

Abstract
Background
This study aims to evaluate the effect of subcutaneous (SC) elamipretide dosing on exercise performance using the 6 min walk test (6MWT), patient‐reported outcomes measuring fatigue, functional assessments, and safety to guide the development of the Phase 3 trial.
Methods
MMPOWER‐2 was a randomized, double‐blind, placebo‐controlled, crossover trial that enrolled participants (N = 30) with genetically confirmed primary mitochondrial myopathy. Participants were randomly assigned (1:1) to 40 mg/day SC elamipretide for 4 weeks followed by placebo SC for 4 weeks, separated by a 4‐week washout period, or the opposite sequence. The primary endpoint was the distance walked on the 6MWT.
Results
The distance walked on the 6MWT by the elamipretide‐treated participants was 398.3 (±134.16) meters compared with 378.5 (±125.10) meters in the placebo‐treated group, a difference of 19.8 m (95% confidence interval, −2.8, 42.5; P = 0.0833). The results of the Primary Mitochondrial Myopathy Symptom Assessment Total Fatigue and Total Fatigue During Activities scores showed that participants treated with elamipretide reported less fatigue and muscle complaints compared with placebo (P = 0.0006 and P = 0.0018, respectively). Additionally, the Neuro‐QoL Fatigue Short Form and Patient Global Assessment showed reductions in symptoms (P = 0.0115 and P = 0.0421, respectively). In this 4‐week treatment period, no statistically significant change was observed in the Physician Global Assessment (P = 0.0636), the Triple Timed Up and Go (P = 0.8423) test, and wrist/hip accelerometry (P = 0.9345 and P = 0.7326, respectively). Injection site reactions were the most commonly reported adverse events with elamipretide (80%), the majority of which were mild. No serious adverse events or deaths were reported.
Conclusions
Participants who received a short‐course treatment of daily SC elamipretide for 4 weeks experienced a clinically meaningful change in the 6MWT, which did not achieve statistical significance as the primary endpoint of the study. Secondary endpoints were suggestive of an elamipretide treatment effect compared with placebo. Nominal statistically significant and clinically meaningful improvements were seen in patient‐reported outcomes. The results of this trial provided an efficacy signal and data to support the initiation of MMPOWER‐3, a 6‐month long, Phase 3 treatment trial in patients with primary mitochondrial myopathy.
Keywords: Myopathy, Primary mitochondrial disease, Elamipretide, Exercise intolerance, Primary mitochondrial myopathy, Crossover trial
Introduction
Patients with primary mitochondrial myopathy (PMM) possess genetic defects impairing normal mitochondrial function that primarily affect skeletal muscle.1, 2 Patients with PMM are less tolerant of physical exercise because of skeletal muscle respiratory chain dysfunction, which leads to muscle weakness, muscle atrophy, limited exercise capacity, and symptoms of fatigue.1, 2 PMM severity is variable, but disease progression significantly compromises daily activity performance.3, 4, 5
There are no US Food and Drug Administration (FDA)‐approved therapies for patients with PMM. Current therapies target symptoms and include physical/occupational therapy, exercise, nutrition, and dietary supplements.6 Elamipretide is an aromatic‐cationic tetrapeptide that readily penetrates cell membranes and localizes to the inner mitochondrial membrane where it associates with cardiolipin and has been demonstrated to restore physiologic cristae architecture.7, 8, 9, 10 Through this mechanism, elamipretide is hypothesized to improve energy production, reduce harmful oxidative stress by decreasing the production of reactive oxygen species, and ultimately increase the energy (adenosine triphosphate) supplied to affected cells and organs.7 In preclinical studies, elamipretide increased the synthesis of adenosine triphosphate and reduced reactive oxygen species production regardless of the specific mitochondrial abnormality causing the impaired mitochondrial respiration; there were no observed effects on normal functioning mitochondria.7, 8, 9, 10
An initial trial (MMPOWER) in human participants with genetically confirmed PMM evaluated three different daily intravenous dosages of elamipretide for 5 days.11 The results showed improvements in the 6 min walk test (6MWT) in participants treated with the highest dose of elamipretide.11 This MMPOWER‐2 trial was designed to evaluate 4‐week dosing of elamipretide with a subcutaneous (SC) formulation comparable with the highest dose studied in MMPOWER. SC bioavailability was calculated (~90%), confirming the sameness of the two formulations.
Methods
Standard protocol approvals, registrations, and consents
MMPOWER‐2 was a multicentre, randomized, double‐blind, placebo‐controlled, crossover trial. The trial was conducted in four US sites and was approved by individual Institutional Review Boards. Informed consent was obtained from each participant or from their legal representatives in accordance with international guidelines, including the Declaration of Helsinki and Council for International Organizations of Medical Sciences International Ethical Guidelines. The study was registered in the ClinicalTrials.gov registry (NCT02805790).
Trial design and subjects
MMPOWER‐2 was open to individuals who participated in the MMPOWER trial.11 Subjects were randomly assigned (1:1) to either 4 weeks of treatment with 40 mg SC elamipretide administered once daily in treatment period 1, followed by 4 weeks of treatment with SC placebo administered once daily in treatment period 2 (separated by a 4‐week washout period) or the opposite sequence (Figure 1A and 1B). The sample size was limited by the number of subjects available from MMPOWER, which enrolled 36 subjects and was designed to include all willing subjects who meet entry criteria. Accordingly, no formal sample size calculations were performed.
Figure 1.
