

Abstract
Primary human fallopian tube secretory epithelial cell (FTSEC) cultures are useful for studying normal fallopian tube epithelial biology as well as developing models of fallopian tube disease, such as cancer. Due to the limited ability of primary human FTSECs to proliferate in vitro, it is necessary to immortalize them in order to establish a cell line that is suitable for long term culture and large scale in vitro experimentation. This protocol describes the isolation of FTSECs from human fallopian tube tissue, conditions for primary FTSEC culture, and techniques for establishing immortal FTSEC lines. The entire process, from primary cell isolation to establishment of an immortal cell line, may take up to 2 months. Once established, immortal FTSECs can typically be maintained for at least 30 passages.
INTRODUCTION
Immortal cell lines derived from healthy human tissue are a fundamental tool for experimental cancer research. They are frequently used to characterize the phenotypic and genotypic differences between normal and malignant cells in vitro and are an excellent tool for cellular transformation studies. Our lab recently published a report describing the immortalization and transformation of primary human fallopian tube secretory epithelial cells1, a cell type that previously received little attention in the cancer research arena. An urgent need for research into fallopian tube-derived cancer has recently been realized with the discovery that many pelvic tumors thought to be ovarian in origin may have actually arisen from the fallopian tube. The most lethal ovarian cancer amongst American women is high-grade serous ovarian carcinoma (HGSOC). This tumor type is conventionally thought to arise from ovarian surface epithelium because the bulk of the tumor typically involves the ovary. However, new data suggests that HGSOC may actually begin in the distal fallopian tube (located immediately next to the ovarian surface) and subsequently spread to the ovary2–5. The presumptive cell of origin for fallopian tube-derived serous tumors is the fallopian tube secretory epithelial cell (FTSEC). In order to study the transformation of this cell type and examine how it may contribute to HGSOC development, it is critical that experimental models of the fallopian tube be developed. Whether all HGSOCs arise from the fallopian tube is not yet clear. Therefore, it is important not to discount the possibility that both the fallopian tube and ovary may serve as sites of serous tumorigenesis. In this context, fallopian tube-based experimental models are an essential complement to pre-existing ovarian-based models for the comprehensive study of pelvic serous cancer development.
The hypothesis that most high-grade pelvic serous tumors begin in the fallopian tube is creating a paradigm shift in our understanding of HGSOC pathogenesis and it has significant clinical implications for screening and prevention4,6–12. Firstly, the methods currently used for early detection of this cancer, which focus primarily on the ovary, are largely ineffective. This has resulted in late-stage diagnoses and strongly contributes to the low 5-year survival rate of only 30% (SEER Cancer Statistics Review, 1975–2004, National Cancer Institute; http://seer.cancer.gov/csr/1975_2004/). It is quickly becoming apparent that screening efforts must be shifted to the fallopian tube in order to reduce HGSOC mortality. Secondly, women with germline BRCA1 mutations, who are genetically predisposed to breast and ovarian malignancies, frequently elect to undergo prophylactic salpingo-oophorectomy (ovary and fallopian tube removal) to reduce their risk of developing cancer. If fallopian tubes are indeed the primary source of HGSOC, it may be possible to reduce risk by removing the fallopian tubes alone, thus preserving fertility and avoiding the morbidity associated with premature menopause11.
These recent advances in our understanding of HGSOC pathogenesis and their potential clinical impact have sparked an unprecedented interest in fallopian tube epithelial biology and a high demand for experimental tools with which to study fallopian tube transformation. To address this need, our lab recently developed two alternate methods for culturing primary human fallopian tube cells; an ex vivo model of the fallopian tube epithelium13,14 and an in vitro model of fallopian tube secretory epithelial cells (FTSECs) that is based on primary cell immortalization1,15 (Fig. 1). The two models differ in that the ex vivo model intends to recapitulate a 3-D polarized epithelium consisting of multiple epithelial cell types, whereas the FTSEC model aims to isolate a pure population of secretory epithelial cells in order to generate immortal FTSEC lines suitable for long term in vitro culture. Our protocol for the ex vivo culture of primary fallopian tube epithelial cells has recently been published elsewhere14. Here, we present our protocol for establishing primary immortal FSTEC lines. Since both of our models require freshly harvested primary human fallopian tube epithelial cells, the initial steps of tissue processing (steps 1-7 of the present protocol) are very similar in both protocols. However, the two protocols diverge significantly following the epithelial dissociation step. Immortal cell lines established by the following procedure may be used to represent “normal” FTSECs in a myriad of in vitro experiments. They may also be used for transformation assays in which immortal FTSECs are transduced with different genetic alterations in order to systematically query the functional consequences of specific genetic changes and assess their respective contributions to cellular transformation1. This is especially useful now that large-scale genomic analyses of HGSOC have been published by The Cancer Genome Atlas and other groups16–22 and tools for the experimental validation of these data are critically needed. Developing immortal cell lines enables us to study the effects of HGSOC genomic aberrations on non-transformed FTSECs and to construct genetically defined human xenograft models of fallopian-derived cancer1. Such models are invaluable for the study of high-grade serous tumor biology as well as for testing novel therapeutic agents.
Figure 1. Methods for culturing primary human fallopian tube epithelial cells.
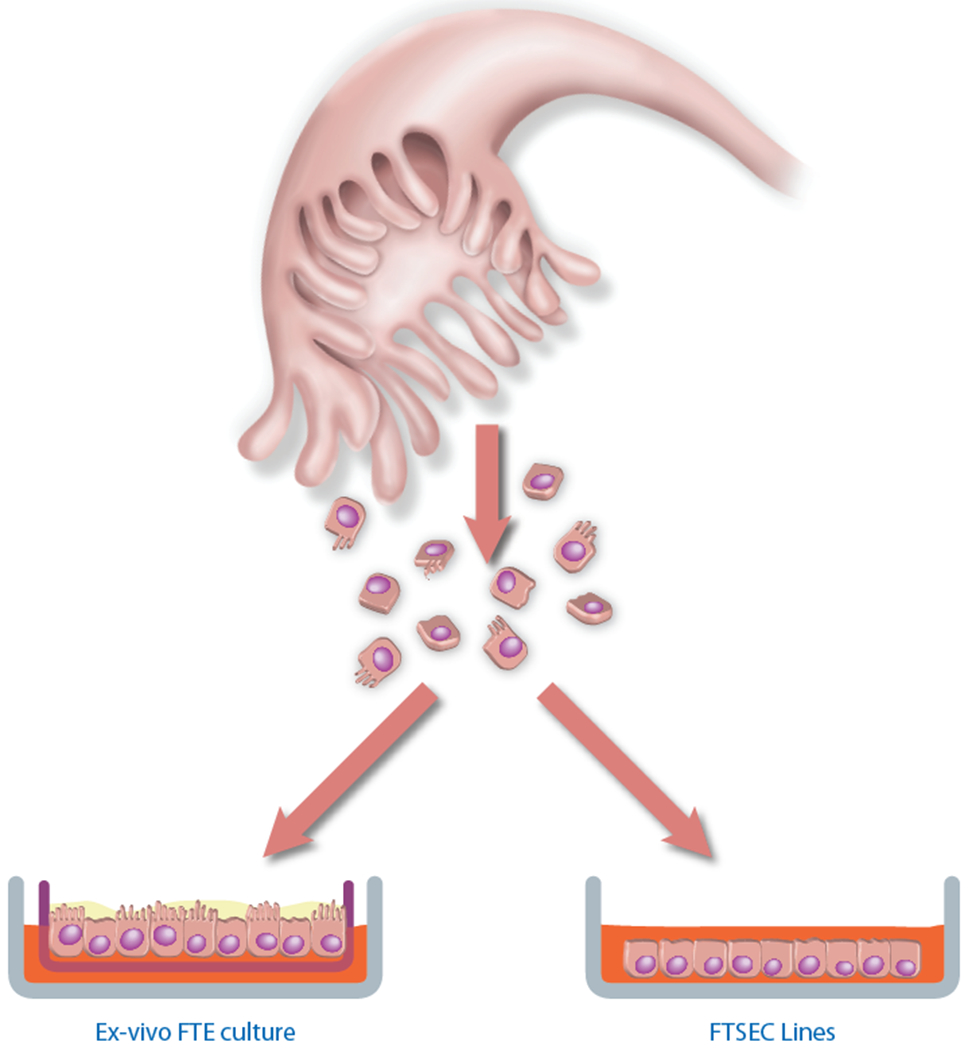
Epithelial cells isolated from primary human fallopian tube tissue may be cultured using one of two methods. Shown on the left, Ex vivo cultures are used to model the fallopian tube epithelium (FTE). Here, epithelial cells are dissociated from fallopian tube tissue and seeded onto a porous membrane, where both the ciliated and secretory cell types assemble in a polarized fashion, thus recreating an intact FTE. This is a small-scale model is best suited for short-term experiments examining epithelial biology. Shown on the right, FTSEC lines are used to study the secretory cell type, which represents the presumptive cell of origin of high-grade pelvic serous carcinomas. For this model, FTSECs are immortalized in vitro to establish cell lines that can be expanded and used for large-scale experiments, such as transformation and tumor xenograft studies.
