

SUMMARY
Mitochondrial outer membrane permeabilization (MOMP) is a core event in apoptosis signaling. However, the underlying mechanism of BAX/BAK pore formation remains incompletely understood. Here, we demonstrate that mitochondria are globally and dynamically targeted by endolysosomes (ELs) during MOMP. In response to pro-apoptotic BH3-only protein signaling and pharmacological MOMP induction, ELs increasingly form transient contacts with mitochondria. Subsequently, ELs rapidly accumulate within the entire mitochondrial compartment. This switch-like accumulation period temporally coincides with mitochondrial BAX clustering and cytochrome c release. Remarkably, interactions of ELs with mitochondria control BAX recruitment and pore formation. Knockdown of Rab5A, Rab5C or USP15 interferes with EL targeting of mitochondria and functionally uncouples BAX clustering from cytochrome c release, while knockdown of the Rab5 exchange factor Rabex-5 impairs both BAX clustering and cytochrome c release. Together, these data reveal that EL-mitochondrial inter-organelle communication is an integral regulatory component of functional MOMP execution during cellular apoptosis signaling.
eTOC Blurb
Wang et al. show that upon activation of regulated cell death, endolysosomal vesicles (ELs) are targeted to mitochondria, prior to and during their permeabilization by BAX-mediated apoptotic pores. These inter-organelle interactions result in a biochemical transformation of mitochondrial membranes and are required for functional in situ apoptotic pore formation.
INTRODUCTION
Apoptosis can be initiated by two main pathways. In the extrinsic pathway, death receptors activate caspase-8, which then activates executioner caspase-mediated proteolytic cell death. The intrinsic pathway is initiated by intracellular stresses which activate pro-apoptotic BH3-only proteins to signal the formation of the BAX and BAK pores responsible for mitochondrial outer membrane permeabilization (MOMP) (Chipuk et al., 2010). BAK resides constitutively at the outer mitochondrial membrane (OMM), whereas inactive BAX is predominantly retained in the cytoplasm. In response to apoptotic stimuli, BAX translocates to the OMM and forms oligomers which generate proteo-lipidic pores (Kushnareva et al., 2012; Luo et al., 2014). These pores release pro-apoptotic intermembrane space (IMS) proteins, including cytochrome c and Smac/DIABLO, over a period of seconds-to-minutes (Munoz-Pinedo et al., 2006; Rehm et al., 2002). Cytoplasmic cytochrome c activates executioner caspases, whereas Smac suppresses the endogenous caspase inhibitor XIAP (Deveraux et al., 1997; Takahashi et al., 1998).
MOMP rapidly commits a cell to proteolytic death (Albeck et al., 2008a; Rehm et al., 2002; Tait and Green, 2010; Thorburn et al., 2014). Deciphering the mechanisms and regulation of BAX recruitment and activation steps remains a priority goal. The current understanding is that, in the absence of BH3-only protein activity, BAX is maintained in the cytosol in its inactive form through the process of retrotranslocation, whereby BCL-XL constantly binds and re-locates loosely-bound mitochondrial BAX to the cytoplasm (Edlich et al., 2011). During apoptosis, BH3-only proteins bind to inhibit anti-apoptotic BCL-2 proteins and/or bind to directly activate BAX (Shamas-Din et al., 2013). Importantly, in the absence of BH3-only proteins, suppression of anti-apoptotic BCL-2 proteins is sufficient to induce mitochondrial BAX accumulation, membrane insertion and activation (O’Neill et al., 2016), thereby identifying the OMM as the critical site of BAX regulation.
An emerging paradigm is that interconnectivity between mitochondria and various other organelle types shapes intrinsic apoptosis signaling. Membrane contacts between mitochondria and the endoplasmic reticulum (ER), also called mitochondria-associated membranes (MAMs), mediate lipid, protein and ion transfer between those two organelle compartments, and thereby impact MOMP (de Brito and Scorrano, 2010; Rowland and Voeltz, 2012). Mitochondrial association with other organelles, including the ER, regulates sphingolipid metabolism to drive BAX pore formation (Chipuk et al., 2012) and MAMs provide a platform for cytochrome c release signaling (Otera et al., 2016; Prudent et al., 2015). In embryonic cells, active BAX can be sequestered at the Golgi and translocated to mitochondria in response to DNA damage (Dumitru et al., 2012).
Associations between apoptotic mitochondria and the endolysosomal (EL) compartment have also been observed. During FasL-induced apoptosis mitochondria acquire Golgi-related endocytic membranes (Ouasti et al., 2007), and ELs form close contacts with mitochondria following apoptosis induction with the Helicobacter pylori toxin VacA (Calore et al., 2010). BH3-only protein signaling triggers mitochondrial targeting by the early endosomal Rab5 GTPase, the Rab5 GDP/GTP exchange factor (GEF) Rabex-5 and the late endosomal Rab7 GTPase, associated with degradation of the mitochondrial pro-apoptotic factor Smac (Hamacher-Brady et al., 2014). During H2O2-induced cell death, Rab5 and Rab5 GEF ALS2/Alsin counteract cytochrome c release (Hsu et al., 2018). Of note, OMMs of apoptotic mitochondria display distinct membrane domains that contain either typical OMM markers or the endocytic regulator Rabex-5 (Hamacher-Brady et al., 2014). BAX clusters were localized at the interface between these membrane domains, prompting our hypothesis that EL-mitochondrial interactions might directly participate in MOMP regulation.
Thus, we sought to investigate the dynamics, mechanisms and functional implications of EL targeting of mitochondria in response to a variety of apoptotic stimuli. Through high-resolution fluorescence imaging, and molecular perturbance of endocytic trafficking-related regulators, we mapped the spatio-temporal dynamics and causal relationships between EL-mitochondrial interactions and MOMP events. We propose that EL targeting of mitochondria during apoptosis is mechanistically integrated with, and required for, full in situ MOMP functionality.
RESULTS
Mitochondria are Globally Targeted by ELs in Response to Diverse Apoptotic Stimuli
To explore whether EL targeting of mitochondria is a general phenomenon during mitochondria-dependent apoptosis, we activated apoptosis extrinsically with the TNF receptor family ligand TNFα, in the presence of actinomycin D (TNFα/AcD), or intrinsically with the DNA topoisomerase-I inhibitor camptothecin (CPT) or the protein kinase inhibitor staurosporine (STS). In MCF-7 cells stably expressing Cytochrome c-GFP (Cyto c-GFP), in response to all treatments, the early EL marker Rab5 (Bucci et al., 1992) colocalized with Cyto c-GFP release+ (MOMP+) TOM70-labeled mitochondria (Figures 1A and S1A). While extent and timepoints of significant population-level MOMP induction differed between treatments (Figure 1B), we noted a striking positive correlation between MOMP induction and mitochondrial localization of Rab5, with mitochondrial Rab5 detected in nearly 100% of MOMP+ cells (Figures 1C and 1D). Similar results were obtained for HeLa cervical cancer cells (Figures 1E and 1F), and MOMP detection using the IMS-RP reporter (Albeck et al., 2008a).
Figure 1. Various Apoptotic Stimuli Trigger Targeting of Rab5+ ELs to Mitochondria.
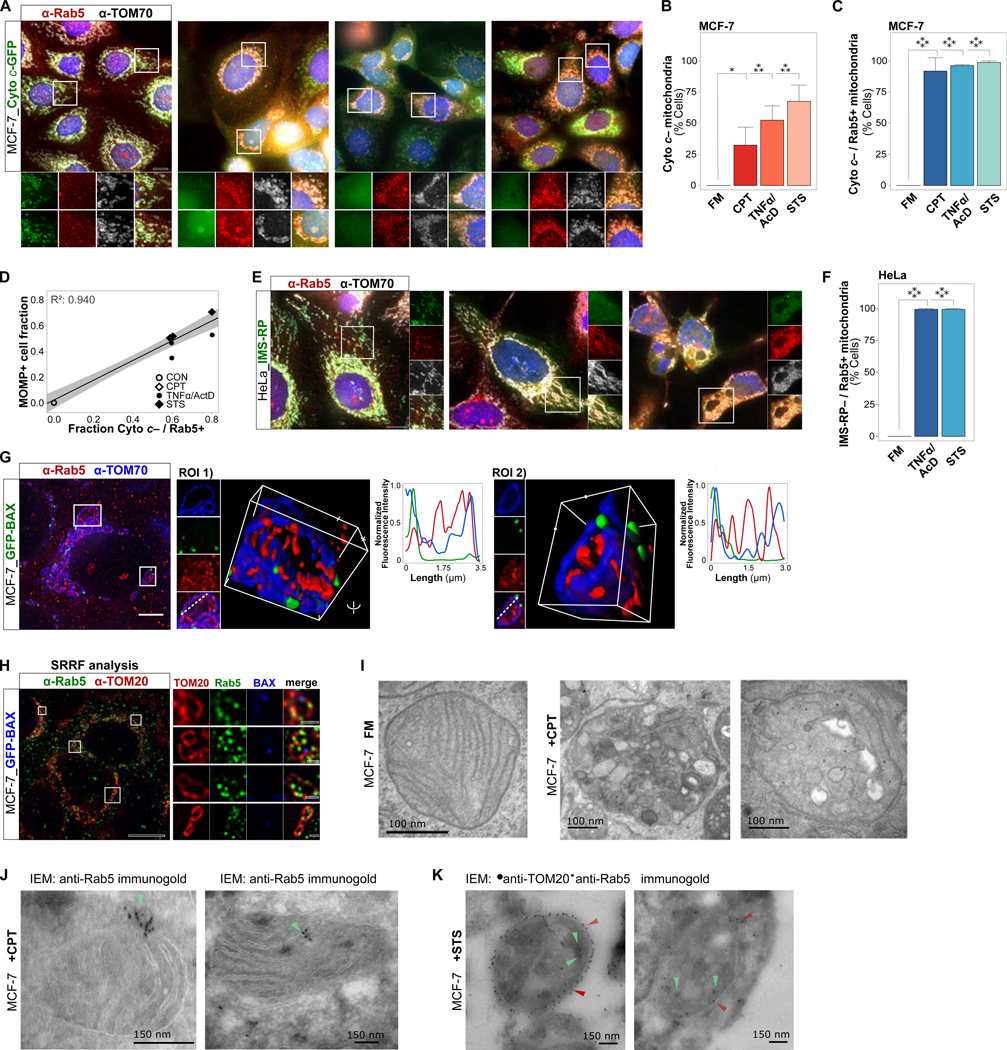
(A) MCF-7_Cyto c-GFP cells, in normal growth medium (full medium, FM) or treated with STS (1 μM) for 4 hrs, TNF α/AcD (50 ng/mL / 1 μg/mL) for 6 hrs, or CPT (20 μM) for 24 hrs. IF of Rab5 and TOM70. Scale bars, 10 μm.
(B) Cells from (A) were scored for mitochondria with released cytochrome c (Cyto c–).
(C) Cells from (A) were scored for Cyto c– mitochondria targeted by Rab5 (Cyto c– / Rab5+).
(D) Correlation between cells positive for MOMP (Cyto c–) and Cyto c– / Rab5+ mitochondria. Symbols represent mean of independent experiments.
(E) HeLa_IMS-RP cells, in FM or treated with STS+zVAD for 4 hrs, or TNFα/AcD+zVAD for 6 hrs. IF of Rab5 and TOM20. Scale bars, 10 μm.
(F) Cells from (E) were scored for mitochondria with released IMS-RP targeted by Rab5 (IMS-RP– / Rab5+).
(G) MCF-7_GFP-BAX cells, treated with TNFα/AcD for 6 hrs, IF of Rab5 and TOM70. 3D reconstruction, single and merged channels of ROIs, and line profile of dotted lines from ROIs. Scale bars, 10 μm. See also Figure S1, and Videos S1 and S2.
(H) SRRF imaging of MCF-7_GFP-BAX cells, treated with TNFα/AcD for 6 hrs, IF of Rab5 and TOM20. Inset scale bar 1 μm.
(I) Electron micrographs of MCF-7 cells in FM or treated with CPT for 24 hrs. Zoom images of single mitochondria. Scale bars, 100 nm. See also Figure S1F.
(J) Immunoelectron microscopy of MCF-7 cells treated with CPT for 24 hrs, immunogold-labeled for Rab5 (green arrowheads). Left, mitochondrion with adjacent Rab5+ vesicle. Right, mitochondrion with internalized Rab5+ vesicle. Scale bars, 150 nm. See also Figures S1G and S1H.
(K) Immunoelectron microscopy of MCF-7 cells treated with STS for 4 hrs, immunogold-labeled for TOM20 (red arrowheads) and Rab5 (green arrowheads).
Graphs, mean of n = 3 experiments. Error bars, standard deviation. ≥ 300 cells per condition. *p < 0.05, ***p ≤ 0.001, ****p ≤ 0.0001
Consistently, Rab5+ vesicles colocalized with GFP-BAX+ mitochondria in response to TNFα/AcD (Figures 1G). High-resolution 3D and line scan analysis revealed that Rab5+ vesicles were localized in close vicinity to BAX clusters. Notably, Rab5+ vesicles both colocalized with the TOM70-labeled OMM and were present inside OMM-confined areas (Video S1). Similarly, Rab5+ vesicles were detected at the OMM and within inner mitochondrial compartments of cells treated with CPT or STS (Figure S1B and Video S2). Super-resolution radial fluctuations (SRRF) imaging (Gustafsson et al., 2016) confirmed Rab5+ vesicle colocalization with the OMM and inside GFP-BAX+ mitochondria (Figure 1H). In support, electron micrographs of MCF-7 cells illustrate severely altered mitochondrial ultrastructure in response to CPT (Figures S1F and S1G), with numerous intra-mitochondrial vesicles (Figures 1I, S1G and S1H). Immunogold-labeling of Rab5 identified Rab5+ ELs targeting mitochondria, and inside mitochondria prior to and following loss of cristae structures (Figures 1J, S1G and S1H). Rab5+ ELs were also present within TOM20 co-labeled mitochondria of STS-treated MCF-7 cells (Figure 1K).
Live Microscopy of Diverse Vesicle Markers Reveals Dynamic EL Targeting of Mitochondria, Pre- and Post-MOMP
We next characterized EL targeting of apoptotic mitochondria by live microscopy. We first asked whether the largescale accumulation of Rab5 in the mitochondrial compartment in MOMP+ cells is preceded by enhanced interactions between ELs and pre-apoptotic mitochondria. Indeed, 5 sec interval recording of MCF-7 cells stably expressing Cyto c-GFP, treated with TNFα/AcD, CPT, or the small-molecule BAX activator BAM7 (Gavathiotis et al., 2012), revealed that, compared to normal growth conditions, both number and duration of transient RFP-Rab5+ EL interactions with mitochondria were increased prior to cytochrome c release (Figures 2A–2C). Similarly, live microscopy of TNFα/AcD-treated HeLa cells stably expressing the MOMP reporter IMS-RP demonstrates targeting of mitochondria by GFP-Rab5+ ELs, 12.9± 2.6 min prior to cell contraction (Figures S2A-S2C), which typically occurs within minutes following MOMP (Albeck et al., 2008a). Prominent interactions between mitochondria and Rab5+ vesicles prior to apoptotic cell contraction were also observed in HeLa cells subjected to H2O2 (Figure S2D), and in HeLa cells treated with BAM7 (Figure S2E).
Figure 2. ELs Targeting Apoptotic Mitochondria are Dynamic and Biochemically Active.
