

Abstract
Background:
CDKN2A mutations confer a substantial risk of cutaneous melanoma, however the magnitude of the risk is uncertain.
Methods:
We estimated the hazard ratio (HR) and the average age-specific cumulative risk (i.e. penetrance) of reported melanoma for CDKN2A mutation carriers in case families using a modified segregation analysis of the first- and higher-degree relatives of 35 population-based cases. The study sample included 223 relatives of 13 early-onset melanoma cases (aged 18-39 years at diagnosis) from Melbourne, Sydney and Brisbane, Australia, and 322 relatives of 22 melanoma cases diagnosed at any age from Yorkshire, United Kingdom (UK).
Results:
The estimated HR for melanoma for mutation carriers relative to the general population decreased with regions of increasing ambient ultraviolet irradiance: being higher for the UK than Australia (87, 95% confidence interval (CI) 50-153 versus 31, 95% CI 20-50, P=0.008), and in Australia, 49 (95% CI 24-98) for Melbourne, 44 (95% CI 22-88) for Sydney, and 9 (95% CI 2-33) for Brisbane (P=0.02). Penetrance did not statistically significantly differ between the two countries or across regions within Australia. For example, we estimated that 16% (95% CI 10%-27%) of UK and 20% (95% CI 13%-30%) of Australian CDKN2A mutation carriers would be diagnosed with melanoma by age 50 years, and 45% (95% CI 29%-65%) and 52% (95% CI 37%-69%), respectively, by age 80 years.
Conclusions:
CDKN2A mutation carriers living in geographic regions with different levels of ambient UV irradiance appear to have the same cumulative risk of melanoma.
Keywords: melanoma, penetrance, CDKN2A, risk, genetic susceptibility
INTRODUCTION
CDKN2A on chromosome 9p21 is the major known melanoma susceptibility gene,[1, 2] encoding two distinct proteins, p16INK4A and p14ARF, which are involved in cell cycle control, tumour suppression and melanocyte senescence.[1] The p16INK4A protein binds to the cyclin-dependent kinases CDK4 and CDK6, inhibiting phosphorylation of the retinoblastoma protein and progression of the cell through the G1 cell cycle checkpoint. The p14ARF protein induces cell cycle arrest or apoptosis via the p53 pathway.
Carriers of a CDKN2A mutation have a substantial risk of developing cutaneous malignant melanoma, however the absolute and relative magnitudes of the risk are uncertain. Two international studies[3, 4] have reported apparently different estimates of age-specific cumulative risk (i.e. penetrance). The Melanoma Genetics Consortium (GenoMEL) study of 80 multiple-case families ascertained predominantly from family cancer clinics estimated that 30% (95% CI: 12% to 62%) of mutation carriers would develop melanoma by age 50 years and 67% (95% CI: 31% to 96%) by age 80 years, with risk varying by geographical region, being higher in Australia (91% by age 80 years) than the United States (76% by age 80 years) and Europe (58% by age 80 years).[4] On the other hand, the Genes, Environment and Melanoma (GEM) study, using data on first-degree relatives of 65 population-based melanoma carriers (probands), estimated penetrance of 14% (95% CI = 8% to 22%) by age 50 years and 28% (95% CI: 18% to 40%) by age 80 years, and no significant differences by region.[3] They also found that the penetrance was lower when estimated from carrier families ascertained through single primary melanoma carrier probands than through multiple primary melanoma carrier probands (P=0.04).
Estimates of penetrance derived from carrier families identified because they have multiple cases of melanoma through clinic-based sampling (as in the GenoMEL study) could be higher than when derived from carriers identified independently of their family history through population-based sampling of cases (as in the GEM study) because carrier families could differ in the prevalence of other genetic or environmental risk factors or the severity of the mutations. That is, penetrance could depend on the context or setting, so there is no single penetrance.[5] For each study, the penetrance being estimated is an average over the mutations causing disease in the probands and the other factors operating in their families.[5, 6, 7]
To provide further insights into penetrance, we estimated the age-specific cumulative risk and hazard ratio (relative risk) of melanoma for carriers of deleterious mutations in CDKN2A by studying the relatives of population-based early-onset melanoma cases (aged 18-39 years at diagnosis) from three major cities in Australia and of population-based melanoma cases diagnosed at any age from Yorkshire, United Kingdom (UK).
METHODS
Subjects
The study sample comprised 35 population-based families (13 from Australia and 22 from the UK) in which probands had histopathologically-confirmed invasive cutaneous melanoma and had been found from mutation screening to carry a pathogenic CDKN2A mutation. Families were obtained from two sources: 1) the Australian Melanoma Family Study,[8] a multi-centre, population-based, case-control-family study of early-onset melanoma; and 2) the Leeds case-control study,[9, 10] a population-based, case-control study of melanoma in the UK. Ethics committee approval for both studies was obtained from all relevant ethics committees in each country, and all participants gave written informed consent.
Australian Melanoma Family Study
The study sample, recruitment methods, data collection and subject characteristics have been described in detail.[8] Briefly, probands were diagnosed between 1st July 2000 and 31st December 2002 at ages 18-39 years with incident, histopathologically-confirmed, first-primary invasive cutaneous melanoma. At the time of recruitment, probands were living in the greater urban areas of Brisbane (27.3°S, high ambient UV), Sydney (33.6°S, intermediate ambient UV) or Melbourne (37.5°S, lower ambient UV), the three largest urban populations in Australia.[8] Probands were identified from population-based state cancer registries; participation was 54% of those eligible and 76% of those contactable. Of the 629 case probands recruited, 596 (95%) gave a blood sample and were screened for mutations in CDKN2A.
