

Abstract
Reticulocytes shed CD71 from the cell membrane and eliminate mitochondria during terminal maturation, but it is unknown whether these two events are coordinated. We demonstrate that timely removal of CD71 is coupled with mitochondrial clearance, which can be disrupted by null mutation of immediate early response gene X-1 (IEX-1), leading to generation of aberrant CD71-positive and mitochondria-negative (CD71+Mito−) reticulocytes. CD71+Mito− reticulocytes were also present in a subset of patients with myelodysplastic syndromes (MDS) in direct proportion to reduced mitochondrial membrane potential (Δψm). Mitochondrial abnormality caused by either IEX-1 deficiency or agents that dissipate Δψm could trigger premature clearance of mitochondria in reticulocytes. Premature clearance of mitochondria or addition of antioxidants lowered intracellular reactive oxygen species (ROS) that in turn hindered CD71 shedding and reticulocyte maturation. In contrast, introduction of ROS accelerated CD71 shedding via release of exosomes that contained a high proportion of Fe3+ over Fe2+, suggesting dual functions of CD71 shedding both in removal of toxic Fe3+ from reticulocytes and in limiting importation of Fe3+ into the cells. These observations emphasize the coordination of mitochondrial and CD71 clearance in erythroid terminal maturation and offer new insights into a role for mitochondrial degeneration in the pathogenesis of some MDS-associated anemia.
Introduction
Myelodysplastic syndromes (MDS) are a heterogeneous group of clonal hematopoietic stem cell (HSC) disorders characterized by dysplastic and ineffective hematopoiesis and resultant peripheral blood cytopenias, especially anemia [1]. Growing evidence suggests that genetic instability of HSCs may be only one of several contributing factors to the disorder. Additional aberrations in myeloid-lineage differentiation may act as a driving force to exhaust the already unstable HSC pool. Clinical studies showed that expression of immediate early response gene X-1 (IEX-1, also known as IER3) was altered in a large portion of early stage/lower-risk MDS patients, in some cases due to translocation of the gene [2, 3]. A severe decrease of the gene expression was correlated with a shorter survival time of the patients [4], but the underlying mechanism by which IEX-1 contributes to disease pathology is poorly understood.
IEX-1 is a stress-inducible gene that modulates F1Fo-ATPase activity [5] and plays an essential role in regulation of mitochondrial functions in a cell-type specific fashion [5, 6]. In addition to clinical observations [3], involvement of IEX-1 deregulation in the etiology of MDS was also suggested by development of MDS-like disease in IEX-1-null mutant mice after exposure to γ-irradiation or bone marrow transplantation [7]. IEX-1 deficiency also impeded proplatelet formation of megakaryocytes, giving rise of 50% fewer platelets than wild type (WT) megakaryocytes [8]. The pivotal role for mitochondria in thrombopoiesis might help explain a higher incidence of MDS occurring in the elderly because mitochondria degenerate with aging, apart from serial acquisition of somatic mutations in leukemia-associated genes throughout life.
Besides irreversible thrombocytopenia, IEX-1 deficient mice also had an abnormally low level of red blood cells (RBCs) following γ-irradiation [7], raising the possibility that IEX-1 deficiency may affect erythroid differentiation as well. Erythroid lineage commitment and differentiation to erythroblasts occur within the bone marrow through several discernible and well described processes [9]. In the circulation, the enucleated reticulocytes gradually and continuously expel mitochondria by autophagy and eliminate surface obsolete proteins by exocytosis [10, 11], while synthesizing a large amount of hemoglobin [10, 12]. Iron used for heme synthesis at mitochondria is transported by the transferrin receptor 1 (TfR1, or CD71) on reticulocytes [13]. CD71 expression declines over time during late erythroid differentiation, and it is eventually eliminated by exocytosis as reticulocytes mature into erythrocytes [12, 14]. However, how CD71 is removed in this timely fashion and what is the significance of the timely removal remain elusive. Understanding of CD71 removal dynamics could shed novel insight into mechanisms underlying inherited and acquired anemia and possibly confer new targets for therapeutics or monitoring of this disorder.
In the present study, we demonstrate that CD71 shedding by reticulocytes is coupled with mitochondrial clearance. This coupling is disrupted by null mutation of IEX-1 or by mitochondrial abnormality, giving rise to an aberrant subset of CD71-positive and mitochondria-negative (CD71+Mito−) reticulocytes not only in IEX-1 deficient mice, but also in a subset of patients with MDS. The presence of the aberrant cells is in direct proportion to decreased mitochondrial membrane potential (Δψm) in MDS patients. These observations support the concept that mitochondrial abnormality, regardless of whether it is caused by IEX-1 deficiency, stress, or aging, could trigger premature clearance of mitochondria that subsequently hinders CD71 shedding and reticulocyte maturation, contributing to the etiology of anemia in MDS.
Materials and methods
Mice
WT and IEX-1 knockout (KO) mice in a 129 Sv/C57BL/6 background were generated in our laboratory [6]. The animal protocol was approved by the subcommittee on Research Animal Care of the Massachusetts General Hospital, according to the National Institutes of Health guidelines for the Care and Use of Laboratory Animals.
Ex vivo maturation of reticulocytes
Mice were injected intraperitoneally with 100 mg/kg phenylhydrazine (PHZ, Sigma). Blood was collected in 3 days after PHZ treatment and stained with FITC-anti-CD71 and PE-anti-Ter119 (Biolegend) for sorting Ter119+CD71high reticulocytes. The sorted cells were cultured in Iscove’s Modified Dulbecco Medium (IMDM, Gibco) supplemented with 30% fetal bovine serum (Hyclone), 1% deionized bovine albumin (Sigma), 100 units/ml penicillin (Gibco), 100 g/ml streptomycin (Sigma) and 0.1 mM α-thioglycerol (Sigma) [15]. In some experiments, 10 μM carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP), 0.1 μM wortmannin, 5 αM N-acetyl-l-cysteine (NAC), 5 mU/ml glucose oxidase (GO), or 1 αM o-phenanthroline (Sigma) was also added to the culture. For iron quantification, 1 × 107 cells were collected from the cultures on indicated days and lysed by NP-40 lysis buffer (1% NP-40, 150 mM NaCl, 50 mM Tris-HCl, pH 7.4). The concentration of Fe2+ and Fe3+ were determined by QuantiChrom Iron Assay Kit (BioAssay Systems) per the manufacturer’ s protocol.
Immunofluorescence staining
After 12-h ex vitro maturation, young reticulocytes were incubated with FITC-anti-CD71 and 5 μM N-rhodamine-labeled phosphatidylethanolamine (NRhPE, Avanti Polar Lipids) for 30 min [16]. The cells were re-suspended in culture medium for additional 4-h ex vivo maturation in a glass bottom culture dish (MatTek) and examined using an Olympus FV1000 confocal microscope (Olympus).
Human samples
The diagnosis of MDS was made according to the 2016 World Health Organization classification. None of the patients had received any disease-modifying therapeutic agents prior to the blood collection. The institutional review board of the Dana-Farber Cancer Institute, USA, and the Affiliated Cancer Hospital of Zhengzhou University, China approved the study protocols and patients provided consent for analysis of a blood sample in accordance with the Declaration of Helsinki.
Statistical analysis
Statistical significance was assessed with 2-tailed Student’s t-test or one way ANOVA using GraphPad Prism 6.0 (GraphPad Software). A value of P < 0.05 was considered statistically significant. The coefficient of determination (R2) was tested by regression and correlation analysis. Extended methods can be found in Supplementary Methods.