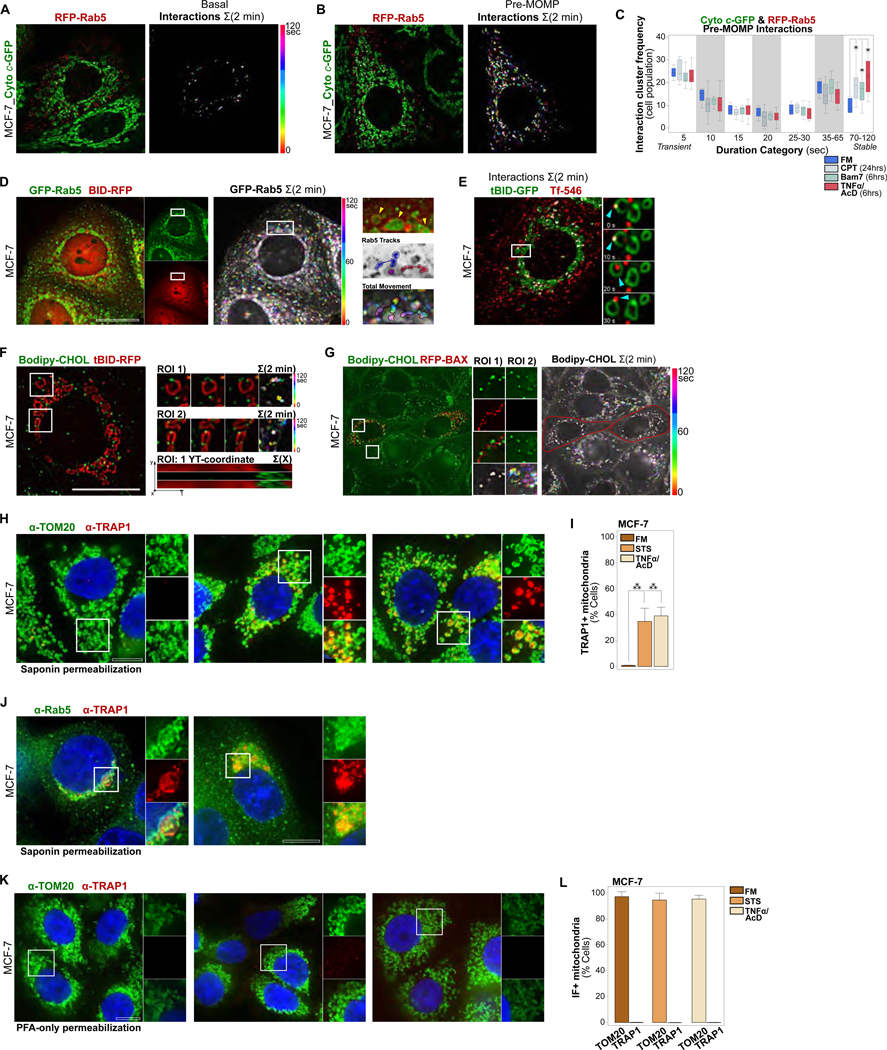
(A) MCF-7_Cyto c-GFP_RFP-Rab5 cells, live imaged every 5 sec over 2 min under FM conditions. Left, merged channel single time-point images with EL-mitochondrial interactions overlaid in white. Right, color-coded EL-mitochondrial interactions, projected over 2 min.
(B) MCF-7_Cyto c-GFP_RFP-Rab5 cells, live imaged every 5 sec over 2 min at 6 hrs of TNFα/AcD treatment. Analyzed as in (A).
(C) Cluster analysis of transient to stable interactions from individual pre-MOMP (Cyto c-GFP+) cells in a population, imaged as in (A), under FM, TNFα/AcD (6 hrs), BAM7 (6 hrs) or CPT (24 hrs). *p ≤ 0.05. 6–19 cells per condition.
(D) MCF-7 cells co-expressing GFP-Rab5 and BID-RFP, treated with TNFα/AcD for 5 hrs and then live imaged every 5 sec for 2 min. Left, single channel images. Middle, GFP-Rab5 color-coded time-steps projected over 2 min. Right, yellow arrows in ROI zoom mark three mitochondria. ROI with tracked vesicle movements (blue and red shades) to show diverse associations of individual Rab5+ ELs with mitochondria. GFP-Rab5+ vesicle movement boundaries are shown in color-coded time-step ROI.
(E) MCF-7 cell expressing tBID-GFP for 18 hrs, loaded with Transferrin-AF546 and imaged live every 5 sec over 2 min. Time-sum merged channel image with interactions overlaid in white. Single time-point ROIs, cyan arrowheads track transferrin-loaded EL. See also Video S3.
(F) MCF-7 cell expressing tBID-RFP for 18 hrs, loaded with Bodipy-CHOL in the presence of u18666a, imaged live every 5 sec over 2 min. Overview image and ROI time-series shown as color-coded time-step 2 min sum. Bottom right, YT-coordinate projection of mitochondrion in ROI 1. See also Video S4.
(G) MCF-7 cells co-expressing tBID-BFP (not shown) and RFP-BAX for 18 hrs, loaded with Bodipy-CHOL in the presence of u18666a, imaged live every 5 sec over 2 min. Left, merged channels of single time point. ROI of channels, and color-coded time-step 2 min sum. Right, overview color-coded time-step 2 min sum of Bodipy-CHOL movement.
(H) MCF-7 cells in FM, or treated with STS (5 hrs) or TNFα/AcD (6 hrs). IF of saponin-permeabilized cells for TOM20 and TRAP1.
(I) Quantifications of cells treated as in (H) scored for TRAP1 saponin-IF+ mitochondria. Graphs, mean of n = 3 experiments. Error bars, standard deviation. > 275 cells per condition. ***p ≥ 0.001.
(J) MCF-7 cells treated with STS (5 hrs) or TNFα/AcD (6 hrs). IF of saponin-permeabilized cells for inner-mitochondrial TRAP1 and Rab5.
(K) MCF-7 cells in FM, or treated with STS (5 hrs) or TNFα/AcD (6 hrs). IF of PFA-permeabilized cells for TOM20 and TRAP1.
(L) Quantifications of cells as in (H) scored for TOM20 and TRAP1 PFA-IF+ mitochondria. Graphs, mean of n = 2 experiments. Error bars, standard deviation. > 90 cells per condition. Scale bars, 10 μm.
We then assessed the dynamics of EL-mitochondrial interactions in MOMP+ cells. In MCF-7 cells expressing BID-RFP, to identify MOMP+ cells via detection of mitochondrial translocated tBID-RFP (Cory and Adams, 2002; Zha et al., 2000), GFP-Rab5 colocalized with tBID-RFP+ mitochondria under TNFα/AcD (Figure 2D). Video microscopy reveals active movements of GFP-Rab5+ vesicles, distal and proximal to tBID-RFP+ mitochondria. As illustrated in the ROI tracking analyses, cytosolic ELs move to mitochondria (blue tracks), remain within a mitochondrion (pink tracks), and move between mitochondria (red tracks). FRAP analysis of RFP-Rab5 in TNFα/AcD-treated MCF-7 cells demonstrates rapid recovery of photobleached RFP-Rab5 fluorescence at mitochondria within seconds, accompanied by Rab5+ vesicular movements (Figures S2F-S2H).
To track another EL vesicle marker, we employed the iron carrier protein Transferrin (Tf). Tf is internalized via endocytosis and then trafficked by early ELs for lysosomal degradation or recycling to the plasma membrane (Gkouvatsos et al., 2012; Mayle et al., 2012). To visualize Tf+ ELs in MOMP+ cells, MCF-7 cells were incubated with fluorescent Tf-AF546 following tBID-GFP expression. Similar to Rab5+ ELs, Tf-AF546-loaded ELs interacted with tBID-GFP+ OMMs (Figure 2E). Strikingly, video microscopy reveals dynamic entrance of Tf-AF546+ vesicles into, and exit from, inner mitochondrial compartments (Video S3), consistent with the localization of Rab5+ vesicles inside MOMP+ mitochondria (Figure 1G). Of note, the observed robust cellular Tf-AF546 uptake indicates that functional endocytic trafficking was maintained in MOMP+ cells, regardless of the mitochondrial diversion of a large fraction of Rab5+ ELs.
We further utilized fluorescently labeled cholesterol, a sterol lipid that marks endosomal, and lysosomal membranes (Kobayashi et al., 1998; Luo et al., 2017). tBID-RFP transfected MCF-7 cells were loaded with Bodipy-CHOL (Wustner et al., 2016), in the presence of u18666A to enrich EL cholesterol content (Lu et al., 2015). In line with the above results, we detected prominent, stable targeting of Bodipy-CHOL+ vesicles to tBID-RFP+ mitochondria (Figure 2F and Video S4). Consistently, in cells co-expressing tBID-BFP with RFP-BAX, Bodipy-CHOL+ vesicle movements were concentrated at the mitochondrial compartment, proximal to RFP-BAX signals (Figure 2G). By comparison, Bodipy-CHOL+ vesicles displayed movement throughout the cytosol of non-transfected neighboring cells.
Remarkably, Bodipy-CHOL+ vesicles predominantly targeted regions of OMM marker discontinuities (Figure 2F and Video S4), previously described as areas of BAX pore localization (Grosse et al., 2016; Hamacher-Brady et al., 2014), prompting us to speculate that EL targeting of mitochondria may result in alterations to OMM lipid composition. To investigate this, we permeabilized cellular membranes with the cholesterol-selective detergent saponin (Seeman et al., 1973). In saponin-permeabilized MCF-7 control cells, co-immunostaining of OMM marker TOM20 and of the inner mitochondrial compartment-localized chaperone TRAP1 (Pridgeon et al., 2007) generated positive signals for TOM20 but not for TRAP1 (Figure 2H and 2I), consistent with the poor cholesterol content of OMMs (Elias et al., 1979). However, following apoptosis induction with TNFα/AcD or STS a significant fraction of cells displayed both TOM20 and TRAP1 fluorescence, indicating saponin permeabilization of OMMs and thus increased OMM cholesterol contents. Notably, TRAP1+ apoptotic cells also stained positive for mitochondrial-localized Rab5 (Figure 2J). TRAP1 antigens were not accessible in cells in which the plasma membrane was permeabilized by paraformaldehyde fixation alone (Figures 2K and 2L), thus excluding the possibility of detergent-independent antibody entry into apoptotic mitochondria through BAX/BAK pores.
We conclude that ELs and mitochondria increasingly interact prior to the onset of MOMP, with continued endocytic trafficking activities following MOMP, and that these interactions alter mitochondrial membrane lipid composition.
Largescale Apoptosis-Related EL Targeting of Mitochondria Occurs Rapidly and Temporally Coincides with MOMP
We next sought to understand the temporal relationship between MOMP and mitochondrial accumulation of ELs during apoptosis signaling. To that end, live cell time-lapse imaging of MOMP dynamics was performed at 1 min time intervals in MCF-7 cells following STS or TNFα/AcD treatment, starting at timepoints at which ~50+% of cells are MOMP+ (Figure 1B). As expected, STS treatment of MCF-7 cells led to MOMP activation, as evidenced by mitochondrial translocation and cluster formation of RFP-BAX, and release of Cyto c-GFP (Figure 3A and Video S5). Plotting single cell kinetics of mitochondrial-localized MOMP reporter fluorescence quantitatively demonstrates that release of Cyto c-GFP was completed rapidly and occurred concurrently with the initial appearance of RFP-BAX at mitochondria (Figure 3B). Notably, and in agreement with recent MOMP measurements (Maes et al., 2017), the majority of detectable RFP-BAX clustering ensues following MOMP, reaching its maximum 30+ min after Cyto c-GFP release.
Figure 3. ELs Accumulate within Apoptotic Mitochondria in a Rapid Manner, Coinciding with Mitochondrial BAX Clustering and Cytochrome c Release.
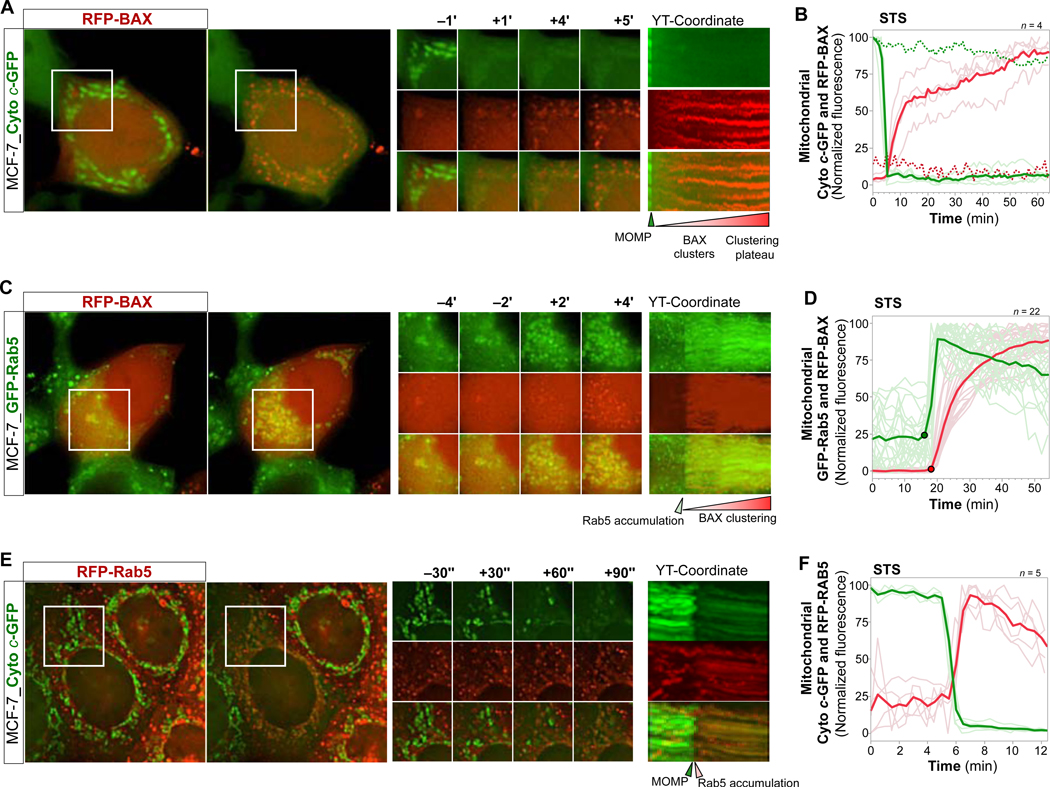
(A) MCF-7_Cyto c-GFP cells, transfected with RFP-BAX, live imaged every 1 min over 40+ min starting at 4 hrs of STS treatment. Overview images, time-series ROI, YT-coordinate of ROI projected over X. See also Video S5.
(B) Graph of mitochondrial Cyto c-GFP (green traces) and RFP-BAX (red traces) intensities of 4 cells treated and imaged as in (A). Bold traces, mean. Pale traces, individual cells. Dotted traces, reference intensities over time.
(C) MCF-7_GFP-Rab5_RFP-BAX cells, live imaged every 2 min over 40+ min at 4 hrs STS. Images displayed as in (A). See also Video S6.
(D) Graph of mitochondrial GFP-Rab5 (green) and RFP-BAX (red) intensities of 22 cells treated and imaged as in (C). Graph displayed as in (B).
(E) MCF-7_Cyto c-GFP_RFP-Rab5 cells, live imaged every 30 sec over 10+ min at 6 hrs TNFα/AcD. Images displayed as in (A). See also Video S7.
(F) Graph of mitochondrial Cyto c-GFP (green) and Rab5-RFP (red) intensities of 5 cells treated with STS for 4 hrs, and imaged as in (C). Graph displayed as in (B).
Scale bars, 10 μm.
We then analyzed the dynamics of GFP-Rab5 targeting to mitochondria in relation to STS-induced RFP-BAX translocation and clustering, at 1 min time intervals (Figure 3C, 3D and Video S6). Strikingly, we observed a robust temporal correlation between RFP-BAX targeting of mitochondria and largescale accumulation of GFP-Rab5 at mitochondria. Similar to Cyto c-GFP release, detection of mitochondrial GFP-Rab5 accumulation coincided with the initial appearance of RFP-BAX at mitochondria. Moreover, GFP-Rab5 targeting of the entire mitochondrial compartment was completed within a few minutes, at a rate similar to Cyto c-GFP release. Consistent with these findings, monitoring at 30 sec intervals following treatment with TNFα/AcD (Figure 3E and Video S7) or STS (Figure 3F) revealed that largescale mitochondrial targeting of RFP-Rab5 coincided with Cyto c-GFP release.
Taken together, we show that upon apoptosis stimulation transient interactions between ELs and mitochondria increase prior to MOMP (Figures 2A–2C), culminating in a global and rapid accumulation of ELs within the entire mitochondrial compartment, at a striking temporal correlation with the onset of MOMP (Figures 3C–3F).
