

Abstract
The skull bones must grow in a coordinated, three-dimensional manner to coalesce and form the head and face. Mammalian skull bones have a dual embryonic origin from cranial neural crest cells (CNCC) and paraxial mesoderm (PM) and ossify through intramembranous ossification. The calvarial bones, the bones of the cranium which cover the brain, are derived from the supraorbital arch (SOA) region mesenchyme. The SOA is the site of frontal and parietal bone morphogenesis and primary center of ossification. The objective of this review is to frame our current in vivo understanding of the morphogenesis of the calvarial bones and the gene networks regulating calvarial bone initiation in the SOA mesenchyme.
Keywords: skull bone, gene regulation, supraorbital arch mesenchyme, bone initiation
Introduction:
Skull bone development involves the formation of complex, three-dimensional structures in a spatially and temporally sensitive manner. The formation of the skull requires the convergence of multiple bones of complex shapes and sizes into a single unit. Cell fate selection, differentiation, and patterning of each bone occurs within close proximity to one another. In addition, two different stem cell progenitor populations contribute to the skull bones. Thus, spatial and temporal genetic regulation is required to coordinate early events in skull bone morphogenesis to ensure normal skull bone formation. Here, we will primarily highlight the morphogenesis of the mammalian calvaria and the genetic and epigenetic mechanisms required for calvarial bone initiation in vivo.
Characteristics of skull bone development:
In the context of bone formation, three traits characterize the development of mammalian skull bones. First, the skull bones are derived from two distinct cranial mesenchymal stem cell (CM) populations; the cranial neural crest cells (CNCC) and the paraxial head mesoderm (PM) (Fan et al., 2016; Hanken and Thorogood, 1993; Jiang et al., 2002; Yoshida et al., 2008). The CNCC is derived from the embryonic ectoderm layer and gives rise to the anterior skull bones. The PM, in contrast, is derived from the embryonic mesoderm germ layer and gives rise to the more posterior skull bones (Jiang et al., 2002; Yoshida et al., 2008) (Figure 1). Both the CNCC- and the PM-derived bones differ in their signaling requirements and timing of development, but ultimately coalesce and contribute to the cohesive structure of the skull (see below). The generation of tissue-origin restricted in vivo animal models are beginning to tease apart the developmental differences between the two stem cell populations (Fan et al., 2016). Second, the skull bones ossify through intramembranous ossification, in which the post-migratory CNCC- and PM-derived mesenchyme give rise to bone by direct ossification. Unlike endochondral ossification, which requires a cartilage template, the skull bones form from condensed mesenchyme that differentiate to mature osteoblasts and form mineralized bone (Dunlop and Hall, 1995; Hall and Miyake, 1992). Thus SRY-box 9 (Sox9), the key determinant of cartilage formation which is required in endochondral ossification, is dispensable for skull bone formation (Bi et al., 1999; Mori-Akiyama et al., 2003). Third, the skull bones must differentiate and ossify in a unique environment within close proximity to other CNCC- and PM- derived tissues such as the dermis, meninges, sutures, and muscle (Goodnough et al., 2014; Kasner et al., 2013; Michailovici et al., 2015; Tran et al., 2010). The simultaneous differentiation of tissues within close proximity to one another poses developmental and genetic constraints for establishing the calvarial bone progenitors. Thus, many of the complex genetic questions pertaining to mammalian skull bone formation, such as tissue-tissue interaction and intramembranous ossification, are difficult to address using in vitro models. Conditional mouse genetics has allowed us to define some of the core set of transcription factors and signaling pathways required during skull bone initiation, and reveal spatial and temporal genetic mechanisms governing the establishment of the skull bone primordia. Disruptions in these genetic pathways can lead to defects in cell fate selection, ossification, and congenital skull bone defects (Bhatt et al., 2013; Fan et al., 2016; Morriss-Kay and Wilkie, 2005; Rice, 2005; Twigg and Wilkie, 2015).
Figure 1: The bones of the mammalian skull are derived from two different stem cell populations.
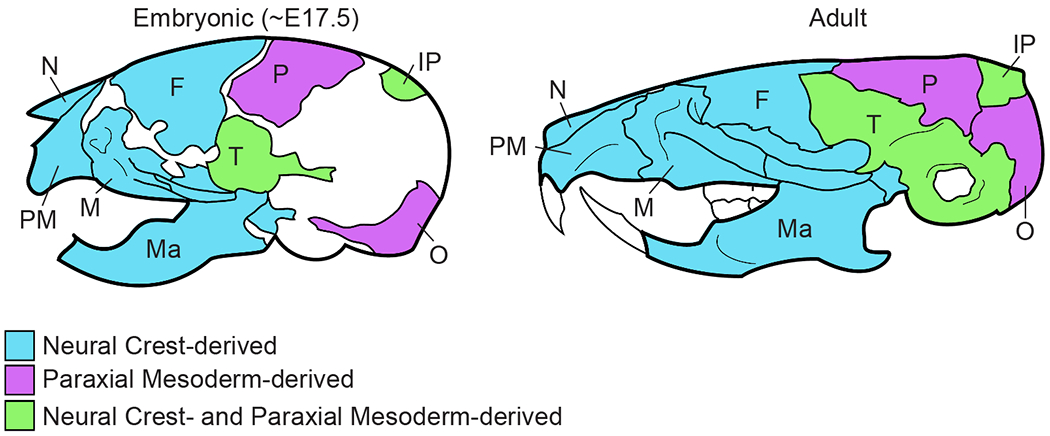
The anterior bones are derived from the CNCC and the posterior bones are derived from the PM. The temporal and parietal bones have contributions from both stem cell populations. N = nasal bone; M = maxilla; PM = premaxilla, Ma = mandible; F = frontal bone; P = parietal bone; IP = interparietal bone; T = temporal bone; O = occipital bone
The dual origin of the skull bones begins with the morphogenesis of the primordia, and we focus on the supraorbital arch (SOA) region which serves as the “organizer” for the calvarial bones (upper part of the skull). Most of the published studies have focused on understanding how the mesenchyme in the SOA (SOA-mesenchyme) is patterned and the signals required for specification of the mammalian frontal and parietal bone progenitors and the other lineages in the SOA (see below).
Embryonic development of the calvaria: morphogenesis.
The origin and site of specification of the mammalian calvarial bone precursors were identified by lineage analysis using lipophilic dye labeling and genetic lineage tracing (Jiang et al., 2002; Morriss-Kay and Wilkie, 2005; Roybal et al., 2010; Tran et al., 2010; Yoshida et al., 2008). Between E8.0-9.5, the CNCC-derived mesenchyme migrate from the mid-hindbrain region beneath the surface ectoderm to the anterior SOA region and give rise to the frontal bone precursors (Morriss-Kay and Wilkie, 2005; Yoshida et al., 2008). The PM from the same region migrates to the posterior SOA to give rise to the parietal bone precursors (Figure 2a,b). The SOA-mesenchyme represents the site of specification and the location of mineralization initiation for the frontal and parietal bones of the calvaria. By E10.5, patterning of the SOA-mesenchyme is established with the calvarial bone precursors located between the meningeal mesenchyme medially and the dermal progenitors laterally under the surface ectoderm (Figure 2b). Between E10.5-12.5, the calvarial bone progenitors in the SOA are specified and express bone lineage-specific markers to form the frontal bone and parietal bone primordia (Figure 2c) (Deckelbaum et al., 2012; Han et al., 2007; Musy et al., 2018; Tran et al., 2010; Yoshida et al., 2008). The mesenchyme apical to the SOA is termed “early migrating mesenchyme” (EMM) and does not contribute to the calvarial bone primordia and does not ossify (Cesario et al., 2018; Roybal et al., 2010).
Figure 2: Origins and morphogenesis of the mammalian calvarial bones.
