

Abstract
Background
Cachexia is a multifactorial syndrome highly associated with specific tumour types, but the causes of variation in cachexia prevalence and severity are unknown. While circulating plasma mediators (soluble cachectic factors) derived from tumours have been implicated with the pathogenesis of the syndrome, these associations were generally based on plasma concentration rather than tissue‐specific gene expression levels. Here, we hypothesized that tumour gene expression profiling of cachexia‐inducing factors (CIFs) in human cancers with different prevalence of cachexia could reveal potential cancer‐specific cachexia mediators and biomarkers of clinical outcome.
Methods
First, we combined uniformly processed RNA sequencing data from The Cancer Genome Atlas and Genotype‐Tissue Expression databases to characterize the expression profile of secretome genes in 12 cancer types (4651 samples) compared with their matched normal tissues (2737 samples). We systematically investigated the transcriptomic data to assess the tumour expression profile of 25 known CIFs and their predictive values for patient survival. We used the Xena Functional Genomics tool to analyse the gene expression of CIFs according to neoplastic cellularity in pancreatic adenocarcinoma, which is known to present the highest prevalence of cachexia.
Results
A comprehensive characterization of the expression profiling of secreted genes in different human cancers revealed pathways and mediators with a potential role in cachexia within the tumour microenvironment. Cytokine‐related and chemokine‐related pathways were enriched in tumour types frequently associated with the syndrome. CIFs presented a tumour‐specific expression profile, in which the number of upregulated genes was correlated with the cachexia prevalence (r 2: 0.80; P value: 0.002) and weight loss (r 2: 0.81; P value: 0.002). The distinct gene expression profile, according to tumour type, was significantly associated with prognosis (P value ≤ 1.96 E‐06). In pancreatic adenocarcinoma, the upregulated CIF genes were associated with tumours presenting low neoplastic cellularity and high leucocyte fraction and not with tumour grade.
Conclusions
Our results present a biological dimension of tumour‐secreted elements that are potentially useful to explain why specific cancer types are more likely to develop cachexia. The tumour‐specific profile of CIFs may help the future development of better targeted therapies to treat cancer types highly associated with the syndrome.
Keywords: Cachexia‐inducing factors, Cancer genomics, GTEx, Omics, Pan‐cancer, TCGA
Introduction
Cancer cachexia is a multifactorial syndrome characterized by muscle wasting, leading to a significant weight loss that impacts patient's quality of life, tolerance to treatment, response to therapy, and survival.1, 2, 3, 4, 5 The syndrome affects up to 80% of advanced cancer patients, and it represents the cause of 20% of all cancer deaths.6 Cachexia is highly associated with specific tumour types such as pancreatic, oesophageal, gastric, lung, and liver, and similarly, patients with these malignancies have the highest degree of weight loss.7, 8, 9, 10, 11, 12 The aetiology of cancer cachexia in different tumour types involves complex and specific tumour–host interactions that remain to be completely elucidated. The combinatorial action of soluble secreted mediators (secretome) by cancer cells and cells within the tumour microenvironment, including many pro‐inflammatory cytokines, contributes to systemic inflammation and directly acts on skeletal muscle to induce wasting.12, 13, 14, 15 Consequently, the efforts to identify mediators and biomarkers have been centred on the levels of cachectic factors from plasma.16, 17, 18, 19, 20, 21 However, it remains unclear whether these factors circulating in the blood are derived from the host or tumour secretome. In addition, the extent to which these secreted factors associated with cachexia are expressed by different tumour types and which are the most ubiquitously expressed in different cancer sites have yet to be determined. A comprehensive characterization of the gene expression profile of cachexia‐inducing factors (CIFs) has the potential to reveal tumour secretome transcriptional patterns, which may help to explain the variation on prevalence and severity of cachexia within and across human cancers.
Transcriptomic approaches have been applied to unravel the secretome of a specific cell or tissue types.22 In a pioneer genome‐wide study based on microarrays, Welsh et al.23 described 74 overexpressed genes encoding secreted proteins in human cancers. Despite the reduced number of genes predicted as encoding secreted proteins and the relatively low number of tissue samples surveyed in this study, a significant fraction of these overexpressed secretome genes in carcinomas was found to be dysregulated in cancer or having demonstrated applications in cancer diagnosis and therapy. The advent of RNA sequencing (RNA‐Seq) has revolutionized the transcriptomic studies and enabled researchers a better understanding of the genetic mechanisms underlying human diseases, especially in cancer.24, 25, 26 Therefore, RNA‐Seq followed by newly emerging algorithms for signal peptide predictions have become useful tools for profiling the secretome22 and have revealed that a larger fraction of human tissue‐enriched proteins is secreted.27 Based on this knowledge, Robinson et al.28 conducted a pan‐cancer analysis of secretome gene expression that resulted in ranked lists of candidate diagnostic biomarkers detectable in biological fluids. This investigation also revealed the patterns and biological functions associated with changes in secreted protein expression in different tumour types, focusing mainly on a ‘core’ secretome. This strategy reduced the complexity of the secretome by narrowing the range of secreted molecules necessary to explore fundamental questions underlying altered secretome expression in different cancer types. However, the secretome complexity is still far from being completely understood and selecting different methodological strategies or specific tumour types might lead to new insights into its biological function.
The pan‐cancer studies integrate different levels of molecular data to comprehensively identify the similarities and differences in single or different tumour types.29 Here, we compared the expression profiles and functions of secreted proteins in 12 tumour types with different prevalence of cachexia. We hypothesized that tumour gene expression profiling of CIFs could reveal potential cancer‐specific mediators and biomarkers of clinical outcome. To test this, we first combined uniformly processed RNA‐Seq data from The Cancer Genome Atlas (TCGA) and Genotype‐Tissue Expression (GTEx) databases to characterize the expression profile of secretome genes. These data sets allowed us to comprehensively characterize the expression landscape of genes encoding predicted secreted proteins in human cancers and revealed potential mediators of cachexia within the tumour microenvironment. Next, we focused on the transcriptomic data to assess the tumour expression profile of 25 known CIFs and their prognostic value in terms of predicting patient survival. Interestingly, we detected a tumour‐specific expression profile of CIF. The number of upregulated cachectic factor genes in tumour compared to normal tissues was strongly correlated with the prevalence of cachexia and weight loss. We identified the expression of tumour cachectic factors genes relevant to cancer biology, which may help to elucidate why specific cancer types are more prone to develop cachexia.
Methods
The Cancer Genome Atlas and Genotype‐Tissue Expression transcriptomics data sets
Transcriptional profiles from TCGA (https://portal.gdc.cancer.gov/) of 12 human cancers were compared with matched normal tissues from TCGA and GTEx30 (http://www.gtexportal.org/). We used RNA‐Seq data uniformly processed and unified by the Toil Pipeline,31 via the web‐based tool Gene Expression Profiling Analysis32 (http://gepia.cancerpku.cn/). A similar strategy was previously described in pan‐cancer studies,33, 34 and their comparability findings demonstrated that TCGA and GTEx expression profiles could be collectively analysed. Differentially expressed genes between tumour and normal samples were determined by one‐way ANOVA applying the statistical cutoffs of log2 fold change > 1 and q value < 0.01.
Transcriptome‐based secretome analysis
Differentially expressed genes were further filtered for secreted protein‐coding genes based on the human secretome list available at The Human Protein Atlas27 (https://www.proteinatlas.org/humanproteome/secretome), which was predicted by a whole‐proteome scan using at least two of the three following methods for signal peptide prediction: SignalP4.0, Phobius, and SPOCTOPUS. Shared upregulated secretome genes among all tumour types were displayed using Circos35 (http://circos.ca/). The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway and Biological Process (Gene Ontology) analyses of the deregulated secretome genes were performed with EnrichR links36, 37 (http://amp.pharm.mssm.edu/Enrichr/), and the enrichment result is represented by P value (Fisher's exact test) and Z score (correction to the test) in a combined score computed by EnrichR.36, 37 The KEGG pathway and Biological Process terms were included in an integrative analysis using the criterion of over‐representation (log2 combined score > 2) in at least one tumour type. The set of CIFs detected and enriched in the top 10 Biological Process terms are displayed through an alluvial graph generated using the online tool http://sankeymatic.com/.
The cachectic soluble factors gene expression profile in 12 tumour types
For messenger RNA profiling of cachectic soluble factors, we selected 25 transcripts coding for soluble factors associated with cachexia. These transcripts were previously selected for blood screening by a multiplex array platform in pancreatic cachectic patients16 (Supporting Information, Table S1). The number of upregulated transcripts was correlated with the prevalence of cachexia and average weight loss for each tumour type obtained from previous studies.8, 11 Although our data set may not represent the entire range of cachexia variation for all the 12 cancer types analysed, these selected data provided numbers with potential correspondence in cachexia studies. We calculated the Pearson's correlations coefficient (r) with corresponding P values for the covariation between the number of differentially expressed CIFs from TCGA data sets (tumour tissues vs. matched normal TCGA and GTEx data) with the prevalence of cachexia and percentage of weight loss. We considered significant correlation when P value < 0.01 and r 2 value ≥ 0.8. The correlation analysis was performed using the software graphpad prism (graphpad prism 6).
Tumour purity analysis
We collected the pancreatic adenocarcinoma (PAAD) clinical and tumour purity information from TCGA Research Network.38 The expression levels of 25 CIF genes were analysed using unsupervised clustering approaches, according to the neoplastic cellularity of PAAD (high‐purity vs. low‐purity tumours). The Xena Functional Genomics Explorer tool was used to collect the gene expression information, using normalized data by the upper quartile method represented as log2 norm_count z+ 1 (http://xenabrowser.net/). Morpheus (https://software.broadinstitute.org/morpheus)39 was used to cluster the expression profile of the 25 CIFs in PAAD samples according to neoplastic cellularity data (leucocyte methylation percentage, tumour DNA hypermethylation, and purity class) and tumour grade.
