

Abstract
The calcitonin receptor-like class B G protein-coupled receptor (CLR) mediates adrenomedullin (AM) and calcitonin gene-related peptide (CGRP) functions including vasodilation, cardioprotection, and nociception. Receptor activity-modifying proteins (RAMP1–3) form heterodimers with CLR and determine its peptide ligand selectivity through an unresolved mechanism. The CGRP (RAMP1:CLR) and AM (RAMP2/3:CLR) receptors are proven or promising drug targets, but short AM and CGRP plasma half-lives limit their therapeutic utility. Here, we used synthetic peptide combinatorial library and rational design approaches to probe the ligand selectivity determinants and develop truncated AM and CGRP antagonist variants with receptor extracellular domain binding affinities that were enhanced ∼1000-fold into the low nanomolar range. Receptor binding studies and a high-resolution crystal structure of a novel library-identified AM variant bound to the RAMP2-CLR extracellular domain complex explained the increased affinities and defined roles for AM Lys46 and RAMP modulation of CLR conformation in the ligand selectivity mechanism. In longer AM and CGRP scaffolds that also bind the CLR transmembrane domain, the variants generated picomolar affinity antagonists, one with an estimated 12.5 h CGRP receptor residence time, and sustained signaling agonists “ss-AM” and “ss-CGRP” that exhibited persistent cAMP signaling after ligand washout. Sustained signaling was demonstrated in primary human umbilical vein endothelial cells and the SK-N-MC cell line, which endogenously express AM and CGRP receptors, respectively. This work clarifies the RAMP-modulated CLR ligand selectivity mechanism and provides AM and CGRP variants that are valuable pharmacological tools and may have potential as long-acting therapeutics.
Keywords: class B GPCR, RAMP, peptide hormone, long residence time ligand, long-acting agonist, combinatorial peptide library
Introduction
The peptides adrenomedullin (AM) and calcitonin gene-related peptide (CGRP) have overlapping and distinct roles in human physiology and pathophysiology.1,2 Their actions are mediated by the calcitonin receptor-like receptor (CLR), which is a class B G protein-coupled receptor (GPCR) that is an important drug target. Both peptides exhibit vasodilator activity and cardioprotective actions.3,4 AM has crucial roles in cardiac and lymphatic development,5 and in adults it promotes endothelial barrier integrity and embryo implantation.6,7 AM showed beneficial effects in treating heart attack and inflammatory bowel disease in pilot clinical trials,8,9 and holds promise for heart failure, sepsis, and infertility.6,10,11 CGRP is a neuropeptide involved in pain transmission and neurogenic inflammation and it has a crucial role in migraine headache pathogenesis.2,12,13 Monoclonal antibodies and small molecule drugs that antagonize CGRP signaling recently obtained regulatory approval for migraine.12 CGRP antagonists also showed promise for treating necrotizing fasciitis in a mouse model,14 and the CGRP agonist inhibited mucosal HIV transmission in a cell model.15 Both AM and CGRP antagonists may be of value for treating various cancers.16,17
There is substantial potential for AM and CGRP agonists and antagonists as novel therapeutics; however, the short plasma half-lives (∼20 min)18,19 of AM and CGRP limit their therapeutic utility. This is particularly problematic for indications for which the agonists are the desired drugs because small molecule CLR agonists are not available. AM or CGRP analogues with PEGylation, acylation, or other modifications to enhance plasma half-life have been developed,20−22 but these have reduced signaling potencies. As an alternative or in addition to prolonging circulatory half-life, there is growing recognition of the power of increasing receptor residence time to achieve efficacious long-lasting drug action.23,24 Although typically applied to inhibitors, this concept can also be of value for GPCR agonists.25,26 Long residence time AM and CGRP analogues might be able to overcome the problem of their short plasma half-lives, but to our knowledge no such analogues have been reported.
AM and CGRP binding to CLR is controlled by three receptor activity-modifying proteins (RAMP1–3) that heterodimerize with CLR.27 RAMP1 favors CGRP binding, giving the CGRP receptor, whereas RAMP2 and RAMP3 favor AM binding, giving the AM1 and AM2 receptors, respectively. A related peptide, adrenomedullin 2/intermedin (AM2/IMD), binds the receptors somewhat nonselectively. CLR and the RAMPs each have extracellular (ECD) and transmembrane (TMD) domains that associate with their counterparts in the heterodimer. Peptide binding to CLR follows the class B GPCR “two-domain” model in which the C-terminal half of the peptide binds the ECD and the N-terminal half binds and activates the TMD.28 Soluble RAMP-CLR ECD fusion proteins exhibited peptide-binding preferences similar to the intact receptors, but with the lower binding affinities expected to accompany lost peptide–TMD contacts.29,30 Crystal structures of C-terminal fragments of a CGRP analogue or AM2/IMD bound to RAMP1-CLR ECD and AM bound to RAMP2-CLR ECD showed that the peptides bind a common site on CLR, adopt relatively unstructured conformations defined by a β-turn structural element, and have minimal RAMP contacts.30,31 Cryo-EM structures of the intact CGRP and AM receptors with bound peptide agonists and Gs heterotrimer showed that the N-terminal half of the peptides occupied the CLR TMD with an α-helical conformation and revealed an absence of peptide-RAMP contacts outside of the ECD complexes.32,33
Understanding how RAMPs determine CLR ligand selectivity is important because this system is a model for accessory membrane protein modulation of GPCR pharmacology and knowledge of the mechanism will aid design of selective therapeutics. The ECD complex is a major selectivity determinant resulting in part from peptide–RAMP contacts.30,31 The C-terminal peptide residues AM Y52 and CGRP F37 anchor in a pocket on the CLR ECD that is augmented on one side by the RAMP subunit. AM Y52 forms a hydrogen bond (H-bond) with RAMP2 E101 that is not available in RAMP1, and CGRP F37 makes hydrophobic contacts with RAMP1 W84 that RAMP2 cannot provide. Unfortunately, beyond this the mechanism is unresolved. In the crystal structure a second AM residue, K46, contacts RAMP2, but it is unclear if this contributes to selectivity because it also has an intramolecular packing role that complicates interpretation of mutagenesis data. In addition, subtle CLR ECD conformational differences observed in the structures with RAMP1 and RAMP2 hinted at an allosteric role for the RAMPs,31 but clear evidence that this is an important component of the selectivity mechanism is lacking.
We previously used rational design to develop truncated ECD-binding AM and CGRP variants with increased affinities by introducing substitutions designed to stabilize the β-turn.34 Here, we used a combinatorial peptide library approach35 to identify novel AM substitutions that further enhance ECD affinity, and we present a high-resolution crystal structure that explains the enhanced affinity of a library-identified variant. Results from the library screen and new rationally designed AM and CGRP variants allowed us to define important roles for AM K46 and RAMP modulation of CLR ECD conformation in the selectivity mechanism. Incorporating key variants in longer peptide scaffolds that also bind the CLR TMD yielded picomolar affinity AM and CGRP antagonists and long-acting, sustained signaling AM and CGRP agonists. These results clarify the ligand selectivity mechanism and provide a suite of novel peptides with immediate value as pharmacological tools and future potential as long residence time therapeutics.
Results and Discussion
Positional Scanning-Synthetic Peptide Combinatorial Library (PS-SPCL) Screen for AM
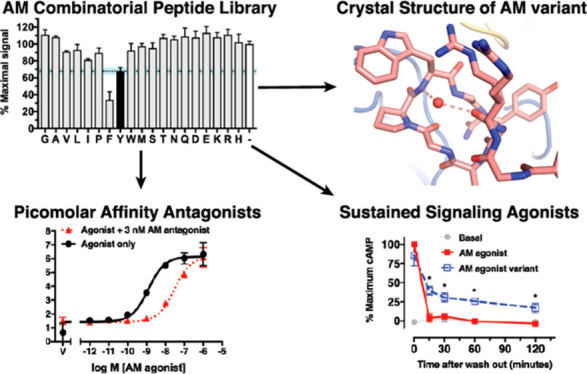
A PS-SPCL35 was designed (see Methods) to identify novel affinity-increasing substitutions in AM and to probe selectivity determinants. The scaffold was the minimal ECD-binding AM(37–52) fragment including the affinity-enhancing Q50W substitution identified by rational design.34 The five positions chosen for substitution were S45, K46, I47, S48, and Y52 (Figure 1B). S45, K46, and Y52 were chosen because our previous rational design effort indicated that substitutions at these positions could enhance affinity and/or alter selectivity as exemplified by the S45W/K46L/Q50W/Y52F variant that had altered preference and increased affinity for RAMP1-CLR ECD34 (Figure 1A). I47 and S48 were chosen as two new positions not previously explored. The AM PS-SPCL, comprising 95 mixtures and nearly 2.5 million unique theoretical peptides, was screened for binding to the three purified RAMP-CLR ECD complexes (Figure 1A–C) in a fluorescence polarization (FP) competition binding assay (Figure 1D).
