

SUMMARY
KDM1A-mediated H3K4 demethylation is a well-established mechanism underlying transcriptional gene repression, but its role in gene activation is less clear. Here, we report a critical function and mechanism of action of KDM1A in glucocorticoid receptor (GR)-mediated gene transcription. Biochemical purification of the nuclear GR complex revealed KDM1A as an integral component. In cell-free assays, GR modulates KDM1A-catalyzed H3K4 progressive demethylation by limiting the loss of H3K4me1. Similarly, in cells, KDM1A binds to most GR binding sites in the genome, where it removes preprogrammed H3K4me2 but leaves H3K4me1 untouched. Blocking KDM1A catalytic activity prevents H3K4me2 removal, severely impairs GR binding to chromatin, and dysregulates GR-targeted genes. Taken together, these data suggest KDM1A-mediated H3K4me2 demethylation at GRBSs promotes GR binding and plays an important role in glucocorticoid-induced gene transcription, broadening the mechanisms that contribute to nuclear receptor-mediated gene activation.
Graphical Abstract
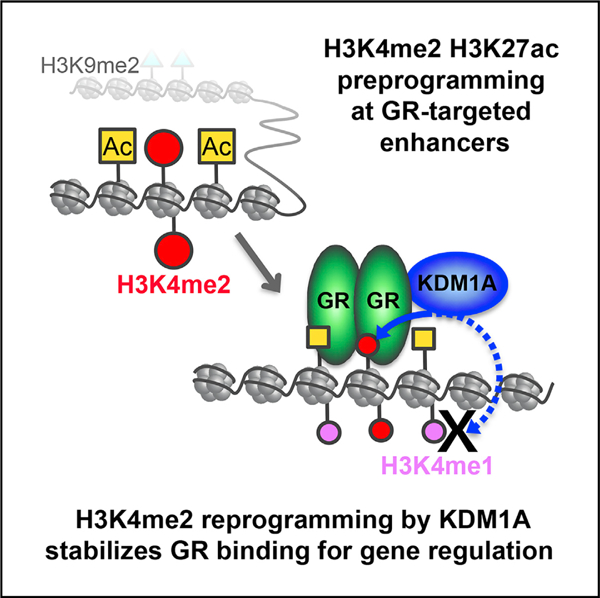
In Brief
Clark et al. dissected the interplay of glucocorticoid receptor (GR) and KDM1A in glucocorticoid-mediated gene regulation. GR recruits KDM1A, which consequently removes preprogrammed H3K4me2 and stabilizes GR-chromatin interaction. KDM1A demethylation of H3K4me2 at GR-targeted enhancers is important for GC-mediated gene transcription, offering a molecular mechanism for H3K4me2 demethylation in gene activation.
INTRODUCTION
Glucocorticoids (GCs) are hormones with essential roles in metabolism, stress response, development, the immune system, and behavior (Kadmiel and Cidlowski, 2013). Clinically, GCs are some of the most potent and widely prescribed anti-inflammatories, and are used in cancer treatment (Kadmiel and Cidlowski, 2013; Silverman and Sternberg, 2012). Many of the biological effects of GCs depend on activation of the glucocorticoid receptor (GR), a hormone-induced transcription factor. Ligand-activated GR binds to distal regulatory sites, like enhancers, in cell-type-specific patterns to induce or repress target genes (Carroll et al., 2005; Clarke and Graham, 2012; Reddy et al., 2009; So et al., 2007). Selective binding of GR throughout the genome is influenced by DNA sequence, chromatin accessibility, and epigenetic modifications such as covalent modification of DNA and histones, as well as other transcription factors. For example, GR binds DNA motifs called GR response elements (GREs), but AP-1 transcription factors also facilitate binding by maintaining an open chromatin structure prior to GR activation (Biddie et al., 2011). These sites with open chromatin are referred to as “preprogrammed,” differing from “reprogrammed” sites where chromatin opens upon GR activation (John et al., 2011). GR also recruits epigenetic regulators such as the histone acetyltransferase CREB binding protein (CBP)/p300, a co-activator that further modifies chromatin (Kato et al., 2011).
A distinct histone code contributes to the epigenetic control of enhancers (Calo and Wysocka, 2013). H3K4me1 marks poised and active enhancers, which are further distinguished by the absence or presence of H3K27ac, respectively. H3K4me3 is also present at enhancers, but at levels significantly lower than transcription start sites (TSSs), a major distinction from gene promoters (Heintzman et al., 2009). H3K4me2 is necessary for genomic localization of FoxA1, a pioneer factor for the androgen receptor (AR) and the estrogen receptor (ER) (Lupien et al., 2008) and, is a distinguishing epigenetic mark at cell-type-specific enhancers (Pekowska et al., 2010; Wang et al., 2014). However, recent studies challenge the canonical view of H3K4 methylation at enhancers by showing that enhancer methylation mediated by MLL3 and MLL4 is dispensable for gene regulation and development (Dhar et al., 2018; Jang et al., 2019; Rahnamoun et al., 2018). Thus, the role of different H3K4-methyl states and their regulation at enhancers is complex and remains poorly understood.
Histone methylation is dynamically regulated by opposing enzymes known as histone methyltransferases (HMTs) and histone demethylases (HDMs). LSD1/KDM1A was the first HDM identified and plays important roles in a broad spectrum of biological processes such as stem cell differentiation, embryonic development, tumorigenesis, and neuronal function (Chen et al., 2012; Rusconi et al., 2015; Wang et al., 2007; Whyte et al., 2012). KDM1A was originally characterized as a co-repressor, demethylating H3K4me2/1 (Shi et al., 2004). More recently, H3K9 demethylation by KDM1A has been proposed to underlie a co-activator function at a subset of genes regulated by ER and AR (Metzger et al., 2005; Nair et al., 2010; Perillo et al., 2008; Wissmann et al., 2007). Paradoxically, it was recently observed that KDM1A also removes H3K4me2 at AR binding sites (Cai et al., 2014). However, the underlying mechanism and role for H3K4me2 demethylation in gene activation is largely unexplored.
Here, we report that KDM1A is a key epigenetic co-regulator for GR-mediated transcriptional regulation. We characterize the GR complex and identify KDM1A as an integral component directly interacting with GR. We show that GR-modulated H3K4me2 demethylation by KDM1A is broad and fundamental to the epigenetic regulation of GR binding to chromatin and resulting gene activation. This mechanism expands the role of KDM1A-mediated H3K4me2 demethylation in gene activation and hormone signaling.
RESULTS
KDM1A Is a Key Transcriptional Co-regulator of Dex Responsive Genes
We were interested to investigate whether KDM1A plays a role in GR-mediated gene regulation and to use GC-induced transcription as a model to explore the function of KDM1A in gene activation. We used A549, a lung adenocarcinoma cell line and established model for studying GR function (McNamara et al., 2018; Schlossmacher et al., 2011; So et al., 2007; Vockley et al., 2016; Wang et al., 2007). KDM1A knockdown (KD) did not significantly affect GR expression or translocation to the nucleus in response to the synthetic GR agonist dexamethasone (Dex) (Figures S1A–S1C). Dex-regulated genes were identified by RNA sequencing (RNA-seq) comparing Dex to vehicle (ethanol [EtOH]) treated cells transduced with scramble control (SC) small hairpin RNA (shRNA). A total of 548 genes were upregulated (Dex-induced [Dex-up]) and 806 were downregulated (Dex-repressed [Dex-dw]) (≥2-fold change, adjusted p value < 0.05). Examining the Dex response in KDM1A shRNA transduced cells (KD), we identified 305 (56%) Dex-induced and 538 (67%) Dex-repressed genes that failed to respond or were significantly impaired (≥2-fold reduction in Dex response) by KDM1A depletion, which we named GR-KDM1A co-regulated genes (Figures 1A and S1D). qRT-PCR of independent experiments confirmed co-regulation in 9 out of 10 Dex-induced, co-regulated genes identified by RNA-seq (Figure S1E). Overall, GR-KDM1A co-regulated genes accounted for 62% of Dex-responsive genes, supporting a significant role for KDM1A in GC signaling. In addition, we identified 184 upregulated and 117 downregulated genes (≥2-fold, adjusted p value < 0.05) comparing EtOH-treated control to KD cells. These genes regulated by KDM1A independent of GCs showed very little overlap (6%) with GR-KDM1A co-regulated genes, and were not examined further. While KDM1A is important for both activation and repression, the large impact on Dex-induced genes supported using GR to further explore co-activation mechanisms.
Figure 1. KDM1A Is a Key GR Co-regulator Recruited to GR Binding Sites.

