

Abstract
Triple-negative breast cancer (TNBC) is an aggressive subset of breast cancer that is more common in African-American and Hispanic women. Early detection followed by intensive treatment is critical to improving poor survival rates. The current standard to diagnose TNBC from histopathology of biopsy samples is invasive and time-consuming. Imaging methods such as mammography and magnetic resonance (MR) imaging, while covering the entire breast, lack the spatial resolution and specificity to capture the molecular features that identify TNBC. Two nonlinear optical modalities of second harmonic generation (SHG) imaging of collagen, and resonance Raman spectroscopy (RRS) potentially offer novel rapid, label-free detection of molecular and morphological features that characterize cancerous breast tissue at subcellular resolution. In this study, we first applied MR methods to measure the whole-tumor characteristics of metastatic TNBC (4T1) and nonmetastatic estrogen receptor positive breast cancer (67NR) models, including tumor lactate concentration and vascularity. Subsequently, we employed for the first time in vivo SHG imaging of collagen and ex vivo RRS of biomolecules to detect different microenvironmental features of these two tumor models. We achieved high sensitivity and accuracy for discrimination between these two cancer types by quantitative morphometric analysis and nonnegative matrix factorization along with support vector machine. Our study proposes a new method to combine SHG and RRS together as a promising novel photonic and optical method for early detection of TNBC.
Graphical Abstract
1 |. INTRODUCTION
Triple-negative breast cancer (TNBC) is an aggressive subtype of breast cancer, characterized by a lack of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor type 2 (HER2) expression [1]. It accounts for 12% of breast cancer cases and for ~22% of cases in African-American women [1, 2]. TNBC carries a poor prognosis when compared with other subtypes of breast cancer [3]. It has been shown to respond well to neoadjuvant chemotherapy; however, the recurrence and death rates are still higher for TNBC than for non-TNBC cancers, especially in the first 2 years of postcancer diagnosis [4]. Thus, early diagnosis of TNBC is essential to improve survival by starting patients early on intensive treatment before the development of micrometastases.
Breast cancer is a heterogeneous disease and the heterogeneity of the tumor microenvironment is now recognized as a critical factor in tumor progression and therapeutic response [5]. The breast cancer microenvironment is composed of extracellular matrix (ECM), newly developed vascular system and various stromal cell types, such as endothelial and immune cells, fibroblasts, and adipocytes [6]. TNBCs have a significant increase in the number of fibroblasts, which drive ECM remodeling by increasing matrix stiffness [7]. Increases in ECM production and breast tissue density reflect a greater abundance of collagen, the most abundant component of the ECM, and have been correlated with increased risk of breast cancer [8]. In patient-derived xenograft models, TNBC exhibited collagen accumulation [9]. ECM remodeling is crucial for tumor malignancy and metastatic progression, which ultimately cause over 90% of deaths from cancer. Besides ECM, microvascular density, a prognostic factor in invasive breast cancer, in both basal-like breast cancer and TNBC is significantly higher than in nonbasal-like breast cancer and non-TNBC [10]. The metabolic profile of TNBC is abnormal and may reveal potential biomarkers [11]. Thus, developing novel diagnostic imaging method and translating our current understanding of breast tissue microenvironment may provide accurate TNBC detection and prediction of prognosis, and understanding the unique characteristics of TNBC may shed light on the heterogeneity of this tumor and provide valuable clinical data.
Current imaging methods used for TNBC detection include mammography, ultrasound, and magnetic resonance imaging (MRI) [12]. While these techniques have a high sensitivity for TNBC detection, they also tend to result in many false positives [13]. Even when TNBC is detected from one of the above imaging modalities, confirmation ultimately requires a biopsy and the removal of tissue from potentially cancerous lesions, followed by histological and immunohistochemical examination by a trained pathologist. A promising alternative method for TNBC diagnosis is based on nonlinear optics and optical spectroscopy [14–17]. The field of using various optical spectroscopy techniques for biomedical samples has been called “optical biopsy” and is becoming a commonplace method to determine the state of tissue ex vivo and in vivo, label-free and with real-time efficiency [18, 19].
To characterize the specific chemical fingerprints associated with breast cancer subtypes, one focus in optical biopsy is to measure Raman spectra of tissues [20]. Raman spectroscopy is a spectroscopic technique based on light scattering to study molecular vibrational modes [16]. Infrared (IR) and Raman spectroscopy have been used to distinguish breast cancer from normal tissue by providing information on molecular metabolism, which can detect early changes of molecular composition and structure that occur in tumor lesions or cancer cell lines [14–17]. They are becoming more common investigative methods to determine the state of tissue in vivo and ex vivo based on metabolic and molecular differences between tissue types [20–22]. For instance, Bi et al. used IR and Raman spectra to differentiate HER2 expression status in HER2+ and HER2− breast cancer cell lines based on spectral differences in nucleic acid, amide III, phenylalanine, tryptophan/tyrosine, and lipids [23]. However, these vibrational modes are not sufficient to accurately classify breast cancer subtypes. Additional vibrational modes of other biomolecules are needed, which will be presented in this study.
Resonance Raman spectroscopy (RRS) provides a potential solution to detect adequate vibration peaks associated with specific chemical and protein secondary structures to distinguish TNBC from other subtypes. The underlying principle of RRS is that the Raman scattering intensity will greatly increase when the incident laser frequency is close to a molecule’s electronic transition frequency, and the sharp peaks from key molecules can be observed from chemical bond vibrations due to RR scattering [20]. Using a 532-nm wavelength laser, which falls in the tail of the absorption spectrum for flavin adenine dinucleotide, RRS enables the simultaneous detection of multiple molecules including collagen, fatty acid, amide, carotenoids, tryptophan, metalloprotein, mitochondrial electron transport protein [20, 24–26], including those that exist at very low concentrations and are otherwise masked by high abundance molecules. The RR scattering signal is not overwhelmed by autofluorescence that is commonly observed in native tissues. In addition, due to the shorter wavelength used for excitation to more local region, there is less signal mixing from deeper tissue when compared with the more conventional 785-nm wavelength [20, 27, 28].
Second harmonic generation (SHG) has been used to image collagen fibers, which provides the visual information on collagen morphology in the tumor environment and greatly enhances our understanding of the ECM [29–32]. SHG has also been employed with coherent antiStokes Raman scattering imaging for detecting colon cancer tissue ex vivo [30].
MR spectroscopy (MRS) and spectroscopic imaging (MRSI) have been used as two preclinical and clinical tools to assess the abnormal lactate metabolism in cancer [33–37]. Dynamic contrast-enhanced MRI (DCE-MRI) provides critical information regarding perfusion/permeability in tumors and normal tissue by dynamic measurement of signal change after injection of a contrast agent (CA) [38–40]. From the changes in the characteristic signal intensity-time curve, the vascular perfusion and permeability information of a tumor can be obtained from tracer kinetic modeling [41–43]. While MRS, MRSI, and DCE-MRI have the ability to image the whole tumor, they also have a much coarser spatial resolution than optical imaging methods [44]. Thus, these methods average out the cell-level microenvironmental characteristics in tumor tissue.
The objective of this study was to employ both in vivo SHG imaging of collagen using multiphoton microscopy and ex vivo RRS of biomolecules as novel, rapid, and label-free techniques for the distinction of metastatic TNBC and nonmetastatic breast cancer. We hypothesized that in vivo whole-tumor characteristics, such as tumor lactate concentration and vascularity that can be measured by MR methods, may reflect observations from optical imaging on a microscopic level at selected tumor locations. We further hypothesized that differences in the microenvironments in TNBC and nonmetastatic tumor tissues would result in different collagen morphology, as well as cellular and chemical composition, which can be detected by SHG imaging and RRS, respectively.
2 |. RESULTS
2.1 |. Tumor lactate and vascularity by in vivo MR
The whole-tumor lactate concentration was found to be similar between metastatic TNBC (4T1) and non-metastatic (67NR) tumors grown in BALB/c mice (Figure 1A) and was independent of tumor size for the size range (VCal < 250 mm3) evaluated (Figure 1B). Lactate is detectable in viable tissue, but barely to not detectable in mainly necrotic voxels (Figures 2 and S1). It is distributed heterogeneously across the tissue in both tumor types. Of note is that the tumor volume determined by MRI methods was consistently larger than that determined by caliper (Figure S2) and more accurately reflected the actual tumor volume as the tumor grew deeper into the body. Thus, the tumor volume determined by MRI, specifically the volume determined from a single-slice reference MRI covering the lactate slice (VMRSI), was used for the quantification of lactate by substitution method [45, 46].