Survival analysis and risk assessment
SurvExpress40 (http://bioinformatica.mty.itesm.mx/SurvExpress) was used to determine the risk assessment and perform a survival analysis of 12 TCGA cancer data sets. This online tool allowed us to assess the tumour gene expression of all 25 pro‐cachectic factors simultaneously and analysed their association with the survival of cancer patients by Cox proportional hazard regression in high‐risk and low‐risk groups, as determined through the SurvExpress optimization algorithm. This analysis was performed without considering other clinical characteristics rather than survival.
Data representation and analysis
Correlation analysis and bar plots were constructed with graphpad prism (graphpad Software). Venn diagrams were plotted using the web server http://bioinformatics.psb.ugent.be/webtools/Venn/. Heat maps and principal component analysis plots were created using the web tools ClustVis41 (http://biit.cs.ut.ee/clustvis/) and Morpheus39 (https://software.broadinstitute.org/morpheus).
Results
The secretome genes show differential expression profiles across human cancers
Gene expression profiles were obtained from 4743 tumours comprising 12 cancer types (TCGA) and 2737 corresponding normal tissues (TCGA and GTEx). These tumours were selected to allow a comparison between cancer types ranging from high to low prevalence of cachexia and weight loss.8, 11, 12 The summary of the number of TCGA and GTEx samples is described in Table S2.
Differentially expressed genes were filtered for those genes previously predicted to have at least one secreted protein by the Human Protein Atlas27 ( Data S1). This analysis revealed 2162 out of 2933 Human Protein Atlas differentially expressed secretome genes in at least one cancer type compared with normal samples (log2 fold change > 1 and q value cutoff = 0.01; Data S2). The clustering analyses based on Euclidian distance revealed that PAAD displays a gene expression profile that is distinct from other cancers (Figure 1A). Also, this analysis grouped cancer types originating from the same anatomical sites, such as lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC), as well as colon adenocarcinoma (COAD) and rectum adenocarcinoma (READ) (Figure 1A). Similarly, oesophageal squamous cell carcinoma (ESCA) and head and neck squamous cell carcinoma (HNSC) also presented similar expression profiles (Figure 1A). To test whether our transcriptome‐based secretome analysis defines tumour types, we performed principal component analysis that revealed that PAAD, acute myeloid leukaemia (LAML), LUSC, and the cluster composed by COAD and READ are distinguished among the 12 cancer types based on the expression profile of the secretory protein‐coding genes (Figure 1B).
Figure 1.
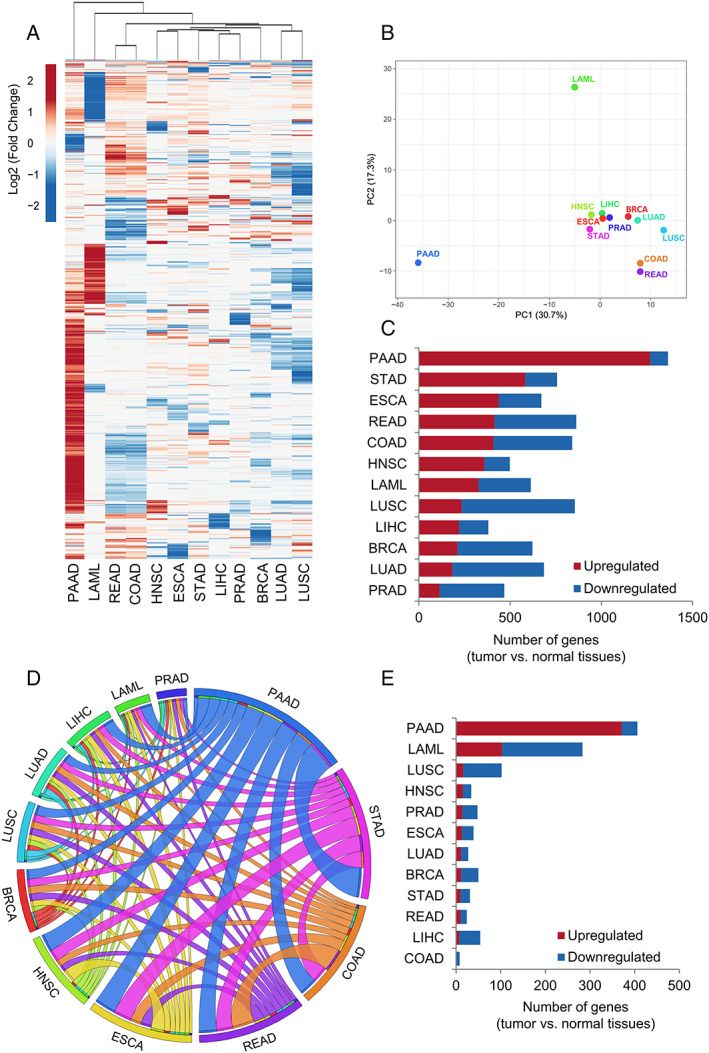
Gene expression analysis of the secretome components in 12 tumour types of The Cancer Genome Atlas (TCGA) compared with corresponding matched normal TCGA and Genotype‐Tissue Expression (GTEx) tissues. (A) Heat map of the mean expression levels [log2 (TPM + 1)] of secretome components in 12 tumour types compared with normal tissues. Both rows (secretome genes) and columns (tumour types) were clustered using Euclidian distance. Differential expression levels were calculated using the web‐based tool Gene Expression Profiling Analysis (http://gepia.cancerpku.cn/).32 The resulting numbers of genes encoding predicted that secreted proteins were filtered based on the Human Protein Atlas secretome data (https://www.proteinatlas.org/humanproteome/secretome).27 The upregulated and downregulated genes with absolute values of fold change > 2.0 and q value < 0.01 (ANOVA) are shown in red and blue, respectively. PAAD shows a clear enrichment of the upregulated secretome genes. (B) Principal component analysis of the secretome gene expression data in 12 tumour types compared with normal tissues. (C) The total number of dysregulated secretome genes in each tumour type (TCGA) compared with corresponding matched normal tissues (TCGA and GTEx). (D) The thickness of each link in the Circos plot represents the number of shared upregulated secretome genes among tumour types. (E) Exclusive upregulated genes within tumour types. BRCA, breast invasive carcinoma; COAD, colon adenocarcinoma; ESCA, oesophageal carcinoma; HNSC, head and neck squamous cell carcinoma; LAML, acute myeloid leukaemia; LIHC, liver hepatocellular carcinoma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; PAAD, pancreatic adenocarcinoma; PRAD, prostate adenocarcinoma; READ, rectum adenocarcinoma; STAD, stomach adenocarcinoma.
We investigated the diversity and complexity of the cancer transcriptome‐based secretome in these selected 12 cancer types. We found that tumours differed highly in the number of dysregulated secretome genes in comparison with normal tissues. For instance, PAAD presented 1365 dysregulated genes, while hepatocellular liver carcinoma (LIHC) had 380 (Figure 1C, Table S3). The proportions of upregulated and downregulated secretome genes also showed wide distribution among malignant tissues. PAAD, stomach adenocarcinoma (STAD), ESCA, and HNSC presented 72% to 93% of upregulated secretome genes, whereas LUSC, LUAD, and prostate adenocarcinoma (PRAD) showed 24% to 27% (Figure 1C, Tables S3–S5).
Secretome genes reveal potential mediators of cachexia within and across human cancers
Given that many secreted soluble factors have been associated with cachexia in different tumour types, significantly upregulated gene coding for secreted proteins shared among these tumours has the potential to reveal key players towards the pathogenesis of the syndrome. PAAD, which is associated with a severe form of cachexia, shared the highest number of upregulated genes in all tumour types tested (Figure 1D, Table S6). Other tumour types highly associated with cachexia, including STAD, READ, COAD, ESCA, and HNSC, had in common a high number of upregulated secretome genes. STAD shared 515 common upregulated genes with PAAD, while ESCA, COAD, READ, and HNSC presented 305–366. Among the cancer types, PRAD had the lowest number (n = 69) of overlapping upregulated genes with PAAD. Figures 1D and S1 are representatives of these findings. Importantly, the shared upregulated genes for secreted proteins revealed potential mediators of cachexia within the tumour microenvironment, especially in tumours highly associated with the syndrome.
Upregulated secretome genes exclusively detected in a single cancer type, particularly in those highly associated with cachexia, are tumour‐specific candidates (Table S7). In PAAD, we found exclusive upregulation of 371 genes encoding mainly for cytokine–cytokine receptor interaction, complement and coagulation cascades, and Rap1 signalling pathways (Figure 1E, Tables S7 and S8). Interestingly, many of these exclusively upregulated genes in PAAD were dysregulated in opposite directions in tumours originating from lung and large intestine (Figure S2, Table S9). A total of 103 genes were exclusively upregulated in LAML, and these genes were related to coagulation cascades and pyruvate metabolism and downregulated 180 unique secretome genes involved in arginine and proline metabolism, ribosome, and metabolic pathways (Tables S10 and S11). Together, these results show that tumour‐specific factors have the potential to explain the variation in the prevalence and severity of cancer cachexia.
Secretome pathways with a potential role in cancer cachexia
Genes significantly deregulated in at least one tumour type were selected to perform enrichment analysis using the KEGG pathways and Gene Ontology terms. Overall, we observed that pathways previously associated with cachexia are over‐represented in highly cachectic tumours (Figure 2A). The upregulated genes enriched pathways associated with chemokine and cytokine in PAAD, STAD, ESCA, HNSC, COAD, and READ (Figures 2 and S3). Terms previously associated with cachexia, such as lysosome, exocytosis, complement and coagulation cascade, extracellular matrix–receptor interaction, PI3K‐Akt signalling, and leucocyte chemotaxis were strongly enriched for upregulated genes in PAAD, STAD, ESCA, and HNSC (Figure 2). PAAD, STAD, ESCA, and HNSC presented a clear pattern of downregulated secretome genes that were less enriched for many pathways highly over‐represented for the upregulated genes (Figure 2A). The pattern of secretome pathways for LUAD and LUSC was distinct from tumours highly associated with cachexia, such as PAAD, STAD, ESCA, and HNSC (Figure 2A). An enrichment for downregulated genes associated with extracellular matrix–receptor interaction, PI3K‐Akt pathway, and protein digestion and absorption was detected in PRAD (Figure 2A).