Interference with the EL Compartment through Rab5 Isoform Knockdown (KD) Uncouples BAX Clustering at the OMM from Cytochrome c and Smac Release
Based on the remarkable spatial and temporal correlation between EL targeting of mitochondria and MOMP, we hypothesized that these inter-organelle interactions might be a regulatory component of MOMP. To test this, we first interfered with the EL compartment through KD of single isoforms of the central GTPase Rab5 (Wandinger-Ness and Zerial, 2014; Zeigerer et al., 2012) and tested the impact on MOMP parameters in response to apoptosis induction with STS or TNFα/AcD. Compared to shRNA control cells, MCF-7 cells with stable KD of either Rab5A or Rab5C displayed unaltered, to enhanced, mitochondrial BAX clustering (Figures 4A–4C and S3A). Cytochrome c release, however, was significantly reduced in Rab5A or Rab5C KD cells (Figures 4D and 4E), resulting in cellular populations with the atypical phenotype of mitochondrial compartments positive for BAX clusters, albeit retained cytochrome c (Figure 4F and Video S8). Similar results were obtained in HeLa cells (Figures 4G–4I). Overexpression of RFP-Rab5A in MCF-7 or HeLa Rab5C KD lines reinstated robust cytochrome c release (Figures 4J and 4K). Enhancing cellular Rab5 protein levels through RFP-Rab5 expression did not increase cytochrome c release sensitivity in response to STS (Figure 4L), indicating that baseline Rab5 levels are not a limiting factor in the here described apoptosis settings.
Figure 4. Interfering with EL Targeting of Mitochondria Through KD of Rab5 Uncouples BAX Clustering at OMMs from Cytochrome c and Smac Release.
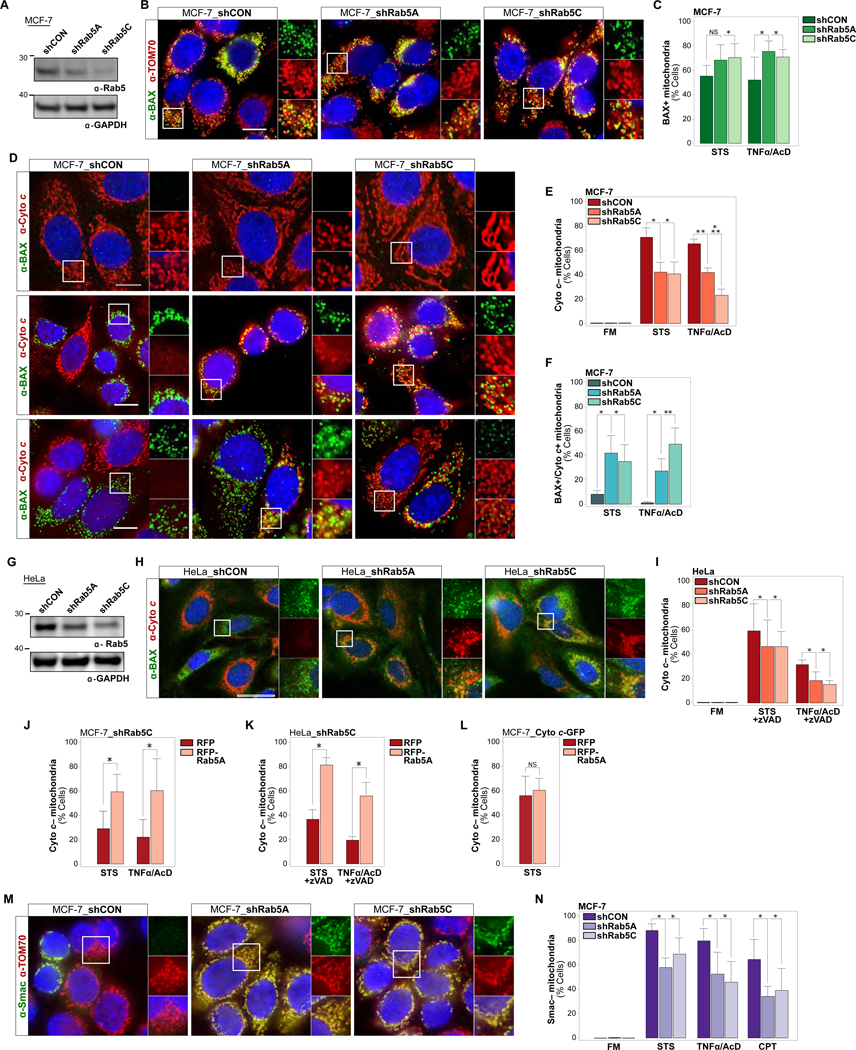
(A) Immunoblot of Rab5 levels in MCF-7 cells stably expressing shCON, shRab5A or shRab5C. GAPDH, loading control.
(B) Cells as in (A), treated with STS for 4 hrs, IF of BAX and TOM70.
(C) Quantifications of cells as in (B) in FM, or treated with STS for 4 hrs, or TNFα/AcD for 6 hrs scored for mitochondrial BAX clusters. > 350 cells per condition.
(D) MCF-7_shCON, MCF-7_shRab5A or MCF-7_shRab5C cells, in FM or treated with STS for 4 hrs, or TNFα/AcD for 6 hrs. IF of BAX and cytochrome c. See also Video S8.
(E) Quantifications of cells as in (D) scored for Cyto c– mitochondria. > 600 cells per condition.
(F) Quantifications of cells as in (D) scored for BAX+ mitochondria retaining cytochrome c (BAX+ / Cyto c+). > 600 cells per condition.
(G) Immunoblot of Rab5 levels in HeLa cells stably expressing shCON, shRab5A or shRab5C.
(H) HeLa_shCON, HeLa_shRab5A or HeLa_shRab5C cells, treated with TNFα/AcD +zVAD for 6 hrs. IF of BAX and cytochrome c.
(I) Quantification of cells as in (H), treated with STS for 5 hrs or TNFα/AcD for 6 hrs in the presence of zVAD, scored for Cyto c– mitochondria. > 300 cells per condition.
(J) MCF-7_shRab5C cells, transfected with RFP or RFP-Rab5A, treated with STS for 4 hrs or TNFα/AcD for 6 hrs, scored for Cyto c– mitochondria. > 200 cells per condition.
(K) HeLa_shCON or HeLa_shRab5C, transfected with RFP or RFP-Rab5A, treated with STS or TNFα/AcD in the presence of zVAD. IF of cytochrome c and TOM20, scored for Cyto c– mitochondria. > 160 cells per condition.
(L) MCF-7_Cyto c-GFP cells, transfected with RFP or RFP-Rab5A, treated with STS for 4 hrs, scored for Cyto c– mitochondria. Graph, mean of n = 5 experiments. > 350 cells per condition.
(M) MCF-7_shCON, MCF-7_shRab5A or MCF-7_shRab5C cells, treated with STS for 4 hrs, TNFα/AcD for 6 hrs, or CPT for 24 hrs. IF of Smac and TOM70. See also Figure S3B.
(N) Quantifications of cells as in (J) scored for Smac–mitochondria. > 600 cells per condition.
Nuclei stained with Hoechst. Scale bars, 10 μm. Graphs, mean of n = 3 experiments. Error bars, standard deviation. NS, not significant, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001
The observed uncoupling of BAX clustering at OMMs from cytochrome c release may be due to either impaired BAX pore formation, or incomplete release of cytochrome c due to effects on MOMP-related remodeling of the inner mitochondrial membrane (IMM) compartment, including opening of crista junctions (Frezza et al., 2006; Tait and Green, 2013). We thus examined the release of Smac, which, unlike cytochrome c, is not associated with the IMM through electrostatic interactions (Uren et al., 2005) and can be released independent of cytochrome c (Estaquier and Arnoult, 2007). Remarkably, stable KD of either Rab5A or Rab5C in MCF-7 cells significantly decreased TNFα/AcD or STS induced Smac release (Figure 4M, 4N and S3B).
Taken together, the above findings indicate that Rab5 KD-related alterations to the EL compartment functionally impact BAX pore formation, suggesting that apoptotic trigger-induced EL targeting of mitochondria actively contributes to MOMP.
Interference with EL Mobility through USP15 KD Impairs MOMP Functionality
The deubiquitinase USP15 is a recently described determinant of EL vesicle trafficking, where it functions to uncouple ubiquitin-binding endocytic adaptors and attached vesicles from ER anchorage, thereby releasing them for dynamic cellular transport (Jongsma et al., 2016). We hypothesized that USP15 may play a role in the dynamic targeting of ELs to mitochondria during apoptosis signaling. Indeed, stable USP15 KD in HeLa cells (Figure 5A) abolished the targeting of Rab5+ ELs to mitochondria in response to TNFα/AcD (Figures 5B and 5C). We thus employed USP15 KD as a Rab GTPase-independent means for pathway perturbation. Remarkably, USP15 KD led to significantly reduced cytochrome c release following treatment with STS or TNFα/AcD, in the presence of zVAD (Figures 5D–5F). However, neither mitochondrial targeting by BAX, nor BAX cluster formation, were impacted and large populations of cells displayed BAX+/cyto c+ mitochondria (Figures 5G-I). Thus, similar to as observed with Rab5 KD, USP15 depletion uncouples BAX aggregation from full MOMP execution.
Figure 5. Reducing EL Mobility Through KD of USP15 Impairs BAX-mediated Cytochrome c Release.

(A) Immunoblot for USP15 protein levels in HeLa_shCON, HeLa_shUSP15_1 or HeLa_shUSP15_2 cells. GAPDH, loading control.
(B) Cells as in (A) treated with TNFα/AcD +zVAD for 6 hrs. IF of Rab5 and TOM20.
(C) Quantification of cells as in (B), in FM or treated with TNFα/AcD +zVAD for 6 hrs, scored for mitochondrial accumulation of endogenous Rab5. ≥ 200 cells counted per condition.
(D) HeLa_shCON or HeLa_shUSP15_1 cells treated with STS +zVAD for 4 hrs. IF of cytochrome c.
(E) Cells as in (D), treated with TNFα/AcD + zVAD for 6 hrs. IF of cytochrome c.
(F) Quantification of HeLa_shCON, HeLa_shUSP15_1 or HeLa_shUSP15_2 cells treated with STS +zVAD for 4 hrs or TNFα/AcD +zVAD for 6 hrs. IF of BAX and cytochrome c. Cells scored for Cyto c– mitochondria. > 200 cells per condition.
(G) HeLa_shCON, HeLa_shUSP15_1 or HeLa_shUSP15_2 cells treated with STS +zVAD for 4 hrs. IF of BAX and cytochrome c.
(H) HeLa_shCON, HeLa_shUSP15_1 or HeLa_shUSP15_2 cells treated with TNFα/AcD + zVAD for 6 hrs. IF of BAX and cytochrome c.
(I) Quantification of cells as in (F) scored for BAX+ / Cyto c+ mitochondria. > 500 cells per condition.
(J) Immunoblot for USP15 protein levels in MCF-7_shCON, MCF-7_shUSP15_1 or MCF-7_shUSP15_2 cells.
(K) MCF-7_shCON, MCF-7_shUSP15_1 or MCF-7_shUSP15_2 cells treated with STS for 4 hrs or TNFα/AcD for 6 hrs. IF of Rab5 and TOM20. Cells scored for mitochondrial accumulation of Rab5. ≥ 300 cells counted per condition.
(L) MCF-7_shCON or MCF-7_shUSP15_1 cells, treated with STS for 4 hrs. IF of cytochrome c.
(M) Cells as in (L), treated with TNFα/AcD for 6 hrs. IF of cytochrome c.
(N) Quantifications of cells as in (L, M), scored for Cyto c– mitochondria. > 500 cells per condition.
(O) MCF-7_shCON or MCF-7_shUSP15_1 cells treated with STS for 4 hrs. IF of BAX and TOM70.
(P) Cells as in (O) treated with TNFα/AcD for 6 hrs. IF of BAX and TOM70.
(Q) Quantifications of cells as in (O) and (P), scored for BAX+ mitochondria. > 500 cells per condition.
(R) Immunoblot of USP15 protein levels in MCF-7_Cyto c-GFP_shCON and MCF-7_Cyto c-GFP_shUSP15_1 cells.
(S) MCF-7_Cyto c-GFP_shCON and MCF-7_Cyto c-GFP_shUSP15_1 cells transfected with RFP-Rab5, treated with TNFα/AcD and live imaged in 1 min intervals for 360 min. Representative time-course analysis of mitochondrial loss of Cyto c-GFP (green traces) and mitochondrial accumulation of RFP-Rab5 (red traces), 4 cells per condition.
(T) MCF-7_Cyto c-GFP_shCON and MCF-7_Cyto c-GFP_shUSP15_1 cells, treated with STS or TNFα/AcD and live imaged in 1 min intervals for 10 hrs. From time lapse datasets (S), the onset of Cyto c release (time to MOMP) was calculated for single cells. Box plot, 26–49 time courses analyzed per condition. Error bars, standard deviation.
Nuclei stained with Hoechst. Scale bars, 10 μm. Graphs, mean of n = 3 experiments. Error bars, standard deviation. NS, not significant, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001
Also in MCF-7 cells, stable USP15 KD (Figure 5J) impaired mitochondrial targeting by Rab5+ ELs (Figure 5K), and strongly reduced cytochrome c release in response to STS or TNFα/AcD (Figures 5L–5N), without impacting mitochondrial BAX clustering (Figures 5O–5Q). Time-lapse imaging of MCF-7_Cyto c-GFP cells further reveals that in the subpopulation of USP15 KD cells that eventually undergo MOMP, the time to MOMP was significantly increased (Figures 5R–5T). Importantly, also here, RFP-Rab5 accumulation in the mitochondrial compartment coincided with the onset of Cyto c-GFP release (Figure 5S).
These data identify USP15 as a regulator of intrinsic cell death signaling and corroborate our conclusions drawn from the effects observed with Rab5 KD, suggesting that during apoptosis signaling, EL interactions with mitochondria are required for full MOMP functionality.
The Rab5 GEF Rabex-5 Functionally Controls MOMP Upstream of Mitochondrial BAX Clustering
We next sought to interfere with EL targeting of mitochondria at the level of Rab5 GEFs, which activate Rab5 on membranes through catalyzing the release of GDP and binding of GTP (Pfeffer, 2013; Zerial and McBride, 2001). Recently, the Rab5 GEF ALS2/Alsin (Otomo et al., 2003) was reported to localize to mitochondria in response to H2O2 and mediate mitochondrial Rab5 recruitment, but counteract cytochrome c release and caspase activation (Hsu et al., 2018). Thus, we here investigated a potential MOMP regulatory role for ALS2 during pharmacological apoptosis induction, using MCF-7 and HeLa cells with stable KD of ALS2 (Figures 6A and 6B). ALS2 KD did not block Rab5 targeting to mitochondria in MCF-7 cells treated with STS or TNFα/AcD (Figure 6C), nor did it affect cytochrome c release (Figure 6E) or cause the BAX+/Cyto c+ phenotype (Figure 6F). However, overexpression of GFP-ALS2 decreased TNFα/AcD-induced cytochrome c release in MCF-7 cells (Figure 6H). Conversely, while stable KD of ALS2 also in HeLa cells failed to affect Rab5 targeting to mitochondria in response to STS+zVAD or TNFα/AcD+zVAD (Figure 6D), it sensitized cells to cytochrome c release under these treatments (Figure 6G). Thus, in line with Hsu et al., ALS2 appears to have a protective potential in the apoptosis models employed here, albeit not correlated with an impact on mitochondrial Rab5 accumulation, and depending on cell type-dependent alterations of ALS2 expression levels.
Figure 6. KD of Rab5 GEF Rabex-5, but not of ALS2, Reduces both BAX Accumulation at OMMs and Cytochrome c release.
