

Abstract
Objective:
Myotonia Congenita (MC) is a hereditary neuromuscular disorder caused by a mutation in chloride voltage-gated channel 1 (CLCN1) gene. The incidence of MC is estimated as 1 in 100.000. The absence of left main coronary artery (LMCA) is a rare coronary anomaly. Here we present a family with four members who have MC variation carrier and cardiovascular risk.
Method:
The demographic features, laboratory findings, anthropometric measurements and cardiological examination of the cases were recorded. In addition, CLCN1 gene was sequenced by NGS (Next Generation Sequencing Method) and possible causes of inherited thrombophilia risk including MTHFR (A1298C), Factor V Leiden (G1691A), Factor II (G20210A), MTHFR (C677T), Factor V Cambridge (G1091C), plasminogen activator inhibitor 1 (PAI-1) 4G/5G, APOE, APOB, ITGB, ACE (ins/del), FVHR2 and FGB gene alterations were evaluated.
Results:
Case 1 had homozygous c.1886T>C (p.Leu629Pro) alteration in CLCN1 gene and also coronary artery disease, myocardial infarction (MI) history, hyperlipidemia, primary hypertension, vertigo, lomber disc herniation and hearing loss. LMCA was not detected in coronary angiography in Case 1. Cases 2, 3 and 4 had heterozygous c.1886T>C (p.Leu629Pro) alteration with normal electrocardiographic and echocardiographic findings. Additionally, all of family members had genetic risk factors for the related gene, which lead to an increased risk of cardiovascular disease.
Conclusion:
Since alteration of chloride channels in cardiomyocytes leads to variable myocardial involvement, cases with MC should be regularly followed for cardiovascular risk. Moreover, the cases with MC and with genetic profile associated with high cardiovascular risk should also be regularly followed up by cardiologists.
Keywords: Myotonia congenita, absence of LMCA, myocardial infarction, genetic risk factors for cardiovascular disease, CLCN1 gene
ÖZ
Amaç:
Myotoni Konjenita (MK), klorür voltaj kapılı kanal 1 (CLCN1) genindeki mutasyonun neden olduğu kalıtsal bir klorür kanalı nöromüsküler bozukluğudur. MK insidansının 100.000’de 1 olduğu tahmin edilmektedir. Sol ana koroner arter yokluğu (LMCA) anomalisi nadir bir koroner anomalidir. MK varyasyonu ve kardiyovasküler risk taşıyan dört üyeli bir aileyi sunuyoruz.
Yöntem:
Olguların demografik özellikleri, laboratuvar bulguları, antropometrik ölçümleri ve kardiyolojik incelemeleri yapıldı. Ayrıca, CLCN1 geni NGS ile dizilendi ve MTHFR (A1298C), Faktör V Leiden (G1691A), Faktör II (G20210A), MTHFR (C677T), Faktör V Cambridge (G1091C), plazminojen aktivatör inhibitör 1 (PAI-1) 4G/5G APOE, APOB, ITGB, ACE (ins / del), FVHR2 ve FGB genlerindeki değişiklikler olası trombofili riski açısından değerlendirildi.
Bulgular:
Olgu 1’de CLCN1 geninde homozigot c.1886T> C (p.Leu629Pro) değişikliği ve ayrıca koroner arter hastalığı, miyokard infarktüsü öyküsü, hiperlipidemi, primer hipertansiyon, vertigo, Lomber disk herniasyonu ve işitme kaybı vardı. Olgu 1’de koroner anjiyografide LMCA saptanmadı. Diğer olgular (2,3 ve 4) heterozigot c.1886T> C (p.Leu629Pro) değişimine sahipti ancak elektrokardiyografi ve transtorasik ekokardiyografileri normaldi. Ek olarak, aile üyelerinin tümü, kardiyovasküler hastalığa yol açan ilgili genler açısından artmış risk faktörlerine sahipti.
Sonuç:
Kardiyomiyositlerdeki klorür kanallarındaki değişikliklerin miyokard tutulumuna yol açabilmesi nedeniyle, MK’li olguların kardiyovasküler risk açısından düzenli olarak incelenmesi gerektiği söylenebilir. Ayrıca, MK’li ve kardiyovasküler hastalık için yüksek genetik risk faktörlerine sahip hastalar düzenli olarak takip edilmelidir.