Figure 2.
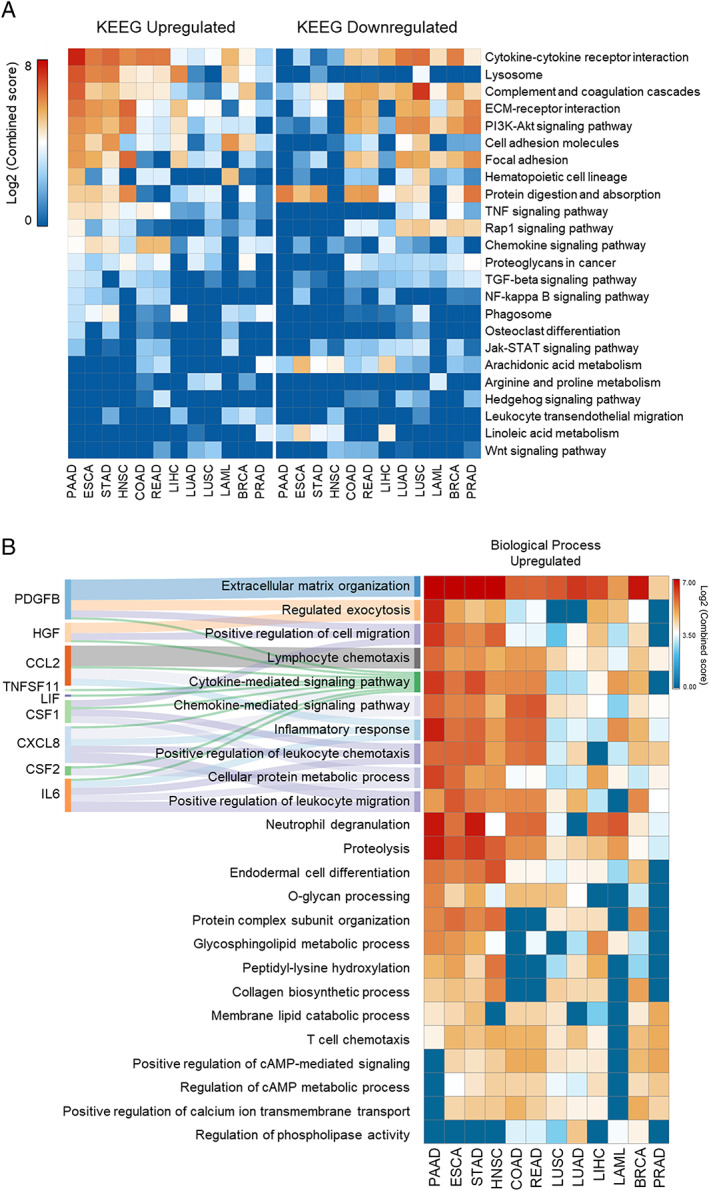
Enriched pathways of the differentially expressed secretome genes. (A) Top‐ranked combined scores for The Kyoto Encyclopedia of Genes and Genomes pathway categories associated with upregulated (right panel) and downregulated (left panel) genes in tumour tissues vs. corresponding matched normal tissues of The Cancer Genome Atlas (TCGA) and Genotype‐Tissue Expression (GTEx), computed by the gene set enrichment analysis tool EnrichR.36, 37 Pathways were included in the analysis when satisfying the criteria of over‐representation (log2 combined score > 2) in, at least, one tumour type (colour intensity codes for combined score). Rows and columns were clustered based on Euclidean distance between log2 combined score values. (B) Right: Top‐ranked combined scores for Gene Ontology categories associated with upregulated genes in tumour tissues vs. corresponding matched normal TCGA and GTEx tissues, computed by the gene set enrichment analysis tool EnrichR.36, 37 Left: alluvial diagram connecting the cachexia‐inducing factors predicted into those biological processes terms. BRCA, breast invasive carcinoma; COAD, colon adenocarcinoma; ESCA, oesophageal carcinoma; HNSC, head and neck squamous cell carcinoma; LAML, acute myeloid leukaemia; LIHC, liver hepatocellular carcinoma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; PAAD, pancreatic adenocarcinoma; PRAD, prostate adenocarcinoma; READ, rectum adenocarcinoma; STAD, stomach adenocarcinoma.
The tumour‐specific expression profile of cachexia‐inducing factors is correlated with the prevalence of cachexia and weight loss
Considering that cytokine and chemokine pathways—enriched in tumours associated with cachexia—include known cachexia mediators (PDGFB, HGF, CCL2, TNFSF11, LIF, CSF1, CXCL8, CSF2, and IL6) (Figure 2B), we further filtered the transcriptome data to 25 cytokines and growth factors. This same set of CIFs was previously used for protein blood screening in pancreatic cachectic patients.16 The list of 25 factors is described in Table S1.
We found that each tumour type shows a specific gene expression profile of CIFs (Figures 3A and S4). PAAD presented the highest number (14) of upregulated CIFs (CXCL8, IL1B, HGF, TNFSF10, LIF, TGFA, TNFSF11, PDGFB, IL6, CCL2, CSF1, IL15, CSF2, and FGF2), while PRAD showed no significant upregulated CIFs. Other tumour types also presented upregulated pro‐cachectic factors, such as breast invasive carcinoma (IL1B and MMP13), LIHC (PDGFB), ESCA (CXCL8, IL1B, LIF, PDGFB, IL6, CSF3, and MMP3), HNSC (CXCL8, IL1B, TNFSF10, PDGFB, CSF2, VEGFA, and MMP3), and STAD (CXCL8, TNFSF10, LIF, TGFAC, and TNFSF11). LUSC and LUAD shared six downregulated CIFs (PDGFB, IL6, HGF, FGF2, CSF3, and CXCL12), whereas only TGFA and MMP13 were upregulated in both lung tumours. Although IL1B, CXCL8 (also known as IL8), TGFA, and LIF were upregulated in at least five tumour types, five molecules (IL10, CD40LG, IFNA1, IL4, and IL17A) showed no alterations (Figure 3a). Additionally, classic cachectic mediators were overexpressed, including IL6 in PAAD and ESCA and TNF in LAML. The transcripts abundance for these 25 cachexia mediators in the 12 tumour types revealed that VEGFA, CCL2, and CXCL8 are expressed at high levels in many tumour tissues, while IL4, IL17A, and CSF2 are expressed at low levels (Figure S5). Our data revealed potential cachexia biomarkers and mediators that are produced at high levels by the tumour tissues.
Figure 3.
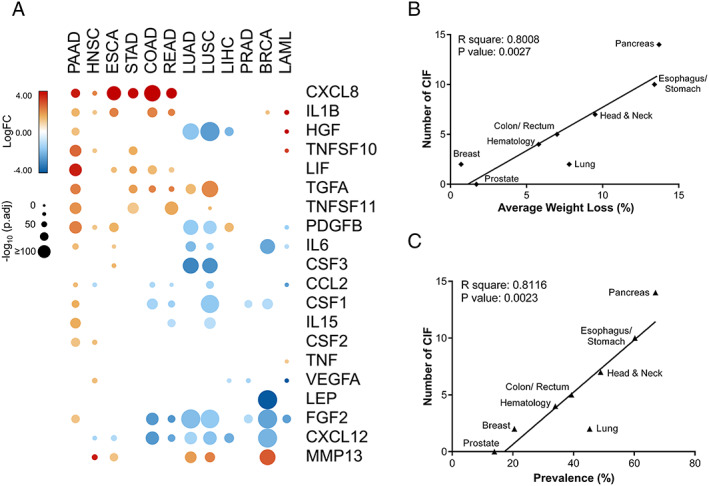
Cachexia‐inducing factors (CIFs) tumour‐specific expression profiles strongly correlate with the prevalence of cachexia and weight loss in different tumour types. (A) Schematic representation of the expression pattern of 25 CIFs in different tumour types of The Cancer Genome Atlas (TCGA). Differential expression levels were calculated using the web‐based tool Gene Expression Profiling Analysis (http://gepia.cancerpku.cn/)32 from tumour tissues vs. matched normal TCGA and Genotype‐Tissue Expression (GTEx) data. Upregulated and downregulated genes with absolute values of fold change > 2.0 and q value < 0.01 (ANOVA) are shown in red and blue, respectively. Five molecules (IL10, CD40LG, IFNA1, IL4, and IL17A) showed no alteration and are not represented in the heat map. (B–C) Pearson's correlations coefficient (r) with corresponding P values for the covariation between the number of differentially expressed CIFs (y‐axis) from TCGA data sets (tumour tissues vs. matched normal TCGA and GTEx data) and the percentage of weight loss (x‐axis; B) or the percentage of cachexia prevalence (x‐axis; C) for specific tumour types from relevant literature data.8, 11, 12 Weight loss and prevalence of cachexia are strongly correlated with the number of CIFs (P < 0.01). BRCA, breast invasive carcinoma; COAD, colon adenocarcinoma; ESCA, oesophageal carcinoma; HNSC, head and neck squamous cell carcinoma; LAML, acute myeloid leukaemia; LIHC, liver hepatocellular carcinoma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; PAAD, pancreatic adenocarcinoma; PRAD, prostate adenocarcinoma; READ, rectum adenocarcinoma; STAD, stomach adenocarcinoma.
