

Abstract
Current influenza vaccines only confer protection against homologous viruses. We synthesized pulmonary surfactant (PS)-biomimetic liposomes encapsulating 2′,3′-cyclic guanosine monophosphate-adenosine monophosphate (cGAMP), an agonist of the interferon gene inducer STING (stimulator of interferon genes). The adjuvant (PS-GAMP) vigorously augmented influenza vaccine-induced humoral and CD8+ T cell immune responses in mice by simulating the early phase of viral infection without concomitant excess inflammation. Two days after intranasal immunization with PS-GAMP-adjuvanted H1N1 vaccine, strong cross-protection was elicited against distant H1N1 and heterosubtypic H3N2, H5N1, and H7N9 viruses for at least 6 months while maintaining lung-resident memory CD8+ T cells. Adjuvanticity was then validated in ferrets. When alveolar epithelial cells (AECs) lacked Sting or gap junctions were blocked, PS-GAMP-mediated adjuvanticity was substantially abrogated in vivo. Thus, AECs play a pivotal role in configuring heterosubtypic immunity.
Graphical Abstract
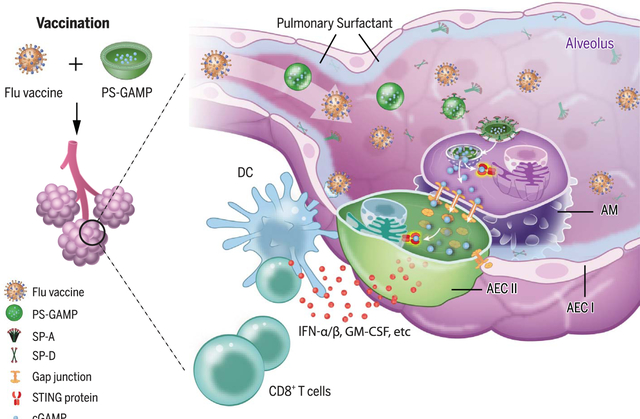
PS-GAMP-mediated adjuvanticity. In alveoli, PS-GAMP associates with SP-A or SP-D before entering AMs by means of SP-A- or SP-D-mediated endocytosis. cGAMP is subsequently released into the cytosol and fluxes into AECs by way of gap junctions. It then activates STING in these cells, resulting in the vigorous production of type 1 immune mediators. These mediators facilitate the recruitment and differentiation of CD11b+ DCs, which in turn direct robust antiviral CD8+ T cell and humoral immune responses.
INTRODUCTION:
Current influenza vaccines must be refreshed annually to address constant mutations of viral hemagglutinin (HA) and neuraminidase (NA) genes because the vaccines induce primarily neutralizing antibodies against these surface antigens. Even with annual updates, there have been years in which influenza vaccines were ineffective because of mismatched HA and/or NA antigenicity between the vaccine viral strains and strains in circulation. Thus, resources have been poured into developing “universal” influenza vaccines that can protect the population from divergent influenza viruses. However, none of these have passed human clinical trials thus far. Broad immunity can be evoked by natural viral infections or live vector-engineered and attenuated influenza vaccines, which all induce lung resident memory T cells (TRM cells) apart from humoral immunity. However, a delicate balance must be struck between safety and immunogenicity of these “replicating” vaccines. Moreover, these vaccines are suitable for only some populations. Thus, safe and potent mucosal adjuvants are urgently needed as part of nonreplicating vaccines in order to stimulate lung TRM cells and engender strong heterosubtypic immunity.
RATIONALE:
Type I interferons (IFN-Is) are the chief immune mediators for protective immunity against viral infections and can be vigorously induced by influenza viral infection of alveolar epithelial cells (AECs) as well as immune cells. Thus, the activation of stimulator of interferon genes (STING) in these two cell types may recapitulate the immune responses provoked by viral infection or replicating vaccines. However, delivery of STING agonists into the cytosol of AECs without breaching the integrity of the pulmonary surfactant (PS) layer remains a substantial challenge because the PS layer forms a strong barrier to prevent nanoparticles and hydrophilic molecules from accessing them. To address this challenge, we encapsulated 2′,3′-cyclicguanosinemonophosphate-adenosine monophosphate (cGAMP), a natural and potent STING agonist, with PS-biomimetic liposomes (PS-GAMP) in an attempt to increase the breadth of nonreplicating influenza vaccines toward universality.
RESULTS:
In mice, PS-GAMP entered alveolar macrophages (AMs) in concert with lung-specific surfactant protein-A (SP-A) and SP-D because of its resemblance to PS. Its cargo was released into the cytosol followed by a flux from AMs into AECs through gap junctions. Disguised as “self,” PS-GAMP escaped immune surveillance after intranasal immunization, activating the STING pathway in both AMs and AECs without breaching PS and alveolar epithelial barriers. Through this mechanism, PS-GAMP averted viral infection-provoked immunopathology while robustly augmenting the recruitment and differentiation of CD11b+ dendritic cells (DCs) and CD8+ T cell and humoral responses of influenza vaccines such as those induced by viral infection in terms of both timing and magnitude. The adjuvant in conjunction with inactivated H1N1 vaccine generated wide-spectrum cross-protection against distant H1N1 and heterosubtypic H3N2, H5N1, and H7N9 viruses as early as 2 days after a single immunization. This cross-protection lasted for at least 6 months, concurrent with durable lung CD8+ TRM cells in mice. The effectiveness of this vaccine approach was also demonstrated in a U.S. Food and Drug Administration-approved ferret model. PS-GAMP-mediated adjuvanticity was abrogated in vivo when AECs were deficient in Sting or when mice were administered gap junction inhibitors.
CONCLUSION:
Nonreplicating influenza vaccines or conventional adjuvants primarily activate immune cells, but this approach appears to be inadequate to induce lung TRM cells, a key element of heterosubtypic immunity. By contrast, PS-GAMP activated immune cells as well as AECs without breaching PS and AEC barriers, effectively averting exaggerated inflammation in the lung. STING activation in both immune cells and AECs resulted in a broad spectrum of immune protection against heterosubtypic influenza viruses. The study sheds light on the pivotal role AECs play in generating broad cross-protection against various influenza viruses. Thus, PS-GAMP is a promising mucosal adjuvant for “universal” influenza vaccines.
Current influenza vaccines protect against viral infections primarily by inducing neutralizing antibodies specific for viral surface hemagglutinin (HA) and neuraminidase (NA). However, these surface proteins undergo constant antigenic drift or shift, greatly limiting the efficacy of these vaccines (1). Studies demonstrating the essential role of lung CD8+ resident memory T cells (TRM cells) in heterosubtypic immunity may provide an explanation to this limitation (2, 3). Induced sufficiently by natural viral infections, these cells not only recognize highly conserved internal proteins that are shared among heterosubtypic influenza viruses but are also capable of clearing viruses at the site of viral entrance when their numbers are low (4–6). Similarly, live vector-engineered and attenuated influenza vaccines can induce lung CD8+ TRM cells (7, 8), but a delicate balance must be struck between their safety and immunogenicity. Moreover, these replicating vaccines are often compromised by preexisting immunity and consequently are suitable in only some populations (9). By contrast, nonreplicating influenza vaccines induce poor T cell immunity in the respiratory tract and require potent mucosal adjuvants to overcome the immunoregulatory mechanisms of the respiratory mucosa. However, there continues to be a dearth of effective mucosal adjuvants despite decades of investigation.
2′,3′-cyclic guanosine monophosphate-adenosine monophosphate (cGAMP), a natural agonist of the stimulator of interferon genes (STING), is a secondary messenger generated in response to DNA viral infections or tissue damage (10, 11). It stimulates the production of type I interferons (IFN-Is), which help determine the magnitude of type 1 immune responses, particularly those of CD8+ T cells (12, 13). STING agonists are potent adjuvants capable of eliciting robust antitumor immunity after intratumoral administration and augmenting intradermal influenza vaccines (13, 14). Using these small, water-soluble agonists as mucosal adjuvants, however, is a challenge. They must be delivered into the cytosol of antigen (Ag)-presenting cells (APCs) and/or alveolar epithelial cells (AECs) without breaching the integrity of the pulmonary surfactant (PS) layer, a mixture of lipids and proteins secreted by type II AECs. This PS layer forms a strong barrier, which separates exterior air from internal alveolar epithelium in alveoli and prevents nanoparticles and hydrophilic molecules from accessing AECs (15, 16).
cGAMP-containing liposomes are fabricated with PS constituents
We synthesized a series of liposomes, based on PS constituents (17), to encapsulate cGAMP (fig. S1A). The negatively charged nano4 was closest to PS in terms of lipid composition and charge. It was the only liposome that, when intranasally administered with whole inactivated A/Vietnam/1203/2004(VN04) H5N1 vaccine, vigorously stimulated the production of serum immunoglobulin G (IgG) and broncho-alveolar lavage fluid (BALF) IgA, concomitant with no body weight loss over vaccine-alone controls (fig. S1, B to E). By contrast, liposomes that were neutral (such as nano1), replaced anionic phosphatidylglycerol (DPPG) with cationic 1,2-dipalmitoyl-3-trimethylammonium-propane (DPTAP) (such as nano3 or naon5), or lacked polyethylene glycol, molecular weight 2000 (PEG-2000) (such as nano2 and nano3) showed substantially less adjuvanticity while causing significant loss in body weight (fig. S1, A to E). This was despite the similar size and encapsulation rate of these preparations to nano4 (fig. S1, F to G). Thus, negative charge and PEG-2000 appear to play an important role in the function and safety of the liposomes. Unexpectedly, bone marrow (BM)-derived dendritic cells (BMDCs) stimulated in vitro with cGAMP encapsulated in positively charged liposomes (nano3 or nano5) expressed higher levels of Ifnb1 than when stimulated with negatively charged liposomes (nano2 and nano4) (fig. S1H). An analogous pattern emerged for BM-derived macrophages (BMMs) when similarly stimulated (fig. S1I). We next added trehalose to the liposome suspension before lyophilization to increase nano4 stability (fig. S1A). The resultant nano6 liposome, which we termed PS-GAMP, was stable at −20°C for at least 6 months and exhibited similar ζ potential, size, function, and safety as those of freshly prepared nano4 (fig. S1, A, B to E, F, and J). Moreover, high Ag-specific IgG titers induced by PS-GAMP-adjuvanted influenza vaccine in wild-type (WT) but not in Sting-deficient mice confirmed that cGAMP, rather than any other constituents, was responsible for PS-GAMP adjuvanticity (fig. S1K).
PS-GAMP uptake by alveolar macrophages requires surfactant proteins A and D
We next studied cellular targets of nano4 and its cargo by labeling nano4 and nano5 membranes with DiD, a fluorescent lipophilic carbocyanine, and packaging another fluorescent dye with a molecular mass and net negative charge comparable with that of cGAMP [sulforhodamine B (SRB)] within the liposomes (Fig. 1A). The nasal tissue, brains, mediastinal lymph nodes (MLNs), and lungs of mice were analyzed by means of flow cytometry at various time points after intranasal administration of these liposomes. The lung was the only tissue in which SRB+ signals were higher than that in controls (Fig. 1B and fig. S2A). There, alveolar macrophages (AMs), recognized as CD11b−CD11c+CD24−, were SRB+DiD+, whereas CD11b−CD11c−EpCAM+MHC II+ AECs were SRB+ but DiD−(Fig. 1, B to D, and fig. S4A) (18). This suggests that only the former took up nano4 directly (Fig. 1E). SRB+AMs represented >95% of CD11c+SRB+ cells (fig. S4A, middle) or 44% of total AMs in the lung (fig. S4B, left). The proportions of SRB+ AMs and SRB+ AECs peaked in the lung at 12 hours and 18 hours, respectively, returning to basal levels within 36 hours (fig. S2B). In marked contrast, very few pulmonary CD103+ dendritic cells (DCs) (<2%) and CD11b+ DCs (<2%) were DiD+ and SRB+, which ruled out the direct uptake of the liposomes by these cells (fig. S4B). The ability of PS-GAMP to deliver cGAMP into AMs was functionally verified through CD40 up-regulation in DiD+ AMs after intranasal inoculation with DiD-labeled and cGAMP-encapsulated nano4 (DiD-PS-GAMP). The same nanoparticle lacking cGAMP (DiD-PS) had no effect on CD40 expression (fig. S5, A and B) (19). In contrast with nano4, nano5 did not significantly associate with either AMs or AECs when compared with free SRB (Fig. 1, B to D, and fig. S2C). Thus, AM activation appears to result directly from PS-GAMP uptake rather than through a bystander effect (fig. S5C).