FIGURE 1.
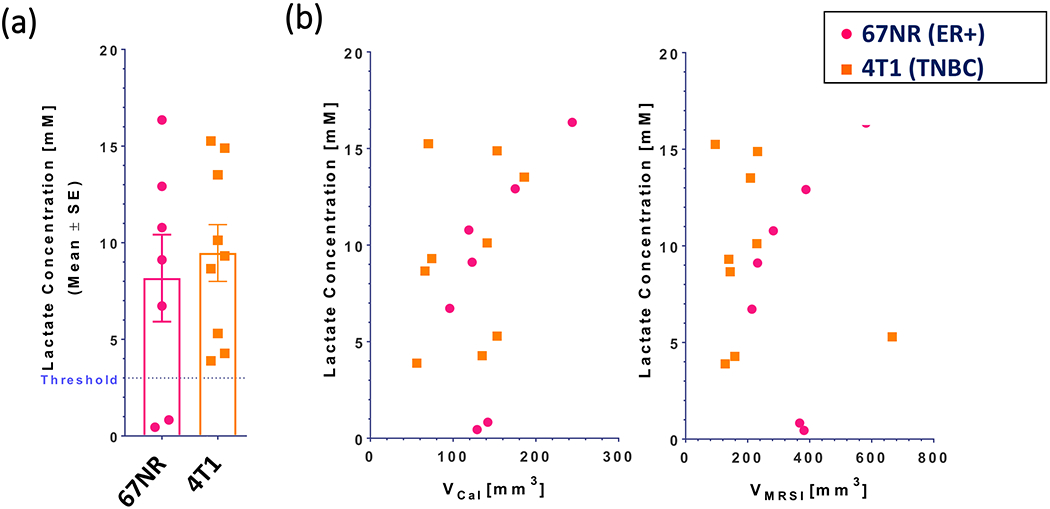
Whole-tumor lactate concentrations for 67NR (n = 7) and 4T1 (n = 9) tumors grown in the mammary fat pad of BALB/c mice. A, Mean lactate concentrations over all tumors. Symbols • and ▪ show the lactate concentrations for single tumors in each group. Threshold depicts the threshold below which the determination of lactate becomes less reliable, due to residual lipid background in the MR spectra. B, Lactate concentration for each tumor as a function of the tumor volume by caliper (VCal, left) and the tumor volume in the lactate slice (VMRSI, right). Of note is, that while 67NR and 4T1 tumors were approximately the same size at time of the MR experiment, the tumor volume determined by MR imaging was consistently larger than the volume by caliper, with the tumors in the two 67NR cohorts (n = 7) were on average slightly larger than the tumors in the two 4T1 cohorts (n = 9) (Figure S2). MR, magnetic resonance; MRSI, MR spectroscopic imaging
FIGURE 2.
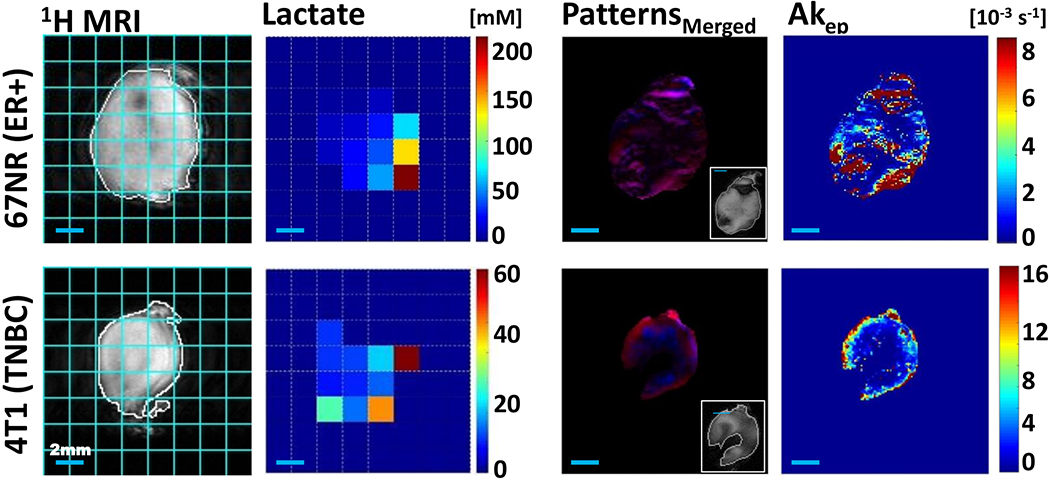
In vivo comparison of the spatial metabolic and vascular structural intra-tumoral heterogeneity of 67NR and 4T1 tumors. Spatially mapped tumor lactate concentrations and tumor vascularity-based characteristics for a representative 67NR (top row) and 4T1 (bottom row) tumor respectively, both grown orthotopically in the mammary fat pad of BALB/c mice (same tumors as shown in Figure 4). 1H MRI: 1H MRI of tumor in the slice used to map lactate for the entire tumor, overlaid with tumor outline (white) and pixel grid, corresponding to the pixels of the lactate map (cyan); Lactate: Lactate concentration map with a 36 and 20 mm3 pixel size for the 67NR and 4T1 tumor respectively (2 mm × 2 mm in-plane spatial resolution; 9- and 5-mm slice thickness for the shown 67NR and 4T1 tumor respectively); Patterns Merged: The spatial distribution of CA uptake patterns in the outermost 1-mm tumor slice of the DCE-MRI acquisition (most closely aligned to the optical imaging slice)—red, well perfused (fast CA washin and washout), blue: necrosis (slow CA washin, with no major washout) with a pixel size of 0.0137 mm3 for the 67NR and 0.0156 mm3 for the 4T1 tumor; the inset shows the corresponding anatomical MRI of the 1-mm tumor slice; Akep: To the pattern map corresponding Akep map. The approximate measure of tumor blood flow and permeability, Akep, consists of the amplitude A, depicting relative MR signal enhancement, and the exchange rate kep, reflecting MR signal increase velocity [40]. CA, contrast agent; DCE-MRI, dynamic contrast-enhanced MRI; MRI, magnetic resonance imaging.
We evaluated tumor vascularity and the associated tumor microenvironment by DCE-MRI as before [47–49]. For the 67NR tumors, well-perfused (P), hypoxic (H) and necrotic (N) tumor areas and areas that contained two or more of these environmental characteristics were present in all tumors (Figure S3A). In contrast, among eight 4T1 tumors, only one displayed all three environmental characteristics, while three did not have detectable necrotic areas by DCE-MRI, one was entirely hypoxic, and three displayed no discernible hypoxic tumor areas (Figure S3A). The respective volume fractions, %VH + N and %VP + H + N, differed significantly (P < .05, unpaired, two-tailed t test) between 67NR and 4T1 tumors, demonstrating the variations of tumor vascular heterogeneity between the two tumor models (Figure S3A). Overall the vascular heterogeneity, as depicted by the different Akep values (a measure of vascular blood flow and permeability) for well-perfused, hypoxic, and necrotic tumor fractions, is lost when assessing the median Akep for the whole-tumor Akep (Akep[whT]) (Figure S3B).
As the optical imaging was performed on the tumor tissue right below the skin, we also analyzed the outermost 1 mm slice of the DCE-MRI data separately as the imaging plane most closely aligned to the optical imaging plane in Figure 2. The in-plane vasculature is spatially less heterogeneous for the 4T1 tumor than for the 67NR tumors, with most of the well-perfused tumor area (high Akep values) located at the rim of the 4T1 tumor. As for the whole tumor (Figure S3), 4T1 tumors have generally fewer voxels that are represented by more than 1 microenvironment than 67NR tumors in the outermost DCE-MRI slice (Figure 3A). Also, the outermost slice tends to have fewer CA uptake patterns reflecting the microenvironment than the whole tumor (Figures 3A and S3A). Overall, the volume fractions and Akep values of the vascular-based microenvironmental features for the entire outermost slice were similar between the two tumor types (Figure 3). This potentially masked spatial differences of the in-plane vascular heterogeneity seen typically between the two tumor types (Figure 2).