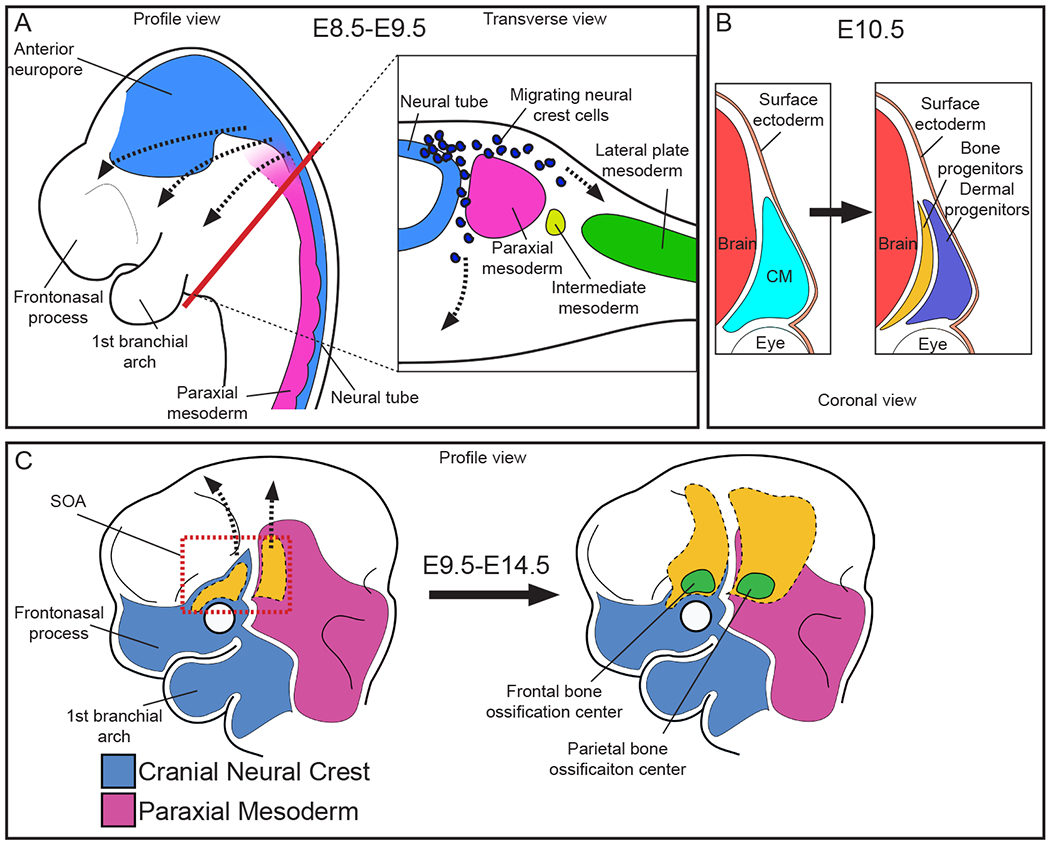
(A) The CNCC migrate from the neural tube to populate the anterior region of the head and face. The paraxial mesoderm flanks the neural tube and populates the posterior region of the head and face. In the head and face, both the CNCC and the PM are considered CM. (B) The CM in the SOA (SOA-mesenchyme) is located between the surface ectoderm and the brain, and gives rise to both the bone and dermal progenitors. (C) As the bone and dermal progenitors develop and differentiate, they grow apically over the brain.
Until recently, the identity and source of the “osteo-inductive” signals for mammalian calvarial bone were not clear in vivo. However, functional mouse genetic experiments have offered some clues. Conditional mutants of well-known signaling pathways of endochondral bone development, such as BMP, FGF, and Wnts, demonstrate that they are required for development and morphogenesis of the skull bones, but they primarily function downstream of the inductive patterning event (reviewed in Fan et al., 2016; Bhatt et al., 1993). However, the Wnt signaling pathway and its effectors, such as TWIST1, are required for calvarial bone fate; thereby qualifying Wnts as a candidate osteo-inductive signal (Day et al., 2005; Goodnough et al., 2012, 2014, 2016; Hill et al., 2005; Tran et al., 2010).
As the bone initiation program (see below) progresses between E9.5 and E13.5, both the CNCC- and PM- derived cranial bone progenitors in the SOA must condense, proliferate, and expand in the baso-apical direction (Figure 2c). Between E10.5-12.5 transcription factors, such as the forkhead domain-containing Fox family and basic-helix-loop-helix transcription factor Twist1, are also expressed throughout the SOA-mesenchyme and are required for mesenchyme condensation and initiation of calvarial bone primordia (Bildsoe et al., 2009; Goodnough et al., 2012; Kume et al., 1998; Sun et al., 2013; Vivatbutsiri et al., 2008). Temporally-inducible knockouts of Twist1 with UbcCre-ERT2 demonstrate the importance of condensation of the SOA-mesenchyme in the morphogenesis of calvarial bones (Fan et al., 2016). Inducible genetic lineage tracing of the SOA-mesenchyme between E10.0-11.5, resulted in the identification of lineage descendants of the SOA-mesenchyme in the apex of the frontal and parietal bones (Deckelbaum et al., 2012; Tran et al., 2010). These experiments are supported by more recent findings that non SOA-mesenchyme cannot be recruited to the calvarial bone progenitor pool (Cesario et al., 2018). After the onset of mineralization in the SOA at E14.0-14.5, frontal and parietal bones continue to grow apically and eventually meet to form the coronal, frontal, and sagittal sutures (Deckelbaum et al., 2012; Goodnough et al., 2012; Kaufman, 1992; Tran et al., 2010; Yoshida et al., 2008). These results together suggest that calvarial bones grow in the baso-apical direction intrinsically and the mechanisms underlying the cellular and tissue-level movements remain to be discovered (Figure 2c).
Transcriptional profiling experiments have identified several factors that are differentially expressed between the CNCC- and the PM-derived mesenchyme in the E9.5 head, highlighting regional signaling differences between the two stem cell populations (Fan et al., 2016). In the next sections, we will focus on the in vivo genetic studies identifying a core set of transcription factors and signaling pathways involved in cranial bone initiation, and how the different genetic landscapes of the CNCC- and PM-derived mesenchyme impact skull bone formation.
The skull bone initiation program: transcription factor cascade for establishing bone progenitors
In mice, the first steps in establishing the skeletal progenitors that will differentiate and ossify into the skull bones occur between E10.5 to E13.5 with the emergence of mineralized ossified bone primordia after E14.0 (Han et al., 2007). The current in vivo data examining the initial establishment of the bone stem cells can be broken down into three tiers; the transcription factors required for the establishment of bone, signaling pathways regulating these factors, and the epigenetic mechanisms contributing to the spatial and temporal coordination of these processes. The transcription factor cascade required for the establishment of skull bone progenitors, which we will refer to as the “bone initiation program”, involves three primary factors; Msh Homeobox 1 (Msx1) and 2 (Msx2), Runt Related Transcription Factor 2 (Runx2), and Osterix (Osx/Sp7) (Baek et al., 2014; Han et al., 2007; Ishii et al., 2003; Nakashima et al., 2002; Nishio et al., 2006).
The activation of the bone initiation program can be monitored with the CNCC- and PM-derived bone precursors expressing Msx1 and Msx2. In the SOA-mesenchyme Msx1 and 2 are expressed broadly at E10.5 and become progressively restricted to calvarial bone progenitors at E12.5 (Holland, 1991). In vivo, Msx1 or Msx2 null mutants, or compound Msx1−/− and Msx2+/− mutants, lead to defects in skull bone patterning and development (Han et al., 2007; Roybal et al., 2010; Satokata and Maas, 1994; Satokata et al., 2000). However, these mutants do not result in a complete loss of skull bones, demonstrating some level of functional redundancy between the two Msx genes. Compound Msx1−/−; Msx2−/− mutants and the inducible deletion of Msx1 and Msx2 in all cells using CaggCreER at E9.5 results in severe calvarial bone agenesis (Han et al., 2007; Roybal et al., 2010). In contrast, inducible deletion of Msx1 and Msx2 at E13.5 using CaggCreER results in little effect on the calvarial bones (Roybal et al., 2010). These functional mouse genetics studies highlight Msx1 and Msx2 as the first factors of the bone initiation program and demonstrate their temporal requirement for early events in bone formation.
The next key step in the program is the expression of Runx2, a transcription factor and a key determinant of bone fate selection required to establish the bone progenitors (Ducy et al., 1997; Komori et al., 1997; Otto et al., 1997). Deletion of Runx2 in mice results in a complete loss of bone throughout the entire body (Komori et al., 1997). Msx1 and Msx2 expression is required for the expression of Runx2 mRNA, as Msx1−/−; Msx2−/− compound mutants lack Runx2 expression in the SOA-mesenchyme (Baek et al., 2014; Han et al., 2007; Nakashima et al., 2002; Nishio et al., 2006). Similar to long bones, RUNX2 protein expression decreases as the calvarial bone matures, further demonstrating its role primarily in early bone development (Kacena and Ciovacco, 2010).
Downstream of RUNX2 is Osx, a zinc finger-containing transcription factor required for intramembranous ossification (Baek et al., 2014; Nishio et al., 2006). Unlike the Runx2 null mice, genetic deletion of Osx leads to a complete loss of ossified intramembranous bone specifically in the head and face. (Fan et al., 2016; Nakashima et al., 2002). In vitro, RUNX2 can bind to the Osx promoter in the 10T1/2 mesenchyme progenitor cell line; however, their interaction in vivo has not been demonstrated (Nishio et al., 2006). Indeed, Runx2 null mice do not express Osx, but further experiments are required to determine if RUNX2 can trans-activate Osx in the skull bones.