Using previously published data,8, 11, 12 we next investigated the total number of upregulated CIFs for each tumour type and their correlation with the prevalence of cachexia and the percentage of weight loss. This data set included the pancreatic, oesophageal/gastric, head and neck, colorectal, lung, haematological, breast, and prostate cancers. These data have the potential to provide relevant correspondence in different cachexia studies, although they do not represent the full variation of cachexia for all the 12 cancer types. Remarkably, the number of upregulated CIF genes in tumour compared with normal tissues was strongly correlated with the prevalence of cachexia and weight loss (average percentage) in the majority of the tumour types available for this analysis (Figure 3B). The only exception was lung cancers, but these results are also in accordance with the gene expression profiles that were detected for LUSC and LUAD, which are distinct from tumours more prominently associated with cachexia. These findings showed specific cachectic tumour signatures with potential to explain why specific cancer types are more likely to develop cachexia.
The expression profile of cachexia‐inducing factors is associated with tumour purity
Tumour microenvironment contains non‐cancerous cells, including a diversity of immune cells that potentially contribute to the secretion of CIFs. We then investigated the expression profile of these 25 CIFs according to tumour purity (neoplastic cellularity and leucocyte fraction) in PAAD. This tumour type—known to present the highest prevalence of cachexia12 and low tumour purity38—showed the highest number of upregulated secretome genes (Figure 3A). We used previously published tumour purity data for PAAD,38 which is constituted by 74 ‘low‐purity’ samples (ABSOLUTE purity < 33%) and 76 ‘high‐purity’ samples (ABSOLUTE purity ≥ 33%). Using hierarchical clustering analysis, we observed that the expression profile of these 25 CIFs discriminates high‐purity and low‐purity tumours, and most of these genes are overexpressed in low‐purity tumours (Figure 4). The overexpression of CIFs in low‐purity tumours was further associated with high leucocytes DNA methylation and low tumour cells DNA hypermethylation mode purity (Figure 4). These data show that leucocyte infiltration correlates with high expression of CIFs in PAAD. Interestingly, no association was observed between the expression levels of CIFs and tumour grade (Figure 4).
Figure 4.
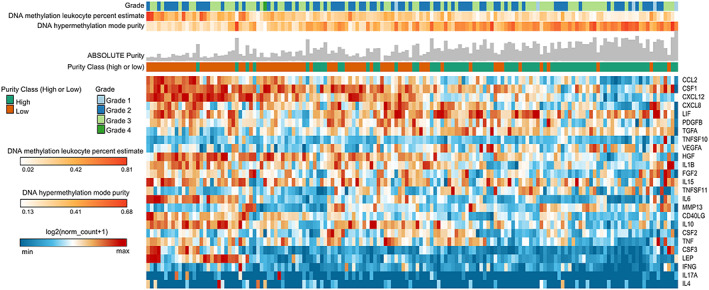
Landscape of cachexia‐inducing factors is associated with low tumour purity. Unsupervised hierarchical clustering of cachexia‐inducing factors is presented in a heat map according to gene expression (log2 norm_count + 1). The integrated epigenomic data for PAAD samples, including DNA hypermethylation mode purity and DNA methylation leucocyte percent, are shown as tracks at the top together with the tumour grade. The absolute tumour purity data for each sample are shown as grey bars at the top. The percentage of patients classified as low or high purity is noted as orange and green tracks, respectively. Rows and columns were clustered based on Euclidean distance between log2 norm_count + 1 values.
The expression landscape of cachexia‐inducing factors predicts cancer outcome
Circulating levels of CIFs such as IL6, CXCL8, IL1B, and MCP‐1 were previously correlated with cachexia development and poor prognosis in cancer.16, 17, 18, 42, 43, 44, 45 Based on this evidence, we compared the tumour expression levels of CIFs with patient prognosis using the platform SurvExpress.40 Table S12 summarizes the TCGA data sets used in our analysis. SurvExpress generated a prognostic index (risk score) based on the gene expression of 25 CIFs and the survival of cancer patients for each of the 12 tumour types. Patients were divided into two groups, high risk and low risk, maximizing the number of patients into risk groups by an optimization algorithm from the ordered prognostic index (Table S12). More than 50% of CIF genes are overexpressed (P < 0.05) in high‐risk groups in HNSC, READ, COAD, LIHC, and BRCA (Figure 5A). This analysis also demonstrated the consistent overexpression of IL4, INF‐γ, and IL6 in at least eight different tumour types (Figure 5A). The heat map generated by clustering analysis differentiating patients into high risk and low risk demonstrated the enrichment of specific CIFs in patients with low survival (high‐risk group) in HNSC, LIHC, and BRCA (Figure 5B). Examples of the CIFs clustered in the heat map and overexpressed in patients with low survival include LIF, IL6, and CSF3 (in HNSC); TGFA, LIF, HGF, CXCL8, and FGF2 (in LIHC); and LEP, FGF2, LIF, TGFA (in BRCA) (Figure 5B). It is also interesting that in PRAD, which is less prone to developing cachexia, only TGFA, IL15, and IL4 presented higher expression in patients with low survival, while IL6, CSF3, LIF clustered in the heat map and were expressed at low levels in patients with high survival (Figure 5B). The heat maps for other tumour types are presented in Figure S6, and the relative expression levels of the 25 CIFs in low‐risk and high‐risk groups of patients are shown in Figure S7. This variability in the expression profiles of CIFs in patients with low and high survival may help to explain the heterogeneity in the clinical manifestation of cachexia in patients with different tumour types.
Figure 5.
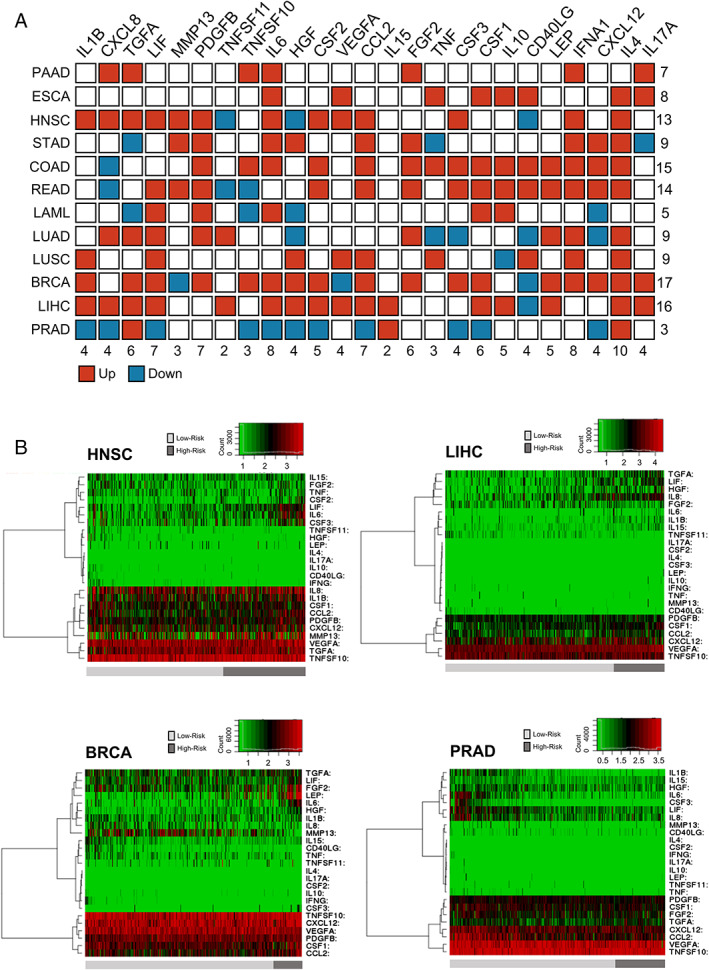
High‐risk groups are correlated with high expression of cachexia‐inducing factors (CIFs) in tumour tissues. (A) Schematic representation summarizing the expression pattern of 25 CIFs in 12 tumour types of The Cancer Genome Atlas (TCGA). Differential expression levels were calculated in the web‐based tool SurvExpress40 by maximizing two risk groups (high risk and low risk) by prognostic index median and Cox fitting. The statistical difference for each messenger RNA expression between high‐risk and low‐risk groups was tested using the t‐test. Upregulated and downregulated genes (high risk vs. low risk) with P value < 0.05 are shown in red and blue, respectively. (B) Representative heat maps are showing the hierarchical clustering analysis of tumour messenger RNA expression of 25 CIFs generated in SurvExpress,40 using TCGA data sets. This analysis demonstrates the considerable variation in tumour expression of some CIFs according to risk groups in HNSC, LIHC, BRCA, and PRAD. The cancer patients were stratified into high‐risk and low‐risk groups, indicated below each heat map as dark‐grey and light‐grey bars, respectively. Risk groups were maximized based on the Prognostic Index assessed by gene expression values multiplied by beta coefficients. BRCA, breast invasive carcinoma; COAD, colon adenocarcinoma; ESCA, oesophageal carcinoma; HNSC, head and neck squamous cell carcinoma; LAML, acute myeloid leukaemia; LIHC, liver hepatocellular carcinoma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; PAAD, pancreatic adenocarcinoma; PRAD, prostate adenocarcinoma; READ, rectum adenocarcinoma; STAD, stomach adenocarcinoma.
Finally, to evaluate whether the CIF genes have prognostic and predictive value in different cancer types, we used 12 TCGA data sets within SurvExpress to analyse the overall survival. Altered expression of CIFs is associated with worse overall survival (Figure 6 and Table S12). The robustness of these 25 genes in stratifying with high‐confidence patients into two risk groups was consistently demonstrated by high hazard ratios in the 12 TCGA cohorts (Figure 6). Using this list of 25 factors, we also detected differentially expressed genes that have higher relevance in predicting worse survival (Table S13). Thus, these findings suggest that CIF genes are predictors of cancer survival outcomes.
Figure 6.