FIGURE 3.
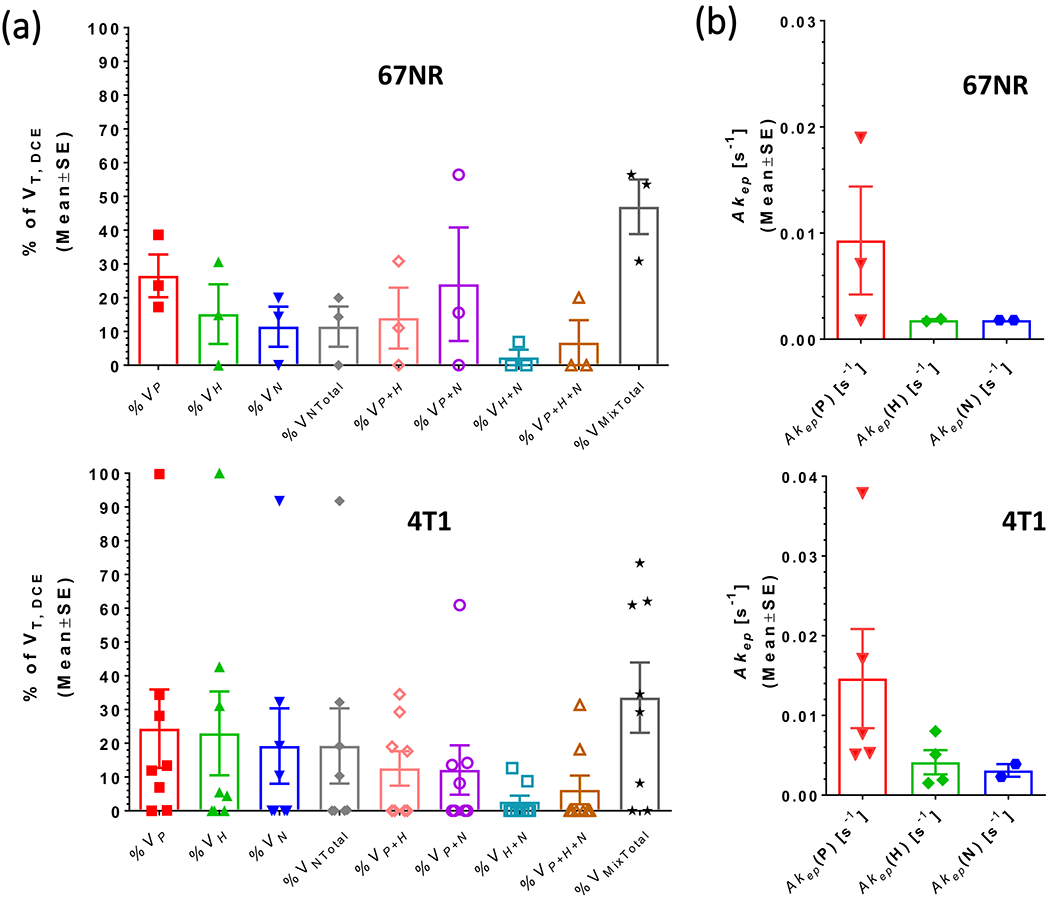
Tumor vascularity in the DCE-MRI slice most closely aligned to the optical imaging slice. A, Percent tumor volumes of well-perfused, hypoxic, and necrotic tumor areas, as well as mixtures of these, for the outermost 1 mm slice of three 67NR and eight 4T1 tumors (abbreviations as in Figure S3A). The outermost slice is more often characterized by fewer vascular patterns than the whole tumor (see Figure S3A) which is reflected by the fewer Akep values displayed in B. Five of eight 4T1 tumors did not have by DCE-MRI detectable necrotic tumor areas in the outermost slice (0% VNTotal) and one of eight 4T1 tumors was entirely well perfused in the outermost slice (100% VP). As in the whole tumor, the fractional tumor area with more than one CA uptake pattern tended to be higher in the 67NR than in the 4T1 tumors; for the latter two of eight tumors were represented by a single signal-time curve pattern (1 with 100% VP and 1 with 100% VH respectively). No significant differences between tumor types could be detected for the outermost slice. CA, contrast agent; DCE-MRI, dynamic contrast-enhanced MRI; MRI, magnetic resonance imaging
2.2 |. In vivo SHG imaging by multiphoton microscopy detects distinct collagen structures in breast cancer subtypes
After the MR measurements, SHG imaging was conducted in vivo on the orthotopically implanted murine 4T1 (TNBC) and 67NR (ER+) lower right mammary fat pad tumors of the mice.
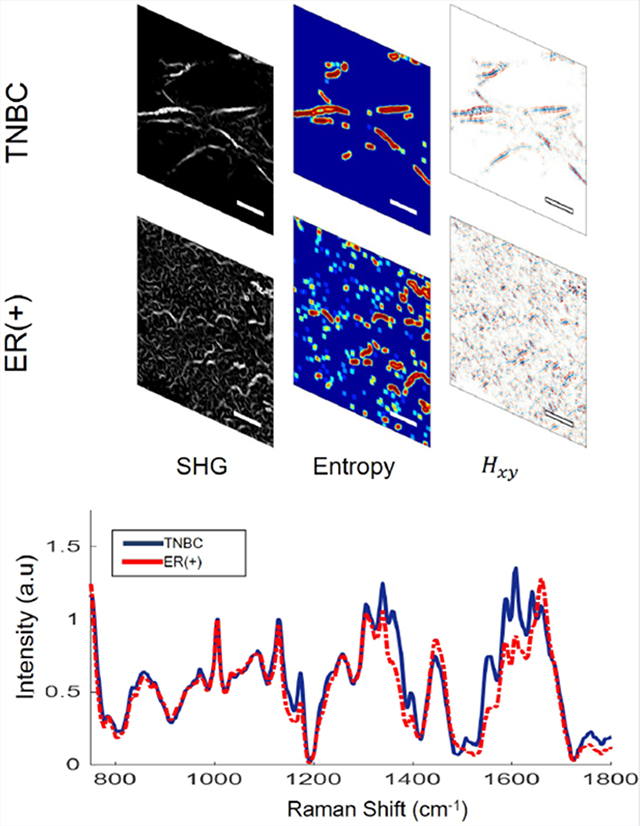
Collagen, which is remodeled during carcinogenesis and tumor progression [40–52], is one of the key molecules of interest in demonstrating the distinct morphological characteristics of tumors. However, current breast cancer classification is based on the histological analysis of ex vivo biopsy samples by a trained pathologist [53]. Classification accuracy is therefore dependent on the experience and skill of the pathologist [54]. In addition, the standard pathology sample thickness of 5 to 10 μm limits the ability to investigate the spatial 3D morphology of ECM components, such as collagen. As demonstrated by hematoxylin and eosin (H&E) staining of 14-μm thick 4T1 and 67NR tumor tissue slices (Figure 4A,E), no obvious difference in collagen morphology could be observed between these tissues. SHG imaging of collagen can provide the submicrometer resolution necessary to study the 3D architecture of collagen in the ECM. Thus, we performed in vivo SHG imaging of collagen on the lower right mammary fat pads of mice and observed that collagen fibers in 67NR tumors have a greater curvature than the long, straight fibers in 4T1 tumors. Furthermore, the fibers in 4T1 tumors are more aligned in groups (bundled) than the more randomly distributed fibers in 67NR tumors (Figure 4B,F).
FIGURE 4.