Both the CNCC- and PM-derived bones use the bone initiation program, with some spatio-temporal differences. In mice, the bone initiation program is activated in the CNCC-derived frontal bone primordia roughly 24 hours prior to the PM-derived parietal bone primordia as indicated by the expression of Msx1 and Msx2. At E10.5, the Msx1 and Msx2 expression can be observed in the frontal bone primordia with ossified bone detectable at E14.0. In contrast, the PM begins to express Msx2 roughly at E11.5 with ossified bone being detectable by E14.5 (Deckelbaum et al., 2012; Han et al., 2007). These results show the regional differences between the CNCC- and the PM-derived calvarial bones and also suggests a difference in developmental timing of early events in bone initiation. In addition, conditional mutants in the CNCC using Wnt1Cre have revealed a level of communication between multiple tissues. Conditional deletion of genes such as Twist1 or Transforming Growth Factor Receptor β II (TGFβRII), or the FGF8 regulator, Specificity Protein 8 (Sp8), in the CNCC using Wnt1Cre results in arrest of both frontal and parietal bone differentiation (Bildsoe et al., 2009; Ito et al., 2003; Kasberg et al., 2013; Sasaki et al., 2006; Yang et al., 2010). Conversely, expression of constitutively-active Fibroblast Growth Factor Receptor 2 (Fgfr2) in the PM does not affect the growth of the parietal bone, but leads to a truncation of the frontal bone and craniosynostosis by an unknown mechanism (Holmes and Basilico, 2012). These phenotypes demonstrate the potential cross-talk between the adjacent tissues during skull bone formation. As more cell-type restricted genetic tools become available, new insights will clarify the interdependence of the neighboring lineages in calvarial bone development.
Signaling factors regulating the calvarial bone initiation program
Many signaling pathways are required during various stages of calvarial bone development and have been reviewed in greater detail previously (Gou et al., 2015; Ishii et al., 2015; Karsenty, 2008; Liu and Lee, 2013; Szabo-Rogers et al., 2010). In the context of the bone initiation program, Bone Morphogenetic Protein (BMP), Fibroblast Growth Factor (FGF), and Wnt/β-catenin signaling have been associated with the regulation of its factors (Cadigan and Nusse, 1997; Fan et al., 2016; Mark et al., 2009; Ornitz and Itoh, 2015; Rhinn and Dolle, 2012; Wang et al., 2014). BMPs are part of the Transforming Growth Factor Beta (TGF-β) super family and transduce intracellular signaling through phosphorylation of SMAD1/5/8. BMP 2/4 ligands and p-Smad1/5/8 are expressed in the SOA-mesenchyme at E12.5 (Roybal et al., 2010; Sun et al., 2013). Msx1 and Msx2 expression can be partially regulated by BMP. In multiple organisms such as zebrafish, Xenopus, and mice, BMP-signaling positively regulates Msx gene expression in vivo. (Bonilla-Claudio et al., 2012; Knecht and Bronner-Fraser, 2002; Suzuki et al., 1997; Tribulo et al., 2003). However, loss of individual BMP ligands is not sufficient to result in a complete loss of Msx1 or 2 gene expression, demonstrating functional redundancy between multiple BMP ligands (Bonilla-Claudio et al., 2012).
Another signaling pathway shown to regulate Msx gene expression is FGF signaling. FGF signaling pathway has 22 ligands and four FGF tyrosine kinase receptors (FGFR) (Ornitz and Itoh, 2015). FGFR2 and the pathway transducer, ERK1/2, are present in SOA mesenchyme, and FGF 8/10 ligands are expressed in the overlying surface ectoderm(Fan et al., 2016; Goodnough et al., 2014; Kim et al., 1998; Rice et al., 2000). Removal of FGFR2 in the SOA-mesenchyme results in decreased proliferation, leading to dome shaped, but ossified, skull (Yu et al., 2003). Similarly, overexpression of the FGF signaling antagonist Sprouty1, or deletion of Sp8 in the CNCC using Wnt1Cre leads to diminished frontal bone (Kasberg et al., 2013; Yang et al., 2010). The presence of ossified bone in these mutants indicates the bone initiation program is not entirely disrupted. However, FGF signaling pathway mutants have loss of Msx1 and Msx2 mRNA expression in branchial arch facial mesenchyme, and FGF2 coated beads can induce Runx2 expression in calvarial explants (Abu-Issa et al., 2002; Choi et al., 2005; Griffin et al., 2013; Kettunen and Thesleff, 1998; Kim et al., 2003; Omoteyama and Takagi, 2009). These results suggest that the calvarial defects observed in FGF mutants may be due to disruptions in Msx1, Msx2, and/or Runx2 expression, but not a complete loss. Relative to BMP signaling, most FGF signaling pathway mutants have less pronounced effects on calvarial bone development, but FGF signaling is often perturbed in calvarial bone agenesis mutants and human craniosynostosis syndromes suggesting it is an integral pathway to calvarial bone development (Abu-Issa et al., 2002; Fan et al., 2016; Griffin et al., 2013; Nie et al., 2006; Rice et al., 2000; Su et al., 2014).
The Wnt/β-catenin signaling pathway has 19 ligands and binds to Frizzled and LRP5/6 receptors. The intracellular signaling pathway is transduced by β-catenin which binds to TCF/LEF transcription factors to regulate gene transcription in diverse contexts during embryonic development (Brugmann et al., 2007; Cadigan and Nusse, 1997; Day et al., 2005; Gaur et al., 2005; Niemann et al., 2004). Of the three signaling pathways discussed in this review, conditional mutants of the Wnt signaling pathway result in the most dramatic skull bone defects with complete agenesis of the CNCC- and PM-derived skull bones (Day et al., 2005; Goodnough et al., 2012, 2014; Hill et al., 2005). During early skull bone development, Wnt/β-catenin signaling is required for the proliferation and survival of craniofacial structures. Deletion of Wnt/β-catenin signaling in the CNCC using Wnt1Cre results in a complete agenesis of the head and face (Brault et al., 2001). Conditional deletion of β-catenin in the SOA mesenchyme by E12.5 using multiple Cre lines leads to a disruption of the Runx2 expression pattern, and subsequent loss of Osx expression in frontal and parietal bone primordia with little impact on Msx1 and 2 expression (Brault et al., 2001; Fan et al., 2016; Goodnough et al., 2012; Hill et al., 2005). Conditional deletion of individual Wnt ligands results in mild effects on bone differentiation, suggesting functional redundancy (Bennett et al., 2005; Später et al., 2006). Deletion of Wntless, which is required for secretion of all Wnt ligands, in the surface ectoderm using CreEct leads to loss of canonical Wnt/β-catenin signaling in the SOA-mesenchyme and agenesis of the skull bones by E12.5 (Bänziger et al., 2006; Goodnough et al., 2014). Unlike other signaling pathway mutants, β-catenin deletion in the SOA and facial mesenchyme leads to ectopic expression of a key chondrogenic determinant, Sox9, and conversion of calvarial bones and non-osteogenic mesenchyme into cartilage. It is currently unclear as to how β-catenin promotes the expression of the bone initiation program and represses the cartilage fate.
One candidate mechanism is that β-catenin signaling function is mediated by TWIST1. (Goodnough et al., 2012, 2014; Hill et al., 2005). The deletion of Twist1 leads to smaller calvarial bones, increased fontanelle size, and ectopic chondrogenesis in the posterior calvarial bones, similar to β-catenin mutants (Bildsoe et al., 2009; Goodnough et al., 2012, 2016; Krawchuk et al., 2010; Loebel et al., 2012). A second candidate mechanism is that β-catenin deletion leads to stabilization of SOX9 protein, and higher SOX9 levels negatively regulate Runx2 transcription and bone formation in vivo (Eames et al., 2004; Zhou et al., 2006) Additional possibilities are shown in Figure 3 (Akiyama et al., 2004; Goodnough et al., 2012; Hoffmeyer et al., 2017; Liu and Lefebvre, 2015; Mead et al., 2013; Reinhold et al., 2006; Weston et al., 2002; Yasuhara et al., 2010).
Figure 3: Proposed mechanism by which β-catenin suppresses chondrogenesis.