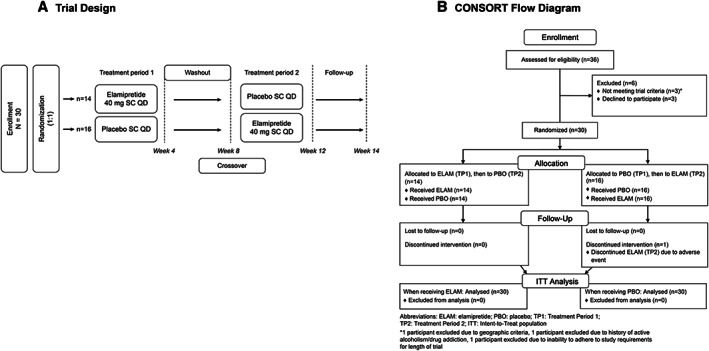
Trial design and CONSORT diagram. Panel (A) highlights the MMPOWER‐2 trial design, including the two different treatment arms and the treatment period timelines. Panel (B) shows the CONSORT flow diagram with the details of participants, enrolment, randomization, and allocation.
Inclusion and exclusion criteria
Subjects were eligible for inclusion in the study if they were willing and able to provide informed consent, completed participation in MMPOWER without a significant protocol deviation, resided in North America for the duration of the study, adhered to the study requirements for the length of the trial, and had not received study drug in MMPOWER within 3 weeks prior to MMPOWER‐2 screening. Subjects were required to have been on stable medications that would not impact the safety or efficacy endpoints of the trial, in the opinion of the investigator, for at least 1 month prior to the baseline visit. Women of childbearing age were required to agree to use a method of birth control from a pre‐specified list from the date of informed consent signing to 2 months after the last dose of study drug.
Subjects were ineligible and excluded from the study if they had any prior/current medical condition that, in the judgement of the investigator, would prevent the subject from safely participating in the study, had received the investigational compound and/or participated in another interventional clinical study within 30 days prior to the baseline visit, were concurrently enrolled in any non‐interventional research that was deemed incompatible with MMPOWER‐2 by the investigator, and had experienced an adverse reaction to study drug in MMPOWER that contraindicated further treatment with elamipretide. Female subjects who were pregnant, planning to become pregnant, or lactating were ineligible for the study. Subjects who had undergone an inpatient hospitalization within 1 month prior to screening or was deemed likely to require inpatient hospitalization/surgical procedure during the course of the study were also excluded. Subjects with a creatinine clearance ≤30 mL/min, a QTc elongation >450 ms in male subjects and > 480 ms in female subjects, uncontrolled hypertension, or a history of active alcoholism/drug addiction the year prior to screening were considered ineligible. If a subject had a history of clinically significant hypersensitivity or allergy to any of the excipient contained in the study drug, they were not permitted to partake in MMPOWER‐2. Any study centre or sponsor company personnel or immediate family of study centre or sponsor company personnel were excluded.
Randomization and masking
Assignment to treatment groups within each cohort was determined by a computer‐generated random sequence using an Interactive Web‐Response System to assign identical glass vials containing either the elamipretide or a placebo, which consisted of the same formulation without elamipretide. The placebo vials were identical in appearance, colour, and viscosity. The pharmacists, the trial staff, the sponsor, and the participants were blinded to the treatment given.
Procedures and assessments
The primary objective of the trial was to evaluate the effect of 4 weeks of a daily dose of SC elamipretide on the distance walked during the 6MWT.11, 12, 13, 14 The 6MWT was completed prior to each treatment period (post‐randomization) and at the end of each treatment period. The 6MWT is measured in meters, and a higher (longer) distance represents better performance. To ensure consistency between various clinical centres, the 6MWT was standardized by way of a systematic clinic–staff training programme.
Additional secondary endpoints included the following:
The Primary Mitochondrial Myopathy Symptom Assessment (PMMSA) is a patient‐reported outcome questionnaire, specifically designed for use in patients with PMM, developed and documented in accordance with measurement–development best practices and FDA guidance.15, 16, 17, 18 The PMMSA includes items that address the most common and relevant symptoms for patients with PMM. These symptoms were identified through a review of the published literature,19, 20 discussions with clinical experts, and interviews with patients. Cognitive interviews were conducted to ensure that patients understood the instructions, items, and response scales of the PMMSA. The PMMSA is completed daily and assesses the severity of 10 of the most common symptoms of PMM using the following 4‐point scale: (1) not at all, (2) mild, (3) moderate, and (4) severe. The results were weekly averages at various timepoints and were analysed using the following pre‐specified fatigue subscales: the PMMSA Total Fatigue score (assessed tiredness and muscle weakness at rest and during activities) and the PMMSA Total Fatigue During Activities score (assessed tiredness and muscle weakness during activities). Additionally, at the first clinic visit, participants were asked to identify which of the symptoms on the PMMSA was their most bothersome symptom. The change in the severity of each participant's most bothersome symptom was analysed. For all PMMSA analyses, lower scores represent less symptom severity.