Figure 1.

AM(37–52) Q50W PS-SPCL screen at purified MBP-RAMP-CLR ECD fusion proteins. (A) Crystal structure of MBP-RAMP1-CLR ECD with a rationally designed altered selectivity and enhanced affinity AM variant [PDB 5V6Y]. (B) Crystal structure of MBP-RAMP2-CLR ECD with AM [PDB 4RWF]. (C) Homology model of the RAMP3-CLR ECD complex with AM.31 In structures A and B MBP is omitted for clarity. (D) Competition FP assays with the AM PS-SPCL mixtures. Black bars indicate the wild-type residue at the indicated position while the white bars represent no competitor control. Data are mean composite of two independent replicates. Error is shown as standard deviation (SD) with the blue shaded region representing one SD of wild-type residue.
Satisfyingly, the library screen confirmed our prior rational design results. W at position 45 increased affinity at all three complexes and L at position 46 improved binding with RAMP1. F at position 52 diminished binding with RAMP2 while it enhanced binding with RAMP1 and had no effect at the RAMP3 complex (Figure 1D). Importantly, the library screen also identified novel and unexpected substitutions. At position 45, F, Y, R, or H also increased affinity, with R perhaps favoring RAMP2/3. Unexpectedly, the library revealed that L at position 46 was tolerated with RAMP2 and indicated that a range of hydrophobic or small polar residues at this position improved binding with RAMP1. Notably, G at position 46 improved binding with RAMP1 and decreased binding with RAMP2/3. These results strongly suggested a significant role for AM K46 in receptor selectivity. For the positions not previously explored, L and M were tolerated at position 47 and G worked surprisingly well at position 48 (Figure 1D).
RAMP-CLR ECD Complex Binding Affinities of Defined AM Variants
The large number of affinity-enhancing substitutions identified made it impractical to test all possible combinations in defined AM variants. Instead, guided by modeling we chose to further examine the effects of the library-identified S45R, K46W/A/G, and S48G substitutions in 10 new defined AM(37–52) variants, several of which also included the previous rationally designed K46L, Q50W, or Y52F substitutions. S45R was chosen because it might make ionic interactions with E101 and/or E105 in RAMP2 (Figure 1B), or E74 in RAMP3 (Figure 1C). The substitutions at position 46 were chosen to test both small and large residues. S48G was chosen because S48 forms intramolecular H-bonds that appear to stabilize the AM β-turn and α-helical turn,31 so it was surprising that loss of this side chain did not diminish binding.
First we characterized the affinities of five prior rational design variants34 for the purified RAMP-CLR ECD complexes in the competition FP assay (Figure 2A and Table S1). This was done because our previous study lacked the RAMP3-CLR ECD complex and the RAMP1- and -2-CLR ECD complexes that were used were produced in E. coli and therefore lacked N-glycosylation. We have since shown that N-glycosylation of the CLR ECD increases peptide-binding affinity.36 Measuring binding at the three N-glycosylated ECD complexes indicated that Q50W and S45W enhanced affinity from the micromolar into the nanomolar range and K46L and Y52F altered selectivity to favor RAMP1 and disfavor RAMP2 (Figure 2A).
Figure 2.

Binding of defined AM(37–52) variants to purified MBP-RAMP-CLR ECD fusion proteins. (A, B) Scatter plot of pKI values from competition FP assays for (A) previously reported, rationally designed AM variants34 or (B) new AM variants incorporating library-identified substitutions. Numbers below the x-axis signify the RAMP1, -2, or -3 complexes. (C) Representative competition binding curves for two key variants compared to wild-type. See Table S1 for pKI values with SEM, selectivity comparisons, and associated statistical analyses.
Next we examined the binding of new variants that incorporated the library-identified substitutions (Figure 2B and Table S1). S45R had little effect in the S45R/Q50W double mutant as compared to Q50W, however, with K46L in the S45R/K46L/Q50W triple mutant S45R increased affinity ∼5-fold at RAMP1/3 and 17-fold at RAMP2 (as compared to K46L/Q50W). A large Trp at position 46 in the S45R/K46W/Q50W and S45W/K46W/Q50W variants yielded somewhat nonselective peptides with lower affinities, whereas a small Gly at position 46 in the S45W/K46G/Q50W/Y52F quadruple mutant yielded a peptide with altered preference and enhanced affinity for RAMP1-CLR ECD equivalent to our prior rationally designed S45W/K46L/Q50W/Y52F variant. The latter result was quite surprising because we had thought that the bulky Leu at position 46 was required for selectivity toward RAMP1.34
The S48G substitution in the S48G/Q50W double mutant enhanced affinity ∼6-fold at RAMP1/3 and 13-fold at RAMP2 as compared to Q50W (Figure 2B and Table S1). Adding S45R to give the S45R/S48G/Q50W triple mutant had no effect, whereas adding K46L favored RAMP1 and disfavored RAMP2 in the K46L/S48G/Q50W variant. The quadruple mutant S45R/K46L/S48G/Q50W exhibited single digit nanomolar affinities for the RAMP2/3 complexes and double digit nanomolar affinity for the RAMP1 complex. Comparing this to the two prior triple mutants indicated that S45R enhanced affinity in the presence of K46L. Ala at position 46 in the S45R/K46A/S48G/Q50W quadruple mutant decreased affinity ∼10-fold at each of the complexes as compared to the K46L-containing version. Representative FP competition binding curves for the S48G/Q50W and S45R/K46L/S48G/Q50W variants highlight their ∼3 orders of magnitude increased affinities (Figure 2C).
Structural Basis for Enhanced RAMP2-CLR ECD Affinity of AM S45R/K46L/S48G/Q50W
To provide insights into how S45R, S48G, and Q50W enhanced affinity, and why S45R only did so in the presence of K46L, we determined a crystal structure of AM S45R/K46L/S48G/Q50W bound to a maltose binding protein (MBP)-RAMP2-CLR ECD fusion at 1.83 Å resolution (Figure 3A; Table S2) and compared it to our prior structure with wild-type AM (Figure 3B). Excellent electron density was observed for the AM variant (Figure S1A); however, crystal packing complicated interpretation of the effects of the S45R and K46L substitutions. R356 from MBP stacked on AM K46L and formed an H-bond/ionic bond with RAMP2 E101 resulting in two alternate E101 conformations (Figure 3A and Figure S1B). In addition, packing of a symmetry mate MBP against the α1−β1 loop in CLR caused shifts in the CLR α1 helix and the β1−β2 loop that forms part of the pocket bordered by the RAMP (Figure S1C,D). The shifting of RAMP2 enabled E105 to form a salt-bridge with CLR R119 that was not observed in our prior structure (Figure 3A,B and Figure S1C). Electron density for the AM variant S45R side chain supported modeling two alternate conformations, neither of which contacted RAMP2 (Figure 3A), possibly because of packing effects.
Figure 3.

Structural basis for enhanced RAMP2-CLR ECD affinity of AM(37–52) S45R/K46L/S48G/Q50W: (A) 1.83 Å resolution crystal structure of the variant bound to MBP-RAMP2-CLR ECD. (B) Crystal structure of AM-bound MBP-RAMP2-CLR ECD [PDB 4RWF] in the same view as panel A for comparison. (C) cAMP signaling antagonism assay in COS-7 cells using RAMP2 E105A:CLR and the indicated concentration of antagonist peptide variants. Right is a scatter plot of mean apparent pKB values determined from three independent experiments with error shown as SEM. (D) Superimposition of the new variant structure and 4RWF showing a detailed view of the peptide β-turn. H-bond distances are shown in angstroms. (E) Competition FP assay for the indicated AM peptides at MBP-RAMP2-CLR ECD. (F) Structural alignment of AM [PDB 4RWF] and AM2/IMD [PDB 6D1U]. (G) Competition FP assay for the indicated AM2/IMD peptides at MBP-RAMP1-CLR ECD. Mean pKI ± SEM values for the H45W and S43G/H45W variants were 6.08 ± 0.06 and 4.75 ± 0.08, respectively.
To test if S45R forms an ionic interaction with RAMP2 E105 we assessed the ability of S45R to enhance affinity in a cell-based functional cAMP antagonism assay using full-length receptor with RAMP2 E105A. In this assay, the S45R/K46L/S48G/Q50W variant retained ∼10-fold higher affinity than the K46L/S48G/Q50W variant (Figure 3C). Thus, RAMP2 E105 does not mediate the AM S45R affinity-enhancing effect. We speculate that S45R sits over K46L similar to MBP R356 in the crystal structure and makes ionic interactions with RAMP2 E101 (Figure 3A) or RAMP3 E74 (Figure 1C). This would explain why S45R had no effect in the presence of the wild-type K46 residue, which would block such an interaction. S45R also enhanced affinity at RAMP1-CLR ECD for reasons that are unclear, but the affinity enhancement was largest with RAMP2, and the library screen suggested that S45R favors RAMP2/3. There may be other S45R-containing variants in the library that retain better selectivity for the AM receptors, perhaps with other K46 substitutions and/or I47L/M, which we did not pursue here.