(A) RNA-seq heatmaps of Dex-regulated genes with ≥2-fold change (adjusted p value [padj] < 0.05) between Dex and EtOH in control shRNA transduced cells (SC Diff). Co-regulated genes were Dex-regulated genes that failed to meet the Dex-regulated threshold or had a ≥2-fold defect in Dex response in KDM1A shRNA-transduced cells (KD Diff). See also Figures S1A–S1E.
(B) ChIP-seq profiles in fragments per kilobase million (FPKM) normalized to input at two representative Dex-induced, GR-KDM1A coregulated genes. See also Figures S1F and S1G.
(C) KDM1A ChIP-seq density. Input-normalized FPKM averaged for all GR peak regions and plotted aligned by peak center. The x axis shows distance in kb from peak center. All density plots generated similarly.
(D) Dex-induced log2 fold change (LFC) in KDM1A density at all GR peak regions (GRBSs). Control (Ctl) regions are annotation-matched DNase I hypersensitive sites that do not overlap a GRBS. p value by Mood’s median test. See also Figure S1H.
(E) KDM1A (blue) and immunoglobulin G (IgG) (gray) ChIP-qPCR at the GRBS nearest each of four Dex-induced genes and an intergenic control region (Interg). Distance to TSS in kb. Bars plot mean + SE of three to five biological replicates. p values, Student’s t test between EtOH and Dex for each antibody. See also Figures S1I–S1K.
KDM1A Is Recruited to GR Binding Sites in the Genome
To gain insight into co-regulation by KDM1A, we mapped GR and KDM1A genome-wide binding using chromatin immunoprecipitation and deep sequencing (ChIP-seq). GR ChIP-seq identified 9,096 high-confidence peaks after Dex treatment (89% overlap between replicates). These GR binding sites (GRBSs) were significantly enriched for the consensus GR motif (Figure S1F). Consistent with previous studies (Reddy et al., 2009; So et al., 2007), only 6% of GRBSs were found at promoters, while the vast majority were distal regulatory elements located in introns and intergenic regions (Figure S1G).
KDM1A ChIP-seq detected binding at promoters, intergenic, and intronic regions in both EtOH- and Dex-treated cells (Figure 1B). To assess whether KDM1A was recruited to GRBSs, we examined its ChIP-seq signal at all GRBSs and observed a significant increase after Dex treatment; median log2-fold change (LFC) of 1.1 (>2-fold increase) (Figures 1C and 1D). In contrast, a control set of annotation matched, GR− DNase I hypersensitive sites (Vockley et al., 2016) showed no overall change in KDM1A binding (median LFC = 0). Plotting KDM1A density at all GRBSs in Dex-versus EtOH-treated cells showed that most sites gained KDM1A rather than a minority with large magnitude changes (Figure S1H). In addition, after Dex treatment KDM1A was ≥2-fold higher than the median control at 63% of GRBSs.
These observations were confirmed at four co-regulated, Dex-induced genes: period circadian regulator 1 (PER1), baculoviral IAP repeat containing 3 (BIRC3), cyclin-dependent kinase inhibitor 1C (CDKN1C), and dual specificity phosphatase 1 (DUSP1). GR-KDM1A co-regulation was validated by two independent shRNAs using quantitative RT-PCR (Figure S1I). Dex induced significant KDM1A binding at all four of the nearest GRBSs in multiple independent ChIP-qPCR experiments (Figures 1E, S1J, and S1K). Together, these data demonstrate that GR activation results in significant KDM1A recruitment to GR binding sites and that the majority of GRBSs are bound by KDM1A in Dex-treated cells, supporting a direct role for KDM1A in GR-mediated gene regulation.
KDM1A Forms a Stable Complex with GR by Direct Interaction with the AF1 Domain
Dex-induced recruitment to GRBSs suggested KDM1A might be part of the GR complex. To test this, we purified the nuclear GR complex from Dex-treated cells using tandem affinity purification (TAP) (Fang et al., 2010; Shi et al., 2005). Silver staining of the FLAG and subsequent hemagglutinin (HA) elutes showed specific enrichment of GR and associated proteins (Figure 2A). Mass spectrometry of the HA elute showed enrichment of transcriptional regulators, along with several chromatin modifiers, including the Swi/Snf complex, the MLL4 H3K4 methyltransferase complex, several histone deacetylases (HDACs), KDM3C/ JMJD1C (an H3K9/36 demethylase), and KDM1A (Figure S2A). To further investigate the interaction between KDM1A and GR, an anti-KDM1A antibody was used for reciprocal immunoprecipitation from mock and GR TAP eluates (Figure 2B). GR remained enriched in the KDM1A reciprocal immunoprecipitation (IP) along with many other regulators, demonstrating that KDM1A and GR form a stable complex (Figure 2C). Glutathione S-transferase (GST) pulldown of purified recombinant proteins showed a direct interaction between GR and KDM1A (Figure 2D, lanes 1–7). GR has three domains: an N-terminal activating function 1 domain (AF1), a central DNA-binding domain, and a C-terminal ligand binding domain with AF2 transactivating activity. GR truncations (Figure 2E) showed that KDM1A directly interacts with the N-terminal AF1 domain (Gm1–419), while the DNA and ligand binding domains (Gm420–777) were not required (Figure 2D). Additional N-terminal truncations showed little KDM1A binding, suggesting the full AF1 domain is necessary (Figure S2B).
Figure 2. GR Forms a Complex with KDM1A and Modulates KDM1A H3K4-Specific Demethylase Activity.

(A) Silver stain of tandem affinity purified (TAP) GR complex and mock control. See also Figure S2A.
(B) Silver stain of anti-KDM1A reciprocal IP of mock or GR complexes.
(C) Western blot of GR complex components identified by tandem mass spectrometry (MS/MS). Lanes 3 and 4 are TAP samples (FLAG then HA purified). Lanes 5 and 6 are KDM1A IP from final TAP samples (underwent FLAG, then HA, then KDM1A IP). Lane 7 is KDM1A IP from the FLAG purification (underwent FLAG then KDM1A IP).
(D) Silver stain of KDM1A co-immunoprecipitation by GR. Purified full-length GR, either FLAG or GST tagged (lanes 2–7), GST-GR truncations, or GST control was used to pull-down KDM1A. Independent replicates loaded in adjacent wells. See also Figure S2B.
(E) Diagram of GR truncation mutants.
(F) Histone demethylase (HDM) assays with TAP complexes.
(G and H) Effect of full-length (G) and GR truncation (H) proteins on KDM1A histone demethylase activity. Recombinant GR protein was added to KDM1A HDM reactions and HDM activities were examined by immunoblot. To control for purification contaminants, a sample of purified GR protein was heat denatured before addition (G; lane 5). See also Figures S2C and S2D.
(I) Quantification of HDM assays by densitometry. Bars plot mean + SE of four to five biological replicates. p values by Student’s t test.
GR Modulates KDM1A Enzymatic Activity to Protect H3K4me1
To investigate KDM1A function in the GR complex, we tested for histone demethylase activity in a cell-free assay, under conditions allowing FAD-dependent demethylation carried out by KDM1A or its homolog KDM1B but not Jumanji domain-containing HDMs such as JMJD1C (also part of the GR complex). Surprisingly, no H3K4 or H3K9 demethylation was detected on bulk histones (Figure 2F). The lack of robust demethylation suggested GR might inhibit KDM1A. Indeed, we observed dose-dependent inhibition of KDM1A by purified recombinant GR protein (Figure 2G, lanes 1–3). In contrast, heat-denatured GR (lane 5) and native GST protein (lane 6) had little to no effect. Consistent with the co-immunoprecipitation results, this inhibitory effect was ligand independent (Figure 2G, lane 4), and the AF1 domain (Gm1–419) was necessary and sufficient to inhibit KDM1A (Figure 2H). No demethylation of H3K9 was detected under any conditions. While both were impacted, the effect of GR on KDM1A-mediated H3K4me1 demethylation appeared to be stronger than H3K4me2. For example, lanes 5–7 of Figure 2G show similar degrees of H3K4me1 and H3K4me2 loss catalyzed by KDM1A alone (compared to the buffer control). Lanes 3 and 4 show more H3K4me1 remained when GR was present, suggesting GR is more effective at blocking H3K4me1 loss. To quantify this effect, fold inhibition defined as H3 normalized (H3K4me with GR)/(H3K4me KDM1A alone) was calculated from densitometry of at least four independent experiments. Full-length GR and the AF1 domain showed 3- to 4-fold inhibition of H3K4me1 demethylase activity, compared to more moderate effects on H3K4me2 demethylation (Figure 2I). Thus, GR inhibits KDM1A H3K4 demethylase activity, creating a preference that largely protects H3K4me1.