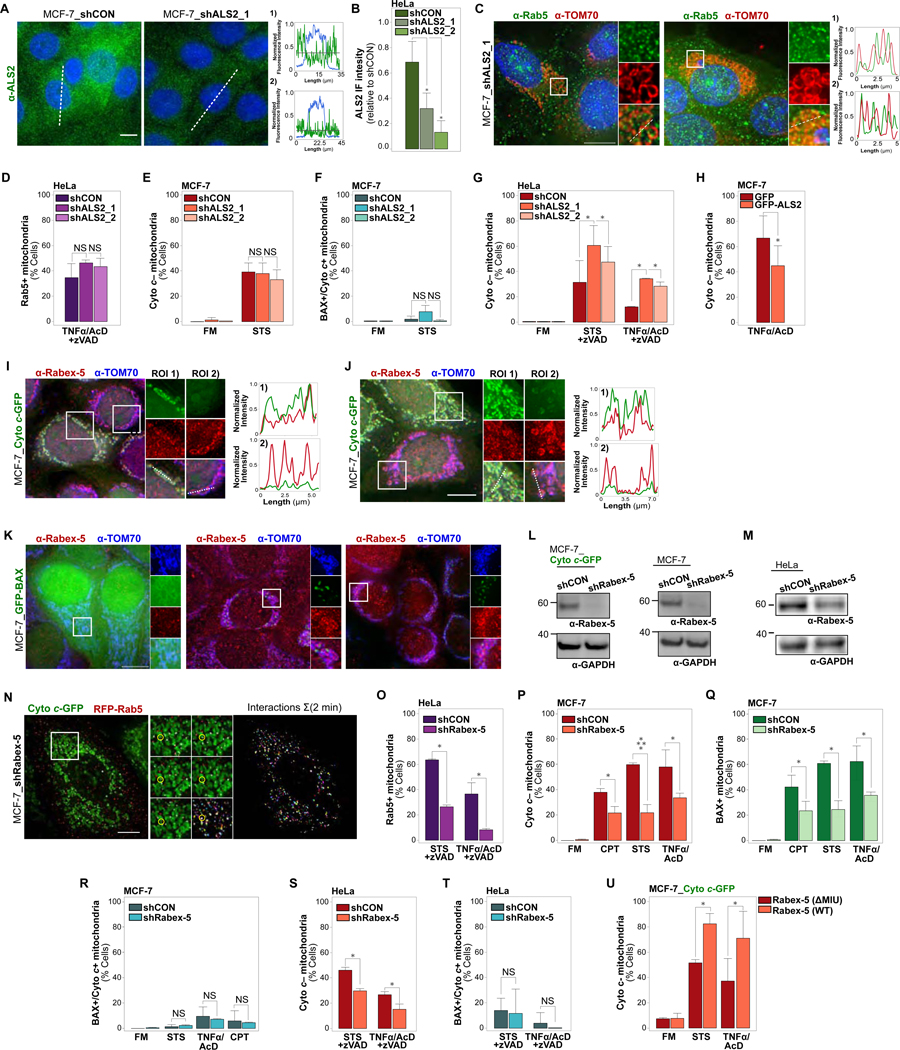
(A) MCF-7_shCON or MCF-7_shALS2_1 cells, IF stained for ALS2. Representative images and intensity profiles over dotted lines. Black lines, mean intensity of ALS2 signal over dotted line.
(B) Quantification of HeLa_shCON, HeLa_shALS2_1 or HeLa_shALS2_2 cells, IF stained for ALS2. Graph, representative analysis of 20 cell mean cytoplasmic ALS2 IF intensities per condition, of n = 3 experiments. Error bars, standard deviation.
(C) MCF-7_shALS2_1 cells treated with STS (4 hrs) or TNFα/AcD (6 hrs). IF of Rab5 and TOM70.
(D) HeLa_shCON, HeLa_shALS2_1 or HeLa_shALS2_2 cells, treated with TNF/AcD +zVAD (6 hrs). IF of Rab5 and TOM70. Cells scored for mitochondrial Rab5. ≥ 800 cells per condition.
(E) MCF-7_shCON, MCF-7_ALS2_1 or MCF-7_shALS2 cells, in FM or treated with STS (4 hrs). IF of cytochrome c, scored for Cyto c– mitochondria. > 800 cells per condition.
(F) MCF-7_shCON, MCF-7_shALS2_1 or MCF-7_shALS2_2 cells, in FM or treated with STS (4 hrs). IF of cytochrome c and BAX. Cells scored for BAX+ mitochondria with retained cytochrome c (BAX+ / Cyto c+). > 500 cells per condition.
(G) HeLa_shCON, HeLa_shALS2_1 or HeLa_shALS2_2 cells, treated with STS (4 hrs) or TNFα/AcD (6 hrs) + zVAD. IF of cytochrome c and TOM20. Cells scored for Cyto c– mitochondria. > 800 cells per condition.
(H) MCF-7 cells transfected with GFP or GFP-ALS2, treated with TNFα/AcD (6 hrs). IF of cytochrome c and TOM20. Cells scored for Cyto c– mitochondria. > 800 cells per condition.
(I) MCF-7_Cyto c-GFP cells, IF of Rabex-5 and TOM70 at 4 hrs of STS. Zoom images and intensity profiles of ROIs. ROI 1), MOMP– cell; ROI 2), MOMP+ cell.
(J) MCF-7_Cyto c-GFP cells, IF of Rabex-5 and TOM70 at 6 hrs of TNFα/AcD. Zoom images and intensity profiles of ROIs. ROI 1), MOMP– cell; ROI 2), MOMP+ cell.
(K) MCF-7_GFP-BAX cells, IF of Rabex-5 and TOM70, in FM or at 6 hrs of TNFα/AcD, or 4 hrs of STS.
(L) Immunoblots of Rabex-5 protein levels in MCF-7_Cyto c-GFP (left) or MCF-7 (right) cells stably expressing shCON or shRabex-5.
(M) Immunoblot of Rabex-5 protein levels in HeLa cells stably expressing shCON or shRabex-5.
(N) MCF-7_Cyto c-GFP_shRabex-5 cells, imaged live every 5 sec for 2 min at 6 hrs TNFα/AcD. Left, merged channel image of single time-point. Middle, ROI time-series of merged channels with interactions overlaid in white. Right, color-coded time-step 2 min projection.
(O) HeLa_shCON or HeLa_shRabex-5 cells, treated with STS +zVAD (4 hrs) or TNFα/AcD +zVAD (6 hrs). IF of Rab5 and TOM20. Cells scored for Rab5+ mitochondria. > 150 cells per condition.
(P) MCF-7_shCON or MCF-7_shRabex-5 cells in FM or treated with STS (4 hrs), TNFα/AcD (6 hrs), or CPT (24 hrs). IF of cytochrome c and BAX. Cells scored for Cyto c+ mitochondria. > 550 cells per condition.
(Q) Cells, treatments, IF and graph representation as in (P). Cells scored for BAX+ mitochondria. > 550 cells per condition.
(R) Cells, treatments, IF and graph representation as in (P). Cells scored for BAX+ mitochondria with retained cytochrome c (BAX+ / Cyto c+). > 550 cells per condition.
(S) HeLa_shCON or HeLa_shRabex-5 cells, in FM or treated with STS +zVAD (4 hrs), or TNFα/AcD +zVAD (6 hrs). IF of cytochrome c and BAX. Cells scored for Cyto c– mitochondria. Graph, mean of n = 3 experiments. Error bars, standard deviation. > 200 cells per condition. *p ≤ 0.05
(T) Cells and treatments as in (S). Cells scored for BAX+ / Cyto c+ mitochondria. > 550 cells per condition.
(U) MCF-7_Cyto c-GFP cells expressing RFP-Rabex-5 or RFP-Rabex-5A58G (∆MIU), treated with STS (4 hrs) or TNFα/AcD (6 hrs). Cells scored for Cyto c-GFP– mitochondria.
Graphs, mean of n = 3 experiments. Error bars, standard deviation. > 150 cells per condition. NS, not significant; *p ≤ 0.05, ****p ≤ 0.0001
As ALS2 KD did not impair Rab5 accumulation at mitochondria under the apoptosis settings employed here, and its impact on cytochrome c release was variable and inverse to Rab5 and USP15 KD impacts, we next turned to the Rab5 GEF Rabex-5/RabGEF1 (Horiuchi et al., 1997). Rabex-5 determines the subcellular membrane localization of Rab5 activity (Blumer et al., 2013) and translocates to OMMs of mitochondria targeted by pro-apoptotic BH3-only proteins tBid or BimEL (Hamacher-Brady et al., 2014). Indeed, endogenous Rabex-5 was prominently recruited to OMMs in response to STS or TNFα/AcD (Figures 6I and 6J). Notably, mitochondrial Rabex-5 was most pronounced in cells that had undergone cytochrome c release. Similarly, Rabex-5 colocalized with GFP-BAX+ mitochondria in response to STS or TNFα/AcD (Figure 6K). For functional investigation, we generated MCF-7, MCF-7_Cyto c-GFP and HeLa cells with stable Rabex-5 KD (Figures 6L and 6M). MCF-7_Cyto c-GFP Rabex-5 KD cells displayed decreased interactions between RFP-Rab5+ ELs and mitochondria during TNFα/AcD treatment, prior to MOMP (Figure 6N), compared to MCF-7_Cyto c-GFP cells (Figure 2B). In HeLa cells, stable Rabex-5 KD reduced Rab5 accumulation at mitochondria following STS or TNFα/AcD, in the presence of zVAD (Figure 6O). Moreover, stable Rabex-5 KD in MCF-7 cells strongly protected against cytochrome c release in response to CPT, STS or TNFα/AcD (Figure 6P). Intriguingly, differing from impacts observed with Rab5 and USP15 KD, Rabex-5 KD also inhibited mitochondrial BAX clustering (Figures 6Q, 6R). Similar effects were observed in HeLa Rabex-5 KD cells in response to STS or TNFα/AcD, in the presence of zVAD (Figures 6S and 6T). Conversely, overexpression of RFP-Rabex-5 sensitized MCF-7_Cyto c-GFP cells to STS or TNFα/AcD induced cytochrome c release, while overexpression of RFP-Rabex-5A58G did not impact cytochrome c release (Figure 6U). RFP-Rabex-5A58G is mutated in its motif interacting with ubiquitin (MIU) (Lee et al., 2006; Penengo et al., 2006), which determines its binding to ELs (Mattera and Bonifacino, 2008) and recruitment to BH3-only protein-targeted mitochondria (Hamacher-Brady et al., 2014).
These data reveal that the potential of ALS2 as a cytochrome c release inhibitory factor varies in a cell type-dependent manner, and does not appear to be correlated with apoptosis-induced Rab5 targeting of mitochondria. Importantly, we identified a general regulatory role for Rabex-5 in driving MOMP upon pharmacological apoptosis induction, in a manner upstream of BAX clustering at OMMs and dependent on its ubiquitin-binding ability.
Impact of EL-Mitochondrial Interactions on BAX Conformational Changes and Oligomerization and Mitochondrial Membrane Transformation
As KD of either Rab5A or Rab5C, or USP15, lead to the uncoupling of BAX cluster formation at OMMs from cytochrome c and Smac release, we investigated a possible impact on known mechanistic parameters of BAX-mediated MOMP. In the course of its activation BAX undergoes a series of conformational changes that are prerequisite to functional pore formation (Leber et al., 2007). Amongst these changes is the membrane interaction-related exposure of an N-terminal epitope of BAX that is detected by 6A7 monoclonal antibodies (Nechushtan et al., 1999; Yethon et al., 2003). To assess whether uncoupling of BAX cluster formation from functional MOMP is associated with alterations to BAX conformational changes, we performed simultaneous immunofluorescence detection of total BAX and of the BAX conformer with 6A7 epitope exposure. Consistent with the above (Figure 4), in response to TNFα/AcD or STS, MCF-7 and HeLa cells with stable KD of either Rab5A or Rab5C displayed unaltered clustering of BAX at OMMs, when detected with conformation-independent α-BAX N-20 antibody (Figures 7A and 7C). Remarkably, BAX clusters in Rab5A or Rab5C KD cells displayed strongly reduced 6A7 immunofluorescence, compared to control cells (Figures 7A-7D). Similarly reduced cellular levels of 6A7-exposed BAX were detected through immunoprecipitation in MCF-7 cells with stable KD of Rab5A, Rab5C, USP15 or Rabex-5 (Figures 7E and 7F).
Figure 7. EL Targeting of Apoptotic Mitochondria Alters OMM Lipid Composition and Impacts BAX Pore Formation and Downstream Cell Death Signaling.
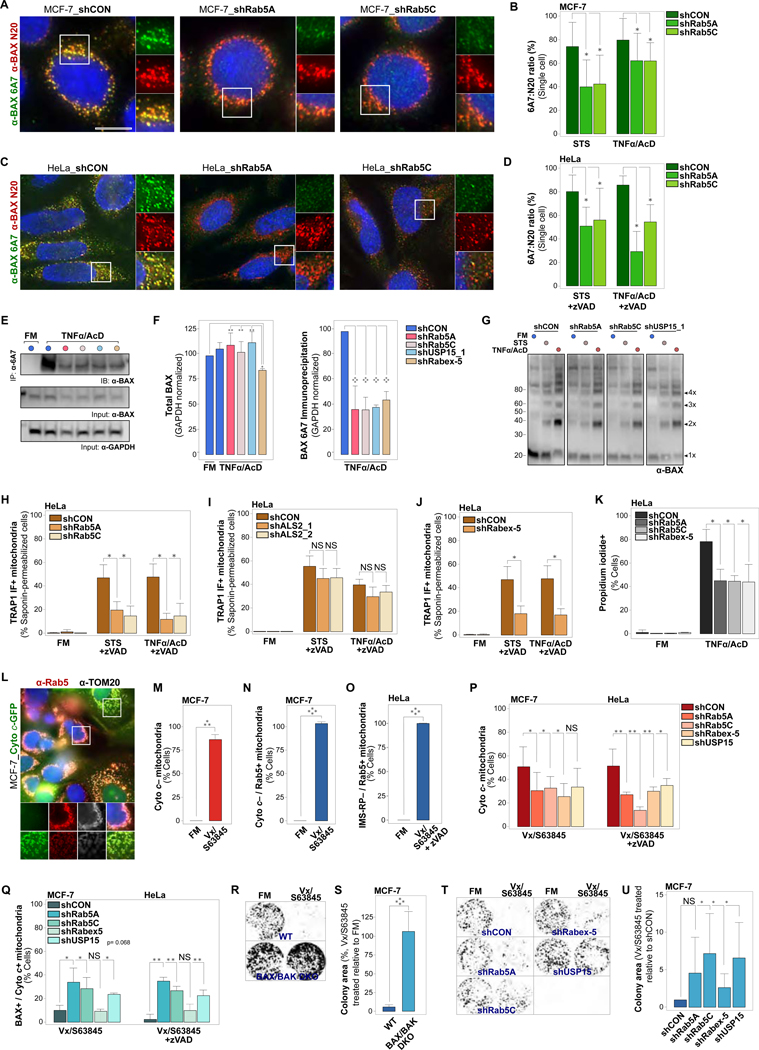
(A) MCF-7_shCON, MCF-7_shRab5A or MCF-7_shRab5C cells, treated with STS (4 hrs), or TNFα/AcD (6 hrs). IF of total cellular BAX (α-BAX N-20) and of BAX in active conformation (α-BAX 6A7).
(B) Quantification of cellular BAX 6A7 over N-20 ratios for cells as in (A). Differences between N20 and 6A7 fluorescence intensities calculated for individual cells. Graphs, plots of 10 cells per condition, from 1 experiment representative of n = 3.
(C) HeLa_shCON, HeLa_shRab5A or HeLa_shRab5C cells, treated with STS (4 hrs), or TNFα/AcD (6 hrs) + zVAD. IF of total cellular BAX (α-BAX N-20) and of BAX in active conformation (α-BAX 6A7).
(D) Quantification of cellular BAX 6A7 over N-20 ratios for cells as in (C), calculated and graphed as in (B). Graphs, plots of 19 cells per condition, from 1 experiment representative of n = 3.
(E) MCF-7 cells stably expressing shCON, shRab5A, shRab5C, shRabex-5 or shUSP15_1, treated with TNFα/AcD (6 hrs). Immunoprecipitation of 6A7 epitope-exposed BAX conformer using α-BAX 6A7. Immunodetection of BAX using α-BAX N20. GAPDH, loading control.
(F) Left, Quantification of total BAX (N-20) in input, normalized to GAPDH, from n = 3 as in(E). Right, Quantification of BAX 6A7 immunoprecipitates, normalized to GAPDH, from n = 3 as in (E).
(G) Immunoblot of MCF-7 cells stably expressing shCON, shRab5A, shRab5C or shUSP15_1, crosslinked with DSS in FM or at 4 hrs STS or 6 hrs TNFα/AcD. Immunodetection of total BAX. All membrane crops stem from the same membrane and were scanned and processed with identical settings.
(H) HeLa_shCON, HeLa_shRab5A or HeLa_shRab5C cells, in FM, or treated with STS (4 hrs) or TNFα /AcD (6 hrs) +zVAD. IF of saponin-permeabilized cells (saponin-IF) for TOM20 and TRAP1. Cells scored for TRAP1 saponin-IF+ mitochondria. Graphs, mean of n = 3 experiments. >150 cells per condition.
(I) HeLa_shCON, HeLa_shALS2_1 or HeLa_shALS2_2 cells, in FM, or treated with STS (4 hrs) or TNFα/AcD (6 hrs) +zVAD. Saponin-IF of TOM20 and TRAP1. Cells scored for TRAP1 saponin-IF+ mitochondria. Graphs, mean of n = 2 experiments. > 100 cells per condition.