Fig. 1. PS-GAMP uptake by AMs requires SP-A and SP-D.
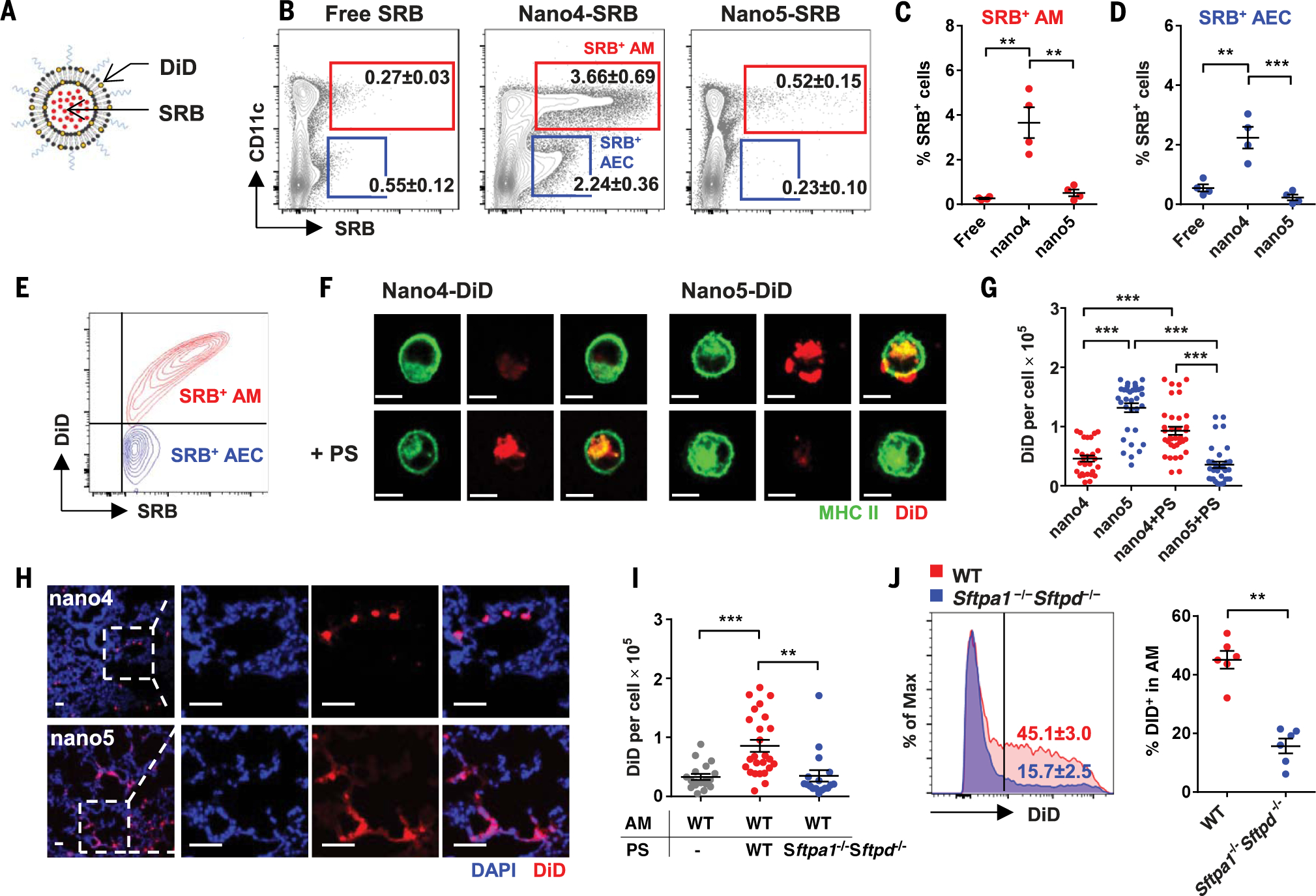
(A) A schematic diagram of PS-liposomes labeled with SRB and DiD. (B to E) Free SRB (20 μg) or SRB-DiD-nano4 or SRB-DiD-nano5 (20 μg SRB) was intranasally administered to mice, followed 12 hours later by flow cytometry analysis of SRB+ and/or DiD+ pulmonary cells. The percentages of SRB+ cells that were also CD11c+ AMs (red) or CD11c− AECs (blue) were analyzed (B) and quantitated [(C) and (D)] (n = 4 mice). (E) A representative overlay flow cytometry plot of AM and AEC staining for DiD and SRB. (F) AMs were isolated from MHC II-GFP mice and incubated with DiD-nano4 or DiD-nano5 for 4 hours after preincubation with (bottom) or without (top) PS for 30 min. Scale bar, 10 μm. (I) Alternatively, AMs were isolated from wild-type (WT) mice and incubated for 4 hours with DiD-nano4 that was pretreated with WT or Sftpa1−/−Sftpd−/− PS for 30 min. (G and I) The cells were imaged by means of fluorescent microscopy and quantified for DiD fluorescence intensity in individual cells with Image J; n = 18 to 36 cells. (H) Lungs were visualized by means of fluorescent microscopy 12 hours after receiving DiD-nano4 or DiD-nano5. Scale bar, 50 μm. (J) DiD-nano4 was intranasally administered to WT or Sftpa1−/−Sftpd−/− mice. CD11c+CD11b−CD24− AMs were analyzed 12 hours later for DiD+; n = 6 mice. Each symbol represents individual mice in (C), (D), and (J) or cells in (G) and (I). The results were presented as means ± SEM. Statistical analysis, one-way ANOVA for (C), (D), (G), and (I) and Student’s t test for (J). **P < 0.01 and ***P < 0.001 between indicated groups. All experiments were repeated three times with similar results.
Surprisingly, AMs isolated from lung lavage did not efficiently ingest nano4 ex vivo. AMs took up more nano5 than nano4, as evidenced by higher DiD fluorescence (Fig. 1, F and G), which complemented our earlier observation that nano5 induced higher Ifnb1 expression in BMDCs and BMMs (fig. S1, H and I). Differences between in vivo and ex vivo uptake of these liposomes may have been due to the lack of PS in ex vivo cultures. We therefore purified PS from BALF and incubated the PS with nanoparticles for 30 min before adding them to AMs. Nano4 uptake increased substantially, whereas nano5 uptake was diminished (Fig. 1, F and G). Positively charged nano5 aggregated on the negatively charged PS, explaining its poor entry into AMs (fig. S6). No such aggregates were formed when PS was incubated with nano4 under similar conditions (fig. S6). Similar results were obtained when AMs and PS were isolated from nonhuman primates (fig. S7). Thus, PS may play an evolutionarily conserved role in PS-GAMP endocytosis.
Consistent with these ex vivo observations, DiD-nano4 localized within individual cells positive for Siglec F, a biomarker for AMs, after intranasal administration (Fig. 1H and fig. S8). By contrast, positively charged nano5 electrostatically interacted and fused with negatively charged PS, exhibiting diffuse staining along the alveolar surface (Fig. 1H). Distinct localizations of nano4 and nano5 were corroborated by means of transmission electron microscopy (TEM) using nanogold-labeled nano5 and nano4 (fig. S9). In vitro validation of effective uptake of nano4 only in the presence of PS hinted that surfactant protein A (SP-A) and SP-D (termed “collectins”) played a role in this uptake. In support, PS isolated from Sftpa1−/− Sftpd−/− mice failed to enhance nano4 uptake by WT AMs in vitro over controls, in marked contrast to PS isolated from WT mice (Fig. 1I). Moreover, nano4 uptake was severely impeded in Sftpa1−/−Sftpd−/− mice (Fig. 1J), which was not due to any defect of Sftpa1−/−Sftpd−/− AMs because Sftpa1−/−Sftpd−/− AMs took up comparable amounts of nano4 as did WT AMs after preincubation in vitro with WT PS (fig. S10).
PS-GAMP transiently activates innate immunity in the lung
Reliance on SP-A and SP-D in nano4 uptake suggested that a natural and molecule-specific mechanism of particle clearance in the lung was involved, which would be the best approach to sustain the integrity of PS and alveolar epithelial barriers (20). Two days after PS-GAMP, whole inactivated VN04H5N1 vaccine, or a combination of both was intranasally administered, mouse lungs, nasal tissue, and brains were indistinguishable, by means of histology, from phosphate-buffered saline (PBS) controls (fig. S11, A and B). There was no cell death, damage to the epithelial barrier, or overt infiltration of inflammatory cells in these tissues (fig. S11). Only modest and transient infiltration of monocytes was found in the lung on day 3, which was markedly less severe than the monocyte response to viral infection (fig. S12E). We also did not observe any significant cytokine production in the brain over controls (fig. S11C). By contrast, administration of a VN04 H5N1 vaccine formulated with cholera toxin (CT) resulted in substantial inflammatory cell infiltration of the lung and measurable cytokine mRNA expression in the brains of some mice (fig. S11).
Although there was a lack of overt lung inflammation over time observed with histology (fig. S13A), PS-GAMP rapidly and robustly, but only transiently, activated innate immunity. Ifnb1, Gmcsf, and Tnf as well as Ccl2, Ccl3, Ccl5, and Cxcl10 expression peaked 12 hours after stimulation and resolved within 48 hours (fig. S14). By contrast, a low-dose infection with CA09 H1N1 influenza virus induced substantially higher levels of these mediators (fig. S14), giving rise to overt lung inflammation that worsened over the course of viral infection, despite robust Il10 expression (figs. S13, B and C, and S14). Transient IFN-β protein was also found in BALF, but tumor necrosis factor-α (TNF-α) and interleukin-10 (IL-10) levels were below the limit of detection (fig. S15). Substantially higher levels of these cytokines were produced on days 2 to 6 as infection proceeded (fig. S15). The transient activation of innate immunity was confined to the lung; serum IFN-β, IFN-γ, IL-6, IL-10, and TNF-α levels did not differ from those of controls (fig. S16, C to G). This accorded with the lack of adjuvant side effects on mouse body weight and temperature (fig. S16, A and B).
PS-GAMP is a potent adjuvant for both humoral and cellular immune responses
Although PS-GAMP only transiently activated innate immunity, it was sufficient to augment both humoral and cellular immune responses, which is consistent with our previous findings that prolonged activation of innate immunity was not necessary for strong adaptive immunity (13, 21, 22). PS-GAMP elevated serum hemagglutination inhibitory (HAI) antibody and BALF IgA titers in a dose-dependent manner (Fig. 2, A and B). The adjuvant had potent effects on both primary and booster immune responses, raising Ag-specific IgG1 10-fold, IgG more than 100-fold, and IgG2c ~1000-fold over VN04 H5N1 vaccine alone in the serum (Fig. 2, C to E). In addition to the whole inactivated VN04 H5N1 vaccine, PS-GAMP also exhibited strong adjuvanticity when combined with split virion (SV) vaccines such as the A/California/7/2009 (CA09) H1N1 vaccine. The adjuvant augmented HAI titers 10-fold, BALF IgA 60-fold, and IgG 10,000-fold over the SV vaccine alone (Fig. 2, F to H). Under similar conditions, polyinosinic: polycytidylic acid [poly(I:C)] showed substantially less efficacy in augmenting HAI titers, BALF IgA, and serum IgG production (Fig. 2, F to H). PS-GAMP not only augmented humoral immune responses but also profoundly enhanced cellular immune responses. PS-GAMP-adjuvanted CA09 H1N1 vaccine increased IFN-γ+CD8+ T cells 24-fold compared with vaccine alone or eightfold over the vaccine formulated with poly(I:C) (Fig. 2I and fig. S20A). This combination also induced the highest proportion of IFN-γ+CD4+ T cells among all vaccination groups (Fig. 2J and fig. S20A). These robust immune responses translated into full protection against 10× the median lethal dose (LD50) CA09 H1N1 viral challenge, concurrent with only mild to no body weight loss (Fig. 2, K and L). By contrast, poly(I:C)-adjuvanted CA09H1N1 vaccine conferred only partial (33%) protection against the viral challenge, with severe body weight loss.