To evaluate whether KDM1A modulation is a general activity of nuclear receptors (NRs), we tested ER, AR, mineralocorticoid receptor (MR) N terminus (1–110aa), thyroid receptor (TR), retinoic acid receptor (RAR), and retinoic X receptor (RXR) in similar cell-free assays (Figures S2C and S2D). Interestingly, RAR-related orphan receptor beta (RORb), lacking a classic N-terminal activation domain, was the only NR that did not inhibit KDM1A. These data support a conserved molecular mechanism across NRs to modulate KDM1A enzymatic activity against different methyl states of the same residue.
GRBSs Show Hormone-Induced H3K4me2 Loss but Maintain H3K4me1
To examine whether GR regulates KDM1A demethylase activity in cells, ChIP-seq was used to determine genome-wide, hormone-induced changes in histone methylation. The LFC of each mark was measured ± 250 bp from the center of each GRBS or control region. H3K4me2 levels were significantly decreased at GRBSs with a median LFC of −0.3, while control regions showed no median change (Figures 3A and 3B). No significant change was observed in H3K4me1, H3K4me3, H3K9me1, or H3K9me2 (median LFC = 0) (Figures 3A and S3A).
Figure 3. Preprogramed H3K4me2 Is Demethylated upon GR Activation at KDM1A-Bound GRBSs.

All histone ChIP-seq densities normalized to input and total H3, quantified ± 250 bp from GRBS or Ctl region center.
(A) Boxplots showing interquartile range and median ChIP-seq LFC at all GRBSs (GR) and Ctl regions. Asterisk indicates p value < 1 E−9 by Mood’s median test. See also Figure S3A.
(B) Average H3K4me2 ChIP-seq density at GRBSs. See also Figure S3B.
(C and D) ChIP-seq LFC in H3K4me2 (C) and H3K4me1 (D) at parsed GRBSs. KDM Neg, GRBSs with KDM1A density < median Ctl (n = 2217); LFC < 1.5, constitutive KDM1A-bound GRBSs with log2(KDM1ADex/KDM1AEtOH) < 1.5 (n = 2917); and LFC ≥ 1.5, GRBSs that recruit KDM1A with log2(KDM1ADex/ KDM1AEtOH) ≥ 1.5 (n = 3962). Asterisk, p < 4 E−12 for indicated pairs by Bonferroni-corrected pairwise Mood’s median test. See also Figures S3C–S3E.
(E) ChIP-qPCR of H3K4me2 (red) and H3K4me1 (magenta) normalized to input and H3 at GRBSs near Dex-induced genes (as in Figure 1E). Bars plot mean + SE of three or four biological replicates; p value, Student’s t test between EtOH and Dex. See also Figure S3F.
(F) H3K4me2 ChIP-seq prior to GR activation (EtOH) at GRBSs parsed as in (C). p value, Mood’s median test for the three indicated groups.
(G) H3K4me2 versus H3K27ac at all GRBSs in EtOH-treated cells. Pearson correlation coefficient = 0.7. See also Figures S3G and S3H.
(H) Average H3K27ac ChIP-seq density at all GRBSs.
(I) Venn diagram of GRBS preprogrammed states. Pre-marked GRBSs had both H3K4me2 and H3K27ac. Unmarked GRBSs lacked both H3K4me2 and H3K27ac. Open GRBSs had high DNase I signal in EtOH.
(J) Boxplots of KDM1A density at GRBSs parsed by preprogrammed state as in (I). p value for indicated pair from Bonferroni-corrected pairwise Mood’s median test. Et, EtOH control; D, Dex treated.
(K) Enrichment of the consensus GR motif. Odds ratio over control regions is plotted.
Examining hormone-induced H3K4me2 loss in more detail, we found the majority (58%) of GRBSs lost H3K4me2 (Figure S3B), suggesting dynamic regulation of this mark is a common feature. To examine the relationship between H3K4me2 loss and KDM1A, GRBSs were parsed into three groups according to KDM1A binding status (Figure S3C). GRBSs with low levels of KDM1A binding (< the median KDM1A level at control regions) in both EtOH- and Dex-treated cells accounted for 24% of all GRBSs (KDM1A negative [KDM Neg]). A total of 32% had moderate changes in KDM1A binding (LFC < 1.5), primarily representing constitutive binding with high levels of KDM1A prior to GR activation. The remaining 44% of GRBSs showed a strong Dex-induced increase in KDM1A with a median LFC of 4.1 (18-fold increase). H3K4me1/2 did not change (LFC = 0.0 for both) at KDM Neg GRBSs (Figures 3C and 3D). GRBSs with constitutive KDM1A binding (LFC < 1.5) showed a modest reduction in both H3K4me1/2 (LFCs of −0.1 and −0.2, respectively). GRBSs with induced KDM1A binding (LFC ≥ 1.5) showed significant loss of H3K4me2 (LFC = −0.6) and an increase in H3K4me1 (LFC = 0.2). KDM1A bound sites also had the highest levels of H3K4me1 after Dex treatment (Figure S3D). To examine this in another way, KDM1A peaks that did not overlap a GRBS (GR−) were compared to KDM1A peaks that did (GR+) (Figure S3E). Validating what we observed at GRBSs, GR+ KDM1A peaks had a strong response to Dex with increased KDM1A binding and H3K4me2 demethylation, whereas GR− KDM1A peaks had significantly less KDM1A recruitment and no H3K4me2 loss. Dex-induced H3K4me2 loss and H3K4me1 enrichment at KDM1A+ GRBSs was further validated by ChIP-qPCR (Figures 3E and S3F). These results indicate that GR recruits KDM1A to demethylate H3K4me2 and protects H3K4me1, consistent with the cell-free assays.
H3K4me2 Is Part of a Preprogrammed Histone Signature at GRBSs
The observation that H3K4me2 decreases in response to Dex suggested it may be an important signature of GRBSs prior to GR activation. Indeed, prior to GR activation, H3K4me2 levels at KDM1A+ GRBSs were significantly higher than the control or KDM Neg GRBSs (Figure 3F). Examining the relationships between pre-activation histone marks, we found a strong correlation between H3K4me2 and H3K27ac (Pearson coefficient = 0.7; Figure 3G). H3K27ac correlation with H3K4me1 and H3K4me3 was weaker (Pearson coefficients = 0.5 and 0.6, respectively; Figure S3G). High pre-activation levels of H3K27ac likely explain the only modest increase with Dex (Figures 3A and 3H). With changes in opposite directions, the correlation between H3K4me2 and H3K27ac weakened after Dex treatment (Figure S3H). Importantly, 71% of GRBSs were pre-marked by both H3K4me2 and H3K27ac (Figure 3I), supporting this dual mark as a pre-activation signature at GRBSs. Comparing GRBSs with this preprogrammed signature (pre-marked) to GRBSs lacking both H3K4me2 and H3K27ac (unmarked) showed more KDM1A is recruited to pre-marked GRBSs (Figure 3J). This suggests KDM1A’s primary role is to regulate preprogrammed H3K4me2.
H3K9 demethylation has been reported to play an important role in ER- and AR-mediated gene activation (Metzger et al., 2005; Nair et al., 2010; Perillo et al., 2008; Wissmann et al., 2007). We observed only 26% of GRBSs pre-marked by H3K9me2, much less than the 83% pre-marked by H3K4me2 (Figures S3I–S3K). While most (85%) of the H3K9me2+ GRBSs lost H3K9me2 after Dex, they showed no difference in KDM1A binding compared to H3K9me2− GRBSs. Ultimately, only 11% of all GRBSs recruited KDM1A (LFC ≥ 1.5) and lost H3K9me2 in response to Dex, while 36% of all GRBSs recruited KDM1A and lost H3K4me2. Together, these data suggest H3K9 demethylation occurs at only a small subset of GR binding sites, whereas H3K4me2 demethylation is a more common feature.
To further understand the features of H3K4me2 pre-marked GRBSs, we examined chromatin accessibility and transcription factor binding potential. We find 60% of all GRBSs have open chromatin prior to GR activation but this only partially overlaps H3K4me2+ H3K27ac+ sites (Figure 3I). Motif analysis showed that unmarked GRBSs were enriched for GR motifs compared to pre-marked GRBSs (Figure 3K). The top enriched motifs (ranked by p value) at pre-marked GRBSs were members of the AP-1 family of transcription factors. Together, these data suggest that H3K4me2 preprogramming is a significant feature of GRBSs and may influence GR binding to chromatin.