Experimental Design
Tissue acquisition: This protocol requires collection of a fresh normal human fallopian tube. Access to fresh tissue should be coordinated through a hospital pathology department or tissue bank service with approval from the Institutional Review Board. It is important to be aware that the approval process may take anywhere from several weeks to months.
Retroviral transduction with hTERT
Primary human FTSECs grown on plastic ware will typically senesce within 2-3 passages and therefore must be immortalized to facilitate expansion and long term culture. FTSEC immortalization can be readily achieved by expressing hTERT and perturbing the p53 and pRb tumor suppressor pathways. In this protocol we use shRNA to silence p53 expression and mutant CDK4R24C to inhibit pRb activity. However, a variety of equivalent genetic elements may be used to obtain similar results. For example SV40 T antigens will perturb both the p53 and pRb pathways1,23. Or, a dominant negative mutant form of p53 may be used instead of p53 shRNA. This protocol uses retroviruses to stably express hTERT, p53 shRNA, and CDK4R24C in primary FTSECs. Retroviral expression vectors available from Addgene can be used to generate retroviruses by standard techniques (see REAGENTS and REAGENT SETUP).
It is important that primary FTSECs be replicating during retroviral infection in order to achieve high efficiency gene transfer24. Primary FTSECs proliferate quickly when first plated, but do not always grow well after they are split. We therefore recommend transducing FTSECs as soon as they reach approximately 50% confluency, before they are passaged. However, primary FTSEC growth dynamics are highly sample-dependent. In some cases, the cells continue to grow well after the first passage and thus the cell population may be expanded before transduction. If a sample exhibits rapid growth and/or becomes 100% confluent before there is an opportunity to transduce the cells, it is reasonable to split the cells 1:2 before performing the transduction, as described in the procedure.
Maintenance of immortal FTSEC lines
We recommend supplementing FTSEC medium with Ultroser G serum substitute throughout this protocol because we find that naive and newly-immortalized FTSECs grow better in the absence of serum. However, once an immortal FTSEC line has been established and low passages have been safely preserved, Ultroser G may be replaced with 10% fetal bovine serum (FBS), which is more economical. In most cases, immortal FTSEC lines adapt well when switched to serum-containing medium. However, it is best to test this on a small plate of cells before making any changes to the culture conditions.
MATERIALS
REAGENTS
Human fallopian tube tissue. CAUTION Human tissue collection requires patient consent and must be approved by the local Institutional Review Board. CAUTION Human tissue and blood are biohazards. Handling and disposal of these materials must be carried out in accordance with institutional biohazard safety regulations.
PBS, sterile (Mediatech, cat. no. 21-040)
Minimum Essential Medium (MEM) with Earle’s salts and L-glutamine (Mediatech, cat. no. 10-090)
Pronase from Streptomyces griseus (Roche, cat. no. 10165921001)
Deoxyribonuclease I (DNAse) from bovine pancreas (Sigma-Aldrich, cat. no. DN25)
Human placental collagen, Bornstein and Traub Type IV (Sigma-Aldrich, cat. no. C7521)
Glacial acetic acid (Fisher Scientific, cat. no. BP2401500). CAUTION Glacial Acetic acid is corrosive. Wear eye and skin protection when handling.
Ultroser G serum substitute (Pall Corporation, cat. no. 15950-017); distributed in the USA by Crescent Chemical Company (cat. no. 67042). CRITICAL Pall Corporation is the sole manufacturer of Ultroser G. Although other serum substitute formulations are commercially available, none have been tested by our lab for use in this protocol.
Water, sterile cell culture grade (Mediatech, cat. no. 25-055)
Water, double distilled (ddH2O)
Dulbecco’s Modification of Eagle’s Medium/Ham’s F-12 50/50 mix (DMEM-Ham’s F12) without L-glutamine (Mediatech, cat. no. 15–090)
Penicillin-streptomycin (Gibco, cat. no. 15140)
Replication-defective VSVG-pseudotyped retroviruses encoding hTERT, p53 shRNA, and CDK4R24C under CMV promoter control. Retroviral expression vectors are available from Addgene (pBabe-neo-hTERT, cat. no. 1774; pMKO.1-puro-p53 shRNA, cat. no. 10671; and pBabe-hygro-CDK4R24C, cat. no. 11254). Standard protocols for retrovirus production are widely available and accessible through the Addgene website.
Polybrene (Hexadimethrine bromide) (Sigma-Aldrich, cat. no. H9268)
Sodium Chloride (Sigma-Aldrich, cat. no. S5886)
0.25% Trypsin-EDTA (Gibco, cat. no. 25200)
Trypsin Neutralizing Solution (TNS) (Lonza, cat. no. CC-5002)
G418 (InvivoGen, cat no. ant-gn-1) optional
Puromycin (InvivoGen, cat no. ant-pr-1)
Hygromycin B (InvivoGen, cat no. ant-hm-1) optional
DMSO (Sigma-Aldrich, cat. no. D2650)
p53 antibody (Santa Cruz Biotechnology, cat. no. sc-81168); use
CDK4 antibody (Epitomics, cat. no. 2341-1)
PAX8 antibody (ProteinTech Group, cat. no. 10336-1-AP)
CK7 antibody (Epitomics, cat. no. 2303-1)
GAPDH antibody (Cell Signaling Technology, cat. no. 2118)
RB antibody (Cell Signaling Technology, cat. no. 9309)
Phospho-RB (Ser 780) antibody (Cell Signaling Technology, cat. no. 8180)
FOXJ1 antibody (Abcam, cat. no. ab40869)
EQUIPMENT
Stainless steel forceps (Roboz, cat. no. RS-5040)
Glass beaker, 200 ml capacity
Petri dish, 100-mm, sterile (BD Falcon, cat. no. 351029)
Disposable scalpel (BD Bard-Parker, cat. no. 372615)
Centrifuge tubes, sterile, 50 ml (Corning, cat. no. 430829)
Centrifuge tubes, sterile, 15 ml (Corning, cat. no. 430829)
Lab rocker
Temperature-controlled centrifuge (Eppendorf model 5180R or equivalent) with a swing bucket rotor suitable for 15 ml tubes, 50 ml tubes, and tissue culture plates.