The Neuro‐QoL Fatigue Item Bank was also completed by participants at the beginning and at the end of each treatment period.18 The Neuro‐QoL is a measurement system that evaluates and monitors the physical, mental, and social effects experienced by adults and children living with neurological conditions. The primary analysis of this item bank, the Neuro‐QoL Fatigue Short Form, the first 8 questions of the item bank (NINDS User Manual, Version 2.0 2015) calculates a T‐score distribution that rescales raw scores into standardized scores with a mean of 50 and a standard deviation of 10; a lower T‐score represents less severity of the concept being measured.18
The Patient Global Assessment (PGA) and Physician Global Assessment (PhGA) scales were completed. This single item scale that captured the participant's and investigator's (or physician designee's) assessment of their general health was completed at the beginning and at the end of each treatment period. For both the PGA and the PhGA, a lower score represents better “perceived general health.21
The Triple Timed Up and Go (3TUG) test time22 was completed by the participants at the beginning and at the end of each treatment period. The 3TUG is the “timed up and go test.” Participants begin by sitting back in a standard armchair, standing up, walking a given distance (3 m/10 feet), returning to a seated position in the chair, and repeating two additional times without pause. The time for the 3TUG is measured in seconds, and a shorter time represents better (faster) performance.
Accelerometry activity was measured using both hip and wrist accelerometers. Participants were instructed to wear the wrist accelerometer daily (24 h per day) and to wear the hip accelerometer daily during waking hours for at least 7 days prior to the beginning and end of each treatment period. Mean vector magnitude per day for wrist and the mean counts per day for hip for seven consecutive days immediately prior to the beginning and the end of each treatment period were analysed. Larger mean vector magnitude per day for wrist and larger mean counts per day for hip represent more activity.
Exploratory biomarkers including fibroblast growth factor 21 (FGF‐21), growth differentiation factor 15 (GDF‐15), and glutathione were also analysed.23, 24, 25
Safety and tolerability of elamipretide were also evaluated as part of the MMPOWER‐2 trial by capturing adverse events (AEs), vital signs, electrocardiograms, and clinical laboratory data.
Statistical methods
A two‐sided hypothesis test was used to evaluate the treatment effect of elamipretide versus placebo at the end of the treatment period using a mixed model with fixed effects for treatment, period and treatment sequence, and patient as a random effect. The results are presented as least square means, which are estimates appropriately adjusted for other analysis effects in the model. Hypothesis tests correspond to differences between least square means, rather than unadjusted means. Conditional upon significance for the primary analysis of the primary endpoint, Type I error control was achieved by testing select secondary efficacy measures sequentially via a pre‐specified hierarchical testing procedure with a two‐sided alpha level of 0.05 for the following measures: PMMSA Total Fatigue score, PMMSA Fatigue During Activities score, 3TUG time, and Neuro‐QoL Fatigue Short Form score. A mixed effect model was performed on these secondary endpoint variables. For the PMMSA, this mixed effect model was performed on each defined timepoint (i.e. week).
Results
Demographic and other baseline characteristics
Of the 36 eligible participants from MMPOWER, 30 were randomized (Figure 1B). These 30 MMPOWER‐2 participants were representative of the 36 participants in MMPOWER. Three participants from MMPOWER did not meet criteria for enrolment; one did not meet geographic criteria, one due to a history of active alcoholism or drug addiction during the year before screening visit, and one did not agree to adhere to the study requirements for the length of the trial. Additionally, three individuals declined to participate. Most participants were White (97%) and female (83%), and the mean age was 45.3 years (Table 1). There were baseline age and sex distribution differences between the treatment sequence groups; however, underlying assumptions and analysis of a crossover design suggest that this would not affect the results because participants effectively serve as their own controls.
Table 1.
Baseline characteristics of the participants in MMPOWER‐2.
| Treatment sequence (elamipretide: placebo) n = 14 | Treatment sequence (placebo: elamipretide) n = 16 | All patients N = 30 | |
|---|---|---|---|
| Age, mean (range), years | 41.5 (17–63) | 48.6 (25–65) | 45.3 (17–65) |
| Sex, n (%) | |||
| Female | 10 (71) | 15 (94) | 25 (83) |
| Male | 4 (29) | 1 (6) | 5 (17) |
| Race/ethnicity, n (%) | |||
| White | 13 (93) | 16 (100) | 29 (97) |
| Multiple (White/Asian/other) | 1 (7) | 0 | 1 (3) |
| Non‐Hispanic or Latino | 14 (100) | 15 (94) | 29 (97) |
| Hispanic or Latino | 0 | 1 (6) | 1 (3) |
| Weight, mean (SD), kg | 60.5 (±10.0) | 69.2 (±16.3) | 65.1 (±14.2) |
| BMI, mean (range), kg/m2 | 22.8 (15.8–33.2) | 25.3 (19.0–36.0) | 24.1 (15.8–36.0) |
| Baseline 6MWT, mean (SD), m | 381.2 (±30.4) | 396.5 (±36.3) | 389.4 (±23.6) |
| Baseline 6MWT, n (%), m | |||
| <450 | 13 (93) | 9 (56) | 22 (73) |
| ≥450 | 1 (7) | 7 (44) | 8 (27) |
| Genotype characteristics: Mitochondrial DNA (mtDNA) | |||
| Disorders involving mtDNA mutations that impair mitochondrial protein synthesis in toto | 19 | ||
| • Mitochondrial deletion syndrome | 11 | ||
| • m.3243A > G | 4 | ||
| • m.8344A > G | 3 | ||
| • Multisystem mitochondrial disorder (MT‐TH and tRNA) | 1 | ||
| Disorders involving mtDNA mutations that affect the subunits of the respiratory chain | 5 | ||
| • Multisystem mitochondrial disorder (MT‐COX1) | 1 | ||
| • Mitochondrial Myopathy (MTCYB) | 1 | ||
| • LHON Plus | 1 | ||
| • Multisystem Mitochondrial Disorder (MT‐ND3) | 1 | ||
| • Leigh syndrome (NDUFV1) | 1 | ||
| Nuclear DNA (nDNA) | |||
| Disorders involving nDNA mutations causing defects of intergenomic signalling | 3 | ||
| • POLG‐related disorder | 3 | ||
| Disorders involving nDNA mutations causing alterations of the lipid milieu of the inner mitochondrial membrane | 1 | ||
| • MEGDEL | 1 | ||
| Disorders involving nDNA mutations causing alterations of mitochondrial motility or fission | 2 | ||
| • Multisystem mitochondrial disorder (OPA1) | 2 | ||
6MWT, 6 min walk test; BMI, body mass index; SD, standard deviation.