Results
Impaired erythropoiesis in IEX-1 KO mice
We previously reported no significant difference in RBC counts in WT and KO mice under steady-state physiological conditions [7]. However, blood smears prepared from KO mice contained more reticulocytes (Fig. 1a, upper right) and polychromasia (Fig. 1a, lower right) compared to WT mice, suggesting perturbation in RBC production. In accordance with this, Ter119+CD71+ reticulocytes were elevated by 7-fold in KO mice, concomitant with a reciprocal decrease of Ter119+CD71− erythrocytes in comparison with WT counterparts (Fig. 1b). Decreases in the number of erythrocytes are known to induce compensatory erythropoiesis in the bone marrow [17]. Indeed, proportions of Ter119lowCD71high early erythroblasts (gate I) and Ter119+CD71high erythroblasts (gate II) were both increased by about 70% (Fig. 1c), and Ter119+CD71− (gate IV) cells declined by more than 200% when compared to WT counterparts (Fig. 1c). The compensatory erythropoiesis corresponded with increased transcription of erythropoietin in the kidney in KO mice (Fig. 1d). The defect of RBC maturation in KO mice may not be noticed under physiological condition because compensatory erythropoiesis continuously replenished the RBC pool. However, upon PHZ treatment, which induces acute hemolytic anemia [11], RBC counts, hemoglobin production, and percentages of hematocrit were significantly lower in the absence compared to the presence of IEX-1 (Figure S1). These results confirm deficient erythropoiesis in the absence of IEX-1.
Fig. 1.
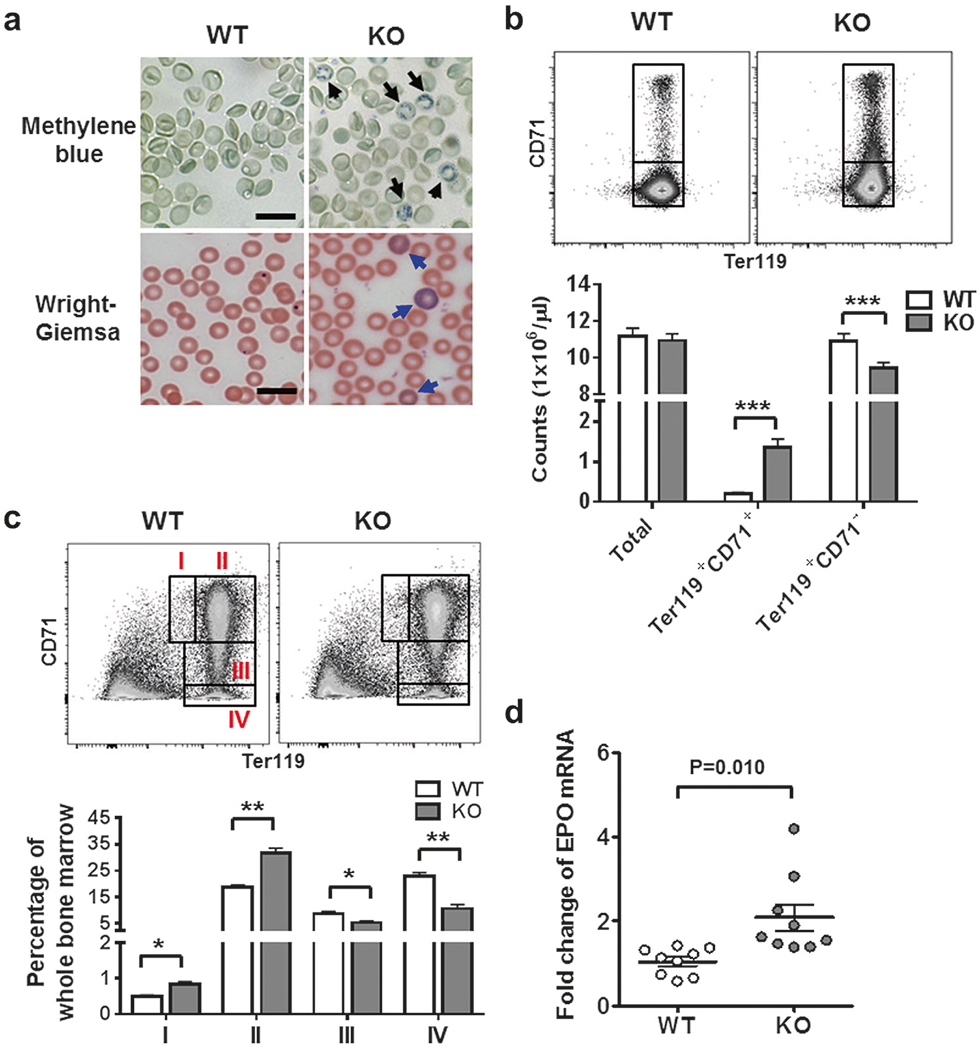
Impaired erythropoiesis in IEX-1 KO mice.
a, b Reticulocytosis in IEX-1 KO mice. Shown in a are representative images of increased reticulocytes marked by Methylene blue (black arrows) and polychromasia stained by Wright-Giemsa (blue arrows) in blood smears from at least 9 mice per group. Scale bar, 20 μm. Peripheral blood cells were stained with Ter119 vs. CD71 and subjected to cell counts by flow cytometry (upper) and statistical analysis of the data (lower) (b).
c, d Compensatory expansion of erythroid precursors in KO mice. Bone marrow cells were stained with Ter119 vs. CD71 and the gates I to IV depict erythroid cell populations with an increasing degree of maturation (c).
Distributions of cells within individual gates, as defined in the upper, were presented as percentages of cells over total bone marrow cells (lower). Erythropoietin (EPO) was quantified by qRT-PCR in mouse kidneys (d). Data are shown as mean ± SEM for all relevant panels. n = 9, *P < 0.05, **P < 0.01, and ***P < 0.001 compared between WT and KO groups
Aberrant CD71+Mito− reticulocytes in IEX-1 KO mice
We went on to examine mitochondrial mass in RBCs in light of the well-described function of IEX-1 in regulation of mitochondrial activity [5, 7, 8]. A significant proportion of KO reticulocytes were found to express CD71 but contain few mitochondria (Fig. 2a, red box), whereas these CD71+Mito− reticulocytes were scarce in WT mice (13 vs 0.4%, p< 0.001). Likewise, confocal microscopy showed that most of KO CD71+ reticulocytes displayed little or weak MitoTracker staining (Fig. 2b, right), in marked contrast to the strong MitoTracker signals in WT counterparts (Fig. 2b, left). The results were also confirmed by transmission electron microscopy showing that most WT reticulocytes contained mitochondria (Fig. 2c, left), whereas KO CD71+ reticulocytes had few intact mitochondria, concomitant with a notable increase of autophagosomes (Fig. 2c, right).
Fig. 2.