Disposable serological pipets, 1 ml (Corning, cat. no. 4485)
Disposable serological pipets, 2 ml (Corning, cat. no. 4486)
Disposable serological pipets, 5 ml (Corning, cat. no. 4487)
Disposable serological pipets, 10 ml (Corning, cat. no. 4488)
0.22 μm bottle-top vacuum filter system, 250 ml capacity (Corning, cat. no. 431096)
0.22 μm bottle-top vacuum filter system, 500 ml capacity (Corning, cat. no. 431097)
Combination hot plate/magnetic stirrer
Magnetic stir bar
Hemocytometer
Light microscope
Cell culture CO2 incubator
Cell culture plates, 24-well (Corning, cat no. 3516)
Cell culture plates, 12-well (Corning, cat no. 3513)
Cell culture plates, 6-well (Corning, cat no. 3526)
Cell culture dish, 100-mm (Corning, cat no. 430167)
Cryogenic vials, 1.8 ml capacity (Thermo Scientific, cat. no. 375418)
REAGENT SETUP
Fallopian tube sample collection.
Collect fallopian tubes from patients undergoing surgery for benign gynecological indications such as fibroids, ovarian cysts, hysterectomy, or alternate conditions not affecting the fallopian tubes. The fallopian tube specimen must be healthy and not associated with gynecological malignancy. Upon surgical excision, the fallopian tube should be immersed in sterile PBS and kept on ice. The most plentiful source of epithelial cells is the distal (fimbrial) region of the fallopian tube. Therefore, it is important that the integrity of the fimbrial region be preserved during specimen collection. The tissue should be picked up and transported to the lab as soon as possible, ideally within a few hours.
Dissociation medium.
Dissolve 350 mg of Pronase and 25 mg of DNAse in 250 ml of MEM. Mix until all solid material is dissolved. Filter-sterilize using a 250 ml capacity 0.22 μm bottle-top vacuum filter. Store at 2-8°C for up to 4 weeks.
Ultroser G (USG).
Ultroser G is supplied as a lyophilized powder and must be reconstituted before use. Add 20 ml of sterile water to one 20 ml bottle of USG and wait until the material is fully dissolved. This may take up to 20 minutes. Store unused reconstituted USG at −20°C for up to 6 weeks. Do not freeze and thaw more than once. USG may be filtered (0.22 μm) if desired without loss of activity, although it is not necessary if prepared using aseptic techniques.
FTSEC medium (FTM).
Under sterile conditions, combine the following reagents: 485 ml of DMEM-Ham’s F12, 10 ml of reconstituted USG, and 5 ml of penicillin-streptomycin. Store at 2-8°C for up to 4 weeks. Warm to 37°C before use.
Human placental collagen solution.
Add 100 ml of ddH2O to a small beaker. Using forceps, weigh out 30 mg of human placental collagen fibers and place them on top of the water. Pipette 100 ul of glacial acetic acid directly onto the collagen to help it dissolve. Add a small magnetic stir bar to the beaker. Using a combination hot plate/magnetic stirrer, warm the solution to 37°C and stir for 30 min or until collagen is completely dissolved. Remove solution from heat and dilute to 500 ml with ddH2O. Filter-sterilize using a 500 ml capacity 0.22 μm bottle-top vacuum filter. Store at 2-8°C for up to 6 months.
Collagen-coated tissue culture plates.
Under sterile conditions, pipet a sufficient volume of human placental collagen solution into the wells of tissue culture plates to cover the bottom of the wells. For a 24-well plate, add 250 ul/well; for a 12-well plate, add 500 ul/well; for a 6-well plate, add 1 ml/well. Ensure that the entire surface of each well is covered. Stack plates in a sealed container to keep them sterile and store at room temperature (20-25°C) for up to 6 months. Immediately before use, rinse the wells with sterile PBS to remove any excess collagen solution.
Polybrene.
Prepare a solution of 1 ug/ml Polybrene in 0.9% NaCl. Filter-sterilize or autoclave the solution prior to use. Store at 2-8°C for up to 1 year.
Retroviruses.
Retroviral supernatants produced using standard techniques (by transfection of 293T cells) typically have titers of 105-106 infectious units/ml. Although unconcentrated viral supernatants may be used to infect primary FTSECs, it is best to concentrate viruses as much as possible in order to achieve higher transduction efficiencies. There are several options including ultracentrifugation, use of a virus concentration kit, or having the virus prepared by a viral vector core. To concentrate by ultracentrifugation, spin the viral supernatant at 20,000 × g for 1 h at 4°C25. Discard the supernatant and resuspend the virus in a small amount of MEM overnight at 2-8°C. To achieve a 500× increase in viral titer, for example, spin down 40 ml of viral supernatant and resuspend the virus in 80 ul of MEM. Alternatively, there are commercial virus concentration kits able to concentrate retroviral supernatants 100–500× or many institutions have viral vector core facilities offering retrovirus production services. Such viral vector cores typically provide purified virus with titers ≥ 108 infectious units/ml, quantified by Southern blot analysis.
PROCEDURE
Isolation of fallopian tube secretory epithelial cells (FTSECs)
-
1|
Rinse the fallopian tube with PBS to wash away excess blood and then transfer it to a 100-mm petri dish using forceps (Fig. 2a–b). CAUTION This and all subsequent steps should be carried out in a Biosafety Level 2 (BL2) cabinet under sterile conditions.
-
2|
Cut off and discard any connective tissue using a scalpel. Then, mince the fallopian tube and transfer the minced tissue to a 50 ml centrifuge tube containing 45 ml of cold dissociation medium (Fig. 2c–d). Cap tightly and place on a lab rocker at 2-8°C. Rock gently for 36-48 h. During this step, epithelial cells are enzymatically dissociated from the bulk tissue.
-
3|
Invert the tube once or twice to resuspend the minced tissue and then hold the tube upright, allowing bulky pieces to settle to the bottom. Immediately decant the supernatant (containing dissociated epithelial cells), into a second 50 ml centrifuge tube. Pellet the decanted epithelial cells by centrifugation at 200 × g for 5 min. Discard the supernatant.
-
4|
To increase the yield of dissociated cells, add 45 ml of PBS to the first tube containing the minced tissue. Cap the tube and, as in Step 3, invert it to resuspend the tissue pieces. Decant the supernatant into the second 50 ml tube containing pelleted cells, thus pooling all of the dissociated epithelial cells. (The minced tissue may now be discarded.) Pellet the pooled cells by centrifugation at 200 × g for 5 min. Discard the supernatant.
-
5|
To ensure that dissociation enzymes are completely removed from the cells, resuspend the cell pellet in 20 ml of PBS, centrifuge again (200 × g for 5 min), and discard the supernatant.
-
6|
Resuspend the cell pellet in 1-2 ml of FTM. To break up any cells clumps, gently pipet up and down at least 10 times using a 1 ml serological pipet.
-
7|
Use a hemocymeter to count the number of secretory cells. There will be three types of cells in the cell suspension; ciliated cells (with beating cilia), secretory cells (non-ciliated), and red blood cells (Fig. 3a). Count only the secretory cells. There will likely be small clumps of epithelial cells (5-10 cells) that did not completely break up during step 6. If you encounter this, try to estimate the number of cells in the clump as best you can.