KDM1A Demethylates H3K4me2 at Active Enhancers
Nuclear receptors, including GR, primarily bind to distal regulatory sites such as enhancers. On the other hand, KDM1A binds broadly in the genome, including at TSSs and enhancers. To better understand how KDM1A contributes to gene activation, we compared the binding pattern and histone modification changes at TSSs and nearby GRBSs of Dex-regulated genes. We first measured the distances between GRBSs and Dex-up and Dex-dw genes, as well as a subset of genes that did not change expression after Dex or KDM1A KD (Ctl). Distances between the nearest GRBS and Dex-repressed gene (Figure 4A, blue plot) were similar to the control genes, while Dex-up genes showed strong enrichment of distances less than 50 kb and depletion of longer distances (Figure 4A, red plot). 55% of Dex-up genes had a GRBS within 25 kb of the TSS and 66% within 50 kb (Figure 4B). In contrast, only 17% of Dex-dw genes had nearby GRBSs, similar to the fraction of control genes. This suggests that GRBSs near Dex-up genes are more likely to play a role in directly inducing transcription. In addition, GRBSs close to Dex-up genes had significantly more GR binding and a larger increase in H3K27ac after Dex (Figure S4A), further supporting their function as active enhancers.
Figure 4. KDM1A Functions at Active Enhancers to Remove H3K4me2.

(A) Binned distances between nearest GRBS and TSSs of Dex-induced (Dex-up), Dex-repressed (Dex-dw), or Ctl genes.
(B) Fraction of genes with a GRBS or control region ± 25 kb or ± 50 kb from the TSS for each gene group. UpCoReg, Dex-induced GR-KDM1A coregulated genes; DwCoReg, Dex-repressed GR-KDM1A coregulated genes.
(C and D) ChIP-seq KDM1A density (C) and H3K4me2 LFC (D) at the TSS of control genes (Ctl TSS), Dex-up genes (Up TSS), Dex-dw genes (Dw TSS), and GRBSs ± 25 kb of a Dex-up gene (Up GRBS). E, EtOH; D, Dex. Density quantified ± 250 bp from TSS or GRBS center. See also Figure S4A.
(E) KDM1A density after Dex at TSSs or Up GRBSs parsed by genes that are co-regulated (CoReg) or not co-regulated (Not) as defined in Figure 1A.
(F and G) H3K27ac ChIP-seq density (F) and LFC (G) at parsed GRBSs as in Figure 3C. TSSs of control genes are included for reference. See also Figures S4B and S4C.
(H) GRBSs ± 25 kb from the TSS of a Dex-responsive gene parsed by KDM1A status, as in Figure 3C. Odds ratio over control regions is plotted.
All ChIP-seq p values from Bonferroni-corrected pairwise Mood’s median test.
To examine the role of KDM1A at putative active enhancers, GRBSs within 25 kb of a Dex-induced gene (Up GRBS) were compared to the TSSs of control, Dex-induced, and Dex-repressed genes. We observed more KDM1A recruitment and more H3K4me2 demethylation at nearby GRBSs than at TSSs (Figures 4C and 4D). It is interesting to note that the TSSs of Dex-induced genes showed more H3K4me2 loss than the TSSs of control or Dex-repressed genes, suggesting concordance between enhancers and promoters. Additionally, GRBSs near Dex-induced, GR-KDM1A co-regulated genes had more KDM1A binding than GRBSs near Dex-induced genes that were not regulated by KDM1A, while their TSSs showed no difference, suggesting that KDM1A bound to nearby enhancers was more likely responsible for co-regulation (Figure 4E). Together, these data support a role for KDM1A in removing H3K4me2 at active enhancers during Dex-induced gene expression.
To further interrogate KDM1A function at putative enhancers, we examined the change in H3K27ac at KDM1A-parsed GRBSs (as in Figure 3C). Sites that recruited KDM1A (LFC ≥ 1.5) and showed the strongest H3K4me2 demethylation not only had the highest levels of H3K27ac (Figure 4F), but also the largest Dex-induced increase (Figure 4G). This was confirmed by independent ChIP-qPCR experiments (Figure S4B). At GRBSs that recruit KDM1A, H3K27ac was similar to active TSSs (Figure 4F, black boxplot). However, H3K4me3 was much lower, supporting enhancer function as opposed to unannotated TSSs (Figure S4C). Lastly, KDM1A+ GRBSs were most enriched near Dex-induced genes, again supporting a role for KDM1A and H3K4me2 demethylation at active enhancers (Figure 4H).
Together, these analyses show that KDM1A-binding and H3K4me2 demethylation occur at a large portion of GRBSs including active enhancers, where KDM1A-mediated H3K4me2 demethylation may play a direct role in GR-mediated gene activation.
KDM1A Enzymatic Activity Is Required for H3K4me2 Demethylation and GR Binding
To unambiguously determine whether KDM1A H3K4 demethylase activity is required for GR-mediated gene regulation, we first tested whether wild-type or catalytically inactive mutants of KDM1A could rescue Dex-induced expression in KDM1A-depleted cells (Figures S4D–S4F). Expression of an RNAi-resistant wild-type KDM1A cDNA rescued GR-induced expression of PER1, CDKN1C, and DUSP1, while two catalytic mutants did not, demonstrating that KDM1A demethylase activity is required for induction of co-regulated genes. Similarly, inhibition of KDM1A enzymatic activity by the monoamine oxidase inhibitor, tranylcypromine (TCP), effectively blocked upregulation by Dex, but did not affect GR expression or translocation to the nucleus (Figures S5A–S5C).
The above functional analysis supported a crucial role for KDM1A in the epigenetic regulation of GR function. We hypothesized that blocking KDM1A enzymatic activity would prevent Dex-induced H3K4me2 demethylation at GRBSs and possibly affect GR binding to the genome. To test this, we treated cells with TCP for 24 h prior to Dex or EtOH treatment. TCP was used instead of KDM1A KD to minimize disruption to the GR complex and to focus on demethylase-dependent mechanisms. TCP significantly increased H3K4me1, H3K4me2, and H3K4me3 at GRBSs compared to control regions (Figure 5A). Importantly, TCP also blocked Dex-induced H3K4me2 demethylation and caused mild loss of H3K4me1 at GRBSs (Figures 5B and S5D). This was confirmed by additional independent ChIP-qPCR experiments with TCP and KDM1A KD (Figure S5E). Moreover, GRBSs near Dex-induced genes showed a stronger TCP-induced increase in H3K4me2 compared to TSSs (Figure 5C). Collectively, these results demonstrate that KDM1A enzymatic activity is required for H3K4me2 demethylation at GRBSs, and further supports a specific role for KDM1A in regulating H3K4me2 levels at active enhancers.
Figure 5. KDM1A-Mediated H3K4me2 Demethylation Promotes GR Binding.

Cells were treated with TCP or DMSO vehicle (Veh) for 24 h prior to Dex or EtOH.
(A and B) TCP-induced (A) and Dex-induced (B) changes in H3K4 methylation ChIP-seq. LFC in (A) calculated as log2(K4meTCP/K4meVeh) after Dex. Ctl, control regions; GR, GRBSs. LFC in (B) calculated as log2(K4meDex/K4meEtOH) in TCP or Veh-treated cells. p value, Mood’s median test. See also Figures S4D–S4F and S5A–S5E.
(C) TCP-induced H3K4me2 ChIP-seq LFC at Ctl TSS, Up TSS, and Up GRBSs (as in Figures 4C and 4D).
(D) Average GR ChIP-seq density at all GRBSs.
(E) Representative GRBSs with loss of GR binding after TCP. Input normalized GR ChIP-seq profiles in FPKM for control (light green) and TCP (dark green).
(F) GR ChIP-qPCR at GRBSs near Dex-induced genes (as in Figure 1E). Gray bars, control shRNA; teal bars, KDM1A shRNA (K-sh1); and brown bars, TCP. Bars plot mean + SE of three to six biological replicates; p value, Student’s t test comparing Dex versus Dex + K-sh1 or Dex + TCP.
(G) GR density at all GRBSs in cells treated with TCP (Dex + TCP, y axis) versus DMSO (Dex + Veh, x axis). Dashed lines indicate thresholds used to define groups in (H) and (I). Mild, LFC > −1 (n = 494); moderate, LFC between −1 and −2.5 (n = 3999); and severe, LFC ≤ −2.5 (n = 4605). See also Figures S5F and S5G.
(H and I) KDM1A ChIP-seq density (H) and TCP-induced H3K4me2 LFC (I) at GRBSs parsed by GR loss as in (G).
p values for indicated groups in ChIP-seq boxplots by Mood’s median test.