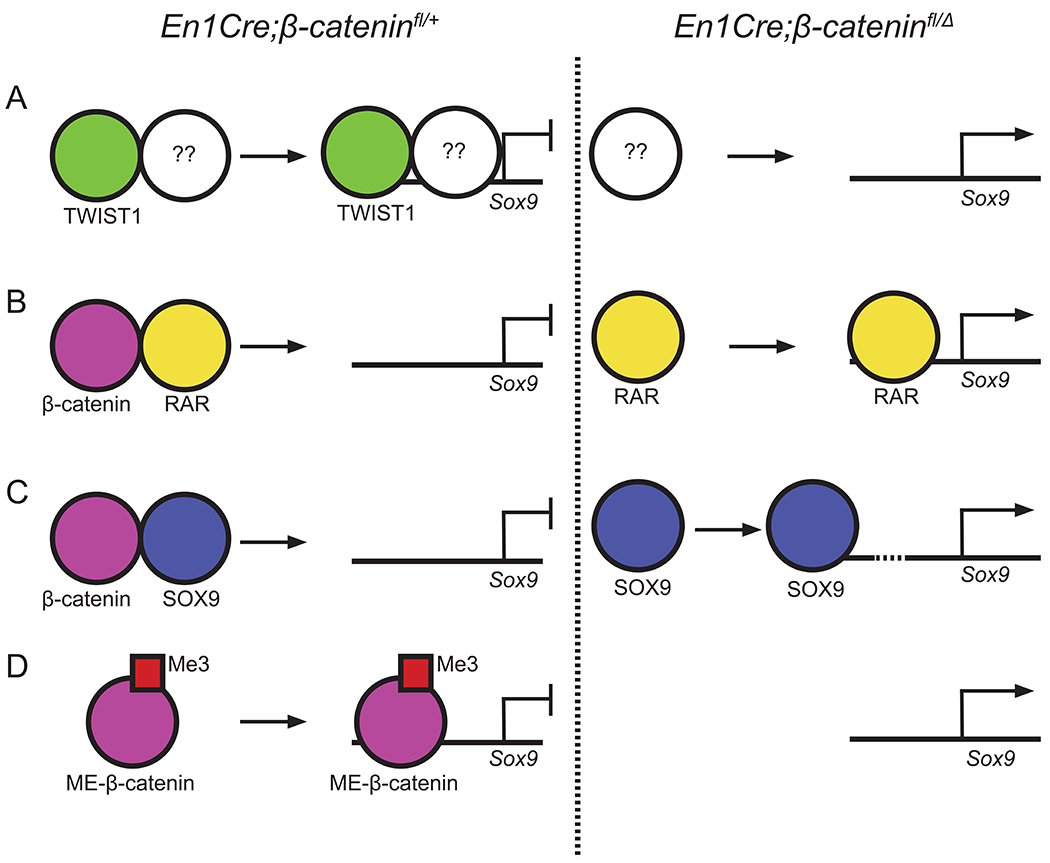
(A) TWIST1 heterodimerizes with an unidentified factor to repress Sox9 transcription. In the absence of β-catenin, Twist1 mRNA is absent leading to the derepression of Sox9 (B) β-catenin dimerizes with retinoic acid receptors (RAR) to prevent the activation of Sox9 transcription. (C) β-catenin dimerizes with SOX9 protein to prevent the auto-regulation of Sox9 transcription. (D) A methylated form of β-catenin acts as a repressor and directly inhibits Sox9 transcription.
Based on these in vivo data, it appears that BMP and FGF signaling establish the Msx1 and 2 positive bone precursors, and Wnt/β-catenin signaling is required for the cell fate selection and commitment to bone. Similar to the spatio-temporal differences in expression observed in the factors of the bone initiation program, the signaling pathways regulating them also exhibit spatio-temporal differences between the CNCC- and the PM-derived mesenchyme. Compared to the PM, the CNCC has increased expression of FGF and Wnt ligands (Fan et al., 2016). In contrast, many BMP signaling components are preferentially expressed in the PM (Fan et al., 2016). However, compound loss of BMP2/4/7 ligands in the CNCC using Wnt1Cre leads to a near complete loss of CNCC-derived bones with little impact on the PM-derived bones (Bonilla-Claudio et al., 2012), showing there is a cell/tissue autonomous role for BMP signaling in CNCC-derived frontal bone despite lower expression compared to the PM. Additional differences in the transcriptional profile of the CNCC and the PM have been identified and reviewed previously (Fan et al., 2016). To date, the majority of the studies examining the bone initiation program are focused on either null mutations or CNCC-specific conditional mutants. A more systematic effort examining the bone initiation program and its regulatory factors in the PM-derived mesenchyme are required to obtain novel mechanistic insights into the regulation of skull bone development.
Epigenetic regulation of the bone initiation program:
Epigenetic modifications allow for the modular regulation of transcription in a reversible and heritable manner (Bracken et al., 2006; Messerschmidt et al., 2014; Rothbart and Strahl, 2014; Schübeler, 2015). During development, the two primary forms of epigenetic regulation are DNA methylation and histone modifications. DNA methylation involves the addition of a methyl group to cytosine residues in cytosine-guanine dense regions (CpG islands) and is linked to transcriptional repression. In mammals, three methyltransferases have been identified; DNA methyltransferase 1 (DNMT1), DNA methyltransferase 3a and 3b (DNMT3a, DNMT3b), with DNTM3a and 3b functioning as the primary DNA methyltransferases during development (Goll and Bestor, 2005; Gopalakrishnan et al., 2008; Okano et al., 1999). Histone modifications are post-translational modifications of histones that affect chromatin structure. In contrast to DNA methylation, which is thought to be established early in development, histone modifications are thought to enable transient transcriptional regulation throughout development (Lynch et al., 2012; Schübeler, 2015; Tanay et al., 2007). The two most studied histone modifications are methylation and acetylation which can be associated with either transcriptional activation or repression (Greer and Shi, 2012; Madrigal and Krajewski, 2015; Roth et al., 2001).
Much of the current understanding regarding epigenetic modifications is based on in vitro data. In various cell types in vitro, epigenetic modifications have been shown to regulate transcription or occupy loci of the bone initiation program factors such as Runx2, Osx, and regulatory factors such as the BMP, Wnt, and FGF signaling pathways (Bracken et al., 2006; Lee et al., 2006; Rosenbloom et al., 2013; Sinha et al., 2010; Yang et al., 2013a; Zhang et al., 2015). However, in vitro, the epigenetic profile of a single cell type can vary based on the specific culture conditions (McEwen et al., 2013). As a result, the extent by which in vitro data accurately represents in vivo conditions is limited. In contrast to in vitro, the role of epigenetic regulation on skull bone development in vivo appears to be relatively limited. Disruptions in many epigenetic modifier pathways result in limited phenotypes (Table 1) (Haberland et al., 2009; Jacques-Fricke et al., 2012; Ueda et al., 2006; Yang et al., 2013b; Zhang et al., 2015). Conditional mouse mutants have identified the Polycomb Repressive Complex 2 (PRC2), a methyltransferase, as the primary regulator of skull bone formation as a loss of PRC2 function results in a loss of multiple bones (Dudakovic et al., 2015; Schwarz et al., 2014).
H3K27me3 is the primary epigenetic modifier during skull bone formation in vivo
PRC2 is a histone modifier that catalyzes the tri-methylation of the 27th lysine on the third histone (H3K27me3) and is typically associated with transcriptional repression. In mammals, PRC2 is a multi-protein complex primarily composed of Enhancer of Zeste 2 (EZH2), Suppressor of Zeste 12 (SUZ12), and Embryonic Ectoderm Development (EED). EZH2 is the methyltransferase catalytic component of PRC2 and is required for the H3K27me3 modification (Figure 4) (Margueron and Reinberg, 2011). In mammals, a homologue of EZH2, EZH1, has also been shown to be associated with the PRC2 complex and possess methyltransferase activity (Shen et al., 2008). However, EZH1 null mice exhibit no phenotype and are viable, indicating that EZH2 is the primary methyltransferase during development (Ezhkova et al., 2011). Thus, most in vivo studies investigating PRC2 have focused on Ezh2 mutants.
Figure 4: Spatial and temporal requirements for Ezh2 in skull bone formation.
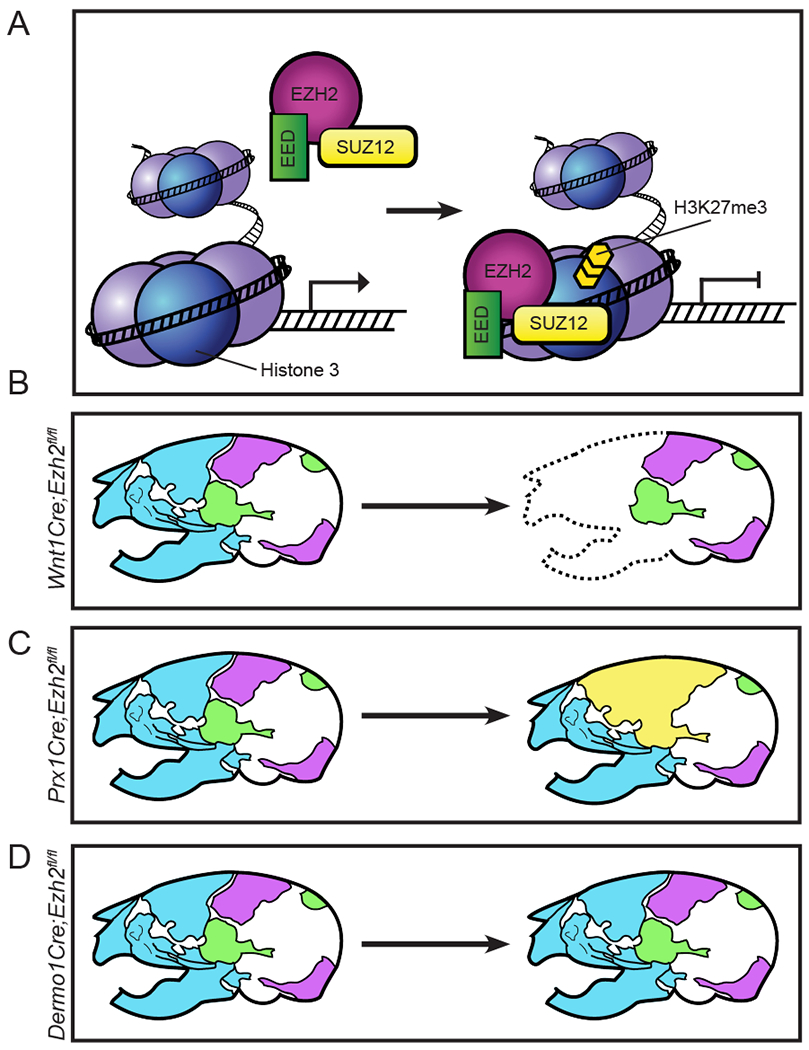
(A) EZH2 is the catalytic component for PRC2. PRC2 binds to DNA and tri-methylates the 27th lysine on the third histone (H3K27me3). H3K27me3 is typically associated with transcriptional repression. (B) Deletion of Ezh2 in the CNCC around E8.5 leads to a complete loss of CNCC-derived skull bones (blue). (C) Deletion of Ezh2 in the PM around E9.5 leads to a fusion of the frontal, parietal, and temporal bones (yellow). (D) Deletion of Ezh2 in the CNCC and PM around E9.5 results in normal skull bone phenotype.