Figure 2. Fallopian tube tissue processing.
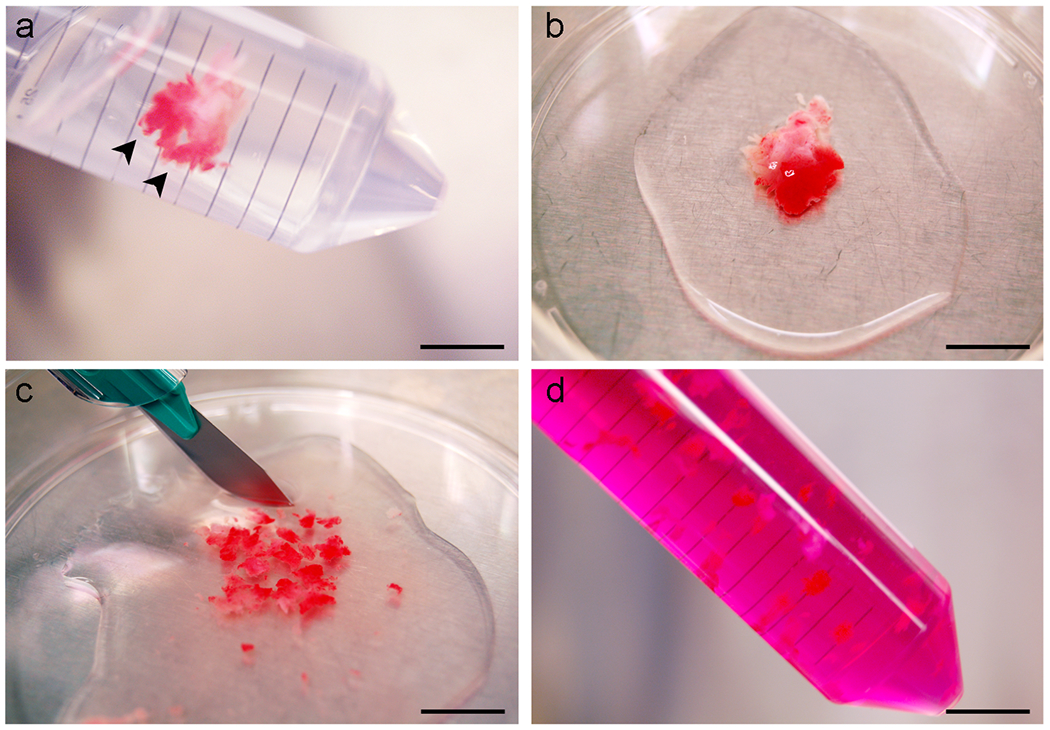
(a) Intact fallopian tube with prominent fimbria (arrowheads), immersed in PBS. (b) Appearance of fallopian tube after transfer to a petri dish. (c) Mincing of fallopian tube tissue. (d) Minced tissue suspended in dissociation medium. Scale bars: a-d, 10 mm.
Figure 3. Seeding and culture of dissociated FTSECs on collagen-coated plastic ware.
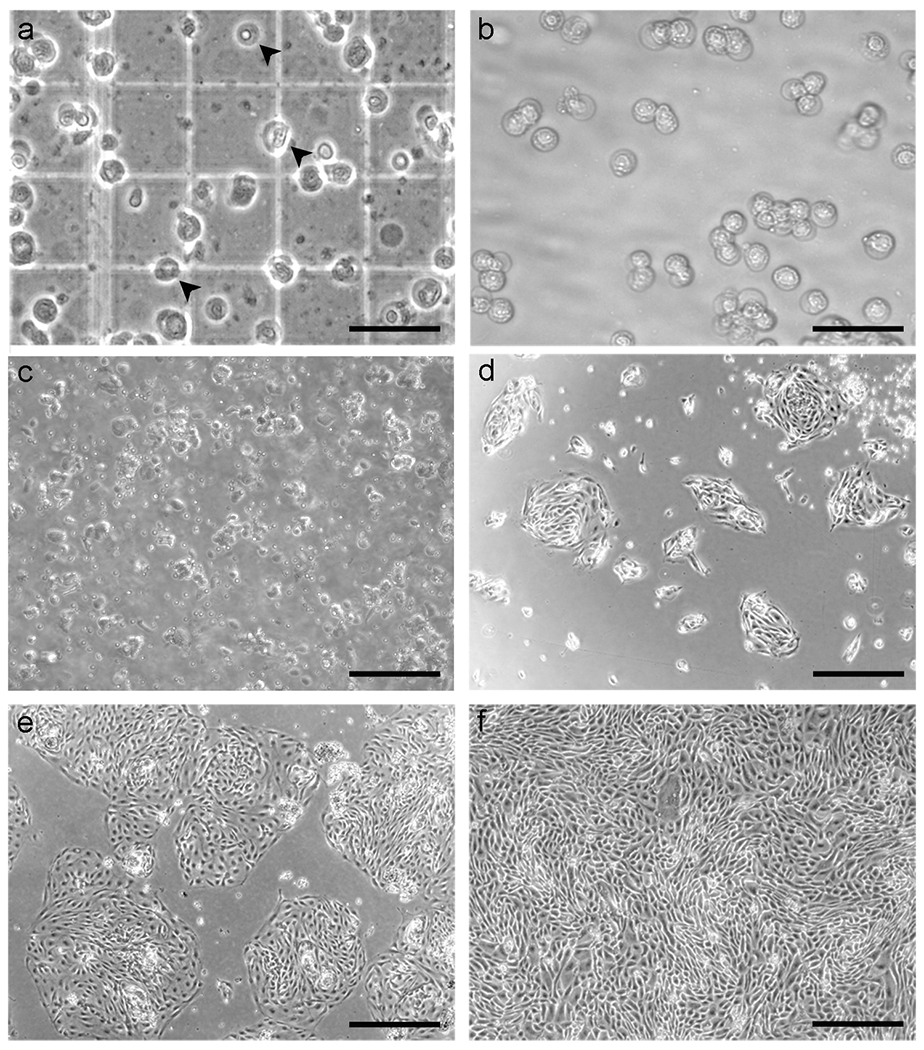
(a) Appearance of dissociated cells on a hemocytometer. The cell mixture obtained from enzymatic dissociation includes red blood cells (upper arrowhead), ciliated cells (middle arrowhead), and secretory cells (lower arrowhead). Ciliated cells are distinguished from secretory cells by the presence of cilia that beats very rapidly and is easily recognizable under the microscope. (b-c) Appearance of FTSECs immediately after plating, shown at 10× and 4× magnification, respectively. (d) 1 d after plating. Secretory cells attach to the plate and form “islands”. The sample shown has been washed with PBS to remove unattached cells and debris. (e) 2 d after plating. The cells are ready for transduction. (f) 3 d after transduction with hTERT. The cells are 100% confluent and exhibit a tight “cobblestone” appearance. with permission from Karst et al, 20111. Scale bars: a-b, 40 μm; c-f, 100 μm.
TROUBLESHOOTING
-
8|
Dilute the cell suspension to 105 secretory cells/ml in FTM and plate 1 ml/well in a 24-well collagen-coated plate (Fig. 3b–c).
-
9|
Incubate the cells overnight in a humidified 37°C tissue culture incubator supplied with 5% CO2.