To test whether KDM1A-mediated H3K4me2 demethylation influences GR binding, we examined the effect of TCP on the genome-wide distribution of GR by ChIP-seq. Strikingly, TCP reduced GR binding by nearly 80% (Figure 5D; representatives shown in Figure 5E). This effect was wide-spread, as 84% of GRBSs were undetectable as peaks in TCP treated cells (Figure S5F), and nearly all GRBSs showed some reduction in GR density (Figure 5G). This result was confirmed by multiple independent ChIP-qPCR experiments with TCP or KDM1A KD, which both significantly reduced GR binding at all four KDM1A+ GRBSs (Figure 5F).
If blocking KDM1A-mediated H3K4me2 demethylation is responsible for impaired GR binding, it predicts that the more severely affected sites should have more KDM1A binding and a larger TCP-induced H3K4me2 increase. To test this prediction, GRBSs were parsed by the TCP-induced defect in GR binding (TCP LFC) calculated as log2 (GRDex TCP/GRDex Veh). Only 5% of GRBSs were mildly affected (TCP LFC > −1), 44% were moderately affected (TCP LFC between −1 and −2.5), and the remaining 51% were severely affected with LFC % −2.5 (GR density reduced by 82% or more) (Figures 5G and S5G). Consistent with TCP inhibition of KDM1A as the cause of GR loss, KDM1A levels were highest at severely affected sites (Figure 5H), which also showed the largest increase in H3K4me2 (Figure 5I). Interestingly, along with loss of GR binding, ChIP-qPCR showed that TCP or KDM1A depletion also blocked recruitment of CBP/ p300, a NR coactivator and H3K27 histone acetyltransferase that promotes enhancer activation (Figure S5H). H3K27ac was also reduced at these GRBSs, likely as a consequence of CBP loss (Figure S5H). This is consistent with Figure 4G, showing that GRBSs with hormone-induced KDM1A binding have the largest increase in H3K27ac.
Together, these analyses demonstrate that KDM1A is responsible for regulating H3K4me2 at KDM1A-targeted GRBSs, including active enhancers. This function is critical for robust GR binding. Without KDM1A activity, H3K4me2 is not removed, which consequently disrupts GR binding and enhancer activation by other co-regulators such as CBP/p300.
DISCUSSION
While initially discovered as a co-repressor acting at gene promoters (Shi et al., 2004), a broader role for KDM1A that includes gene activation has been increasingly appreciated. Here, we report KDM1A is an essential co-regulator for GR. KDM1A forms a stable complex with GR and significantly co-localizes at GRBSs in the genome. As a result, most Dex-responsive genes require functional KDM1A for normal hormone regulation. KDM1A regulates H3K4me2 levels at a large portion of GRBSs, including active enhancers. The demethylase activity of KDM1A is required for GC-induced H3K4me2 loss and robust GR binding, providing another mechanism by which KDM1A functions in gene regulation. We propose a model for KDM1A’s contribution to GC-mediated gene activation in which H3K4me2 removal is an early step in enhancer activation that facilitates transcription factor binding (Figure 6). This differs significantly from previous work, which focused mainly on H3K9 demethylation during gene activation or removal of H3K4me1 during enhancer silencing (Shen et al., 2016; Whyte et al., 2012).
Figure 6. A Model of KDM1A-Mediated H3K4me2 Demethylation Contributing to GR-Mediated Gene Activation.

GRBSs, including enhancers potentially bound by pioneer factors, are premarked by H3K4me2 and other modifications, such as H3K27ac. Upon Dex activation, GR is targeted and transiently binds to the GRBS, where it recruits KDM1A and other cofactors such as CBP, for epigenetic reprogramming of the region. As an initial key step, KDM1A actively and preferentially demethylates H3K4me2 but leaves H3K4me1 intact due to inhibition from GR. H3K4me2 demethylation by KDM1A is required to stabilize GR binding. Other cofactors recruited by stabilized GR, such as CBP, increase H3K27ac at GRBSs, and this coordinated epigenetic reprogramming activates enhancers, thereby inducing GR-target gene expression, a process that can be blocked by the KDM1A inhibitors such as TCP.
Our finding that GR limits progressive H3K4 demethylation by KDM1A is interesting. GR is not the first protein to influence KDM1A activity. CoREST and PHF21A/BHC80 can enhance or inhibit KDM1A-mediated H3K4 demethylation, respectively (Shi et al., 2005), and AR or PELP1 have been reported to help in H3K4 to H3K9 substrate switching (Nair et al., 2010). In contrast, GR creates a preference between methylated states of the same residue. We note that this bias is most obvious in cells where H3K4me2 loss is readily observed, while H3K4me1 is un-changed or increased. The mechanism by which GR influences KDM1A activity is not clear. Changes in substrate binding or catalytic kinetics, as well as the interplay between HDMs and HMTs in the GR complex, are potential mechanisms that warrant further investigation.
Our study also showed that the majority of GRBSs are preprogrammed with H3K4me2, consistent with a known role for this mark in the binding of other NR pioneer factors (Lupien et al., 2008). Blocking H3K4me2 demethylation greatly reduced GR binding to chromatin, suggesting reduction of this mark upon GR activation is also crucial. The major influence of H3K4me2 loss seems to be in the transition to robust GR binding. This is consistent with transient and highly dynamic GR binding observed in live imaging (Groeneweg et al., 2014) and an “assisted loading” model in which initial GR binding assists rather than competes for additional binding at the same DNA motif (Voss et al., 2011). Consistent with this model, KDM1A recruited by GR may mediate epigenetic reprogramming and consequently stabilize GR binding to chromatin. We speculate that this assisted loading model may play a more important role at GRBSs that lack a strong GRE and are thus more dependent on pioneering factors.
It is noted that nucleosome eviction is an important energetic barrier to transcription factor (TF) binding (Tillo et al., 2010). Interestingly, we observe H3K4me2 enrichment at the center of GRBSs overlapping maximal GR and KDM1A binding, while other marks are enriched at flanking nucleosomes (Figures 3B, 3H, and S3A). Further investigation into the mechanism of H3K4me2 influence on GR chromatin loading may shed light on the relationship between the histone code and enhancer regulation.
Our findings have broadened the role of dynamic H3K4me2 and highlighted an important distinction between active regulation of H3K4me1 and H3K4me2 at enhancers. The presence of H3K4 methylation is canonically associated with gene activation, and KDM1A is known to remove both H3K4me1 and H3K4me2. Furthermore, H3K4me1 is a hallmark of enhancers, and removal by KDM1A inactivates enhancers during development or if artificially targeted (Kearns et al., 2015; Whyte et al., 2012). We find that GR, and many other NRs, can modulate KDM1A activity to protect H3K4me1. Surveying the steady-state genome for enhancer marks finds H3K4me1 and H3K27ac (Calo and Wysocka, 2013; Sakabe and Nobrega, 2013). Our data are consistent with this as H3K4me1 remains enriched after GR activation and KDM1A recruitment. Additionally, demethylation of H3K4me3 at enhancers by KDM5C acts as a break to prevent overactivation (Shen et al., 2016). Thus, all three H3K4 methyl states appear to be precisely regulated and have important roles in enhancer function. This fine-tuned control suggests each methyl state may contribute differently to gene transcription and should be considered individually.
KDM1A co-activator function has been suggested to rely on an enzymatic switch from H3K4 to H3K9 demethylation, as demonstrated at a subset of ER and AR target genes (Metzger et al., 2005; Nair et al., 2010; Perillo et al., 2008; Wissmann et al., 2007). However, a recent study reported KDM1A demethylated H3K4me2 at AR binding sites, but concluded gene activation occurred despite H3K4 loss, and that H3K4 demethylation may suppress cryptic enhancers (Cai et al., 2014). By investigating GR-mediated gene regulation, we show that not only does KDM1A-mediated H3K4me2 demethylation play a direct role in GR target gene activation, but this role is broad and fundamental. KDM1A inhibition impairs GR binding and consequently, a large portion of GC-mediated gene regulation fails to occur. Thus, GR-mediated gene regulation, including gene activation, is dependent on H3K4me2 demethylation. While we do observe Dex-induced loss of H3K9me2 at a small fraction of KDM1A-associated GRBSs where KDM1A may play a role in H3K9me2 demethylation, H3K4me2 demethylation occurs at a majority of GRBSs. Thus, KDM1A-mediated H3K4me2 demethylation at GR binding sites is an important new mechanism in gene regulation.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Yujian G. Shi (yujiang_shi@hms.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
HeLa-S cells (female) were optimized for large-scale suspension culture and were used for complex purification. A549 male lung adenocarcinoma cell line (ATCC CCL-185) was use for studying GR-KDM1A co-regulation of gene expression.