The bold text describes the overall number of patients within the headlined category. i.e there are 19 patients overall in the disorders involving mtDNA mutations that impair mitochondrial protein synthesis in toto.
Each participant had genetic confirmation of their mitochondrial disease and a clinical diagnosis of PMM. In total, there were 12 different genetic diagnoses (Table 1), the most common of which were mitochondrial deletion syndrome (n = 11), tRNA mutations (n = 8), and POLG (polymerase gamma)‐related disorders (n = 3).
Efficacy findings
Primary endpoint
An analysis of the primary endpoint at the end of treatment showed that the distance walked in the 6MWT was 398.3 (±134.16) meters for participants that received elamipretide and 378.5 (±125.10) meters for participants that received placebo, a 19.8 m difference between the two groups [95% confidence interval (CI), −2.8, 42.5; P = 0.0833]. A pre‐specified subgroup analysis showed that participants treated with elamipretide who walked <450 m at baseline (n = 22) had a greater incremental change in distance walked at the end of elamipretide treatment compared with placebo (24.3 m; 95% CI, −6.2, 54.7; P = 0.1118). By contrast, participants who walked ≥450 m at baseline (n = 8) walked only 8.5 m more at the end of treatment compared with placebo (95% CI, −28.0, 45.2; P = 0.5729). These results are consistent with the purported mechanism of action for elamipretide as detailed in the Discussion section.
Secondary endpoints
Participants treated with elamipretide reported less total fatigue as assessed by the four‐question PMMSA Total Fatigue score throughout the treatment period (Figure 2A). At the end of the 4‐week treatment period, participants reported a 1.7‐point relative reduction in symptom severity while on elamipretide compared with placebo (95% CI, −2.6, −0.8; P = 0.0006). Similarly, participants reported less fatigue during activities as assessed by the two‐question PMMSA Fatigue During Activities score throughout the treatment period (Figure 2B), as suggested by a 0.8‐point relative reduction in symptom severity while on elamipretide compared with placebo (95% CI, −1.2, −0.3; P = 0.0018; Figure 2B). For PMMSA Total Fatigue and PMMSA Fatigue During Activities, the treatment benefit was not sustained upon discontinuation of elamipretide therapy, with subjects returning to pre‐dose severity 2 weeks after the end of treatment. While receiving elamipretide (versus placebo), participants reported improvements in individual myopathy‐related symptoms on the PMMSA: tiredness at rest (P = 0.0008), tiredness during activities (P = 0.0046), muscle weakness at rest (P = 0.0007), muscle weakness during activities (P = 0.0019), and muscle pain (P = 0.0079). There was no statistically significant treatment difference observed at the end of the treatment period in the individual PMMSA symptoms of balance problems, vision problems, abdominal discomfort, numbness, or headache. In a separate analysis of the “most bothersome” symptom, which was established at study entry, participants reported greater improvement on elamipretide, compared with placebo, in the severity of their individually chosen “most bothersome” symptoms of the PMMSA (P = 0.0111).
Figure 2.
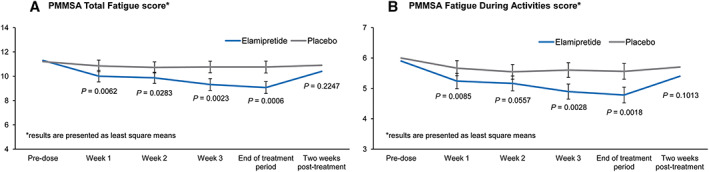
Primary Mitochondrial Myopathy Symptom Assessment (PMMSA) during the MMPOWER‐2 trial. Panel (A) shows the participants' total fatigue scores throughout the trial period. During the treatment with elamipretide, participants exhibited statistically significantly less total fatigue throughout the elamipretide treatment period (blue line) compared with the period while being treated with placebo (grey line) (95% CI, −2.6, −0.8; P = 0.0006). Panel (B) shows the participants' fatigue during activity scores throughout the trial period. During treatment with elamipretide, participants exhibited statistically significantly less fatigue during activities throughout the treatment period (blue line) compared with the period while being treated with placebo (grey line) (95% CI, −1.2, −0.3; P = 0.0018). For both scores, there was a steady improvement throughout the treatment period and a return to baseline score upon discontinuation of elamipretide therapy.
Participants who received elamipretide therapy also had an improved Neuro‐QoL Fatigue Short Form T‐score and PGA. At the end of treatment, there was a 4‐point reduction in the Neuro‐QoL Fatigue Short Form T‐score of participants treated with elamipretide compared with placebo (95% CI, −7.0, −1.0; P = 0.0115) and a 0.3‐point reduction on the PGA (95% CI, −0.6, −0.0; P = 0.0421). There was a 0.3‐point reduction on the PhGA (95% CI, −0.5, 0.0; P = 0.0636).