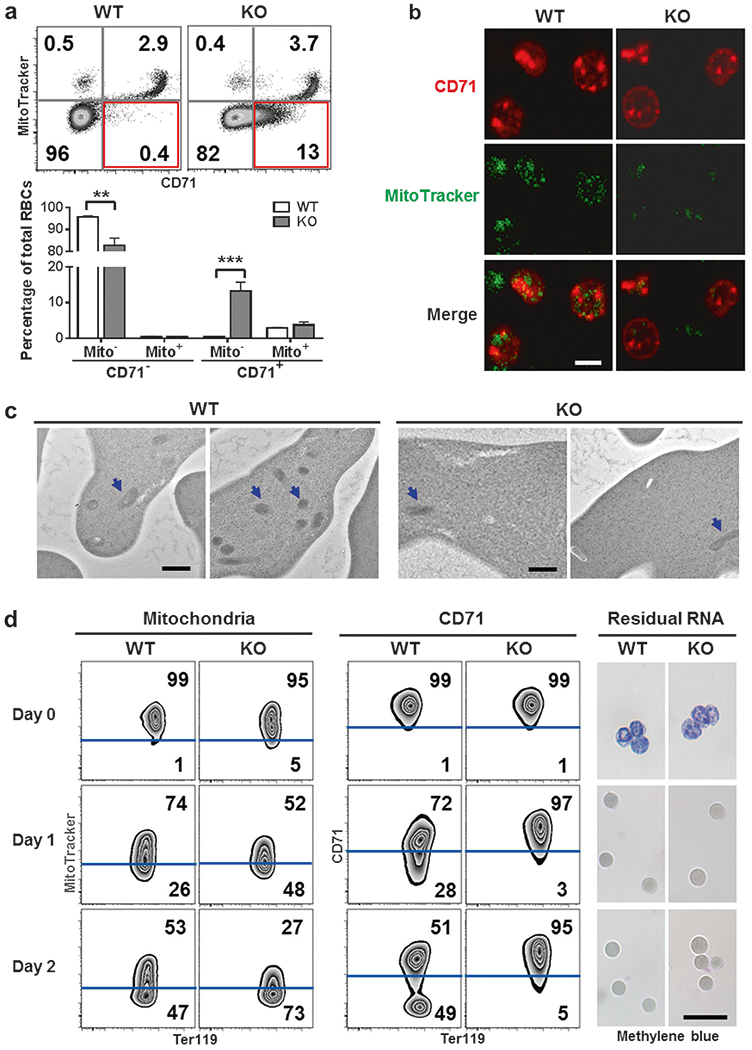
Loss of mitochondria in IEX-1 KO reticulocytes while retaining CD71 on the cells. a–c IEX-1 KO CD71+ reticulocytes were absent of mitochondria. Peripheral blood cells of WT and KO mice were stained with Ter119, CD71, and MitoTracker, counted by flow cytometry (upper) and statistically analyzed (lower) (a). IEX-1 KO mice had an abnormal population of CD71+Mito− cells highlighted in red, within Ter119+ RBCs. Data were presented as mean ± SEM (n = 12), **P < 0.01 and ***P < 0.001 compared between WT and KO cells. Ter119+CD71+ reticulocytes were sorted from WT and KO peripheral blood cells for analyzing mitochondrial content by confocal microscopy (b; scale bar, 5 μm) or transmission electron microscopy (c; mitochondria, blue arrows; scale bar, 500 nm). d Accelerated mitochondrial clearance and delayed CD71 shedding in KO reticulocytes during maturation. Ter119+ CD71high reticulocytes were sorted from WT and KO mice 3 days after PHZ treatment and cultured for 2 days. MitoTracker, anti-CD71 antibody, and Methylene blue were then used to stain mitochondria, CD71, and residual RNA, respectively. Scale bar, 20 μm.
CD71+Mito− reticulocytes might result from premature clearance of mitochondria or delayed removal of CD71 during KO reticulocyte maturation, since CD71 transcription was not elevated in the absence of IEX-1 (Figure S2a). To determine which process is responsible, we treated mice with PHZ to induce a high number of young reticulocytes released into the circulation [11]. Young reticulocytes were sorted on the basis of Ter119+CD71high (Figure S2b) [18] on day 3 after PHZ injection and matured ex vivo as previously described [11]. There were no differences in mitochondrial content and CD71 level between WT and KO young reticulocytes when they were freshly isolated (Fig. 2d, day 0). During 2 days of ex vivo differentiation, WT reticulocytes gradually eliminated mitochondria by 26% on day 1 and 47% on day 2, concomitant with a similar proportion of cells devoid of CD71 (Fig. 2d, Figure S2c and S2d). The coordinated elimination of CD71 and mitochondria was disrupted in the absence of IEX-1. IEX-1 deficient reticulocytes expulsed mitochondria almost twice as fast as those in WT reticulocytes (Fig. 2d), but CD71 shedding from cell surface was severely retarded, generating only 3% CD71− cells on day 1 and 5% on day 2 (Fig. 2d). Despite premature removal of mitochondria and delayed shedding of CD71, KO reticulocytes underwent enucleation and loss of reticular staining similarly to WT reticulocytes (Fig. 2d, right). The results suggest that although all cell organelles and obsolete proteins are eliminated by autophagosomes and exocytosis, respectively, CD71 shedding appears to be specifically coordinated with mitochondrial clearance.
Increased vulnerability to autophagy of mitochondria in reticulocytes
We next measured mitochondrial mass and Δψm in Ter119+CD71+ bone marrow erythroblasts and peripheral reticulocytes, in an attempt to explore the mechanism behind the premature mitochondrial loss in IEX-1 deficient reticulocytes. We observed no difference in mitochondrial content between WT and KO erythroblasts (Fig. 3a, upper left), in spite of a slight decrease of Δψm in KO erythroblasts (Fig. 3a, lower left). A severe loss of mitochondria and Δψm was observed in KO reticulocytes compared to WT controls (Fig. 3a, p < 0.001). Moreover, a reduced amount of mitochondrial complex IV (COX IV) and heightened autophagy in KO young reticulocytes were also corroborated by immunostaining of COX IV and microtubule-associated protein light chain 3(LC3), a marker for autophagesomes (Figure S3). The observations suggest increased vulnerability to mitophagy in reticulocytes, since IEX-1 deficient mitochondria were not taken up by autophagosomes in erythroblasts.
Fig. 3.
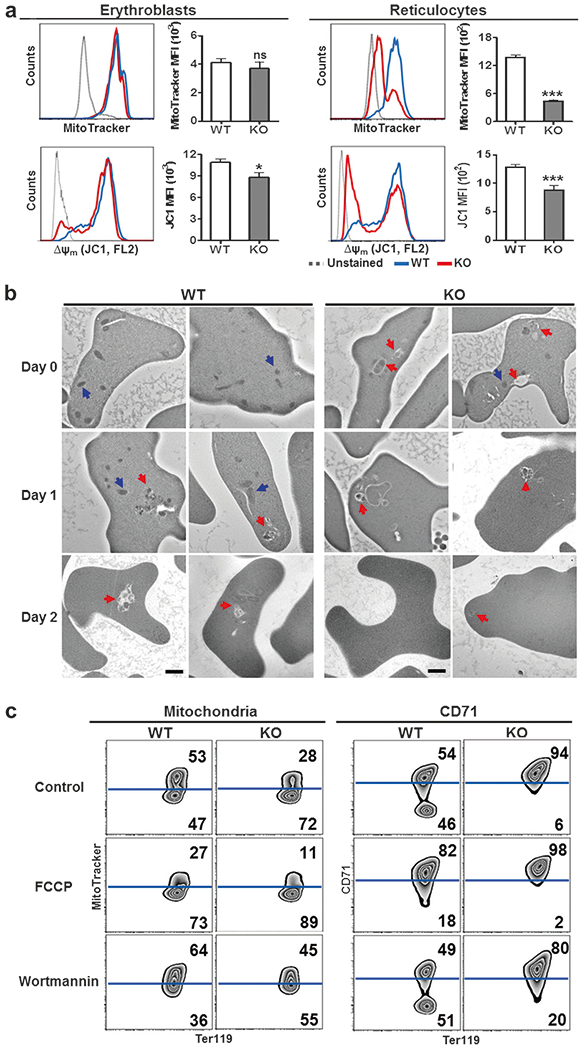
Premature clearance of mitochondria and impaired CD71 shedding only in KO reticulocytes, not erythroblasts. a Reduced mitochondrial membrane potential in the bone marrow erythroblasts and peripheral reticulocytes of IEX-1 KO mice. Ter119+CD71+ cells in the bone marrow (erythroblasts, left panels) or peripheral blood (reticulocytes, right panels) were stained with MitoTracker and JC1 to detect mitochondrial content and membrane potential, respectively. The mean fluorescence intensity (MFI) of MitoTracker and JC1 (red J-aggregate) was analyzed by flow cytometry and presented as mean ± SEM. ns no significance. *P < 0.05 and ***P < 0.001 compared with WT group. b Enhanced clearance of mitochondria by autophagy in KO reticulocytes. Young reticulocytes were matured ex vivo as in Fig. 2d and analyzed by transmission electron microscopy at indicated days: intact mitochondria, blue arrows; mitochondria phagocytosed by autophagosomes, red arrows; scale bar, 1 μm. c Effects of FCCP and wortmannin on mitochondrial clearance and CD71 shedding in WT and KO reticulocytes. Ter119+CD71high reticulocytes were cultured in the presence of 10 μM FCCP or 100 nM wortmannin, followed by flow cytometric analysis of mitochondria and CD71 at indicated days. Data were representative for at least 12 samples per group in all relevant panels
Transmission electron microscopy was employed to further analyze reticulocyte maturation as Fig. 2d. Mitochondria appeared morphologically normal and were mostly located outside of the autophagic vacuoles in WT young reticulocytes (Fig. 3b, upper left, blue arrows). Although some mitochondria were also observed similarly in the cytoplasm in KO young reticulocytes, these cytoplasmic mitochondria were relatively small in number and numerous mitochondria were observed within autophagic vacuoles (Fig. 3b, upper right, red arrows). The number of mitochondrion-containing autophagic vacuoles decreased significantly on day 2 of the culture, probably owing to few mitochondria left in KO cells. The autophagic vacuoles started to show up on days 1 and 2 in WT reticulocytes, concomitant with the progressive decline of mitochondria in the cytoplasm (Fig. 3b).