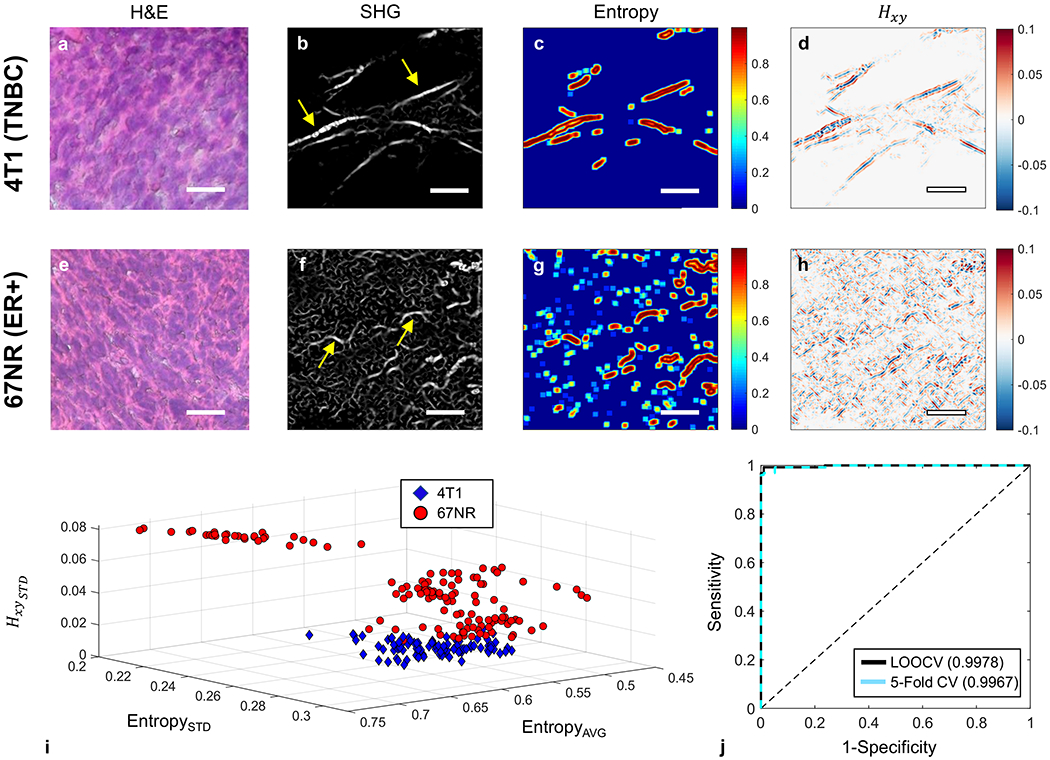
Comparison of the structural, that is, collagen fibers, intra-tumoral heterogeneity of 67NR and 4T1 tumors. A,E, H&E staining of a representative tumor section; B,F, Representative collagen images of the same 67NR and 4T1 tumors as shown in Figure 2, captured by SHG imaging (920 nm excitation, 460 nm emission). The 67NR tumor showed curly, less densely packed fibers than the 4T1 tumor with its long, straight, and bundled fibers. The yellow arrows indicate the collagen fibers; C,G, Local entropy calculated for both images. D,H, The 2D Hessian cross-term (Hxy) result obtained post-convolution with B,F. Scale bars are 20 μm. I, 3D scatter plot of the averaged entropy, SD of entropy, and SD of the 2D Hessian cross-terms for each extracted SHG image, calculated across all nonzero (intensity) spatial locations, for both 4T1 and 67NR. J, AUROC curves and corresponding AUC scores obtained via two separate forms of cross-validated SVM separation using the three features listed above. AUC, area under the curve; AUROC, area under the receiver operating characteristic curve; H&E, hematoxylin and eosin; SHG, second harmonic generation; SVM, support vector machine
To quantify whether 67NR fibrils exhibited this morphological feature at a significantly greater degree, the local entropy of each SHG image was obtained. Entropy is often used as a metric of randomness or information content [55]. The calculated entropy value at any given region of interest is highly dependent on the amount of information contained in it. Redundant values are multiplied by a scaled fraction and are thus weighted less. Given 67NR’s highly unstructured fibril morphology, one would expect a correspondingly large variation in the entropy values across the image plane compared with those obtained from 4T1 (Figure 4C,G).
Furthermore, 2D Hessian matrix operations have provided highly sensitive feature extraction capabilities in numerous applications on note, such as blood vessel segmentation [56], automated detection of vessel tree abnormalities for ophthalmoscopy [57] among others. The second-order information obtained via the Hessian operation and the corresponding eigenvalues provide sensitive and highly interpretable information regarding fibril morphology [58]. These features include orientation, curvature, and edge strength and thus, we employed 2D Hessian to characterize further the structural collagen characteristics of these two tumor types (Figure 4D,H). Further analysis related to the eigenvalue decomposition can be found in Figure S4.
To evaluate how well SHG images can be used to distinguish between 4T1 and 67NR tissues, a support vector machine (SVM) was used along with two entropy-derived values (average and SD) and Hessian cross-term (Figure 4I, each data point corresponding to a value across entire nonzero image plane). By evaluating the SVM’s performance, we were able to identify correctly 219 out of total 224 images of 4T1 and 67NR all together, in which 95 out of 97 of 4T1 were correctly identified (2 incorrect) and 124 out of 127 of 67NR were correctly identified (3 incorrect), respectively.
Our SHG imaging and image analysis confirmed that literalized and stiffened collagen morphology in 4T1 tumor can be used as a marker for metastatic potential [52]. Unlike these more aggressive tumors, nonmetastatic 67NR tumors lack this more organized structure and fibril density.
2.3 |. Ex vivo RRS on breast cancer tissue
After in vivo SHG imaging, we excised the tumors from the mice and performed ex vivo RRS. Using RRS, we obtained biochemical information in a near-realistic environment containing proteins with the full range of amino acids, fatty acids and lipids, DNA and RNA, saccharides, primary metabolites, and other important components in the biological environment, such as carotenoids, ascorbic acid, riboflavin, and glutamine. Figure 5A compares the RR spectra averaged over all 4T1 and 67NR tumors. The most significant differences between the two spectra are in the peaks at 1090, 1339, 1358, 1395, 1444, 1557, 1588, 1608, 140, and 1658 cm−1. Each spectral peak is attributed to the vibrations of a specific chemical bond or functional group and is assigned in Table 1. The main spectral differences are due to lipid modes, including fatty acids, triglycerides, and phospholipids at 1090, 1339, 1444, and 1658 cm−1, proteins at 1358 cm−1, CH rocking at 1395 cm−1, CH2 bending in protein and lipids at 1444 cm−1, amide II at 1557 cm−1, C—C mode of phenylalanine or hydroxyproline at 1588 cm−1, cytosine at 1608 cm−1, amide I at 1640 and 1658 cm−1. Lipids are the primary component of the cell membrane, and thus, altered lipid composition and metabolism of the cell membranes combined with active increased cell proliferation have been observed to be related to aggressive cancers [60–63]. Collagen is the most abundant protein in the body [64] and is a major component of breast tissue ECM, with hydroxyproline being one of the primary amino acids in collagen [64]. The amide I bands peaks are also associated with collagen [65, 66]. Changes in the collagen structure of the tumor environment are associated with increased metastatic potential [67]. The differences between the RR spectra for peaks associated with lipids and collagen indicate different metabolic activities and structures in metastatic 4T1 and nonmetastatic 67NR tumors.
FIGURE 5.