First- and second-degree relatives were recruited where possible via the probands. One mutation-carrying proband was excluded from this analysis because no information could be collected for their relatives. The Cannings-Thompson sequential ascertainment scheme[11] was used to extend recruitment to higher-degree relatives in an unbiased way, such that any additional diagnosis of melanoma (either in situ or invasive) for any eligible relative subsequently made eligible all first-degree relatives of the newly identified melanoma case. A blood sample and information on lifetime sun exposure, phenotype and residence history were requested from probands and participating living relatives, and proxy-reported demographic and cancer information was collected for deceased relatives. Verification of reported melanomas in relatives was sought from cancer registries, hospital and pathology records, treating clinicians, general practitioners and death certificates after participant or next-of-kin (if participant deceased) consent had been obtained. For the relatives in this analysis, 48% of melanoma reports were confirmed.
Leeds case-control study
The study sample and methods have been previously described.[9, 10] Briefly, between September 2000 and December 2006, 1043 probands diagnosed between 18 and 82 years with incident, histopathologically-confirmed, invasive cutaneous melanoma living in a geographically defined area of Yorkshire and the Northern region of the UK (53.8° N) were recruited. Proband participation was 67%. Between September 2000 and June 2003 all patients with invasive melanoma were invited to participate, but from July 2003 to December 2006 patients with Breslow thickness less than 0.75mm were not invited. The cases were identified through clinicians, pathology registers and the Northern and Yorkshire cancer registry to ensure maximal ascertainment. First- and second-degree relatives of probands, and higher-degree relatives with complete ascertainment were included irrespective of melanoma status. Probands were asked to report their personal and family history of melanoma. Melanomas were verified by obtaining histopathology reports where possible or by Cancer Registry for probands and where relatives were able or willing to consent to this process. In the absence of consent to confirm a relative’s melanoma then the best judgement of the interviewers was reported. For the relatives in this analysis, 31% of melanoma reports were confirmed. Germline DNA was screened for mutations in CDKN2A for the first 1000 probands.
Screening for CDKN2A mutations
Probands were screened for variants in the CDKN2A coding region, including exons 1α, 1β, 2, and 3, splice junctions, and the 5′ and 3′ UTRs. Mutation screening was also conducted for exon 2 (the p16INK4A binding domain) of CDK4. All available samples in relatives of the identified pathogenic mutation carriers (see below) were then screened for the mutation identified in the proband. Australian samples were screened at the University of Sydney, using bidirectional DNA sequencing (CDKN2A exons 1α, 1β, 3 and CDK4 exon 2) and denaturing high performance liquid chromatography (DHPLC) (CDKN2A exon 2 only). UK samples were screened at St James’s University Hospital, Leeds, using either DHPLC or high resolution melting curve analysis (HRMCA). Primers, protocols and reaction conditions were as previously described for DHPLC[12] and HRMCA[13, 14] These methods for identifying mutations has been shown to be equivalent.[12] All amplicons that displayed an aberrant SSCP, DHPLC or HRMCA profile were bi-directionally sequenced to identify the underlying nucleotide change, as previously described.[12]
Variants were designated as pathogenic if they had been previously reported to cosegregate with melanoma in multiple-case kindreds, if there was functional evidence of impaired protein function, or if these data were not available, if the mutation satisfied bioinformatic criteria (SIFT, http://blocks.fhcrc.org/sift/SIFT.html; Polyphen, http://genetics.bwh.harvard.edu/pph/; pMut, http://mmb2.pcb.ub.es:8080/PMut/) predicting loss of function and the variants were never or rarely observed in previous reports of control series (Harland et al, in preparation).
Statistical analysis
Hazard ratio (HR; the ratio of age-specific incidence of melanoma for mutation carriers to that for non-carriers) and age-specific cumulative risk (penetrance) were estimated from data on carrier families using modified segregation analysis. This method incorporates both genotyped and ungenotyped relatives and conditions on ascertainment to produce unbiased estimates. Models were fitted and statistical inference conducted under maximum likelihood theory using MENDEL version 3.2.[15] The joint log-likelihood was calculated for each carrier family, based on information for each member on their observed melanoma status, age at last contact, death or diagnosis, family relationships and carrier status (carrier, non-carrier or unknown). To adjust for ascertainment, the joint likelihood for each pedigree was conditioned on the proband’s melanoma status, mutation status and age at diagnosis. The log HR, and hence the age-specific cumulative risk, was estimated by maximizing the sum of the logs of the conditioned likelihoods, assuming families were independent. Likelihood ratio tests were used to test whether the HR and penetrance estimates differed by sex, region or age.
The population CDKN2A allele frequency was assumed to be 0.001 and mutations were assumed to act on disease risk in a dominant fashion. As CDKN2A mutations are rare, we assumed the incidence of melanoma in non-carriers was equal to that for the general population. Average sex- and age-specific population incidences of melanoma were obtained from ‘Cancer Incidence in Five Continents’[16] for each geographic region from which probands were recruited (Queensland, New South Wales, and Victoria in Australia, and Yorkshire in the UK). Population incidences were selected for the period corresponding to the relatives’ average year of melanoma diagnosis (1980 for Australia, 1994 for the UK); thus for Australia volume 5 (1978-1982) was used and for the UK volume 8 (1993-1997) was used.[16] Region and sex-specific HR and penetrance estimates were estimated using region and sex-specific population melanoma incidences as the baseline. To derive estimates of penetrance for all Australian carriers combined, we constructed an average population incidence curve using the incidence curves for each state and sex, weighted in proportion to the number of probands and relatives that were male and female recruited in that state. Similarly, to derive estimates of penetrance for UK males and females combined, we constructed an average population incidence curve using the incidence curves for each sex, weighted in proportion to the number of males and females recruited in the UK. Ten-year cumulative risk estimates of melanoma for CDKN2A mutation carriers who have no melanoma at the beginning of the 10-year period were calculated as [R(t + 10) − R(t)]/[(1 − R(t)], where t is the carrier’s age in years and R(t) and R(t + 10) are the relevant cumulative risks to ages t and t +10 years, respectively. Confidence intervals for the 10-year estimates were calculated in the same way, using the lower and upper bounds of the 95% confidence intervals of the cumulative risks.