Fig. 2. Adjuvanticity of PS-GAMP.
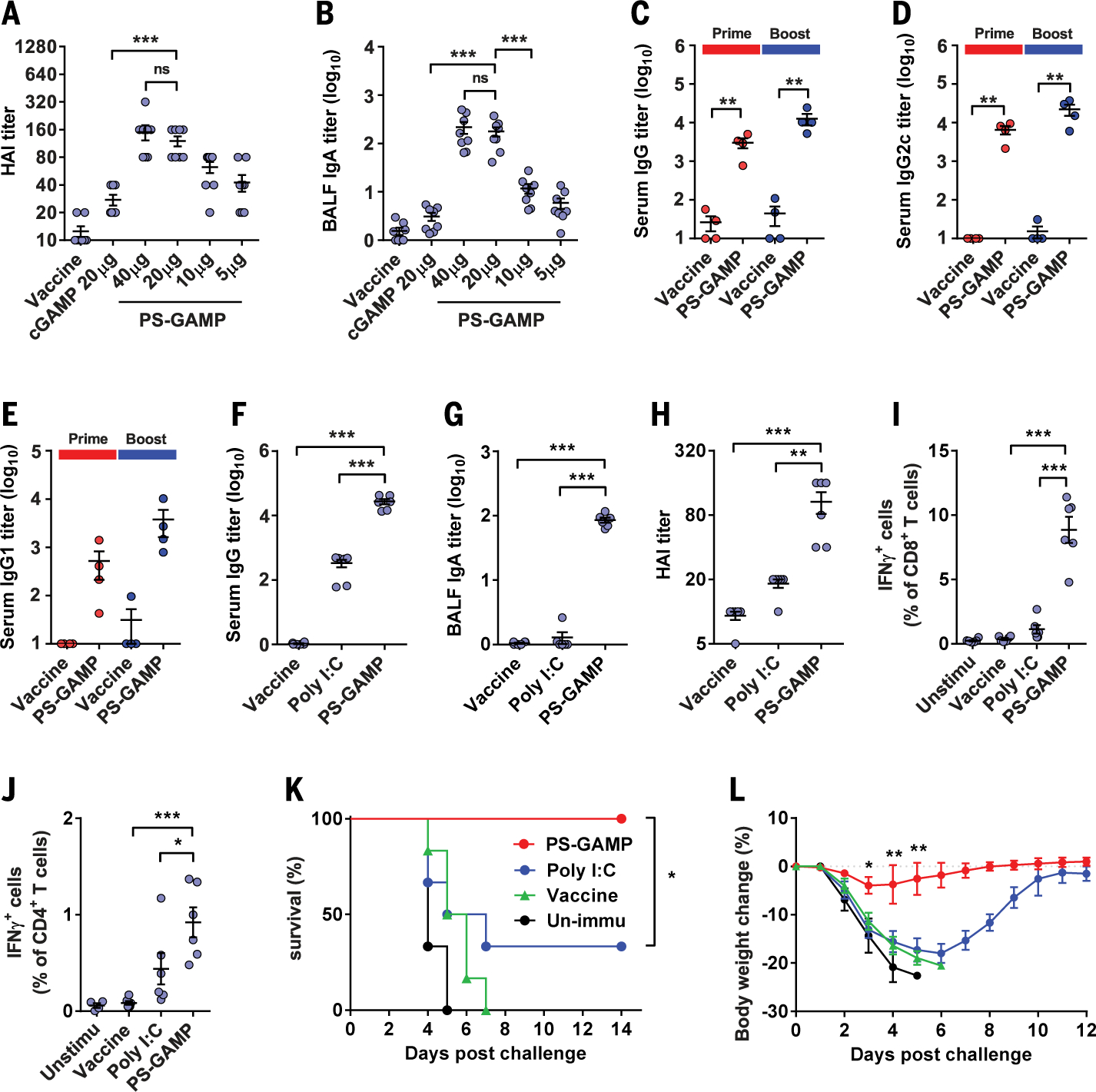
(A and B) Swiss Webster mice were intranasally immunized with VN04 H5N1 vaccine plus 20 μg of free cGAMP or PS-GAMP containing an indicated amount of cGAMP. Ag-specific serum HAI (A) and BALF IgA (B) titers were measured 2 weeks later; n = 8 mice. (C to E) C57BL/6 mice were intranasally immunized with VN04 H5N1 vaccine in presence or absence of PS-GAMP (20 μg cGAMP) on day 0 and boosted on day 14. Sera were collected on day 14 (Prime) or 21 (Boost) and measured for Ag-specific IgG (C), IgG2c (D), and IgG1 (E) titers; n = 4 mice. (F to L) C57BL/6 mice were intranasally immunized with CA09 H1N1 vaccine with or without 20 μg of PS-GAMP or poly(I:C). Serum IgG (F), BALF IgA (G), and serum HAI (H) titers were measured 2 weeks later. [(I) to (J)] Splenocytes were isolated 7 days after immunization and stimulated with the CA09 H1N1 vaccine. CD8+ (I) and CD4+ (J) T cells producing IFN-γ after viral Ag stimulation were determined by means of flow cytometry. [(K) and (L)] Survival curves (K) and changes in body weight (L) of unimmunized mice (black) or mice receiving a single immunization of vaccine alone (green), the vaccine combined with poly(I:C) (blue), or PS-GAMP (red), which were challenged 28 days later with 10 × LD50 CA09 H1N1 virus; n = 6 mice. The results are presented as means ± SEM. Each symbol represents individual mice in (A) to (J). Statistical analysis, one-way ANOVA for (A) to (J), two-way ANOVA for (L), and log-rank test for (K). *P < 0.05, **P < 0.01, and ***P < 0.001 in the presence or absence of PS-GAMP. ns, no significance. All experiments were repeated twice with similar results.
PS-GAMP elicits robust CD8+ T cell responses
After intranasal administration of PS-GAMP, CD11b+ DCs but not CD11b− DCs were elevated 14-fold and 36-fold on day 3 relative to day 0 in the lung (Fig. 3A, top) and MLN (Fig. 3A, bottom), respectively. Among CD11b+ DCs, monocyte-derived CD11b+ DCs (Mono-DCs), and tissue-resident CD11b+ DCs (tDCs) were distinguished through major histocompatibility complex II (MHC II) and Ly6C expression (fig. S3B) (23, 24). MHC IIhiCD11b+ tDCs are the main lung DC population that cross-presents to CD8+ T cells during influenza viral infection (23). After PS-GAMP administration, tDCs expanded to levels matching those seen during the first 3 days of viral infection. By contrast, pro-inflammatory mono-DCs increased only slightly during the same experimental period (Fig. 3B). CD11b+ tDCs declined thereafter in the lung and MLN, in marked contrast to the continued increase of these cells in both the lung and MLN during the 6 days of infection (Fig. 3B). Various other immune cell populations were characterized in the lungs, MLNs, nasal tissue, and brains after immunization or infection (fig. S12). In addition to DCs, only natural killer (NK) cell and CD4+ T cell numbers were briefly elevated for 1 or 2 days in the lung (Fig. 3A).
Fig. 3. CD8+ T cell responses are induced by PS-GAMP.
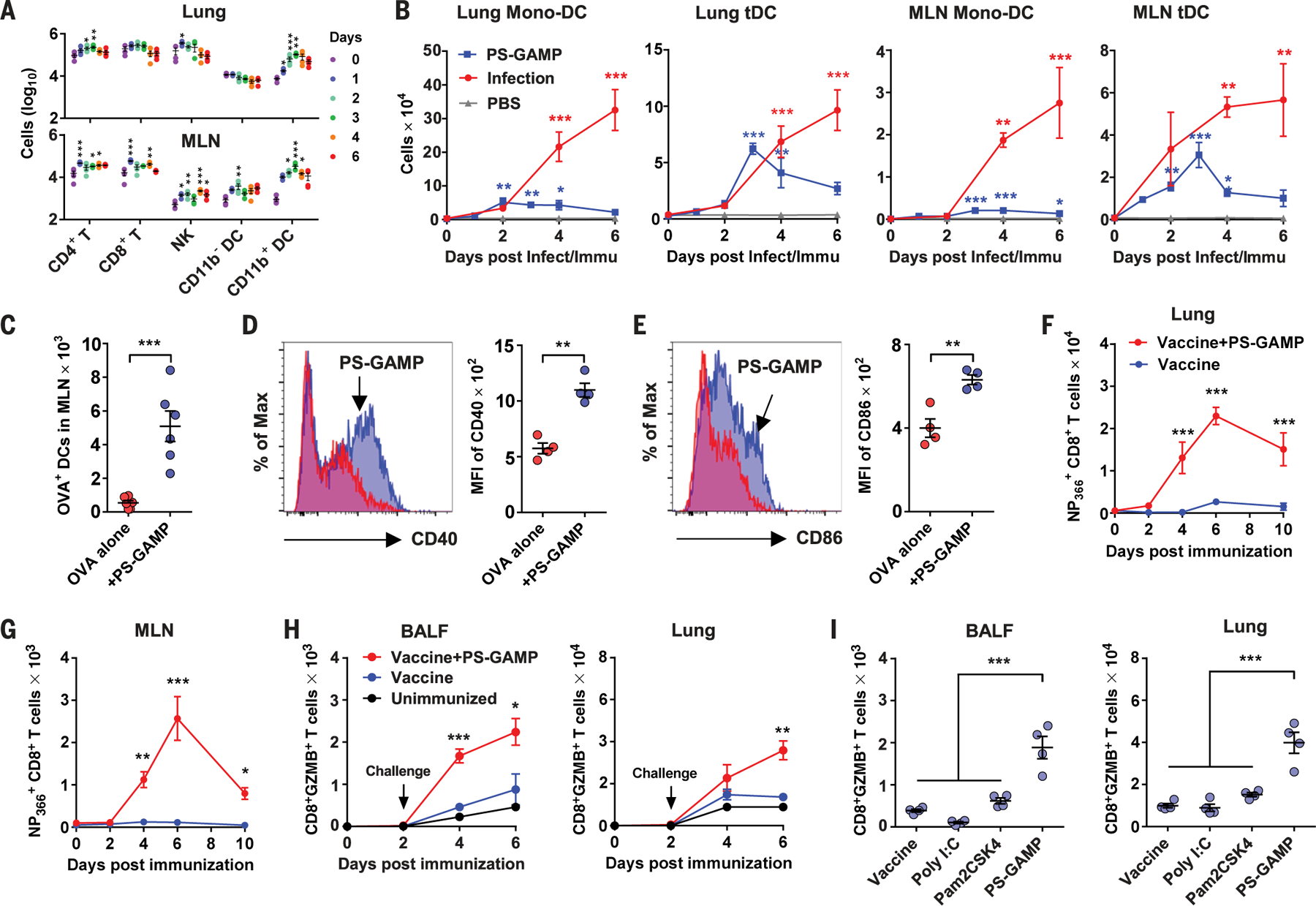
(A) Numbers of CD4+ and CD8+ T cells, NK cells, and CD11b+ and CD11b− DCs in the lung (top) and MLN (bottom) were analyzed by means of flow cytometry at an indicated day after mice were intranasally administered with PS-GAMP; n = 4 mice. (B) CD11b+ mono-DCs and CD11b+ tDC were quantified by means of flow cytometry in the lung and MLN at an indicated day after mice were intranasally immunized with PS-GAMP or infected with 1 × LD50 CA09 H1N1 virus; n = 4 mice. (C to E) Mice were intranasally vaccinated with OVA-AF647 with or without PS-GAMP. (C) DCs capturing OVA were enumerated in the MLN 36 hours after immunization; n = 6 mice. The mean fluorescence intensity (MFI) of CD40 (D) or CD86 (E) on these DCs was quantified by means of flow cytometry; n = 4 mice. (F and G) Mice were intranasally immunized with CA09 H1N1 vaccine with or without PS-GAMP or PBS alone as unimmunized controls. CD8+ T cells in the lung and MLN were analyzed on the indicated day after immunization for their Ag-specificity by staining with NP366–374 tetramer; n = 4 to 8 mice. Representative flow cytometry plots are shown in fig. S20A. (H) Mice were immunized as described in (F) and (G) and challenged 2 days later with 10 × LD50 CA09 H1N1 virus. BALF and lung cells were enumerated for GZMB+CD8+ T cells at indicated days after immunization; n = 4 mice. (I) Mice were similarly immunized and challenged as in (H), except that 20 μg of poly(I:C) or Pam2CSK4 was used in place of PS-GAMP for immunization. GZMB+CD8+ T cells were counted 4 days after challenge as in (H); n = 4 mice. Each symbol represents individual mice in (A), (C) to (E), and (I). The results were presented as means ± SEM. Statistical analysis, one-way ANOVA for (A), (B), and (I); two-way ANOVA for (F) to (H); and Student’s t test for (C) to (E). *P < 0.05; **P < 0.01, and ***P < 0.001 compared with day 0 [(A) and (B)], influenza vaccine alone [(F) to (H)], or between indicated groups. All experiments were repeated twice with similar results.