-
10|
Observe cells under the microscope. The secretory cells should adhere to the plate overnight and appear as nest-like “islands” of cells (Fig. 3d). Ideally, cell confluency should be at least ~30%. However, due to the variable quality of fresh tissue samples, there may be some non-viable secretory cells, resulting in lower confluency. Ciliated cells often do not adhere to the plate, but if a ciliated cell does adhere (usually because it is still attached to a secretory cell) this is not a concern. Ciliated cells do not persist in 2-D culture, although the reason for this is unclear. One possibility is that ciliated cells are terminally differentiated and therefore cannot proliferate. Another possibility is that ciliated cells undergo transdifferentiation to the secretory phenotype when cultured in vitro.
TROUBLESHOOTING
-
11)
Aspirate medium from cells to remove blood cells and debris. Gently rinse the adherent cells with 500 ul of PBS using a serological pipet. If debris appears to be stuck on top of the cells, obscuring your view of them, do not rinse vigorously to remove it, as this may result in the detachment and loss of underlying secretory cells. Cell debris will usually detach on its own as the cells proliferate and can then be rinsed away. Aspirate the PBS and replace it with 500 ul of fresh FTM.
-
12|
Continue to culture the cells until they reach ~50% confluency (Fig. 3e). This usually takes 1-2 days, depending upon the sample. Change the medium every 24 h during this period to remove debris and prevent acidification of the medium.
CRITICAL If a sample grows rapidly and/or becomes 100% confluent before the cells can be transduced, split 1:2 before transduction. To split cells, follow steps 19-26. In steps 25-26, however, plate cells in 2 wells of a 24-well plate instead of 1 well of a 12-well plate, and halve the volumes of medium used.
TROUBLESHOOTING
Retroviral transduction with hTERT
-
13|
When cells are ~50% confluent in a 24-well plate and ready for transduction (Fig. 3e), aspirate the medium and replace it with 1 ml of FTM supplemented with 8 ug/ml polybrene. Polybrene is a cationic polymer used to increase the efficiency of retrovirus-mediated gene transfer.
-
14|
Add sufficient hTERT retrovirus to achieve a dose of 2.5 × 105 infectious units/well. For example, if the viral titer is ~108 infectious units/ml, add 2.5 ul of virus/well. Swirl to mix.
-
15|
Centrifuge the 24-well plate at 1000 × g for 30 min at 37°C.
-
16|
Return cells to the incubator overnight.
-
17|
Aspirate the retrovirus-containing medium and replace it with 500 ul of FTM.
-
18|
Return cells to the incubator for a further 2 days.
CRITICAL We generally do not perform antibiotic selection for hTERT expression at this point because FTSECs expressing hTERT alone do not always grow well in our experience. Our immortalization success rates have been higher with hTERT-transduced cells, even when selection is not performed. If one wishes to eliminate hTERT-negative cells from the immortal cell population, it can easily be done at a later point in time, after the cells have achieved a more robust growth rate. To select an immortal FTSEC line for hTERT expression, culture the cells in FTM containing 200-400 ug/ml G418.
Retroviral transduction with p53 shRNA and CDK4R24C
-
19|
At 72h post-infection FTSEC cultures should be 100% confluent (Fig. 3f). The cells must therefore be re-plated in larger wells to enable proliferation during the next round of retroviral transduction. To do this, aspirate the medium and rinse the cells with 500 ul of PBS.
TROUBLESHOOTING
-
20|
Aspirate the PBS and replace it with 250 ul of pre-warmed trypsin.
-
21|
Place the plate of cells in the incubator and wait until the cells detach. This may take up to 10 min. Watch the cells closely to avoid over trypsinization.
TROUBLESHOOTING
-
22|
When the cells have detached, add 500 ul of TNS to neutralize the trypsin. Pipet up and down to completely dislodge the cells from the plate.
-
23|
Transfer all of the cells into one 15 ml conical tube. Rinse each well with 500 ul of PBS to collect residual cells and add them to the 15 ml tube as well.
-
24|
Pellet the cells by centrifugation at 200 × g for 5 min. Discard the supernatant.
-
25|
Resuspend the cell pellet in 1 ml of FTM for each well trypsinized. Plate the cells in a 12-well collagen-coated plate, adding 1 ml of cell suspension/well.
-
26|
Collect residual cells by rinsing the tube with FTM (1 ml for each trypsinized well) and pipetting 1 ml into each well so that each well now contains 2 ml of cell suspension.
-
27|
Add 16 ul of polybrene solution to each well (final concentration = 8 ug/ml).
-
28|
Add sufficient p53 shRNA virus to achieve a dose of 2.5 × 105 infectious units/well. Repeat with the CDK4R24C virus. Swirl to mix. Each well should now contain a total retroviral dose of 5.0 × 105 infectious units.
-
29|
Centrifuge the 12-well plate at 1100 × g for 30 min at 37°C.
-
30|
Return cells to the incubator overnight.
-
31|
Aspirate the retrovirus-containing medium and replace it with 1 ml of FTM.
-
32|
Return the cells to the incubator for a further 2 days.
Antibiotic selection of transduced cells
-
33|
At 72h post-infection, aspirate the medium from the cells and replace it with 1 ml of FTM containing 0.5 ug/ml puromycin to select for cells expressing p53 shRNA.
-
34|
Culture cells for 1 week in puromycin-containing medium, replacing it every 2 days (Fig. 4a). If the cells become over confluent, they may be split 1:2 during this time. As described in steps 19-26. If the cells are sub-confluent, do not split them.
Figure 4. Emergence of FTSECs colonies under selective pressure.
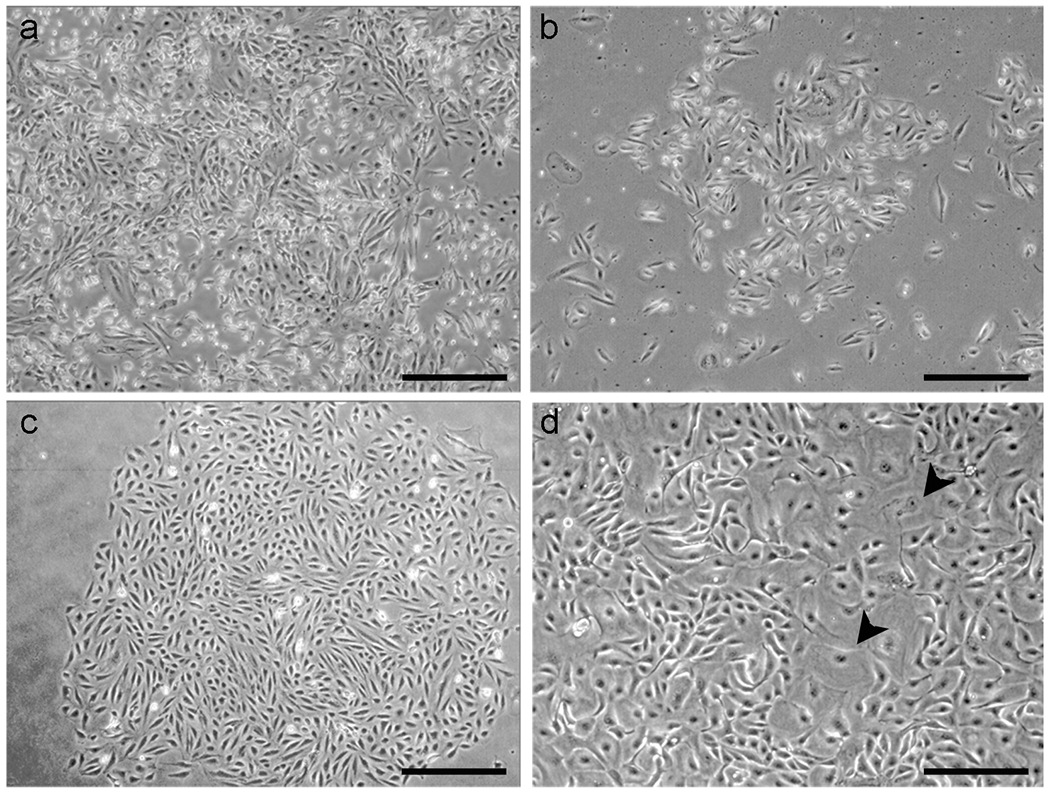
(a) FTSECs tranduced with neo-hTERT, puro-p53 shRNA, and hygro-CDK4R24C under puromycin selection for p53 shRNA expression. The cells have been exposed to selective medium for 2 d. Dying cells, which appear bright white, represent cells that were not successfully transduced with puro-p53 shRNA. (b) 7 d after the addition of selective medium. A puromycin-resistant colony of “immortal” cells slowly begins to emerge. (c) An expanding colony of immortal FTSECs. (d) Senescent FTSECs, which appear as large flattened cells (arrows), are commonly seen in primary and immortal cultures, even after antibiotic selection. Scale bars: a-c, 100 μm ; d, 40 μm.
