl Physiol 92: 1964–1969, 2002. [DOI] [PubMed] [Google Scholar]
- 654.Wagner EM, Foster WM. Interdependence of bronchial circulation and clearance of 99mTc-DTPA from the airway surface. J Appl Physiol 90: 1275–1281, 2001. [DOI] [PubMed] [Google Scholar]
- 655.Wagner EM, Foster WM. The role of the bronchial vasculature in soluble particle clearance. Environ Health Perspect 109(Suppl 4): 563–565, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 656.Wagner EM, Mitzner W, Brown RH. Site of functional bronchopulmonary anastomoses in sheep. Anatom Rec 254: 360–366, 1999. [DOI] [PubMed] [Google Scholar]
- 657.Wagner JG, Roth RA. Neutrophil migration mechanisms, with an emphasis on the pulmonary vasculature. Pharmacol Rev 52: 349–374, 2000. [PubMed] [Google Scholar]
- 658.Walch L, Norel X, Gascard JP, Brink C. Functional studies of leukotriene receptors in vascular tissues. Am J Respir Crit Care Med 161: S107–S111, 2000. [DOI] [PubMed] [Google Scholar]
- 659.Walker BR, Voelkel NF, Reeves JT. Pulmonary pressor response after prostaglandin synthesis inhibition in conscious dogs. J Appl Physiol Respir Environ Exerc Physiol 52: 705–709, 1982. [DOI] [PubMed] [Google Scholar]
- 660.Walsh-Sukys MC, Tyson JE, Wright LL, Bauer CR, Korones SB, Stevenson DK, Verter J, Stoll BJ, Lemons JA, Papile LA, Shankaran S, Donovan EF, Oh W, Ehrenkranz RA, Fanaroff AA. Persistent pulmonary hypertension of the newborn in the era before nitric oxide: Practice variation and outcomes. Pediatrics 105: 14–20, 2000. [DOI] [PubMed] [Google Scholar]
- 661.Walther SM, Domino KB, Glenny RW, Hlastala MP. Positive end-expiratory pressure redistributes perfusion to dependent lung regions in supine but not in prone lambs. Crit Care Med 27: 37–45, 1999. [DOI] [PubMed] [Google Scholar]
- 662.Wang J, Juhaszova M, Rubin LJ, Yuan XJ. Hypoxia inhibits gene expression of voltage-gated K+ channel a subunits in pulmonary artery smooth muscle cells. J Clin Invest 100: 2347–2353, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 663.Wang J, Shimoda LA, Sylvester JT. Ca2+ responses of pulmonary arterial myocytes to acute hypoxia require release from ryanodine and inositol trisphosphate receptors in sarcoplasmic reticulum. Am J Physiol Lung Cell Mol Physiol 303: L161–L168, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 664.Wang J, Shimoda LA, Weigand L, Wang W, Sun D, Sylvester JT. Acute hypoxia increases intracellular [Ca2+] in pulmonary arterial smooth muscle by enhancing capacitative Ca2+ entry. Am J Physiol Lung Cell Mol Physiol 288: L1059–L1069, 2005. [DOI] [PubMed] [Google Scholar]
- 665.Wang J, Weigand L, Foxson J, Shimoda LA, Sylvester JT. Ca2+ signaling in hypoxic pulmonary vasoconstriction: effects of myosin light chain and Rho kinase antagonists. Am J Physiol Lung Cell Mol Physiol 293: L674–L685, 2007. [DOI] [PubMed] [Google Scholar]
- 666.Wang J, Weigand L, Lu W, Sylvester JT, Semenza GL, Shimoda LA. Hypoxia inducible factor 1 mediates hypoxia-induced TRPC expression and elevated intracellular Ca2+ in pulmonary arterial smooth muscle cells. Circ Res 98: 1528–1537, 2006. [DOI] [PubMed] [Google Scholar]