(J) HeLa_shCON or HeLa_Rabex-5 cells, in FM, or treated with STS (4 hrs), or TNFα/AcD (6 hrs) +zVAD. Saponin-IF of TOM20 and TRAP1. Cells scored for TRAP1 saponin-IF+ mitochondria. Graphs, mean of n = 3 experiments. > 550 cells per condition.
(K) HeLa_shCON, Hela_shRab5A, HeLa_shRab5C and HeLa_shRabex-5, treated with TNF α/AcD (6 hrs), stained with Hoechst and propidium iodide (PI), scored for PI+ cells. Graphs, mean of n = 3 experiments. > 175 cells per condition.
(L) MCF-7_Cyto c-GFP cells, in FM or treated with Vx/S63845 (10 μM/5 μM) for 6 hrs, IF of Rab5 and TOM20.
(M) Cells from (L) were scored for mitochondria with released cytochrome c (Cyto c–). Graph, mean of n = 3 experiments. ≥ 175 cells per condition.
(N) Cells from (L) were scored for Cyto c– mitochondria targeted by Rab5 (Cyto c– / Rab5+). Graph, mean of n = 3 experiments. ≥ 175 cells per condition.
(O) HeLa_IMS-RP cells, in FM or treated with Vx/S63845 +zVAD for 6 hrs. IF of Rab5 and TOM20. Cells scored for mitochondria with released IMS-RP targeted by Rab5 (IMS-RP– / Rab5+). Graphs, mean of n = 3 experiments. ≥ 175 cells per condition.
(P) MCF-7 and HeLa shCON, shRab5A, shRab5C, shRabex-5 and shUSP15 KD cells in FM or treated for 6 hrs with Vx/S63845 or Vx/S63845 +zVAD, respectively. IF of BAX and cytochrome c. Cells scored for Cyto c– mitochondria. Graphs, mean of n = 3 experiments. > 170 cells per condition.
(Q) Cells as in (P), scored for BAX+ mitochondria with retained cytochrome c (BAX+ / Cyto c+). ≥ 170 cells per condition.
(R) MCF-7 WT and BAX/BAK DKO cells were treated with Vx/S63845 for 6 hrs. Representative images of colony formation at 7 days following treatment.
(S) Quantification of colony area for experiment in (R). n = 3 experiments.
(T) MCF-7_shCON, shRab5A, shRab5C, shRabex-5 and shUSP15 cells were treated with Vx/S63845 for 6 hrs. Representative images of colony formation at 7 days following treatment.
(U) Quantification of colony area for experiment in (T). n = 5 experiments.
Nuclei stained with Hoechst. Scale bars, 10 μm. Graphs: Error bars, standard deviation. NS, not significant, *p < 0.05, **p ≤ 0.01, ****p ≤ 0.0001
Following BAX conformational changes, BAX forms dimers and then higher order oligomers, a process that is thought to be related to its membrane perforation activity (Birkinshaw and Czabotar, 2017; Volkmann et al., 2014). We tested whether BAX oligomerization states are altered under conditions of uncoupled BAX clustering and cytochrome c release through chemical crosslinking. Compared to control cells, MCF-7 cells with stable KD of Rab5A, Rab5C or USP15 contained slightly reduced, but not abolished, levels of BAX oligomerization in response to STS or TNFα/AcD (Figure 7G).
BAX pore formation is impacted by membrane lipid composition (Christenson et al., 2008; Lucken-Ardjomande et al., 2008), and we showed that EL-mitochondrial interactions are associated with increased OMM cholesterol content (Figures 2H–2K). We thus tested whether the endocytic regulators, which in the above we identified to exert control over MOMP, also govern the biochemical transformation of OMMs of apoptotic mitochondria. Indeed, KD of Rab5A, Rab5C or Rabex-5, but not of ALS2, diminished the increased sensitivity of OMMs to the cholesterol-based detergent saponin in HeLa cells treated with STS+zVAD or TNFα/AcD+zVAD (Figures 7H-7J and S4A-S4C). Furthermore, HeLa cells with stable KD of Rab5A, Rab5C or Rabex-5 displayed significantly reduced levels of cell death in response to treatment with TNFα/AcD (Figure 7K), indicating that the observed inhibitory impacts on MOMP are of functional relevance for downstream cell death signaling. Together, these data suggest a mechanism wherein, upon apoptosis induction, EL targeting of mitochondria establishes an OMM lipid environment that is conducive to BAX accumulation and pore formation-associated conformational changes.
ELs Target Mitochondria in Response to Combinatorial Treatment with Venetoclax and S63845, and Impact MOMP and Clonogenic Survival
To test whether interference with EL targeting of mitochondria manifests in improved cellular long-term survival, and thus sustained impairment of pro-death signaling of MOMP, we induced MOMP through treatment with the BCL-2 inhibitor venetoclax/ABT-199 (Vx) (Souers et al., 2013) in combination with the MCL-1 inhibitor S63845 (Kotschy et al., 2016). Similar to the above treatment strategies, in both MCF-7 and HeLa cells, Vx/S63845-induced MOMP was associated with Rab5 targeting to mitochondria (Figures 7L-7O). Moreover, KD of Rab5A, Rab5C and USP15 reduced cytochrome c release, including from BAX+ mitochondria, while Rabex-5 KD impaired both cytochrome c release and mitochondrial BAX accumulation (Figures 7P and 7Q). Long-term clonogenic survival of MCF-7 cells following Vx/S63845 treatment was rescued by CRISPR/Cas9-mediated BAX/BAK double knockout (DKO) (Figures 7R and 7S), and significantly improved by Rab5C, Rabex-5 and USP15 KD (Figures 7T and 7U).
Of note, while Rabex-5 KD blocked MOMP upstream of BAX accumulation at OMMs, it was less effective in rescuing long-term survival compared to Rab5 and USP15 KD. It is important to consider that KD of key endocytic regulators such as Rab5, Rabex-5 and USP15, in addition to interfering with apoptosis-related EL targeting of mitochondria, will to a degree impact overall functioning of the endocytic compartment. Indeed, while all KD lines were still active in endocytic uptake and trafficking of transferrin, mild alterations were detectable (Figures S5A-S5D). Thus, it is conceivable that colony outgrowth following cellular stress would be differentially impacted by MOMP-independent, distinct roles of Rab5, USP15 and Rabex-5 in cellular survival and proliferation (Homma et al., 2019; Peng et al., 2019; Tam et al., 2015; Zou et al., 2014).
Taken together, these data underscore the relevance of EL regulation of MOMP and cellular survival in response to the clinically promising drug combination Vx/S63845 (Kotschy et al., 2016; Letai, 2016; Li et al., 2019; Prukova et al., 2019; Roberts et al., 2016).
DISCUSSION
We here demonstrate that EL targeting of mitochondria is fundamentally integrated into the process of MOMP. Live cell microscopy recording of MOMP-related events at high spatial and temporal resolution revealed that various pharmacological inducers of intrinsic apoptosis signaling trigger increased transient interactions between ELs and mitochondria, culminating in an accumulation of ELs within the entire mitochondrial compartment at the onset of MOMP. Functionally, we found that EL targeting of mitochondria contributes to full MOMP execution during intrinsic apoptosis signaling.
We investigated the active participation of EL-mitochondrial interactions in the process of MOMP through complementary approaches, in response to apoptosis induction with STS, TNFα/AcD or CPT (MCF-7 cells) or STS+zVAD or TNFα/AcD+zVAD (HeLa cells). We perturbed cellular EL dynamics via KD of the early EL regulators Rab5A or Rab5C (Bucci et al., 1992; Zeigerer et al., 2012), or of the EL mobility regulator USP15 (Jongsma et al., 2016). Additionally, we interfered with EL signaling through KD of the Rab5 GEFs ALS2 (Otomo et al., 2003) or Rabex-5 (Horiuchi et al., 1997). Of note, ALS2 contributes to Rab5 targeting of mitochondria in response to H2O2 or laser irradiation (Hsu et al., 2018), and Rabex-5 localizes to OMMs upon BH3-only protein-activated intrinsic apoptosis signaling (Hamacher-Brady et al., 2014). Strikingly, while KD of either Rab5A, Rab5C, or USP15 did not block BAX clustering at OMMs, it significantly reduced STS(+zVAD)- or TNFα/AcD(+zVAD)-induced cytochrome c release. Similar to BAX clustering results, we did not detect impacts on BAX oligomerization in response KD of Rab5A, Rab5C, or USP15. However, we found that pore forming activity-related BAX 6A7 epitope exposure was significantly reduced, in line with the observed uncoupling of BAX recruitment to and oligomerization at OMMs from functional BAX-mediated MOMP.
KD of Rabex-5 also impaired Rab5 targeting to apoptotic mitochondria and had a strong inhibitory effect on CPT-, STS- or TNFα/AcD-induced cytochrome c release. Differing from the inhibition of cytochrome c release downstream of BAX clustering as seen with USP15 and Rab5 KD, Rabex-5 KD phenocopied BCL-2 overexpression in that it potently blocked mitochondrial BAX accumulation and clustering, and thus interfered with MOMP execution upstream of BAX. We therefore speculate that Rabex-5 is particularly important in recruiting EL subpopulations and factors to the OMM that are important for BAX pore formation. Based on the BAX clustering and cytochrome c release uncoupling potential, as seen with USP15 and Rab5 KD, we further speculate that there are subsequent Rabex-5-independent regulatory steps, controlled by distinct EL-mitochondrial interactions, that are required for functional MOMP, i.e. cytochrome c release.
In contrast, KD of ALS2, albeit without inhibiting Rab5 targeting to mitochondria, sensitized to cytochrome c release in HeLa but not MCF-7 cells, while ALS2 overexpression was cytoprotective in MCF-7 cells. Similarly, overexpression of ALS2 reduced caspase activation in H2O2-treated HeLa cells, and depletion of ALS2 impaired mitochondrial Rab5 recruitment and sensitized spinal motor neurons to H2O2-induced cytochrome c release (Hsu et al., 2018). Thus, ALS2 holds the potential to negatively regulate MOMP signaling, in a cell type- and condition-dependent manner. We here identified Rabex-5 as a critical positive regulator of BAX-mediated MOMP in response to CPT-, STS- and TNFα/AcD-mediated apoptosis signaling. Of note, in the high-grade oxidative stress condition of 100–500 μM H 2O2 (Hsu et al., 2018), membrane alterations caused by lipid peroxidation and the related impacts on endomembrane integrity (Dingjan et al., 2016; Wong-Ekkabut et al., 2007) are likely affecting membrane-based organelle interactions. This might in part explain the observed differences to our findings. In line with this assumption, H2O2 treatment impaired endocytic trafficking of transferrin (Hsu et al., 2018), while transferrin trafficking remained functional in response to the here employed pharmacological apoptosis inducers. Furthermore, we propose the existence of multiple, stress-specific modes of EL-mitochondrial interactions that are characterized by the engagement of specific EL regulators and subpopulations.
Notably, we detected interactions of Bodipy-CHOL+ ELs with MOMP+ mitochondria. Live microscopy revealed that cholesterol+ vesicles targeted regions of OMM protein marker discontinuities, previously identified as Rabex-5-marked OMM subdomains (Hamacher-Brady et al., 2014). Intriguingly, BAX cluster localization was confined to areas of OMM marker discontinuities, and thus presumably to the interface between apoptotic OMM subdomains (Hamacher-Brady et al., 2014). We therefore speculated that the observed interactions between ELs and mitochondria might result in local alterations to OMM lipid composition, through lipid transfer at membrane contact sites (Chen et al., 2018; Prinz, 2014; Wilhelm et al., 2017). While mitochondrial membranes are primarily comprised of phospholipids (Osman et al., 2011), late endosomal and lysosomal membranes contain the sterol lipid cholesterol (Luo et al., 2017). Indeed, apoptosis induction increased the sensitivity of OMMs to the cholesterol-dependent detergent saponin, in a Rab5 and Rabex-5 dependent manner. Remarkably, several studies have described alterations to BAX activities as a result of altered membrane cholesterol contents. These studies reported that enhanced membrane cholesterol contents increased BAX association, but reduced BAX membrane insertion (Christenson et al., 2008; Lucken-Ardjomande et al., 2008). Another report described a positive correlation between mitochondrial accumulation of cholesterol, and cytochrome c release, in rat hearts subjected to cardiac ischemia/reperfusion injury (Martinez-Abundis et al., 2009). Furthermore, cholesterol can promote phase-separated domains (Stepanyants et al., 2015), and the localization of endocytic effectors to specific OMM regions (Hamacher-Brady et al., 2014) is supportive of the formation of OMM microdomains with distinct lipid composition. It is perceivable that the observed membrane alterations positively contribute to in situ apoptotic BAX activation and functional pore formation.
ELs have been described as a BAX reservoir (Calore et al., 2010). Moreover, BAX/BAK-dependent targeting of XIAP to depolarized mitochondria is associated with EL and proteasomal degradation of Smac and, when pre-activated, confers protection from TNFα-induced caspase activation (Hamacher-Brady and Brady, 2015; Hamacher-Brady et al., 2014). We thus suggest that, in addition to the here uncovered role of EL-mitochondrial interactions in apoptotic pore formation, the observed sustained post-MOMP targeting of mitochondria by ELs may function to fine-tune various MOMP outcomes (Bock and Tait, 2019). Future work will be aimed at the uncovering of pathway-specific regulators to enable discrimination between indirect effects of the endocytic compartment and direct impacts through EL-mitochondrial interactions, and further in-depth investigation of the intricate mechanistic details underlying the multi-layered EL control of MOMP.
Taken together, we show that ELs globally target mitochondria in response to various pharmacological inducers of intrinsic apoptosis signaling. This inter-organelle communication is spatially and temporally integrated with BAX-mediated MOMP, and EL-mitochondrial interactions actively contribute to functional in situ MOMP execution and downstream cell death signaling. Thus, we propose EL-mitochondrial interactions constitute a previously unknown fundamental layer of intrinsic apoptosis regulation.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Anne Hamacher-Brady (abrady9@jhu.edu).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement negotiated as governed by the Johns Hopkins University and state requirements and, where applicable, covering costs associated with preparation and shipping.
Data and Code Availability
This study did not generate unique code or dataset.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Culture and Stable Cell Line Generation
Human MCF-7 breast cancer cells (CLS Cell Lines Service) and HeLa cervical cancer cells (ATCC) were maintained in DMEM (1 g/L D-glucose, 0.11 g/L sodium pyruvate). Lenti-X embryonic kidney 293T (Clontech) cells were maintained in DMEM (4.5 g/L D-glucose). Media were supplemented with 10% FBS (Sigma), GlutaMAX (Gibco), non-essential amino acids (Gibco), penicillin/streptomycin/amphotericin B (Gibco), and 100 μg/mL Normocin (Invivogen). Cell lines were routinely controlled for mycoplasma contamination using Hoechst 33342 (1 μg/mL; Thermo Fisher Scientific). Transient transfections were performed using jetPRIME (Polyplus) culturing media renewed 4–6 hrs following transfection.
MCF-7 cells stably expressing cytochrome c-GFP (MCF-7_Cyto c-GFP), GFP-BAX (MCF-7_GFP-BAX), and RFP-BAX (MCF-7_RFP-BAX) were generated from single cell colonies via transient transfection with the respective construct. Double stable cell lines MCF-7_Cyto c-GFP_IMS-RP and MCF-7_GFP-Rab5_RFP-BAX were generated through lentiviral transduction of MCF-7_Cyto c-GFP cells with pLJM1-IMS-RP and of MCF-7_RFP-BAX cells with pLJM1-GFP-Rab5, respectively. HeLa cells stably expressing IMS-RP (HeLa_IMS-RP) were generated via lentiviral transduction with pLJM1-IMS-RP.