Long term culture
-
35|
After 1 week of selection, when cell death has subsided, remove puromycin from the cell culture medium. Continue to regularly check the cells. In approximately 2-4 weeks, small colonies of immortal cells should emerge and give rise to tightly packed colonies (Fig. 4b–c). Keep culturing the cells until the immortal colonies become confluent. A small proportion of surviving cells (~10%) may exhibit a senescent morphology (Fig 4d). This commonly occurs in early immortal FTSEC cultures.
TROUBLESHOOTING
-
36|
Trypsinize the immortal cells and split them at a ratio of 1:2. Representative examples of cell morphology after splitting are shown in Fig. 5. As the cell population expands, trypsinize and replate the cells in larger wells, moving from 12-well plates, to 6-well plates, to 100-mm plates during the expansion phase. Always split the cells at a ratio of 1:2-l :3. Once you reach the 100-mm plates, it is no longer necessary to coat the plates with collagen. Nor is it necessary to use TNS. Instead, collect trypsinized cells in FTM, pellet them, rinse with PBS, pellet cells again, and re-suspend in FTM for plating.
-
37|
When the cells have been expanded to two 100-mm plates, you may wish to culture them in hygromycin-containing medium (200-400 ug/ml) for 1 week to eliminate any cells that do not express CDK4R24C. It is prudent to do this with only one plate at a time, in case the cells do not express high levels of CDK4R24C and a significant degree of cell death occurs. Once selection is complete, it is not necessary to maintain the cells in selective medium.
Figure 5. Expansion of immortal FTSECs.
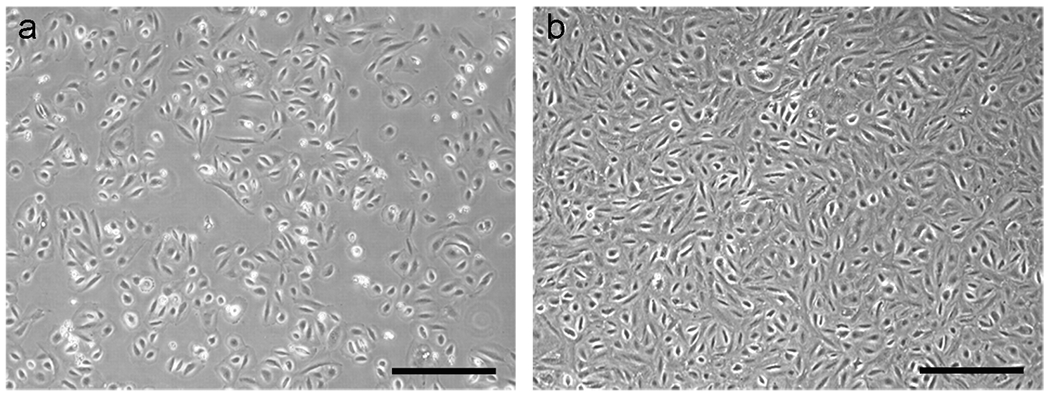
(a) Immortal FTSECs after their first passage from 24-well to 12-well plates. (b) A confluent layer of immortal FTSECs. The cells exhibit contact inhibition and can be cultured without splitting for a few weeks at a time. Scale bars: a-b, 100 μm.
Freezing immortal FTSECs
-
38)
Once immortal FTSECs have been expanded to 3 or more 100-mm plates, some cells should be frozen down and stored in liquid nitrogen to preserve a low-passage population. First trypsinize the cells, collect them in FTM, and pellet them by centrifugation at 200 × g for 5 min.
-
39|
Discard supernatant and resuspend pelleted cells in 2-3 ml of PBS to rinse away residual trypsin. Re-pellet cells by centrifugation at 200 × g for 5 min.
-
40|
Discard supernatant and resuspend cells in FTM containing 10% DMSO at a concentration of 106 cells/ml.
-
41|
Aliquot 1 ml each into cryogenic vials and store at −80°C overnight.
-
42|
The next day, transfer the vials to a liquid nitrogen storage tank. Early passage immortal cells (up to p10) tolerate freezing fairly well and typically maintain ≥ 70% viability upon thawing. Later passages (p10-p30) are more tolerant and show very little cell death upon thawing.
PAUSEPOINT Immortal FTSEC lines may be stored in liquid nitrogen for at least 5 years.
Thawing immortal FTSECs
-
43)
To thaw cells, remove a vial from liquid nitrogen and place it immediately into a 37°C water bath, taking care not to immerse the cap. CAUTION Wear a protective face shield when removing vials from liquid nitrogen. Improperly sealed vials containing liquid nitrogen can explode due to rapid expansion at room temperature.
-
44|
Once the ice crystals melt, promptly remove the vial from the water bath and use a pipet to transfer the contents to a 10 cm tissue culture dish containing 9 ml of pre-warmed FTM.
-
45|
Change the medium within 24 h to minimize exposure of cells to DMSO and remove dead cells.
Timing
Day 1: Collect fallopian tube sample (Reagent Setup) and begin epithelial dissociation (steps 1-2)
Day 2: No action required
Day 3: Quantify and plate dissociated FTSECs (steps 3-9)
Days 4-5: Maintain primary FTSEC cultures (steps 10-12)
Day 6: Transduce primary cultures with hTERT retrovirus (steps 13-16)
Day 7: Replace culture medium (steps 17-18)
Day 8: No action required
Day 9: Transduce cultures with p53 shRNA and CDK4R24C retroviruses (steps 19-30)
Day 10: Replace culture medium (steps 31-32)
Day 11: No action required
Day 12: Begin antibiotic selection of transduced FTSECs (step 33)
Days 13-18: Continue antibiotic selection (step 34)
Next 2-4 weeks: Culture surviving cells and monitor cultures for immortal colony growth (steps 35-37)
As required: Freezing (steps 38-41) and thawing (steps 42-44) immortal FTSEC lines.
TROUBLE SHOOTING
Troubleshooting advice is shown in Table 1.