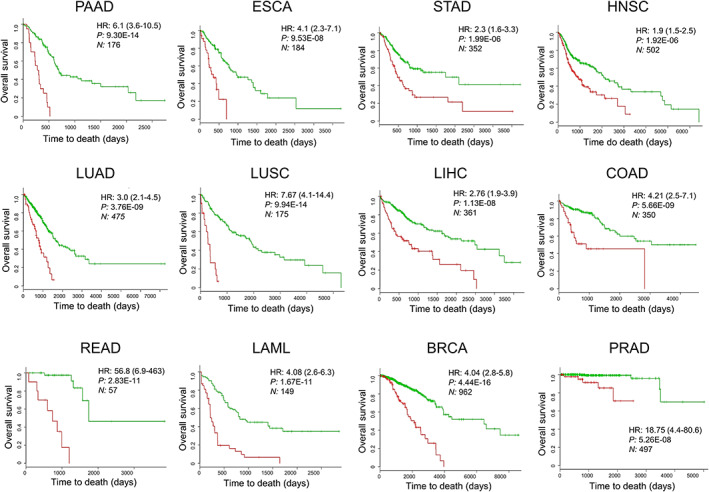
The expression landscape of cachexia‐inducing factors (CIFs) predicts cancer outcome. Survival analysis based on the tumour messenger RNA expression of 25 CIFs. The data were calculated using the data sets of The Cancer Genome Atlas of 12 tumour types in the web‐based tool SurvExpress,40 which stratified the cancer patients in high‐risk or low‐risk groups (red and green, respectively). The adjusted hazard ratio (HR) with corresponding 95% confidence intervals, log‐rank P value (P), and the number of patients successfully stratified (N) determined by univariate Cox regression analyses are shown on each survival Kaplan–Meier curve. BRCA, breast invasive carcinoma; COAD, colon adenocarcinoma; ESCA, oesophageal carcinoma; HNSC, head and neck squamous cell carcinoma; LAML, acute myeloid leukaemia; LIHC, liver hepatocellular carcinoma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; PAAD, pancreatic adenocarcinoma; PRAD, prostate adenocarcinoma; READ, rectum adenocarcinoma; STAD, stomach adenocarcinoma.
Discussion
Cachexia prevalence and severity vary depending on tumour types. Pancreatic, oesophageal, gastric, lung, and liver cancers have the highest prevalence, while breast or prostate cancers are not commonly associated with the syndrome.12 The causes of such variations are still mostly unknown, and it is possible that each cancer type present tumour‐specific biomarkers and mediators. We sought to characterize the molecular landscape of CIFs in human cancers with different prevalence of cachexia, using the RNA‐Seq data set from TCGA and GTEx. Our results revealed tumour‐specific secretome transcriptional patterns, potential mediators, and pathways associated with the syndrome. We also showed that a set of 25 CIFs presented a tumour‐specific expression profile, which was significantly associated with poor prognosis and correlated with the prevalence of cachexia and weight loss in cancer patients. In PAAD, these upregulated pro‐cachectic factors were also associated with tumours that present low neoplastic cellularity and high leucocyte fraction. These correlations are plausible explanations of the variation found in the prevalence and severity of cachexia in human cancers and showed that specific therapeutic strategies, according to tumour types, must be considered to treat these patients.
Our comprehensive characterization of the secretome based on transcriptome revealed that the number of upregulated secreted protein‐coding genes, compared with normal tissue, correlates with cachexia prevalence. PAAD presented the highest number of upregulated secretome genes (1267) across all tumour types. This result is particularly expected for the pancreatic tissues, which present more than 70% of the transcripts encoding secreted proteins in the normal tissues.27 However, a substantial fraction of upregulated secretome genes in PAAD is also significantly increased in tumour types commonly associated with cachexia, such as STAD (515 genes), COAD (308 genes), READ (308 genes), ESCA (368 genes), and HNSC (305 genes) (Figure S1). The majority of these shared secretome genes include proteins associated with a cytokine‐mediated signalling pathway, inflammatory response, cytokine–cytokine receptor interaction, and positive regulation of leucocyte chemotaxis. Several of these secreted proteins were previously investigated as potential blood biomarkers of cachexia in cancer patients.16, 17, 18, 19, 20, 21 For example, we found high expression levels of CXCL8 in six tumour types with a high prevalence of cachexia (PAAD, STAD, COAD, READ, ESCA, and HNSC). This cytokine has been reported as an independent predictor of survival in pancreatic and colorectal cancer patients.42, 46 CXCL8 has also been positively associated with weight loss in pancreatic and gastric cancers.42, 47, 48 Interestingly, TNF expression levels, a cytokine commonly associated with cancer cachexia,18 were not changed in cancer types with a high prevalence of the syndrome. This result is similar to that found in resectable pancreatic cancer patients, in which cachexia was not associated with increased plasma levels of canonical pro‐inflammatory cytokines.16
Subsequently, we focused on the tumour expression profile of 25 potential CIFs in 12 human cancers with different risks to develop cachexia. These factors have been previously investigated in pancreatic cancer cachexia16; however, the synergistic expression profile of these molecules in different tumour types needed to be clarified. Cancer literature data describing cachexia prevalence and weight loss8, 11 were associated with the number of these upregulated CIFs. We found that CIFs present a tumour‐specific expression profile. Also, the number of CIF genes upregulated in the tumour is strongly associated with average weight loss and prevalence of cachexia. This is particularly clear in patients with pancreatic cancer, as they have a body weight loss of approximately 13.7 kg,11 which was correlated with the upregulation of 14 CIFs (CXCL8, IL1B, HGF, TNFSF10, LIF, TGFA, TNFSF11, PDGFB, IL6, CCL2, CSF1, IL15, CSF2, and FGF2) that we found in PAAD. By contrast, prostate cancer patients have a body weight loss of around 1.7 kg,11 and PRAD does not upregulate CIF genes when compared with normal tissues. These findings suggest a simple explanation for the discrepancy in average weight loss and cachexia prevalence across cancer patients.
The prognostic value of the expression of these 25 selected pro‐cachectic factors revealed an association with shorter overall survival for the predicted high‐risk groups in the 12 cancer types. The molecular landscape of these CIFs in each cancer type presented a distinct expression profile between high‐risk and low‐risk patients. Our TCGA data reanalysis showed that patients with shorter survival presented upregulation of a large number of CIF. A previous study showed that interleukin 6 increases gradually during the early stages of cachexia but then shows a sudden and steep rise just before death.49 Thus, it is possible that other CIFs present a temporal pattern of regulation that influences the course and severity of the syndrome.
Currently, the source of these cytokines into the tumour microenvironment (from tumour cells or from infiltrating immune cells) is unexplored. PAAD, the cancer type with the highest prevalence of cachexia, often demonstrates only 5–20% neoplastic cellularity (low‐purity tumours) and high leucocyte infiltration.38, 50 Thus, we hypothesized that the set of upregulated CIFs in PAAD is associated with low‐purity tumours. Our clustering analysis showed that CIFs are highly expressed in low‐purity tumours. Infiltrating immune cells have been associated with tumour growth, invasion, and metastasis in several cancer types.51 However, this is the first study indicating the potential influence of infiltrating immune cells in a tumour type highly associated with cachexia. Further investigation of the source of these molecules will contribute to our understanding of cachexia and will allow us to probe specific and accurate therapeutic strategies for the syndrome.
Most studies focusing on silico‐based methods suffer from limitations. Tumour gene expression profiling of CIFs for each tumour type presented herein should be experimentally validated in a larger cohort of cancer patients to circumvent these limitations. Furthermore, we were not able to validate the diagnostic of cachexia due to the absence of clinical data in several patients from the 12 TCGA tumour types. Thus, it remains to be tested whether tumour expression levels of CIF genes can stratify patients according to the cachectic status in these tumour types. However, it is challenging to obtain an independent cohort of individuals as used in our study, which is consisted not only by a high number of tumour samples but also by several non‐diseased tissues (from TCGA and GTEx). Our strategy collectively analysed TCGA and GTEx expression profiles adding higher precision, sensitivity, and robustness to our transcriptomic analysis. Further single‐cell RNA‐Seq investigation is an alternative to differentiate the specific contributions of different cell types in the tumour microenvironment to the tumour expression profile of cachectic patients. Despite such limitations, we used a systematic investigation to verify the expression of CIF genes in cancer cells using a large number of human samples and tumour types, allowing the identification of new potential biomarkers and mediators of cachexia. Overall, we provided insights that open new perspectives in cancer cachexia scenario.
In conclusion, the synergic expression of the CIFs in several tumour types highlights the importance of this group of soluble factors in cancer pathophysiology and presents a strong case for their targeting in specific anti‐cachexia therapeutic development in different tumour contexts. In addition, the tumour‐specific profile of CIFs will facilitate the development of better targeted therapies for clinical decisions.
Conflict of interests
The authors Paula Paccielli Freire, Geysson Javier Fernandez, Diogo de Moraes, Sarah Santiloni Cury, Maeli Dal‐Pai‐Silva, Patricia Pintor dos Reis, Silvia Regina Rogatto, and Robson Francisco Carvalho declare that they have no conflict of interest.