Age was recorded at diagnosis of first melanoma, death, or last known age, whichever occurred first. For relatives known to be alive but missing a date of last contact, we assigned the date of proband interview (Australia) or the 1st January 2005 (UK). Relatives whose age was unknown were assigned an age of 0 years and therefore did not contribute to penetrance estimates. All confirmed and unconfirmed reports of melanomas for relatives were included in the analysis.
To test the robustness of our assumptions we conducted several sensitivity analyses: 1) using imputed ages where possible for relatives with missing ages; 2) including confirmed melanomas only and assuming that those with an unconfirmed report of melanoma did not have melanoma; 3) using more recent population incidences of melanoma from ‘Cancer Incidence in Five Continents’ volume 9, 1998-2002;[17] and 4) using a population allele frequency of 0.0001. For 1) we used the following methods to impute missing ages: for relatives missing a date of birth, the year of birth was assigned in the following order, as either i) the average known birth year of siblings if available, or ii) as 20 years (women) or 22 years (men) earlier than their first offspring’s year of birth, or iii) based on the spouse’s year of birth, assuming the wife was two years younger than the husband. If none of the above were available then the relative was assigned age 0. For relatives assigned a year of birth and assumed to be alive at the time of the proband’s interview, age was calculated as year of proband interview (Australia) or the 1st January 2005 (UK) minus assigned year of birth, and age was censored at age 80 years.
To estimate the number of mutation carriers in the identified carrier families, we calculated the probability of being a CDKN2A mutation carrier for each ungenotyped relative, based on the genetic relatedness to known or inferred carriers and non-carriers (but ignoring melanoma status). This calculation assumed Mendel’s laws of inheritance and the population allele frequency noted above, and was conducted by modifying MENDEL 3.2. The total number of mutation carriers was estimated by summing the carrier probabilities of the ungenotyped relatives and adding this to the number of known carriers.
RESULTS
A total of 20 different CDKN2A pathogenic mutations were identified in 35 probands. These mutations and their predicted effects on the p16INK4A and p14ARF protein coding sequences are presented in Table 1. The mutations included three small exonic insertions or deletions affecting p16INK4A only found in seven probands, seven missense mutations predicted to affect p16INK4A only found in nine probands, three missense mutations affecting p14ARF only found in three probands, one small exonic deletion affecting both p16INK4A and p14ARF found in two probands, four missense mutations affecting both p16INK4A and p14ARF found in seven probands, and two mutations outside the coding region (c.−34G>T false start mutation affecting p16INK4A only, and the intronic c.458-105A>G mutation likely affecting both p16INK4A and p14ARF) found in seven probands. In addition, a nonsense mutation p.W34X was identified in exon 1β of one proband, but was excluded from this analysis because no information could be collected for the proband’s relatives. No mutations were identified in CDKN2A exon 3 or CDK4 exon 2.
Table 1.
CDKN2A mutations identified in population-based families in Australia and the United Kingdom
| Gene element |
CDK2NA gene variant (protein variant) | Families by Region | ||||||
|---|---|---|---|---|---|---|---|---|
| p16INK4A | p14ARF | Australia | UK | |||||
| Nucleotide change1 | Effect on protein |
Nucleotide change1 | Effect on protein2 |
Sydney | Melbourne | Brisbane | Leeds | |
| 5' UTR | c.−34G>T | r.−34_−32>p.M1 | _ | 1 | 1 | |||
| Exon 1α | c.9_32del24 | p.A4_10Edel7 | _ | 1 | ||||
| c.32_33ins9_32 | p.M1_S8dup | _ | 1 | 3 | ||||
| c.52_57dup6 | p.20T_21Adup | _ | 1 | 1 | ||||
| c.68G>A | p.G23D | _ | 1 | |||||
| c.95T>C | p.L32P | _ | 1 | |||||
| c.104G>C | p.G35A | _ | 1 | |||||
| Exon 1β | _ | c.81C>G | p.I27M | 1 | ||||
| _ | c.193G>A | p.G65S | 1 | |||||
| _ | c.193G>C | p.G65R | 1 | |||||
| Exon 2 | c.159G>C | p.M53I | c.202G>C | p.D68H | 4 | |||
| c.188T>C | p.L63P | c.231T>C | p.A77A (silent) | 1 | ||||
| c.194T>C | p.L65P | c.237T>C | p.A79A (silent) | 1 | ||||
| c.202_203GC>TT | p.A68L | c.245_246GC>TT | p.R82L | 1 | ||||
| c.206A>G | p.E69G | c.249A>G | p.G83G (silent) | 1 | 2 | |||
| c.212A>C | p.N71T | c.255A>C | p.Q85H | 1 | ||||
| c.228_246del19 | p.L77TfsX62 | c.271_289del19 | p.R90VfsX75 | 2 | ||||
| c.251A>C | p.D84A | c.294A>C | p.R84R (silent) | 1 | ||||
| c.331G>A | p.G111S | c.374G>A | p.G125E | 1 | ||||
| Intron 2 | c.458-105A>G | altered splicing | _ | altered splicing | 1 | 4 | ||
| Total | 5 | 4 | 4 | 22 | ||||
Nucleotides are numbered from the first A of the initiation codon of p16INK4A in the standard nomenclature for mutations employed.[23]
Synonymous variants are presumed ‘silent’.
A description of the Australian and UK carrier families are shown in Table 2. In the 13 Australian carrier families, 17 (52%) of 33 genotyped relatives (32 first-degree, one second-degree) were found to be CDKN2A mutation carriers. In the 22 UK carrier families, 10 (56%) of 18 genotyped relatives (17 first-degree, one third-degree) were found to be CDKN2A mutation carriers. We estimated a total of 72 mutation carriers in the Australian carrier families and 115 mutation carriers in the UK carrier families.
Table 2.