Crystal packing did not affect the S48G and Q50W substitutions. Q50W contacted the CLR turret loop and intramolecularly packed against P49, presumably stabilizing the β-turn (Figure 3D). Flexibility enabled by S48G appeared to allow tighter packing of P49 with Q50W than would be possible with S48. Supporting this, S48G alone enhanced affinity only ∼5-fold at RAMP2-CLR ECD (Figure 3E and Table S1), rather than the 13-fold enhancement observed with Q50W. In addition, in the variant structure with S48G, a water molecule occupied the position of the S48 hydroxyl where it formed bridging H-bonds with the main chains of Q50W in the β-turn and S45R in the α-helical turn similar to the S48 side chain, but with shorter bond distances (Figure 3D). Notably, we would never have arrived at the S48G substitution by rational design because the S48 side chain H-bonds appeared to be critical for stabilizing the AM turns and previous work showed that the S48A substitution decreased AM binding to the purified RAMP2-CLR ECD complex and the full-length AM1 receptor.29,37 It was thus interesting that the structure suggested that a bridging water stabilized the AM turns better than S48.
AM2/IMD shares the AM β-turn structure and contains S43 at the position equivalent to AM S48, but it lacks the α-helical turn (Figure 3F). We previously showed that the AM2/IMD H45W substitution at the position equivalent to AM Q50 increased affinity.30 In contrast to our results with AM, however, the S43G substitution in AM2/IMD H45W decreased its ECD affinity ∼20-fold (Figure 3G). This result supported the hypothesis that the bridging water enabled by S48G in AM enhanced affinity by bridging and stabilizing the two AM turns. Thus, we propose that the increased AM affinity resulting from S48G and Q50W is largely due to turn stabilization and the accompanying reduction of the entropic penalty associated with peptide ordering upon receptor binding.
Probing the Ligand Selectivity Mechanism by AM K46 and CGRP F37 Mutagenesis
The role of AM K46 in selectivity has been difficult to define because it has several structural functions. The aliphatic portion of the side chain intramolecularly packs against Y52 and intermolecularly contacts CLR W72, while the side chain amino group is within H-bond/ionic bond distance of RAMP2 E101 and E105 (Figure 4A). In mutagenesis studies RAMP2 E101A decreased AM cAMP signaling potency 25-fold, but surprisingly E105A had no effect.31 It was suggested that the K46 aliphatic contacts were most important, and that K46L alteration of selectivity to favor RAMP1 resulted from steric effects rather than lost RAMP2 contact(s).34 K46L was thought to push the position 52 side chain into a position such as CGRP F37 to improve shape complementarity with the pocket in the RAMP1 complex while decreasing complementarity with the RAMP2 complex pocket (Figure 4A). The AM library position 46 results revealed that a variety of small polar or nonpolar residues at position 46 increased binding with RAMP1 (Figure 1D). This result and crystal packing limitations on interpretation of K46L shape complementarity effects in the new structure called for revisiting the roles of AM K46 and RAMP-mediated allosteric modulation of the CLR ECD in ligand selectivity.
Figure 4.

Probing the ligand selectivity mechanism by AM K46 and CGRP F37 mutagenesis. (A) Detailed view of the pocket over the W72 Trp shelf in superimposed AM-bound RAMP2-CLR ECD [4RWF] and CGRP N31D/S34P/K35F-bound RAMP1-CLR ECD [4RWG] crystal structures. AM position 46 substitutions K46Nle and K46L are modeled and key residues are shown as sticks. (B,C) Scatter plots of mean pKI values from competition FP assays with purified MBP-RAMP-CLR ECD complexes and the indicated AM(37–52) K46 variants in the (B) wild-type or (C) Q50W backgrounds. (D) Crystal structure of CGRP N31D/S34P/K35F-bound MBP-RAMP1-CLR ECD [PDB 4RWG] with MBP omitted. (E) CGRP K35W, A36S, and F37P substitutions are modeled and shown with the indicated RAMP1–3 residues that augment the pocket. RAMP3 is a homology model. (F) Scatter plot of mean pKI values from competition FP assays using purified MBP-RAMP-CLR ECD complexes with the indicated CGRP F37 variants in the N31D/S34P/K35W/A36S background. The high-affinity detection limit of the assay prevented unambiguous determination of the affinity of the N31D/S34P/K35W/A36S variant at the RAMP1 complex. See Table S3 for the pKI values with SEM, selectivity comparisons, and associated statistical analyses.
AM K46 function was probed by determining the ECD complex affinities of AM(37–52) peptides containing diverse substitutions at position 46 in an otherwise wild-type background (Figure 4B and Table S3). K46R selectively decreased binding with RAMP2/3. The nonstandard amino acid norleucine (Nle) was used to remove the amino group while maintaining aliphatic packing ability (Figure 4A). K46Nle diminished binding with RAMP2/3 and increased binding with RAMP1 (Figure 4B). K46A, which further removes some of the intramolecular packing against Y52, was similar to K46Nle, but with slightly decreased affinity at all three complexes. K46G, which removes all side chain intra- and intermolecular contacts had little effect with RAMP1, but decreased binding at RAMP2/3. K46L and K46M increased binding with RAMP1 and decreased binding with RAMP2 while having little or no effect with RAMP3. W, F, and Y selectively decreased binding with RAMP2/3, and I and V decreased binding at all three complexes. The K46Nle, K46A, and K46L substitutions were also tested in the Q50W background in which they yielded results similar to those observed in the WT background (Figure 4C).
These results revealed a crucial role for K46 in selectivity and indicated that its side chain amino group is important for this function. AM K46 has a positive role to contact RAMP2 E101 or RAMP3 E74, and a negative role to diminish binding at the RAMP1 complex as evidenced by the K46Nle and K46A substitutions decreasing binding to the RAMP2 and -3 complexes and increasing binding with RAMP1. Electrostatics can explain these results because RAMP2/3 have more favorable charge complementarity with K46 than RAMP1 (Figure S2D–G), and this is supported by RAMP mutagenesis data.38,39 Thus, AM K46 contacts with RAMP2 E101 and RAMP3 E74 are important for ligand selectivity in addition to the previously identified contacts of the C-terminal residue of the peptides with RAMP1 W84, RAMP2 E101, and RAMP3 E74/W84.30,31,34 These contacts are summarized in Figure S2 panels A–C, which highlight the importance of the K46 side chain amino group for discriminating RAMP2/3 from RAMP1 (Figure 1D and Figure 4B,C).
The small number of RAMP-peptide contacts in the structures suggested that RAMPs may also have an allosteric role in selectivity,30−32 but evidence for this has been limited. To examine RAMP allosteric effects we turned to CGRP because it has only a single residue, F37, that contacts the RAMP subunit (Figure 4D). We reasoned that CGRP peptides that cannot contact the RAMPs would likely still exhibit different ECD complex affinities if the RAMPs stabilize different CLR conformations. Starting with our prior rational design variant CGRP(27–37) N31D/S34P/K35W/A36S that has greatly increased ECD complex affinity,34 we added the F37P or F37A substitutions to remove the possibility of contact with the conserved cis-Pro in RAMP1–3 and W84 unique to RAMP1/3 (Figure 4E). Both variants strongly preferred the RAMP1/3 complexes and the F37P version had higher affinities, presumably due to Pro packing against the Trp shelf (Figure 4E,F, Table S3).
These data suggested that RAMP1/3 stabilize different CLR ECD conformations than RAMP2. This is consistent with the RAMP1- and RAMP2-CLR ECD crystal structures for which subtle movement of the CLR β1-β2 loop (Figure 4A) and more dramatic movement of the CLR R119 side chain (Figure S2A,B) resulted in altered pocket shapes that appeared to be RAMP-dependent.31 The ECD complex binding data seem to suggest that RAMP3 and RAMP1 stabilize similar CLR ECD conformations (Figure 4F), but a crystal structure of the RAMP3-CLR ECD complex is needed to verify this. The data now available support a model in which RAMPs modulate CLR ligand selectivity at the level of the ECD complexes through both direct RAMP-peptide contacts and allosteric modulation of CLR.