IEX-1 deficiency increases the amount of ATPase Inhibitory Factor 1 (IF1) in mitochondria because of diminished degradation of the inhibitor [5], which lowers Δψm [19, 20]. O-phenanthroline, an inhibitor of ATP-dependent proteases, prevents IF1 degradation, mimicking the effect of IEX-1 deficiency [5]. Incubation of WT young reticulocytes with O-phenanthroline not only reduced Δψm by 33% (Figure S4a), but also accelerated mitochondrial removal during ex vivo maturation (Figure S4b and S4c). Apparently, premature clearance of mitochondria in reticulocytes can be triggered by altered functions of mitochondria, besides null mutation of IEX-1.
To extend this observation to mitochondrial abnormalities caused by other means, Ter119+CD71high reticulocytes were treated with FCCP, an uncoupling agent that abolishes mitochondrial inner membrane potential [21]. As can be seen in Fig. 3c, upper and Figure S5, 47% of non-treated WT controls lost mitochondria, which increased to 73% following FCCP treatment, confirming a linkage of mitochondrial abnormality with their vulnerability to clearance in reticulocytes. As expected, the increased mitochondrial clearance was associated with decreased CD71 shedding to 18%, from 46% in WT reticulocytes (Fig. 3c, middle and Figure S5). FCCP also exaggerated mitophagy and further delayed CD17 shedding in KO reticulocytes. On the contrary, wortmannin, an inhibitor of phosphatidylinositol 3-kinase (PI3K) that blocks autophagy at an early stage [22], slowed mitochondrial clearance and reciprocally accelerated CD71 shedding in both WT and KO reticulocytes with predominant effects on the latter (Fig. 3c, low and Figure S5). The finding that mitophagy is inversely correlated with CD71 shedding during reticulocyte terminal maturation supports the concept that CD71 shedding is coupled to mitochondrial clearance.
Premature clearance of mitochondria hinders CD71 shedding via exocytosis
We went on to investigate how mitophagy was coordinated with CD71 shedding. As shown in Fig. 4a, while young reticulocytes were undergoing ex vivo maturation, WT cells decreased in size by 15% on day 1 and 28% on day 2, but KO cells decreased in size only by 8% on day 1 and 15% on day 2 (P < 0.01). The observation suggests the possibility of a deficit in the multivesicular body (MVB)/endosome-exosome pathway in KO reticulocytes, a major pathway by which reticulocytes shed plasma membrane and shrink cell volumes to achieve the biconcave shape of mature RBCs [23]. Overall activity of exocytosis can be quantified by acetylcholinesterase (AChE) activity in exosomes [24]. KO reticulocytes excreted ~50% fewer exosomes than WT controls during 2 days of the ex vivo culture (Fig. 4b). Immunochemical staining along with intracellular MVB-labeling with NRhPE [16] was then carried out to determine whether IEX-1 deficiency specifically impaired CD71-containing MVBs or instead altered overall activity of exocytosis. In comparison with WT controls, KO reticulocytes had significantly lower levels of CD71-containing vesicles (Fig. 4c, CD71 dots per cell: WT, 9.1 ± 1.2; KO, 4.3 ± 1.4; P < 0.001), as well as total MVBs (Fig. 4c, NRhPE dots per cell: WT, 11.0 ± 1.8; KO, 6.5 ± 1.3; P < 0.01). However, the ratio of CD71-containing MVBs to total MVBs diminished significantly to 66%, from 82% in the absence compared to the presence of IEX-1. Under similar conditions, excretion of CD29 or CD47, both of which are known to be released by the MVB pathway [25], was comparable in the presence vs. absence of IEX-1 (Figure S6). The results suggest that premature clearance of mitochondria preferably hampers formation of CD71-containing MVBs.
Fig. 4.
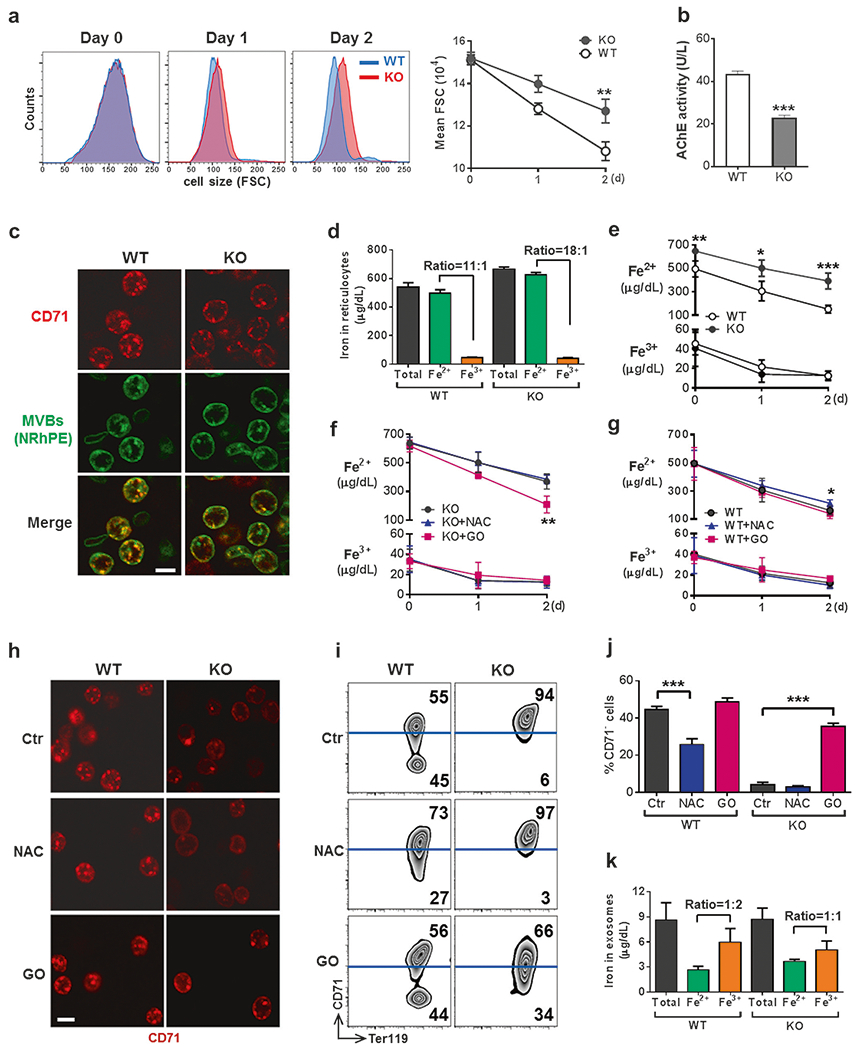
CD71 shedding is coupled with mitochondrial clearance in a ROS-dependent manner. a–c Exosome release is impaired in IEX-1 KO reticulocytes. Ter119+CD71high reticulocytes were sorted from PHZ-treated mice as above and cultured for 2 days. The size of reticulocytes was analyzed daily by flow cytometry on the basis of forward scatter (a). Exosomes released from 1 × 107 reticulocytes were assessed by the activity of AChE (b). After culturing for 12 h, cells were stained with FITC-anti-CD71 and N-Rh-PE that labeled MVBs to demonstrate their co-localization (c). Scale bar, 5 μm. Images were representative for at least 9 samples per group. d–g Changes of cellular iron concentrations during reticulocyte maturation. Concentrations of total iron, Fe2+, and Fe3+ were measured in reticulocytes before culturing (d) and daily during ex vivo maturation (e). f–i Modulation of ROS regulates CD71 shedding via exosome release. Ter119+CD71high reticulocytes as in a were cultured in the presence of 5 μM antioxidant NAC or 5 mU/mL glucose oxidase (GO), followed by daily measurement of cellular iron concentrations (f, g). Also shown are anti-CD71 staining for visualizing MVBs at 12 h after initial culture (h; scale bar, 5 μm), Ter119 and CD71 expression profiles by flow cytometry (i) with statistical analysis (j) on day 2, and concentrations of indicated irons and ratios of Fe2+ to Fe3+ measured in exosomes released from WT and KO reticulocytes 2 days after ex vivo maturation (k). Data represent as mean ± SEM (n = 12), *P < 0.05, **P < 0.01 and ***P < 0.001 compared between WT and KO cells or between indicated groups
We thought to determine whether perturbation of CD71 shedding resulted from inadequate heme synthesis owing to premature clearance of mitochondria. WT reticulocytes were incubated with succinylacetone, an inhibitor of the enzyme 6-aminolevulinic acid dehydratase required for heme synthesis [26]. The inhibitor did not slow down CD71 elimination (Figure S7), arguing against a role for heme in the regulation of CD71 shedding. Considering that the major function of CD71 is to transport transferrin-bound iron, we next assessed iron content during reticulocyte maturation. Fe2+ that is needed for heme synthesis is the major form of iron in reticulocytes: Fe2 outnumbered Fe3+ by a ratio more than 10:1, irrespective of IEX-1 expression (Fig. 4d). However, Fe2+ levels were consistently higher in KO reticulocytes than in WT ones during 2 days of the ex vivo maturation, while both WT and KO young reticulocytes had similarly low amounts of Fe3+ (Fig. 4e). An increase of Fe2+, along with a high level of surface CD71 in KO reticulocytes, challenges the long-held view that CD71 elimination results from a decreasing demand for iron as hemoglobin accumulates in reticulocytes [23, 27]. Nevertheless, the increased Fe2+ was consistent with a high level of CD71 on the cells which could continuously transport Fe3+ into reticulocytes.