Lentiviral stable transduction was performed by transfecting pUMVC, pCMV-VSV-G and the respective pLKO.1 or lentiCRISPR v2 construct into Lenti-X 293T cells (Sanjana et al., 2014; Stewart et al., 2003). Virus particle-containing supernatants were harvested and used to infect MCF-7 and HeLa cells. Selection of stably shRNA-expressing polyclonal cell lines was achieved using puromycin (1 μg/mL; Gibco). pLKO.1 with non-targeting sequence 5’-AATTGCCAGCTGGTTCCATCA-3’ was used to generate shRNA control (shCON) cell lines. For lentiviral gene knockdown, indicated cell lines were transduced with pLKO.1 shRNA directed against the following human target sequences:
5’-GCAGGATTCCAGTTCTTGTTA-3’ (ALS2 #1),
5’-CGACTAAATAAGCAGCCAGAT-3’ (ALS2 #2),
5’-CCAGGAATCAGTGTTGTAGTA-3’ (Rab5A),
5’-CATCACCAACACAGATACATT-3’ (Rab5C),
5’-CGTCAAGCAAATGTATAAGAA-3’ (Rabex-5),
5’-CCTTGGAAGTTTACTTAGTTA-3’ (USP15 #1),
5’-CCCATTGATAACTCTGGACTT-3’ (USP15 #2).
For CRISPR/Cas9-mediated genome editing, single cell colonies were selected from cells transduced with the following sequences targeting human genes cloned into lentiCRISPR v2:
5’-ACGGCAGCTCGCCATCATCG-3’ (BAK),
5’-CTGCAGGATGATTGCCGCCG-3’ (BAX),
Stable shRNA cell lines were maintained in selection media. Experiments were performed following 16–24 hrs of expression and/or culture in selection-free medium.
METHOD DETAILS
Plasmids
Fluorescent protein constructs encoding fusion proteins previously described: GFP-Rab5A, tagRFP-Rabex-5, tagRFP-Rabex-5(∆MIU), tBID-GFP, tBID-mCherry, (Hamacher-Brady and Brady, 2015; Hamacher-Brady et al., 2014), and GFP-ALS2/Alsin (Lai et al., 2006), Cytochrome c-GFP (Goldstein et al., 2000).
Fluorescent protein constructs generated for this study: PCR-amplified from cDNA, BID-mCherry, iRFP670-Rab5A, and iRFP670 or GFP fused to the OMM-targeting transmembrane domain (amino acids 101–152) of Fis1 (Hailey et al., 2010), referred to in text as irFP-OMM or GFP-OMM. tagRFP-BAX was subcloned from GFP-BAX WT (Wolter et al., 1997). pLJM1-IMS-RP and pLJM1-GFP-Rab5A were subcloned from pLJM1-GFP (Sancak et al., 2008), pBabe-puro-IMS-RP (Albeck et al., 2008b) and GFP-Rab5A. All constructs generated in this study were sequenced verified. In text and figure legends, tagRFP and mCherry are referred to as RFP, iRFP670 is referred to as irFP.
Drug Treatments
For drug treatments, compounds were added to phenol red-free fully supplemented media (FM) at the following concentrations: tumor necrosis factor alpha (TNFa; 50 ng/mL; R&D Systems), actinomycin D (AcD; 1 μg/ml; Sigma), staurosporine (STS; 1 μM; Cell Signaling), camptothecin (CPT; 20 μM; Cell Signaling); u18666a (10 μg/ml; Calbiochem); zVAD(OMe)-FMK (zVAD; 10 μM; Cell Signaling); BAM7 (10 μM; Sigma-Aldrich); venetoclax/ABT-199 (Vx; 10 μM; LC Laboratories); S63845 (5 μM; Selleck Chemicals).
Fluorescent Probes
To monitor lipid peroxidation, cells were incubated with 1 μM Bodipy ™ 581/591 C11 (Thermo Fisher Scientific) for 1 hr. To visualize endosomal cargo, cells were incubated with 5 μg/mL fluorescent Transferrin-AlexaFluor546 (Tf-AF546, Thermo Fisher Scientific), at 20 hrs following tBid transfection, for 2 hrs at 37ºC. For endocytic pulse-chase experiments, cells were placed on ice for 20 min to arrest endocytic trafficking, then incubated with pre-chilled 5 μg/mL Tf-AF 546 for 10 min on ice. Following two washes with dye-free FM, cells were incubated with dye-free FM to allow for Tf-AF546 internalization for 10 min at 37°C and then fixed with paraformaldehyde (PFA; EMS; 4% in PBS, pH 7.4) for 15 min. To visualize EL membranes, cells were incubated with 0.5 μM Bodipy-Cholesterol (Bodipy-CHOL/TopFluor-Cholesterol; Avanti), in the presence of 10 μg/ml u18666A (Calbiochem) to enrich EL cholesterol content through block of NPC1-mediated cholesterol export from lysosomes (Lu et al., 2015), for 18 hrs at 37ºC. Following loading with fluorescent probes, cells were placed in dye-free fresh media.
Immunofluorescence
For standard immunofluorescence (IF) detection of endogenous proteins, cells were plated in 8-well microscopy μ-slides (Ibidi), transfected and treated as indicated, and fixed in 4% PFA for 15 min. Following, cells were permeabilized with 0.3% Triton X-100 in PBS for 10 min and blocked with 3% BSA for 45 min. Cells were then incubated with primary antibodies against BAX (Santa Cruz Biotechnology; ABclonal), cytochrome c (BD Biosciences), Rabex-5 (Santa Cruz Biotechnology), Smac/DIABLO (Santa Cruz Biotechnology), TOM20 (Santa Cruz Biotechnology), TOM70 (Santa Cruz Biotechnology), at room temperature for 1 hr, or with Rab5 (Cell Signaling) or ALS2 (Sigma-Aldrich; ABclonal) antibodies at 4°C overnight. Fluorescence staining was performed for 30 min at room temperature using highly cross-adsorbed Alexa Fluor 488, 546 or 647 secondary antibodies (Thermo Fisher Scientific). Following primary and secondary antibody incubations, cells were washed three times in PBS. To visualize nuclei, cells were stained with 1 μg/mL Hoechst 33342 in PBS for 10 min.
Saponin-based Selective Permeabilization of Cholesterol-rich Membranes
Mitochondrial membranes are poor in cholesterol content, as opposed to plasma membrane and endocytic membrane compartments (Elias et al., 1979). To assess transformation of mitochondrial membrane lipid composition we utilized saponin, a detergent that selectively permeabilizes cholesterol-containing membranes (Goldenthal et al., 1985; Seeman et al., 1973). Following indicated treatments, cells were fixed with 2% PFA for 10 min at room temperature. Cholesterol-rich membrane permeabilization was achieved through incubation with 0.1% saponin (EMD Millipore) in PBS (pH 7.4) for 10 min at room temperature. Cells were then blocked with 3% BSA in PBS/0.1% Saponin. Immunofluorescence staining was performed in PBS. OMMs were stained using antibodies against OMM protein TOM20 (Santa Cruz Biotechnology). Accessibility of antigenic epitopes inside OMM-enclosed mitochondrial compartments was assessed using antibodies against the mitochondrial chaperone TRAP1 (Novus Biologicals), which resides in inner mitochondrial compartments (Pridgeon et al., 2007). Nuclei were stained with Hoechst 33342 in PBS for 10 min.
Fluorescence Imaging
Cells were plated in 8-well microscopy μ-slides (Ibidi), transfected and treated as indicated, and imaged live or within 48 hrs following fixation in electron microscopy-grade methanol-free paraformaldehyde (PFA; EMS; 4% in PBS, pH 7.4). Widefield fluorescence microscopy was performed with a DeltaVision Elite microscope system (GE Healthcare), equipped with a Scientific CMOS camera (Chip size: 2560 × 2160 pixels), an UltraFast solid-state illumination, an environmental chamber for live cell imaging at 37°C and 5% CO 2, a 60x (N.A. 1.42) oil immersion objective, the UltimateFocus module, and 488 nm and 568 nm laser modules. Single image slices, 2D Z-stacks using optical axis integration (OAI), or Z-stacks using 0.2 μm step increments were acquired and deconvolved (Softworx, Applied Precision). Single slices, or Z-stacks of representative cells were captured with 0.2 μm increments and deconvolved.
Image Analysis, Quantification and Presentation
Image preparation and analysis was performed using Fiji (Schindelin et al., 2012). Image pseudocolors correspond to font colors of protein labels. 3D surface renderings were built from Z-stacks captured in 0.2 μm steps. Deconvolved slices were contrast adjusted in Fiji before implementation of the ImageJ 3D Viewer plugin. Blue TOM20 or TOM70 immunofluorescence signals are set to 30–40% transparency in 3D surface renderings displaying three channels.
Stack projections of live time series imaging were generated to represent 3D and 4D dynamics. Stacks of calculated inter-organelle interactions were combined, sum-projected, and pseudo-colored as indicated. Resulting images are sometimes merged with raw or filtered images of whole channels. Sum projection and pseudo-coloring of time-steps was performed using the ‘Image -> Hyperstacks -> Temporal-Color Code’ function. Alternatively, ROIs from combined stacks of mitochondria, ELs, and interactions were re-ordered by swapping the X (space) and Z (time) axes using ‘Image -> Hyperstacks -> Re-Order Hyperstacks…’ in Fiji. The resulting YT-coordinate sum images demonstrate the stability of interactions over time.
Intensity profiles were measured using ‘Analyze -> Plot Profile’ on images contrast adjusted in Fiji. Fluorescence intensities were normalized and plotted in colors corresponding to displayed images.
Movies were prepared from live cell time series images. Contrast adjusted channels or interactions were merged as shown. Scale bars, and labels were generated and combined in Fiji.
Pipeline for Detecting and Quantifying Frequencies and Durations of Transient EL Interactions with Mitochondria
To monitor transient proximity interactions between ELs and mitochondria, we imaged live cells with mitochondrial (Cyto c-GFP, GFP-OMM or irFP-OMM) and endosomal markers (RFP-Rab5, irFP-Rab5 or Tf-AF546) in 1 μm thick optical axis integration (OAI) Z-slices in 5 sec intervals for 2 min under indicated conditions. Using Fiji, segmentation masks for mitochondrial markers were generated from thresholded, Minimum filter-corrected images. Segmentation masks of endosomal vesicles were generated from thresholded, Gaussian-filter corrected images. For each timepoint, the overlap between mitochondrial and endosomal masks was calculated using the Boolean AND function, yielding an image stack of proximity interactions.
Next, from the image stack, the durations of interactions were quantified using the TrackMate (Tinevez et al., 2017) plugin (settings vesicle diameter = 1 μm; 0.01 threshold; linking and gap closing distances = 1 μm). With these settings, an interaction is considered maintained if it remains within a radius of 1 μm between time points, and track length durations correspond to the duration of interaction. For each analyzed cell, we obtain the total (i) number of interactions, and (ii) the duration of those interactions.
Per cell, the distribution of interaction durations is represented as histograms with overlaid dot and violin plots. To compare durations between different cells under the same condition, or between different conditions, Normal Mixtures clustering was performed on MCF-7 interactions, under full medium conditions. The 7 clusters represent different interaction durations, ranging from most transient (5 sec) to stable (2 min), and were used to compare cell types, and conditions.
To represent the response of multiple cells under the same conditions, the fractional contribution for each interaction duration of individual cells was plotted (ie. data points within clusters represent single cells). Each cluster represents cell-to-cell variability for the indicated interaction duration. Clusters can be compared between cell types or conditions.
Measuring Dynamics of Mitochondrial Rab5 Accumulation in Relation to MOMP
MOMP dynamics were detected in live cells at high temporal and spatial resolution, using both HeLa and MCF-7 cells. Of note, MCF-7 cells lack caspase 3 (Janicke et al.) and thus do not undergo caspase-driven contraction rapidly following MOMP (Albeck et al., 2008a; Thorburn et al., 2014), rendering them especially amenable to dynamic detection. In cells expressing combinations of Cyto c-GFP, IMS-RP, GFP-Rab5 or RFP-Rab5 and/or GFP-BAX or RFP-BAX, the Fiji ‘find edges’ function was used to detect Cyto c -GFP release, and mitochondrial Rab5 and BAX accumulation. From processed time-series, total intensities of individual cells were measured. Values obtained from time course experiments were scaled between 0–1 and plotted using JMP.
Timepoint Assessment of MOMP
Following indicated treatments and durations, MCF-7 or HeLa cells were fixed and immunostained for endogenous TOM20 or TOM70, cytochrome c, Smac/DIABLO and/or BAX. Alternatively, to simultaneously detect MOMP and co-stain mitochondrial and EL markers, MCF-7 or HeLa cells stably expressing established MOMP markers cytochrome c-GFP (Goldstein et al., 2000) or IMS-RP (Albeck et al., 2008a) were subjected to the indicated treatments and immunostained for endogenous TOM20 and Rab5. To prevent detachment of HeLa cells from imaging chambers, zVAD-FMK was included in apoptotic drug treatments, without pre-incubation and at low concentration (10 μM), to attenuate caspase activity. Nuclei were labeled with Hoechst. Imaging was performed with a large field of view (207.47 μm x 207.47 μm). Total number of cells, and cells positive for BAX accumulation at mitochondria and/or cytochrome c or Smac release were scored using the ImageJ Cell Counter Plugin.
Fluorescence Recovery After Photobleaching (FRAP)
Photobleaching of GFP or RFP fluorescence within the defined ROI was performed using the 488 nm and 568 nm laser modules of the DeltaVision microscope, respectively. Prior to photobleaching, 3–4 images were acquired every 5 sec. Following photobleaching, cells were imaged every 5 sec for 2 min to capture fluorescence recovery. ROI fluorescence intensities were measured and analyzed for all timepoints using Fiji.
Western Blotting
Whole-cell lysates were prepared with RIPA lysis buffer (Millipore) containing cOmplete EDTA free protease inhibitor cocktail (Roche). Protein concentrations were determined using Coomassie reagent (Sigma-Aldrich). Samples were denatured in LDS sample reducing buffer (Thermo Fisher Scientific), electrophoresed using Bis-Tris NuPAGE gels (Thermo Fisher Scientific) and proteins were transferred to nitrocellulose using the iBlot 2 dry blotting system (Thermo Fisher Scientific). Immunodetection was performed using antibodies against BAX (Santa Cruz Biotechnology; ABclonal), BAK (Santa Cruz Biotechnology), GAPDH (Developmental Studies Hybridoma Bank), Rab5 (Cell Signaling), Rabex-5 (Santa Cruz Biotechnology), USP15 (Cell Signaling). Horseradish peroxidase-conjugated secondary antibodies (Rockland) and home-made ECL substrate were used to visualize immunoreactions and chemiluminescence signals recorded digitally (LI-COR Biosciences). Blots shown are representative of at least three independent experiments.
Detection of BAX Conformational Changes
For immunofluorescence detection of the BAX activation-associated conformational change leading to exposure of the epitope 6A7 (Hsu and Youle, 1997; Nechushtan et al.), cells were fixed in 4% PFA and permeabilized with 0.2% CHAPS in PBS for 2 min at room temperature, washed once in 0.02% Tween 20/PBS and blocked in 0.02% Tween 20/PBS containing 1% BSA for 5 min. Following, cells were incubated with a conformation-specific BAX antibody 6A7 (Abcam) and an antibody detecting total cellular BAX (Santa Cruz Biotechnology) at 37°C for 1 hr.
Alternatively, BAX 6A7 exposure was detected via immunoprecipitation (Yethon et al., 2003). Protein G-coupled Dynabeads (Thermo Fisher Scientific) were incubated with 2 μg α-BAX 6A7 (Abcam) for 30 min at room temperature, followed by three washing steps. Cells subjected to the indicated treatments were lysed on ice for 15 min in CHAPS lysis buffer (2% (w/v) CHAPS, 137 mM NaCl, 10% (v/v) glycerol, 50 mM Tris-EDTA pH 8.0) supplemented with protease inhibitors (Roche). Lysates were clarified by centrifugation and incubated with anti-BAX 6A7 antibody-conjugated Dynabeads overnight at 4°C. Dynabeads with immunoprecipitated complexes were concentrated by magnetization, subjected to three washing steps in ice-cold CHAPS buffer and eluted in 2X LDS sample reducing buffer at 70°C for 10 min. Following, samples were subjected to Western blot analysis.
Detection of BAX Oligomerization
BAX dimers and higher order oligomers were detected through chemical crosslinking experiments (Antonsson et al., 2001; Peng et al., 2013). Following the indicated treatments, cells were lysed in protease inhibitor-containing CHAPS lysis buffer on ice for 15 min. Clarified lysates were dosed and concentrations equalized to the lysate with lowest protein concentration using CHAPS lysis buffer. Equalized lysates were crosslinked with fresh disuccinimidyl suberate (DSS; Thermo Fisher Scientific) dissolved in PBS to a final concentration of 2 mM for 15 min at room temperature with gentle, continuous inversions. Crosslinking reactions were quenched by addition of Tris-HCl (pH 8.0) to a final concentration of 20 mM for 5 min. Quenched crosslinking reactions were pelleted at 4ºC and reconstituted in 2X LDS sample reducing buffer. 30% of the final product was subjected to Western blot analysis for the detection of BAX oligomers, using anti-BAX antibody (ABclonal). Band intensities were measured in Image Studio Lite (LI-COR Biosciences), and data analyzed and graphed with JMP.