Keywords: Miyotoni konjenita, LMCA yokluğu, miyokard infarktüsü, kardiyovasküler hastalık için genetik risk faktörleri, CLCN1 geni
Introduction
Myotonia Congenita (MC) is a hereditary neuromuscular disorder related with mutation in chloride voltage-gated channel 1 (CLCN1) gene that encodes a chloride channel of the CLC channel/transporter family CLC-1 and classified as either autosomal dominant Thomsen disease or recessive Becker type1,2. Signs and symptoms of MC include, impaired muscle relaxation, muscular hypertrophy, transient or permanent muscle weakness, muscle pain and sensitivity to cold with a little variance3. The MC is estimated to affect 1 in 100.000 population around the world4.
The left main coronary artery (LMCA) and the right coronary artery (RCA) supply oxygenated blood to the myocardium. While the RCA originates from the right sinus of Valsalva (SV), the LMCA from the left SV. The LMCA has two branches; left anterior descending (LAD) artery and the left circumflex artery (LCX). Also the intermediate artery (IMA) may root from the bifurcation of the LMCA, forming a trifurcation5. The rate of vascular anomaly in coronary arteries ranges between 0.6% and 1.3% in routine angiographic series. The LMCA anomaly has an incidence of 0.02%-0.07%. The left main coronary artery is absent in 0.41% of patients with LMCA anomalies. In that condition, the LAD and LCX arteries bifurcate separately6.
In the current study, we report a family with four members who have MC and coronary artery anomalies. To the best of our knowledge, there is no study about MC cases with absence of LMCA in the literature.
Material and Methods
Four cases of suspected MC syndrome including a case with coronary artery anomalies were enrolled to the current study after informed consent of the parents were obtained. The study protocol was approved by local Ethics Committee. Demographic features of the participants, laboratory findings (hemogram, fasting blood glucose, Na, K, Cl, Ca, Mg, creatinine, ALT, AST, TSH, LDL, HDL, total cholesterol, triglyceride) were recorded (Table 1). Anthropometric measurements and findings in physical examination were evaluated. Additionally, cardiological examination of the cases were carried out, carefully. Twelve-lead electrocardiograms (ECG) (NIHON KOHDEN CARDIOFAX ECG 1250K MODEL) were obtained for each case. Routine measurements were taken into consideration. Also, all of the patients in the study were evaluated using a transthoracic echocardiograph (Siemens Acuson SC 2000). Transthoracic two dimensional (2D) guided, (M. mode), color Doppler echocardiogram, and continuous wave Doppler CWs were obtained using suitable probes. Cardiac anatomy, ventricular function and valvular competence were assessed using standardized projections, and measurements were done according to the recommendations of the American Society of Echocardiography7.
Table 1.
Demographical, laboratory and clinical features of cases.
| Case 1 | Case 2 | Case 3 | Case 4 | |
|---|---|---|---|---|
| Age (years) | 50 | 33 | 30 | 27 |
| Sex | M | M | F | F |
| Weight in Diagnosis (kg) | 80 | 70 | 71 | 75 |
| Length in Diagnosis (cm) | 175 | 180 | 170 | 178 |
| VKI in Diagnosis (kg/m2) | 26.1 | 21.6 | 23.7 | 23.6 |
| DM | - | - | - | - |
| HT | + | - | - | - |
| Cigarette using | + | - | - | - |
| Other diseases and operation | Coronary heart disease and myocardial infarction history, vertigo, Lomber disc herniation, hearing loss, myringoplasty operation, | Lomber disc herniation | Nasal septum deviation | |
| Fasting Blood Glucose in Diagnosis (mg/dl) | 92 | 79.1 | 84 | 88 |
| WBC (mm3) | 10000 | 5600 | 6400 | 7200 |
| Hemoglobine (g/dL) | 10 | 15 | 13 | 12 |
| Platelets | 339X103 | 263X103 | 245X103 | 324X103 |
| Creatinin (mg/dL) | 1 | 0.9 | 0.8 | 0.61 |
| Na (mmol/L) | 137.8 | 140.2 | 139 | 136 |
| K (mmol/L) | 4.97 | 4.63 | 4.4 | 4.23 |
| Cl (mmol/L) | 103 | 99 | 101 | 99.3 |
| Ca (mg/dL) | 10.2 | 9.5 | 9.8 | 9.98 |
| Mg (mg/dL) | 2 | 2.1 | 1.9 | 2 |
| ALT (IU/L) | 29.3 | 22.4 | 32 | 12.4 |
| AST (IU/L) | 24.9 | 22 | 31 | 14.2 |
| TSH (mIU / L) | 2.2 | 2.22 | 2.1 | 2 |
| LDL (mg/dL) | 95 | 147 | 102 | 106 |
| HDL (mg/dL) | 50 | 57 | 55 | 48 |
| Total Cholesterol (mg/dL) | 170 | 253 | 180 | 178 |
| Triglyceride (mg/dL) | 121 | 243 | 112 | 120 |
HDL: High-density lipoprotein, LDL: Low-density lipoprotein, TSH: BMI: Body mass index, Min-Max: Minimum-Maximum, SD: Standard deviation, FBG: Fasting Blood Glucose