In comparison to other epigenetic mechanisms, Ezh2 mutants can result in the most severe skull phenotypes. In mice, conditional deletion of Ezh2 can result in a complete loss of multiple skull bones. However, the severity of the phenotype is highly dependent on the specific timing of the Cre line. For example, loss of Ezh2 in the CNCC at E8.5 using Wnt1Cre leads to a decrease in Runx2 and Osx expression in the first branchial arch and a complete loss of the CNCC derived skull bones (Schwarz et al., 2014). In contrast, loss of Ezh2 primarily in the PM at E9.5 using Prx1Cre leaves the skull bones intact, but results in fusion of multiple sutures at 3 weeks of age (Dudakovic et al., 2015). Furthermore, deletion of Ezh2 in both the CNCC and the PM at E9.5 or E10.5 using Dermo1Cre or En1Cre results in no skull bone phenotype (Ferguson et al., 2017; Snitow et al., 2016).
The mechanism by which PRC2 regulates the skull bone in the CNCC has not been fully elucidated. Deletion of Ezh2 using Wnt1Cre results in the down regulation of both Runx2 and Osx in the first branchial arch (Schwarz et al., 2014). Because H3K27me3 is typically associated with transcriptional repression, an anti-osteogenic intermediate factor may be involved. One potential group of factors proposed to inhibit the skull bone in the Ezh2 mutants are the Hox genes. In both the CNCC- and the PM-specific Ezh2 mutants, multiple Hox genes are upregulated (Dudakovic et al., 2015; Schwarz et al., 2014). Hox genes have been associated with repression of Runx2 in skull bone in vivo (Carroll and Capecchi, 2015; Santagati et al., 2005). In addition, the SOA-mesenchyme lacks expression of all Hox genes, resulting in a potential sensitivity to ectopic Hox gene expression. Functional studies demonstrating that the ectopic Hox gene expression in the Ezh2 mutants leads to the disruption in the bone initiation program and craniofacial defects are still required.
The specific cause of the dramatic phenotypic differences between each Ezh2 mutant is not fully clear. One possibility is that Ezh2 is required preceding the induction of the bone initiation program. The phenotypic differences observed between Ezh2 conditional mutants are due to the temporal differences between the Cre drivers. Wnt1Cre mutants are a result of deletion of Ezh2 by E8.5, and Dermo1Cre mutants are a result of deletion of Ezh2 at E9.5. Using Wnt1Cre, the Ezh2 target genes may be sufficiently dysregulated prior to the onset of the bone initiation program around E10.5 in the CNCC-derived mesenchyme. In contrast, Ezh2 activity is not sufficiently lost by E10.5 in Dermo1Cre mutants resulting in normal activation of the bone initiation program in the CM (Ferguson et al., 2017). Temporally inducible genetic clonal analysis will be required to understand when calvarial bone precursors become lineage-restricted and if Ezh2 has a role in this process. A second possibility is due to inherent differences between the CNCC- and the PM-derived mesenchyme. Loss of Ezh2 in the CNCC with Wnt1Cre lead to a loss of skull bone, but loss of Ezh2 in the PM with Prx1Cre does not lead to loss of skull bone. Multiple histone modifications have been shown to work in tandem to coordinate stem cell identity and position (Minoux et al., 2017). The variations in the other methylation modifications between the CNCC and the PM could account for the differences in phenotype.
The Ezh2 mutants highlight a level of complexity by which epigenetics may function during development. PRC2 has been associated with the regulation of multiple signaling factors in a spatial and temporal manner. As a result, the specific mechanism by which Ezh2 regulates skull bones has not been fully demonstrated. Genetic studies utilizing tissue-restricted and inducible conditional mutants will provide new insights into the complex dynamics of PRC2 and epigenetic modifications in skull bone development.
Conclusions:
The formation of the skull is a complex process requiring the coordination of multiple genetic mechanisms and tissues. The mechanisms governing the establishment of the stem cells, the bone initiation program, and the epigenetics coordinating these processes require spatial and temporal regulation. As a result, skull bone development is susceptible to congenital and environmental perturbations (Trainor, 2007). A greater understanding of the cellular and genetic mechanisms controlling these processes will provide insights into the pathogenesis of birth defects, tissue engineering, and wound/fracture healing.
To this end, in vivo studies are required to provide a greater picture into the dynamics governing skull bone formation. With the loss of spatial and temporal information in vitro, advancements in the available in vivo tools are providing a greater understanding of these developmental processes. New Cre-recombinase drivers specific to either the CNCC or the PM will help tease apart the genetics and cross-talk between the two tissues enabling skull bone formation. New in vivo cell reporters and live imaging technology will provide advancements in the understanding of the formation of the SOA and the mechanism underlying the apical expansion of skull bones. Sequencing technology such as Hi-ChIP, Assay for Transposase Accessible Chromatin (ATAC), and single cell RNA sequencing, which all facilitate the generation of large data sets from relatively few cells will enable the identification of new sub population and relevant target genes in vivo (Mumbach et al., 2016). As the genetic mechanisms governing the formation of the skull bones are uncovered, a greater picture of the complex processes of skull bone formation will begin to become clear.
Acknowledgments:
Thanks to previous and current members of the Atit laboratory for excellent contributions and discussion, in particular L.Henry Goodnough, Gregg DiNuoscio, and Thu Tran. Figures were modified from: Ferguson, J.W. (2018) The Spatial and Temporal Role of Ezh2 in Skull Bone Formation (doctoral dissertation). Retrieved from OhioLINK (Accession Number: case1530898825341447)
Funding: This work was supported by the following grants: NIH National Institute of Dental and Craniofacial Research R01 DE-01870 (R.P.A.), National Institutes of Health (NIH) T32 AR-007505 (J.F.)