- 667.Wang J, Yang K, Xu L, Zhang Y, Lai N, Jiang H, Zhang Y, Zhong N, Ran P, Lu W. Sildenafil inhibits hypoxia-induced transient receptor potential canonical protein expression in pulmonary arterial smooth muscle via cGMP-PKG-PPARgamma axis. Am J Respir Cell Mol Biol 49: 231–240, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 668.Wang L, Dudek SMC. Regulation of vascular permeability by sphingosine 1-phosphate. Microvasc Res 77: 39–45, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 669.Wang L, Yin J, Nickles HT, Ranke H, Tabuchi A, Hoffmann J, Tabeling C, Barbosa-Sicard E, Chanson M, Kwak BR, Shin HS, Wu S, Isakson BE, Witzenrath M, de Wit C, Fleming I, Kuppe H, Kuebler WM. Hypoxic pulmonary vasoconstriction requires connexin 40-mediated endothelial signal conduction. J Clin Invest 122: 4218–4230, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 670.Wang le F, Patel M, Razavi HM, Weicker S, Joseph MG, McCormack DG, Mehta S. Role of inducible nitric oxide synthase in pulmonary microvascular protein leak in murine sepsis. Am J Resp Crit Care Med 165: 1634–1639, 2002. [DOI] [PubMed] [Google Scholar]
- 671.Ward JPT, Mcmurtry IF. Mechanisms of hypoxic pulmonary vasoconstriction and their roles in pulmonary hypertension: New findings for an old problem. Cur Opin Pharmacol 9: 287–296, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 672.Warrell DA, Evans JW, Clarke RO, Kingaby GP, West JBC. Pattern of filling in the pulmonary capillary bed. J Appl Physiol 32: 346–356, 1972. [DOI] [PubMed] [Google Scholar]
- 673.Warren RL, Powell WJ Jr. Acute alveolar hypoxia increases bronchopulmonary shunt flow in the dog. J Clin Invest 77: 1515–1524, 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 674.Waypa GB, Chandel NS, Schumacker PT. Model for hypoxic pulmonary vasoconstriction involving mitochondrial oxygen sensing. Circ Res 88: 1259–1266, 2001. [DOI] [PubMed] [Google Scholar]
- 675.Waypa GB, Guzy R, Mungai PT, Mack MM, Marks JD, Roe MW, Schumacker PT. Increases in mitochondrial reactive oxygen species trigger hypoxia-induced calcium responses in pulmonary artery smooth muscle cells. Circ Res 99: 970–978, 2006. [DOI] [PubMed] [Google Scholar]
- 676.Waypa GB, Marks JD, Guzy R, Mungai PT, Schriewer J, Dokic D, Schumacker PT. Hypoxia triggers subcellular compartmental redox signaling in vascular smooth muscle cells. Circ Res 106: 526–535, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 677.Waypa GB, Marks JD, Mack MM, Boriboun C, Mungai PT, Schumacker PT. Mitochondrial reactive oxygen species trigger calcium increases during hypoxia in pulmonary arterial myocytes. Circ Res 91: 719–726, 2002. [DOI] [PubMed] [Google Scholar]
- 678.Weibel ER. Morphological basis of alveolar-capillary gas exchange. Physiol Rev 53: 419–495, 1973. [DOI] [PubMed] [Google Scholar]
- 679.Weigand L, Foxson J, Wang J, Shimoda LA, Sylvester JT. Inhibition of hypoxic pulmonary vasoconstriction by antagonists of store-operated Ca2+ and nonselective cation channels. Am J Physiol Lung Cell Mol Physiol 289: L5–L13, 2005. [DOI] [PubMed] [Google Scholar]
- 680.Weir EK, McMurtry IF, Tucker A, Reeves JT, Grover RF. Prostaglandin synthetase inhibitors do not decrease hypoxic pulmonary vasoconstriction. J Appl Physiol 41: 714–718, 1976. [DOI] [PubMed] [Google Scholar]
- 681.Weir EK, Tucker A, Reeves JT, Will DH, Grover RF. The genetic factor influencing pulmonary hypertension in cattle at high altitude. Cardiovasc Res 8: 745–749, 1974. [DOI] [PubMed] [Google Scholar]
- 682.Weissmann N, Voswinckel R, Hardebusch T, Rosseau S, Ghofrani HA, Schermuly R, Seeger W, Grimminger F. Evidence for a role of protein kinase C in hypoxic pulmonary vasoconstriction. Am J Physiol 276: L90–L95, 1999. [DOI] [PubMed] [Google Scholar]