Total DNA was isolated via Magnesia 16 Complete Blood Genomic DNA Isolation Kit-102 (Anatolia Diagnostics and Biotechnology Products Inc.) from peripheral blood samples of the all cases and PCR pools generated prior to the Next Generation Sequence (NGS) reaction were purified using the NucleoFast 96 PCR (MACHEREY-NAGEL GmbH) kit. Subsequently, the quantification of the PCR products was standardized and these samples were prepared for NGS using the Nextera XT sample preparation kit from Illumina and CLCN1 gene was sequenced by NGS (MISEQ-Illumina method). The variants were verified with NGS. Pathogenicity of the variants in CLCN1 gene was detected using bioinformatic tools, which examine functional effects of single nucleotide variants in humans Provean (Protein Variation Effect Analayzer) (http://provean.jcvi.org Mutation Taster (http://www.mutationtaster.org) and SIFT (Sorting Intolerant From Tolerant) (http://sift.jcvi.org/www/SIFT_enst_submit.html). Also the genetic susceptibility to cardiovascular disease in our cases was evaluated via MTHFR (A1298C), Factor V Leiden (G1691A), Factor II (G20210A), MTHFR (C677T), Factor V Cambridge (G1091C), plasminogen activator inhibitor 1 (PAI-1) 4G/5G, APOE, APOB, ITGB, ACE (ins/del), FVHR2 and FGB gene alterations.
Statistical analysis
Statistical analyses were performed using IBM SPSS Statistics for Windows, version 23.0 (IBM Corp., Armonk, NY, USA). The data were expressed as mean±SD, min, max and percentages. Descriptive statistical methods were carried out.
Results
The demographic, laboratory and clinical findings of cases are given in Table 1. Also alteration in CLCN1 gene, inheritance, zygosity and phenotype of cases are expressed in Table 2. Additionally, genetic risk factors for cardiovascular disease of cases are summarized in Table 3.
Table 2.
Alteration in CLCN1 gene, inheritance, zygosity and phenptype of cases.
| Sex | Age | Alteration in CLCN1 gene | Exon | Zygosity | Class | Inheritance | Phenotype | |
|---|---|---|---|---|---|---|---|---|
| Case 1 | M | 50 | c.1886T>C/p.Leu629Pro rs1009716258 | 16 | Homozygous | Class3 | OR/OD | Myotonia congenita |
| Case 2 | M | 33 | c.1886T>C/p.Leu629Pro rs1009716258 | 16 | Heterozygous | Class3 | OR/OD | Myotonia congenita |
| Case 3 | F | 30 | c.1886T>C/p.Leu629Pro rs1009716258 | 16 | Heterozygous | Class3 | OR/OD | Myotonia congenita |
| Case 4 | F | 27 | c.1886T>C/p.Leu629Pro rs1009716258 | 16 | Heterozygous | Class3 | OR/OD | Myotonia congenita |
Table 3.
Genetic risk factors for cardiovascular disease of cases.
| C | MTHFR A1298C | FII G20210A | FVL G1691A | MTHFR C677T | F VC G1091C | PAI | APOE | APOB | ITGB | ACE ins/del | FVHR2 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| C1 | Heterozygous | N | N | N | N | 4G/5G | E2/E3 | N | N | del/del | N |
| C2 | Heterozygous | N | N | N | N | 4G/5G | E2/E3 | N | N | del/del | N |
| C3 | Homozygous | N | N | N | N | 5G/5G | E2/E3 | N | Heterozygous | ins/del | N |
| C4 | Homozygous | N | N | N | N | 5G/5G | E2/E3 | N | N | del/del | N |
N: Normal, FII: Factor II, FVL: Factor V Leiden, FVC: Factor V Cambridge, PAI: Plasminogen activator inhibitör, C: Case
Case 1: A 51-year-old male patient carried c.1886T>C (p.Leu629Pro)/rs1009716258 alteration in exon 16 of CLCN1 gene as homozygous (Figure 1) and also he had coronary artery disease, myocardial infarction history, hyperlipidemia, primary hypertension, vertigo, lumbar disc herniation, hearing loss and myringoplasty operation. He was taking acetylsalicylic acid, beta-blocker, statin and angiotensin receptor blocker. His blood pressure was 140/100 mmHg, and pulse rate 71 bpm. Electrocardiography demonstrated sinus rhythm with pathological q in D3 and AVF derivations. Transthoracic echocardiography revealed inferior wall hypokinesis, septal hypertrophy (1.3 cm) and mild mitral valve regurgitation. Pulmonary artery pressure was normal. Left ventricular ejection fraction was measured as 52% with modified Simpson’s method. Seven years ago, he underwent coronary angiography with the diagnosis of acute inferoposterior myocardial infarction. LMCA was not detected in coronary angiography (Figure 2). LAD and LCX each stemmed directly from the left sinus of Valsalva. There were plaques in proximal and mid regions of LAD and RCA. Also there was a plaque in mid region of LCX and the distal of LCX was fully occluded. Cardiac stent was applied to LCX. The patient is currently being followed medically.