Table 1:
troubleshooting
| Step | Problem | Possible Causes | Possible Solution |
|---|---|---|---|
| 7 | Low cell number | Insufficient mincing of tissue mincing, resulting in suboptimal enzyme access. Tissue sample was too small. |
Chop the tissue into very small pieces, < 1 mm3. Repeat protocol with a larger sample. |
| 10 | No cells adhered to the plate | Cells in tissue sample were not viable. Tissue sample was left sitting out too long before it was immersed in PBS. Too much time elapsed between tissue collection and tissue processing. |
Poor sample quality. Non-motile cilia (step 7) may indicate poor sample quality. Repeat protocol with a new sample. Minimize the time that the tissue is exposed to air during sample collection. Process the tissue (steps 1-2) as soon as possible after receiving the sample |
| 12 | Cells stop growing before reaching 50% confluency | Cells in tissue sample were not proliferative enough. Cells were seeded too sparsely in step 8. |
Cell proliferation rates vary considerably with each sample and are sometimes insufficient for in vitro culture. Repeat protocol with a new sample. Increase the number of cells/well when plating cells. |
| 19 | Cells appear static and do not grow to 100% confluency. |
Virus titer was too high and cells underwent arrest. | Decrease the amount of virus added to cells in step 14. |
| 21 | Cells will not detach during trypsinization. | The cells have formed very strong attachments to the collagen. | Extend trypsinization time until ~50% of the cells lift off the plate. Gently pipet the trypsin up and down using with a 1000 ul pipet to promote cell detachment. Transfer the cells to a clean tube containing 500 ul of TNS. Add 250 ul more trypsin to the well and repeat this process. It may take a few trypsinization-neutralization cycles to collect all of the cells. If a few stubborn cells refuse to detach, you can simply leave them behind and proceed with the protocol, or you can try transducing them in situ, in parallel with the detached cells. This will increase the total amount of infection medium and virus needed for steps 13-14. |
| 35 | No colonies have emerged after 2-3 weeks | Inadequate transduction efficiency. Immortal cells grow very slowly. |
Increase the viral dose in step 28. In some cases it takes several weeks for colonies to emerge. Therefore, it is best not to discard the cells prematurely. Keep the cells in culture, changing the medium every 2-3 days and wait for colonies to appear. In the mean time, repeat the protocol with a new sample. |
ANTICIPATED RESULTS
Before using immortal FTSECs for experiments, a Western blot should be performed to verify that immortal cells maintain the Mü1lerian phenotype, which is a fundamental characteristic of FTSECs. Expression of PAX8, a Mü1lerian lineage marker1,13,26,27, and CK7, a specific cytokeratin expressed by FTSECs, indicates that cells have not dedifferentiated in culture (Fig. 6a). Western blotting can also be used to validate the presence of genetic elements introduced into the cells (Fig. 6a). Cell lysates for Western blot may be prepared using any standard cell lysis buffer. To detect hTERT expression, which can sometimes be problematic by Western blot, RT-PCR can alternatively be used, as previously described1. In addition to validating the presence of genetic alterations, it is useful to assess their activity. For example, CDK4R24C activity should induce phosphorylation of pRb at Serine 780, which is a specific CDK4 target in pRb pathway activation (Fig. 6b). Similarly, telomerase activity resulting from hTERT expression may be measured using the telomeric repeat amplification protocol (TRAP)28. Lastly, it is a good idea to perform immunofluorescent staining for PAX8 (Fig. 6c) in order to detect any PAX8 negative cells. Large numbers of PAX8 negative cells may indicate that a contaminating cell type (for example, fibroblasts) may have been inadvertently immortalized along with FTSECs. Conditions for Western blotting and immunofluorescent staining of FTSECs (including antibody specifications), have been previously described1.
Figure 6. Validation of immortal FTSECs.
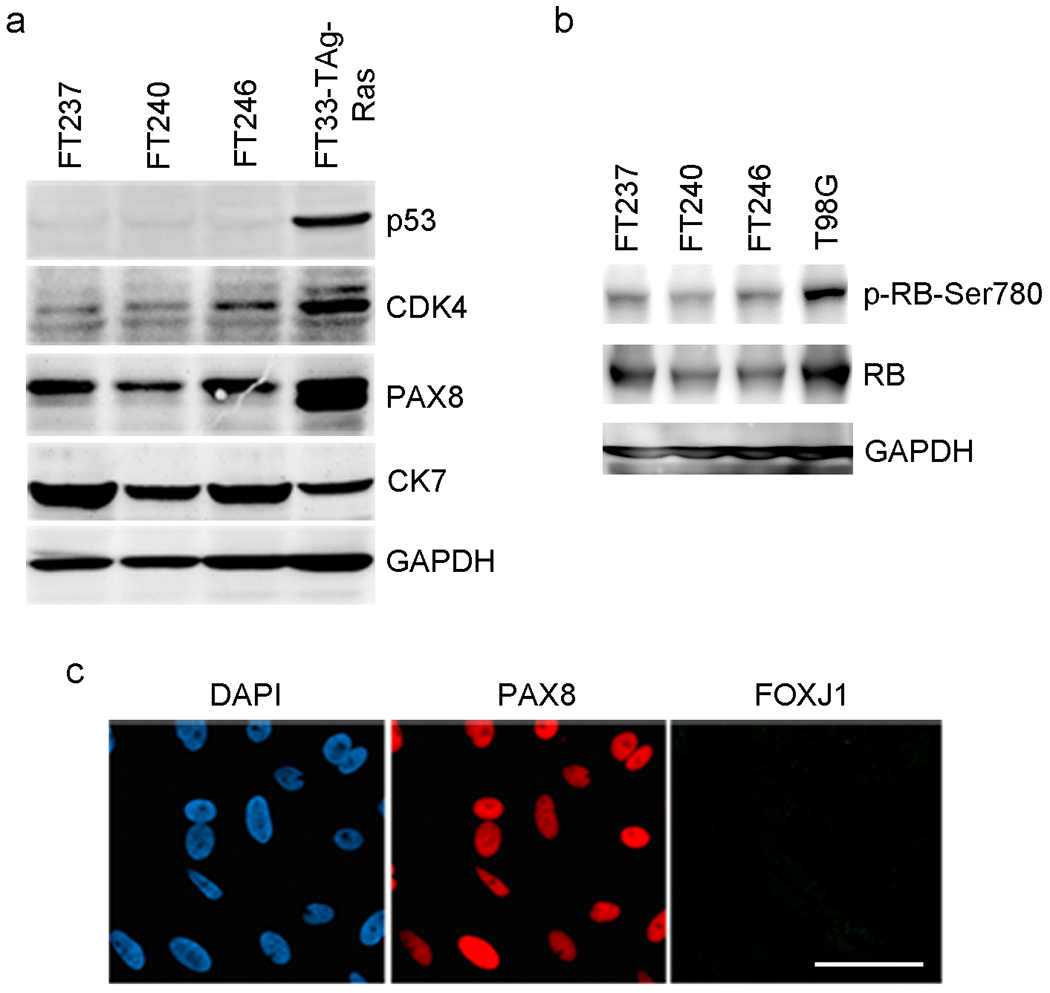
(a) Western blot analysis of immortal FTSEC lines derived from three different patients (FT237, FT240, and FT246). The cells express PAX8 and CK7, indicating that they maintain a Mullerian phenotype in vitro. They also express the genetic alterations used to immortalize the; namely, p53 knockdown and CDK4R24C. The positive control (FT33-Tag-Ras) is a Ras-transformed FTSEC line, previosuly described1. Antibody dilution: p53, CDK4, PAX8 and CK7, 1:1000; GAPDH, 1:2000. (b) CDK4R24C kinase activity results in hyperphosphorylation of pRb at Ser780. The positive control (T98G) is a glioblastoma cell line. Antibody dilution: p-RB-Ser780 and RB, 1:1000; GAPDH, 1:2000. (c) Immunofluorescent staining of immortal FTSECs for PAX8 confirms that all cells are of the secretory cell type. They are negative for FOXJ1, a marker of FTE ciliated cells. Reproduced with permission from Karst et al, 20111. Antibody dilution: PAX8, 1000; FOXJ1, 1:750. Scale bar: 30 μm.