Upon endocytosis, Fe3+ is converted into Fe2+ by six-transmembrane epithelial antigen of the prostate 3 (Steap3) [28] in the endosomes prior to its transportation into the cytoplasm by divalent metal transporter 1 (DMT1) [29]. The conversion of Fe3+ to Fe2+ is likely to be facilitated by lower ROS in the cells (Figure S8a). It is thus possible that CD71 shedding is regulated by excess Fe3+ so as to minimize its toxicity. To test this, we treated KO reticulocytes with GO that catalyzed oxidation of glucose to hydrogen peroxide that could oxidize Fe2+ to Fe3+ iron [30]. GO treatment increased ROS production substantially (Figure S8b, P < 0.001) and also reduced Fe2+ level in KO reticulocytes, which however did not concur with a corresponding rise of Fe3+ (Fig. 4f). As controls, KO cells were also treated with anti-oxidant, NAC, a ROS scavenger. The anti-oxidant did not affect Fe2+ level, probably because Fe2+/Fe3+ already reached a low threshold in spite of further reducing ROS production in the cells (Fig. 4f and Figure S8b). Under similar conditions, NAC elevated Fe2+ level slightly in WT reticulocytes, while GO had minimal effects on Fe2+ level in WT reticulocytes (Fig. 4g).
We questioned whether a diminished level of Fe2+, concomitant with no corresponding increase of Fe3+, was ascribed to acceleration of CD71 shedding in IEX-1 KO reticulocytes after GO treatment. In support of this possibility, GO-treated KO reticulocytes increased exocytosis of CD71-containing MVBs by 51% compared to controls at 12 h after initial culture (Fig. 4h; CD71 dots per cell: KO control, 4.3 ± 1.4; KO GO, 6.5 ± 1.2; p < 0.001). At the end of ex vivo maturation, CD71− reticulocytes were raised to 34%, from 6% in the absence of IEX-1 following GO treatment, as a result of accelerated removal of CD71 (Fig. 4i, j). Once again, NAC exhibited little influence on CD71 shedding in KO reticulocytes. In contrast, NAC-treated WT reticulocytes reduced CD71 dots by 40% compared to controls (Fig. 4h; CD71 dots per cell: WT control, 9.1 ± 1.2; WT NAC, 6.5 ± 1.0; P < 0.001), concomitant with reduced CD71 elimination to 27%, from 45% (Fig. 4i,j, P <0.001). Interestingly, neither NAC nor GO affected mitochondrial content significantly in reticulocytes regardless of IEX-1 expression (Figure S8c). On the contrary, the overall exocytosis was augmented by GO in IEX-1 deficient reticulocytes, whereas a decrease in exocytosis was seen in WT reticulocytes treated with NAC (Figure S9), underscoring an importance of adequate ROS in the late stage of reticulocyte differentiation.
Finally, if part of the function of CD71 shedding was to eliminate Fe3+ as we proposed, exosomes released from reticulocytes in response to Fe3+-mediated stress should contain a relatively higher amount of Fe3+ than Fe2+. We subsequently corroborated this by examination of iron content of the exosomes released by reticulocytes; we observed a 2:1 ratio of Fe3+ to Fe2+ in WT exosomes and a 1:1 ratio in KO exosomes (Fig. 4k). Because Fe3+ levels were 11-fold lower than Fe2+ in WT reticulocytes and 18-fold lower in KO cells (Fig. 4d), it meant the efficiency of Fe3+ exocytosis was at least 22-fold or 18-fold higher than that of Fe2+ in WT and KO reticulocytes, respectively. Hence, in addition to control of intracellular ROS via mitochondrial clearance, CD71 shedding acts as another safeguard to minimize iron toxicity during reticulocyte maturation by reducing Fe3+ entrance as well as removing excess Fe3+ from the cells via exocytosis.
CD71+Mito− reticulocytes in MDS patients are proportionate to decreased Δψm
To determine a clinical significance of these observations, we collected peripheral blood samples from 23 MDS patients (Table 1) and 10 healthy donors, and isolated RNA from mononuclear cells for IEX-1 quantification. Among the 23 MDS patients, five of them had ring sideroblasts and 13 of them showed cytogenetic abnormalities. The MDS patients were divided into two groups on the basis of IEX-1 levels: 5 MDS patients with at least 2-fold less IEX-1 expression than healthy controls were assigned to MDS with low IEX-1; 18 MDS patients with normal or increased IEX-1 levels were assigned to other MDS (Fig. 5a). Anemia appeared more severe in patients with low IEX-1 compared to other MDS, concomitant with reduced hemoglobin (Figure S10a and S10b), although low IEX-1 did not significantly affect white blood cells or platelets in the patients (Figure S10c and S10d). Almost all of MDS patients showed defective erythropoiesis characterized by a significantly increased amount of CD235a+CD71+ reticulocytes regardless of IEX-1 levels (Fig. 5b), in agreement with multiple factors contributing to the defective erythropoiesis in the patients. Strikingly, about 70% or 25% of CD235a+CD71+ reticulocytes were devoid of mitochondria in MDS patients with low IEX-1 or other MDS, respectively, both of which were significantly higher than those in healthy controls (5%, p < 0.001, Fig. 5c), indicating direct involvement of IEX-1 in severe anemia in MDS. Apparently, the high percentages of CD71+Mito− reticulocytes were not related with ring sideroblasts or any specific cytogenetic abnormalities in these MDS patients tested. The ring sideroblasts appear to begin to develop in the early stage of erythroid precursors in the bone marrow [31, 32], whereas CD71+Mito− reticulocytes are generated in the periphery during the final differentiation stage of RBCs. Although both of them are associated with iron metabolism alterations and mitochondria, no correlation between ring sideroblasts and CD71+Mito− reticulocytes underscores specific molecular mechanisms involved in distinct differentiation stages during erythroid maturation.