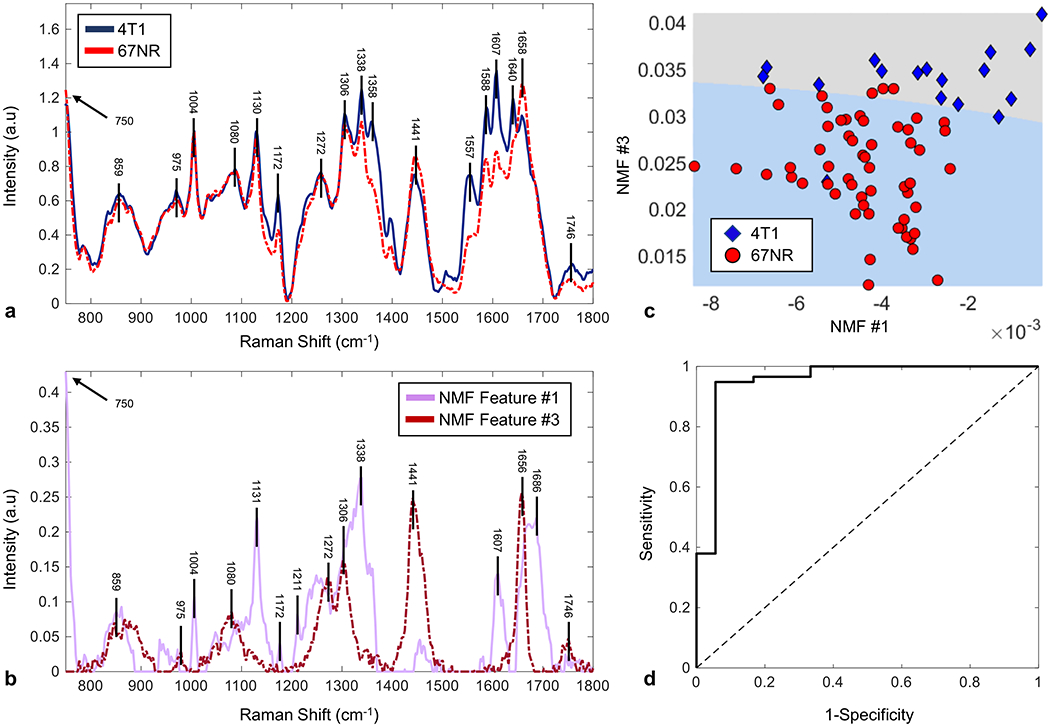
NMF analysis of resonance Raman spectra identifies distinguishing features between 67NR and 4T1 tumors. A, Representative resonance Raman spectra of 4T1 (solid, blue) and 67NR tissues (dotted, red), background subtracted and normalized to the intensity at 1006 cm−1. Prominent peaks in both spectra have wavenumbers indicated above 4T1 spectra. Differences in peak intensity are most obvious for the peaks at 1090, 1339, 1358, 1395, 1446, 1554, 1587, 1610, 1639, and 1658 cm−1. B, NMF-extracted feature spectra showing the two feature spectra used to obtain the highest accuracy and SVM separation. Feature spectrum #1 is the solid, pink line and Feature spectrum #3 is the dotted, red line. C, 2D SVM separation using the feature spectra displayed in B. Weights of the given feature spectra 1 and 3 for 67NR (red circle) and 4T1 (blue diamond) indicate degree to which a given RR spectrum is composed of a given feature spectrum. The shallow slope of the line indicated that feature spectrum #3 is the primary separator between the two tumor types. D, AUROC separation demonstrates a cross-validated characterization accuracy of 92.1% along with the sensitivity of 96.6% and specificity of 77.8% to distinguish 67NR (n = 5) from 4T1 tumors (n = 9). AUROC, area under the receiver operating curve; NMF, nonnegative matrix factorization; RR, resonance Raman; SVM, support vector machine
TABLE 1.
Wavenumber peaks and corresponding biomolecules
| Observed peak | Assignment |
|---|---|
| (1,3) ≈859 cm−1 | Collagen, tyrosine |
| (3) 975 cm−1 | =C—H out-of-plane deformation (collagen) |
| (1) 1004 cm−1 | Symmetric CC aromatic ring breathing (phenylalanine) |
| (3) 1080 cm−1 | Typical phospholipids |
| (1) 1131 cm−1 | Fatty acid, phospholipid structural change, etc. |
| (1) 1172 cm−1 | CH3 rocking |
| (1) 1211 cm−1 | C—C6H5 stretching mode in tyrosine and phenylalanine |
| (3) ≈1272 cm−1 | Amide III, CHα′ rocking |
| (3) 1306 cm−1 | Amide III (N—H), α-helix, C—C stretching, and C—H bending |
| (1) 1338 cm−1 | Tryptophan, CH2/CH3 wagging, twisting, and bending mode of collagens and lipids |
| (3) 1441 cm−1 | CH2 scissoring and CH3 bending (lipids) |
| (1) 1607 cm−1 | CO stretching, C=C bending (tyrosine, phenylalanine ring vibration |
| (3) 1656 cm−1 | Amide I unordered |
| (1) ≈1686 cm−1 | Amide I (disordered structure; nonhydrogen bonded) |
To evaluate the performance of the classification between the spectra of 4T1 and 67NR tissues, we performed nonnegative matrix factorization (NMF) [68, 69] analyses on RRS results. NMF was performed using an extracted wavenumber region (750-2200 cm−1) in a similar fashion to a prior work that utilized it to achieve high discriminative performance between normal and cancerous breast tissue samples [70]. Differences between groups of data can be determined by an algorithm analyzing how the weights of each feature spectrum tend to differ between groups. Total 80 spectra of 4T1 and 67NR were analyzed and 73 were correctly identified. Among these, 57 out of 59 spectra of 4T1 were corrected identified (2 incorrect) and 16 out of 21 spectra 67NR were correctly identified (5 incorrect), respectively. The significant peaks of the feature spectra are correlated with tyrosine/collagen at 859 cm−1, C—H out-of-plane deformation at 975 cm−1, lipids at 1338 cm−1, CH2 in lipids at 1441 cm−1, tyrosine/phenylalanine at 1607 cm−1, amide I/proteins at 1656 and 1658 cm−1. Figure 5B displays the NMF extracted feature spectra. The 2D scatter plot in Figure 5C shows the weights corresponding to both NMF extracted components and the corresponding SVM separation line. The area under the receiver operating characteristic curve (AUROC) in Figure 5D measures the prediction power of the characterization method (see Section 4) [20, 70]. Most of the modes found to be significantly different between the cancer subtypes were consistent with previous breast cancer studies [20, 70].
The NMF extracted spectra (Figure 5C) possess much higher sparsity than the processed input spectra (Figure 5A), with a few distinct wavenumber peaks. The relatively shallow slope of the separation line in Figure 5C suggests that NMF feature 3 contributes most strongly to the differentiation between 4T1 and 67NR RR spectra. The modes that contribute to differences between 4T1 and 67NR spectra suggest that most of the differences in signal are due to collagen and lipids (Table 1). These results are consistent with the analysis on SHG images, which confirms the distinct collagen signatures for the two breast cancer models (Figures 4 and S4). The classification accuracy obtained through the chosen workflow indicates that RR spectra are potential new method to differentiate TNBC from non-TNBC cancers.
3 |. DISCUSSION
The microenvironment of tumors significantly influences their malignant behavior and growth, as well as participation in reprogramming of stromal cells, which might otherwise counteract the progression of cancer cells [71–74]. Thus, understanding the characterization of the tumor microenvironment may provide accurate prediction of breast cancer prognosis, and studying the unique characteristics of TNBC may shed light on the heterogeneity of this tumor type, treatment options, and prognosis.
We used a 532-nm excitation RRS for TNBC and non-TNBC cancer tissues and found different vibrational modes than previously observed [19]. Previous studies [20, 75] have compared the vibrational modes by IR Raman spectroscopy and RRS. Liu et al. [20] stated that the methyl group peaks in the spectra collected from breast cancer tissue were identified with enhanced intensity at 1378 cm−1, and another methyl group on an aromatic ring with one peak near 2935 cm−1 was due to the in-phase symmetric methyl (—CH3) stretching vibration and a vibration in Fermi resonance with the —CH3 1460 cm−1 deformation peak overtone—a weaker vibration in the spectra collected from breast cancer tissues. Surmacki’s work [75] showed that peaks for carotenoids were significantly enhanced with 532 nm excitation, while its peaks were hardly seen with 785 nm excitation. Peaks at 745, 877, and 958 cm−1 became significantly detectable with 532 nm excitation. Surmacki’s study also repeated Liu’s results that 1304, 1378, 1440, 1460, 1520 cm−1 peaks were in noise level with 785 nm excitation but were significantly enhanced with 532 nm excitation. RRS process for flavins in tissue arises from the in band transitions (in the wings of absorption) and out band transitions (arising from emission) resonance [76]. The Raman resonance effect for nicotinamide adenine dinucleotide + hydrogen might be due to the laser wavelength falling in the tail of its emission spectrum. Besides the resonance effect, another enhancement in the RRS signal compared to near infrared radiation (NIR) nonresonant Raman is due to the use of a shorter excitation wavelength which also increases the Raman scattering cross section. The enhancement in tryptophan is unclear. One hypothesized mechanism was two-photon absorption in ref. [76]. Since we used 532 nm, a shorter wavelength compared to 785 nm NIR wavelength, all Raman signals could be enhanced due to the increase in the Raman scattering cross section, which could be part of or the main reason for the observed enhancement in collagen and tryptophan peaks.