Figure 1.
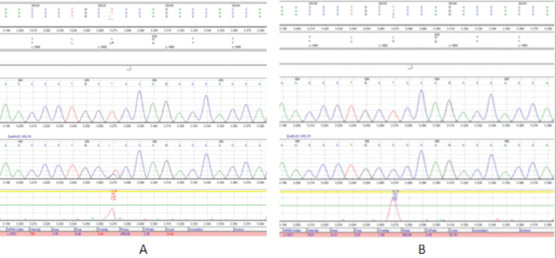
Heterozygous (A) and Homozygous (B) c.1886T>C (p.Leu629Pro)/rs1009716258 alteration in exon 16 of CLCN1 gene.
Figure 2.
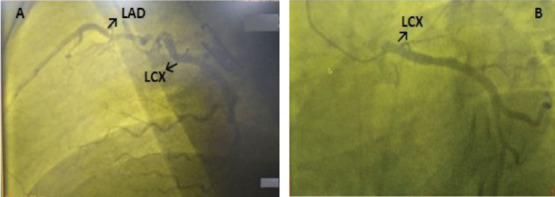
Lateral (A) and spider(B) views of coronary angiography.
Case 2: A 33-year-old male patient carried c.1886T>C (p.Leu629Pro)/rs1009716258 alteration in exon 16 of CLCN1 gene as heterozygous. He had lumbar disc herniation but not any other disease. His blood pressure was 120/80 mmHg, and pulse rate 74 bpm. Electrocardiography demonstrated sinus rhythm without any pathological findings. Transthoracic echocardiography revealed mild tricuspid regurgitation.
Case 3: A 30-year-old female patient carried c.1886T>C (p.Leu629Pro)/rs1009716258 alteration in exon 16 of CLCN1 gene as heterozygous. He had no diseases except nasal septum deviation. His blood pressure was 110/80 mmHg, and pulse rate 80 bpm. Electrocardiography demonstrated sinus rhythm without any pathological findings. Transthoracic echocardiography was reported as normal.
Case 4: A 27-year-old female patient carried 1886T>C (p.Leu629Pro)/rs1009716258 alteration in exon 16 of CLCN1 gene as heterozygous. She had no comorbidities. Her blood pressure was 110/70 mmHg, and pulse rate 76 bpm. Electrocardiography demonstrated sinus rhythm without any pathological findings. Transthoracic echocardiography was reported as normal.
Discussion
Genetic studies have shown that mutations in CLCN-1gene are related with MC (prevalence<1:100 000). According to inheritance pattern of the disease, MC is divided into Thomsen’s autosomal dominant myotonia (DMC) and Becker’s recessive generalized myotonia (RMC). Patients with MC have classic myotonia on examination, “warm-up” phenomenon, and muscular hypertrophy8. Miryounesi M et al.9 described a family with a novel missense mutation in CLCN1 gene (c.1886T>C, p.Leu629Pro). This mutation will impair the function of protein according to in silico analyses with multiple software as well as segregation analysis. The case and his affected children had transient generalized myotonia, which started in early childhood, and amelioration of stiffness after repeated activity but without any muscle weakness. In our cases, we detected also missense mutation in CLCN1 gene (c.1886T>C, p.Leu629Pro). Thus, this is the second report in the literature about the related mutation in CLCN1 gene.
While some researchers reported that cardiac abnormalities such as accompanying arrhythmia, pre-excitation syndrome, or left ventricular enlargement were observed in cases with MC10 but others reported that there was no cardiac problem in cases with MC11,12. According to inheritance pattern of the disease, family members of our case also had c.1886T>C, (p.Leu629Pro) alteration in CLCN1 gene with autosomal recessive congenital myotonia, too. Our proband (Case 1) had amelioration of stiffness after repeated activity but there was no muscle weakness. Also our proband had coronary heart disease and myocardial infarction history, vertigo, lumbar disc herniation and hearing loss. Additionally our proband had absence of LMCA that is very rare in the general population. To the best of our knowledge, this is the first patient reported so far with absence of LMCA, MI story and MC in combination.