These CD11b+ DCs appeared to efficiently cross-prime CD8 T cells and induce robust proliferation. When fluorescently labeled ovalbumin (OVA) was intranasally administered, very few lung CD11b+ DCs (0.3%) showed OVA uptake. The proportion of these DCs ingesting OVA, however, rose substantially from 3% at 12 hours to 26% at 36 hours after immunization in the presence of PS-GAMP (fig. S17A). This translated into a 10-fold increase in MLN OVA+ (mostly CD11b+) DCs compared with that in mice receiving OVA alone (Fig. 3C and fig. S17B). These DCs had matured and were activated, as suggested by CD40 and CD86 up-regulation (Fig. 3, D and E). This effect was presumably secondary to AEC and AM activation because most MLN DCs were PS-GAMP-negative (fig. S17, C and D). The increase in Ag-specific CD11b+ DCs did not result from altered Ag processing or uptake because OVA uptake or its proteolytic cleavage was unaffected by PS-GAMP (fig. S18, D and E). Thus, the enhanced proliferation of transferred OVA-specific OT-I cells in the presence of PS-GAMP was likely due to the augmented differentiation and maturation of CD11b+ DCs. These cells, in turn, gave rise to a greater than sixfold increase in highly proliferating OT-I cells in both the lungs and MLNs (fig. S19, A to D).
A large number of nucleoprotein (NP)366–374-specific CD8+ T cells were observed in the lung and, to a lesser extent, in the MLN, as early as 4 days after immunization with PS-GAMP-adjuvanted influenza vaccine (Fig. 3, F and G, and fig. S20B). NP366–374 was the dominant CD8+ T cell epitope, and CD8+ T cells specific for other epitopes, such as PA224–233 or PB1703–711, were undetectable in these animals, probably because of a low copy number of these proteins in inactivated influenza vaccine (fig. S20C) (25). These virus-specific CD8+ T cells expressed the early activation biomarker, granzyme B (GZMB), upon viral challenge (fig. S21A) (26). The proportion of GZMB+CD8+ T cells rose significantly 4 days in BALF and 6 days in the lung after receiving PS-GAMP-adjuvanted CA09 H1N1 vaccine (Fig. 3H). More than 65% of these GZMB+CD8+ T cells were also positive for NP366–374, whereas only ~3% of the cells were positive for PA224–233 or PB1703–711 (fig. S21B). Under similar conditions, vaccine alone failed to expand GZMB+CD8+ T cells significantly (Fig. 3H). The CD8+ T cell response evoked by PS-GAMP was superior to poly(I:C) or Toll-like receptor 2 (TLR2) agonist Pam2CSK4 (Fig. 3I) (27, 28). Although T cell immune responses were induced soon after immunization, Ag-specific BALF IgA and IgM were undetectable on day 6 after immunization (fig. S21C). Thus, PS-GAMP mimics crucial events of viral infection such as CD8+ T cell induction without provoking excessive lung inflammation or immunopathology (figs. S11 to S16).
PS-GAMP offers robust protection as early as 2 days after immunization
The rapid induction of CD8+ T cells prompted us to determine how quickly protection could be achieved with PS-GAMP. To this end, mice were challenged on day 0, 2, 4, 6, 8, or 14 after immunization (fig. S22A). Inclusion of PS-GAMP in the vaccination fully protected mice from homologous viral challenges as early as 2 days after immunization (Fig. 4A). At all early challenge time points (days −2, −4, and −6), mice experienced only minor reductions in body weight (<10%) (fig. S22B), and all mice survived (Fig. 4A). When challenged 8 days after immunization, mice showed no body weight loss, with 100% survival (Fig. 4A and fig. S22B). This early protection did not result directly from innate immunity because PS-GAMP alone given on day 0 or 2 prior did not confer any protection (Fig. 4B and fig. S22C). To determine whether CD8+ T cells were responsible for the early protection, CD8+ T cells were depleted by intraperitoneal injections of antibodies to CD8 every other day starting 2 days before and ending 4 days after immunization. Depletion of CD8+ T cells abolished the early protection, as evidenced by a precipitous body weight loss and 100% mortality similar to those of mice receiving vaccine alone (Fig. 4C and fig. S22D). To ensure that this early protection was not specific to the CA09 H1N1 vaccine, we administered the H5N1 vaccine, which is an immunogenically weak vaccine by comparison. PS-GAMP conferred 75 to 100% protection against rgVN04 H5N1 viral challenge for mice immunized 2 to 8 days prior in an adaptive-immune-dependent manner (Fig. 4D and fig. S22E) because no protection was attained with PS-GAMP alone (Fig. 4E and fig. S22F). Under similar conditions, the vaccine combined with CT provided no early protection (Fig. 4E and fig. S22F), arguing persuasively that the heightened inflammation is not necessarily required for strong adaptive immune responses (fig. S11). Mice were also significantly or fully protected from a lethal challenge of a clinical isolate of pre-pandemic A/Shanghai/4664T/2013 (SH13) H7N9 virus 2 or 14 days after immunization with PS-GAMP-adjuvanted inactivated H7N9 vaccine (H7-Re1) (Fig. 4F and fig. S22, G to I). Vaccine adjuvanted by poly(I:C) provided no benefit over vaccine alone under similar conditions (Fig. 4F and fig. S22I).
Fig. 4. PS-GAMP mediates early protection.
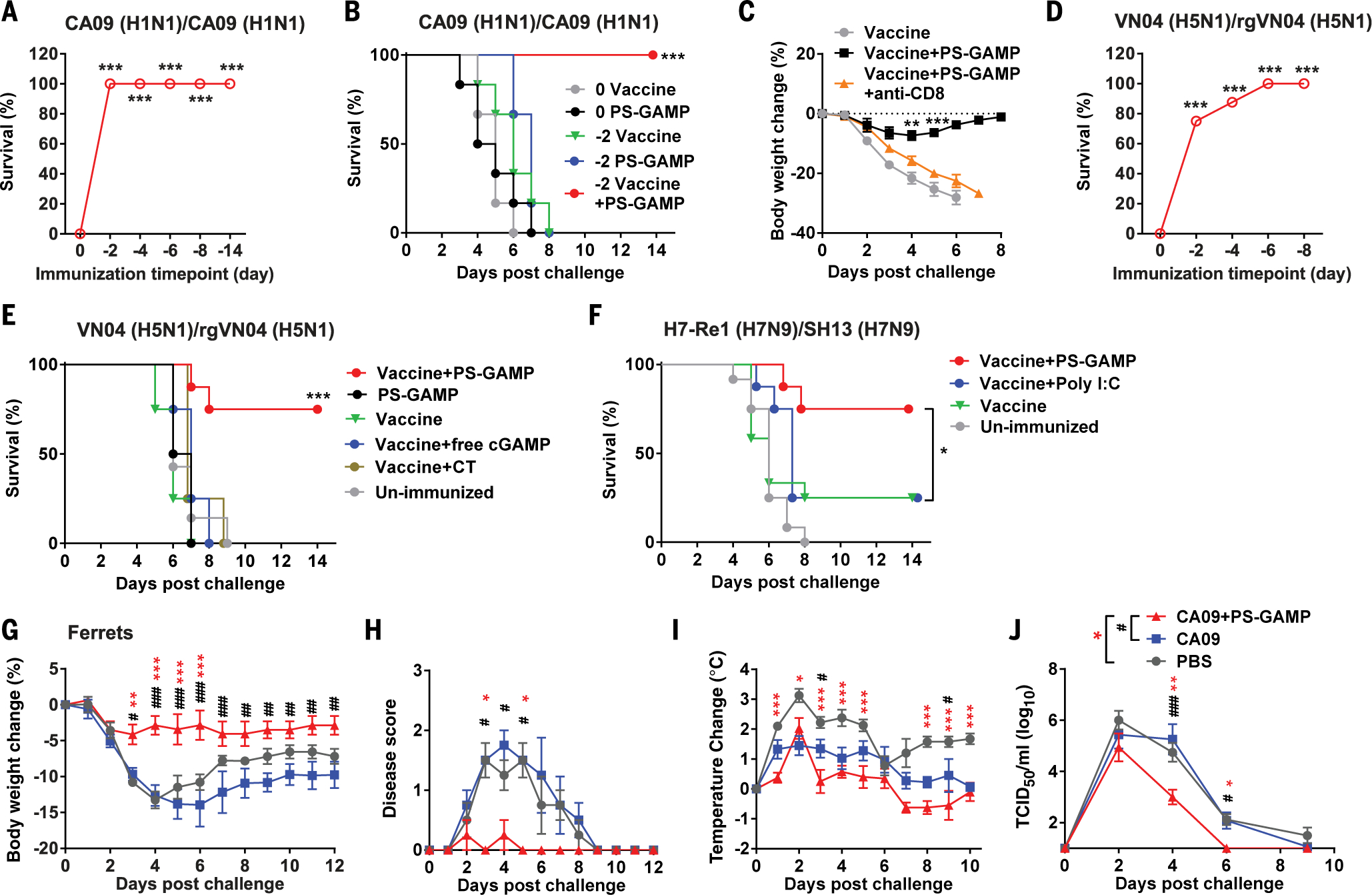
(A and B) Survival rates of immunized C57BL/6 mice after 10 × LD50 CA09 H1N1 viral challenge. (A) The mice were intranasally immunized with CA09 H1N1 vaccine (0.5 μg HA) and PS-GAMP (20 μg cGAMP) on day 2, 4, 6, 8, or 14 before viral challenging (fig. S22A); n = 6 to 11 mice. (B) Mice were immunized and challenged either on the same day (0) or 2 days (−2) after immunization; n = 6 mice. (C) Mice were immunized and challenged 2 days later as in (A). CD8+ T cells were depleted in some mice through injections of antibody to CD8 2 days before and 0, 2, and 4 days after vaccination; n = 4 mice. (D) Survival rates of mice immunized with VN04 H5N1 vaccine plus PS-GAMP at indicated day before challenge on day 0 with 10 × LD50 rgVN04 H5N1 virus (fig. S22A); n = 4 to 8 mice. (E) Survival rates of mice immunized with VN04 H5N1 vaccine, PS-GAMP, or the vaccine plus free cGAMP, CT, or PS-GAMP, followed with rgVN04 H5N1 viral challenge 2 days later; n = 4 to 8 mice. (F) Mice were intranasally immunized with H7-Re1 H7N9 vaccine and 20 μg of PS-GAMP or poly(I:C) and challenged 2 days later with a clinically isolated SH13 H7N9 virus at 40 × LD50; n = 8 to 12 mice. (G to J) Ferrets were intranasally immunized with CA09 H1N1 vaccine (9 μg) with or without 200 μg of PS-GAMP and challenged with 106 TCID50 CA09 H1N1 virus 2 days later. Body weight (G), disease score (H), temperature (I), and nasal wash viral titers (J) were monitored for 12 days; n = 4 ferrets. The results were presented as means ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001 compared with day 0 [(A) and (D)], vaccine alone [(B), (E), and (F)], or in the presence or absence of antibody to CD8 (C). Mouse experiments were repeated twice with similar results. For ferrets, asterisks indicate significance between PBS and vaccine+PS-GAMP, and pound signs (#) indicate significance between vaccine and vaccine+PS-GAMP. *,#P < 0.05; **,##P < 0.01; and ***,###P < 0.001. Statistical analysis, two-way ANOVA for (C), (G), (I), and (J); Kruskal-Wallis test for (H); and the log-rank test for (A), (B), and (D) to (F).