Early detection of TNBC relies on the ability to discern the microenvironmental molecular, morphological, and metabolic features that distinguish this subtype from other types of breast cancer. Previous studies have shown that the lactate concentration in a central slice of 67NR and 4T1 tumors grown in athymic nude mice differed significantly and were dependent on tumor size [37]. Here, no such difference could be detected for the whole tumor. The main differences between the two studies were (a) the host, (b) interrogating slice versus whole tumor, and (c) the specific implementation of the lactate substitution method for quantitation, most notably the use of an updated transverse and longitudinal magnetic resonance relaxation times correction factor [45, 46]. The previous study [37] used tumors implanted in mice with a compromised immune system, while this study used a syngeneic model. This might impact metabolic cooperation with stromal cells [44, 77, 78] as well as vasculature, which in turn would impact the washout of lactate. Specifically, even at small tumor sizes, 4T1 tumors develop a central surface necrotic patch when grown in BALB/c mice with regrowth at larger tumor sizes [79], while they develop a necrotic core at tumor sizes above 150 mm3 when grown in athymic nude mice [37]. Secondly, in this study we used the tumor volume determined by MR instead of the caliper volume to quantify lactate and acquired data for the entire tumor, instead of a 5-mm central slice which would cover the whole tumor for small tumors (<150 mm3 by caliper), but not for medium (150-400 mm3) or large (>400 mm3) tumors. This resulted in a more accurate determination of tumor volume and quantification of whole-tumor lactate, in contrast to the previous study. These differences, together with an updated relaxation time correction factor [45, 46], may account for the different results when compared to the previous study by Serganova et al. [37] In BALB/c mice, both tumor types displayed heterogeneous vascular CA uptake. In the imaging plane similar to the optical imaging plane, however, 67NR tumors appeared to be better perfused across the imaging plane, while 4T1 tumors tended to have higher rim perfusion. This was associated with distinct collagen signatures for these two tumor types.
Beyond in vivo MR, which has the advantages of being non-invasive, imaging the entire tumor, and easy clinical translation, our approach novelly uses SHG imaging and RRS, two optical methods that characterize biological samples, to distinguish between metastatic TNBC and nonmetastatic breast cancer. SHG and RRS are particularly well suited to the task of more accurate diagnosis; they are fast and label-free, and generate subcellular-resolution images. RRS provides an advantage over IR spectroscopy, another vibrational spectroscopy method, in that it requires no sample preparation and, though not done in this study, can be performed in vivo. Our study shows that in vivo SHG imaging and ex vivo RRS are able to differentiate the collagen, molecular and metabolic features between the metastatic 4T1 tumors and the nonmetastatic 67NR tumors, which can be analyzed both quantitatively and qualitatively to determine differences between the tumor subtypes with high sensitivity and accuracy.
Analyses using various methods, such as those well-suited for structural feature extraction and spectral decomposition, allowed for effective characterization of a given tumor subtype. This is important for early detection of this aggressive, metastatic form of cancer to improve patient outcomes. SVM analysis of morphological features obtained from SHG images generates a very high capability (95 out of 97 and 124 out of 127 images were correctly identified for 4T1 and 67NR, respectively) to distinguish 4T1 and 67NR tumors. Similarly, RRS characterization based on NMF displayed low probabilities of TNBC misdiagnosis, but relatively higher probabilities of incorrectly diagnosing a 67NR tumor as a 4T1 tumor. This may be due to fewer spectra obtained for 67NR than for 4T1 (21 and 59 spectra, respectively). It is likely that the misdiagnosis of 67NR as 4T1 would decrease with higher 67NR spectrum count coupled with a more sophisticated supervised classification approach. The use of both SHG and RRS in conjunction would be beneficial to ensure that samples are not misclassified.
In conclusion, our study proposes SHG imaging of collagen and RRS as two rapid and label-free optical techniques for successful detection of TNBC, by differentiating the morphological and molecular features between TNBC and nonmetastatic breast cancer tissues at subcellular resolution. The use of SHG imaging and RRS in cancer for optical biopsy offers exciting new opportunities for accurate diagnostic and therapeutic approaches to detect TNBC. Further feature analysis of the spectral components and research into the characteristics of TNBC that result in its collagen phenotype will be conducted. In addition, careful measurements of SHG images and Raman spectra from TNBC, non-TNBC, and healthy breast tissues may lead to a better understanding of the molecular and morphological differences between TNBC and non-TNBC breast cancer subtypes.
4 |. MATERIALS AND METHODS
All experiments on mice were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee (IACUC) of Memorial Sloan Kettering Cancer Center and the IACUC of the City College of New York.
4.1 |. Cell lines and culture
The 67NR (ER+, PR−, HER2−, displays mixed luminal/basal phenotype) and 4T1 breast cancer cells (both cell lines kindly provided by Fred Miller, Karmanos Cancer Institute, Detroit, MI) were derived from a spontaneous breast tumor in a BALB/c mouse [80] and cultured as described by Simoes et al. [81]. Briefly, cells were cultured in Dulbecco’s modified Eagle’s medium containing 25 mM glucose and 6 mM glutamine, and supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin. Cells were grown at 37°C, 5% CO2 in a humidified atmosphere and subcultured every 2 to 3 days. For tumor implantation, cells were harvested by trypsinization, counted by Trypan Blue exclusion assay, and suspended at a concentration of 2 × 106 67NR and 1 × 106 4T1 cells per 100 μL culture medium.
4.2 |. Tumor model and growth
The 67NR and 4T1 cells were injected orthotopically into the lower right mammary fat pad of 6 to 8 weeks old mice (BALB/cAnNCrl, Charles River Laboratory, Wilmington, MA). Twenty mice were purchased in 2 cohorts with 10 mice each, and half of each cohort (5 mice) were selected for injection with either 67NR or 4T1 cells. Mouse age at tumor cell injection was 6 weeks (67NR, cohort 1), 6.3 weeks (67NR, cohort 2), 7.1 weeks (4T1, cohort 1), and 6.9 weeks (4T1, cohort 2), respectively. Tumor size was calculated using the ellipsoid formula (l × w × d × π/6) with the three orthogonal tumor diameters l, w, d determined by sliding jaw caliper [82]. Tumors were used for experiments when reaching between 50 and 250 mm3 caliper volume (VCal). Based on available literature, tumors were projected to reach 150 ± 50 mm in 14 to 21 days (67NR) and 9 to 22 days (4T1) [83], that is, 67NR and 4T1 tumors reaching imaging tumor size at approximately the same time post tumor implantation by injection of double the number of 67NR than 4T1 cells. Tumors were imaged by MR post tumor cell implantation at days 10 to 13 for 67NR and at days 11 to 14 for 4T1 tumors, respectively. Mice with tumor growing outside the mammary fat pad or being moribund and requiring sacrifice were excluded from the imaging studies.
4.3 |. Animal preparation for imaging studies
For in vivo MR experiments, the mouse tail vein was catheterized to facilitate the administration of the CA (gadopentetate dimeglumine [Gd-DTPA], Magnevist®, Bayer Health Care, Leverkusen, Germany) via a home-built catheter line assembly. During the MR experiment, mice were anesthetized with <2% isoflurane in 100% oxygen and the breathing rate was kept at 50 to 90 per minute by adjusting the isoflurane percentage. The rodent core temperature was maintained at 31.5°C to 36° C (Figure S5), using a home-built, water-circulating heating pad. A suture was placed through the skin, without touching the tumor, to ensure steady tumor positioning in the 1H MRI coil during the MR experiment.
For in vivo SHG imaging, five 67NR mice and nine 4–T1 mice were used. Some mice were excluded from optical imaging due to timing constraints on the number of mice being able to be imaged each day. Prior to optical imaging, the mice were anesthetized using isoflurane in 100% oxygen, transported to the imaging stage, and fitted with a nose-cone to supply isoflurane in 100% oxygen throughout imaging. Reflexes were tested periodically to ensure deep sedation. For each tumor, the skin was incised and removed from the tumor to expose the tumor from surrounding tissue, while not severing any blood vessels leading to the tumor. Exposed tumors were secured to the mounting stage to reduce the effect of breathing motion on imaging. For the water-immersion objective lens, a polymer coat was applied around the perimeter of the tumor to hold the water.