Characteristics of population-based families carrying CDKN2A mutations, in Australia and the United Kingdom
| Country | Number of families |
Number of probands’ relatives | Relatives, % male |
Relatives with melanoma1 |
Family history of melanoma for proband | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Number (%) of relatives with melanoma in each family | |||||||||||
| Total (average per proband) |
1st degree | 2nd degree | 3rd or higher degree |
0 | 1 | 2 | ≥ 3 | ||||
| United Kingdom | 22 | 322 (14.6) | 122 (38%) | 193 (60%) | 7 (2%) | 56% | 16 (5%) | 14 (64%) | 2 (9%) | 4 (18%) | 2 (9%) |
| Australia | 13 | 223 (17.2) | 67 (30%) | 136 (61%) | 20 (9%) | 54% | 21 (9%) | 3 (23%) | 5 (38%) | 3 (23%) | 2 (15%) |
Includes confirmed and unconfirmed reports of melanoma in relatives
Table 3 shows, for descriptive purposes, the characteristics of all 35 probands and the 419 relatives who either were known to carry a CDKN2A mutation or had an estimated 25% or higher probability of being a mutation carrier. Although all relatives were included in the analyses, regardless of their probability of carrying a mutation, relatives whose probability of being a mutation carrier was less than 25% are not shown in Table 3 because of their minor influence on the penetrance estimates in comparison with known or more likely mutation carriers. The mean age at diagnosis of melanoma was 34 years for Australian probands (reflecting the recruitment criteria of early-onset melanoma) and 47 years for UK probands (who were unselected for age at diagnosis). Affected relatives who were known or likely carriers in the Australian and UK carrier families had a similar mean age at diagnosis of melanoma (47 years).
Table 3.
Characteristics of probands and relatives who are known carriers of a CDKN2A mutation or with a predicted 25% or higher probability1 of being a carrier, by region of the proband and sex
| Geographic region and sex |
Number of families |
Number of relatives with ≥ 25% probability of being a carrier1 |
Mean age (range) at diagnosis of melanoma of proband |
Mean last known age (range) of relatives without melanoma2 |
Number (%) of relatives with melanoma2 |
Mean age (range) at diagnosis of melanoma2 in relatives |
|---|---|---|---|---|---|---|
| United Kingdom | ||||||
| All | 22 | 276 | 47 (24-72) | 50 (0-97) | 13 (5) | 47 (17-75) |
| Females | - | 119 | 46 (24-72) | 49 (0-97) | 6 (5) | 43 (17-66) |
| Males | - | 157 | 50 (41-67) | 50 (6-88) | 7 (4) | 51 (39-75) |
| Australia | ||||||
| All | 13 | 143 | 34 (27-40) | 57 (18-91) | 20 (14) | 47 (18-87) |
| Females | - | 69 | 31 (27-37) | 62 (26-91) | 13 (19) | 46 (18-84) |
| Males | - | 74 | 36 (31-40) | 53 (18-86) | 7 (9) | 50 (22-87) |
| Brisbane | 4 | 41 | 31 (27-36) | 61 (26-91) | 3 (7) | 48 (22-67) |
| Sydney | 5 | 45 | 35 (29-40) | 58 (18-85) | 9 (20) | 49 (18-87) |
| Melbourne | 4 | 57 | 34 (30-39) | 55 (25-86) | 8 (14) | 45 (27-77) |
Carrier probability was calculated from the genetic relatedness to known or inferred carriers, and not from melanoma status. The calculation assumes Mendel’s laws of inheritance, Hardy-Weinberg equilibrium and a population allele frequency of 0.001. Excludes 4 UK individuals whose sex was unknown.
Includes confirmed and unconfirmed reports of melanoma in relatives
Estimates of HRs and cumulative risk of melanoma for CDKN2A mutation carriers are shown in Table 4. The incidence of melanoma for carriers of a CDKN2A mutation relative to the general population was higher in the UK (HR 87, 95% CI 50-153) than in Australia (HR 31, 95% CI 20-50), P for heterogeneity (Phet)=0.008. There was modest evidence that the HR for melanoma was greater for female carriers than male carriers in Australia (HR = 48 and 18, respectively, Phet=0.07) but not in the UK (HR = 78 for females and 98 for males, Phet=0.20). The HRs for Australian carriers differed by geographical region (Phet=0.02) with the HR for melanoma being lowest for carrier families based in Brisbane (HR 9, 95% CI 2-33), the most northerly city of recruitment (i.e. high ambient ultraviolet irradiance), and the HR highest for those based in Melbourne (HR 49, 95% CI 24-98), the most southerly. There was no evidence that the HR differed by age of the carrier (Phet=0.9).
Table 4.
Estimates of hazard ratios and age-specific cumulative risk of melanoma from birth, for carriers of a CDKN2A mutation, by region and sex
| Age-specific cumulative risk % (95% CI) for mutation carriers |
||||||
|---|---|---|---|---|---|---|
| Geographic region and sex |
Hazard ratio1 (95% CI) |
To age 40 | To age 50 | To age 60 | To age 70 | To age 80 |
| United Kingdom | ||||||
| All | 87 (50-153) | 9 (5-15) | 16 (10-27) | 26 (16-41) | 35 (22-53) | 45 (29-65) |
| Females | 78 (33-181) | 11 (5-23) | 20 (9-40) | 29 (14-55) | 37 (18-66) | 45 (23-75) |
| Males | 98 (44-215) | 7 (3-15) | 13 (6-26) | 23 (11-43) | 33 (16-58) | 45 (24-73) |
| Australia | ||||||
| All | 31 (20-50) | 11 (7-17) | 20 (13-30) | 30 (20-44) | 41 (28-57) | 52 (37-69) |
| Females | 48 (25-89) | 19 (11-33) | 33 (19-52) | 44 (26-66) | 53 (33-76) | 64 (42-85) |
| Males | 18 (8-41) | 5 (3-12) | 11 (5-23) | 19 (9-37) | 29 (14-53) | 38 (19-66) |
| Brisbane | 9 (2-33) | 4 (1-15) | 9 (3-29) | 14 (4-44) | 21 (6-57) | 28 (9-70) |
| Sydney | 44 (22-88) | 14 (7-26) | 24 (13-43) | 35 (19-58) | 47 (27-72) | 57 (35-82) |
| Melbourne | 49 (24-98) | 15 (8-28) | 24 (13-43) | 36 (20-59) | 47 (27-72) | 58 (35-83) |
CI = confidence interval
Hazard ratio for melanoma for mutation carriers compared to the general population. P-values for hazard ratio differences between subgroups are: UK all versus Australia all: 0.008; UK females versus UK males: 0.20; Australian females versus Australian males: 0.07; Brisbane versus Sydney versus Melbourne: 0.02
We estimated that 45% (95% CI 29%-65%) of UK and 52% (95% CI 37%-69%) of Australian CDKN2A mutation carriers would be diagnosed with melanoma by age 80 years (Table 4). Cumulative risk estimates are shown separately for males and females and for different geographic regions (Table 4 and Figure 1). Penetrance did not statistically significantly differ by sex or geographic region. By age 80 years, the estimated proportion of mutation carriers that would be diagnosed with melanoma was 64% (95% CI 42%-85%) for Australian females, 38% (95% CI 19%-66%) for Australian males, 45% (95% CI 23%-75%) for UK females, and 45% (95% CI 24%-73%) for UK males. For a 60-year old CDKN2A mutation carrier without a previous melanoma diagnosis, the 10-year risk of melanoma was estimated to be 16% (95% CI 10%-24%) in Australia, and 12% (7%-20%) in the UK (Table 5).