Supporting information
Figure S1. Upregulated genes shared among 12 tumor types. Venn diagrams showing shared upregulated secretome genes in 12 tumor types. Pancreatic adenocarcinomas (PAAD) presented the highest number of upregulated secretome genes and have the highest prevalence of cachexia8,11,12. Based on these data, PAAD was included in all comparisons. PAAD: Pancreatic adenocarcinoma; ESCA: Esophageal carcinoma; STAD: Stomach adenocarcinoma; HNSC: Head and neck squamous cell carcinoma; LUAD: Lung adenocarcinoma; LUSC: Lung squamous cell carcinoma; LIHC: Liver hepatocellular carcinoma; COAD: Colon adenocarcinoma; READ: Rectum adenocarcinoma; LAML: Acute myeloid leukemia; PRAD: Prostate adenocarcinoma; BRCA: Breast invasive carcinoma
Figure S2. Exclusive upregulated genes within tumor types. Heatmap of the mean expression levels [log2 (TPM+1)] of specifically upregulated (a) or downregulated (b) secretome genes in each tumor type compared with corresponding matched normal TCGA and GTEx tissues. Differential expression levels were calculated using the web‐based tool Gene Expression Profiling Analysis (GEPIA, http://gepia.cancerpku.cn/)32. The resulting numbers of genes encoding predicted secreted proteins were filtered based on the Human Protein Atlas secretome data27 (https://www.proteinatlas.org/humanproteome/secretome). Genes that are specifically upregulated or downregulated genes in one tumor type (absolute values of fold‐change > 2.0 and q‐value < 0.01; ANOVA) are shown in red and blue, respectively. PAAD: Pancreatic adenocarcinoma; ESCA: Esophageal carcinoma; STAD: Stomach adenocarcinoma; HNSC: Head and neck squamous cell carcinoma; LUAD: Lung adenocarcinoma; LUSC: Lung squamous cell carcinoma; LIHC: Liver hepatocellular carcinoma; COAD: Colon adenocarcinoma; READ: Rectum adenocarcinoma; LAML: Acute myeloid leukemia; PRAD: Prostate adenocarcinoma; BRCA: Breast invasive carcinoma
Figure S3. The cytokine‐cytokine receptor interaction pathway is enriched in pancreatic adenocarcinoma. Heatmap of the mean expression levels [log2 (TPM+1)] of specifically upregulated (a) or downregulated (b) genes from the cytokine‐cytokine receptor interaction pathway (The Kyoto Encyclopedia of Genes and Genomes, KEGG) in each tumor type (TCGA) compared with corresponding matched normal tissues (TCGA and GTEx). Differential expression levels were calculated using the web‐based tool Gene Expression Profiling Analysis (GEPIA, http://gepia.cancerpku.cn/)32. Genes that are specifically upregulated or downregulated genes in each tumor type (absolute values of fold‐change > 2.0 and q‐value < 0.01; ANOVA) are shown in red and blue, respectively. PAAD: Pancreatic adenocarcinoma; ESCA: Esophageal carcinoma; STAD: Stomach adenocarcinoma; HNSC: Head and neck squamous cell carcinoma; LUAD: Lung adenocarcinoma; LUSC: Lung squamous cell carcinoma; LIHC: Liver hepatocellular carcinoma; COAD: Colon adenocarcinoma; READ: Rectum adenocarcinoma; LAML: Acute myeloid leukemia; PRAD: Prostate adenocarcinoma; BRCA: Breast invasive carcinoma
Figure S4. The expression landscape of cachexia‐inducing factors in 12 tumor types. Heatmap of the mean expression levels [log2 (TPM+1)] of 25 cachexia‐inducing factors (CIF) in 12 TCGA tumor types compared with corresponding matched normal tissues (TCGA and normal GTEx). Differential expression levels were calculated using the web‐based tool Gene Expression Profiling Analysis (GEPIA, http://gepia.cancerpku.cn/)32. Genes that are specifically upregulated or downregulated genes in each tumor type (absolute values of fold‐change > 2.0 and q‐value < 0.01; ANOVA) are shown in red and blue, respectively. PAAD: Pancreatic adenocarcinoma; ESCA: Esophageal carcinoma; STAD: Stomach adenocarcinoma; HNSC: Head and neck squamous cell carcinoma; LUAD: Lung adenocarcinoma; LUSC: Lung squamous cell carcinoma; LIHC: Liver hepatocellular carcinoma; COAD: Colon adenocarcinoma; READ: Rectum adenocarcinoma; LAML: Acute myeloid leukemia; PRAD: Prostate adenocarcinoma; BRCA: Breast invasive carcinoma
Figure S5. Gene expression profile of cachexia‐inducing factors in 12 tumor types. Dot plots of the expression levels [log2 (TPM+1)] of 25 cachexia‐inducing factors (CIF) in different 12 TCGA tumor types. Differential expression levels were calculated using the web‐based tool Gene Expression Profiling Analysis (GEPIA, http://gepia.cancerpku.cn/)32 from the tumor (TCGA; red dots) vs. matched normal (TCGA and GTEx; green dots) tissues. Upregulated and downregulated genes with absolute values of fold‐change > 2.0 and q‐value < 0.01 (ANOVA) are highlighted in red and green for the TCGA abbreviations, respectively. PAAD: Pancreatic adenocarcinoma; ESCA: Esophageal carcinoma; STAD: Stomach adenocarcinoma; HNSC: Head and neck squamous cell carcinoma; LUAD: Lung adenocarcinoma; LUSC: Lung squamous cell carcinoma; LIHC: Liver hepatocellular carcinoma; COAD: Colon adenocarcinoma; READ: Rectum adenocarcinoma; LAML: Acute myeloid leukemia; PRAD: Prostate adenocarcinoma; BRCA: Breast invasive carcinoma
Figure S6. Tumor‐specific expression profile of cachexia‐inducing factors stratified patients into low‐ and high‐risk groups. Heatmaps are representing non‐hierarchical clustering analysis of tumor mRNA expression of 25 cachexia‐inducing factors (CIF) generated in SurvExpress40, using 12 TCGA tumor types. The cancer patients were stratified into high‐ and low‐risk groups, indicated below each heat map as dark‐grey and light‐grey bars, respectively. Risk groups were maximized based on the Prognostic Index assessed by gene expression values multiplied by beta coefficients. PAAD: Pancreatic adenocarcinoma; ESCA: Esophageal carcinoma; STAD: Stomach adenocarcinoma; HNSC: Head and neck squamous cell carcinoma; LUAD: Lung adenocarcinoma; LUSC: Lung squamous cell carcinoma; LIHC: Liver hepatocellular carcinoma; COAD: Colon adenocarcinoma; READ: Rectum adenocarcinoma; LAML: Acute myeloid leukemia; PRAD: Prostate adenocarcinoma; BRCA: Breast invasive carcinoma
Figure S7. Cachexia‐inducing factors tumor‐specific expression profile from patients with low‐risk and high‐risk groups. Box plots representing tumor mRNA expression of 25 cachexia‐inducing factors (CIF) generated by SurvExpress40, using 12 TCGA datasets. The cancer patients were stratified into high‐ and low ‐risk groups, and are indicated as red and green, respectively. The statistical difference for each mRNA expression between high‐ and low‐risk groups was tested using a t‐test, and the resulting p‐value is indicated. PAAD: Pancreatic adenocarcinoma; ESCA: Esophageal carcinoma; STAD: Stomach adenocarcinoma; HNSC: Head and neck squamous cell carcinoma; LUAD: Lung adenocarcinoma; LUSC: Lung squamous cell carcinoma; LIHC: Liver hepatocellular carcinoma; COAD: Colon adenocarcinoma; READ: Rectum adenocarcinoma; LAML: Acute myeloid leukemia; PRAD: Prostate adenocarcinoma; BRCA: Breast invasive carcinoma
Data S1. Genes enconding a predicted secreted protein according to The human Protein Altas (https://www.proteinatlas.org/)
Data S2. Secretome genes differentially expressed in 12 tumor types
Table S1. Cachectic soluble factors genes selected in tumor and normal tissues samples.
Table S2. The Cancer Genome Atlas (TCGA) and The Genotype‐Tissue Expression (GTEx) samples used for gene expression analyses.
Table S3. Number (n) and porcentage (%) of dysregulated secretome genes in 12 cancer types.
Table S4. Upregulated secretome genes in each tumor type (TCGA) matched with the normal tissues (TCGA and GTEx).
Table S5. Downregulated secretome genes in each tumor type compared with the corresponding matched normal tissues ( TCGA and GTEx ).
Table S6. Upregulated secretome genes (Log2 Fold‐Change) common in pancreatic adenocarcinomas and other tumor types.
Table S7. Secretome genes exclusively upregulated in a single cancer type.
Table S8. Top‐ranked Kyoto Encyclopedia of Genes and Genomes pathways associated with upregulated secretome genes detected exclusively in pancreatic adenocarcinoma
Table S9. Secretome genes downregulated exclusively in a single cancer type.
Table S10. Top‐ranked combined scores for Kyoto Encyclopedia of Genes and Genomes pathways categories associated with upregulated secretome genes specifically in acute myeloid leukemia.
Table S11. Top‐ranked combined scores for Kyoto Encyclopedia of Genes and Genomes pathways categories associated with exclusive downregulated secretome genes in acute myeloid leukemia.
Table S12. The expression of cachectic‐inducing factors based on The Cancer Genome Atlas (TCGA) tumor samples predicts poor overall survival.
Table S13. Differentially expressed genes (DEGs) among the list of 25 factors, which have higher relevance in predicting worse survival.
Acknowledgements
This study was supported by the São Paulo Research Foundation—FAPESP (grants 12/13961‐6, 13/50343‐1, 14/13941‐0, and 18/19695‐2) and by the National Council for Scientific and Technological Development, CNPq (Process 311530/2019‐2 to RFC). The results shown here are in whole based upon data generated by the TCGA Research Network (http://cancergenome.nih.gov/) and by the Genotype‐Tissue Expression project (GTEx) (https://gtexportal.org/). The authors of this manuscript certify that they comply with the ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia, and Muscle.52
Freire P. P., Fernandez G. J., de Moraes D., Cury S. S., Dal Pai‐Silva M., dos Reis P. P., Rogatto S. R., and Carvalho R. F. (2020) The expression landscape of cachexia‐inducing factors in human cancers, Journal of Cachexia, Sarcopenia and Muscle, 11, 947–961. 10.1002/jcsm.12565.
Paula Paccielli Freire and Geysson Javier Fernandez contributed equally to this study
Contributor Information
Silvia Regina Rogatto, Email: silvia.regina.rogatto@rsyd.dk.
Robson Francisco Carvalho, Email: robson.carvalho@unesp.br.