METHOD DETAILS
Tandem Affinity Purification of GR Complex
Tandem affinity purification of the GR complex was performed as previously described (Shi et al., 2003). Briefly, Flag-HA-GR expressing HeLa-S were cultured in DMEM supplemented with 10% FBS, and changed to DMEM with 5% charcoal stripped FBS for 24 hours prior to 50 nM Dex treatment for 6 hours. Nuclear extract was sequentially immunoprecipitated using anti-FLAG and anti-HA antibody conjugated agarose beads, and enriched complexes were eluted using FLAG and HA peptides, respectively. The mock purification was carried out in parallel under identical conditions from cells expressing FLAG-HA only. Purified proteins were sequenced by tandem mass spectrometry (MS/MS) at the Harvard Medical School Taplin Biological Mass Spectrometry Facility. The FLAG elute after a single step of anti-FLAG immunoprecipitation or the doubly purified GR complexes in the final HA elute were used for reciprocal IP using a bead-conjugated anti-KDM1A antibody.
GST-Pulldown
GR and GR truncation mutants were expressed and purified from the pGEX-4T-1 vector in E. coli. The homogeneity of the purified protein was determined using SDS–PAGE followed by Coomassie blue staining. For GST pulldown, GR proteins were incubated with 6xHIS tagged KDM1A overnight at 4°C in buffer containing 50 mM Tris-HCl pH 8.0, 200 mM NaCl, and 0.1% IGEPAL CA-630. Pull-down complexes were washed five times in the same buffer before elution by boiling in SDS sample buffer and analyzed by SDS-PAGE and Coomassie blue staining.
Histone Demethylase Assays
Histone demethylase assays were carried out as described previously (Shi et al., 2005). Briefly, 6xHIS-KDM1A was incubated with purified NR proteins for 1h at 4°C prior to overnight incubation at 32°C with bulk histones or purified nucleosomes in demethylase buffer (50mM Tris, pH 8.0, 0.1 Units formaldehyde dehydrogenase and 1mM NAD+). In a typical reaction, 4 µg of calf thymus histones or 6 µg of purified nucleosomes were incubated with 0.5 µg KDM1A and 0.5–2.5 µg NR in a total reaction volume of 50 ml. Reactions were analyzed by SDS–PAGE and western blotting using methyl-specific antibodies. Purified TR, ER, MR (1–110aa), and ROR were purchased from Abnova (catalog ID: H00007067-P01, H00002100-P01, H00004306-Q01, H00006096-P01). AR was purchased from RayBiotech (RB-14–0003P). GST-RAR and RXR was a gift from Dr. Anders Näär.
RNA Interference, RNA Sequencing, and RT-qPCR
Retroviral shRNA targeting human KDM1A/LSD1 (Sigma, L-sh1:TRCN0000046071, L-sh2:TRCN0000046072) and control scramble (SC) shRNA was used to transduce A549 cells. The knockdown efficiency was determined by RT–qPCR and western blot. A549 cells transduced with KDM1A shRNA or a scramble control shRNA were grown in DMEM, 5% stripped serum, 2 µg/ml puromycin for 5 day prior to treatment with 100nM Dex or an equivalent volume of ethanol. RNA was extracted from treated A549 cells by Trizol (Life Science) after a 24-hour Dex or ethanol treatment. For RNA-seq, poly-A enriched mRNA was subjected to 2×150 bp pair-end Illumina sequencing per manufacturer’s instructions. 24 ± 0.7 million (mean ± SE) reads were obtained for each sample. For RT-qPCR validation, cDNA from independent biological replicates were produced using SuperScript III RT kit (Invitrogen) according to manufacturer’s instructions. Sequences of RT-qPCR primers are available upon request.
Immunofluorescence Staining
Cells were seeded on glass coverslips and grown in DMEM, 5% charcoal stripped FBS for 3 days. After treatment with 100nM Dex or ethanol for 1 hour, cells were fixed with 4% paraformaldehyde for 10 min, permeabilize with 0.2% Triton X-100, PBS for 20 minutes, stained with anti-GR antibody (Santa Cruz, sc-8992), and visualized with anti-rabbit AF549 antibody (Invitrogen). Similar results were obtained using a second anti-GR antibody (Santa Cruz, sc-1003).
Cellular Fractionation
Cells grown in DMEM, 5% charcoal stripped FBS were treated with vehicle or Dex for 1 hour. After trypsinization and washing with PBS, cells were resuspended in cell lysis buffer (15mM Tris-HCl pH 7.5, 60mM 0.3 M sucrose, 0.2% IGEPAL CA-630, 60mM KCl, 15mM NaCl, 5mM MgCl2, 0.1mM EGTA, 0.5mM DTT, 0.1mM PMSF) and incubated on ice for 10 min. The cytosolic fraction and the nuclei were separated by centrifugation at 4000 rpm for 5 minutes. The nuclei pellet was washed once in cell lysis buffer to remove traces of cytosolic proteins and lysed in 15mM Tris pH 7.5, 1% SDS, 0.1mM PMSF. For each sample, equal amount of protein quantified by Bradford assays (Bio-Rad) was analyzed by SDS-PAGE and immunoblot with indicated antibodies.
ChIP-qPCR and ChIP Sequencing
Conventional ChIP was performed as previously described (Shi et al., 2003) using formaldehyde-crosslinked chromatin. Cells were treated with 100nM Dex for 2 h. Antibodies KDM1A/LSD1 (Abcam ab17721), GR-H300 (Santa Cruz sc-8992), H3K4me1 (Abcam ab8895), H3K4me2 (Millipore 07–030), H3K4me3 (Millipore 07–473), H3K9me2 (Abcam ab1220), H3K27ac (Millipore 17–683), and H3 (Abcam ab1791) were used. All histone modification antibodies have been validated by ENCODE for ChIP-seq (Egelhofer et al., 2011). ChIP-seq was performed on two biological replicates, each with 35 ± 4 million (mean ± SE) reads sequenced by the Illumina Genome Analyzer II (GA II). Quantitative PCR (qPCR) of independent ChIP samples was used for validation. ChIP-qPCR primer sets were as follows: BIRC3_TSS, 5′-GGTTATTACCGCTGGAGTTC-3′ and 5′-AAATGCGTCACCCAAATCC-3′; BIRC3_GRB, 5′-GATGGCCAGTAATGGAACTG-3′ and 5′-ATGCATCTCATCAGGGCATC-3′; CDKN1C_TSS, 5′-ACTAGTACTGGGAAGGTCC-3′ and 5′-TTCTTCTCGCTGTCCTCTC-3′; CDKN1C_GRB, 5′-AGGTCAGCTCACAGGATTG-3′ and 5′-CCCTTGCGCAAAGAGAAAG-3′; DUSP1_TSS, 5′-GTCAGACCACTTAACTGTGG-3′ and 5′-GCAAAGGCATGGAAGAGTAG-3′; DUSP1_GRB, 5′-CCAGGTGCATTACAGGTATC-3′ and 5′-CTTAGGCATGTGACCTTTGG-3′; PER1_TSS, 5′-CATCATGTTCTCTTGGCTGGTGG-3′ and 5′-AGGACGGCTGTCGTTTTGTTG-3′; PER1_GRE, 5′-CATCAGATTGGAAGTGGCAG-3′ and 5′-CGACCAGGTAGGCATCTC-3′.
QUANTIFICATION AND STATISTICAL ANALYSIS
For RNA-seq analysis, Salmon was used to map and quantify counts (Patro et al., 2017), and DESeq2 was used for differential gene expression analysis (Love et al., 2014). Dex-responsive genes were defined by ≥ 2-fold change, adjusted p value < 0.05 comparing scramble-Dex to scramble-ethanol. Dex-responsive genes also regulated by KDM1A (co-regulated) were defined as either failing to be regulated by Dex according to the above thresholds, or as having at least a 2-fold reduction in the magnitude of Dex response. Genes regulated by KDM1A at the basal level were defined by ≥ 2-fold change, adjusted p value < 0.05 comparing KDM1A KD-ethanol to scramble-ethanol. MA plots show genes with transcript counts > 10. The x axis plots the mean variance stabilizing transformed (vst) counts as expression. The y axis plots log2 fold change transformed by adaptive shrinkage estimator (Stephens, 2017).