- 683.Weitzberg E, Hemsen A, Rudehill A, Modin A, Wanecek M, Lundberg JM. Bosentan-improved cardiopulmonary vascular performance and increased plasma levels of endothelin-1 in porcine endotoxin shock. Br J Pharmacol 118: 617–626, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 684.West J, Fagan K, Steudel W, Fouty B, Lane K, Harral J, Hoedt-Miller M, Tada Y, Ozimek J, Tuder R, Rodman DM. Pulmonary hypertension in transgenic mice expressing a dominant-negative BMPRII gene in smooth muscle. Circ Res 94: 1109–1114, 2004. [DOI] [PubMed] [Google Scholar]
- 685.West J, Hemnes A. Experimental and transgenic models of pulmonary hypertension. Compr Physiol 1: 769–782, 2011. [DOI] [PubMed] [Google Scholar]
- 686.West JB. Comparative physiology of the pulmonary circulation. Compr Physiol 1: 1525–1539, 2011. [DOI] [PubMed] [Google Scholar]
- 687.West JB. Fragility of pulmonary capillaries. J Appl Physiol 115: 1–15, 2013. [DOI] [PubMed] [Google Scholar]
- 688.West JB, Dollery CT, Naimark A. Distribution of blood flow in isolated lung; relation to vascular and alveolar pressures. J Appl Physiol 19: 713–724, 1964. [DOI] [PubMed] [Google Scholar]
- 689.Wharton J, Strange JW, Moller GM, Growcott EJ, Ren X, Franklyn AP, Phillips SC, Wilkins MR. Antiproliferative effects of phosphodiesterase type 5 inhibition in human pulmonary artery cells. Am J Respir Crit Care Med 172: 105–113, 2005. [DOI] [PubMed] [Google Scholar]
- 690.White K, Dempsie Y, Nilsen M, Wright AF, Loughlin L, MacLean MR. The serotonin transporter, gender, and 17β oestradiol in the development of pulmonary arterial hypertension. Cardiovasc Res 90: 373–382, 2011. [DOI] [PubMed] [Google Scholar]
- 691.Whitman EM, Pisarcik S, Luke T, Fallon M, Wang J, Sylvester JT, Semenza GL, Shimoda LA. Endothelin-1 mediates hypoxia-induced inhibition of voltage-gated K+ channel expression in pulmonary arterial myocytes. Am J Physiol Lung Cell Mol Physiol 294: L309–L318, 2008. [DOI] [PubMed] [Google Scholar]
- 692.Wicks TC, Rose JC, Johnson M, Ramwell PW, Kot PA. Vascular responses to arachidonic acid in the perfused canine lung. Circ Res 38: 167–171, 1976. [DOI] [PubMed] [Google Scholar]
- 693.Wiener CM, Kirk W, Albert RK. Prone position reverses gravitational distribution of perfusion in dog lungs with oleic acid-induced injury. J Appl Physiol 68: 1386–1392, 1990. [DOI] [PubMed] [Google Scholar]
- 694.Wiener F, Morkin E, Skalak R, Fishman AP. Wave propagation in the pulmonary circulation. Circ Res 19: 834–850, 1966. [DOI] [PubMed] [Google Scholar]
- 695.Wiklund NP, Cederqvist B, Matsuda H, Gustafsson LE. Adenosine can excite pulmonary artery. Acta Physiol Scand 131: 477–478, 1987. [DOI] [PubMed] [Google Scholar]
- 696.Wilkins MR, Ali O, Bradlow W, Wharton J, Taegtmeyer A, Rhodes CJ, Ghofrani HA, Howard L, Nihoyannopoulos P, Mohiaddin RH, Gibbs JS, Simvastatin Pulmonary Hypertension Trial Study G. Simvastatin as a treatment for pulmonary hypertension trial. Am J Resp Crit Care Med 181: 1106–1113, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 697.Wilkinson M, Langhorne CA, Heath D, Barer GR, Howard P. A patho-physiological study of 10 cases of hypoxic cor pulmonale. Q J Med 66: 65–85, 1988. [PubMed] [Google Scholar]
- 698.Will DH, Hicks JL, Card CS, Alexander AF. Inherited susceptibility of cattle to high-altitude pulmonary hypertension. J Appl Physiol 38: 491–494, 1975. [DOI] [PubMed] [Google Scholar]