Figure 1. Age-specific cumulative risk of melanoma for carriers of a CDKN2A mutation, by country and sex.
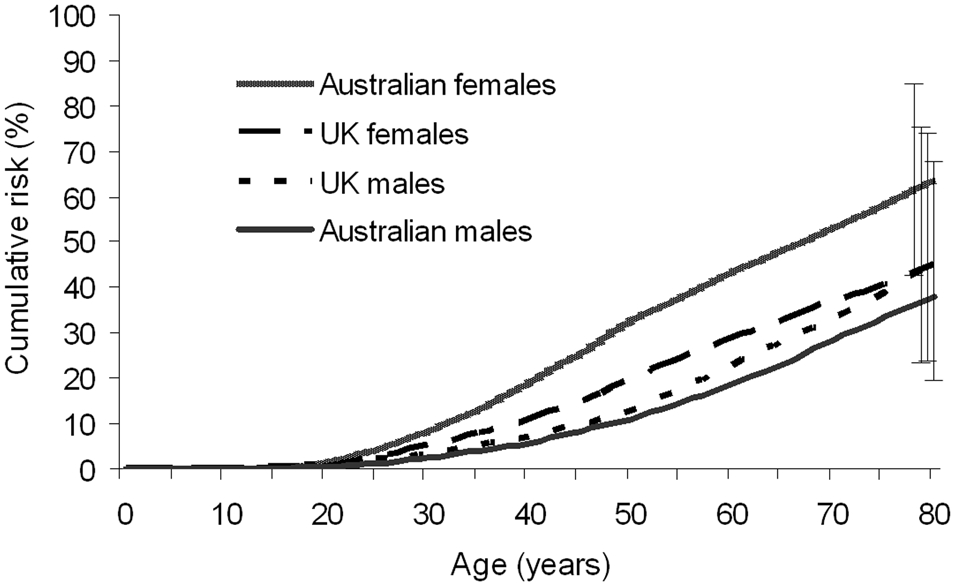
The bars at age 80 indicate the 95% confidence intervals for the penetrance at age 80 for each of the four groups.
Table 5.
10-year cumulative risk estimates of melanoma for CDKN2A mutation carriers, by region and sex
| Geographic region and sex |
Cumulative risk of melanoma (%, 95% confidence interval) in the next 10-years for CDKN2A mutation carriers who are unaffected at the beginning of the 10-year period |
|||
|---|---|---|---|---|
| For a 40 year- old |
For a 50 year- old |
For a 60 year- old |
For a 70 year- old |
|
| United Kingdom | ||||
| All | 8 (5-14) | 12 (7-19) | 12 (7-20) | 15 (9-26) |
| Females | 10 (4-21) | 11 (5-25) | 12 (5-25) | 13 (6-27) |
| Males | 6 (3-13) | 12 (5-24) | 13 (6-26) | 19 (9-36) |
| Australia | ||||
| All | 10 (7-16) | 13 (8-19) | 16 (10-24) | 18 (12-28) |
| Females | 17 (9-29) | 16 (9-28) | 17 (10-29) | 23 (13-38) |
| Males | 6 (3-12) | 9 (4-19) | 12 (6-25) | 13 (6-27) |
| Brisbane | 5 (1-17) | 6 (2-20) | 7 (2-24) | 9 (3-31) |
| Sydney | 12 (6-22) | 14 (7-27) | 19 (10-34) | 19 (10-34) |
| Melbourne | 11 (6-21) | 15 (8-27) | 18 (9-33) | 21 (11-38) |
Several sensitivity analyses were conducted. We were able to impute ages for 16 of the 20 Australian relatives whose ages were not known, and for 111 of the 170 UK relatives whose ages were not known. When using imputed ages, the melanoma risk estimates for Australian carriers were very similar to that based on known ages only (HR, and penetrance at age 80, respectively, for Brisbane = 9, 28%; Sydney = 42, 55%; Melbourne = 49, 58%), but for UK carriers were lower than that based on known ages (HR = 46, 95% CI 26-81, and penetrance at age 80 = 27%, 95% CI 16%-43%). When only confirmed reports of melanoma in relatives were considered as affected, the melanoma risk estimates were approximately halved for Australian carriers (HR = 14, 95% CI 7-27; penetrance at age 80 = 28%, 95% CI 16%-47%) and were two-thirds lower for UK carriers (HR= 23, 95% CI 8-65; penetrance at age 80 = 15%, 95% CI 5%-36%). The estimated HRs were also lower when we used the more recent (higher) population incidences of melanoma rather than the incidences corresponding to relatives’ average melanoma diagnosis dates, although this resulted in minimal change to penetrance estimates for Brisbane and UK carriers, and slightly higher penetrance estimates for Sydney and Melbourne carriers (results not shown). Changing the CDKN2A allele frequency from 0.001 to 0.0001 had minimal influence on the HR or penetrance estimates (results not shown).