Table 1.
Clinical and laboratory characteristics of MDS patients
| No. | % BM blasts | 2016 WHO classification | Karyotype | IPSS risk | IEX-1 mRNA | % CD71+ reticulocytes | % Mito− reticulocytes | |
|---|---|---|---|---|---|---|---|---|
| Other MDS | 1 | <5 | MDS-MLD | Del(11q23), + 8 | Int-1 | 0.7 | 0.29 | 13.7 |
| 2 | <5 | MDS-MLD | Add(1)(p23) | Int-1 | 1.5 | 4.42 | 4.5 | |
| 3 | <5 | MDS-MLD | Normal | Int-1 | 0.8 | 2.33 | 34.3 | |
| 4 | 15 | t-MDS | −7 | High | 0.6 | 0.33 | 22.8 | |
| 6 | <5 | t-MDS | Normal | Int-1 | 0.7 | 2.27 | 24.3 | |
| 9 | 6 | CMML-1 (low WBC dysplastic subtype) | Normal | Int-1 | 3.0 | 1.13 | 33 | |
| 10 | 6 | MDS-EB1 (rings 30%) | Complex | Int-2 | 2.6 | 1.52 | 39.2 | |
| 11 | <5 | MDS/MPN-RS-T | Normal | Low | 4.5 | 0.40 | 24.4 | |
| 12 | <5 | MDS-MLD | Normal | Int-1 | 1.4 | 2.13 | 34 | |
| 13 | 7 | MDS-EB1 | Complex | Int-2 | 1.0 | 0.72 | 2.7 | |
| 14 | <5 | MDS-SLD-RS | Normal | Low | 2.5 | 0.68 | 32.4 | |
| 15 | 8 | MDS-EB1 | Complex | Int-2 | 0.7 | 0.96 | 37.4 | |
| 16 | 9 | MDS-EB1 | Normal | Int-1 | 1.8 | 3.04 | 29.6 | |
| 18 | <5 | MDS-MLD | Del(20) (q12q13) | Int-1 | 0.9 | 0.50 | 43.8 | |
| 19 | <5 | t-MDS | Der(1;7), + 1 | Int-1 | 0.6 | 0.30 | 44.7 | |
| 20 | <5 | MDS-MLD-RS | Normal | Low | 1.0 | 4.49 | 1.7 | |
| 21 | <5 | MDS-SLD-RS | Normal | Low | 0.8 | 0.10 | 4.7 | |
| 22 | 8 | t-MDS | +13 | Int-2 | 0.8 | 0.01 | 17.1 | |
| Low IEX-1 | 5 | 18 | MDS-EB2 | Del(5)(q13q33) | Int-2 | 0.3 | 1.41 | 69.0 |
| 7 | 14 | MDS-EB2 | Del(9)(q) | High | 0.2 | 1.84 | 70.8 | |
| 8 | <5 | t-MDS | Der(1;7), +1 | Int-1 | 0.3 | 1.02 | 73.4 | |
| 17 | 9 | MDS-EB1 | Normal | Int-1 | 0.3 | 1.03 | 67.9 | |
| 23 | <5 | MDS-MLD | Del(20) (q12q13) | Low | 0.3 | 1.68 | 62.8 | |
| Average value of healthy donors (mean ± SD): | 1.0 ± 0.4 | 0.06 ± 0.04 | 5.4 ± 3.2 | |||||
Fig. 5.
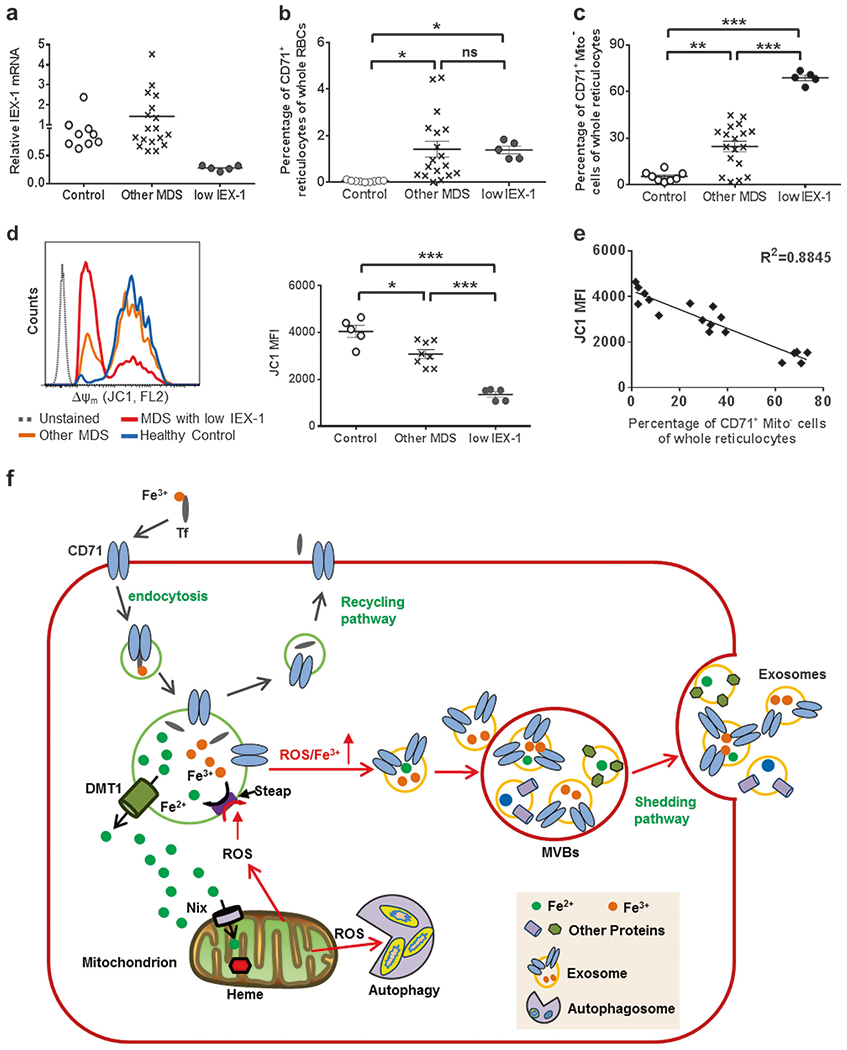
CD71+Mito− reticulocytes are presented in a majority of MDS patients. a Relative levels of IEX-1 mRNA in human peripheral blood cells in patients and healthy donors. MDS patients with at least a twofold decrease of IEX-1 mRNA compared with the median of healthy controls were defined as MDS patients with low IEX-1. b Reticulocytosis in MDS patients. Peripheral blood cells of healthy donors and MDS patients were stained with CD235a and CD71, followed by flow cytometric analysis. MDS patients exhibited a significantly increased proportion of CD235a+CD71+ reticulocytes compared to healthy controls independent of IEX-1. c Increased CD71+Mito− reticulocytes in MDS patients with a highest level in MDS patients with low IEX-1. d, e Reduced mitochondrial membrane potential Δψm in reticulocytes of MDS patients. Mitochondrial membrane potential Δψm of CD235a+CD71+ reticulocytes was analyzed by flow cytometry using JC1 as Fig. 3a (d). Inverse correlations between mitochondrial membrane potential Δψm and the percentages of CD71+Mito− reticulocytes are analyzed in all healthy donors and MDS patients by coefficient of determination (e). Data represent mean ± SEM, *P < 0.05, **P < 0.01, and ***P < 0.001 compared between indicated groups. (f) A model for CD71 shedding in couple with mitochondrial clearance. Fe3+ released from the CD71/Tf complex by a low pH in the endosomes after endocytosis of CD71/Tf-bounded Fe3+ is converted to Fe2+ by a metalloreductase Steap3 before transported to the cytoplasm by DMT1. An increasing ROS modulates Steap3 activity resulting in a shift of the reductive reaction toward Fe3+. A rise of ROS/Fe3+ would trigger fusion of the endosomes with MVBs followed by exosome excretion. An increase of ROS would also trigger mitophagy to limit ROS production. Altogether, a couple of CD71 shedding with mitochondrial clearance can effectively minimize the toxicity of Fe3+ and ROS during reticulocyte maturation
In addition to IEX-1, other factors causing mitochondrial dysfunction can also trigger premature clearance of mitochondria in reticulocytes. Consistent with this and prior observations in MDS cohorts, MDS patients consistently exhibited a decline in Δψm, with the most severe decline in MDS patients expressing the lowest levels of IEX-1 (Fig. 5d) and concomitant decreases in ROS production (Figure S10e). The decreased Δψm was highly correlated with a rising number of CD71+Mito− reticulocytes, with coefficient of determination R2 = 0.8845 in all healthy and MDS samples regardless of IEX-1 expression (Fig. 5e). The finding, along with uncoupling of CD71 shedding with mitochondrial clearance demonstrated in IEX-1 KO reticulocytes, suggests that abnormal mitochondria may trigger their premature clearance in MDS patients, disrupt CD71 shedding, and thus impede RBC maturation, contributing to the pathobiology of anemia in MDS.