The end of treatment results of the 3TUG test showed a test time of 34.7 s for subjects that received elamipretide and a test time of 35.0 s for subjects that received placebo. No statistically significant difference was observed between elamipretide and placebo (P = 0.8423). Additionally, there was no statistically significant difference observed at the end of the treatment period in mean vector magnitude per day for wrist accelerometers (P = 0.9345) and mean counts per day for hip accelerometers (P = 0.7326).
Because of the potential for Type 1 error due to multiple comparisons, the findings for analyses of secondary endpoints should be interpreted as exploratory.
There were no treatment differences observed in exploratory biomarkers, which consisted of the analysis of levels of serum GDF‐15 (P = 0.5713), FGF‐21 (P = 0.3112), or glutathione (P = 0.8646).
Overall, the majority of endpoints suggest the potential for a treatment effect of elamipretide in participants with PMM (Figure 3 ).
Figure 3.
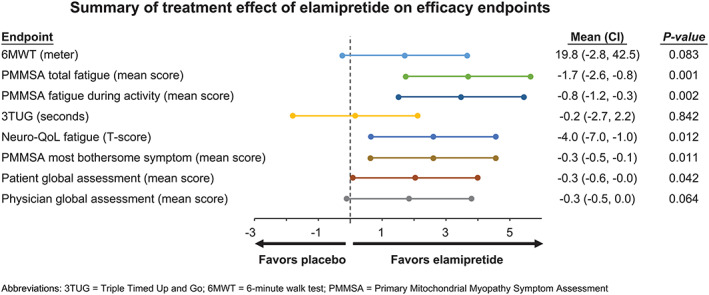
Summary of treatment effect of elamipretide on efficacy endpoints. Outcome measures used in the MMPOWER‐2 trial are highlighted in this forest plot. Analyses of functional tests and of patient‐reported as well as physician‐reported outcomes suggest potential with elamipretide in participants with primary mitochondrial myopathy.
Safety evaluation
Over the duration of the MMPOWER‐2 trial, elamipretide was generally well tolerated. There were no serious AEs or deaths reported. Sixty per cent of subjects experienced only mild AEs, and 40% experienced at least one AE of moderate severity. No severe AEs were reported. Injection site reactions were the most commonly reported AEs with elamipretide therapy (Table 2). These reactions were most commonly characterized by erythema (57%), pruritus (47%), pain (20%), urticaria (20%), and irritation (10%). Of the participants experiencing injection site reactions with elamipretide therapy, the majority were reported to be mild, though moderate bruising, discomfort, erythema, induration, irritation, and/or pain were reported in a few participants. Injection site erythema, pain, bruising, and irritation were also reported with placebo, but at a lesser frequency (<10% each). Among participants reporting injection site reactions with elamipretide therapy, 67% did so upon the first administration and 46% continued to experience them daily. There was a single discontinuation during the study, which occurred during elamipretide therapy due to moderate injection site pain. Excluding injection site reactions, the only AE reported in ≥10% of subjects on elamipretide therapy was dizziness (10%). Falls were the most commonly reported AE with placebo (10% vs. 3.3% in elamipretide) (see Table 2).
Table 2.
Adverse events (AEs) (≥2 participants).
| Event, n (%) | Elamipretide (n = 30) | Placebo (n = 30) |
| Injection site reactions | ||
| Erythema | 17 (56.7) | 1 (3.3) |
| Pruritus | 14 (46.7) | 0 (0) |
| Pain | 6 (20.0) | 1 (3.3) |
| Urticaria | 6 (20.0) | 0 (0) |
| Irritation | 3 (10.0) | 1 (3.3) |
| Bruising | 2 (6.7) | 2 (6.7) |
| Dizziness | 3 (10.0) | 1 (3.3) |
| Abdominal pain | 2 (6.7) | 1 (3.3) |
| Dysarthria | 2 (6.7) | 0 (0) |
| Urinary tract infection | 2 (6.7) | 0 (0) |
| Viral upper respiratory tract infection | 2 (6.7) | 0 (0) |
| Diarrhoea | 1 (3.3) | 2 (6.7) |
| Fall | 1 (3.3) | 3 (10.0) |
| Muscle spasms | 1 (3.3) | 2 (6.7) |
| Back pain | 0 (0) | 2 (6.7) |
| Headache | 0 (0) | 2 (6.7) |
Discussion
Patients with PMM have predominant symptoms of skeletal muscle dysfunction that commonly include exercise intolerance, fatigue, and muscle weakness.1, 2 In fact, the presentation of these symptoms has been reported to be the primary reasons for which patients with PMM would be motivated to participate in clinical trials.26 These hallmark symptoms of PMM led to the 6MWT being identified as the primary efficacy assessment in the MMPOWER‐2 trial. The 6MWT has been used to evaluate exercise performance in clinical trials for other diseases affecting muscle function27 and has been accepted as a clinical endpoint by regulatory authorities.12, 13, 14 However, the 6MWT results can be variable due to the presence of other co‐morbidities (cardiovascular and neurological) affecting performance or due to technician coaching.