Also the genetic risk factors for cardiovascular disease of cases including MTHFR (A1298C), Factor V Leiden (G1691A), Factor II (G20210A), MTHFR (C677T), Factor V Cambridge (G1091C), Plasminogen activator inhibitor 1 (PAI-1) 4G/5G, APOE, APOB, ITGB, ACE (ins/del), FVHR2 and FGB genes were more marked in our cases (Table 3). To our knowledge, this is also the first report about the genetic risk factors for cardiovascular disease in MC cases, too.
Conclusion
Because chloride channels in cardiomyocytes may contribute to the variable myocardial involvement, it may be said that cases with MC should be examined for cardiovascular risk. When we take frequent hospitalization of our proband into consideration, it can be said that cases MC together with absence of LMCA and higher genetic risk factors for coronary artery disease have increased risk for cardiovascular disease. So these cases should be regularly followed up for cardiovascular disease. To obtain most accurate knowledge about the current topic, additional studies including large series should be carried out.
Current study is the first report not only about the absence of LMCA together with MC but also about the genetic risk factors for cardiovascular disease in MC patients. So we thought that our manuscript make important contributions to the literature concerning this current topic.
References
- 1.Matthews E, Fialho D, Tan SV et al. The non-dystrophic myotonias: molecular pathogenesis, diagnosis and treatment. Brain. 2010;133:9–22. doi: 10.1093/brain/awp294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lehmann-Horn F, Ruedel R, Jurkat-Rott K. Myology. 3rd ed. McGraw-Hill: 2004. Nondystrophic myotonias and periodic paralysis; pp. 1257–1300. Engel AG, Franzini-Armstrong C (Eds.) [Google Scholar]
- 3.Jentsch TJ, Stein V, Weinreich F, Zdebik AA. Molecular structure and physiological function of chloride channels. Physiol Rev. 2002;82:503–68. doi: 10.1152/physrev.00029.2001. [DOI] [PubMed] [Google Scholar]
- 4. https://ghr.nlm.nih.gov/condition/myotonia-congenita#statistics.
- 5.Altin C, Kanyilmaz S, Koc S et al. Coronary anatomy, anatomic variations and anomalies: a retrospective coronary angiography study. Singapore Med J. 2015;56:339–45. doi: 10.11622/smedj.2014193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yilmaz-Cankaya B, Kantarci M, Yalcin A, Durur-Karakaya A, Yuce I. Absence of the Left Main Coronary Artery: MDCT Coronary Angiographic Imaging. Eurasian J Med. 2009;41:56–8. [PMC free article] [PubMed] [Google Scholar]
- 7.Lang RM, Badano LP, Mor-Avi V et al. Recommendations for cardiac chamber quantification by echocardiography in adults: an update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. Eur Heart J Cardiovasc Imaging. 2015;16:233–70. doi: 10.1093/ehjci/jev014. [DOI] [PubMed] [Google Scholar]
- 8.Cardani R, Giagnacovo M, Botta A et al. Co-segregation of DM2 with a recessive CLCN1 mutation in juvenile onset of myotonic dystrophy type 2. J Neurol. 2012;259:2090–9. doi: 10.1007/s00415-012-6462-1. [DOI] [PubMed] [Google Scholar]
- 9.Miryounesi M, Ghafouri-Fard S, Fardaei M. A Novel Missense Mutation in CLCN1 Gene in a Family with Autosomal Recessive Congenital Myotonia. Iran J Med Sci. 2016;41:456–8. [PMC free article] [PubMed] [Google Scholar]
- 10.Meng YX, Zhao Z, Shen HR, Bing Q, Hu J. Identification of novel mutations of the CLCN1 gene for myotonia congenital in China. Neurol Res. 2016;38(1):40–4. doi: 10.1080/01616412.2015.1114741. [DOI] [PubMed] [Google Scholar]
- 11.Morales F, Cuenca P, del Valle G et al. Clinical and molecular diagnosis of a Costa Rican family with autosomal recessive myotonia congenita (Becker disease) carrying a new mutation in the CLCN1 gene. Rev Biol Trop. 2008;56:1–11. doi: 10.15517/rbt.v56i1.5505. [DOI] [PubMed] [Google Scholar]
- 12.Ferradini V, Cassone M, Nuovo S et al. Targeted Next Generation Sequencing in patients with Myotonia Congenita. Clin Chim Acta. 2017;470:1–7. doi: 10.1016/j.cca.2017.04.012. [DOI] [PubMed] [Google Scholar]