Antagonism of cAMP Signaling by Truncated Single Site ECD-Binding and Dual Site ECD/TMD-Binding AM and CGRP Variants with Enhanced ECD Affinities
Using a COS-7 cell-based cAMP signaling assay with transiently expressed RAMP:CLR complexes, we examined the enhanced affinity AM and CGRP variants in the context of truncated antagonist peptide scaffolds that lack the N-terminal disulfide-linked loop required for receptor activation (Figure 5A,B). In the short ECD-binding fragments we characterized the library-identified AM variants S48G/Q50W, S45R/K46L/Q50W, K46L/S48G/Q50W, and S45R/K46L/S48G/Q50W and the new rationally designed CGRP variant N31D/S34P/K35W/A36S/F37P. Representative assays for AM(37–52) S48G/Q50W are shown in Figure 5C, and the receptor affinities (apparent pKB values) derived from the functional data are summarized in Figure 5F,G and Table S4. The AM and CGRP ECD-binding antagonist variants exhibited surmountable antagonism as expected for competitive antagonists and they had nanomolar apparent KB values that were comparable to their KI values for ECD complex binding. Plots comparing the pKBapp and pKI values for the AM variants at each of the receptors showed good correlations, which indicated that the purified MBP-ECD fusion protein constructs used for the FP assays were suitable surrogates for the ECD complexes in the full-length receptors (Figure S3).
Figure 5.

Antagonism of cAMP signaling by single site ECD and dual site ECD/TMD-binding truncated AM and CGRP variants. (A) Cryo-EM structure of CGRP-bound full-length RAMP1:CLR [PDB 6E3Y] with the superimposed AM ECD-binding fragment from 4RWF. (B) Amino acid sequence alignment of AM and CGRP and cartoon depictions of their agonist and N-terminally truncated ECD/TMD-binding and ECD-binding antagonist forms. (C–E) Representative cAMP signaling antagonism assays for (C) AM(37–52) S48G/Q50W, (D) AM(22–52) S48G/Q50W, or (E) CGRP(8–37) N31D/S34P/K35W/A36S at transiently expressed RAMP:CLR complexes in COS-7 cells. (F,G) Scatter plots of mean apparent pKB values for the indicated (F) AM or (G) CGRP antagonist peptides from at least three independent experiments performed on different days. Values for the peptides with open symbols are from Booe et al. for comparison.34 See Table S4 for the pKBapp values with SEM, selectivity comparisons, and associated statistical analyses and Table S5 for the hemiequilibrium model parameters derived from fitting the CGRP(8–37) N31D/S34P/K35W/A36S data.
In the dual site ECD/TMD-binding fragments we tested the AM S48G/Q50W and CGRP N31D/S34P/K35W/A36S variants because these had the highest ECD affinities while retaining selectivities comparable to the wild-type peptides. AM(22–52) S48G/Q50W exhibited surmountable antagonism at all three receptors and a striking 80 pM apparent KB for the AM1 receptor while having 170-fold and 12-fold lower affinities at the CGRP and AM2 receptors, respectively (Figure 5D,F and Table S4). Interestingly, CGRP(8–37) N31D/S34P/K35W/A36S antagonism was insurmountable at all three receptors (Figure 5E). We previously showed that the ECD-binding 27–37 version of this variant exhibited surmountable antagonism and had nanomolar apparent KB values for each receptor34 (Figure 5G). The insurmountable behavior of the ECD/TMD-binding 8–37 version could be caused by a very slow off-rate leading to hemiequilibrium conditions or possibly arise from complex allosteric mechanisms.25,40 To provide insight into this we analyzed CGRP(8–37) N31D/S34P/K35W/A36S antagonism using a simultaneous antagonist/agonist addition format rather than the antagonist preincubation format used thus far. Under these conditions little or no depression of the Emax was observed (Figure S4), which suggested that the insurmountable behavior reflected hemiequilibrium conditions due to a slow antagonist off rate.25 Assuming that the antagonist and agonist cannot simultaneously occupy the receptors we fit the insurmountable antagonism data to a hemiequilibrium operational model to estimate the antagonist pKB and dissociation rate values.41 This analysis yielded KB values of 141, 468, and 158 pM and off rates of 0.0014, 0.0036, and 0.0018 min–1 for the RAMP1, -2, and -3 complexes, respectively (Figure 5G and Tables S4 and S5). These off rates corresponded to residence times of 12.5, 5.2, and 11 h at the CGRP, AM1, and AM2 receptors, respectively (Table S5).
The results from the functional antagonism assays suggested that the CGRP(8–37) N31D/S34P/K35W/A36S peptide is a slow offset antagonist with very long receptor residence times, particularly at the CGRP receptor. It is notable that despite the AM(22–52) S48G/Q50W antagonist exhibiting strong pM binding affinity at the AM1 receptor its antagonism was surmountable. This suggested that its kinetic properties were different from those of the CGRP variant. We emphasize that the parameters derived from the hemiequilibrium operational model for CGRP(8–37) N31D/S34P/K35W/A36S are only estimates and future studies using direct receptor binding assays will be needed to further evaluate the binding kinetics of the CGRP and AM antagonist variants and accurately determine their receptor residence times. Nonetheless, these antagonists have immediate value as pharmacological tools and they may have future potential as high-affinity, long residence-time therapeutics.
AM and CGRP Agonist Variants with Dramatically Enhanced ECD Affinities Exhibit Long-Acting, Sustained cAMP Signaling in Model Cell Lines and Primary Cells
Next, we characterized the AM(13–52) S48G/Q50W and CGRP(1–37) N31D/S34P/K35W/A36S agonist variants in cAMP accumulation assays using COS-7 cells transiently expressing the receptors. Both variants had significantly enhanced potencies at their noncognate receptors, but showed only minor potency increases at their cognate receptors (Figure 6A,B and Table S6). We reasoned that slow kinetics at the cognate receptors prevented equilibrium from being reached, so we turned to an alternative assay format to measure sustained cAMP signaling after exposure of the cells to 100 nM agonist followed by ligand washout (see Methods). In this format, AM(13–52) S48G/Q50W and CGRP(1–37) N31D/S34P/K35W/A36S yielded significantly enhanced sustained signaling at their cognate AM1 and CGRP receptors in COS-7 cells, respectively (Figure 6C,D). To lesser extents, both variants also had enhanced sustained signaling at the AM2 receptor (Figure S5).
Figure 6.

cAMP signaling properties of AM and CGRP agonist variants with enhanced ECD affinities. (A,B) Concentration–response cAMP accumulation assays in COS-7 cells transiently expressing the indicated receptors with the indicated (A) AM or (B) CGRP peptides. (C,D) Sustained cAMP signaling after ligand washout assays using 100 nM of the indicated (C) AM or (D) CGRP peptides at the indicated receptors transiently expressed in COS-7 cells. (E,F) Concentration–response cAMP accumulation assays for the indicated AM and CGRP peptides in (E) primary HUVECs or (F) SK-N-MC cells. (G,H) Sustained cAMP signaling after ligand washout assays with 100 nM of the indicated AM and CGRP peptides in (G) primary HUVECs or (H) SK-N-MC cells. Representative accumulation assays from three independent replicates performed on different days are shown, whereas the washout assays are presented as a normalized composite of three independent experiments. See Table S6 for the pEC50 values with SEM from the accumulation assays and associated statistical analyses.
Last, we characterized the agonist variants using primary human umbilical vein endothelial cells (HUVECs) that express the AM1 receptor42 and the SK-N-MC neuroblastoma cell line that expresses the CGRP receptor.43 In standard cAMP accumulation assays the AM and CGRP agonist variants had only slightly enhanced potencies at their cognate receptors in HUVECs or SK-N-MC cells, respectively, and exhibited significantly enhanced potencies at their noncognate receptors (Figure 6E,F), similar to the results in COS-7 cells. In washout assays with HUVECs, the wild-type agonists and the CGRP variant failed to elicit sustained cAMP signaling, whereas the AM variant exhibited a significant sustained response with a cAMP level 2 h postwashout equivalent to ∼20% of the prewashout level for wild-type AM (Figure 6G). In SK-N-MC cells the wild-type agonists did not exhibit sustained signaling, the AM variant had a low level of sustained signaling, and the CGRP variant elicited a dramatic sustained response with a cAMP level 2 h postwashout equivalent to ∼55% of the prewashout level for wild-type CGRP (Figure 6H). We term these long-acting, sustained signaling AM(13–52) S48G/Q50W and CGRP(1–37) N31D/S34P/K35W/A36S agonist variants “ss-AM” and “ss-CGRP”.