DISCUSSION
Our results are based on CDKN2A mutation carriers who are relatives of population-based melanoma cases from Australia and the UK who were unselected for family history. We estimated that the relative increase in the risk of melanoma for CDKN2A mutation carriers in this context was higher for carriers living in the UK (87 times that of the general population) than for carriers living in Australia (31 times that of the general population), and in Australia the relative risk of melanoma for mutation carriers differed by region and marginally by sex. However, there was no evidence that the cumulative risk estimates were different between Australia and the UK, with 20% of Australian and 16% of UK CDKN2A mutation carriers estimated to be diagnosed with melanoma by age 50 years, and 52% and 45%, respectively, by age 80 years.
Our findings suggest that ambient UV irradiance might not further increase cumulative risk of melanoma for CDKN2A mutation carriers as the penetrance was similar whether living in Australia or the UK. Similarly, in Australia, the penetrance was not higher for carriers in Brisbane, the city with the highest ambient UV irradiance. In previous studies, the GenoMEL penetrance estimates differed substantially by geographical location, and were highest in regions with the highest population melanoma incidence,[4] whereas the GEM penetrance estimates did not differ by country.[3]
In Australia, the risk of melanoma was marginally higher for female carriers than for male carriers, but there was no evidence of any sex differences in the UK. The GenoMEL study observed a 24% lower penetrance of melanoma for male carriers than female carriers although this difference was not statistically significant.[4] Although these sex differences might reflect the small numbers in these subgroups, it is possible that there is a more complex association between CDKN2A mutations, sun exposure, and sex.
Given the similar penetrances by region, the differences in the relative risk of melanoma for carriers compared with non-carriers that we observed between regions reflects differences in the baseline age-standardised melanoma incidences, with the relative risk increasing as the level of ambient UV irradiance decreases. Our relative risk estimates of melanoma (87 in the UK and 31 in Australia) for CDKN2A mutation carriers are similar to a study of 13 multiple-case families from the United States,[18] which reported a relative risk of melanoma of 57 (95% CI 17-259) for CDKN2A mutation carriers compared with non-carriers. A much lower relative risk of 4.3 (95% CI 2.3-7.7) was observed by the GEM study,[19] but this was for relative risk of subsequent melanoma for CDKN2A mutation carriers compared with those without a CDKN2A mutation among individuals already diagnosed with a single primary melanoma, who are known to be at higher baseline risk than the general population.
Penetrance estimates of melanoma for carriers of a CDKN2A mutation in the population-based GEM study were 14% by age 50 years and 28% by age 80 years,[3] and in the multiple-case, predominantly clinic-based GenoMEL study were 30% by age 50 years and 67% by age 80 years.[4] Our penetrance estimates sit roughly in between these two previous studies. There are several reasons why risk estimates might differ between studies. First, they may depend on the populations studied, and on characteristics of the study sample and the selection criteria; for example, there may be other genetic or environmental risk factors shared by family members with multiple cases. This rationale is supported by findings from the GEM study, in which the penetrance estimated from the families of carrier probands with multiple primary melanoma was higher than that for families of carrier probands with single primary melanoma.[3] In our study, the Australian and UK study populations also differed in that all of the Australian probands had early-onset melanoma (aged 18-39 years at diagnosis) whereas the UK probands were unselected for age at diagnosis. Differences in age at diagnosis might be related to the type of mutation or to the presence of other melanoma risk factors, and it is possible that risk estimates derived from families with early-onset melanoma might be higher than from families with melanoma diagnosed at any age. In addition, risk estimates derived from population-based sources including our study might still be higher than for mutation carriers in the general population without a family history of melanoma, since all relatives in our study had at least one relative (the proband) diagnosed with melanoma.
Second, there is some evidence that different mutations on the CDKN2A gene confer different risks of melanoma.[4, 19] In the GenoMEL study, individuals with mutations that affect both p16INK4A and p14ARF had a risk of melanoma 1.8 times that of carriers of a mutation affecting p16INK4A only.[4] In the GEM study, missense mutations affecting both p16INK4A and p14ARF and the non-coding mutation at c.−34G>T conferred statistically significantly greater relative risk of melanoma than mutations that were insertions or deletions or affecting only p16INK4A or only p14ARF.[19] In our study, nine (26%) of the 35 families had missense mutations affecting both p16INK4a and p14ARF, or the c.−34G>T mutation, compared to 40% of mutations in the GEM study and 41% of mutations in the GenoMEL study. Furthermore, neither the GEM nor GenoMEL studies genotyped exon 1β, which is unique to p14ARF, and in which 3 missense mutations were found in three (9%) families. The GEM study also did not screen for the deep intronic c.458-105A>G mutation that was present in 5 (14%) of the families described here. Also in contrast to the GEM and GenoMEL studies, we designated the c.373G>C variant as non-causal, due to its presence in control probands and lack of cosegregation in a number of melanoma pedigrees.
Each study has also used a different statistical method. An advantage of the modified segregation analysis used here is that this method maximizes power by incorporating data on ungenotyped as well as genotyped relatives, by considering all possible genotype combinations and, essentially, weighting them by their likelihood of occurring. Another advantage is that the method incorporates a rigorous method of adjusting for ascertainment, thereby reducing bias.
This study is supported by a robust population-based design in which we systematically recruited first and higher-degree relatives of probands who were unselected for family history. We also attempted to verify reports of melanoma for relatives, in contrast to the GEM study.[3] The melanoma risk estimates were lower when confined to confirmed reports of melanoma than when all reported melanomas were included. The true risk estimates likely lie between the two, since accuracy of self-reports of melanoma is relatively poor compared with other cancers, especially in Australia[20, 21] however a proportion of reports unable to be verified would be genuine. There was a lack of statistical power to examine the associations with melanoma risk for different types of CDKN2A mutations or to examine the influence of phenotype (e.g. number of nevi) or personal sun exposure on the risk estimates. It has been shown that MC1R variants, pigmentation and nevus phenotype influence melanoma risk for CDKN2A carriers.[22] Another potential limitation was that about half of the UK relatives and 9% of the Australian relatives had an unknown age and thus were not included in the main penetrance analysis. The lower risk estimates for the UK from the sensitivity analysis that included imputed ages suggests two possibilities. On the one hand, if melanoma was under-reported in those with missing ages but the true incidence was equal to that of relatives with known ages then our main estimate would be unbiased for UK carriers. On the other hand, if recording of age information was less diligent for unaffected relatives, then our main estimate of penetrance may biased upwards for UK carriers.