In an effort to minimize the effect of these variables, the investigators excluded participants with severe co‐morbidities and used a standardized protocol across sites for performing the test.11 In this trial, there was a 19.8 m estimated difference in distance walked during the 6MWT for participants while on elamipretide compared with placebo. The 6MWT results from MMPOWER‐2 are in line with distances that have been determined to be clinically meaningful as reported in the literature. A systematic review of studies was conducted to interpret the clinical relevance and the minimal clinically important differences (MCID) for the 6MWT in various disease states (respiratory, cardiovascular, or musculoskeletal diseases).27 The review analysed the results of 19 publications that utilized either of the two approaches, anchor‐based method or distribution‐based method, to estimate the MCID for the 6MWT. The MCIDs, as measured in meters walked, ranged from 11 to 54 m for studies using the anchor‐based approach. Those studies using a distribution‐based analysis were similar to what was observed in studies using the anchor‐based approach.27 The 19.8 m (5% relative change) increase with elamipretide versus placebo reported in this current trial falls within the MCID range for the 6MWT and is considered by the investigators/authors to be a clinically important observation. In the MMPOWER‐2 study, participants that walked less than 450 m at baseline, which can be viewed as suffering more impairment, experienced a greater incremental improvement in distance walked during the 6MWT with elamipretide (versus placebo) than participants that walked ≥450 m at baseline. This group of patients were identified as having a higher relative change in MCID of 24.3 m (7.23% relative change).
In the post hoc analysis of MMPOWER,11 results were similar, with participants who walked <450 m at baseline showing greater improvement during the 6MWT while on elamipretide as compared with placebo. These observations are consistent with the purported mechanism of elamipretide, which has been shown to restore cellular bioenergetics in dysfunctional in vitro and in vivo systems; however, elamipretide has little effect in normally functioning systems.
Nominal significance levels associated with secondary endpoint analyses were observed with elamipretide for participants with PMM. In particular, participants receiving elamipretide had improvements in PMMSA Total Fatigue, PMMSA Total Fatigue During Activities, and their “most bothersome” symptom scores on the PMMSA, compared with their symptoms while on placebo. The results of these patient‐reported outcomes have real‐world implications for patients with PMM who have difficulty with daily activities because of exercise intolerance, fatigue, and/or muscle weakness. Of note, the PMMSA was developed specifically for use in the PMM population and documented in accordance with measurement–development best practices and followed the FDA guidance for development of a patient‐reported outcome survey.15 As part of the development and validation work, anchor‐based and distribution‐based responder analyses were completed for the PMMSA. The results of this work suggest that an approximate 1.6‐point change from baseline on the Total Fatigue domain is considered a clinically important improvement. In the MMPOWER‐2 study, participants receiving elamipretide reported less total fatigue on a weekly basis, culminating in a clinically meaningful 1.7‐point relative reduction in symptom severity compared with placebo (P = 0.0006).
The individualized assessment–severity change of the participant's “most bothersome” symptom on the PMMSA (as identified at the first clinic visit) showed improvements in severity of each participants' selected symptom, when receiving elamipretide compared with placebo. In this individualized approach, all participants had a “symptom change” endpoint, but the individual symptoms may have been different across participants. A similar endpoint approach is endorsed, for example, in the FDA's Guidance on Developing Drugs for Acute Treatment of Migraine (2014). Because symptoms of fatigue, exercise intolerance, and muscle weakness are the primary symptoms for which patients seek treatment, especially in clinical trials,26 there is inherent clinical relevance in focusing on these most bothersome symptoms for each participant. In addition to improvements in PMMSA outcomes, elamipretide‐treated participants also had improvements in their Neuro‐QoL Fatigue Short Form and PGA scores, compared with placebo.
Consistent with what was observed in the MMPOWER trial, there were no serious AEs in the MMPOWER‐2 trial. However, the SC formulation used in MMPOWER‐2 resulted in the occurrence of local injection site reactions, an AE found in both arms, which were reported as being mild to moderate, but generally well tolerated by most participants.
Serum FGF‐2123 and GDF‐1524 have been identified as potential biomarkers of mitochondrial respiratory chain deficiency and may be complementary indicators to the conventional biomarkers, such as lactate, creatine kinase, or alanine. In addition, glutathione is an indicator of cellular health and can be used as a biomarker of reduction/oxidation to quantify mitochondrial dysfunction.25 Our results showed no statistical significant with elamipretide on FGF‐21, GDF‐15, or glutathione. This may be related to the small number of participants in our trial and the molecular heterogeneity of their PMM.
This study is limited by a small sample size (limited by the number of participants available for enrolment from the initial MMPOWER trial), which prevented an assessment of differences in efficacy responses in specific genetic groups. The choice of using the same participants from the MMPOWER trial into the MMPOWER‐2 trial is not believed to have influenced the outcomes. MMPOWER was a short‐term, IV dose finding study that has informed dose selection based on a functional endpoint, but the study was not designed to be an outcome trial unlike the MMPOWER‐2 trial using the subcutaneous formulation of elamipretide. It is important to note that only nine participants received the highest elamipretide dose in MMPOWER. Therefore, it was decided that the same patient population should be studied in this MMPOWER‐2 trial.
During the conduct of the trial, there existed a theoretical potential for an unblinding effect due to the injection site reactions, an AE found in both treatment arms. It should be noted that the placebo was an identical formulation to the active drug and was buffered to mimic the active drug as closely as possible. Further, if there was unblinding, it conceivably would have been present more in treatment period 2 than treatment period 1. This theoretical potential would be due to patients “comparing” their experience in treatment period 2 to that of treatment period 1. Of greatest concern would be a potential bias in treatment period 2 leading to an inflated estimate of treatment benefit from the crossover as a whole. Accordingly, elamipretide treatment effects were only evaluated in treatment period 1 to further assess potential treatment benefit. This finding suggests that any potential unblinding effect (particularly if present in treatment period 2) would not have increased the likelihood of a spurious conclusion of effectiveness (i.e. a Type I error).