References
- 1. Vaughan VC, Martin P, Lewandowski PA. Cancer cachexia: impact, mechanisms and emerging treatments. J Cachexia Sarcopenia Muscle 2013;4:95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. von Haehling S, Anker MS, Anker SD. Prevalence and clinical impact of cachexia in chronic illness in Europe, USA, and Japan: facts and numbers update 2016. J Cachexia Sarcopenia Muscle 2016;7:507–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dewys WD, Begg C, Lavin PT, Band PR, Bennett JM, Bertino JR, et al. Prognostic effect of weight loss prior to chemotherapy in cancer patients. Eastern Cooperative Oncology Group. Am J Med 1980;69:491–497. [DOI] [PubMed] [Google Scholar]
- 4. Martin L, Birdsell L, MacDonald N, Reiman T, Clandinin MT, McCargar LJ, et al. Cancer cachexia in the age of obesity: skeletal muscle depletion is a powerful prognostic factor, independent of body mass index. J Clin Oncol 2013;31:1539–1547. [DOI] [PubMed] [Google Scholar]
- 5. Fearon K, Arends J, Baracos V. Understanding the mechanisms and treatment options in cancer cachexia. Nat Rev Clin Oncol 2013;10:90–99. [DOI] [PubMed] [Google Scholar]
- 6. Fearon KCH, Glass DJ, Guttridge DC. Cancer cachexia: mediators, signaling, and metabolic pathways. Cell Metab 2012;16:153–166. [DOI] [PubMed] [Google Scholar]
- 7. Harimoto N, Shirabe K, Yamashita YI, Ikegami T, Yoshizumi T, Soejima Y, et al. Sarcopenia as a predictor of prognosis in patients following hepatectomy for hepatocellular carcinoma. Br J Surg 2013;100:1523–1530. [DOI] [PubMed] [Google Scholar]
- 8. Pressoir M, Desné S, Berchery D, Rossignol G, Poiree B, Meslier M, et al. Prevalence, risk factors and clinical implications of malnutrition in French comprehensive cancer centres. Br J Cancer 2010;102:966–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bozzetti F. Screening the nutritional status in oncology: a preliminary report on 1,000 outpatients. Support Care Cancer 2009;17:279–284. [DOI] [PubMed] [Google Scholar]
- 10. Segura A, Pardo J, Jara C, Zugazabeitia L, Carulla J, de las Peñas R, et al. An epidemiological evaluation of the prevalence of malnutrition in Spanish patients with locally advanced or metastatic cancer. Clin Nutr 2005;24:801–814. [DOI] [PubMed] [Google Scholar]
- 11. Hébuterne X, Lemarié E, Michallet M, de Montreuil CB, Schneider SM, Goldwasser F. Prevalence of malnutrition and current use of nutrition support in patients with cancer. J Parenter Enteral Nutr 2014;38:196–204. [DOI] [PubMed] [Google Scholar]
- 12. Baracos VE, Martin L, Korc M, Guttridge DC, Fearon KCH. Cancer‐associated cachexia. Nat Rev Dis Primers 2018;4:17105. [DOI] [PubMed] [Google Scholar]
- 13. Tsoli M, Robertson G. Cancer cachexia: malignant inflammation, tumorkines, and metabolic mayhem. Trends Endocrinol Metab 2013;24:174–183. [DOI] [PubMed] [Google Scholar]
- 14. Twelkmeyer B, Tardif N, Rooyackers O. Omics and cachexia. Curr Opin Clin Nutr Metab Care 2017;20:181–185. [DOI] [PubMed] [Google Scholar]
- 15. Argilés JM, Stemmler B, López‐Soriano FJ, Busquets S. Inter‐tissue communication in cancer cachexia. Nat Rev Endocrinol 2018;15:9–20. [DOI] [PubMed] [Google Scholar]
- 16. Talbert EE, Lewis HL, Farren MR, Ramsey ML, Chakedis JM, Rajasekera P, et al. Circulating monocyte chemoattractant protein‐1 (MCP‐1) is associated with cachexia in treatment‐naïve pancreatic cancer patients. J Cachexia Sarcopenia Muscle 2018;9:358–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Penafuerte CA, Gagnon B, Sirois J, Murphy J, MacDonald N, Tremblay ML. Identification of neutrophil‐derived proteases and angiotensin II as biomarkers of cancer cachexia. Br J Cancer 2016;114:680–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lerner L, Hayes TG, Tao N, Krieger B, Feng B, Wu Z, et al. Plasma growth differentiation factor 15 is associated with weight loss and mortality in cancer patients. J Cachexia Sarcopenia Muscle 2015;6:317–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lerner L, Tao J, Liu Q, Nicoletti R, Feng B, Krieger B, et al. MAP 3K11/GDF15 axis is a critical driver of cancer cachexia. J Cachexia Sarcopenia Muscle 2016;7:467–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fukawa T, Yan‐Jiang BC, Min‐Wen JC, Jun‐Hao ET, Huang D, Qian CN, et al. Excessive fatty acid oxidation induces muscle atrophy in cancer cachexia. Nat Med 2016;22:666–671. [DOI] [PubMed] [Google Scholar]
- 21. McLean JB, Moylan JS, Horrell EMW, Andrade FH. Proteomic analysis of media from lung cancer cells reveals role of 14‐3‐3 proteins in cachexia. Front Physiol 2015;6:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mukherjee P, Mani S. Methodologies to decipher the cell secretome. Biochim Biophys Acta Proteins Proteomics 2013;1834:2226–2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Welsh JB, Sapinoso LM, Kern SG, Brown DA, Liu T, Bauskin AR, et al. Large‐scale delineation of secreted protein biomarkers overexpressed in cancer tissue and serum. Proc Natl Acad Sci U S A 2003;100:3410–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang Z, Gerstein M, Snyder M. RNA‐Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 2009;10:57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Da Costa Martins PA, De Windt LJ. MicroRNAs in control of cardiac hypertrophy. Cardiovasc Res 2012;93:563–572. [DOI] [PubMed] [Google Scholar]
- 26. Ozsolak F, Milos PM. RNA sequencing: advances, challenges and opportunities. Nat Publ Gr 2010;12:87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Proteomics. Tissue‐based map of the human proteome. Science 2015;347:1260419. [DOI] [PubMed] [Google Scholar]
- 28. Robinson JL, Feizi A, Uhlén M, Nielsen J. A systematic investigation of the malignant functions and diagnostic potential of the cancer secretome. Cell Rep 2019;26:2622–2635.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K, et al. The Cancer Genome Atlas Pan‐Cancer analysis project. Nat Genet 2013;45:1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S, et al. The Genotype‐Tissue Expression (GTEx) project. Nat Genet 2013;45:580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vivian J, Rao AA, Nothaft FA, Ketchum C, Armstrong J, Novak A, et al. Toil enables reproducible, open source, big biomedical data analyses. Nat Biotechnol 2017;35:314–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res 2017;45:W98–W102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Aran D, Sirota M, Butte AJ. Systematic pan‐cancer analysis of tumour purity. Nat Commun 2015;6:8971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Aran D, Camarda R, Odegaard J, Paik H, Oskotsky B, Krings G, et al. Comprehensive analysis of normal adjacent to tumor transcriptomes. Nat Commun 2017;8:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, et al. Circos: an information aesthetic for comparative genomics. Genome Res 2009;19:1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 2016;44:W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 2013;14:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Raphael BJ, Hruban RH, Aguirre AJ, Moffitt RA, Yeh JJ, Stewart C, et al. Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell 2017;32:185–203.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Starruß J, de Back W, Brusch L, Deutsch A. Morpheus: a user‐friendly modeling environment for multiscale and multicellular systems biology. Bioinformatics 2014;30:1331–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Aguirre‐Gamboa R, Gomez‐Rueda H, Martínez‐Ledesma E, Martínez‐Torteya A, Chacolla‐Huaringa R, Rodriguez‐Barrientos A, et al. SurvExpress: an online biomarker validation tool and database for cancer gene expression data using survival analysis. PLoS ONE 2013;8:e74250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Metsalu T, Vilo J. ClustVis: a web tool for visualizing clustering of multivariate data using principal component analysis and heatmap. Nucleic Acids Res 2015;43:W566–W570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hou Y‐C, Wang CJ, Chao YJ, Chen HY, Wang HC, Tung HL, et al. Elevated serum interleukin‐8 level correlates with cancer‐related cachexia and sarcopenia: an indicator for pancreatic cancer outcomes. J Clin Med 2018;7:502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kuroda K, Nakashima J, Kanao K, Kikuchi E, Miyajima A, Horiguchi Y, et al. Interleukin 6 is associated with cachexia in patients with prostate cancer. Urology 2007;69:113–117. [DOI] [PubMed] [Google Scholar]
- 44. Zhang D, Song B, Wang S, Zheng H, Wang X. Association of interleukin‐8 with cachexia from patients with low‐third gastric cancer. Comp Funct Genomics 2009;2009:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Richey LM, George JR, Couch ME, Kanapkey BK, Yin X, Cannon T, et al. Defining cancer cachexia in head and neck squamous cell carcinoma. Clin Cancer Res 2007;13:6561–6567. [DOI] [PubMed] [Google Scholar]
- 46. Xia W, Chen W, Zhang Z, Wu D, Wu P, Chen Z, et al. Prognostic value, clinicopathologic features and diagnostic accuracy of interleukin‐8 in colorectal cancer: a meta‐analysis. PLoS ONE 2015;10:e0123484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Song B, Zhang D, Wang S, Zheng H, Wang X. Association of interleukin‐8 with cachexia from patients with low‐third gastric cancer. Comp Funct Genomics 2009;2009:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bo S, Dianliang Z, Hongmei Z, Xinxiang W, Yanbing Z, Xiaobo L. Association of interleukin‐8 gene polymorphism with cachexia from patients with gastric cancer. J Interferon Cytokine Res 2010;30:9–14. [DOI] [PubMed] [Google Scholar]