Citations:
- Abu-Issa R, Smyth G, Smoak I, Yamamura K, and Meyers EN (2002). Fgf8 is required for pharyngeal arch and cardiovascular development in the mouse. Development 129, 4613–4625. [DOI] [PubMed] [Google Scholar]
- Akiyama H, Lyons JP, Mori-Akiyama Y, Yang X, Zhang R, Zhang Z, Deng JM, Taketo MM, Nakamura T, Behringer RR, et al. (2004). Interactions between Sox9 and β-catenin control chondrocyte differentiation. Genes Dev. 18, 1072–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek JE, Choi JY, and Kim JE (2014). Skeletal analysis and differential gene expression in Runx2/Osterix double heterozygous embryos. Biochem. Biophys. Res. Commun 451, 442–448. [DOI] [PubMed] [Google Scholar]
- Bänziger C, Soldini D, Schütt C, Zipperlen P, Hausmann G, and Basler K (2006). Wntless, a Conserved Membrane Protein Dedicated to the Secretion of Wnt Proteins from Signaling Cells. Cell 125, 509–522. [DOI] [PubMed] [Google Scholar]
- Bennett CN, Longo KA, Wright WS, Suva LJ, Lane TF, Hankenson KD, and MacDougald OA (2005). Regulation of osteoblastogenesis and bone mass by Wnt10b. Proc. Natl. Acad. Sci 102, 3324–3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt S, Diaz R, Trainor P. a, Wu DK, Kelley MW, Tam PL, Nichols J, and Smith A (2013). Signals and Switches in Mammalian Neural Crest Cell Differentiation Signals and Switches in Mammalian Neural Crest Cell Differentiation. 1–20. [Google Scholar]
- Bi W, Deng JM, Zhang Z, Behringer RR, and De Crombrugghe B (1999). Sox9 is required for cartilage formation. Nat. Genet 22, 85–89. [DOI] [PubMed] [Google Scholar]
- Bildsoe H, Loebel DAF, Jones VJ, Chen YT, Behringer RR, and Tam PPL (2009). Requirement for Twist1 in frontonasal and skull vault development in the mouse embryo. Dev. Biol 331, 176–188. [DOI] [PubMed] [Google Scholar]
- Bonilla-Claudio M, Wang J, Bai Y, Klysik E, Selever J, and Martin JF (2012). Bmp signaling regulates a dose-dependent transcriptional program to control facial skeletal development. Development 139, 709–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracken AP, Dietrich N, Pasini D, Hansen KH, and Helin K (2006). Genome-wide mapping of polycomb target genes unravels their roles in cell fate transitions. Genes Dev. 20, 1123–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brault V, Moore R, Kutsch S, Ishibashi M, Rowitch DH, McMahon a P., Sommer L, Boussadia O, and Kemler R (2001). Inactivation of the β-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development 128, 1253–1264. [DOI] [PubMed] [Google Scholar]
- Brugmann SA, Goodnough LH, Gregorieff A, Leucht P, ten Berge D, Fuerer C, Clevers H, Nusse R, and Helms JA (2007). Wnt signaling mediates regional specification in the vertebrate face. Development 134, 3283–3295. [DOI] [PubMed] [Google Scholar]
- Cadigan KM, and Nusse R (1997). Wnt signalling: a common theme in animal development. Genes Dev. 11, 3286–3305. [DOI] [PubMed] [Google Scholar]
- Carroll LS, and Capecchi MR (2015). HOXC8 initiates an ectopic mammary program by regulating Fgf10 and Tbx3 expression, and Wnt/β-catenin signaling. Development dev.128298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesario JM, Landin Malt A, Chung JU, Khairallah MP, Dasgupta K, Asam K, Deacon LJ, Choi V, Almaidhan AA, Darwiche NA, et al. (2018). Anti-osteogenic function of a LIM-homeodomain transcription factor LMX1B is essential to early patterning of the calvaria. Dev. Biol [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi KY, Kim HJ, Lee MH, Kwon TG, Nah HD, Furuichi T, Komori T, Nam SH, Kim YJ, Kim HJ, et al. (2005). Runx2 regulates FGF2-induced Bmp2 expression during cranial bone development. Dev. Dyn 233, 115–121. [DOI] [PubMed] [Google Scholar]
- Day TF, Guo X, Garrett-Beal L, and Yang Y (2005). Wnt/β-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev. Cell 8, 739–750. [DOI] [PubMed] [Google Scholar]
- Deckelbaum RA, Holmes G, Zhao Z, Tong C, Basilico C, and Loomis CA (2012). Regulation of cranial morphogenesis and cell fate at the neural crest-mesoderm boundary by engrailed 1. Development 139, 1346–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducy P, Zhang R, Geoffroy V, Ridall AL, and Karsenty G (1997). Osf2/Cbfa1: A transcriptional activator of osteoblast differentiation. Cell 89, 747–754. [DOI] [PubMed] [Google Scholar]
- Dudakovic A, Camilleri ET, Xu F, Riester SM, McGee-Lawrence ME, Bradley EW, Paradise CR, Lewallen EA, Thaler R, Deyle DR, et al. (2015). Epigenetic control of skeletal development by the histone methyltransferase Ezh2. J. Biol. Chem 290, 27604–27617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlop LLT, and Hall BK (1995). Relationships between cellular condensation, preosteoblast formation and epithelial-mesenchymal interactions in initiation of osteogenesis. Int. J. Dev. Biol 39, 357–371. [PubMed] [Google Scholar]
- Eames BF, Sharpe PT, and Helms JA (2004). Hierarchy revealed in the specification of three skeletal fates by Sox9 and Runx2. Dev. Biol 274, 188–200. [DOI] [PubMed] [Google Scholar]
- Ezhkova E, Lien WH, Stokes N, Pasolli HA, Silva JM, and Fuchs E (2011). EZH1 and EZH2 cogovern histone H3K27 trimethylation and are essential for hair follicle homeostasis and wound repair. Genes Dev. 25, 485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X, A F Loebel D, Bildsoe H, E Wilkie E, P L Tam P, Qin J, and Wang J (2016). Tissue interactions, cell signaling and transcriptional control in the cranial mesoderm during craniofacial development. AIMS Genet. 3, 74–98. [Google Scholar]
- Ferguson J, Devarajan M, DiNuoscio G, Saiakhova A, Liu C-F, Lefebvre V, Scacheri P, and Atit RP (2017). PRC2 is Dispensable in Vivo for β-Catenin-Mediated Repression of Chondrogenesis in Mouse Embryonic Cranial Mesenchyme. G3: Genes|Genomes|Genetics 8, g3.300311.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaur T, Lengner CJ, Hovhannisyan H, Bhat RA, Bodine PVN, Komm BS, Javed A, Van Wijnen AJ, Stein JL, Stein GS, et al. (2005). Canonical WNT signaling promotes osteogenesis by directly stimulating Runx2 gene expression. J. Biol. Chem 280, 33132–33140. [DOI] [PubMed] [Google Scholar]
- Goll MG, and Bestor TH (2005). Eukaryotic Cytosine Methyltransferases. Annu. Rev. Biochem 74, 481–514. [DOI] [PubMed] [Google Scholar]
- Goodnough LH, Chang A, Treloar C, Yang J, Scacheri PC, and Atit RP (2012). Twist1 mediates repression of chondrogenesis by β-catenin to promote cranial bone progenitor specification. Development 139, 4428–4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodnough LH, DiNuoscio GJ, Ferguson JW, Williams T, Lang RA, and Atit RP (2014). Distinct Requirements for Cranial Ectoderm and Mesenchyme-Derived Wnts in Specification and Differentiation of Osteoblast and Dermal Progenitors. PLoS Genet. 10, 12–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodnough LH, Dinuoscio GJ, and Atit RP (2016). Twist1 contributes to cranial bone initiation and dermal condensation by maintaining wnt signaling responsiveness. Dev. Dyn 245, 144–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalakrishnan S, Van Emburgh BO, and Robertson KD (2008). DNA methylation in development and human disease. Mutat. Res. - Fundam. Mol. Mech. Mutagen 647, 30–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gou Y, Zhang T, and Xu J (2015). Transcription Factors in Craniofacial Development. From Receptor Signaling to Transcriptional and Epigenetic Regulation (Elsevier Inc.). [DOI] [PubMed] [Google Scholar]
- Greer EL, and Shi Y (2012). Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet 13, 343–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin JN, Compagnucci C, Hu D, Fish J, Klein O, Marcucio R, and Depew MJ (2013). Fgf8 dosage determines midfacial integration and polarity within the nasal and optic capsules. Dev. Biol 374, 185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberland M, Mokalled MH, Montgomery RL, and Olson EN (2009). Epigenetic control of skull morphogenesis by histone deacetylase 8. Genes Dev. 23, 1625–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall BK, and Miyake T (1992). The membranous skeleton: the role of cell condensations in vertebrate skeletogenesis. Anat. Embryol. (Berl) 186, 107–124. [DOI] [PubMed] [Google Scholar]
- Han J, Ishii M, Bringas P, Maas RL, Maxson RE, and Chai Y (2007). Concerted action of Msx1 and Msx2 in regulating cranial neural crest cell differentiation during frontal bone development. Mech. Dev 124, 729–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanken J, and Thorogood P (1993). Evolution and development of the veterbrate skull: the role of pattern formation. Tree 8, 9–15. [DOI] [PubMed] [Google Scholar]
- Hill TP, Später D, Taketo MM, Birchmeier W, and Hartmann C (2005). Canonical Wnt/β-Catenin Signaling Prevents Osteoblasts from Differentiating into Chondrocytes. Dev. Cell 8, 727–738. [DOI] [PubMed] [Google Scholar]
- Hoffmeyer K, Junghans D, Kanzler B, and Kemler R (2017). Trimethylation and Acetylation of β-Catenin at Lysine 49 Represent Key Elements in ESC Pluripotency. Cell Rep. 18, 2815–2824. [DOI] [PubMed] [Google Scholar]