The ability of PS-GAMP to quickly establish protection was then validated in a U.S. Food and Drug Administration-approved ferret model. Ferrets receiving PS-GAMP-adjuvanted CA09 H1N1 vaccine 2 days prior experienced <5% body weight loss when infected with homologous CA09H1N1 virus, which is concomitant with mild to no clinical symptoms and only a brief and limited spike in body temperature on day 2 after viral challenges (Fig. 4, G to I). Viral shedding was significantly blunted from day 4 onward (Fig. 4J). By contrast, CA09H1N1 vaccine alone failed to prevent substantial weight loss after a similar viral challenge and did not improve clinical symptoms or reduce viral shedding over controls, despite showing milder increases in body temperature compared with that of controls (Fig. 4, G to J).
AECs are indispensable for PS-GAMP-mediated adjuvanticity
cGAMP can be readily transferred by way of gap junctions presented between AMs and AECs (29, 30). A dynamic flux from AMs to AECs was marked by the gradual loss of SRB in AMs, concurrent with the continuous gain of SRB in AECs from 12 to 18 hours after intranasal administration of SRB-nano4 (fig. S23A). The loss of SRB in AMs could not be ascribed to a loss of the liposomes because the number of DiD+ cells was unaltered up to 18 hours later (fig. S23B). The entry of SRB into AECs was blocked by carbenoxolone (CBX) (Fig. 5, A and C), a gap-junction blocker (29), which did not affect SRB uptake by AMs (Fig. 5, A and B). In AMs and AECs sorted from lungs receiving PS-GAMP, CBX greatly diminished the transcription of Ifnb1 and Gmcsf in AECs while increasing Ifnb1 transcription in AMs (fig. S24, B and C). This was most likely a consequence of elevated cGAMP levels in the cells. Thus, there is a gap junction-mediated flux of cGAMP to AECs from AMs. By contrast, poly(I:C) remained primarily within AMs (>97%) after intranasal immunization (fig. S25A). Only 0.4% of total AECs and 4% of total DCs took up poly(I:C) in the lung (fig. S25, B and C). MLN and nasal tissue DCs as well as nasal epithelial cells rarely internalized poly(I:C) (fig. S25, D and E).
Fig. 5. AECs make an indispensable contribution to PS-GAMP adjuvanticity.
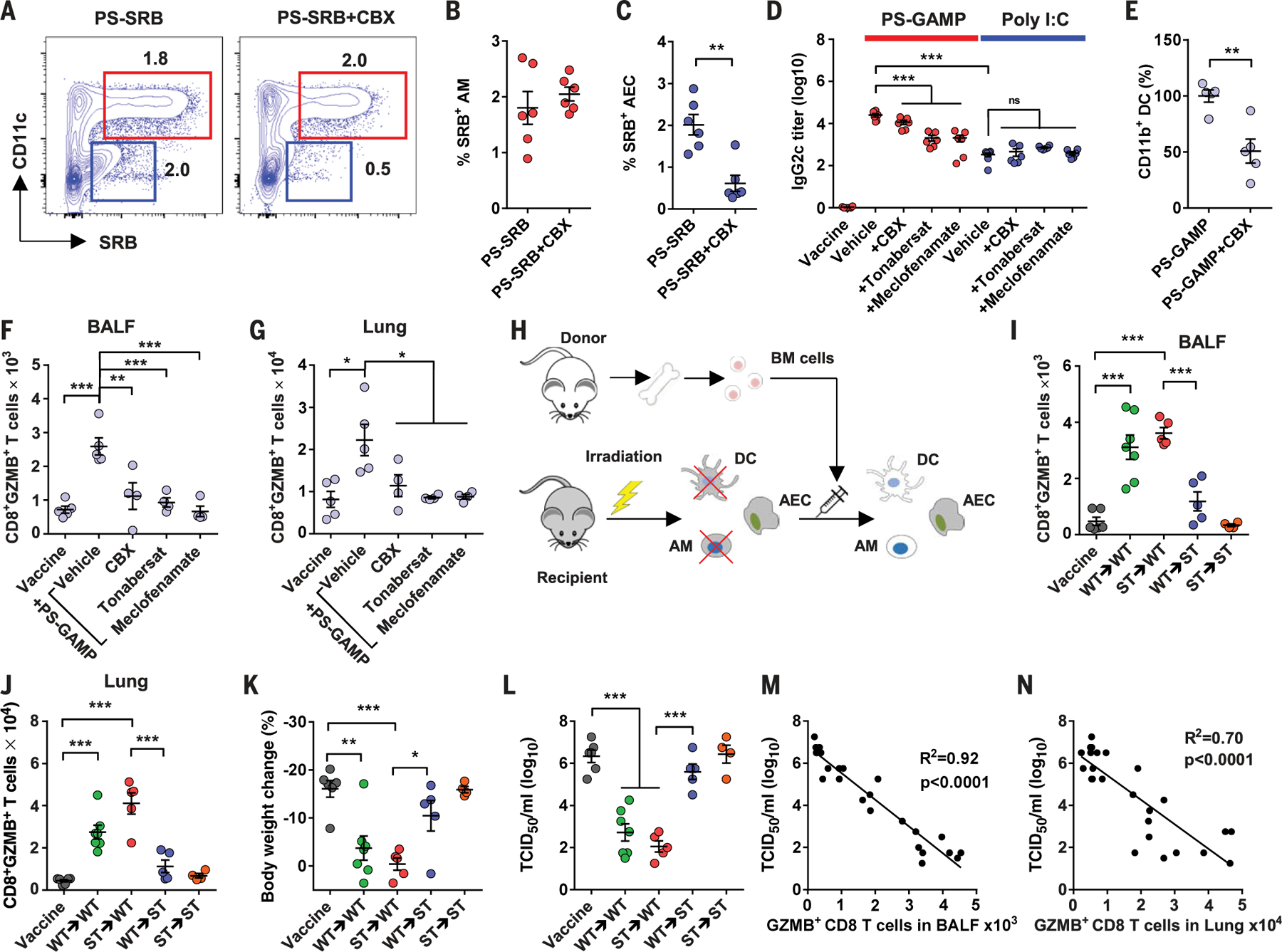
(A) Mice were intraperitoneally administered CBX once a day for 3 consecutive days, after which SRB-nano4 was intranasally administered to the mice. (B and C) Percentages of SRB+ AMs (red) and AECs (blue) that were analyzed 12 hours after (A); n = 6 mice. (D) Mice were intraperitoneally administered CBX, tonabersat, or meclofenamate and intranasally immunized with CA09 H1N1 vaccine with or without 20 μg of poly(I:C) or PS-GAMP. Sera were collected 14 days later and analyzed for IgG2c; n = 6 mice. (E) Mice were immunized with CA09 H1N1 vaccine and PS-GAMP in the presence or absence of CBX as in (D). Lung CD11b+ DCs were counted 24 hours later; n = 4 mice. (F and G) Mice receiving an indicated gap junction inhibitor were immunized as in (D) and challenged with 10 × LD50 CA09 H1N1 virus 2 days later. GZMB+CD8+ T cells in BALF (F) and the lung (G) were analyzed by means of flow cytometry; n = 4 mice. (H) A schematic diagram of generating chimeric mice. Mice were administered lethal irradiation before BM cell transfer. Chimeras were confirmed after 3 months (fig. S26), immunized, and challenged as in (F). (I and J) Four days after challenge, GZMB+CD8+ T cells were enumerated by means of flow cytometry in BALF (I) and lung (J). (K and L) Changes in body weight relative to day 0 (K) and lung viral titers (L) were also measured; n = 4 to 7 mice. (M and N) A correlation between the number of GZMB+CD8+ T cells and viral titers was determined by means of regression analysis. The results were presented as means ± SEM. Each symbol represents individual mice. Statistical analysis, one-way ANOVA for (D), (F), (G), and (I) to (L); Student’s t test for (B), (C), and (E). *P < 0.05, **P < 0.01, and ***P < 0.001. All experiments were repeated twice with similar results.
PS-GAMP induced 100-fold-higher IgG2c titers than did poly(I:C) (Fig. 5D). However, the adjuvanticity was blunted substantially when mice were treated with CBX or two other gap junction inhibitors, tonabersat and meclofenamate, before and during immunization (Fig. 5D) (31, 32). By contrast, these inhibitors had few effects on poly(I:C)-mediated adjuvanticity (Fig. 5D), which is consistent with the inability of poly(I:C), a large molecule, to enter the neighbor cells by way of gap junctions (fig. S25A). The blockade on the entry of cGAMP into AECs reduced recruitment of CD11b+ DCs by 50% (Fig. 5E) and exhibited more profound effects on the early CD8+ T cell responses in both the BALF and lung (Fig. 5, F and G). Moreover, chimeric mice (ST→WT) comprising Sting-deficient (Sting−/− or ST) BM cells and WT AECs had similar BALF and lung CD8+ T cell numbers as those of WT→WT mice (Fig. 5, H to J, and fig. S26) (33). By contrast, significantly lower numbers of Ag-specific CD8+ T cells were recovered from the BALF and lungs of chimeras with Sting-deficient AECs and a WT hematopoietic compartment (WT→ST mice) (Fig. 5, I and J). WT→ST mice showed poor protection by PS-GAMP-adjuvanted CA09 H1N1 vaccine, as suggested by body weight loss and high lung viral titers, in contrast to the similarly observed protection between ST→WT and WT→WT mice (Fig. 5, K and L). There was an inverse correlation between viral titers and the number of GZMB+CD8+ T cells in the BALF and lung, supporting the notion that GZMB+CD8+ T cells play a pivotal role in the control of viral infections (Fig. 5, M and N). Thus, AECs rather than AMs appear to be essential for determining the potency of PS-GAMP, which is consistent with their critical role in orchestrating innate and adaptive immune responses in the respiratory system during viral infection (24, 34–36).
PS-GAMP broadens protection against heterosubtypic influenza viruses
Mice that received CA09H1N1 vaccine (Fig. 6, A to F) or A/Shanghai/37T/2009 (SH09) H1N1 vaccine (Fig. 6, G and H), together with PS-GAMP, were highly protected from lethal challenges with distinct PR8 H1N1 virus and heterosubtypic A/Aichi/2/1968 (Aichi) H3N2, rgVN04 H5N1, or the highly pathogenic SH13 H7N9 virus, irrespective of whether the animals were infected at either 2 days (Fig. 6, A, C, E, and G) or 14 days (Fig. 6, B, D, F, and H) after immunization (fig. S27, A to H). This vaccination strategy also protected mice from an oseltamivir-resistant A/North Carolina/39/2009 H1N1 virus with an H275Y mutation (NC09) [in which histidine (H) at position 275 was replaced with tyrosine (Y)] (Fig. 6I and fig. S27I), which emerged during 2009 H1N1 pandemic and H7N9 epidemic (37, 38). The resistance of this virus to oseltamivir was verified by the ability of oseltamivir to effectively control CA09H1N1, but not NC09, viral infection (Fig. 6I). Under similar conditions, H1N1 vaccines alone provided little to no protection against challenges with these heterosubtypic variants (Fig. 6, A to I, and fig. S27, A to I). In contrast to PS-GAMP, poly(I:C)-adjuvanted SH09 H1N1 vaccine failed to generate significant heterosubtypic protection against H7N9 virus (Fig. 6, G and H, and fig. S27, G and H). In addition to monovalent vaccines, PS-GAMP enhanced the breadth of immune responses induced by trivalent 2018–2019 seasonal influenza vaccines (SIV18–19) against mismatched reassortant A/Guizhou/54/1989 H3N2 (rgGZ89) virus (Fig. 6J and fig. S27J) or Florida/4/2006 influenza B virus from Yamagata-lineage (fig. S28). Thus, PS-GAMP appears to be similarly effective for both influenza A and B viral vaccines.
Fig. 6. PS-GAMP broadens cross-protection against heterosubtypic influenza A viruses.