This study provides refinement of information on age-specific cumulative risk and relative risk of melanoma for individuals with a CDKN2A mutation living in sunny or cooler regions, which will be useful to genetic counsellors and clinicians. These risk estimates are most relevant to mutation carriers with a weak family history of melanoma.
Acknowledgments:
We gratefully acknowledge the research interviewers, examiners and data management staff from on the Australian Melanoma Family Study, including Judith Maskiell, Jackie Arbuckle, Steven Columbus, Michaela Lang, Helen Rodais, Caroline Ellis (Victoria); Carol El Hayek, Lynne Morgan, Joanne Roland, Emma Tyler, Jodi Barton, Caroline Watts and Lesley Porter (NSW); Jodie Jetann, Megan Ferguson, Michelle Hillcoat, Kellie Holland, Pamela Saunders, Joan Roberts and Sheree Tait (Queensland). We are extremely grateful to Birute Karpavicius, Susan Leake, Susan Haynes and Elaine Fitzgibbon for their efforts in recruiting the participants from the Leeds case-control study.
Funding: The Australian Melanoma Family Study received funding from the National Health and Medical Research Council of Australia (NHMRC) (project grants 107359, 211172, 566946 and Program Grant 402761 to GJM and RFK); project grants from the Cancer Councils New South Wales (77/00, 06/10), Victoria and Queensland (371); and the US National Institutes of Health (via RO1 grant CA-83115-01A2 to the international Melanoma Genetics Consortium (GenoMEL)). AEC is the recipient of a NHMRC public health postdoctoral fellowship (520018) and received a Victorian Cancer Agency Early Career Seed Grant (ECSG07_010). BKA’s research was supported by a University of Sydney Medical Foundation Program Grant and JLH is an Australia Fellow of the NHMRC. This research was funded in part by Cancer Research UK (C588/A4994, C588/A10589 C8216/A6129) and by the National Institute of Health (CA83115). UK study recruitment was facilitated by the UK National Cancer Research Network.
Abbreviations:
- AMFS
Australian Melanoma Family Study
Footnotes
Competing interests: There are no competing interests
Copyright licence statement: The Corresponding Author has the right to grant on behalf of all authors and does grant on behalf of all authors, an exclusive licence (or non-exclusive for government employees) on a worldwide basis to the BMJ Publishing Group Ltd and its Licensees to permit this article (if accepted) to be published in Journal of Medical Genetics and any other BMJPGL products to exploit all subsidiary rights, as set out in our licence (http://group.bmj.com/products/journals/instructions-for-authors/licence-forms).
REFERENCES
- 1.Meyle KD, Guldberg P. Genetic risk factors for melanoma. Hum Genet 2009;126:499–510. [DOI] [PubMed] [Google Scholar]
- 2.Kamb A, Shattuck-Eidens D, Eeles R, Liu Q, Gruis NA, Ding W, Hussey C, Tran T, Miki Y, Weaver-Feldhaus J, et al. Analysis of the p16 gene (CDKN2) as a candidate for the chromosome 9p melanoma susceptibility locus. Nat Genet 1994;8:23–6. [DOI] [PubMed] [Google Scholar]
- 3.Begg CB, Orlow I, Hummer AJ, Armstrong BK, Kricker A, Marrett LD, Millikan RC, Gruber SB, Anton-Culver H, Zanetti R, Gallagher RP, Dwyer T, Rebbeck TR, Mitra N, Busam K, From L, Berwick M. Lifetime risk of melanoma in CDKN2A mutation carriers in a population-based sample. J Natl Cancer Inst 2005;97:1507–15. [DOI] [PubMed] [Google Scholar]
- 4.Bishop DT, Demenais F, Goldstein AM, Bergman W, Bishop JN, Bressac-de Paillerets B, Chompret A, Ghiorzo P, Gruis N, Hansson J, Harland M, Hayward N, Holland EA, Mann GJ, Mantelli M, Nancarrow D, Platz A, Tucker MA. Geographical variation in the penetrance of CDKN2A mutations for melanoma. J Natl Cancer Inst 2002;94:894–903. [DOI] [PubMed] [Google Scholar]
- 5.Antoniou A, Pharoah PD, Narod S, Risch HA, Eyfjord JE, Hopper JL, Loman N, Olsson H, Johannsson O, Borg A, Pasini B, Radice P, Manoukian S, Eccles DM, Tang N, Olah E, Anton-Culver H, Warner E, Lubinski J, Gronwald J, Gorski B, Tulinius H, Thorlacius S, Eerola H, Nevanlinna H, Syrjakoski K, Kallioniemi OP, Thompson D, Evans C, Peto J, Lalloo F, Evans DG, Easton DF. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet 2003;72:1117–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hopper JL, Chenevix-Trench G, Jolley DJ, Dite GS, Jenkins MA, Venter DJ, McCredie MR, Giles GG. Design and analysis issues in a population-based, case-control-family study of the genetic epidemiology of breast cancer and the Co-operative Family Registry for Breast Cancer Studies (CFRBCS). J Natl Cancer Inst Monogr 1999:95–100. [DOI] [PubMed] [Google Scholar]
- 7.Scott CL, Jenkins MA, Southey MC, Davis TA, Leary JA, Easton DF, Phillips KA, Hopper JL. Average age-specific cumulative risk of breast cancer according to type and site of germline mutations in BRCA1 and BRCA2 estimated from multiple-case breast cancer families attending Australian family cancer clinics. Hum Genet 2003;112:542–51. [DOI] [PubMed] [Google Scholar]