- 699.Williams AE, Chambers RCCP. The mercurial nature of neutrophils: Still an enigma in ARDS? Am J Physiol Lung Cell Mol Physiol 306: L217–L230, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 700.Winters JW, Wong J, Van Dyke D, Johengen M, Heymann MA, Fineman JR. Endothelin receptor blockade does not alter the increase in pulmonary blood flow due to oxygen ventilation in fetal lambs. Pediatr Res 40: 152–157, 1996. [DOI] [PubMed] [Google Scholar]
- 701.Wojciak-Stothard B, Haworth SG. Perinatal changes in pulmonary vascular endothelial function. Pharmacol Ther 109: 78–91, 2006. [DOI] [PubMed] [Google Scholar]
- 702.Wojciak-Stothard B, Zhao L, Oliver E, Dubois O, Wu Y, Kardassis D, Vasilaki E, Huang M, Mitchell JA, Harrington LS, Prendergast GC, Wilkins MR. Role of RhoB in the regulation of pulmonary endothelial and smooth muscle cell responses to hypoxia. Circ Res 110: 1423–1434, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 703.Wright JL, Petty T, Thurlbeck WM. Analysis of the structure of the muscular pulmonary arteries in patients with pulmonary hypertension and COPD: National Institutes of Health nocturnal oxygen therapy trial. Lung 170: 109–124, 1992. [DOI] [PubMed] [Google Scholar]
- 704.Xu W, Koeck T, Lara AR, Neumann D, DiFilippo FP, Koo M, Janocha AJ, Masri FA, Arroliga AC, Jennings C, Dweik RA, Tuder RM, Stuehr DJ, Erzurum SC. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc Nat Acad Sci U S A 104: 1342–1347, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 705.Yadav VR, Song T, Joseph L, Mei L, Zheng YM, Wang YX. Important role of PLC-gamma1 in hypoxic increase in intracellular calcium in pulmonary arterial smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 304: L143–L151, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 706.Yaghi A, Webb CD, Scott JA, Mehta S, Bend JR, McCormack DG. Cytochrome P450 metabolites of arachidonic acid but not cyclooxygenase-2 metabolites contribute to the pulmonary vascular hyporeactivity in rats with acute Pseudomonas pneumonia. J Pharmacol Exp Ther 297: 479–488, 2001. [PubMed] [Google Scholar]
- 707.Yamamoto H, Yun EJ, Gerber HP, Ferrara N, Whitsett JA, Vu TH. Epithelial-vascular cross talk mediated by VEGF-A and HGF signaling directs primary septae formation during distal lung morphogenesis. Dev Biol 308: 44–53, 2007. [DOI] [PubMed] [Google Scholar]
- 708.Yan W, Wu F, Morser J, Wu Q. Corin, a transmembrane cardiac serine protease, acts as a pro-atrial natriuretic peptide-converting enzyme. Proc Nat Acad Sci U S A 97: 8525–8529, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 709.Yanagisawa M, Kurihara H, Kimura S, Goto K, Masaki T. A novel peptide vasoconstrictor, endothelin, is produced by vascular endothelium and modulates smooth muscle Ca2+ channels. J Hypertens Suppl 6: S188–S191, 1988. [DOI] [PubMed] [Google Scholar]
- 710.Yang C, Jiang L, Zhang H, Shimoda LA, DeBerardinis RJ, Semenza GL. Analysis of hypoxia-induced metabolic reprogramming. Methods Enzymol 542: 425–455, 2014. [DOI] [PubMed] [Google Scholar]
- 711.Yang J, Li X, Al-Lamki R, Wu C, Weiss A, Berk J, Schermuly RT, Morrell NW. Sildenafil potentiates bone morphogenetic protein signaling in pulmonary arterial smooth muscle cells and in experimental pulmonary hypertension. Arterioscler Thromb Vasc Biol 33: 34–42, 2013. [DOI] [PubMed] [Google Scholar]
- 712.Yang X, Castilla LH, Xu X, Li C, Gotay J, Weinstein M, Liu PP, Deng CX. Angiogenesis defects and mesenchymal apoptosis in mice lacking SMAD5. Development 126: 1571–1580, 1999. [DOI] [PubMed] [Google Scholar]