- Holland PWH (1991). Cloning and evolutionary analysis of msh-like homeobox genes from mouse, zebrafish and ascidian. Gene 98, 253–257. [DOI] [PubMed] [Google Scholar]
- Holmes G, and Basilico C (2012). Mesodermal expression of Fgfr2S252W is necessary and sufficient to induce craniosynostosis in a mouse model of Apert syndrome. Dev. Biol 368, 283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii M, Merrill AE, Chan Y-S, Gitelman I, Rice DPC, Sucov HM, and Maxson RE (2003). Msx2 and Twist cooperatively control the development of the neural crest-derived skeletogenic mesenchyme of the murine skull vault. Development 130, 6131–6142. [DOI] [PubMed] [Google Scholar]
- Ishii M, Sun J, Ting MC, and Maxson RE (2015). The Development of the Calvarial Bones and Sutures and the Pathophysiology of Craniosynostosis (Elsevier Inc.). [DOI] [PubMed] [Google Scholar]
- Ito Y, Yeo JY, Chytil A, Han J, Bringas P, Nakajima A, Shuler CF, Moses HL, and Chai Y (2003). Conditional inactivation of Tgfbr2 in cranial neural crest causes cleft palate and calvaria defects. Development 130, 5269–5280. [DOI] [PubMed] [Google Scholar]
- Jacques-Fricke BT, Roffers-Agarwal J, and Gammill LS (2012). DNA Methyltransferase 3b Is Dispensable for Mouse Neural Crest Development. PLoS One 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Iseki S, Maxson RE, Sucov HM, and Morriss-Kay GM (2002). Tissue origins and interactions in the mammalian skull vault. Dev. Biol 241, 106–116. [DOI] [PubMed] [Google Scholar]
- Kacena MA, and Ciovacco WA (2010). Megakaryocyte-bone cell interactions. [DOI] [PubMed] [Google Scholar]
- Karsenty G (2008). Transcriptional Control of Skeletogenesis. Annu. Rev. Genomics Hum. Genet 9, 183–196. [DOI] [PubMed] [Google Scholar]
- Kasberg AD, Brunskill EW, and Steven Potter S (2013). SP8 regulates signaling centers during craniofacial development. Dev. Biol 381, 312–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasner E, Hunter CA, Ph D, Kariko K, Ph D, Richtsmeier JT, and Flaherty K (2013). NIH Public Access. Acta Neuropathol. Commun 125, 469–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman MH (1992). The atlas of mouse development. Acad. London 428. [Google Scholar]
- Kettunen P, and Thesleff I (1998). Expression and function of FGFs-4, -8, and -9 suggest functional redundancy and repetitive use as epithelial signals during tooth morphogenesis. Dev. Dyn 211, 256–268. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Rice DP, Kettunen PJ, and Thesleff I (1998). FGF-, BMP- and Shh-mediated signalling pathways in the regulation of cranial suture morphogenesis and calvarial bone development. Development 125, 1241–1251. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Kim JH, Bae SC, Choi JY, Kim HJ, and Ryoo HM (2003). The protein kinase C pathway plays a central role in the fibroblast growth factor-stimulated expression and transactivation activity of Runx2. J. Biol. Chem 278, 319–326. [DOI] [PubMed] [Google Scholar]
- Knecht AK, and Bronner-Fraser M (2002). Induction of the neural crest: a multigene process. Nat. Rev. Genet 3, 453–461. [DOI] [PubMed] [Google Scholar]
- Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, Shimizu Y, Bronson R., Gao Y-H, Inada M, et al. (1997). Targeted Disruption of Cbfa1 Results in a Complete Lack of Bone Formation owing to Maturational Arrest of Osteoblasts. Cell 89, 755–764. [DOI] [PubMed] [Google Scholar]
- Krawchuk D, Weiner SJ, Chen YT, Lu BC, Costantini F, Behringer RR, and Laufer E (2010). Twist1 activity thresholds define multiple functions in limb development. Dev. Biol 347, 133–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kume T, Deng KY, Winfrey V, Gould DB, Walter MA, and Hogan BLM (1998). The forkhead/winged helix gene Mf1 is disrupted in the pleiotropic mouse mutation congenital hydrocephalus. Cell 93, 985–996. [DOI] [PubMed] [Google Scholar]
- Lee JY, Lee YM, Kim MJ, Choi JY, Park EK, Kim SY, Lee SP, Yang JS, and Kim DS (2006). Methylation of the mouse DIx5 and Osx gene promoters regulates cell type-specific gene expression. Mol. Cells 22, 182–188. [PubMed] [Google Scholar]
- Liu CF, and Lefebvre V (2015). The transcription factors SOX9 and SOX5/SOX6 cooperate genome-wide through super-enhancers to drive chondrogenesis. Nucleic Acids Res. 43, 8183–8203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu TM, and Lee EH (2013). Transcriptional regulatory cascades in Runx2-dependent bone development. Tissue Eng. Part B. Rev 19, 254–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loebel DAF, Hor ACC, Bildsoe H, Jones V, Chen YT, Behringer RR, and Tam PPL (2012). Regionalized Twist1 activity in the forelimb bud drives the morphogenesis of the proximal and preaxial skeleton. Dev. Biol 362, 132–140. [DOI] [PubMed] [Google Scholar]
- Lynch MD, Smith AJH, De Gobbi M, Flenley M, Hughes JR, Vernimmen D, Ayyub H, Sharpe JA, Sloane-Stanley JA, Sutherland L, et al. (2012). An interspecies analysis reveals a key role for unmethylated CpG dinucleotides in vertebrate Polycomb complex recruitment. EMBO J. 31, 317–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madrigal P, and Krajewski P (2015). Uncovering correlated variabilityin epigenomic datasets usingthe Karhunen-Loeve transform. BioData Min. 8, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, and Reinberg D (2011). The Polycomb complex PRC2 and its mark in life. Nature 469, 343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark M, Ghyselinck NB, and Chambon P (2009). Function of retinoic acid receptors during embryonic development. Nucl.Recept.Signal 7, e002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen KR, Leitch HG, Amouroux R, and Hajkova P (2013). The impact of culture on epigenetic properties of pluripotent stem cells and pre-implantation embryos. Biochem Soc Trans 41, 711–719. [DOI] [PubMed] [Google Scholar]
- Mead TJ, Wang Q, Bhattaram P, Dy P, Afelik S, Jensen J, and Lefebvre V (2013). A far-upstream (-70 kb) enhancer mediates Sox9 auto-regulation in somatic tissues during development and adult regeneration. Nucleic Acids Res. 41, 4459–4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messerschmidt DM, Knowles BB, and Solter D (2014). DNA methylation dynamics during epigenetic reprogramming in the germline and preimplantation embryos. Genes Dev. 28, 812–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michailovici I, Eigler T, and Tzahor E (2015). Craniofacial Muscle Development (Elsevier Inc.). [DOI] [PubMed] [Google Scholar]
- Minoux M, Holwerda S, Vitobello A, Kitazawa T, Kohler H, Stadler MB, and Rijli FM (2017). Gene bivalency at Polycomb domains regulates cranial neural crest positional identity. Science (80-. ). 355, eaal2913. [DOI] [PubMed] [Google Scholar]
- Mori-Akiyama Y, Akiyama H, Rowitch DH, and de Crombrugghe B (2003). Sox9 is required for determination of the chondrogenic cell lineage in the cranial neural crest. Proc. Natl. Acad. Sci 100, 9360–9365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morriss-Kay GM, and Wilkie AOM (2005). Growth of the normal skull vault and its alteration in craniosynostosis: insights from human genetics and experimental studies. J. Anat 207, 637–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumbach MR, Rubin AJ, Flynn RA, Dai C, Khavari PA, Greenleaf WJ, and Chang HY (2016). HiChIP: Efficient and sensitive analysis of protein-directed genome architecture. Nat. Methods 13, 919–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musy M, Flaherty K, Raspopovic J, Robert-Moreno A, Richtsmeier J, and Sharpe J (2018). A quantitative method for staging mouse embryos based on limb morphometry. Development dev.154856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima K, Zhou X, and Al GK. et (2002). The novel zinc fingercontaining transcription factor Osterix is required for osteoblast 12 BioMed Research International differentiation and bone formation. Cell 108, 17–29. [DOI] [PubMed] [Google Scholar]
- Nie X, Luukko K, and Kettunen P (2006). FGF signalling in craniofacial development and developmental disorders. Oral Dis. 12, 102–111. [DOI] [PubMed] [Google Scholar]
- Niemann S, Zhao C, Pascu F, Stahl U, Aulepp U, Niswander L, Weber JL, and Müller U (2004). Homozygous WNT3 mutation causes tetra-amelia in a large consanguineous family. Am. J. Hum. Genet 74, 558–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishio Y, Dong Y, Paris M, O'Keefe RJ., Schwarz EM, and Driss i, H. (2006). Runx2-mediated regulation of the zinc finger Osterix/Sp7 gene. Gene 372, 62–70. [DOI] [PubMed] [Google Scholar]
- Okano M, Bell DW, Haber DA, and Li E (1999). DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99, 247–257. [DOI] [PubMed] [Google Scholar]
- Omoteyama K, and Takagi M (2009). FGF8 regulates myogenesis and induces Runx2 expression and osteoblast differentiation in cultured cells. J. Cell. Biochem 106, 546–552. [DOI] [PubMed] [Google Scholar]