- 8.Cust AE, Schmid H, Maskiell JA, Jetann J, Ferguson M, Holland EA, Agha-Hamilton C, Jenkins MA, Kelly J, Kefford RF, Giles GG, Armstrong BK, Aitken JF, Hopper JL, Mann GJ. Population-based, Case-Control-Family Design to Investigate Genetic and Environmental Influences on Melanoma Risk: Australian Melanoma Family Study. Am J Epidemiol 2009;170:1541–54. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Randerson-Moor JA, Taylor JC, Elliott F, Chang YM, Beswick S, Kukalizch K, Affleck P, Leake S, Haynes S, Karpavicius B, Marsden J, Gerry E, Bale L, Bertram C, Field H, Barth JH, Silva Idos S, Swerdlow A, Kanetsky PA, Barrett JH, Bishop DT, Bishop JA. Vitamin D receptor gene polymorphisms, serum 25-hydroxyvitamin D levels, and melanoma: UK case-control comparisons and a meta-analysis of published VDR data. Eur J Cancer 2009;45:3271–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Newton-Bishop JA, Chang YM, Iles MM, Taylor JC, Bakker B, Chan M, Leake S, Karpavicius B, Haynes S, Fitzgibbon E, Elliott F, Kanetsky PA, Harland M, Barrett JH, Bishop DT. Melanocytic nevi, nevus genes, and melanoma risk in a large case-control study in the United Kingdom. Cancer Epidemiol Biomarkers Prev 2010;19:2043–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cannings C, Thompson EA. Ascertainment in the sequential sampling of pedigrees. Clin Genet 1977;12:208–12. [DOI] [PubMed] [Google Scholar]
- 12.Harland M, Goldstein AM, Kukalizch K, Taylor C, Hogg D, Puig S, Badenas C, Gruis N, ter Huurne J, Bergman W, Hayward NK, Stark M, Tsao H, Tucker MA, Landi MT, Scarra GB, Ghiorzo P, Kanetsky PA, Elder D, Mann GJ, Holland EA, Bishop DT, Bishop JN. A comparison of CDKN2A mutation detection within the Melanoma Genetics Consortium (GenoMEL). Eur J Cancer 2008;44:1269–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pjanova D, Engele L, Randerson-Moor JA, Harland M, Bishop DT, Newton Bishop JA, Taylor C, Debniak T, Lubinski J, Kleina R, Heisele O. CDKN2A and CDK4 variants in Latvian melanoma patients: analysis of a clinic-based population. Melanoma Res 2007;17:185–91. [DOI] [PubMed] [Google Scholar]
- 14.Taylor CF. Mutation scanning using high-resolution melting. Biochem Soc Trans 2009;37:433–7. [DOI] [PubMed] [Google Scholar]
- 15.Lange K, Cantor R, Horvath S, Perola M, Sabatti C, Sinsheimer J, Sobel E. Mendel version 4.0: A complete package for the exact genetic analysis of discrete traits in pedigree and population data sets. . Amer J Hum Genetics 2001;69(supplement):504.11462172 [Google Scholar]
- 16.Parkin DM, Whelan SL, Ferlay J, H. S Cancer Incidence in Five Continents, Vol. I to VIII Lyon, IARC; 2005. [Google Scholar]
- 17.Curado MP, Edwards B, Shin HR, Storm H, Ferlay J, Heanue M, Boyle P. Cancer Incidence in Five Continents, Vol. IX Lyon, IARC; 2007. [Google Scholar]
- 18.Goldstein AM, Falk RT, Fraser MC, Dracopoli NC, Sikorski RS, Clark WH, Jr., Tucker MA. Sun-related risk factors in melanoma-prone families with CDKN2A mutations. J Natl Cancer Inst 1998;90:709–11. [DOI] [PubMed] [Google Scholar]
- 19.Berwick M, Orlow I, Hummer AJ, Armstrong BK, Kricker A, Marrett LD, Millikan RC, Gruber SB, Anton-Culver H, Zanetti R, Gallagher RP, Dwyer T, Rebbeck TR, Kanetsky PA, Busam K, From L, Mujumdar U, Wilcox H, Begg CB. The prevalence of CDKN2A germ-line mutations and relative risk for cutaneous malignant melanoma: an international population-based study. Cancer Epidemiol Biomarkers Prev 2006;15:1520–5. [DOI] [PubMed] [Google Scholar]
- 20.Aitken JF, Youl P, Green A, MacLennan R, Martin NG. Accuracy of case-reported family history of melanoma in Queensland, Australia. Melanoma Res 1996;6:313–7. [DOI] [PubMed] [Google Scholar]
- 21.Green A, Battistutta D. Incidence and determinants of skin cancer in a high-risk Australian population. Int J Cancer 1990;46:356–61. [DOI] [PubMed] [Google Scholar]
- 22.Demenais F, Mohamdi H, Chaudru V, Goldstein AM, Newton Bishop JA, Bishop DT, Kanetsky PA, Hayward NK, Gillanders E, Elder DE, Avril MF, Azizi E, van Belle P, Bergman W, Bianchi-Scarra G, Bressac-de Paillerets B, Calista D, Carrera C, Hansson J, Harland M, Hogg D, Hoiom V, Holland EA, Ingvar C, Landi MT, Lang JM, Mackie RM, Mann GJ, Ming ME, Njauw CJ, Olsson H, Palmer J, Pastorino L, Puig S, Randerson-Moor J, Stark M, Tsao H, Tucker MA, van der Velden P, Yang XR, Gruis N. Association of MC1R Variants and Host Phenotypes With Melanoma Risk in CDKN2A Mutation Carriers: A GenoMEL Study. J Natl Cancer Inst. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Antonarakis SE. Recommendations for a nomenclature system for human gene mutations. Nomenclature Working Group. Hum Mutat 1998;11:1–3. [DOI] [PubMed] [Google Scholar]