4.4 |. In vivo MR experiments
We assessed the tumor volume, lactate concentration, as well as vascular perfusion (blood flow and vascular permeability) in vivo using MR for seven 67NR and nine 4T1 tumors. The MR experiments were performed on a horizontal-bore Bruker 7T MR scanner using a home-built, solenoid 1HMR coil. After positioning of the setup in the scanner, the coil was tuned and matched to 300 MHz; the line width of the tumor water signal was minimized by field-map-based shimming.
For each tumor, we assessed tumor anatomy and volume by multislice 1HMR imaging (MRI). The tumor volume, obtained by outlining tumor tissue on each image and adding the tumor areas for all image slices, was compared to the tumor volume obtained from the single-slice reference image corresponding to the lactate slice and the volume of tumor tissue assessed for tumor vascularity (Figure S2).
We used the SelMQC sequence [33, 37] to measure whole-tumor lactate by single-slice MRS, and mapped in selected tumors with easily detectable lactate (three each for 67NR and 4T1, respectively) the spatial lactate distribution by MRSI, as described previously [45, 46]. The slice thickness was adjusted to cover the entire tumor. Lactate spectra were processed and the lactate signal fitted using XsOsNMR (kindly provided by Dr Dikoma Shungu and Ms. Xiangling Mao). Absolute quantification of lactate was performed by phantom replacement method with correction factors as described previously [37, 45, 46]. Following the lactate spectra acquisition, we assessed tumor vascular perfusion (blood flow and permeability) by DCE-MRI, as described previously [48]. Only three of seven 67NR tumors had successful DCE-MRI scans, due to experimental issues or catheter failure, while DCE-MRI was successfully acquired for eight out of nine 4T1 tumors. Briefly, DCE-MRI data were acquired using FLASH with 15 mm × 15 mm or 16 mm × 16 mm field of view, 128 × 128 matrix, four to seven slices (# of slices depending on tumor size) of 1 mm each. DCE-MRI data were analyzed using principal component analysis followed by constrained NMF, thus, estimating the spatial distributions of well-perfused (fast CA washin/washout, well oxygenated) tumor areas, and tumor areas characterized by delayed CA washin/washout (typically associated with hypoxic tissue) and by slow or no accumulation of CA (no functional vasculature, typically associated with necrosis) [47–49].
4.5 |. In vivo SHG imaging
The multiphoton imaging system consisted of a mode-locked femtosecond Titanium: sapphire (Ti:S) laser (Coherent Chameleon Ultra II, Coherent Inc., Santa Clara, CA) with a pulse width of 140 ± 20 ficoseconds, repetition rate of 80 MHz, energy of pulses are in nj range and tunable from 680 to 1080 nm. The objective lens collected SHG signals, which were split into two photomultiplier tube channels, corresponding to red and green wavelengths respectively. As all emissions of interest fell below the cutoff wavelength for channel 1, only signals from channel 2 were considered in this study (green wavelengths). The wavelength for SHG of collagen was 920 nm. Band-pass filters around 460 nm were used to filter the multiphoton signals. The pulse energies density is below J/cm [2] to reduce molecular changes from damage. Compared to conventional optical imaging techniques, multiphoton imaging has been known to have reduced photodamage and photobleaching due to the use of highly localized excitation energy.
Images were acquired by PrairieView (Prairie Technologies). The in-plane resolution for all images was 512 pixels × 512 pixels, with a pixel area of 0.47 μm × 0.47 μm, and a z-axis step size of 3 μm. For each sample, six to eight regions on the surface of the tumors were imaged. Following selection of a region of interest, the highest and lowest imaging depths were defined in PrairieView and imaging was performed at a given location by vertical sectioning, creating a “z-stack.” Laser intensity and/or PMT sensitivity was occasionally increased or decreased as required to obtain a clear signal. Mice were sacrificed immediately following imaging and tumors were excised for ex vivo Raman spectroscopy. The SHG imaging data were analyzed using entropy and Hessian operation (Section 4.7).
4.6 |. Ex vivo RRS
Immediately after SHG imaging, each tumor was excised and ex vivo resonant Raman spectroscopy was performed using Ocean Optics ID Raman micro Raman spectrometer (Model# IDR-MICRO532). Prior to measurement of the tumor, the spectrometer was calibrated to the 992 cm−1 benzene signal. For RR, an excitation wavelength of 532 nm was used—a wavelength that is near the absorption wavelength of many biological tissue components, and thus, provides an enhanced signal. The laser beam was focused to a spot of ~2 μm with a power of 3.5 mW using a ×40 objective. The spectra were acquired using 1-second acquisition time, and 30 spectra averages. We did not fix the tissue with formaldehyde for RR measurements because fixation may cause significant Raman spectral changes in the amino acid fingerprint region of proteins, as formaldehyde promotes the cross-linkage of amino groups in collagen [84, 85].
For each sample, a small section of the surface of the tumor, estimated 1 to 2 mm thick, was sliced off to avoid contaminating signal from the polymer gel used during SHG imaging. Between four and six regions on the exposed surface of the tumor were analyzed, with two to four acquisitions performed in each region to qualitatively assess reproducibility. Each spectrum was normalized to its own phenylalanine peak at 1006 cm−1. After discarding noisy and anomalous data, analysis of RRS data was performed using 21 spectra from 5 67NR tumors and 59 spectra from 9 4T1 tumors. The RRS data were analyzed by NMF (Section 4.8) as described in the following sections to recover unsupervised, spectral characteristics separating metastatic TNBC from non-metastatic, ER+ breast cancer.
4.7 |. SHG image processing
In this study, it was apparent that the collagen fibril structure varied between 4T1 and 67NR tumors. The obtained SHG data (22 and 10 volumetric images for 4T1 and 67NR respectively) each possessed between 20 and 100 image planes along the z-axis. Due to breathing-related motion artifacts, volumetric processing was infeasible. A median filter of size 3 × 3, followed by a background threshold of 300 photon count was used to eliminate background noise. Many z-stacks contained either (a) much fibril information on a small fraction of the total image plane or (b) nothing of interest and it would be nonsensical to treat every 2D image plane as viable for analyses. Thus, the data were further segmented into fourths (corners) and candidate images were obtained by extracting the two images from each stack that possessed the highest summed intensity.
After the preprocessing step, both entropy and second-order properties were obtained for discriminative quantification. Application of the 2D Hessian for the localized second-order features and eigenvalues at every location on the image plane was performed using MATLAB (Mathworks, Natick, MA) [86]. Entropy was calculated using a 7 × 7 kernel and MATLAB’s image processing toolbox.
4.8 |. Nonnegative matrix factorization
NMF [68, 87] is an alternative matrix decomposition method which allows one to choose an internal dimensionality (r value) that is problem specific [68, 69]. The details of this method, as applied to optical imaging, are introduced by Wu et al. [70] Briefly, the problem that NMF attempts to solve is a multiplicative reconstruction of a given input matrix by minimizing the Frobenius norm, reaching a convergence in the process. Once the Frobenius norm reached convergence, a modified version of the input matrix, containing the normalized spectral data without background subtraction, was used to obtain a matrix of size r (characteristic spectra) × n (spectra) that related directly to the presence of the extracted characteristic spectra. In this study we used r = 10, so the spectral feature count was reduced from 1450 (the range of wavenumbers per spectrum) to 10 values. NMF is appropriate for analysis of Raman spectra because their data are strictly nonnegative and the resulting feature spectra contain only nonnegative values and are easily interpretable.
4.9 |. Performance validation (SVM, leave-one-out cross-validation, AUROC)
In this study, SVM determined the line (2D) or plane (3D) which resulted in the highest separation accuracy given an input of features. Leave-one-out cross-validation (LOOCV) was used to validate the robustness of the separation between a seemingly high-performance feature combination. LOOCV is an iterative method which begins by leaving out the first feature combination, fitting an SVM to the remaining data, then determines and stores whether the not included data point would be classified correctly using the newly defined SVM separation. This process is performed on every data point, resulting in a much higher performance for feature combinations which have a high degree of separability, but will result in much lower performance in datasets which possess many points which lie on the boundary of the separation. AUROC is a commonly used graphical representation of classification performance in which an area under the resulting curve calculated to be 1.0 represents perfect separation and 0.5 represents random separation.