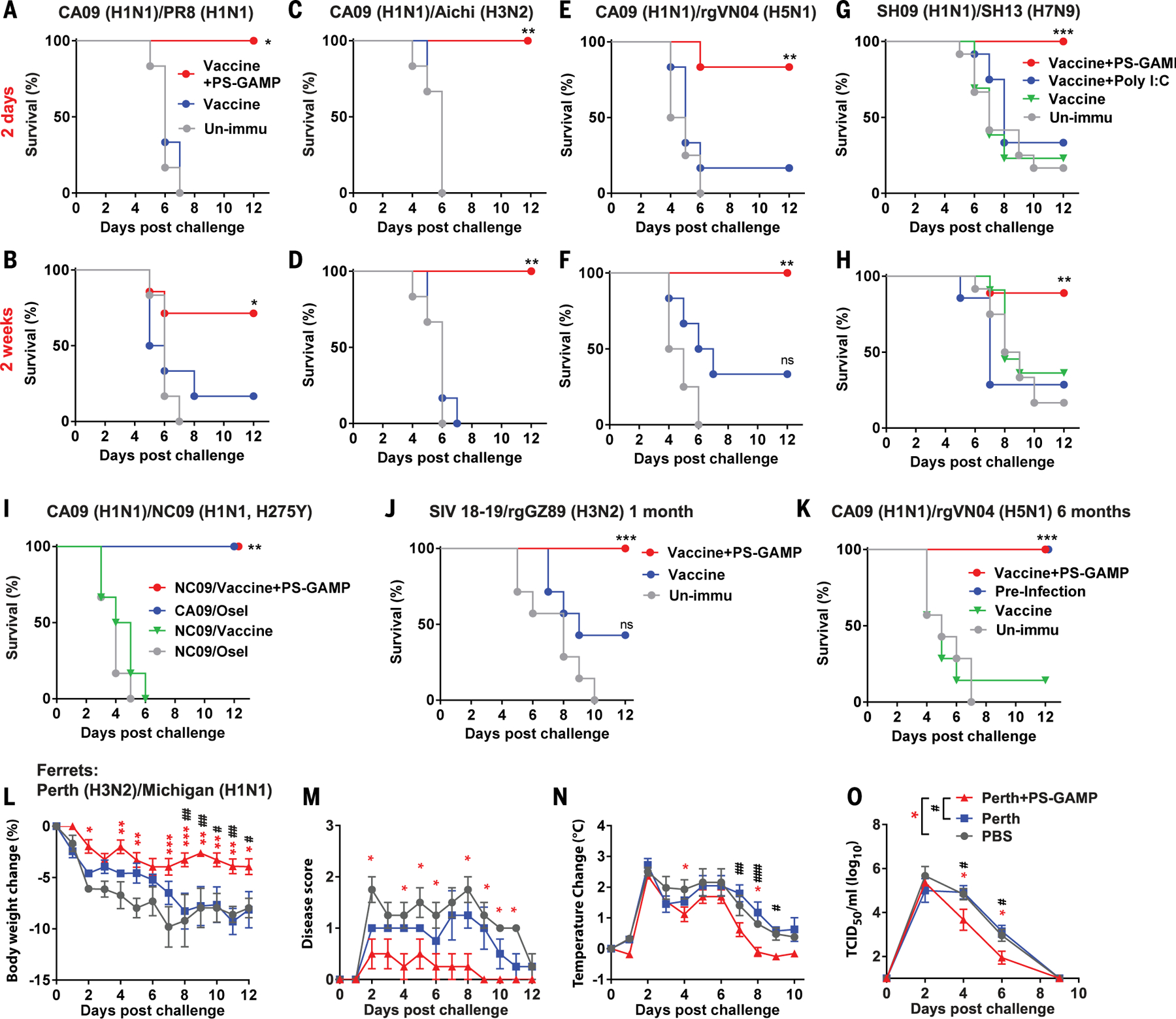
(A to H) Mice were intranasally immunized with CA09 H1N1 vaccine except for SH09 H1N1 vaccine in (G) and (H) or the vaccine plus PS-GAMP and challenged 2 days (top row) or 2 weeks (bottom row) later with 5 × LD50 distant PR8 H1N1 virus [(A) and (B)] and heterosubtypic Aichi H3N2 [(C) and (D)], rgVN04 H5N1 [(E) and (F)], or SH13 H7N9 virus [(G) and (H)]; n = 6 to 7 mice for (A) to (F) and n = 8 to 13 mice for (G) and (H). (I) Mice were immunized as in (A) and challenged 2 days later with 10 × LD50 oseltamivir-resistant NC09 H1N1 virus. Unimmunized mice were treated with oseltamivir (20 mg/kg/day) 6 hours before challenge and then daily after viral challenge until the end of the study. The treated mice were challenged with either 10 × LD50 CA09 H1N1 or NC09 H1N1 virus; n = 6 mice. (J) Mice were immunized with 2018–2019 trivalent seasonal influenza vaccine (SIV18–19) alone or alongside PS-GAMP and challenged 1 month later with 5 × LD50 mismatched GZ89 H3N2 virus; n = 6 to 12 mice. (K) Mice were immunized with CA09 H1N1 vaccine alone or together with PS-GAMP and challenged 6 months later with 5 × LD50 heterosubtypic rgVN04 H5N1 virus. Alternatively, mice were infected with 1 × LD50 PR8 H1N1 virus, and the mice that survived the infection were challenged again 6 months later with 5 × LD50 rgVN04 H5N1 virus for comparison (preinfection); n = 6 to 7 mice. (L to O) Ferrets were intranasally immunized with inactivated Perth H3N2 vaccine (15 μg) with or without PS-GAMP (200 μg). Thirty days after immunization, ferrets were challenged with 106 TCID50 heterosubtypic Michigan15 H1N1 virus. Body weight (L), disease score (M), temperature (N), and nasal wash viral titers (O) were monitored for 12 days; n = 4 ferrets. The results were presented as means ± SEM. Mice, *P < 0.05, **P < 0.01, and ***P < 0.001 compared with unimmunized mice. Experiments with mice were repeated twice with similar results. For ferrets, asterisks indicate significance between PBS and Vaccine+PS-GAMP, and pound signs (#) indicate significance between Vaccine and Vaccine+PS-GAMP. *,#P < 0.05; **,##P < 0.01, and ***,###P < 0.001. Statistical analysis, two-way ANOVA for (L), (N), and (O); Kruskal-Wallis test for (M); and the log-rank test for (A) to (K).
Long-lived Ag-specific memory CD8+ T cells capable of rapid recall upon viral infection are critical for the control of viral replication in the lung (2, 3). In mice that received OT-I cells, the number of lung CD8+CD103+CD49a+CD69+ TRM cells rose 20-fold after immunization with OVA combined with PS-GAMP relative to OVA alone (fig. S29, A to C). Moreover, PS-GAMP-adjuvanted CA09 H1N1 vaccine fully protected mice from heterosubtypic rgVN04 H5N1 viral challenge 6 months after a single immunization (Fig. 6K and fig. S27K). This long-term cross-protection concurred with durable influenza-specific CD8+ TRM cells in the lung, which could also be readily detected 6 months after immunization (fig. S29, D and E). These CD8+ TRM cells, rather than circulating memory CD8+ T cells, contributed to the long-term protection observed because their function was not compromised by T cell egress inhibitor FTY720 (fig. S30) (3).
Heterosubtypic immunity was further corroborated in ferrets by means of immunization with PS-GAMP alongside inactivated rgPerthH3N2 vaccine. The body weight and temperature of these animals did not differ from those receiving PBS or the vaccine alone, demonstrating a good safety profile for PS-GAMP in ferrets (fig. S31, A and B). The immunization induced 40-fold higher serum IgG titers and fivefold higher HAI titers against homologous Perth H3N2 virus than vaccine alone did 28 days after immunization (fig. S31, C and D), but no HAI antibody was detected against heterosubtypic A/Michigan/45/2015 H1N1 (Michigan H1N1) virus, as anticipated (fig. S31E). Ferrets receiving the vaccine and PS-GAMP, when challenged with Michigan H1N1 virus, showed significantly less body weight loss and milder clinical symptoms, especially in the late phase (>7 days) of the infection, and normalized their temperature more quickly compared with animals that received PBS or vaccine alone (Fig. 6, L to N). PS-GAMP-treated ferrets also shed significantly lower amounts of virus 2 days after infection (Fig. 6O). Thus, the ability to induce heterosubtyptic immunity in ferrets by PS-GAMP-adjuvanted inactivated influenza vaccines suggests the potential for PS-GAMP use in humans.
Discussion
The development of a universal influenza vaccine that confers protection against not only intrasubtypic variants but other subtypes of influenza viruses as well would be highly desirable. However, despite decades of extensive investigation, whether such universal influenza vaccines are achievable remains unclear. It has been long recognized in both humans and animal models that viral infection can stimulate heterosubtypic immunity primarily mediated by CD8+ T cells (2, 3, 6). In this work, a single immunization with inactivated H1N1 vaccine adjuvanted with PS-GAMP confers protection against lethal challenges with H1N1, H3N2, H5N1, or H7N9 viruses as early as 2 days after immunization. This cross-protection was sustained for at least 6 months, concurrent with durable virus-specific CD8+ TRM cells in the lung. This was largely due to the fact that PS-GAMP-adjuvant influenza vaccine simulated viral infection-induced immunity, characterized by AEC activation, rapid CD11b+ DC recruitment and differentiation, and robust CD8+ T cell responses in the respiratory system. PS-GAMP is a standalone adjuvant, compatible with not only inactivated influenza viral vaccines but also vaccines comprising cocktails of multiple B and T cell epitopes or influenza vaccine subunits. The ability of PS-GAMP to potentiate nonreplicating influenza vaccines for strong heterosubtypic immunity makes it a promising adjuvant for “universal” influenza vaccines if its efficacy can be shown in humans. As such, it would offer a substantial advantage over “replicating” vaccines.
Distinct from conventional vaccine adjuvants that target primarily APCs, PS-GAMP activated both AMs and AECs, with activation of the latter appearing to be crucial for its adjuvanticity. Pharmacological inhibition of gap junctions and Sting deficiency in AECs both diminished the adjuvanticity of this preparation considerably. By contrast, Sting deficiency in myeloid cells did not. The pivotal role played by AECs over AMs in orchestrating innate and adaptive immune responses is in agreement with what has been described during the early phase of influenza viral infection (24). The ability of cGAMP to enter AECs without breaching the PS layer was ascribed to SP-A/D-receptor-mediated endocytosis after incorporation of SP-A and SP-D into PS-biomimetic liposomes, which is not feasible in any non-PS-biomimetic liposomes (39–41). Additionally, this adjuvant could induce robust protection within just 2 days after immunization, which is in sharp contrast to current influenza vaccines, which require at least 10 to 14 days to be effective. Early cross-protection is extremely important to protect first responders and high-risk individuals, especially when antiviral drug-resistant viruses or highly pathogenic viruses such as H5N1 and H7N9 viruses emerge to become pandemics. Because viral spreading can accelerate exponentially after a transition from an epidemic to pandemic, early protection would be the most effective means to confine viral spreading during an epidemic phase and prevent pandemics, potentially saving millions of lives (42).