Discussion
The present study unravels an unappreciated link between CD71 shedding and mitochondrial removal. Uncoupling of these two events hampers RBC maturation and results in an aberrant subset of CD71+Mito− reticulocytes in some MDS patients, the generation of which correlates with decreased Δψm irrespective of IEX-1 expression. Based on the current observations and previously published data, a model of CD71 shedding in couple of mitochondrial clearance is depicted in Fig. 5f. Upon endocytosis, Fe3+ is released from the CD71/Tf-Fe3+complex and then converted to Fe2+ via Steap3 [28]. A rise of Fe3+ in the endosomes as a result of increased ROS may trigger their fusion with MVBs followed by excretion, leading to relatively high levels of Fe3+ excretion in association with CD71 shedding. Too much ROS would also trigger mitophagy to limit their production. Altogether, a couple of CD71 shedding with mitochondrial clearance can effectively keep Fe3+ and ROS under check during reticulocyte maturation.
The coordinated removal of CD71 and mitochondria can be manipulated physiologically or pharmacologically. In this regard, induction of mitophagy by FCCP hindered CD71 shedding in WT reticulocytes, whereas inhibition of mitophagy by wortmannin restored CD71 clearance in KO reticulocytes. Moreover, induction of ROS normalized CD71 shedding in KO reticulocytes. Premature clearance of mitochondria hampering CD71 elimination was induced by antioxidant not only in cultured WT reticulocytes but also in WT mice fed with NAC, as evidenced by an increased proportion of CD71+Mito− cells (Figure S11). The investigation demonstrates an importance of ROS in regulation of CD71 shedding.
Alterations of mitochondrial function have been shown to play a pathological role in a variety of hematopoietic disorders, especially those affecting erythroid lineage [33]. Delayed mitophagy and resulting anemia have been well documented in mice lacking the Bcl-2 family member Nix [11, 34] or autophagy-related protein Atg7 [35], and in mice with mitochondrial DNA mutations [36]. In those scenarios, mitochondrial retention can render the cells to apoptosis, triggering their clearance by macrophages [11, 35, 36]. Structurally abnormal mitochondria [37], decreased Δψm [38, 39], or caspase overactivation [40] have been widely reported in MDS patients and may contribute to ineffective hematopoiesis characteristic of the disease via multiple mechanisms. In addition to excessive apoptosis, the abnormal mitochondria may also trigger their premature clearance, which is consistent with the observation of CD71+Mito− reticulocytes in a significant portion of MDS patients tested. A previous study of erythroid precursors from lower-risk MDS patients demonstrated that mitophagy was more pronounced and occurred earlier in the differentiation program than in controls [41]. Moreover, an increasing number of CD71+Mito− cells were seen in both WT and IEX-1 KO mice as they aged or after irradiation (Figure S12).
Mitochondrial dysfunction and impaired erythroid maturation are important characteristics of early stage/lower-risk MDS [41]. Uncoupling of mitochondrial clearance with CD71 shedding is likely to be one of the mechanisms by which mitochondrial abnormalities cause insufficient erythropoiesis, contributing to MDS-associated anemia, because CD71+Mito− reticulocytes were detected in MDS patients in direct proportion to decreased Δψm. The abnormal reticulocytes can potentially serve as another biomarker for MDS, although further study with a large sample size would be needed to determine the clinical utility. The finding of coordinated regulation of mitochondrial and CD71 clearance sheds novel light onto the pathogenesis of MDS in a subgroup of patients with defective mitochondria.
Supplementary Material
Acknowledgements
We thank the staff at the photopathology core at Wellman Center for Photomedicine for FACS and electron microscopy assistance. This research was supported in part by the National Institute of Health (NIH) grant CA158756 and Wellman Department fund to M.X.W., the Edward P. Evans Foundation for research in MDS to D.P.S., and a Bullock-Wellman postdoctoral fellowship to Q.Z.
Footnotes
Electronic supplementary material The online version of this article (https://doi.org/10.1038/s41375-018-0204-z) contains supplementary material, which is available to authorized users.
Conflict of interest The authors declare that they have no conflict of interest.