- 713.Yang X, Lee PJ, Long L, Trembath RC, Morrell NW. BMP4 induces HO-1 via a Smad-independent, p38MAPK-dependent pathway in pulmonary artery myocytes. Am J Respir Cell Mol Biol 37: 598–605, 2007. [DOI] [PubMed] [Google Scholar]
- 714.Yang X, Long L, Southwood M, Rudarakanchana N, Upton PD, Jeffery TK, Atkinson C, Chen H, Trembath RC, Morrell NW. Dysfunctional Smad signaling contributes to abnormal smooth muscle cell proliferation in familial pulmonary arterial hypertension. Circ Res 96: 1053–1063, 2005. [DOI] [PubMed] [Google Scholar]
- 715.Yang Y, Schmidt EPC. The endothelial glycocalyx: An important regulator of the pulmonary vascular barrier. Tissue Barriers 1: pii: 23494, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 716.Yi X, Liang Y, Huerta-Sanchez E, Jin X, Cuo ZX, Pool JE, Xu X, Jiang H, Vinckenbosch N, Korneliussen TS, Zheng H, Liu T, He W, Li K, Luo R, Nie X, Wu H, Zhao M, Cao H, Zou J, Shan Y, Li S, Yang Q, Asan Ni P, Tian G, Xu J, Liu X, Jiang T, Wu R, Zhou G, Tang M, Qin J, Wang T, Feng S, Li G, Huasang, Luosang J, Wang W, Chen F, Wang Y, Zheng X, Li Z, Bianba Z, Yang G, Wang X, Tang S, Gao G, Chen Y, Luo Z, Gusang L, Cao Z, Zhang Q, Ouyang W, Ren X, Liang H, Zheng H, Huang Y, Li J, Bolund L, Kristiansen K, Li Y, Zhang Y, Zhang X, Li R, Li S, Yang H, Nielsen R, Wang J, Wang. Sequencing of 50 human exomes reveals adaptation to high altitude. Science 329: 75–78, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 717.Ying X, Minamiya Y, Fu C, Bhattacharya J. Ca2+ waves in lung capillary endothelium. Circ Res 79: 898, 1996. [DOI] [PubMed] [Google Scholar]
- 718.Yoon W, Kim YH, Kim JK, Kim YC, Park JG, Kang HK. Massive hemoptysis: Prediction of nonbronchial systemic arterial supply with chest CT. Radiology 227: 232–238, 2003. [DOI] [PubMed] [Google Scholar]
- 719.Yu AY, Shimoda LA, Iyer NV, Huso DL, Sun X, McWilliams R, Beaty T, Sham JS, Wiener CM, Sylvester JT, Semenza GL. Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1α. J Clin Invest 103: 691–696, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 720.Yu L, Hales CA. Silencing of NHE1 attenuates PASMC proliferation, hypertrophy and migration via E2F1. Am J Respir Cell Mol Biol 45: 923–930, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 721.Yu L, Quinn DA, Garg HG, Hales CA. Deficiency of the NHE1 gene prevents hypoxia-induced pulmonary hypertension and vascular remodeling. Am J Respir Crit Care Med 177: 1276–1284, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 722.Yu M, McAndrew RP, Al-Saghir R, Maier KG, Medhora M, Roman RJ, Jacobs ER. Nitric oxide contributes to 20-HETE-induced relaxation of pulmonary arteries. J Appl Physiol 93: 1391–1399, 2002. [DOI] [PubMed] [Google Scholar]
- 723.Yu Y, Fantozzi I, Remillard CV, Landsberg JW, Kunichika N, Platoshyn O, Tigno DD, Thistlethwaite PA, Rubin LJ, Yuan JX. Enhanced expression of transient receptor potential channels in idiopathic pulmonary arterial hypertension. Proc Natl Acad Sci U S A 101: 13861–13866, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 724.Yu Y, Keller SH, Remillard CV, Safrina O, Nicholson A, Zhang SL, Jiang W, Vangala N, Landsberg JW, Wang JY, Thistlethwaite PA, Channick RN, Robbins IM, Loyd JE, Ghofrani HA, Grimminger F, Schermuly RT, Cahalan MD, Rubin LJ, Yuan JX. A functional single-nucleotide polymorphism in the TRPC6 gene promoter associated with idiopathic pulmonary arterial hypertension. Circulation 119: 2313–2322, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 725.Yuan JX, Aldinger AM, Juhaszova M, Wang J, Conte JV Jr., Gaine SP, Orens JB, Rubin LJ. Dysfunctional voltage-gated K+ channels in pulmonary artery smooth muscle cells of patients with primary pulmonary hypertension. Circulation 98: 1400–1406, 1998. [DOI] [PubMed] [Google Scholar]