Materials and methods
PS-GAMP synthesis
All lipids were purchased from Avanti Polar Lipids, including 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), 1,2-dipalmitoyl-sn-glycero-3-phospho-(1’-rac-glycerol) (DPPG), 1,2-dipalmitoyl-3-trimethylammonium-propane (DPTAP), and 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DPPE-PEG2000). Cholesterol was obtained from Sigma Aldrich. The mass ratio of nano4 and nano6 was DPPC/DPPG/DPPE-PEG/Chol at 10:1:1:1. The lipids were dissolved in 3 ml of chloroform and mixed with 1 ml cGAMP solution (200μg cGAMP, 13.7 mM NaCl, 0.27 mM KCl, 0.43 mM Na2HPO4, and 0.147 mM KH2PO4). Alternatively, cGAMP was replaced with SRB (Sigma Aldrich) and/or 0.5 μmol DiD dye (Life Technologies) was added to the lipid mixture to label cargo or liposome membrane, respectively. The liposomes were synthesized by reverse-phase evaporation (43). In brief, the mixture of lipids and cGAMP was sonicated to achieve a water-in-oil emulsion under N2 for 30 min at 50°C, followed by gentle removal of the solvent via rotary evaporation at a speed of 220 rpm. An excess amount of buffer was added to the mixture and continuously rotated for another 5 min at 50°C. Resultant liposomes were extruded through 400- and 200-nm membranes (Avanti Polar Lipids) at 50°C. The size and zeta potential of liposomes were measured by Zetasizer (Malvern). Encapsulation efficiency was determined by UV absorption of cGAMP at 260 nm in Nanodrop (Life Technologies) and confirmed by liquid chromatography-mass spectrometry (LC-MS) (Agilent). Free cGAMP was removed by a size-exclusion column G-50 (GE Healthcare). To stabilize the liposomes, trehalose was added to the liposome suspension at a final concentration of 2.5%. The resultant suspension was frozen in dry ice/ethanol bath and then lyophilized at −45°C under vacuum by Freezone 4.5 (Labconco). The lyophilized liposome (PS-GAMP) was stored at −20°C until use and used in all in vivo studies unless otherwise specified.
Animals
C57BL/6J and BALB/c mice were purchased from Jackson Laboratories or Shanghai SLAC Laboratory Animal Co. Sting-deficient mice (C57BL/6J-Tmem173 gt/J), Sftpa1−/−Sftpd−/− mice (B6.Cg-Sftpa1tm2Haw Sftpdtm2Haw/J), C57BL/6 CD45.1 mice (B6.SJL-Ptprca Pepcb/BoyJ), and Swiss Webster mice were attained from Jackson Laboratories or Charles River Laboratories. MHC II-EGFP mice expressing MHC class II molecule infused into enhanced green fluorescent protein (EGFP) was a kind gift of H. Ploegh, Massachusetts Institute of Technology. Influenza-free 4-month-old female ferrets were purchased from Marshall Bio-Resources. Healthy naïve 6-year-old male rhesus macaques were obtained from Beijing Institute of Xieerxin Biology Resource, China. The animals were housed in the pathogen-free animal facilities of Massachusetts General Hospital (MGH) or Fudan University in compliance with institutional, hospital, and NIH guidelines. The studies were reviewed and approved by the MGH or Fudan University Institutional Animal Care and Use Committee.
Influenza viruses and vaccines
SH13 H7N9 virus (A/Shanghai/4664T/2013), SH09 H1N1 virus (A/Shanghai/37T/2009), and reverse-genetically (rg) modified GZ89 H3N2 (rgGZ89 H3N2) virus consisting of H3 and N2 of A/Guizhou/54/1989 H3N2 virus and A/Puerto Rico/8/1934 (PR8) viral backbone were obtained from Fudan University. Pandemic CA09 H1N1 virus was requested from the American Type Culture Collection (ATCC, #FR-201). PR8 (NR-348), A/Aichi/2/68 H3N2 (Aichi, NR-3177), rgPerth H3N2 (NR-41803), and B/Florida/4/2006 (Florida06, NR-9696) viral strains were obtained from BEI Resources, NIAID. rgVN04 H5N1 virus was a kind gift of R. Webby, St. Jude Children’s Research Hospital, which comprised H5 and N1 genes from A/Vietnam/1203/2004 H5N1 virus and a PR8 viral backbone. A/Michigan/45/2015 H1N1 (Michigan15, FR-1483) and antiviral drug-resistant A/North Carolina/39/2009 H1N1 viruses (NC09, FR-488) were acquired from International Reagent Resources, CDC. Viruses were expanded in 10-day-old embryonated chicken eggs (Charles River Laboratories) at 35°C for 3 d, harvested, purified by sucrose gradient ultracentrifugation, and frozen at −80°C. To challenge mice, the virus was adapted in mice for three cycles of intranasal instillation-lung homogenate preparation, and their infectivity in mice was assayed by the LD50 following a standard protocol.
Monovalent CA09 H1N1 vaccine (NR-20347, Sanofi Pasteur) and whole inactivated H5N1 vaccine (NR-12148, Baxter AG) were obtained from BEI Resources, NIAID. H7-Re1 H7N9 whole inactivated vaccine was a kind gift from Harbin Veterinary Research Institute, Chinese Academy of Agricultural Sciences. Trivalent seasonal influenza vaccine 2018–2019 (SIV 18–19) was attained from Hualan Biological Bacterin Co., China. SH09 H1N1 and Perth H3N2 inactivated vaccines were made by inactivation of the viruses with 0.02% formalin for 24 hours at 37°C and purified as above. Ag concentration was quantified by the BCA protein assay and SDS-polyacrylamide gel electrophoresis based on HA content.
Mouse immunizations and challenges
Mice were sedated with ketamine/xylazine and intranasally inoculated with 30 μl (15 μl per nostril) of an indicated influenza vaccine or a mixture of the vaccine and an adjuvant. VN04 H5N1, SIV 18–19, and CA09 H1N1 SV vaccines were employed at a corresponding dose of 1 μg (HA content), 1 μg, or 0.5 μg per mouse, respectively, whereas H7-Re1 and SH09 H1N1 vaccines each were administered at 0.25 μg or 3 μg per dose, respectively. Poly(I:C) (Invivogen), Pam2CSK4 (Invivogen), and cholera toxin (Sigma) each were administered at 20, 20, or 10 μg per mouse, respectively. To block gap junctions, CBX, tonabersat, and meclofenamate were obtained from Sigma Aldrich and intraperitoneally injected into individual mice for 4 consecutive days (from 2 days prior to 1 day after immunization) at corresponding dosages of 25, 10, or 20 mg/kg/day, respectively (31, 32). To deplete CD8+ T cells during vaccination and challenge, mice were administered anti-CD8α (53–6.7, BioLegend) antibody 2 days prior and in 0, 2, and 4 days after immunization at a dose of 200 μg/day. C57BL/6 mice were used for the challenge studies, except for Aichi H3N2, Florida06 influenza B, and GZ89 viruses which challenged Swiss Webster mice or BALB/c mice instead unless otherwise indicated, because C57BL/6 mice were relatively less susceptible to these viruses. To verify antiviral drug resistance of the NC09 virus, unimmunized mice were treated with oseltamivir (20 mg/kg/day) at 6 hours before the challenge and then daily until the end of the study. Immunized and control mice were challenged with intranasal instillation of 10 × LD50 mouse-adapted homologous virus at an indicated day after immunization, except for H7N9 virus at 40 × LD50. However, heterologous viruses each at 5 × LD50 were utilized for challenges except for Florida06 influenza B virus at a dose of 4 ×105 median tissue culture infectious dose (TCID50) as this virus is not lethal to mice. Body weight and survival were monitored daily for 12 days after the challenge.
Ferret immunizations and challenges
Four-month-old female ferrets negative to antiinfluenza virus antibody were anesthetized by ketamine/xylazine/atropine and intranasally immunized with a vehicle, an influenza vaccine, or a mixture of the vaccine and PS-GAMP. To assay early protection, each ferret receiving 9 μg of CA09 H1N1 vaccine alone or alongside 200 μg of PS-GAMP was challenged with 106 TCID50 CA09 H1N1 viruses 2 days post-immunization. To evaluate cross-protection, each ferret was intranasally immunized with 15 μg of PerthH3N2 vaccine in the presence or absence of 200 μg of PS-GAMP and challenged with 106 TCID50 heterosubtypic Michigan15 H1N1 viruses 30 days after immunization. Body temperature was monitored by two microchips implanted in each animal (BioMedic Data Systems) and clinical symptoms were scored according to a published protocol (table S1) (44). Animals were euthanized humanely 2 weeks after viral challenge by sedation and injection of 0.5 ml of Euthanasia-III into the heart.
Tissue processing and flow cytometry
Lungs, nasal tissues, MLNs, and spleens were dissected from indicated mice and processed into single-cell suspensions for analysis by flow cytometry. Specifically, the lung and nasal tissues were minced into 1-mm2 pieces, digested with 1 ml of collagenase D (2 mg/ml)/DNase I (5 mg/ml), both from Roche, at 37°C for 60 min, and then passed through 40-μm cell strainers (18). To collect BALF, mice were first perfused thoroughly with ice-cold PBS followed by intratracheal lavage with 0.5% BSA in PBS. Single-cell suspensions of the spleen and MLN were prepared by passing the tissues through 40-μm cell strainers directly. After removal of red blood cells in ACK buffer, the remaining cells were washed, blocked by anti-CD16/CD32 antibody (clone 93,10 μg/ml, BioLegend) for 20 min, and stained with fluorescently conjugated antibodies for 30 min on ice or NP366–374, PA224–233, PB1703–711 MHC I tetramers for 1 hour on ice. Activated T cells were fixed and permeabilized after surface staining, followed by intracellular staining with anti-granzyme B antibody at 4°C overnight. Stained cells were acquired on a FACSAria II (BD) and analyzed using FlowJo software (Tree Star). Cell populations and subsets in the mouse respiratory system were gated and analyzed as described (18). The information of various antibodies is provided in table S2.
Cytokine and chemokine measurements
C57BL/6 mice were intranasally administered 20 μg of PS-GAMP or infected with 1 × LD50 CA09 H1N1 virus. Lungs were harvested at indicated times and prepared for total RNA extraction with an RNA purification kit (Roche). To measure cytokines in brains, mice were intranasally administered VN04 H5N1 vaccine (1 μg HA) alone or together with PS-GAMP (20 μg) or CT (10 μg) and sacrificed 48 hours later to collect the brain tissue for RNA extraction as above. The RNA was reverse-transcribed (Life technologies) and amplified by real-time PCR using an SYBR Green PCR kit (Roche). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) served as an internal control. All primers used are listed in table S3. Mouse GM-CSF (eBioscience), IFN-β (Invivogen), TNF-α (BioLegend), IFN-γ (eBioscience), IL-6 (eBioscience), and IL-10 (BioLegend) levels in BALF and serum were measured by specific ELISA kits.
Histology
Swiss Webster mice were intranasally administered PBS, PS-GAMP (20 μg), H5N1 vaccine (1 μg HA), or the vaccine plus PS-GAMP or CT (5 μg). Some mice were infected by CA09 H1N1 virus (250 PFU) as positive controls. Lungs, nasal tissue, and brains were dissected at indicated days after immunization or infection, fixed, and stained using a standard H&E procedure. The slides were scanned and analyzed using a NanoZoomer (Hamamatsu).
Confocal microscopy
To track DiD-labeled liposomes in the lung, C57BL/6 mice were intranasally administered an equal amount of DiD-nano4 or DiD-nano5. Lungs were excised after 12 hours, embedded in an optimal cutting temperature (OCT) compound (Sakura Finetek), and cut into 5-μm frozen sections. The slides were mounted with a ProLong Antifade Mountant containing DAPI (Life Technologies) and imaged by confocal microscopy (Olympus FV1000, UPLSAPO 60XW). To visualize AM uptake of nanoparticles ex vivo, mouse lungs were lavaged six times with 1 ml of PBS containing 0.5% BSA and 5 mM EDTA. The lung lavage was pooled and centrifuged at 220 × g. The cells were collected, washed thoroughly by PBS, and cultured in RPMI 1640 medium for 45 min, followed by removal of nonadherent cells. The adherent cells were collected as AMs, suspended at 2 × 105 cells/ml in medium, and added to 96-well-plates at 200 μl/well. To purify PS, lung lavage was prepared by washing the lung for six times with 1 ml of PBS and centrifuged at 220 × g for 10 min to remove cell debris and then at 100,000 × g for 1 hour to pellet PS. The supernatant (6 ml) was concentrated to 200 μl by 3-kDa Amicon Ultra Centrifugal Filter Units (Merk Millipore) and mixed with PS pellet prepared above. The resultant PS (100 μg total protein) was then mixed with DiD-nano4 or DiD-nano5 (12 μg lipid content in nanoparticles) for 30 min before added to AM cell culture with 4 × 104 cells in 200 μl of medium. After 4 hours incubation under 5% CO2 at 37°C, cells were stained with a vital dye Calcein-AM (Life Technologies). Uptake of liposomes was quantified by confocal microscopy (Olympus FV1000, UPLSAPO 60XW) followed by ImageJ software analysis.