In the standard cAMP accumulation assays ss-AM and ss-CGRP appeared to be nonselective (Figure 6A,B); however, the washout assays in HUVECs and SK-N-MC cells revealed that they selectively promoted sustained cAMP signaling at their cognate receptors (Figure 6G,H). The weak sustained signaling observed with ss-AM in SK-N-MC cells may result from low-level AM1 expression because RAMP2 mRNA has been detected in this cell line.43,44 The sustained signaling could result from long residence time at cell surface receptors or internalized receptors.26 Interestingly, in the context of pain transmission CGRP receptor signaling through PKC and ERK occurred from endosomes,45 and sustained cAMP signaling from endosomes is well documented for the class B parathyroid hormone receptor.46 Internalization of the CGRP receptor occurs on a time scale relevant to our washout assay,47 so we speculate that the sustained signaling arises from internalized receptors, at least in part. Deciphering the mechanism of the sustained signaling and its effects on cell responses is an important area for future studies, particularly because the altered temporal characteristics of ss-AM and ss-CGRP could have positive or negative effects for potential future therapeutic applications. Nonetheless, in theory ss-AM and ss-CGRP might be able to overcome the short plasma half-life problem of the wild-type agonists in vivo.24 Encouragingly, an acylated CGRP analogue with prolonged circulatory half-life exhibited protective effects in mouse models of hypertension, cardiac hypertrophy, and heart failure.48 ss-AM and ss-CGRP may have potential as novel therapeutics with long-acting properties resulting from long receptor residence times rather than prolonged circulatory half-lives.
Methods
Cell Culture
COS-7 (CRL 1651), SK-N-MC (HTB-10), and primary human umbilical vein endothelial cells (HUVECs) (PCS-100–010) were from American Type Culture Collection (Manassas, VA, USA). Dulbecco’s modified eagle media (DMEM) with 4.5 g/L glucose and l-glutamine was from Lonza (Basel, Switzerland). Minimal Essential Media (MEM) was from Thermo Fisher Scientific. Vascular Cell Basal Media (PCS-100-030) and Endothelial Cell Growth Kit-BBE (PCS-100-040) were from American Type Culture Collection (Manassas, VA, USA). Fetal bovine serum was from Life Technologies (Carlsbad, CA). COS-7 cells were cultured in DMEM supplemented with 10% fetal bovine serum. SK-N-MC cells were cultured in MEM supplemented with 10% fetal bovine serum. HUVECs were cultured in Vascular Cell Basal Media supplemented with Endothelial Cell Growth Kit-BBE according to manufacturer’s instructions (American Type Culture Collection). All cells were grown at 37 °C, 5% CO2 in a humidified CO2 incubator.
Plasmids
The mammalian expression plasmids for the tethered ECD fusion proteins maltose binding protein (MBP)-RAMP1.24–111-(GS)5-CLR.29-144-H6, MBP-RAMP2.55-140[L106R]-(GS)5-CLR.29-144-H6, MBP-RAMP3.25-111-(GS)5-CLR.29.-144-H6, and the bacterial expression plasmid for MBP-RAMP2.55-140[L106R]-(GSA)3-CLR.29-144-H6 were previously described.30,31 Mammalian expression plasmids encoding full-length RAMP1, RAMP2, RAMP3, CLR, FLAG21-RAMP2 [E105A], and HA-CLR have been described elsewhere.30,31 All plasmids used the human RAMP and CLR sequences.
Synthetic Peptides
The AM(37–52) Q50W positional scanning-synthetic peptide combinatorial library was custom synthesized by RS Synthesis (Louisville, KY, USA). The amino acid sequences for the five positional libraries were: (1) DKDNVAPROXXXPWGX-NH2, (2) DKDNVAPRXOXXPWGX-NH2, (3) DKDNVAPRXXOXPWGX-NH2, (4) DKDNVAPRXXXOPWGX-NH2, and (5) DKDNVAPRXXXXPWGO-NH2, where O is the defined position with one of 19 natural amino acids (no cysteine) and X is a variable position containing these 19 amino acids in an equimolar mixture. Each positional library comprised 19 distinct mixtures that each contained 130 321 theoretical peptides (194). There were 2 476 099 theoretical peptides total in the library (130 321 × 19). Each of the 95 crude lyophilized peptide mixtures was reconstituted at 10 mg/mL in 10% (v/v) DMSO. Peptide mixture concentrations were determined by UV absorbance at 280 nm using molar absorptivity calculated based on Trp and Tyr residues at defined positions, ignoring the contribution from Trp or Tyr residues at the randomized positions.
Defined AM, CGRP, and AM2/IMD variant peptides were custom synthesized and HPLC purified by RS Synthesis (Louisville, KY, USA). Peptides containing the nonstandard amino acid norleucine (Nle) were custom synthesized and HPLC purified by New England Peptide (Gardner, MA, USA). Wild-type agonists αCGRP(1–37) and AM(13–52) were from Bachem (Bubendorf, Switzerland). The lyophilized powders were reconstituted at 10 mg/mL in sterile ultrapure water. Peptide concentrations were determined by UV absorbance at 280 nm using the molar absorptivity calculated based on Tyr, Trp, and cystine residues. The concentration of the FITC-(Ahx)-AM(37–52) S45W/Q50W peptide was determined as previously described.34 If a peptide lacked Tyr or Trp residues, its concentration was determined by assuming 80% peptide content. Peptides were stored as aliquots at −80 °C. Table S7 lists the sequences of all defined peptides used in this study.
Purified Proteins
The MBP-RAMP1.24-111-(GS)5-CLR.29-144-H6, MBP-RAMP2.55-140[L106R]-(GS)5-CLR.29-144-H6, and MBP-RAMP3.25-111-(GS)5-CLR.29-144-H6 tethered ECD fusion proteins for FP binding assays were expressed in HEK293T cells and purified as described.30 The MBP-RAMP2.55-140[L106R]-(GSA)3-CLR.29-144-H6 fusion protein for crystallization was expressed in E. coli and purified as previously described.31 MBP facilitates crystallization of the fusion protein. Purified proteins were stored at −80 °C and their concentrations were determined by Bradford assay with a BSA standard curve.
FP Peptide-Binding Assay
Fluorescence polarization/anisotropy (FP) peptide binding assays using FITC-labeled AM(37–52) S45W/Q50W probe and the three purified, HEK293T cell-produced MBP-RAMP-CLR ECD fusion proteins were performed at room temperature as previously described30 except using 100 mM sodium HEPES pH 7.4. For library screening, 10 μL of peptide mixtures at 5X (prediluted in reaction buffer) were mixed with 20 μL of a master mix containing the FITC-AM probe followed by addition of 20 μL of a master mix containing purified ECD fusion protein. The final concentration of each unique peptide in the screening assay was 500 pM for RAMP1- and RAMP3-CLR ECD fusion proteins and 1.5 nM for RAMP2-CLR ECD. Raw data were normalized as percent of mean of the no-competitor control performed in each experiment. The data shown are a composite of normalized data from two independent experiments each performed with duplicate technical replicates.
For defined peptides, competition binding assays were performed using 7 nM FITC-AM probe and 60 nM MBP-RAMP1-CLR ECD, 40 nM MBP-RAMP2-CLR ECD, or 7 nM MBP-RAMP3-CLR ECD and increasing amounts of unlabeled competitor peptides. Equilibrium dissociation constants (KD) of the probe for the fusion proteins expressed in HEK293T cells were previously reported.30 Equilibrium dissociation constants of the unlabeled peptides (KI) were determined using nonlinear regression curve fitting to user-defined exact analytical equations expressed in terms of the total ligand and receptor concentrations in Prism v. 7.0d as previously described.30 For low affinity peptides for which the entire competition curve could not be defined, the bottom was constrained to be the same as that of a high-affinity peptide assayed within the same experiment. A Polarstar Omega plate reader was used for the FP measurements (BMG Labtech, Germany).
Crystallization, Diffraction Data Collection, Structure Solution, and Refinement
Purified, bacterially expressed MBP-RAMP2 ECD-(GSA)3-CLR ECD fusion protein was dialyzed to buffer containing 10 mM Tris-HCl pH 7.5, 50 mM NaCl, 1 mM maltose, 1 mM EDTA, and mixed with AM(37–52) S45R/K46L/S48G/Q50W in a 1:1.2 protein:peptide molar ratio, incubated on ice for 1 h, and then concentrated to 30 mg/mL using a 3000 Da molecular weight cutoff spin concentrator device (Millipore). Crystals were grown by hanging drop vapor diffusion in 20% (w/v) PEG monomethyl ether (MME) 5000, 0.1 M sodium HEPES pH 8.2, 150 mM sodium formate, and 3% (v/v) DMSO. Crystals were cryoprotected by overnight dialysis into a solution containing mother liquor supplemented with 12% (w/v) sucrose then flash frozen in liquid nitrogen. Initial crystals were checked for diffraction on a home X-ray source, and then high-resolution diffraction data were remotely collected from a single crystal at 100 K at LS-CAT 21-ID-G (λ = 0.9786 Å) of the Advanced Photon Source (Argonne, IL). The data were processed using HKL2000 v. 71249 and CCP4 v. 7.0.066.50 The structure was solved by molecular replacement (MR) with Phaser v. 2.8.251 using MBP with maltose removed (PDB: 3C4M) and RAMP2-CLR ECD with MBP and peptide removed (PDB: 4RWF) as search inputs. The MR solution contained one molecule in the asymmetric unit. The MR solution was rigid body refined with REFMAC5 v. 5.8.023852 by treating MBP, RAMP2 ECD, and CLR ECD as separate rigid bodies. The final model was completed by iterative rounds of manual model building using COOT53 and TLS restrained refinement in REFMAC5 v. 5.8.0238.52 The Ramachandran plot had no outliers and 1.6% and 98.4% of residues were in the allowed and preferred regions, respectively.