- 726.Yuan SY. New insights into eNOS signaling in microvascular permeability. Am J Physiol Heart Circ Physiol 291:H1029–1031, 2006. [DOI] [PubMed] [Google Scholar]
- 727.Yuan SY, Rigor RR. Methods for measuring permeability In: Yuan SY, Rigor RR, editors. Regulation of Endothelial Barrier Function. San Rafael (CA): Morgan and Claypool Lifesciences, 2010. [PubMed] [Google Scholar]
- 728.Yuan XJ, Goldman WF, Tod ML, Rubin LJ, Blaustein MP. Hypoxia reduces potassium currents in cultured rat pulmonary but not mesenteric arterial myocytes. Am J Physiol 264: L116–L123, 1993. [DOI] [PubMed] [Google Scholar]
- 729.Yuan XJ, Tod ML, Rubin LJ, Blaustein MP. Hypoxic and metabolic regulation of voltage-gated K+ channels in rat pulmonary artery smooth muscle cells. Exp Physiol 80: 803–813, 1995. [DOI] [PubMed] [Google Scholar]
- 730.Yuan XJ, Wang J, Juhaszova M, Gaine SP, Rubin LJ. Attenuated K+ channel gene transcription in primary pulmonary hypertension. Lancet 351: 726–727, 1998. [DOI] [PubMed] [Google Scholar]
- 731.Zeldin DC, Foley J, Ma J, Boyle JE, Pascual JM, Moomaw CR, Tomer KB, Steenbergen C, Wu S. CYP2J subfamily P450s in the lung: Expression, localization, and potential functional significance. Mol Pharmacol 50: 1111–1117, 1996. [PubMed] [Google Scholar]
- 732.Zhang RZ, Yang Q, Yim AP, Huang Y, He GW. Role of NO and EDHF-mediated endothelial function in the porcine pulmonary circulation: Comparison between pulmonary artery and vein. Vascul Pharmacol 44: 183–191, 2006. [DOI] [PubMed] [Google Scholar]
- 733.Zhao L, Sebkhi A, Ali O, Wojciak-Stothard B, Mamanova L, Yang Q, Wharton J, Wilkins MR. Simvastatin and sildenafil combine to attenuate pulmonary hypertension. Eur Respir J 34: 948–957, 2009. [DOI] [PubMed] [Google Scholar]
- 734.Zhao Y, Young SL. Expression of transforming growth factor-beta type II receptor in rat lung is regulated during development. Am J Physiol 269: L419–L426, 1995. [DOI] [PubMed] [Google Scholar]
- 735.Zhou H, Gao Y, Raj JU. Antenatal betamethasone therapy augments nitric oxide-mediated relaxation of preterm ovine pulmonary veins. J Appl Physiol 80: 390–396, 1996. [DOI] [PubMed] [Google Scholar]
- 736.Zhu D, Bousamra M II, Zeldin DC, Falck JR, Townsley M, Harder DR, Roman RJ, Jacobs ER. Epoxyeicosatrienoic acids constrict isolated pressurized rabbit pulmonary arteries. Am J Physiol Lung Cell Mol Physiol 278: L335–L343, 2000. [DOI] [PubMed] [Google Scholar]
- 737.Zhu D, Zhang C, Medhora M, Jacobs ER. CYP4A mRNA, protein, and product in rat lungs: novel localization in vascular endothelium. J Appl Physiol 93: 330–337, 2002. [DOI] [PubMed] [Google Scholar]
- 738.Zuckerbraun BS, George P, Gladwin MT. Nitrite in pulmonary arterial hypertension: Therapeutic avenues in the setting of dysregulated arginine/nitric oxide synthase signalling. Cardiovas Res 89: 542–552, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 739.Zuckerbraun BS, Shiva S, Ifedigbo E, Mathier MA, Mollen KP, Rao J, Bauer PM, Choi JJ, Curtis E, Choi AM, Gladwin MT. Nitrite potently inhibits hypoxic and inflammatory pulmonary arterial hypertension and smooth muscle proliferation via xanthine oxidoreductase-dependent nitric oxide generation. Circulation 121: 98–109, 2010. [DOI] [PubMed] [Google Scholar]