Clonogenic Survival Assay
10 × 103 cells were plated per well of a 12-well plate one day prior to treatments. Cells were then treated with 10 μM venetoclax in combination with 5 μM S63845, or with DMSO as a solvent control. 6 hrs later, media were replaced with fresh media and surviving cells left in culture for 7 days. Following, cells were fixed in ice-cold methanol (Sigma-Aldrich) for 10 min and stained with 0.5% crystal violet (Alfa Aesar) in 25% methanol for 10 min to visualize colonies formed by proliferating cells. Monochromatic images of wells were captured using a FluorChem Q (Alpha Innotech) imager. Colonies were quantified using the ImageJ plugin ColonyArea (Guzman et al., 2014) from at least 3 independent experiments.
Ultrathin-Section Transmission Electron Microscopy (TEM)
MCF-7 cells were subjected to the indicated treatments and fixed in 2.5% glutaraldehyde (EMS) in 0.1 M sodium cacodylate buffer (pH 7.4) for 1 hr at room temperature, followed by three washes in 0.1 M cacodylate buffer (Sigma). Subsequently, cells were postfixed for 1 hr in 1% osmium tetroxide (EMS) in the same buffer at room temperature. Next, samples were washed in water and stained for 1 hr at room temperature in 2% uranyl acetate (Polyscience), and then washed again in water and dehydrated in a graded series of ethanol. The samples were then embedded in Embed-812 epoxy resin (EMS). Ultrathin (50–60-nm) sections were cut using an Ultracut ultramicrotome (Reichart-Jung) and collected on formvar- and carbon-coated nickel grids (Ted Pella), stained with 2% uranyl acetate and lead citrate (Ted Pella) before examination with a Philips/FEI BioTwin CM120 transmission electron microscope at 80 kV. Images were captured with a high-resolution, high-speed 8 megapixel AMT CCD XR80 camera. Mitochondria were identified based on a surrounding double-membrane, cristae or cristae remnants, and oftentimes contained calcium phosphate crystals.
Immuno-Electron Microscopy (IEM)
MCF-7 cell monolayers, untreated or treated as indicated, were fixed in 4% PFA (EMS) in 0.25 M HEPES (pH 7.4) for 1 hr at room temperature, then in 8% PFA in the same buffer overnight at 4oC. Samples were infiltrated, frozen and sectioned as previously described (Folsch et al., 2001). The sections were immunolabeled with the following primary antibodies diluted in PBS/1% fish skin gelatin. For single stainings, sections were incubated with rabbit anti-Rab5 antibody (Cell Signaling) at 1/10. For co-staining, sections were incubated with rabbit anti-TOM20 (Santa Cruz Biotechnology) at 1/10 and mouse anti-Rab5 (Cell Signaling) at 1/20. The sections were then incubated with IgG antibodies, followed directly by 10 nm (for rabbit anti-Rab5 or rabbit anti-TOM20) or 5 nm (mouse anti-Rab5) protein A-gold particles before examination with a Philips CM120 Electron Microscope (Eindhoven, the Netherlands) under 80 kV.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data were analyzed in JMP or Microsoft Excel. Asterisks represent p-value classes as shown. p-values were calculated using two-tailed students t-test, other than for quantifications shown in Figures 2C, 7B and 7D, which represent one-tailed t-test p-values. Graphs were compiled in JMP and typeset using Inkscape. Data are representative of n = 3 biological replicates, unless stated otherwise in the figure legend.
Supplementary Material
Video S1. Rab5+ Early ELs Target Apoptotic Mitochondria, Related to Figure 1
3D reconstruction video of ROI from MCF-7_GFP-BAX cell (green), treated with TNFα/AcD for 6 hrs and immunostained for Rab5 (red) and TOM70 (blue), imaged in 0.2 μm steps.
Video S2. Mitochondria Targeted by Rab5+ Early ELs in Response to Proapoptotic Stimuli, Related to Figures 1G and S1D-E
3D reconstruction videos of ROI from MCF-7 cells treated with camptothecin (CPT, left) for 24 hrs or staurosporine (STS, right) for 4 hrs and immunostained for TOM70 (blue) and Rab5 (red), imaged in 0.2 μm steps.
Video S3. Transferrin-loaded ELs Interact with tBID-GFP+ Apoptotic Mitochondria,Related to Figure 2E
Representative video of MCF-7 cell transfected with tBID-GFP (green), live imaged every 5 sec for 2 min following loading with fluorescently-labeled Transferrin, Tf-AF546 (red). Membrane interactions between tBid-GFP labeled OMMs and Tf+ ELs are shown in white.
Video S4. Bodipy-CHOL+ ELs Interact with tBid-RFP+ Apoptotic Mitochondria, Relatedto Figure 2F
Representative video of MCF-7 cell transfected with tBID-RFP (red), live imaged every 5 sec for 2 min following loading, in the presence of u18666a, with fluorescently-labeled Cholesterol, Bodipy-CHOL (green). Membrane interactions between tBid-GFP labeled OMMs and Bodipy-CHOL+ ELs are shown in white.
Video S5. Mitochondrial Cytochrome c Release Precedes BAX Clustering, Related to Figure 3A
Representative video of MCF-7_Cyto c-GFP cells (green) transfected with RFP-BAX (red), live imaged every 1 min for 40+ min, starting at 4 hrs of STS treatment to capture MOMP events. Zero time-point is set to depletion of mitochondrial cytochrome c.
Video S6. GFP-Rab5 Relocation to Apoptotic Mitochondria Precedes RFP-BAX Clustering at OMMs, Related to Figure 3C
Representative video of MCF-7_GFP-Rab5_RFP-BAX, live imaged every 2 min for 60+ min, starting at 4 hrs of STS treatment to capture relationship of endosomal targeting and BAX clustering. Zero time-point is set to onset of RFP-BAX clustering.
Video S7. RFP-Rab5 Targeting to Apoptotic Mitochondria Coincides with Cytochrome c Release, Related to Figure 3E
Representative video of MCF-7_Cyto c-GFP_RFP-Rab5 cells, live imaged every 30 sec for 10+ min, starting at 6 hrs of TNFα/AcD treatment to capture relationship of endosomal targeting of mitochondria and cytochrome c release. Zero time-point is set to depletion of mitochondrial cytochrome c.
Video S8. Knockdown of Rab5C Uncouples BAX Clustering at the OMM from Cytochrome c Release, Related to Figure 4D
3D reconstruction video of ROIs from MCF-7_shRab5C cells treated with TNFα/AcD for 6 hrs and immunostained for BAX (green) and cytochrome c (red), imaged in 0.2 μm steps. Note presence of BAX clusters at cytochrome c-containing mitochondria.
Highlights.
Endolysosomal targeting of mitochondria is a common feature during apoptosis signaling
Knockdown of Rab5 or USP15 uncouples BAX clustering from MOMP
Rab5 GEF Rabex-5 regulates MOMP upstream of BAX recruitment
EL transformation of mitochondria positively regulates BAX-mediated MOMP
ACKNOWLEDGMENTS
For plasmids we gratefully acknowledge Drs. H Cai for GFP-ALS2/Alsin, DR Green for cytochrome c-GFP, RJ Youle for GFP-BAX WT, D Sabatini for pLJM1-EGFP, and P Sorger for IMS-RP. We thank the technical staff of the Electron Microscopy Core Facility at Yale School of Medicine and the Johns Hopkins School of Medicine Microscopy Facility for their excellent services.
This work was supported through Startup funds No. 1606500086 of the W. Harry Feinstone Department of Molecular Microbiology & Immunology, Johns Hopkins Bloomberg School of Public Health (AH-B); e:Bio Grant No. 0316191 (LysoSys) of the Federal Ministry of Education and Research (BMBF), Germany (AH-B). TSW was supported by the Biochemistry, Cellular, and Molecular Biology graduate program of the Johns Hopkins School of Medicine, and by the National Institutes of Health (NIH) through Grant No. T32AI007417 (to the Department of Molecular Microbiology & Immunology, Bloomberg School of Public Health).
AS was supported by the Longmuir Endowment (to the Department of Molecular Microbiology & Immunology, Bloomberg School of Public Health).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes Experimental Procedures and 5 figures and 8 videos.
DECLARATION OF INTERESTS
The authors declare no competing interests. TSW is currently affiliated with Oncology Research and Development, Pfizer Inc., San Diego, California, USA.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Albeck JG, Burke JM, Aldridge BB, Zhang M, Lauffenburger DA, and Sorger PK (2008a). Quantitative analysis of pathways controlling extrinsic apoptosis in single cells. Mol Cell 30, 11–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albeck JG, Burke JM, Spencer SL, Lauffenburger DA, and Sorger PK (2008b). Modeling a snap-action, variable-delay switch controlling extrinsic cell death. PLoS Biol 6, 2831–2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonsson B, Montessuit S, Sanchez B, and Martinou JC (2001). Bax is present as a high molecular weight oligomer/complex in the mitochondrial membrane of apoptotic cells. J Biol Chem 276, 11615–11623. [DOI] [PubMed] [Google Scholar]
- Birkinshaw RW, and Czabotar PE (2017). The BCL-2 family of proteins and mitochondrial outer membrane permeabilisation. Semin Cell Dev Biol 72, 152–162. [DOI] [PubMed] [Google Scholar]
- Blumer J, Rey J, Dehmelt L, Mazel T, Wu YW, Bastiaens P, Goody RS, and Itzen A. (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- RabGEFs are a major determinant for specific Rab membrane targeting. J Cell Biol 200, 287–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock FJ, and Tait SWG (2019). Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol. [DOI] [PubMed] [Google Scholar]
- Bucci C, Parton RG, Mather IH, Stunnenberg H, Simons K, Hoflack B, and Zerial M. (1992). The small GTPase rab5 functions as a regulatory factor in the early endocytic pathway. Cell 70, 715–728. [DOI] [PubMed] [Google Scholar]
- Calore F, Genisset C, Casellato A, Rossato M, Codolo G, Esposti MD, Scorrano L, and de Bernard M. (2010). EL-mitochondria juxtaposition during apoptosis induced by H. pylori VacA. Cell Death Differ 17, 1707–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Qiu T, Wu J, Yang Y, Wright GD, Wu M, and Ge R. (2018). Extracellular anti-angiogenic proteins augment an endosomal protein trafficking pathway to reach mitochondria and execute apoptosis in HUVECs. Cell Death Differ. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipuk JE, McStay GP, Bharti A, Kuwana T, Clarke CJ, Siskind LJ, Obeid LM, and Green DR (2012). Sphingolipid metabolism cooperates with BAK and BAX to promote the mitochondrial pathway of apoptosis. Cell 148, 988–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, and Green DR (2010). The BCL-2 family reunion. Mol Cell 37, 299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christenson E, Merlin S, Saito M, and Schlesinger P. (2008). Cholesterol effects on BAX pore activation. J Mol Biol 381, 1168–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cory S, and Adams JM (2002). The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer 2, 647–656. [DOI] [PubMed] [Google Scholar]
- de Brito OM, and Scorrano L. (2010). An intimate liaison: spatial organization of the endoplasmic reticulum-mitochondria relationship. EMBO J 29, 2715–2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deveraux QL, Takahashi R, Salvesen GS, and Reed JC (1997). X-linked IAP is a direct inhibitor of cell-death proteases. Nature 388, 300–304. [DOI] [PubMed] [Google Scholar]
- Dingjan I, Verboogen DR, Paardekooper LM, Revelo NH, Sittig SP, Visser LJ, Mollard GF, Henriet SS, Figdor CG, Ter Beest M, et al. (2016). Lipid peroxidation causes endosomal antigen release for cross-presentation. Sci Rep 6, 22064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumitru R, Gama V, Fagan BM, Bower JJ, Swahari V, Pevny LH, and Deshmukh M. (2012). Human embryonic stem cells have constitutively active Bax at the Golgi and are primed to undergo rapid apoptosis. Mol Cell 46, 573–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edlich F, Banerjee S, Suzuki M, Cleland MM, Arnoult D, Wang C, Neutzner A, Tjandra N, and Youle RJ (2011). Bcl-x(L) retrotranslocates Bax from the mitochondria into the cytosol. Cell 145, 104–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias PM, Friend DS, and Goerke J. (1979). Membrane sterol heterogeneity. Freeze-fracture detection with saponins and filipin. J Histochem Cytochem 27, 1247–1260. [DOI] [PubMed] [Google Scholar]
- Estaquier J, and Arnoult D. (2007). Inhibiting Drp1-mediated mitochondrial fission selectively prevents the release of cytochrome c during apoptosis. Cell Death Differ 14, 1086–1094. [DOI] [PubMed] [Google Scholar]
- Folsch H, Pypaert M, Schu P, and Mellman I. (2001). Distribution and function of AP-1 clathrin adaptor complexes in polarized epithelial cells. J Cell Biol 152, 595–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, Bartoli D, Polishuck RS, Danial NN, De Strooper B, et al. (2006). OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 126, 177–189. [DOI] [PubMed] [Google Scholar]
- Gavathiotis E, Reyna DE, Bellairs JA, Leshchiner ES, and Walensky LD (2012). Direct and selective small-molecule activation of proapoptotic BAX. Nat Chem Biol 8, 639–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gkouvatsos K, Papanikolaou G, and Pantopoulos K. (2012). Regulation of iron transport and the role of transferrin. Biochim Biophys Acta 1820, 188–202. [DOI] [PubMed] [Google Scholar]
- Goldenthal KL, Hedman K, Chen JW, August JT, and Willingham MC (1985). Postfixation detergent treatment for immunofluorescence suppresses localization of some integral membrane proteins. J Histochem Cytochem 33, 813–820. [DOI] [PubMed] [Google Scholar]
- Goldstein JC, Waterhouse NJ, Juin P, Evan GI, and Green DR (2000). The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nat Cell Biol 2, 156–162. [DOI] [PubMed] [Google Scholar]
- Grosse L, Wurm CA, Bruser C, Neumann D, Jans DC, and Jakobs S. (2016). Bax assembles into large ring-like structures remodeling the mitochondrial outer membrane in apoptosis. EMBO J 35, 402–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson N, Culley S, Ashdown G, Owen DM, Pereira PM, and Henriques R. (2016). Fast live-cell conventional fluorophore nanoscopy with ImageJ through super-resolution radial fluctuations. Nat Commun 7, 12471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman C, Bagga M, Kaur A, Westermarck J, and Abankwa D. (2014). ColonyArea: an ImageJ plugin to automatically quantify colony formation in clonogenic assays. PLoS One 9, e92444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, and Lippincott-Schwartz J. (2010). Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 141, 656–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamacher-Brady A, and Brady NR (2015). Bax/Bak-dependent, Drp1-independent Targeting of X-linked Inhibitor of Apoptosis Protein (XIAP) into Inner Mitochondrial Compartments Counteracts Smac/DIABLO-dependent Effector Caspase Activation. J Biol Chem 290, 22005–22018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamacher-Brady A, Choe SC, Krijnse-Locker J, and Brady NR (2014). Intramitochondrial recruitment of ELs mediates Smac degradation and constitutes a novel intrinsic apoptosis antagonizing function of XIAP E3 ligase. Cell Death Differ 21, 1862–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homma Y, Kinoshita R, Kuchitsu Y, Wawro PS, Marubashi S, Oguchi ME, Ishida M, Fujita N, and Fukuda M. (2019). Comprehensive knockout analysis of the Rab family GTPases in epithelial cells. J Cell Biol 218, 2035–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu F, Spannl S, Ferguson C, Hyman AA, Parton RG, and Zerial M. (2018). Rab5 and Alsin regulate stress-activated cytoprotective signaling on mitochondria. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu YT, and Youle RJ (1997). Nonionic detergents induce dimerization among members of the Bcl-2 family. J Biol Chem 272, 13829–13834. [DOI] [PubMed] [Google Scholar]
- Janicke RU, Sprengart ML, Wati MR, and Porter AG (1998). Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J Biol Chem 273, 9357–9360. [DOI] [PubMed] [Google Scholar]