Concentration–Response cAMP Accumulation Assays
COS-7 cells were seeded into 96-well plates (Corning, NY, USA) at a cell density of 20 000 cells/well and cotransfected with human RAMP1-, RAMP2-, or RAMP3-, and CLR-encoding plasmids using PEI as previously described.34 These assays were performed at 37 °C. At 48 h after transfection, the cells were preincubated in assay buffer consisting of DMEM supplemented with 0.1% (w/v) fatty acid-free bovine serum albumin (BSA) and 1 mM 3-isobutyl-1-methylxanthine (IBMX) for 30 min, followed by continuous exposure to the indicated agonist concentration in assay buffer for 15 min. For antagonism experiments, fixed concentrations of antagonists were preincubated with cells in assay buffer for 30 min followed by continuous exposure to the indicated agonist concentration in the presence of the antagonist for 30 min, unless otherwise noted.
SK-N-MC cells and HUVECs were seeded into 96-well plates at cell densities of 15 000 cells/well and 10 000 cells/well, respectively. HUVECs were used at passage four or lower. At 48 h after seeding, SK-N-MC cells and HUVECs were preincubated for 30 min in Krebs-Ringer-HEPES (KRH) buffer (25 mM HEPES pH 7.4, 104 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1.2 mM MgSO4, and 1.2 mM KH2PO4) supplemented with 0.1% (w/v) fatty acid-free BSA and 1 mM IBMX. Cells were then stimulated by continuous exposure to the indicated concentrations of agonist peptides in the same buffer for 15 min.
In all cases the cells were lysed with 40 μL of 6% (v/v) perchloric acid and neutralized with sodium bicarbonate and sodium HEPES pH 7.4 for a total lysate volume of 91 μL. A LANCE cAMP detection kit (PerkinElmer) was used to quantify cAMP in the lysates according to the manufacturer’s directions as previously described.34 Six microliters of lysate was used in the 24 μL total assay volume, and the data are presented as nM cAMP in this assay volume. A Polarstar Omega plate reader was used for the LANCE measurements (BMG Labtech, Germany).
Concentration–response data were fit by nonlinear regression in Prism v. 7.0d (GraphPad) using the log(agonist) versus response model with a standard Hill slope of 1 to determine agonist potency, pEC50. For antagonism assays where surmountable antagonism was observed, the apparent affinity of the antagonist (pKBapp) was determined using the Gaddum/Schild EC50 shift model with the Hill and Schild slopes constrained to 1 assuming competitive antagonism. For insurmountable antagonism, the data were fit to a user-defined hemiequilibrium operational model as previously described:41
where:
These data were normalized to percent maximum cAMP (Em) of the agonist curve in the absence of antagonist. Stimulation time (t) was constrained as 30 min, and the parameters for the equilibrium dissociation constant of the antagonist (KB), the equilibrium dissociation constant of the agonist (KA), the operational efficacy (τ), and the antagonist dissociation rate (koff) were globally fit.
Sustained cAMP Signaling after Agonist Washout Assay
This assay was based on one previously described with modifications.54 Cells were cultured and seeded into 96-well plates as described above. These assays were performed at room temperature. The cells were washed twice with 100 μL assay buffer (serum-free DMEM + 0.1% fatty acid-free BSA for COS-7 cells or KRH buffer + 0.1% fatty acid-free BSA for HUVECs and SK-N-MC cells), then preincubated in assay buffer for 30 min followed by stimulation with 100 nM agonist in 100 μL assay buffer for 10 min. The agonist-containing buffer was aspirated and the cells were washed with 100 μL of assay buffer three times over a period of ∼3 min to wash away unbound agonist. The cells were then incubated in assay buffer for the indicated times. At the indicated time point the buffer was aspirated and replaced with assay buffer supplemented with 2 mM IBMX for 10 min followed by lysate preparation as described above. For the t = 0 time point, 100 nM agonist was added to cells in assay buffer containing 2 mM IBMX, and the cells were lysed after 10 min. The cAMP concentration in the cell lysates was determined as described above. Data were normalized as percent maximum cAMP of the wild-type AM agonist (HUVECs or RAMP2-CLR in COS-7) or wild-type CGRP agonist (SK-N-MC or RAMP1-CLR in COS-7) at the t = 0 time point with baseline as 0%.
Statistical Analysis
The pKI, pKBapp, and pEC50 values are reported as means ± SEM from at least three independent experiments performed on separate days. Each independent experiment was performed with duplicate technical replicates. Statistical comparisons of mean pKI, pKBapp, or pEC50 values were performed using an unpaired student’s t test for two groups or one-way ANOVA with Tukey’s or Dunnet’s posthoc test for three or more groups using Prism v. 7.0d (GraphPad). Selectivity was reported as Δlog of mean values between indicated pairs, and error was reported as 95% confidence interval (CI). For the agonist wash-out assays statistical comparisons of wild-type and mutants were performed for each time point using unpaired student’s t test. No weighting or outlier detection was used.
Data Availability
The coordinates and structure factors for the AM S45R/K46L/S48G/Q50W-bound MBP-RAMP2-CLR ECD crystal structure were deposited to the worldwide protein databank (wwPDB) under accession code 6V2E.
Acknowledgments
The authors thank Drs. Paul Weigel and Paul DeAngelis for suggesting a peptide library approach to identify enhanced affinity variants, Dr. Debbie Hay for helpful suggestions on working with the SK-N-MC cell line, and Dr. Chris Langmead for kindly providing a Prism file for fitting data to the hemiequilibrium operational model for slow off-rate antagonists and advice on its use. Use of the Advanced Photon Source LS-CAT Sector 21 beamlines was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor Grant 085P1000817. We thank Zdzislaw Wawrzak for assistance with remote data collection at APS beamline 21-1D-G. Use of the home X-ray source and microscope for crystal imaging at the Biomolecular Structure Core-OKC was supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under Grant P20GM103640. This work was supported by Grants NIH R01GM104251 and OCAST HR16-005 (AAP) and a predoctoral fellowship from the American Heart Association 18PRE33990152 (J.M.B.).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.0c00031.
Tables S1–S7 summarizing binding and signaling properties of AM and CGRP peptide variants obtained in the binding and signaling assays, X-ray crystallographic data collection and refinement statistics, and a complete list of peptide sequences; Figures S1–S5 showing electron density and crystal packing relevant to the structure determination, a summary of RAMP-peptide contacts and electrostatics, pKI vs pKBapp correlation plots, and additional signaling assay controls (PDF)
Author Contributions
J.M.B. and M.L.W. performed experiments. J.M.B. and A.A.P. analyzed data and wrote the manuscript. A.A.P. conceived and managed the project and acquired funding.
The authors declare the following competing financial interest(s): A.A.P. is the inventor on a patent for AM and CGRP variants described herein.