References
- 1.Heaney ML, Golde DW. Myelodysplasia. N Engl J Med. 1999;340:1649–60. May/27/1999 [DOI] [PubMed] [Google Scholar]
- 2.Hofmann WK, de Vos S, Komor M, Hoelzer D, Wachsman W, Koeffler HP. Characterization of gene expression of CD34+cells from normal and myelodysplastic bone marrow. Blood. 2002;100: 3553–60. November/15/2002 [DOI] [PubMed] [Google Scholar]
- 3.Steensma DP, Neiger JD, Porcher JC, Keats JJ, Bergsagel PL, Dennis TR, et al. Rearrangements and amplification of IER3 (IEX-1) represent a novel and recurrent molecular abnormality in myelodysplastic syndromes. Cancer Res. 2009;69:7518–23. October/1/2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prall WC, Czibere A, Grall F, Spentzos D, Steidl U, Giagounidis AA, et al. Differential gene expression of bone marrow-derived CD34+cells is associated with survival of patients suffering from myelodysplastic syndrome. Int J Hematol. 2009;89:173–87. March/2009 [DOI] [PubMed] [Google Scholar]
- 5.Shen L, Zhi L, Hu W, Wu MX. IEX-1 targets mitochondrial F1Fo-ATPase inhibitor for degradation. Cell Death Differ. 2009;16:603–12. April/2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang Q, Zhou C, Hamblin MR, Wu MX. Low-level laser therapy effectively prevents secondary brain injury induced by immediate early responsive gene X-1 deficiency. J Cereb Blood Flow Metab. 2014;34:1391–401. August/2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramsey H, Zhang Q, Brown DE, Steensma DP, Lin CP, Wu MX. Stress-induced hematopoietic failure in the absence of immediate early response gene X-1 (IEX-1, IER3). Haematologica. 2014;99: 282–91. February/2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang Q, Dong T, Li P, Wu MX. Noninvasive low-level laser therapy for thrombocytopenia. Sci Transl Med 2016;8:349ra101 July/27/2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Holm TM, Braun A, Trigatti BL, Brugnara C, Sakamoto M, Krieger M, et al. Failure of red blood cell maturation in mice with defects in the high-density lipoprotein receptor SR-BI. Blood. 2002;99:1817–24. March/1/2002 [DOI] [PubMed] [Google Scholar]
- 10.Kundu M, Lindsten T, Yang CY, Wu J, Zhao F, Zhang J, et al. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood. 2008;112:1493–502. August/15/2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, et al. Essential role for Nix in autophagic maturation of erythroid cells. Nature. 2008;454:232–5. July/10/2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Geminard C, de Gassart A, Vidal M. Reticulocyte maturation: mitoptosis and exosome release. Biocell 2002;26:205–15. August/2002 [PubMed] [Google Scholar]
- 13.Kanias T, Acker JP. Biopreservation of red blood cells-the struggle with hemoglobin oxidation. FEBS J. 2010;277:343–56. January/2010 [DOI] [PubMed] [Google Scholar]
- 14.Johnstone RM, Adam M, Pan BT. The fate of the transferrin receptor during maturation of sheep reticulocytes in vitro. Can J Biochem Cell Biol. 1984;62:1246–54. November/1984 [DOI] [PubMed] [Google Scholar]
- 15.Koury MJ, Koury ST, Kopsombut P, Bondurant MC. In vitro maturation of nascent reticulocytes to erythrocytes. Blood. 2005;105:2168–74. March/1/2005 [DOI] [PubMed] [Google Scholar]
- 16.Gibbings DJ, Ciaudo C, Erhardt M, Voinnet O. Multivesicular bodies associate with components of miRNA effector complexes and modulate miRNA activity. Nat Cell Biol. 2009;11:1143–9. September/2009 [DOI] [PubMed] [Google Scholar]
- 17.Socolovsky M, Nam H, Fleming MD, Haase VH, Brugnara C, Lodish HF. Ineffective erythropoiesis in Stat5a(−/−)5b(−/−) mice due to decreased survival of early erythroblasts. Blood. 2001;98:3261–73. December/1/2001 [DOI] [PubMed] [Google Scholar]
- 18.Liu J, Guo X, Mohandas N, Chasis JA, An X. Membrane remodeling during reticulocyte maturation. Blood. 2010;115:2021–7. March/11/2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Campanella M, Casswell E, Chong S, Farah Z, Wieckowski MR, Abramov AY, et al. Regulation of mitochondrial structure and function by the F1Fo-ATPase inhibitor protein, IF1. Cell Metab. 2008;8:13–25. July/2008 [DOI] [PubMed] [Google Scholar]
- 20.Ramsey H, Zhang Q, Wu MX. Mitoquinone restores platelet production in irradiation-induced thrombocytopenia. Platelets 2015;26:459–66. 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Benz R, McLaughlin S. The molecular mechanism of action of the proton ionophore FCCP (carbonylcyanide ptrifluoromethoxyphenylhydrazone). Biophys J. 1983;41:381–98. March/1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blommaart EF, Krause U, Schellens JP, Vreeling-Sindelarova H, Meijer AJ. The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur J Biochem. 1997;243:240–6. January/15/1997 [DOI] [PubMed] [Google Scholar]
- 23.Griffiths RE, Kupzig S, Cogan N, Mankelow TJ, Betin VM, Trakarnsanga K, et al. Maturing reticulocytes internalize plasma membrane in glycophorin A-containing vesicles that fuse with autophagosomes before exocytosis. Blood. 2012;119:6296–306. June/28/2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Savina A, Furlan M, Vidal M, Colombo MI. Exosome release is regulated by a calcium-dependent mechanism in K562 cells. J Biol Chem. 2003;278:20083–90. May/30/2003 [DOI] [PubMed] [Google Scholar]
- 25.Blanc L, Liu J, Vidal M, Chasis JA, An X, Mohandas N. The water channel aquaporin-1 partitions into exosomes during reticulocyte maturation: implication for the regulation of cell volume. Blood. 2009;114:3928–34. October 29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Richardson DR, Ponka P, Vyoral D. Distribution of iron in reticulocytes after inhibition of heme synthesis with succinylacetone: examination of the intermediates involved in iron metabolism. Blood. 1996;87:3477–88. April 15 [PubMed] [Google Scholar]
- 27.Harding CV, Heuser JE, Stahl PD. Exosomes: looking back three decades and into the future. J Cell Biol. 2013;200:367–71. February/18/2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ohgami RS, Campagna DR, Greer EL, Antiochos B, McDonald A, Chen J, et al. Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nat Genet. 2005;37:1264–9. November [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Canonne-Hergaux F, Levy JE, Fleming MD, Montross LK, Andrews NC, Gros P. Expression of the DMT1 (NRAMP2/DCT1) iron transporter in mice with genetic iron overload disorders. Blood. 2001;97:1138–40. February 15 [DOI] [PubMed] [Google Scholar]
- 30.Chiu DT, Liu TZ. Free radical and oxidative damage in human blood cells. J Biomed Sci. 1997;4:256–9. 1997 [DOI] [PubMed] [Google Scholar]
- 31.Cazzola M, Invernizzi R, Bergamaschi G, Levi S, Corsi B, Travaglino E, et al. Mitochondrial ferritin expression in erythroid cells from patients with sideroblastic anemia. Blood. 2003;101: 1996–2000. March 1 [DOI] [PubMed] [Google Scholar]
- 32.Cuijpers ML, Raymakers RA, Mackenzie MA, de Witte TJ, Swinkels DW. Recent advances in the understanding of iron overload in sideroblastic myelodysplastic syndrome. Br J Haematol. 2010;149:322–33. May [DOI] [PubMed] [Google Scholar]
- 33.Fontenay M, Cathelin S, Amiot M, Gyan E, Solary E. Mitochondria in hematopoiesis and hematological diseases. Oncogene. 2006;25:4757–67. August/7/2006 [DOI] [PubMed] [Google Scholar]
- 34.Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci USA. 2007; 104:19500–5. December/4/2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mortensen M, Ferguson DJ, Edelmann M, Kessler B, Morten KJ, Komatsu M, et al. Loss of autophagy in erythroid cells leads to defective removal of mitochondria and severe anemia in vivo. Proc Natl Acad Sci USA. 2010;107:832–7. January/12/2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ahlqvist KJ, Leoncini S, Pecorelli A, Wortmann SB, Ahola S, Forsstrom S, et al. MtDNA mutagenesis impairs elimination of mitochondria during erythroid maturation leading to enhanced erythrocyte destruction. Nat Commun. 2015;6:6494 March/9/2015 [DOI] [PubMed] [Google Scholar]
- 37.van de Loosdrecht AA, Brada SJ, Blom NR, Hendriks DW, Smit JW, van den BE, et al. Mitochondrial disruption and limited apoptosis of erythroblasts are associated with high risk myelodysplasia. An ultrastructural analysis. Leuk Res. 2001;25:385–93. May/2001 [DOI] [PubMed] [Google Scholar]
- 38.Berger G, Hunault-Berger M, Rachieru P, Fontenay-Roupie M, Baranger L, Ifrah N, et al. Increased apoptosis in mononucleated cells but not in CD34+cells in blastic forms of myelodysplastic syndromes. Hematol J. 2001;2:87–96. 2001 [DOI] [PubMed] [Google Scholar]
- 39.Matthes TW, Meyer G, Samii K, Beris P. Increased apoptosis in acquired sideroblastic anaemia. Br J Haematol. 2000;111:843–52. December/2000 [PubMed] [Google Scholar]
- 40.Lin CW, Manshouri T, Jilani I, Neuberg D, Patel K, Kantarjian H, et al. Proliferation and apoptosis in acute and chronic leukemias and myelodysplastic syndrome. Leuk Res. 2002;26:551–9. June/2002 [DOI] [PubMed] [Google Scholar]
- 41.Houwerzijl EJ, Pol HW, Blom NR, van der Want JJ, de Wolf JT, Vellenga E. Erythroid precursors from patients with low-risk myelodysplasia demonstrate ultrastructural features of enhanced autophagy of mitochondria. Leukemia. 2009;23:886–91. May/2009 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.