- Jongsma ML, Berlin I, Wijdeven RH, Janssen L, Janssen GM, Garstka MA, Janssen H, Mensink M, van Veelen PA, Spaapen RM, et al. (2016). An ER-Associated Pathway Defines Endosomal Architecture for Controlled Cargo Transport. Cell 166, 152–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Stang E, Fang KS, de Moerloose P, Parton RG, and Gruenberg J. (1998). A lipid associated with the antiphospholipid syndrome regulates EL structure and function. Nature 392, 193–197. [DOI] [PubMed] [Google Scholar]
- Kotschy A, Szlavik Z, Murray J, Davidson J, Maragno AL, Le Toumelin-Braizat G, Chanrion M, Kelly GL, Gong JN, Moujalled DM, et al. (2016). The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 538, 477–482. [DOI] [PubMed] [Google Scholar]
- Kushnareva Y, Andreyev AY, Kuwana T, and Newmeyer DD (2012). Bax activation initiates the assembly of a multimeric catalyst that facilitates Bax pore formation in mitochondrial outer membranes. PLoS Biol 10, e1001394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai C, Xie C, McCormack SG, Chiang HC, Michalak MK, Lin X, Chandran J, Shim H, Shimoji M, Cookson MR, et al. (2006). Amyotrophic lateral sclerosis 2-deficiency leads to neuronal degeneration in amyotrophic lateral sclerosis through altered AMPA receptor trafficking. J Neurosci 26, 11798–11806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leber B, Lin J, and Andrews DW (2007). Embedded together: the life and death consequences of interaction of the Bcl-2 family with membranes. Apoptosis 12, 897–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Tsai YC, Mattera R, Smith WJ, Kostelansky MS, Weissman AM, Bonifacino JS, and Hurley JH (2006). Structural basis for ubiquitin recognition and autoubiquitination by Rabex-5. Nat Struct Mol Biol 13, 264–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letai A. (2016). S63845, an MCL-1 Selective BH3 Mimetic: Another Arrow in Our Quiver. Cancer Cell 30, 834–835. [DOI] [PubMed] [Google Scholar]
- Li Z, He S, and Look AT (2019). The MCL1-specific inhibitor S63845 acts synergistically with venetoclax/ABT-199 to induce apoptosis in T-cell acute lymphoblastic leukemia cells. Leukemia 33, 262–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu F, Liang Q, Abi-Mosleh L, Das A, De Brabander JK, Goldstein JL, and Brown MS (2015). Identification of NPC1 as the target of U18666A, an inhibitor of lysosomal cholesterol export and Ebola infection. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucken-Ardjomande S, Montessuit S, and Martinou JC (2008). Bax activation and stress-induced apoptosis delayed by the accumulation of cholesterol in mitochondrial membranes. Cell Death Differ 15, 484–493. [DOI] [PubMed] [Google Scholar]
- Luo J, Jiang L, Yang H, and Song BL (2017). Routes and mechanisms of post-endosomal cholesterol trafficking: A story that never ends. Traffic 18, 209–217. [DOI] [PubMed] [Google Scholar]
- Luo L, Yang J, and Liu D. (2014). Integration and oligomerization of Bax protein in lipid bilayers characterized by single molecule fluorescence study. J Biol Chem 289, 31708–31718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes ME, Schlamp CL, and Nickells RW (2017). Live-cell imaging to measure BAX recruitment kinetics to mitochondria during apoptosis. PLoS One 12, e0184434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Abundis E, Correa F, Pavon N, and Zazueta C. (2009). Bax distribution into mitochondrial detergent-resistant microdomains is related to ceramide and cholesterol content in postischemic hearts. FEBS J 276, 5579–5588. [DOI] [PubMed] [Google Scholar]
- Mattera R, and Bonifacino JS (2008). Ubiquitin binding and conjugation regulate the recruitment of Rabex-5 to early ELs. EMBO J 27, 2484–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayle KM, Le AM, and Kamei DT (2012). The intracellular trafficking pathway of transferrin. Biochim Biophys Acta 1820, 264–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Pinedo C, Guio-Carrion A, Goldstein JC, Fitzgerald P, Newmeyer DD, and Green DR (2006). Different mitochondrial intermembrane space proteins are released during apoptosis in a manner that is coordinately initiated but can vary in duration. Proc Natl Acad Sci U S A 103, 11573–11578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nechushtan A, Smith CL, Hsu YT, and Youle RJ (1999). Conformation of the Bax C-terminus regulates subcellular location and cell death. EMBO J 18, 2330–2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill KL, Huang K, Zhang J, Chen Y, and Luo X. (2016). Inactivation of prosurvival Bcl-2 proteins activates Bax/Bak through the outer mitochondrial membrane. Genes Dev 30, 973–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osman C, Voelker DR, and Langer T. (2011). Making heads or tails of phospholipids in mitochondria. J Cell Biol 192, 7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otera H, Miyata N, Kuge O, and Mihara K. (2016). Drp1-dependent mitochondrial fission via MiD49/51 is essential for apoptotic cristae remodeling. J Cell Biol 212, 531–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouasti S, Matarrese P, Paddon R, Khosravi-Far R, Sorice M, Tinari A, Malorni W, and Degli Esposti M. (2007). Death receptor ligation triggers membrane scrambling between Golgi and mitochondria. Cell Death Differ 14, 453–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penengo L, Mapelli M, Murachelli AG, Confalonieri S, Magri L, Musacchio A, Di Fiore PP, Polo S, and Schneider TR (2006). Crystal structure of the ubiquitin binding domains of rabex-5 reveals two modes of interaction with ubiquitin. Cell 124, 1183–1195. [DOI] [PubMed] [Google Scholar]
- Peng R, Tong JS, Li H, Yue B, Zou F, Yu J, and Zhang L. (2013). Targeting Bax interaction sites reveals that only homo-oligomerization sites are essential for its activation. Cell Death Differ 20, 744–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y, Liao Q, Tan W, Peng C, Hu Z, Chen Y, Li Z, Li J, Zhen B, Zhu W, et al. (2019). The deubiquitylating enzyme USP15 regulates homologous recombination repair and cancer cell response to PARP inhibitors. Nat Commun 10, 1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer SR (2013). Rab GTPase regulation of membrane identity. Curr Opin Cell Biol 25, 414–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pridgeon JW, Olzmann JA, Chin LS, and Li L. (2007). PINK1 protects against oxidative stress by phosphorylating mitochondrial chaperone TRAP1. PLoS Biol 5, e172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz WA (2014). Bridging the gap: membrane contact sites in signaling, metabolism, and organelle dynamics. J Cell Biol 205, 759–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prudent J, Zunino R, Sugiura A, Mattie S, Shore GC, and McBride HM (2015). MAPL SUMOylation of Drp1 Stabilizes an ER/Mitochondrial Platform Required for Cell Death. Mol Cell 59, 941–955. [DOI] [PubMed] [Google Scholar]
- Prukova D, Andera L, Nahacka Z, Karolova J, Svaton M, Klanova M, Havranek O, Soukup J, Svobodova K, Zemanova Z, et al. (2019). Cotargeting of BCL2 with Venetoclax and MCL1 with S63845 Is Synthetically Lethal In Vivo in Relapsed Mantle Cell Lymphoma. Clin Cancer Res 25, 4455–4465. [DOI] [PubMed] [Google Scholar]
- Rehm M, Dussmann H, Janicke RU, Tavare JM, Kogel D, and Prehn JH (2002). Single-cell fluorescence resonance energy transfer analysis demonstrates that caspase activation during apoptosis is a rapid process. Role of caspase-3. J Biol Chem 277, 24506–24514. [DOI] [PubMed] [Google Scholar]
- Roberts AW, Davids MS, Pagel JM, Kahl BS, Puvvada SD, Gerecitano JF, Kipps TJ, Anderson MA, Brown JR, Gressick L, et al. (2016). Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N Engl J Med 374, 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland AA, and Voeltz GK (2012). Endoplasmic reticulum-mitochondria contacts: function of the junction. Nat Rev Mol Cell Biol 13, 607–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, and Sabatini DM (2008). The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320, 1496–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F. (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeman P, Cheng D, and Iles GH (1973). Structure of membrane holes in osmotic and saponin hemolysis. J Cell Biol 56, 519–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamas-Din A, Kale J, Leber B, and Andrews DW (2013). Mechanisms of action of Bcl-2 family proteins. Cold Spring Harb Perspect Biol 5, a008714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, et al. (2013). ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med 19, 202–208. [DOI] [PubMed] [Google Scholar]
- Stepanyants N, Macdonald PJ, Francy CA, Mears JA, Qi X, and Ramachandran R. (2015). Cardiolipin’s propensity for phase transition and its reorganization by dynamin-related protein 1 form a basis for mitochondrial membrane fission. Mol Biol Cell 26, 3104–3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, Sabatini DM, Chen IS, Hahn WC, Sharp PA, et al. (2003). Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 9, 493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tait SW, and Green DR (2010). Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol 11, 621–632. [DOI] [PubMed] [Google Scholar]
- Tait SW, and Green DR (2013). Mitochondrial regulation of cell death. Cold Spring Harb Perspect Biol 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi R, Deveraux Q, Tamm I, Welsh K, Assa-Munt N, Salvesen GS, and Reed JC (1998). A single BIR domain of XIAP sufficient for inhibiting caspases. J Biol Chem 273, 7787–7790. [DOI] [PubMed] [Google Scholar]
- Tam SY, Lilla JN, Chen CC, Kalesnikoff J, and Tsai M. (2015). RabGEF1/Rabex-5 Regulates TrkA-Mediated Neurite Outgrowth and NMDA-Induced Signaling Activation in NGF-Differentiated PC12 Cells. PLoS One 10, e0142935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorburn J, Andrysik Z, Staskiewicz L, Gump J, Maycotte P, Oberst A, Green DR, Espinosa JM, and Thorburn A. (2014). Autophagy controls the kinetics and extent of mitochondrial apoptosis by regulating PUMA levels. Cell Rep 7, 45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinevez JY, Perry N, Schindelin J, Hoopes GM, Reynolds GD, Laplantine E, Bednarek SY, Shorte SL, and Eliceiri KW (2017). TrackMate: An open and extensible platform for single-particle tracking. Methods 115, 80–90. [DOI] [PubMed] [Google Scholar]
- Uren RT, Dewson G, Bonzon C, Lithgow T, Newmeyer DD, and Kluck RM (2005). Mitochondrial release of pro-apoptotic proteins: electrostatic interactions can hold cytochrome c but not Smac/DIABLO to mitochondrial membranes. J Biol Chem 280, 2266–2274. [DOI] [PubMed] [Google Scholar]
- Volkmann N, Marassi FM, Newmeyer DD, and Hanein D. (2014). The rheostat in the membrane: BCL-2 family proteins and apoptosis. Cell Death Differ 21, 206–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wandinger-Ness A, and Zerial M. (2014). Rab proteins and the compartmentalization of the endosomal system. Cold Spring Harb Perspect Biol 6, a022616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm LP, Wendling C, Vedie B, Kobayashi T, Chenard MP, Tomasetto C, Drin G, and Alpy F. (2017). STARD3 mediates endoplasmic reticulum-to-EL cholesterol transport at membrane contact sites. EMBO J 36, 1412–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolter KG, Hsu YT, Smith CL, Nechushtan A, Xi XG, and Youle RJ (1997). Movement of Bax from the cytosol to mitochondria during apoptosis. J Cell Biol 139, 1281–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong-Ekkabut J, Xu Z, Triampo W, Tang IM, Tieleman DP, and Monticelli L. (2007). Effect of lipid peroxidation on the properties of lipid bilayers: a molecular dynamics study. Biophys J 93, 4225–4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wustner D, Lund FW, Rohrl C, and Stangl H. (2016). Potential of BODIPY-cholesterol for analysis of cholesterol transport and diffusion in living cells. Chem Phys Lipids 194, 12–28. [DOI] [PubMed] [Google Scholar]
- Yethon JA, Epand RF, Leber B, Epand RM, and Andrews DW (2003). Interaction with a membrane surface triggers a reversible conformational change in Bax normally associated with induction of apoptosis. J Biol Chem 278, 48935–48941. [DOI] [PubMed] [Google Scholar]
- Zeigerer A, Gilleron J, Bogorad RL, Marsico G, Nonaka H, Seifert S, Epstein-Barash H, Kuchimanchi S, Peng CG, Ruda VM, et al. (2012). Rab5 is necessary for the biogenesis of the EL system in vivo. Nature 485, 465–470. [DOI] [PubMed] [Google Scholar]
- Zerial M, and McBride H. (2001). Rab proteins as membrane organizers. Nat Rev Mol Cell Biol 2, 107–117. [DOI] [PubMed] [Google Scholar]
- Zha J, Weiler S, Oh KJ, Wei MC, and Korsmeyer SJ (2000). Posttranslational N-myristoylation of BID as a molecular switch for targeting mitochondria and apoptosis. Science 290, 1761–1765. [DOI] [PubMed] [Google Scholar]
- Zou Q, Jin J, Hu H, Li HS, Romano S, Xiao Y, Nakaya M, Zhou X, Cheng X, Yang P, et al. (2014). USP15 stabilizes MDM2 to mediate cancer-cell survival and inhibit antitumor T cell responses. Nat Immunol 15, 562–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. Rab5+ Early ELs Target Apoptotic Mitochondria, Related to Figure 1
3D reconstruction video of ROI from MCF-7_GFP-BAX cell (green), treated with TNFα/AcD for 6 hrs and immunostained for Rab5 (red) and TOM70 (blue), imaged in 0.2 μm steps.
Video S2. Mitochondria Targeted by Rab5+ Early ELs in Response to Proapoptotic Stimuli, Related to Figures 1G and S1D-E
3D reconstruction videos of ROI from MCF-7 cells treated with camptothecin (CPT, left) for 24 hrs or staurosporine (STS, right) for 4 hrs and immunostained for TOM70 (blue) and Rab5 (red), imaged in 0.2 μm steps.
Video S3. Transferrin-loaded ELs Interact with tBID-GFP+ Apoptotic Mitochondria,Related to Figure 2E
Representative video of MCF-7 cell transfected with tBID-GFP (green), live imaged every 5 sec for 2 min following loading with fluorescently-labeled Transferrin, Tf-AF546 (red). Membrane interactions between tBid-GFP labeled OMMs and Tf+ ELs are shown in white.
Video S4. Bodipy-CHOL+ ELs Interact with tBid-RFP+ Apoptotic Mitochondria, Relatedto Figure 2F
Representative video of MCF-7 cell transfected with tBID-RFP (red), live imaged every 5 sec for 2 min following loading, in the presence of u18666a, with fluorescently-labeled Cholesterol, Bodipy-CHOL (green). Membrane interactions between tBid-GFP labeled OMMs and Bodipy-CHOL+ ELs are shown in white.
Video S5. Mitochondrial Cytochrome c Release Precedes BAX Clustering, Related to Figure 3A
Representative video of MCF-7_Cyto c-GFP cells (green) transfected with RFP-BAX (red), live imaged every 1 min for 40+ min, starting at 4 hrs of STS treatment to capture MOMP events. Zero time-point is set to depletion of mitochondrial cytochrome c.
Video S6. GFP-Rab5 Relocation to Apoptotic Mitochondria Precedes RFP-BAX Clustering at OMMs, Related to Figure 3C
Representative video of MCF-7_GFP-Rab5_RFP-BAX, live imaged every 2 min for 60+ min, starting at 4 hrs of STS treatment to capture relationship of endosomal targeting and BAX clustering. Zero time-point is set to onset of RFP-BAX clustering.
Video S7. RFP-Rab5 Targeting to Apoptotic Mitochondria Coincides with Cytochrome c Release, Related to Figure 3E
Representative video of MCF-7_Cyto c-GFP_RFP-Rab5 cells, live imaged every 30 sec for 10+ min, starting at 6 hrs of TNFα/AcD treatment to capture relationship of endosomal targeting of mitochondria and cytochrome c release. Zero time-point is set to depletion of mitochondrial cytochrome c.
Video S8. Knockdown of Rab5C Uncouples BAX Clustering at the OMM from Cytochrome c Release, Related to Figure 4D
3D reconstruction video of ROIs from MCF-7_shRab5C cells treated with TNFα/AcD for 6 hrs and immunostained for BAX (green) and cytochrome c (red), imaged in 0.2 μm steps. Note presence of BAX clusters at cytochrome c-containing mitochondria.
Data Availability Statement
This study did not generate unique code or dataset.