Supplementary Material
References
- Kato J.; Kitamura K. (2015) Bench-to-bedside pharmacology of adrenomedullin. Eur. J. Pharmacol. 764, 140–148. 10.1016/j.ejphar.2015.06.061. [DOI] [PubMed] [Google Scholar]
- Russell F. A.; King R.; Smillie S. J.; Kodji X.; Brain S. D. (2014) Calcitonin gene-related peptide: physiology and pathophysiology. Physiol. Rev. 94, 1099–1142. 10.1152/physrev.00034.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kee Z.; Kodji X.; Brain S. D. (2018) The Role of Calcitonin Gene Related Peptide (CGRP) in Neurogenic Vasodilation and Its Cardioprotective Effects. Front. Physiol. 9, 1249. 10.3389/fphys.2018.01249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuruda T.; Kato J.; Kuwasako K.; Kitamura K. (2019) Adrenomedullin: Continuing to explore cardioprotection. Peptides 111, 47–54. 10.1016/j.peptides.2018.03.012. [DOI] [PubMed] [Google Scholar]
- Caron K. M.; Smithies O. (2001) Extreme hydrops fetalis and cardiovascular abnormalities in mice lacking a functional Adrenomedullin gene. Proc. Natl. Acad. Sci. U. S. A. 98, 615–619. 10.1073/pnas.98.2.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matson B. C.; Pierce S. L.; Espenschied S. T.; Holle E.; Sweatt I. H.; Davis E. S.; Tarran R.; Young S. L.; Kohout T. A.; van Duin M.; Caron K. M. (2017) Adrenomedullin improves fertility and promotes pinopodes and cell junctions in the peri-implantation endometrium. Biol. Reprod. 97, 466–477. 10.1093/biolre/iox101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Ponce A.; Chanez Paredes S.; Castro Ochoa K. F.; Schnoor M. (2016) Regulation of endothelial and epithelial barrier functions by peptide hormones of the adrenomedullin family. Tissue Barriers 4, e1228439 10.1080/21688370.2016.1228439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashizuka S.; Inatsu H.; Kita T.; Kitamura K. (2016) Adrenomedullin Therapy in Patients with Refractory Ulcerative Colitis: A Case Series. Dig. Dis. Sci. 61, 872–880. 10.1007/s10620-015-3917-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataoka Y.; Miyazaki S.; Yasuda S.; Nagaya N.; Noguchi T.; Yamada N.; Morii I.; Kawamura A.; Doi K.; Miyatake K.; Tomoike H.; Kangawa K. (2010) The first clinical pilot study of intravenous adrenomedullin administration in patients with acute myocardial infarction. J. Cardiovasc. Pharmacol. 56, 413–419. 10.1097/FJC.0b013e3181f15b45. [DOI] [PubMed] [Google Scholar]
- Geven C.; Kox M.; Pickkers P. (2018) Adrenomedullin and Adrenomedullin-Targeted Therapy As Treatment Strategies Relevant for Sepsis. Front. Immunol. 9, 292. 10.3389/fimmu.2018.00292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voors A. A.; Kremer D.; Geven C.; Ter Maaten J. M.; Struck J.; Bergmann A.; Pickkers P.; Metra M.; Mebazaa A.; Dungen H. D.; Butler J. (2019) Adrenomedullin in heart failure: pathophysiology and therapeutic application. Eur. J. Heart Failure 21, 163–171. 10.1002/ejhf.1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edvinsson L.; Haanes K. A.; Warfvinge K.; Krause D. N. (2018) CGRP as the target of new migraine therapies - successful translation from bench to clinic. Nat. Rev. Neurol. 14, 338–350. 10.1038/s41582-018-0003-1. [DOI] [PubMed] [Google Scholar]
- Iyengar S.; Ossipov M. H.; Johnson K. W. (2017) The role of calcitonin gene-related peptide in peripheral and central pain mechanisms including migraine. Pain 158, 543–559. 10.1097/j.pain.0000000000000831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinho-Ribeiro F. A.; Baddal B.; Haarsma R.; O’Seaghdha M.; Yang N. J.; Blake K. J.; Portley M.; Verri W. A.; Dale J. B.; Wessels M. R.; Chiu I. M. (2018) Blocking Neuronal Signaling to Immune Cells Treats Streptococcal Invasive Infection. Cell 173 (1083–1097), e1022 10.1016/j.cell.2018.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganor Y.; Drillet-Dangeard A. S.; Lopalco L.; Tudor D.; Tambussi G.; Delongchamps N. B.; Zerbib M.; Bomsel M. (2013) Calcitonin gene-related peptide inhibits Langerhans cell-mediated HIV-1 transmission. J. Exp. Med. 210, 2161–2170. 10.1084/jem.20122349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benyahia Z.; Dussault N.; Cayol M.; Sigaud R.; Berenguer-Daize C.; Delfino C.; Tounsi A.; Garcia S.; Martin P. M.; Mabrouk K.; Ouafik L. (2017) Stromal fibroblasts present in breast carcinomas promote tumor growth and angiogenesis through adrenomedullin secretion. Oncotarget 8, 15744–15762. 10.18632/oncotarget.14999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallmayer M.; Li J.; Ohmura S.; Alba Rubio R.; Baldauf M. C.; Holting T. L. B.; Musa J.; Knott M. M. L.; Stein S.; Cidre-Aranaz F.; Wehweck F. S.; Romero-Perez L.; Gerke J. S.; Orth M. F.; Marchetto A.; Kirchner T.; Bach H.; Sannino G.; Grunewald T. G. P. (2019) Targeting the CALCB/RAMP1 axis inhibits growth of Ewing sarcoma. Cell Death Dis. 10, 116. 10.1038/s41419-019-1372-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeran K.; O’Shea D.; Upton P. D.; Small C. J.; Ghatei M. A.; Byfield P. H.; Bloom S. R. (1997) Circulating adrenomedullin does not regulate systemic blood pressure but increases plasma prolactin after intravenous infusion in humans: a pharmacokinetic study. J. Clin. Endocrinol. Metab. 82, 95–100. 10.1210/jcem.82.1.3656. [DOI] [PubMed] [Google Scholar]
- Kraenzlin M. E.; Ch’ng J. L.; Mulderry P. K.; Ghatei M. A.; Bloom S. R. (1985) Infusion of a novel peptide, calcitonin gene-related peptide (CGRP) in man. Pharmacokinetics and effects on gastric acid secretion and on gastrointestinal hormones. Regul. Pept. 10, 189–197. 10.1016/0167-0115(85)90013-8. [DOI] [PubMed] [Google Scholar]
- Kubo K.; Tokashiki M.; Kuwasako K.; Tamura M.; Tsuda S.; Kubo S.; Yoshizawa-Kumagaye K.; Kato J.; Kitamura K. (2014) Biological properties of adrenomedullin conjugated with polyethylene glycol. Peptides 57, 118–121. 10.1016/j.peptides.2014.05.005. [DOI] [PubMed] [Google Scholar]
- Nilsson C.; Hansen T. K.; Rosenquist C.; Hartmann B.; Kodra J. T.; Lau J. F.; Clausen T. R.; Raun K.; Sams A. (2016) Long acting analogue of the calcitonin gene-related peptide induces positive metabolic effects and secretion of the glucagon-like peptide-1. Eur. J. Pharmacol. 773, 24–31. 10.1016/j.ejphar.2016.01.003. [DOI] [PubMed] [Google Scholar]
- Schonauer R.; Els-Heindl S.; Fischer J. P.; Kobberling J.; Riedl B.; Beck-Sickinger A. G. (2016) Adrenomedullin 2.0: Adjusting Key Levers for Metabolic Stability. J. Med. Chem. 59, 5695–5705. 10.1021/acs.jmedchem.6b00126. [DOI] [PubMed] [Google Scholar]
- Copeland R. A.; Pompliano D. L.; Meek T. D. (2006) Drug-target residence time and its implications for lead optimization. Nat. Rev. Drug Discovery 5, 730–739. 10.1038/nrd2082. [DOI] [PubMed] [Google Scholar]
- Vauquelin G.; Charlton S. J. (2010) Long-lasting target binding and rebinding as mechanisms to prolong in vivo drug action. Br. J. Pharmacol. 161, 488–508. 10.1111/j.1476-5381.2010.00936.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo D.; Hillger J. M.; IJzerman A. P.; Heitman L. H. (2014) Drug-target residence time--a case for G protein-coupled receptors. Med. Res. Rev. 34, 856–892. 10.1002/med.21307. [DOI] [PubMed] [Google Scholar]
- Hothersall J. D.; Brown A. J.; Dale I.; Rawlins P. (2016) Can. residence time offer a useful strategy to target agonist drugs for sustained GPCR responses?. Drug Discovery Today 21, 90–96. 10.1016/j.drudis.2015.07.015. [DOI] [PubMed] [Google Scholar]
- Hay D. L.; Garelja M. L.; Poyner D. R.; Walker C. S. (2018) Update on the pharmacology of calcitonin/CGRP family of peptides: IUPHAR Review 25. Br. J. Pharmacol. 175, 3–17. 10.1111/bph.14075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoare S. R. (2005) Mechanisms of peptide and nonpeptide ligand binding to Class B G-protein-coupled receptors. Drug Discovery Today 10, 417–427. 10.1016/S1359-6446(05)03370-2. [DOI] [PubMed] [Google Scholar]