- Ornitz DM, and Itoh N (2015). The fibroblast growth factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol 4, 215–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto F, Thornell AP, Crompton T, Denzel A, Gilmour KC, Rosewell IR, Stamp GWH, Beddington RSP, Mundlos S, Olsen BR, et al. (1997). Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell 89, 765–771. [DOI] [PubMed] [Google Scholar]
- Reinhold MI, Kapadia RM, Liao Z, and Naski MC (2006). The Wnt-inducible transcription factor Twist1 inhibits chondrogenesis. J. Biol. Chem 281, 1381–1388. [DOI] [PubMed] [Google Scholar]
- Rhinn M, and Dolle P (2012). Retinoic acid signalling during development. Development 139, 843–858. [DOI] [PubMed] [Google Scholar]
- Rice DPC (2005). Craniofacial Anomalies: From Development to Molecular Pathogenesis. Curr. Mol. Med 5, 699–722. [DOI] [PubMed] [Google Scholar]
- Rice DP, Aberg T, Chan Y, Tang Z, Kettunen PJ, Pakarinen L, Maxson RE, and Thesleff I (2000). Integration of FGF and TWIST in calvarial bone and suture. Development 127, 1845–1855. [DOI] [PubMed] [Google Scholar]
- Rosenbloom KR, Sloan CA, Malladi VS, Dreszer TR, Learned K, Kirkup VM, Wong MC, Maddren M, Fang R, Heitner SG, et al. (2013). ENCODE Data in the UCSC Genome Browser: Year 5 update. Nucleic Acids Res. 41, 56–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth SY, Denu JM, and Allis CD (2001). Histone acetyltransferases. Annu. Rev. Biochem 70, 81–120. [DOI] [PubMed] [Google Scholar]
- Rothbart SB, and Strahl BD (2014). Interpreting the language of histone and DNA modifications. Biochim. Biophys. Acta - Gene Regul. Mech 1839, 627–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roybal PG, Wu NL, Sun J, Ting M chun, Schafer CA, and Maxson RE (2010). Inactivation of Msx1 and Msx2 in neural crest reveals an unexpected role in suppressing heterotopic bone formation in the head. Dev. Biol 343, 28–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santagati F, Minoux M, Ren S-Y, and Rijli FM (2005). Temporal requirement of Hoxa2 in cranial neural crest skeletal morphogenesis. Development 132, 4927–4936. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Ito Y, Bringas P, Chou S, Urata MM, Slavkin H, and Chai Y (2006). TGFbeta-mediated FGF signaling is crucial for regulating cranial neural crest cell proliferation during frontal bone development. Development 133, 371–381. [DOI] [PubMed] [Google Scholar]
- Satokata I, and Maas R (1994). Msx1 deficient mice exhibit cleft palate and abnormalities of craniofacial and tooth development. Nat. Genet 6, 348–356. [DOI] [PubMed] [Google Scholar]
- Satokata I, Ma L, Ohshima H, Bei M, Ian W, Nishizawa K, Maeda T, Takano Y, Uchiyama M, Heaney S, et al. (2000). Msx2 deficiency in mice causes pleiotropic defects in bone growth and ectodermal organ formation. Nat. Genet 24, 391–395. [DOI] [PubMed] [Google Scholar]
- Schübeler D (2015). Function and information content of DNA methylation. Nature 517, 321–326. [DOI] [PubMed] [Google Scholar]
- Schwarz D, Varum S, Zemke M, Schöler A, Baggiolini A, Draganova K, Koseki H, Schübeler D, and Sommer L (2014). Ezh2 is required for neural crest-derived cartilage and bone formation. Development 141, 867–877. [DOI] [PubMed] [Google Scholar]
- Shen X, Liu Y, Hsu YJ, Fujiwara Y, Kim J, Mao X, Yuan GC, and Orkin SH (2008). EZH1 Mediates Methylation on Histone H3 Lysine 27 and Complements EZH2 in Maintaining Stem Cell Identity and Executing Pluripotency. Mol. Cell 32, 491–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha KM, Yasuda H, Coombes MM, Yr Dent S, and De Crombrugghe B (2010). Regulation of the osteoblast-specific transcription factor Osterix by NO66, a Jumonji family histone demethylase. EMBO J. 29, 68–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snitow M, Lu M, Cheng L, Zhou S, and Morrisey EE (2016). Ezh2 restricts the smooth muscle lineage during mouse lung mesothelial development. Development dev.134932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Später D, Hill TP, Gruber M, and Hartmann C (2006). Role of canonical Wnt-signalling in joint formation. Eur. Cells Mater 12, 71–80. [DOI] [PubMed] [Google Scholar]
- Su N, Jin M, and Chen L (2014). Role of FGF/FGFR signaling in skeletal development and homeostasis: Learning from mouse models. Bone Res. 2, 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Ishii M, Ting M-C, and Maxson R (2013). Foxc1 controls the growth of the murine frontal bone rudiment by direct regulation of a Bmp response threshold of Msx2. Development 140, 1034–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Ueno N, and Hemmati-Brivanlou A (1997). Xenopus msx1 mediates epidermal induction and neural inhibition by BMP4. Development 124, 3037–3044. [DOI] [PubMed] [Google Scholar]
- Szabo-Rogers HL, Smithers LE, Yakob W, and Liu KJ (2010). New directions in craniofacial morphogenesis. Dev. Biol 341, 84–94. [DOI] [PubMed] [Google Scholar]
- Tanay A, O’Donnell AH, Damelin M, and Bestor TH (2007). Hyperconserved CpG domains underlie Polycomb-binding sites. Proc. Natl. Acad. Sci 104, 5521–5526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trainor PA (2007). Craniofacial birth defects: the role of neural crest cells in the etiology and pathogenesis of treacher collins syndrome and the potential for prevention. Am. J. Med. Genet 2, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran TH, Jarrell A, Zentner GE, Welsh A, Brownell I, Scacheri PC, and Atit R (2010). Role of canonical Wnt signaling/β-catenin via Dermo1 in cranial dermal cell development. Development 137, 3973–3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tribulo C, Aybar MJ, Nguyen VH, Mullins MC, and Mayor R (2003). Regulation of Msx genes by a Bmp gradient is essential for neural crest specification. Development 130, 6441–6452. [DOI] [PubMed] [Google Scholar]
- Twigg SRF, and Wilkie AOM (2015). A Genetic-Pathophysiological Framework for Craniosynostosis. Am. J. Hum. Genet 97, 359–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda Y, Okano M, Williams C, Chen T, Georgopoulos K, and Li E (2006). Roles for Dnmt3b in mammalian development: a mouse model for the ICF syndrome. Development 133, 1183–1192. [DOI] [PubMed] [Google Scholar]
- Vivatbutsiri P, Ichinose S, Hytönen M, Sainio K, Eto K, and Iseki S (2008). Impaired meningeal development in association with apical expansion of calvarial bone osteogenesis in the Foxc1 mutant. J. Anat 212, 603–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RN, Green J, Wang Z, Deng Y, Qiao M, Peabody M, Zhang Q, Ye J, Yan Z, Denduluri S, et al. (2014). Bone Morphogenetic Protein (BMP) signaling in development and human diseases. Genes Dis. 1, 87–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston AD, Chandraratna RAS, Torchia J, and Underhill TM (2002). Requirement for RAR-mediated gene repression in skeletal progenitor differentiation. J. Cell Biol 158, 39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Lawson KA, Teteak CJ, Zou J, Hacquebord J, Patterson D, Ghatan AC, Mei Q, Zielinska-Kwiatkowska A, Bain SD, et al. (2013a). ESET histone methyltransferase is essential to hypertrophic differentiation of growth plate chondrocytes and formation of epiphyseal plates. Dev. Biol 380, 99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Lawson KA, Teteak CJ, Zou J, Hacquebord J, Patterson D, Ghatan AC, Mei Q, Zielinska-Kwiatkowska A, Bain SD, et al. (2013b). ESET histone methyltransferase is essential to hypertrophic differentiation of growth plate chondrocytes and formation of epiphyseal plates. Dev. Biol 380, 99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Kilgallen S, Andreeva V, Spicer DB, Pinz I, and Friesel R (2010). Conditional expression of Spry1 in neural crest causes craniofacial and cardiac defects. BMC Dev. Biol 10, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuhara R, Yuasa T, Williams JA, Byers SW, Shah S, Pacifici M, Iwamoto M, and Enomoto-Iwamoto M (2010). Wnt/β-Catenin and retinoic acid receptor signaling pathways interact to regulate chondrocyte function and matrix turnover. J. Biol. Chem 285, 317–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T, Vivatbutsiri P, Morriss-Kay G, Saga Y, and Iseki S (2008). Cell lineage in mammalian craniofacial mesenchyme. Mech. Dev 125, 797–808. [DOI] [PubMed] [Google Scholar]
- Yu K, Xu J, Liu Z, Sosic D, Shao J, Olson EN, Towler DA, and Ornitz DM (2003). Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development 130, 3063–3074. [DOI] [PubMed] [Google Scholar]
- Zhang F, Xu L, Xu L, Xu Q, Karsenty G, and Chen CD (2015). Histone demethylase JMJD3 is required for osteoblast differentiation in mice. Sci. Rep 5, 13418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G, Zheng Q, Engin F, Munivez E, Chen Y, Sebald E, Krakow D, and Lee B (2006). Dominance of SOX9 function over RUNX2 during skeletogenesis. Proc. Natl. Acad. Sci. U. S. A 103, 19004–19009. [DOI] [PMC free article] [PubMed] [Google Scholar]