Statistical analysis
A two-tailed Student’s t test was used to analyze differences between two groups. analysis of variance (ANOVA) or Kruskal-Wallis test was used to analyze differences among multiple groups by PRISM software (GraphPad). A P value of <0.05 was considered statistically significant. Sample sizes were determined on the basis of preliminary experiments to give a statistical power of 0.8. Most experiments were repeated at least twice with similar results. The investigators were not blinded to the experiments which were carried out under highly standardized and predefined conditions, except for microscopy images and H&E slide examinations, which were evaluated in an investigator-blind manner.
Supplementary Material
ACKNOWLEDGMENTS
We thank R. J. Webby at St. Jude Children’s Research Hospital for rgVN04 H5N1 virus and D. J. Irvine at MIT for consultation of liposome preparation; M. Hogan and S. Andrea at MGH CCM for their assistance in ferret study; Q. Wang at FDU SHMC for technical support; J. H. Wu, M. Ishii, and L. Leanse for editing; and staff in Wellman photopathology core for histology and flow cytometry. We are grateful to BEI Resources, NIH, and International Reagent Resources, CDC, for their generous support in providing various influenza viruses and vaccines.
Funding: This work is supported by NIH grants AI089779, AI070785, and AI097696; MGH ECOR 230002; and a department fund to M.X.W.; by China National Megaprojects for Major Infectious Diseases 2018ZX10301403 and National Natural Science Foundation 81822045 to L.L.; and by Hundred-Talents program of Sun Yat-sen University to J.W.
Footnotes
Competing interests: M.X.W. has filed a provisional patent application entitled “Pulmonary surfactant-mimetic liposomes for safe delivery of small molecules for therapeutics and prophylaxis of respiratory diseases or infections.” The authors declare that they have no other competing interests. S.J. is affiliated with Lindsley F. Kimball Research Institute, New York Blood Center, New York, NY 10065, USA, and P.L. is affiliated with the Key Laboratory of Endogenous Infection, Shenzhen Nanshan People’s Hospital, Guangdong Medical University, Shenzhen 518052, China.
Data and material availability: All data are available in the main text or the supplementary materials.
SUPPLEMENTARY MATERIALS
science.sciencemag.org/content/367/6480/eaau0810/suppl/DC1
Materials and Methods
Figs. S1 to S31
Tables S1 to S3
References (45–49)
REFERENCES AND NOTES
- 1.Paules CI, Sullivan SG, Subbarao K, Fauci AS, Chasing seasonal influenza-The need for a universal influenza vaccine. N. Engl. J. Med 378, 7–9 (2018). doi: 10.1056/NEJMp1714916; [DOI] [PubMed] [Google Scholar]
- 2.Pizzolla A et al. , Influenza-specific lung-resident memory T cells are proliferative and polyfunctional and maintain diverse TCR profiles. J. Clin. Invest 128, 721–733 (2018). doi: 10.1172/JCI96957; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zens KD, Chen JK, Farber DL, Vaccine-generated lung tissue-resident memory T cells provide heterosubtypic protection to influenza infection. JCI Insight 1, 85832 (2016). doi: 10.1172/jci.insight.85832; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sridhar S et al. , Cellular immune correlates of protection against symptomatic pandemic influenza. Nat. Med 19, 1305–1312 (2013). doi: 10.1038/nm.3350; [DOI] [PubMed] [Google Scholar]
- 5.Weinfurter JT et al. , Cross-reactive T cells are involved in rapid clearance of 2009 pandemic H1N1 influenza virus in nonhuman primates. PLOS Pathog. 7, e1002381 (2011). doi: 10.1371/journal.ppat.1002381; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koutsakos M et al. , Human CD8+ T cell cross-reactivity across influenza A, B and C viruses. Nat. Immunol 20, 613–625 (2019). doi: 10.1038/s41590-019-0320-6; [DOI] [PubMed] [Google Scholar]
- 7.Si L et al. , Generation of influenza A viruses as live but replication-incompetent virus vaccines. Science 354, 1170–1173 (2016). doi: 10.1126/science.aah5869; [DOI] [PubMed] [Google Scholar]
- 8.Wang L et al. , Generation of a live attenuated influenza vaccine that elicits broad protection in mice and ferrets. Cell Host Microbe 21, 334–343 (2017). doi: 10.1016/j.chom.2017.02.007; [DOI] [PubMed] [Google Scholar]
- 9.Hoft DF et al. , Live and inactivated influenza vaccines induce similar humoral responses, but only live vaccines induce diverse T-cell responses in young children. J. Infect. Dis 204, 845–853 (2011). doi: 10.1093/infdis/jir436; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ablasser A et al. , cGAS produces a 2′−5′-linked cyclic dinucleotide second messenger that activates STING. Nature 498, 380–384 (2013). doi: 10.1038/nature12306; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu J et al. , Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339, 826–830 (2013). doi: 10.1126/science.1229963; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li XD et al. , Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science 341, 1390–1394 (2013). doi: 10.1126/science.1244040; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang J, Li P, Wu MX, Natural STING Agonist as an “Ideal” adjuvant for cutaneous vaccination. J. Invest. Dermatol 136, 2183–2191 (2016). doi: 10.1016/j.jid.2016.05.105; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Corrales L et al. , Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell Reports 11, 1018–1030 (2015). doi: 10.1016/j.celrep.2015.04.031; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Whitsett JA, Wert SE, Weaver TE, Alveolar surfactant homeostasis and the pathogenesis of pulmonary disease. Annu. Rev. Med 61, 105–119 (2010). doi: 10.1146/annurev.med.60.041807.123500; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Woodrow KA, Bennett KM, Lo DD, Mucosal vaccine design and delivery. Annu. Rev. Biomed. Eng 14, 17–46 (2012). doi: 10.1146/annurev-bioeng-071811-150054; [DOI] [PubMed] [Google Scholar]
- 17.Parra E, Pérez-Gil J, Composition, structure and mechanical properties define performance of pulmonary surfactant membranes and films. Chem. Phys. Lipids 185, 153–175 (2015). doi: 10.1016/j.chemphyslip.2014.09.002; [DOI] [PubMed] [Google Scholar]
- 18.Misharin AV, Morales-Nebreda L, Mutlu GM, Budinger GR, Perlman H, Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am. J. Respir. Cell Mol. Biol 49, 503–510 (2013). doi: 10.1165/rcmb.2013-0086MA; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qin H, Wilson CA, Lee SJ, Zhao X, Benveniste EN, LPS induces CD40 gene expression through the activation of NF-kB and STAT-1a in macrophages and microglia. Blood 106, 3114–3122 (2005). doi: 10.1182/blood-2005-02-0759; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haczku A, Protective role of the lung collectins surfactant protein A and surfactant protein D in airway inflammation. J. Allergy Clin. Immunol 122, 861–881 (2008). doi: 10.1016/j.jaci.2008.10.014; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang J, Li B, Wu MX, Effective and lesion-free cutaneous influenza vaccination. Proc. Natl. Acad. Sci. U.S.A 112, 5005–5010 (2015). doi: 10.1073/pnas.1500408112; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang J, Shah D, Chen X, Anderson RR, Wu MX, A micro-sterile inflammation array as an adjuvant for influenza vaccines. Nat. Commun 5, 4447 (2014). doi: 10.1038/ncomms5447; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ballesteros-Tato A, León B, Lund FE, Randall TD, Temporal changes in dendritic cell subsets, cross-priming and costimulation via CD70 control CD8+ T cell responses to influenza. Nat. Immunol 11, 216–224 (2010). doi: 10.1038/ni.1838; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Unkel B et al. , Alveolar epithelial cells orchestrate DC function in murine viral pneumonia. J. Clin. Invest 122, 3652–3664 (2012). doi: 10.1172/JCI62139; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sullivan JS et al. , Heterosubtypic anti-avian H5N1 influenza antibodies in intravenous immunoglobulins from globally separate populations protect against H5N1 infection in cell culture. J. Mol. Genet. Med 3, 217–224 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lawrence CW, Ream RM, Braciale TJ, Frequency, specificity, and sites of expansion of CD8+ T cells during primary pulmonary influenza virus infection. J. Immunol 174, 5332–5340 (2005). doi: 10.4049/jimmunol.174.9.5332; [DOI] [PubMed] [Google Scholar]
- 27.Ichinohe T et al. , Synthetic double-stranded RNA poly(I:C) combined with mucosal vaccine protects against influenza virus infection. J. Virol 79, 2910–2919 (2005). doi: 10.1128/JVI.79.5.2910-2919.2005; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kang JY et al. , Recognition of lipopeptide patterns by Toll-like receptor 2-Toll-like receptor 6 heterodimer. Immunity 31, 873–884 (2009). doi: 10.1016/j.immuni.2009.09.018; [DOI] [PubMed] [Google Scholar]
- 29.Ablasser A et al. , Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature 503, 530–534 (2013). doi: 10.1038/nature12640; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Westphalen K et al. , Sessile alveolar macrophages communicate with alveolar epithelium to modulate immunity. Nature 506, 503–506 (2014). doi: 10.1038/nature12902; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Furlow PW et al. , Mechanosensitive pannexin-1 channels mediate microvascular metastatic cell survival. Nat. Cell Biol 17, 943–952 (2015). doi: 10.1038/ncb3194; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen Q et al. , Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 533, 493–498 (2016). doi: 10.1038/nature18268; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ramsey H et al. , Stress-induced hematopoietic failure in the absence of immediate early response gene X-1 (IEX-1, IER3). Haematologica 99, 282–291 (2014). doi: 10.3324/haematol.2013.092452; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fritz JH, Le Bourhis L, Magalhaes JG, Philpott DJ, Innate immune recognition at the epithelial barrier drives adaptive immunity: APCs take the back seat. Trends Immunol. 29, 41–49 (2008). doi: 10.1016/j.it.2007.10.002; [DOI] [PubMed] [Google Scholar]
- 35.Saenz SA, Taylor BC, Artis D, Welcome to the neighborhood: Epithelial cell-derived cytokines license innate and adaptive immune responses at mucosal sites. Immunol. Rev 226, 172–190 (2008). doi: 10.1111/j.1600-065X.2008.00713.x; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Whitsett JA, Alenghat T, Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat. Immunol 16, 27–35 (2015). doi: 10.1038/ni.3045; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hai R et al. , Influenza A(H7N9) virus gains neuraminidase inhibitor resistance without loss of in vivo virulence or transmissibility. Nat. Commun 4, 2854 (2013). doi: 10.1038/ncomms3854; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moore C et al. , Evidence of person-to-person transmission of oseltamivir-resistant pandemic influenza A(H1N1) 2009 virus in a hematology unit. J. Infect. Dis 203, 18–24 (2011). doi: 10.1093/infdis/jiq007; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hidalgo A, Cruz A, Pérez-Gil J, Barrier or carrier? Pulmonary surfactant and drug delivery. Eur. J. Pharm. Biopharm 95 (Pt A), 117–127 (2015). doi: 10.1016/j.ejpb.2015.02.014; [DOI] [PubMed] [Google Scholar]
- 40.Olsson B et al. , in Controlled Pulmonary Drug Delivery, Hugh AJH Smyth DC, Ed. (Springer-Verlag, 2011), pp. 21–50. [Google Scholar]
- 41.Sung JC, Pulliam BL, Edwards DA, Nanoparticles for drug delivery to the lungs. Trends Biotechnol. 25, 563–570 (2007). doi: 10.1016/j.tibtech.2007.09.005; [DOI] [PubMed] [Google Scholar]
- 42.Bakken IJ et al. , Febrile seizures after 2009 influenza A (H1N1) vaccination and infection: A nationwide registry-based study. BMC Infect. Dis 15, 506 (2015). doi: 10.1186/s12879-015-1263-7; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Szoka F Jr., Papahadjopoulos D, Procedure for preparation of liposomes with large internal aqueous space and high capture by reverse-phase evaporation. Proc. Natl. Acad. Sci. U.S.A 75, 4194–4198 (1978). doi: 10.1073/pnas.75.9.4194; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Matsuoka Y, Lamirande EW, Subbarao K, The ferret model for influenza. Curr. Protoc. Microbiol 13, 15G.2.1–15G.2.29 (2009). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.