Because multiple antibiotic resistance markers are employed to generate immortal cells lines, it is not possible to keep immortal FTSECs under long term selective pressure for all of the genetic alterations introduced. An immortal FTSEC line will normally maintain high level expression of any genetic alteration on which it depends for viability or which confers a significant growth advantage. However, other genetic elements may not be critically required after immortalization and consequently their expression may be lost at some point in the absence of selective pressure. In our experience, immortal FTSECs typically maintain p53 perturbations (such as p53 shRNA, mutant p53 or SV40 Large T Antigen) whereas CDK4R24C levels sometimes decrease over time. In this context it is possible that CDK4R24C plays a greater role in the immortalization process than in sustaining FTSEC proliferation.
Most immortal FTSEC lines can be passaged continuously for at least 1 year. However, their growth rate and cell morphology will likely change over time. Therefore, we recommend using low passage cells whenever possible. It is a good idea to periodically perform Western blot analysis of immortal cell lines to ensure that the cells still express PAX8, CK7, and other relevant markers. Additionally, long term culture of immortal cells may eventually lead to the acquisition of genetic defects, either spontaneously or as a direct consequence of the loss of p53 and pRb tumor suppressor function. Immortal cells should not exhibit anchorage independent growth in soft agar. If cells acquire the ability to grow in soft agar, they have likely undergone a significant genomic change. To assess the genomic integrity of an immortal FTSEC line, we recommend that array comparative genomic hybridization (aCGH) analysis be performed.
Acknowledgments
Special thanks to the faculty and staff of the BWH Department of Pathology for allocation of tissues. This work was supported by a Canadian Institutes of Health Research Fellowship (AMK), NIH - P50 CA105009 (SPORE), Ovarian Cancer Research Fund (RD); Robert and Debra First Fund (RD), Randi and Joel Cutler Ovarian Cancer Research Fund (RD), The Mary Kay Foundation (RD), The Sandy Rollman Ovarian Cancer Foundation (RD), and the Susan Smith Center for Women’s Cancers and the Dana-Farber Cancer Institute (RD).
Footnotes
Competing financial interests
The authors declare that they have no competing financial interests.
REFERENCES
- 1.Karst AM, Levanon K & Drapkin R Modeling high-grade serous ovarian carcinogenesis from the fallopian tube. Proc Natl Acad Sci U S A 108, 7547–7552 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Medeiros F et al. The tubal fimbria is a preferred site for early adenocarcinoma in women with familial ovarian cancer syndrome. Am J Surg Pathol 30, 230–236 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Lee Y et al. A candidate precursor to serous carcinoma that originates in the distal fallopian tube. J Pathol 211, 26–35 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Crum CP Intercepting pelvic cancer in the distal fallopian tube: theories and realities. Mol Oncol 3, 165–170 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Przybycin CG, Kurman RJ, Ronnett BM, Shih Ie M & Vang R Are all pelvic (nonuterine) serous carcinomas of tubal origin? Am J Surg Pathol 34, 1407–1416 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Crum CP et al. The distal fallopian tube: a new model for pelvic serous carcinogenesis. Curr Opin Obstet Gynecol 19, 3–9 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Kurman RJ & Shih Ie M Molecular pathogenesis and extraovarian origin of epithelial ovarian cancer--shifting the paradigm. Hum Pathol 42, 918–931 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Salvador S et al. The fallopian tube: primary site of most pelvic high-grade serous carcinomas. Int J Gynecol Cancer 19, 58–64 (2009). [DOI] [PubMed] [Google Scholar]
- 9.Karst AM & Drapkin R Ovarian cancer pathogenesis: a model in evolution. J Oncol 2010, 932371 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levanon K, Crum C & Drapkin R New insights into the pathogenesis of serous ovarian cancer and its clinical impact. J Clin Oncol 26, 5284–5293 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Greene MH, Mai PL & Schwartz PE Does bilateral salpingectomy with ovarian retention warrant consideration as a temporary bridge to risk-reducing bilateral oophorectomy in BRCA1/2 mutation carriers? Am J Obstet Gynecol 204, 19 e11–16 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Folkins AK, Jarboe EA, Roh MH & Crum CP Precursors to pelvic serous carcinoma and their clinical implications. Gynecol Oncol 113, 391–396 (2009). [DOI] [PubMed] [Google Scholar]
- 13.Levanon K et al. Primary ex vivo cultures of human fallopian tube epithelium as a model for serous ovarian carcinogenesis. Oncogene 29, 1103–1113 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fotheringham S, Levanon K & Drapkin R Ex Vivo Culture of Primary Human Fallopian Tube Epithelial Cells. JoVE 51, http://www.jove.com/index/Details.stp?ID=2728, doi: 2710.3791/2728 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karst AM & Drapkin R The new face of ovarian cancer modeling: better prospects for detection and treatment. F1000 Med Rep 3, 22 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cancer Genome Atlas Research Network: Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pearce CL et al. Validating genetic risk associations for ovarian cancer through the international Ovarian Cancer Association Consortium. Br J Cancer 100, 412–420 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fasching PA et al. Role of genetic polymorphisms and ovarian cancer susceptibility. Mol Oncol 3, 171–181 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goode EL et al. A genome-wide association study identifies susceptibility loci for ovarian cancer at 2q31 and 8q24. Nat Genet 42, 874–879 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Etemadmoghadam D et al. Integrated genome-wide DNA copy number and expression analysis identifies distinct mechanisms of primary chemoresistance in ovarian carcinomas. Clin Cancer Res 15, 1417–1427 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Notaridou M et al. Common alleles in candidate susceptibility genes associated with risk and development of epithelial ovarian cancer. Int J Cancer 128, 2063–2074 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bolton KL et al. Common variants at 19p13 are associated with susceptibility to ovarian cancer. Nat Genet 42, 880–884 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jazaeri AA et al. Molecular requirements for transformation of fallopian tube epithelial cells into serous carcinoma. Neoplasia 13, 899–911 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miller DG, Adam MA & Miller AD Gene transfer by retrovirus vectors occurs only in cells that are actively replicating at the time of infection. Mol Cell Biol 10, 4239–4242 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kanbe E & Zhang DE A simple and quick method to concentrate MSCV retrovirus. Blood Cells Mol Dis 33, 64–67 (2004). [DOI] [PubMed] [Google Scholar]
- 26.Laury AR et al. PAX8 reliably distinguishes ovarian serous tumors from malignant mesothelioma. Am JSurg Pathol 34, 627–635 (2010). [DOI] [PubMed] [Google Scholar]
- 27.Bowen NJ et al. Emerging roles for PAX8 in ovarian cancer and endosalpingeal development. Gynecol Oncol 104, 331–337 (2007). [DOI] [PubMed] [Google Scholar]
- 28.Herbert BS, Hochreiter AE, Wright WE & Shay JW Nonradioactive detection of telomerase activity using the telomeric repeat amplification protocol. Nat Protoc 1, 1583–1590 (2006). [DOI] [PubMed] [Google Scholar]