For ChIP-seq, sequenced reads were mapped onto the reference human genome (NCBI Build UCSC hg19) using Bowtie (v0.12.7) (Langmead et al., 2009). The hg19 genome was download from the UCSC website (http://genome.ucsc.edu). PCR induced duplicate reads were removed with samtools. Unique mapped reads per library was 23 ± 3 million (mean ± SE). Using the Model-based Analysis of ChIP-Seq (MACS v1.4) package (Zhang et al., 2008), regions of significant enrichment were determined against sample input with p < 1E-5. Few GR peaks were identified in ethanol treated samples, consistent with hormone depletion prior to Dex. Regions with GR peaks in ethanol were filtered from the final set to remove possible fragmentation artifacts (< 2% of regions were removed). GR peaks overlapped 89% between replicate ChIP-seq experiments.
ChIP-seq of all histone modifications were carried out under the same conditions. All were normalized to anti-H3 ChIP-seq in addition to normalization with input. Normalization to H3 is necessary to account for potential nucleosome occupation changes, which can indirectly alter histone modification levels. ChIP-seq density plots were centered on scaled GR peak regions and generated using the DeepTools package (Ramírez et al., 2016). ChIP-seq and DNase-seq quantification was carried out using KentUtils (Kent et al., 2010). GR and KDM1A were quantified on each GR peak region (averaged 400 bp in size). The only exception was where KDM1A density was compared between GRBSs and TSS regions. For this comparison, regions were size matched by quantifying KDM1A density ± 250 bp from the center of each GR peak region. All TSS region densities were quantified ± 250 bp from the +1 nucleotide. Similarly, all histone modification densities were quantified ± 250bp from the region center to capture flanking nucleosomes. All bed region analysis was carried out using BEDtools (Quinlan and Hall, 2010). Genomic distance and overlap analysis used the GenomicRanges package (Lawrence et al., 2013). Control regions were generated by subsampling DNase I hypersensitive sites that did not overlap a GRBS. Because the set of DNase I HS sites contained a much larger proportion of transcription start sites than the GRBS set, we proportionally sampled promoter and non-promoter DNase I HS sites to generate a control set of open regions with the same annotation mix as GRBSs. Thresholds for DNase I signal (distinguishing open and closed) and for H3K4me2 and H3K27ac (distinguishing pre-marked and unmarked) at GRBSs were defined as the minimum between the two modes of the bimodal distribution of density at all GRBSs. The HOMER software package and MEME Suit were used for motif analysis and validation (Bailey et al., 2009; Heinz et al., 2010).
Violin/boxplots, Venn diagrams, and heatmaps were generated using the R packages ggplot2 and ComplexHeatmap (Gu et al., 2016; Wickham, 2009). P values were also calculated in R. For ChIP-seq, p values > 0.001 are not reported. The nonparametric Mood’s median test and Kolmogorov–Smirnov test were used, as the ChIP-seq data was not normally distributed. For all reported Mood’s median p values, Kolmogorov–Smirnov p value was also < 0.001. Post hoc pairwise testing of groups with more than one comparison were adjusted for multiple tests using a Bonferroni correction.
DATA AND SOFTWARE AVAILABILITY
Raw fastq files of RNA-seq and ChIP-seq, processed ChIP-seq bigwig and bedgraph files, and bed files containing GR peak regions or control regions are available at GEO (https://www.ncbi.nlm.nih.gov/geo/), accession number GEO: GSE108588.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| KDM1A/LSD1 | Abcam | Cat#ab17721; RRID: AB_443964 |
| GR-H300 | Santa Cruz | Cat#sc-8992; RRID: AB_2155784 |
| H3K4me1 | Abcam | Cat#ab8895; RRID: AB_306847 |
| H3K4me2 | Millipore | Cat#07–030; RRID: AB_310342 |
| H3K4me3 | Millipore | Cat#07–473; RRID: AB_1977252 |
| H3K9me2 | Abcam | Cat#ab1220; RRID: AB_449854 |
| H3K27ac | Millipore | Cat#17–683; RRID: AB_1977529 |
| H3 | Abcam | Cat#ab1791; RRID: AB_302613 |
| Bacterial and Virus Strains | ||
| KDM1A shRNA (L-sh1) | Sigma | TRCN0000046071 |
| KDM1A shRNA (L-sh2) | Sigma | TRCN0000046072 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| GR protein | This paper | N/A |
| KDM1A protein | This paper | N/A |
| Dexamethasone | Sigma | Cat#D1756 |
| Tranylcypromine | Sigma | Cat#P8511 |
| Deposited Data | ||
| RNA-seq and ChIP-seq | This paper | GEO: GSE108588 |
| Experimental Models: Cell Lines | ||
| A549 | ATCC | ATCC CCL-185; RRID: CVCL_0023 |
| Recombinant DNA | ||
| GR-pGEX-4T-1 | This paper | available upon request |
| Software and Algorithms | ||
| Salmon | Patro et al., 2017 | https://combine-lab.github.io/salmon/ |
| DESeq2 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Bowtie (v0.12.7) | Langmead et al., 2009 | http://bowtie-bio.sourceforge.net/index.shtml |
| MACS v1.4 | Zhang et al., 2008 | http://liulab.dfci.harvard.edu/MACS/ |
| Samtools | http://samtools.sourceforge.net | |
| DeepTools | Ramírez et al., 2016 | https://deeptools.readthedocs.io/en/develop/ |
| KentUtils | Kent et al., 2010 | https://github.com/ENCODE-DCC/kentUtils |
| BEDtools | Quinlan and Hall, 2010 | https://bedtools.readthedocs.io/en/latest/content/bedtools-suite.html |
| GenomicRanges | Lawrence et al., 2013 | https://bioconductor.org/packages/release/bioc/html/GenomicRanges.html |
| HOMER (v4.10) | Heinz et al., 2010 | http://homer.ucsd.edu/homer/motif/ |
| MEME Suit | Bailey et al., 2009 | http://homer.ucsd.edu/homer/motif/ |
| ggplot2 | Wickham, 2009 | https://ggplot2.tidyverse.org |
| ComplexHeatmap | Gu et al., 2016 | https://bioconductor.org/packages/release/bioc/html/ComplexHeatmap.html |
Highlights.
KDM1A forms a complex with GR to regulate glucocorticoid hormone signaling
Removal of H3K4me2 by KDM1A is crucial for stable GR genomic localization
The GR-KDM1A complex preferentially demethylates H3K4me2 but not H3K4me1
H3K4me2 demethylation by KDM1A at GR-bound enhancers is key for gene activation
ACKNOWLEDGMENTS
We thank Anders Näär for purified RAR and RXR protein. This work was partly supported by NIH grants CA194302 and GM112062 to Y.G.S. and the Fund to Sustain Research Excellence to R.F. The contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. Y.G.S. is a Pew Scholar.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.05.062.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, and Noble WS (2009). MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37, W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biddie SC, John S, Sabo PJ, Thurman RE, Johnson TA, Schiltz RL, Miranda TB, Sung M-H, Trump S, Lightman SL, et al. (2011). Transcription factor AP1 potentiates chromatin accessibility and glucocorticoid receptor binding. Mol. Cell 43, 145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai C, He HH, Gao S, Chen S, Yu Z, Gao Y, Chen S, Chen MW, Zhang J, Ahmed M, et al. (2014). Lysine-specific demethylase 1 has dual functions as a major regulator of androgen receptor transcriptional activity. Cell Rep 9, 1618–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calo E, and Wysocka J (2013). Modification of enhancer chromatin: what, how, and why? Mol. Cell 49, 825–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll JS, Liu XS, Brodsky AS, Li W, Meyer CA, Szary AJ, Eeckhoute J, Shao W, Hestermann EV, Geistlinger TR, et al. (2005). Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell 122, 33–43. [DOI] [PubMed] [Google Scholar]
- Chen Y, Jie W, Yan W, Zhou K, and Xiao Y (2012). Lysine-specific histone demethylase 1 (LSD1): A potential molecular target for tumor therapy. Crit. Rev. Eukaryot. Gene Expr 22, 53–59. [DOI] [PubMed] [Google Scholar]
- Clarke CL, and Graham JD (2012). Non-overlapping progesterone receptor cistromes contribute to cell-specific transcriptional outcomes. PLoS One 7, e35859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar SS, Zhao D, Lin T, Gu B, Pal K, Wu SJ, Alam H, Lv J, Yun K, Gopalakrishnan V, et al. (2018). MLL4 Is Required to Maintain Broad H3K4me3 Peaks and Super-Enhancers at Tumor Suppressor Genes. Mol. Cell 70, 825–841.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egelhofer TA, Minoda A, Klugman S, Lee K, Kolasinska-Zwierz P, Alek-seyenko AA, Cheung M-S, Day DS, Gadel S, Gorchakov AA, et al. (2011). An assessment of histone-modification antibody quality. Nat. Struct. Mol. Biol 18, 91–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang R, Barbera AJ, Xu Y, Rutenberg M, Leonor T, Bi Q, Lan F, Mei P, Yuan G-C, Lian C, et al. (2010). Human LSD2/KDM1b/AOF1 regulates gene transcription by modulating intragenic H3K4me2 methylation. Mol. Cell 39, 222–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groeneweg FL, van Royen ME, Fenz S, Keizer VIP, Geverts B, Prins J, de Kloet ER, Houtsmuller AB, Schmidt TS, and Schaaf MJM (2014). Quantitation of glucocorticoid receptor DNA-binding dynamics by single-molecule microscopy and FRAP. PLoS One 9, e90532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Eils R, and Schlesner M (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. [DOI] [PubMed] [Google Scholar]
- Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, et al. (2009). Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459, 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang Y, Broun A, Wang C, Park Y-K, Zhuang L, Lee J-E, Froimchuk E, Liu C, and Ge K (2019). H3.3K4M destabilizes enhancer H3K4 methyltransferases MLL3/MLL4 and impairs adipose tissue development. Nucleic Acids Res 47, 607–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John S, Sabo PJ, Thurman RE, Sung M-H, Biddie SC, Johnson TA, Hager GL, and Stamatoyannopoulos JA (2011). Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat. Genet 43, 264–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadmiel M, and Cidlowski JA (2013). Glucocorticoid receptor signaling in health and disease. Trends Pharmacol. Sci 34, 518–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato S, Yokoyama A, and Fujiki R (2011). Nuclear receptor coregulators merge transcriptional coregulation with epigenetic regulation. Trends Bio-chem. Sci 36, 272–281. [DOI] [PubMed] [Google Scholar]
- Kearns NA, Pham H, Tabak B, Genga RM, Silverstein NJ, Garber M, and Maehr R (2015). Functional annotation of native enhancers with a Cas9-histone demethylase fusion. Nat. Methods 12, 401–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Zweig AS, Barber G, Hinrichs AS, and Karolchik D (2010). BigWig and BigBed: enabling browsing of large distributed datasets. Bioinformatics 26, 2204–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Huber W, Pagès H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, and Carey VJ (2013). Software for computing and annotating genomic ranges. PLoS Comput. Biol 9, e1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupien M, Eeckhoute J, Meyer CA, Wang Q, Zhang Y, Li W, Carroll JS, Liu XS, and Brown M (2008). FoxA1 translates epigenetic signatures into enhancer-driven lineage-specific transcription. Cell 132, 958–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara KM, Kannai A, and Sasano H (2018). Possible roles for glucocorticoid signalling in breast cancer. Mol. Cell. Endocrinol 466, 38–50. [DOI] [PubMed] [Google Scholar]
- Metzger E, Wissmann M, Yin N, Müller JM, Schneider R, Peters AHFM, Günther T, Buettner R, and Schüle R (2005). LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 437, 436–439. [DOI] [PubMed] [Google Scholar]
- Nair SS, Nair BC, Cortez V, Chakravarty D, Metzger E, Schüle R, Brann DW, Tekmal RR, and Vadlamudi RK (2010). PELP1 is a reader of histone H3 methylation that facilitates oestrogen receptor-alpha target gene activation by regulating lysine demethylase 1 specificity. EMBO Rep 11, 438–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, and Kingsford C (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekowska A, Benoukraf T, Ferrier P, and Spicuglia S (2010). A unique H3K4me2 profile marks tissue-specific gene regulation. Genome Res 20, 1493–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perillo B, Ombra MN, Bertoni A, Cuozzo C, Sacchetti S, Sasso A, Chiariotti L, Malorni A, Abbondanza C, and Avvedimento EV (2008). DNA oxidation as triggered by H3K9me2 demethylation drives estrogen-induced gene expression. Science 319, 202–206. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahnamoun H, Hong J, Sun Z, Lee J, Lu H, and Lauberth SM (2018). Mutant p53 regulates enhancer-associated H3K4 monomethylation through interactions with the methyltransferase MLL4. J. Biol. Chem 293, 13234–13246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44 (W1), W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy TE, Pauli F, Sprouse RO, Neff NF, Newberry KM, Garabedian MJ, and Myers RM (2009). Genomic determination of the glucocorticoid response reveals unexpected mechanisms of gene regulation. Genome Res 19, 2163–2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusconi F, Paganini L, Braida D, Ponzoni L, Toffolo E, Maroli A, Landsberger N, Bedogni F, Turco E, Pattini L, et al. (2015). LSD1 Neurospecific Alternative Splicing Controls Neuronal Excitability in Mouse Models of Epilepsy. Cereb. Cortex 25, 2729–2740. [DOI] [PubMed] [Google Scholar]
- Sakabe NJ, and Nobrega MA (2013). Beyond the ENCODE project: using genomics and epigenomics strategies to study enhancer evolution. Philos. Trans. R. Soc. Lond. B Biol. Sci 368, 20130022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlossmacher G, Stevens A, and White A (2011). Glucocorticoid receptor-mediated apoptosis: mechanisms of resistance in cancer cells. J. Endocrinol 211, 17–25. [DOI] [PubMed] [Google Scholar]
- Shen H, Xu W, Guo R, Rong B, Gu L, Wang Z, He C, Zheng L, Hu X, Hu Z, et al. (2016). Suppression of Enhancer Overactivation by a RACK7-Histone Demethylase Complex. Cell 165, 331–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Sawada J, Sui G, Affar B, Whetstine JR, Lan F, Ogawa H, Luke MP-S, Nakatani Y, and Shi Y (2003). Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature 422, 735–738. [DOI] [PubMed] [Google Scholar]
- Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, and Shi Y (2004). Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119, 941–953. [DOI] [PubMed] [Google Scholar]
- Shi Y-J, Matson C, Lan F, Iwase S, Baba T, and Shi Y (2005). Regulation of LSD1 histone demethylase activity by its associated factors. Mol. Cell 19, 857–864. [DOI] [PubMed] [Google Scholar]
- Silverman MN, and Sternberg EM (2012). Glucocorticoid regulation of inflammation and its functional correlates: from HPA axis to glucocorticoid receptor dysfunction. Ann. N Y Acad. Sci 1261, 55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- So AY-L, Chaivorapol C, Bolton EC, Li H, and Yamamoto KR (2007). Determinants of cell- and gene-specific transcriptional regulation by the glucocorticoid receptor. PLoS Genet 3, e94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens M (2017). False discovery rates: a new deal. Biostatistics 18, 275–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillo D, Kaplan N, Moore IK, Fondufe-Mittendorf Y, Gossett AJ, Field Y, Lieb JD, Widom J, Segal E, and Hughes TR (2010). High nucleosome occupancy is encoded at human regulatory sequences. PLoS One 5, e9129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vockley CM, D’Ippolito AM, McDowell IC, Majoros WH, Safi A, Song L, Crawford GE, and Reddy TE (2016). Direct GR Binding Sites Potentiate Clusters of TF Binding across the Human Genome. Cell 166, 1269–1281.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss TC, Schiltz RL, Sung M-H, Yen PM, Stamatoyannopoulos JA, Biddie SC, Johnson TA, Miranda TB, John S, and Hager GL (2011). Dynamic exchange at regulatory elements during chromatin remodeling underlies assisted loading mechanism. Cell 146, 544–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Scully K, Zhu X, Cai L, Zhang J, Prefontaine GG, Krones A, Ohgi KA, Zhu P, Garcia-Bassets I, et al. (2007). Opposing LSD1 complexes function in developmental gene activation and repression programmes. Nature 446, 882–887. [DOI] [PubMed] [Google Scholar]
- Wang Y, Li X, and Hu H (2014). H3K4me2 reliably defines transcription factor binding regions in different cells. Genomics 103, 222–228. [DOI] [PubMed] [Google Scholar]
- Whyte WA, Bilodeau S, Orlando DA, Hoke HA, Frampton GM, Foster CT, Cowley SM, and Young RA (2012). Enhancer decommissioning by LSD1 during embryonic stem cell differentiation. Nature 482, 221–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H (2009). ggplot2 (Springer Science+Business Media; ). [Google Scholar]
- Wissmann M, Yin N, Müller JM, Greschik H, Fodor BD, Jenuwein T, Vogler C, Schneider R, Günther T, Buettner R, et al. (2007). Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nat. Cell Biol 9, 347–353. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw fastq files of RNA-seq and ChIP-seq, processed ChIP-seq bigwig and bedgraph files, and bed files containing GR peak regions or control regions are available at GEO (https://www.ncbi.nlm.nih.gov/geo/), accession number GEO: GSE108588.