- Moad H. E.; Pioszak A. A. (2013) Selective CGRP and adrenomedullin peptide binding by tethered RAMP-calcitonin receptor-like receptor extracellular domain fusion proteins. Protein science: a publication of the Protein Society 22, 1775–1785. 10.1002/pro.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roehrkasse A. M.; Booe J. M.; Lee S. M.; Warner M. L.; Pioszak A. A. (2018) Structure-function analyses reveal a triple beta-turn receptor-bound conformation of adrenomedullin 2/intermedin and enable peptide antagonist design. J. Biol. Chem. 293, 15840–15854. 10.1074/jbc.RA118.005062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booe J. M.; Walker C. S.; Barwell J.; Kuteyi G.; Simms J.; Jamaluddin M. A.; Warner M. L.; Bill R. M.; Harris P. W.; Brimble M. A.; Poyner D. R.; Hay D. L.; Pioszak A. A. (2015) Structural Basis for Receptor Activity-Modifying Protein-Dependent Selective Peptide Recognition by a G Protein-Coupled Receptor. Mol. Cell 58, 1040–1052. 10.1016/j.molcel.2015.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y. L.; Khoshouei M.; Deganutti G.; Glukhova A.; Koole C.; Peat T. S.; Radjainia M.; Plitzko J. M.; Baumeister W.; Miller L. J.; Hay D. L.; Christopoulos A.; Reynolds C. A.; Wootten D.; Sexton P. M. (2018) Cryo-EM structure of the active, Gs-protein complexed, human CGRP receptor. Nature 561, 492–497. 10.1038/s41586-018-0535-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y. L.; Belousoff M. J.; Fletcher M. M.; Zhang X.; Khoshouei M.; Deganutti G.; Koole C.; Furness S. G. B.; Miller L. J.; Hay D. L.; Christopoulos A.; Reynolds C. A.; Danev R.; Wootten D.; Sexton P. M. (2020) Structure and Dynamics of Adrenomedullin Receptors AM1 and AM2 Reveal Key Mechanisms in the Control of Receptor Phenotype by Receptor Activity-Modifying Proteins. ACS Pharmacol Transl Sci. 3, 263–284. 10.1021/acsptsci.9b00080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booe J. M.; Warner M. L.; Roehrkasse A. M.; Hay D. L.; Pioszak A. A. (2018) Probing the Mechanism of Receptor Activity-Modifying Protein Modulation of GPCR Ligand Selectivity through Rational Design of Potent Adrenomedullin and Calcitonin Gene-Related Peptide Antagonists. Mol. Pharmacol. 93, 355–367. 10.1124/mol.117.110916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinilla C.; Appel J. R.; Blanc P.; Houghten R. A. (1992) Rapid identification of high affinity peptide ligands using positional scanning synthetic peptide combinatorial libraries. Biotechniques 13, 901–905. [PubMed] [Google Scholar]
- Lee S. M.; Jeong Y.; Simms J.; Warner M. L.; Poyner D. R.; Chung K. Y.; Pioszak A. A. (2020) Calcitonin Receptor N-Glycosylation Enhances Peptide Hormone Affinity by Controlling Receptor Dynamics. J. Mol. Biol. 432, 1996–2014. 10.1016/j.jmb.2020.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins H. A.; Au M.; Bobby R.; Archbold J. K.; Abdul-Manan N.; Moore J. M.; Middleditch M. J.; Williams G. M.; Brimble M. A.; Dingley A. J.; Hay D. L. (2013) Identification of key residues involved in adrenomedullin binding to the AM1 receptor. Br. J. Pharmacol. 169, 143–155. 10.1111/bph.12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay D. L.; Christopoulos G.; Christopoulos A.; Sexton P. M. (2006) Determinants of 1-piperidinecarboxamide, N-[2-[[5-amino-l-[[4-(4-pyridinyl)-l-piperazinyl]carbonyl]pentyl]amino]-1-[(3,5-d ibromo-4-hydroxyphenyl)methyl]-2-oxoethyl]-4-(1,4-dihydro-2-oxo-3(2H)-quinazoliny l) (BIBN4096BS) affinity for calcitonin gene-related peptide and amylin receptors--the role of receptor activity modifying protein 1. Mol. Pharmacol. 70, 1984–1991. 10.1124/mol.106.027953. [DOI] [PubMed] [Google Scholar]
- Qi T.; Ly K.; Poyner D. R.; Christopoulos G.; Sexton P. M.; Hay D. L. (2011) Structure-function analysis of amino acid 74 of human RAMP1 and RAMP3 and its role in peptide interactions with adrenomedullin and calcitonin gene-related peptide receptors. Peptides 32, 1060–1067. 10.1016/j.peptides.2011.03.004. [DOI] [PubMed] [Google Scholar]
- Vauquelin G.; Szczuka A. (2007) Kinetic versus allosteric mechanisms to explain insurmountable antagonism and delayed ligand dissociation. Neurochem. Int. 51, 254–260. 10.1016/j.neuint.2007.05.005. [DOI] [PubMed] [Google Scholar]
- Riddy D. M.; Valant C.; Rueda P.; Charman W. N.; Sexton P. M.; Summers R. J.; Christopoulos A.; Langmead C. J. (2015) Label-Free Kinetics: Exploiting Functional Hemi-Equilibrium to Derive Rate Constants for Muscarinic Receptor Antagonists. Mol. Pharmacol. 88, 779–790. 10.1124/mol.115.100545. [DOI] [PubMed] [Google Scholar]
- Kamitani S.; Asakawa M.; Shimekake Y.; Kuwasako K.; Nakahara K.; Sakata T. (1999) The RAMP2/CRLR complex is a functional adrenomedullin receptor in human endothelial and vascular smooth muscle cells. FEBS Lett. 448, 111–114. 10.1016/S0014-5793(99)00358-0. [DOI] [PubMed] [Google Scholar]
- Choksi T.; Hay D. L.; Legon S.; Poyner D. R.; Hagner S.; Bloom S. R.; Smith D. M. (2002) Comparison of the expression of calcitonin receptor-like receptor (CRLR) and receptor activity modifying proteins (RAMPs) with CGRP and adrenomedullin binding in cell lines. Br. J. Pharmacol. 136, 784–792. 10.1038/sj.bjp.0704761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLatchie L. M.; Fraser N. J.; Main M. J.; Wise A.; Brown J.; Thompson N.; Solari R.; Lee M. G.; Foord S. M. (1998) RAMPs regulate the transport and ligand specificity of the calcitonin-receptor-like receptor. Nature 393, 333–339. 10.1038/30666. [DOI] [PubMed] [Google Scholar]
- Yarwood R. E.; Imlach W. L.; Lieu T.; Veldhuis N. A.; Jensen D. D.; Klein Herenbrink C.; Aurelio L.; Cai Z.; Christie M. J.; Poole D. P.; Porter C. J. H.; McLean P.; Hicks G. A.; Geppetti P.; Halls M. L.; Canals M.; Bunnett N. W. (2017) Endosomal signaling of the receptor for calcitonin gene-related peptide mediates pain transmission. Proc. Natl. Acad. Sci. U. S. A. 114, 12309–12314. 10.1073/pnas.1706656114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrandon S.; Feinstein T. N.; Castro M.; Wang B.; Bouley R.; Potts J. T.; Gardella T. J.; Vilardaga J. P. (2009) Sustained cyclic AMP production by parathyroid hormone receptor endocytosis. Nat. Chem. Biol. 5, 734–742. 10.1038/nchembio.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingell J. J.; Rees T. A.; Hendrikse E. R.; Siow A.; Rennison D.; Scotter J.; Harris P. W. R.; Brimble M. A.; Walker C. S.; Hay D. L. (2020) Distinct Patterns of Internalization of Different Calcitonin Gene-Related Peptide Receptors. ACS Pharmacol Transl Sci. 3, 296–304. 10.1021/acsptsci.9b00089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubdool A. A.; Thakore P.; Argunhan F.; Smillie S. J.; Schnelle M.; Srivastava S.; Alawi K. M.; Wilde E.; Mitchell J.; Farrell-Dillon K.; Richards D. A.; Maltese G.; Siow R. C.; Nandi M.; Clark J. E.; Shah A. M.; Sams A.; Brain S. D. (2017) A Novel alpha-Calcitonin Gene-Related Peptide Analogue Protects Against End-Organ Damage in Experimental Hypertension, Cardiac Hypertrophy, and Heart Failure. Circulation 136, 367–383. 10.1161/CIRCULATIONAHA.117.028388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z.; Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326. 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr., Sect. D: Biol. Crystallogr. 67, 235–242. 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674. 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., Sect. D: Biol. Crystallogr. 53, 240–255. 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. (2010) Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean T.; Vilardaga J. P.; Potts J. T. Jr.; Gardella T. J. (2008) Altered selectivity of parathyroid hormone (PTH) and PTH-related protein (PTHrP) for distinct conformations of the PTH/PTHrP receptor. Mol. Endocrinol. 22, 156–166. 10.1210/me.2007-0274. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The coordinates and structure factors for the AM S45R/K46L/S48G/Q50W-bound MBP-RAMP2-CLR ECD crystal structure were deposited to the worldwide protein databank (wwPDB) under accession code 6V2E.