Mitochondrial DNA mutant heteroplasmy levels for those participants with mtDNA disorders were not available in all participants. When heteroplasmy data were available, they were assessed in various tissues (e.g. muscle and blood) making meaningful comparisons challenging. Participants were instructed to maintain their normal diet, daily caffeine and fibre intake, and activity/exercise level throughout the trial period; however, changes in these parameters were neither captured nor analysed. Additionally, participants were allowed to continue any supplements and antioxidants that they were taking at study entry, which may have had an effect on the current results. There were too few participants in this trial to determine any differential effect resulting from the use of these supplements.
Conclusions
Our results showed that elamipretide was generally well tolerated and that participants who received short‐course daily elamipretide for 4 weeks had clinically meaningful improvements in various outcome measures, despite the primary endpoint not reaching statistical significance. There is an unmet need for effective treatment options for patients with PMM. Novel therapies that target the disease instead of the symptoms need to be developed. Accordingly, although the primary endpoint was not met from a statistical perspective, the authors believe that the totality of the study data provides encouraging and directional results suggestive of a clinical benefit from administering SC elamipretide to patients with PMM. Consequently, the authors support the initiation of a Phase 3 trial to investigate the long‐term dosing of elamipretide in a larger population of PMM patients.
Funding
The trial was funded by Stealth BioTherapeutics, Newton, MA.
Conflict of interest
Amel Karaa received research grant, reimbursement for travel, and consulting payments from Stealth BT, Sanofi Genzyme, and Shire; received research grant and reimbursement for travel from Protalix and REATA; received consulting payments from MitoBridge; and is on the medical advisory board of MitoAction and on the scientific and medical advisory board of the United Mitochondrial Disease Foundation; is a board member of Rare New England and the Mitochondrial Medicine Society; and is an investigator in the North American Mitochondrial Disease Consortium. Richard Haas received research grant, reimbursement for travel, and consulting payments from Stealth BT; is on the scientific and medical advisory board of the United Mitochondrial Disease Foundation and the advisory board for MitoBridge; received clinical trial funding from Edison Pharmaceuticals, Stealth BioTherapeutics, Horizon Pharma (previously Raptor), and Sarepta; and received grant funding through the FDA Orphan Products Clinical Trials Grants Program (previously Orphan Products Grants; 1RO1FD004147) and the NIH (U54 NS078059). Amy Goldstein received research grant, reimbursement for travel, and consulting payments from Stealth BT; is on the scientific and medical advisory board of the United Mitochondrial Disease Foundation, DSMB at the University of Pittsburgh, and the editorial boards for the Journal of Child Neurology and Pediatric Neurology; and is a consultant for Biomarin; an investigator in the North American Mitochondrial Disease Consortium; and the president of the Mitochondrial Medicine Society. Jerry Vockley received research grant, reimbursement for travel, and consulting payments from Stealth BT. Bruce H. Cohen received research grant, reimbursement for travel, and consulting payments from Stealth BT; received research grants from Reata Pharmaceuticals, BioElectron Technology (Edison Pharma), and Horizon Pharma (Raptor Pharma); received travel support from Reata; serves on the UMDF Board of Trustees; and is an investigator for the NAMDC. All payments from Stealth BT were directly pertaining to travel for investigator meetings and the conduct of this clinical trial. As part of the trial, the investigators and coordinators have also received a mini iPad to conduct trial procedures.
Ethical Guidelines
All human studies described in this trial have been approved by the appropriate Ethics Committee and have therefore been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments. All participating subjects in this trial gave their informed consent prior to their inclusion in the study, and no details that might disclose the identity of these subjects under this study have been used.
Acknowledgements
We would like to thank the participants and their families for their participation, as well as MitoAction and the United Mitochondrial Disease Foundation for helping with recruitment. We also thank Harvard Catalyst (http://catalyst.harvard.edu/about/citingsupport.html). Medical writing assistance was provided by James A. Shiffer, RPh, Write On Time Medical Communications, LLC, which included assistance to the authors in drafting the outline and first draft of the manuscript, responding to reviewer comments, and medical editing. Anthony Aiudi, PharmD (Stealth BT), edited the manuscript and tables for nonintellectual content. Jeffrey S. Finman, PhD, Jupiter Point Pharma Consulting, LLC, were used for statistical consultation. Statistical analysis was conducted by Dean Rutty, Everest Clinical Research Corp, Markham, ON. All authors declare that the submitted work has not been published before (neither in English nor in any other language) and that the work is not under consideration for publication elsewhere. The authors of this manuscript certify that they comply with the ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle.28
Karaa A., Haas R., Goldstein A., Vockley J., and Cohen B. H. (2020) A randomized crossover trial of elamipretide in adults with primary mitochondrial myopathy, Journal of Cachexia, Sarcopenia and Muscle, 11, 909–918. 10.1002/jcsm.12559.