- 49. Iwase S, Murakami T, Saito Y, Nakagawa K. Steep elevation of blood interleukin‐6 (IL‐6) associated only with late stages of cachexia in cancer patients. Eur Cytokine Netw 2004;15:312–316. [PubMed] [Google Scholar]
- 50. Wood LD, Hruban RH. Pathology and molecular genetics of pancreatic neoplasms. Cancer J 2012;18:492–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science 2011;331:1565–1570. [DOI] [PubMed] [Google Scholar]
- 52. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2019. J Cachexia Sarcopenia Muscle 2019;10:1143–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Upregulated genes shared among 12 tumor types. Venn diagrams showing shared upregulated secretome genes in 12 tumor types. Pancreatic adenocarcinomas (PAAD) presented the highest number of upregulated secretome genes and have the highest prevalence of cachexia8,11,12. Based on these data, PAAD was included in all comparisons. PAAD: Pancreatic adenocarcinoma; ESCA: Esophageal carcinoma; STAD: Stomach adenocarcinoma; HNSC: Head and neck squamous cell carcinoma; LUAD: Lung adenocarcinoma; LUSC: Lung squamous cell carcinoma; LIHC: Liver hepatocellular carcinoma; COAD: Colon adenocarcinoma; READ: Rectum adenocarcinoma; LAML: Acute myeloid leukemia; PRAD: Prostate adenocarcinoma; BRCA: Breast invasive carcinoma
Figure S2. Exclusive upregulated genes within tumor types. Heatmap of the mean expression levels [log2 (TPM+1)] of specifically upregulated (a) or downregulated (b) secretome genes in each tumor type compared with corresponding matched normal TCGA and GTEx tissues. Differential expression levels were calculated using the web‐based tool Gene Expression Profiling Analysis (GEPIA, http://gepia.cancerpku.cn/)32. The resulting numbers of genes encoding predicted secreted proteins were filtered based on the Human Protein Atlas secretome data27 (https://www.proteinatlas.org/humanproteome/secretome). Genes that are specifically upregulated or downregulated genes in one tumor type (absolute values of fold‐change > 2.0 and q‐value < 0.01; ANOVA) are shown in red and blue, respectively. PAAD: Pancreatic adenocarcinoma; ESCA: Esophageal carcinoma; STAD: Stomach adenocarcinoma; HNSC: Head and neck squamous cell carcinoma; LUAD: Lung adenocarcinoma; LUSC: Lung squamous cell carcinoma; LIHC: Liver hepatocellular carcinoma; COAD: Colon adenocarcinoma; READ: Rectum adenocarcinoma; LAML: Acute myeloid leukemia; PRAD: Prostate adenocarcinoma; BRCA: Breast invasive carcinoma
Figure S3. The cytokine‐cytokine receptor interaction pathway is enriched in pancreatic adenocarcinoma. Heatmap of the mean expression levels [log2 (TPM+1)] of specifically upregulated (a) or downregulated (b) genes from the cytokine‐cytokine receptor interaction pathway (The Kyoto Encyclopedia of Genes and Genomes, KEGG) in each tumor type (TCGA) compared with corresponding matched normal tissues (TCGA and GTEx). Differential expression levels were calculated using the web‐based tool Gene Expression Profiling Analysis (GEPIA, http://gepia.cancerpku.cn/)32. Genes that are specifically upregulated or downregulated genes in each tumor type (absolute values of fold‐change > 2.0 and q‐value < 0.01; ANOVA) are shown in red and blue, respectively. PAAD: Pancreatic adenocarcinoma; ESCA: Esophageal carcinoma; STAD: Stomach adenocarcinoma; HNSC: Head and neck squamous cell carcinoma; LUAD: Lung adenocarcinoma; LUSC: Lung squamous cell carcinoma; LIHC: Liver hepatocellular carcinoma; COAD: Colon adenocarcinoma; READ: Rectum adenocarcinoma; LAML: Acute myeloid leukemia; PRAD: Prostate adenocarcinoma; BRCA: Breast invasive carcinoma
Figure S4. The expression landscape of cachexia‐inducing factors in 12 tumor types. Heatmap of the mean expression levels [log2 (TPM+1)] of 25 cachexia‐inducing factors (CIF) in 12 TCGA tumor types compared with corresponding matched normal tissues (TCGA and normal GTEx). Differential expression levels were calculated using the web‐based tool Gene Expression Profiling Analysis (GEPIA, http://gepia.cancerpku.cn/)32. Genes that are specifically upregulated or downregulated genes in each tumor type (absolute values of fold‐change > 2.0 and q‐value < 0.01; ANOVA) are shown in red and blue, respectively. PAAD: Pancreatic adenocarcinoma; ESCA: Esophageal carcinoma; STAD: Stomach adenocarcinoma; HNSC: Head and neck squamous cell carcinoma; LUAD: Lung adenocarcinoma; LUSC: Lung squamous cell carcinoma; LIHC: Liver hepatocellular carcinoma; COAD: Colon adenocarcinoma; READ: Rectum adenocarcinoma; LAML: Acute myeloid leukemia; PRAD: Prostate adenocarcinoma; BRCA: Breast invasive carcinoma
Figure S5. Gene expression profile of cachexia‐inducing factors in 12 tumor types. Dot plots of the expression levels [log2 (TPM+1)] of 25 cachexia‐inducing factors (CIF) in different 12 TCGA tumor types. Differential expression levels were calculated using the web‐based tool Gene Expression Profiling Analysis (GEPIA, http://gepia.cancerpku.cn/)32 from the tumor (TCGA; red dots) vs. matched normal (TCGA and GTEx; green dots) tissues. Upregulated and downregulated genes with absolute values of fold‐change > 2.0 and q‐value < 0.01 (ANOVA) are highlighted in red and green for the TCGA abbreviations, respectively. PAAD: Pancreatic adenocarcinoma; ESCA: Esophageal carcinoma; STAD: Stomach adenocarcinoma; HNSC: Head and neck squamous cell carcinoma; LUAD: Lung adenocarcinoma; LUSC: Lung squamous cell carcinoma; LIHC: Liver hepatocellular carcinoma; COAD: Colon adenocarcinoma; READ: Rectum adenocarcinoma; LAML: Acute myeloid leukemia; PRAD: Prostate adenocarcinoma; BRCA: Breast invasive carcinoma
Figure S6. Tumor‐specific expression profile of cachexia‐inducing factors stratified patients into low‐ and high‐risk groups. Heatmaps are representing non‐hierarchical clustering analysis of tumor mRNA expression of 25 cachexia‐inducing factors (CIF) generated in SurvExpress40, using 12 TCGA tumor types. The cancer patients were stratified into high‐ and low‐risk groups, indicated below each heat map as dark‐grey and light‐grey bars, respectively. Risk groups were maximized based on the Prognostic Index assessed by gene expression values multiplied by beta coefficients. PAAD: Pancreatic adenocarcinoma; ESCA: Esophageal carcinoma; STAD: Stomach adenocarcinoma; HNSC: Head and neck squamous cell carcinoma; LUAD: Lung adenocarcinoma; LUSC: Lung squamous cell carcinoma; LIHC: Liver hepatocellular carcinoma; COAD: Colon adenocarcinoma; READ: Rectum adenocarcinoma; LAML: Acute myeloid leukemia; PRAD: Prostate adenocarcinoma; BRCA: Breast invasive carcinoma
Figure S7. Cachexia‐inducing factors tumor‐specific expression profile from patients with low‐risk and high‐risk groups. Box plots representing tumor mRNA expression of 25 cachexia‐inducing factors (CIF) generated by SurvExpress40, using 12 TCGA datasets. The cancer patients were stratified into high‐ and low ‐risk groups, and are indicated as red and green, respectively. The statistical difference for each mRNA expression between high‐ and low‐risk groups was tested using a t‐test, and the resulting p‐value is indicated. PAAD: Pancreatic adenocarcinoma; ESCA: Esophageal carcinoma; STAD: Stomach adenocarcinoma; HNSC: Head and neck squamous cell carcinoma; LUAD: Lung adenocarcinoma; LUSC: Lung squamous cell carcinoma; LIHC: Liver hepatocellular carcinoma; COAD: Colon adenocarcinoma; READ: Rectum adenocarcinoma; LAML: Acute myeloid leukemia; PRAD: Prostate adenocarcinoma; BRCA: Breast invasive carcinoma
Data S1. Genes enconding a predicted secreted protein according to The human Protein Altas (https://www.proteinatlas.org/)
Data S2. Secretome genes differentially expressed in 12 tumor types
Table S1. Cachectic soluble factors genes selected in tumor and normal tissues samples.
Table S2. The Cancer Genome Atlas (TCGA) and The Genotype‐Tissue Expression (GTEx) samples used for gene expression analyses.
Table S3. Number (n) and porcentage (%) of dysregulated secretome genes in 12 cancer types.
Table S4. Upregulated secretome genes in each tumor type (TCGA) matched with the normal tissues (TCGA and GTEx).
Table S5. Downregulated secretome genes in each tumor type compared with the corresponding matched normal tissues ( TCGA and GTEx ).
Table S6. Upregulated secretome genes (Log2 Fold‐Change) common in pancreatic adenocarcinomas and other tumor types.
Table S7. Secretome genes exclusively upregulated in a single cancer type.
Table S8. Top‐ranked Kyoto Encyclopedia of Genes and Genomes pathways associated with upregulated secretome genes detected exclusively in pancreatic adenocarcinoma
Table S9. Secretome genes downregulated exclusively in a single cancer type.
Table S10. Top‐ranked combined scores for Kyoto Encyclopedia of Genes and Genomes pathways categories associated with upregulated secretome genes specifically in acute myeloid leukemia.
Table S11. Top‐ranked combined scores for Kyoto Encyclopedia of Genes and Genomes pathways categories associated with exclusive downregulated secretome genes in acute myeloid leukemia.
Table S12. The expression of cachectic‐inducing factors based on The Cancer Genome Atlas (TCGA) tumor samples predicts poor overall survival.
Table S13. Differentially expressed genes (DEGs) among the list of 25 factors, which have higher relevance in predicting worse survival.