References
- 1. Mancuso M, Hirano M. NORD physician guide to mitochondrial myopathies (MM). Nat Org Rare Disord 2016;1–8. [Google Scholar]
- 2. Mancuso M, McFarland R, Klopstock T, Hirano M. International workshop: outcome measures and clinical trial readiness in primary mitochondrial myopathies in children and adults. Neuromuscul Disord 2017;27:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pfeffer G, Horvath R, Klopstock T, Mootha VK, Suomalainen A, Koene S, et al. New treatments for mitochondrial disease—no time to drop our standards. Nat Rev Neurol 2013;9:474–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tarnopolsky M. Exercise testing as a diagnostic entity in mitochondrial myopathies. Mitochondrion 2004;4:529–542. [DOI] [PubMed] [Google Scholar]
- 5. DiMauro S, Mancuso M, Naini A. Mitochondrial encephalomyopathies: therapeutic approach. Ann N Y Acad Sci 2004;1011:232–245. [DOI] [PubMed] [Google Scholar]
- 6. Pfeffer G, Chinnery PF. Diagnosis and treatment of mitochondrial myopathies. Ann Med 2013;45:4–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Siegel MP, Kruse SE, Percival JM, Goh J, White CC, Hopkins HC, et al. Mitochondrial‐targeted peptide rapidly improves mitochondrial energetics and skeletal muscle performance in aged mice. Aging Cell 2013;12:763–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhao K, Luo G, Giannelli S, Szeto HH. Mitochondria‐targeted peptide prevents mitochondrial depolarization and apoptosis induced by tert‐butyl hydroperoxide in neuronal cell lines. Biochem Pharmacol 2005;70:1796–1806. [DOI] [PubMed] [Google Scholar]
- 9. Brown DA, Hale SL, Baines CP, del Rio CL, Hamlin RL, Yueyama Y, et al. Reduction of early reperfusion injury with the mitochondria‐targeting peptide bendavia. J Cardiovasc Pharmacol Ther 2014;19:121–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nickel A, Kohlhaas M, Maack C. Mitochondrial reactive oxygen species production and elimination. J Mol Cell Cardiol 2014;73:1–8. [DOI] [PubMed] [Google Scholar]
- 11. Karaa A, Haas R, Goldstein A, Vockley J, Weaver WD, Cohen B. Randomized dose‐escalation trial of elamipretide in adults with primary mitochondrial myopathy. Neurology 2018;90:e1212–e1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Crapo RO, Casaburi R, Coates AL, Enright PL, Hankinson JL, Irvin CG, et al. ATS statement: guidelines for the six‐minute walk test. Am J Respir Crit Care Med 2002;166:111–117. [DOI] [PubMed] [Google Scholar]
- 13. Tarnopolsky M. Exercise testing in metabolic myopathies. Phys Med Rehabil Clin N Am 2012;23:173–186. [DOI] [PubMed] [Google Scholar]
- 14. Mancuso M, Angelini C, Bertini E, Carelli V, Comi GP, Minetti C, et al. Fatigue and exercise intolerance in mitochondrial diseases. Literature revision and experience of the Italian network of mitochondrial diseases. Neuromuscul Disord 2012;22:S226–S229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. FDA . Guidance for industry patient‐reported outcome measures: use in medical product development to support labeling claims: draft guidance. Clin Fed Regist 2006;1–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Patrick DL, Burke LB, Gwaltney CJ, Leidy NK, Martin ML, Molsen E, et al. Content validity—establishing and reporting the evidence in newly developed patient‐reported outcomes (PRO) instruments for medical product evaluation: ISPOR PRO good research practices task force report: part 1—eliciting concepts for a new PRO instrument. Value Health 2011;14:967–977. [DOI] [PubMed] [Google Scholar]
- 17. Patrick DL, Burke LB, Gwaltney CJ, Leidy NK, Martin ML, Molsen E, et al. Content validity—establishing and reporting the evidence in newly developed patient‐reported outcomes (PRO) instruments for medical product evaluation: ISPOR PRO good research practices task force report: part 2—assessing respondent understanding. Value Health 2011;14:978–988. [DOI] [PubMed] [Google Scholar]
- 18. Cella D, Lai JS, Nowinski CJ, Victorson D, Peterman A, Miller D, et al. Neuro‐QOL: brief measures of health‐related quality of life for clinical research in neurology. Neurology 2012;78:1860–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Elson JL, Cadogan M, Apabhai S, Whittaker RG, Phillips A, Trennell MI, et al. Initial development and validation of a mitochondrial disease quality of life scale. Neuromuscul Disord 2013;23:324–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gorman GS, Elson JL, Newman J, Payne B, McFarland R, Newton JL, et al. Perceived fatigue is highly prevalent and debilitating in patients with mitochondrial disease. Neuromuscul Disord 2015;25:563–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Idler EL, Benyamini Y. Self‐rated health and mortality: a review of twenty‐seven community studies. J Health Soc Behav 1997;38:21–37. [PubMed] [Google Scholar]
- 22. Podsiadlo D, Richardson S. The timed “up & go”: a test of basic functional mobility for frail elderly persons. J Am Geriatr So 1991;39:142–148. [DOI] [PubMed] [Google Scholar]
- 23. Suomalainen A, Elo JM, Pietiläinen KH, Hakonen AH, Sevastianova K, Korpela M, et al. FGF‐21 as a biomarker for muscle‐manifesting mitochondrial respiratory chain deficiencies: a diagnostic study. Lancet Neurol 2011;10:806–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yatsuga S, Fujita Y, Ishii A, Fukumoto Y, Arahata H, Kakuma T, et al. Growth differentiation factor 15 as a useful biomarker for mitochondrial disorders. Ann Neurol 2015;78:814–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Enns GM, Cowan TM. Glutathione as a redox biomarker in mitochondrial disease—implications for therapy. J Clin Med 2017;6:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zolkipli‐Cunningham A, Xiao R, Stoddart A, McCormick EM, Holberts A, Burrill N, et al. Mitochondrial disease patient motivations and barriers to participate in clinical trials. PLoS ONE 2018;13:e0197513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schrover R, Evans K, Giugliani R, Noble I, Bhattacharya K. Minimal clinically important difference for the 6‐min walk test: literature review and application to Morquio A syndrome. Orphanet J Rare Dis 2017;12:5–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2019. J Cachexia Sarcopenia Muscle 2019;10:1143–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]