4.10 |. H&E staining
Tissue samples were collected from freshly excised specimens, frozen, and embedded in Optimal cutting temperature compound. The bulk tissue was cryosectioned at ~25°C with a Cryostat (CM1850, Leica Biosystems, Buffalo Grove, IL) into 15-μm thick sections for histopathological examination by H&E staining.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge support by the NIH grants U54 CA 132378 and U54 CA137788 (L. S., E. A., J. A. K.), L. S. acknowledges the support of start-up funds from UCSD, and NIH U54 pilot 2U54CA132378-11A1 (CM00004266-00), P30 CA008748 (J. A. K.), R01 CA172846, R01 CA163980 (J. A. K.), and the Breast and Molecular Imaging Fund at the MSKCC Evelyn H. Lauder Breast Center. We thank Professor Karen Hubbard for discussions and support of this line of research. We thank Dr Dikoma Shungu and Ms Xiangling Mao for the software package XsOsNMR as well as Mr Dov P. Winkleman for his technical support.
Funding information
MSKCC Evelyn H. Lauder Breast Center; Breast and Molecular Imaging Fund; UCSD, Grant/Award Number: UCSD Start-up funding, and NIH U54 pilot 2U54CA132378-11A1 (CM00004266-00); NIH, Grant/Award Number: 2U54CA 132378-11A1, U54 CA132378 and U54 CA 137788, NIH R01 CA163980, R01 CA172846, P30 CA008748
Footnotes
CONFLICT OF INTEREST
The authors declare no potential conflict of interest.
DATA AVAILABILITY STATEMENT
The data sets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of this article.
REFERENCES
- [1].Foulkes WD, Smith IE, Reis-Filho JS, New England Journal of Medicine 2010, 363, 1938 10.1056/NEJMra1001389. [DOI] [PubMed] [Google Scholar]
- [2].DeSantis CE, Fedewa SA, Sauer AG, Kramer JL, Smith RA, Jemal A, CA Cancer J. Clin 2016, 66, 31 10.3322/caac.21320. [DOI] [PubMed] [Google Scholar]
- [3].Boyle P, Ann. Oncol 2012, 23(Suppl 6), vi7 10.1093/annonc/mds187. [DOI] [PubMed] [Google Scholar]
- [4].Lin NU et al. , Cancer 2012, 118, 5463 10.1002/cncr.27581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Duechler M, Peczek L, Zuk K, Zelesna I, Jaziorski A, Czyz M, Immunobiology 2014, 219, 158 10.1016/j.imbio.2013.09.003. [DOI] [PubMed] [Google Scholar]
- [6].Place AE, Jin Huh S, Polyak K, Breast Cancer Res. 2011, 13, 227 10.1186/bcr2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kaushik S, Pickup MW, Weaver VM, Cancer Metastasis Rev. 2016, 35, 655 10.1007/s10555-016-9650-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Boyd NF, Martin LJ, Yaffe MJ, Minkin S, Breast Cancer Res. 2011,13, 223 10.1186/bcr2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Takai K, Le A, Weaver VM, Werb Z, Oncotarget 2016, 7, 82889 10.18632/oncotarget.12658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Mohammed RA et al. , Mod. Pathol 2011, 24, 774 10.1038/modpathol.2011.4. [DOI] [PubMed] [Google Scholar]
- [11].Kanaan YM et al. , Cancer Genom. Proteom 2014,11, 279. [Google Scholar]
- [12].Dogan BE, Turnbull LW, Ann. Oncol 2012, 23(6), vi23–vi29. 10.1093/annonc/mds191. [DOI] [PubMed] [Google Scholar]
- [13].Schmadeka R, Harmon BE, Singh M, Am. J. Clin. Pathol 2014, 141, 462. [DOI] [PubMed] [Google Scholar]
- [14].Alfano R et al. , IEEE J. Quantum Electron 1984, 20, 1507 10.1109/JQE.1984.1072322. [DOI] [Google Scholar]
- [15].Alfano R, Das B, Cleary J, Prudente R, Celmer E, Bull. NY. Acad. Med 1991, 67, 143. [PMC free article] [PubMed] [Google Scholar]
- [16].Liu CH et al. , J. Photochem. Photobiol. B Biol 1992,16, 187. [DOI] [PubMed] [Google Scholar]
- [17].Zhou Y, Liu CH, Sun Y, Pu Y, Boydston-White S, Liu Y, Alfano RR, J. Biomed. Opt 2012, 17, 116021 10.1117/1.JBO.17.11.116021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Croce AC, Ferrigno A, Bottiroli G, Vairetti M, Liver Int. 2018, 38, 1160 10.1111/liv.13753. [DOI] [PubMed] [Google Scholar]
- [19].Kennedy GT et al. , Ann. Surg 2015, 262, 602 10.1097/SLA.0000000000001452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Liu C-H, Zhou Y, Sun Y, Li JY, Zhou LX, Boydston-White S, Masilamani V, Zhu K, Pu Y, Alfano RR, Technol. Cancer Res. Treat 2013,12, 371. [DOI] [PubMed] [Google Scholar]
- [21].Brozek-Pluska B, Musial J, Kordek R, Bailo E, Dieing T, Abramczyk H, Analyst 2012, 137, 3773 10.1039/c2an16179f. [DOI] [PubMed] [Google Scholar]
- [22].Gao P, Han B, du Y, Zhao G, Yu Z, Xu W, Zheng C, Fan Z, J. Spectrosc 2017, 2017, 1 10.1155/2017/5383948. [DOI] [Google Scholar]
- [23].Bi X, Rexer B, Arteaga CL, Guo M, Mahadevan-Jansen A, J. Biomed. Opt 2014, 19, 025001 10.1117/1.JBO.19.2.025001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Liu CH, Boydston-White S, Weisberg A, Wang W, Sordillo LA, Perotte A, Tomaselli VP, Sordillo PP, Pei Z, Shi L, Alfano RR, J. Biomed. Opt 2016, 21, 127006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Liu C.-h. et al. , J. Cancer Metastasis Treat 2019, 5, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zhou Y, Liu CH, Wu B, Zhang C, Yu X, Cheng G, Chen H, Li S, Liang Q, Zhang M, Zhu K, Shi L, Alfano RR, APL Photonics 2018, 3, 120802. [Google Scholar]
- [27].Alimova A, Chakraverty R, Muthukattil R, Elder S, Katz A, Sriramoju V, Lipper S, Alfano RR, J. Photochem. Photobiol. B Biol 2009, 96, 178 10.1016/j.jphotobiol.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wang L, Zhang G, Luo JC, Zeng F, Wang QZ, Alfano SA, Katz A, Zevallos M, Alfano RR, Biomed. Microdevices 2005, 7, 111 10.1007/s10544-005-1588-x. [DOI] [PubMed] [Google Scholar]
- [29].Tilbury K, Campagnola PJ, Perspect. Med. Chem 2015, 7, 21 10.4137/PMC.S13214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Campagnola PJ, Dong CY, Laser Photon. Rev 2011, 5(1), 13e26. [Google Scholar]
- [31].Zipfel WR, Williams RM, Christie R, Nikitin AY, Hyman BT, Webb WW, Proc. Natl. Acad. Sci. USA 2003, 100, 7075 10.1073/pnas.0832308100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Williams RM, Zipfel WR, Webb WW, Biophys. J 2005, 88, 1377 10.1529/biophysj.104.047308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].He Q, Shungu DC, van Zijl PC, Bhujwalla ZM, Glickson JD, J. Magn. Reson 1995,106, 203. [DOI] [PubMed] [Google Scholar]
- [34].Yaligar J, Thakur SB, Bokacheva L, Carlin S, Thaler HT, Rizwan A, Lupu ME, Wang Y, Matei CC, Zakian KL, Koutcher JA, NMR Biomed. 2012, 25, 113 10.1002/nbm.1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Thakur SB, Yaligar J, Koutcher JA, Magn. Reson. Med 2009, 62, 591 10.1002/mrm.22065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Rizwan A, Serganova I, Khanin R, Karabeber H, Ni X, Thakur S, Zakian KL, Blasberg R, Koutcher JA, Clin. Cancer Res 2013, 19, 5158 10.1158/1078-0432.CCR-12-3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Serganova I et al. , Clin. Cancer Res 2011,17, 6250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Barrett T, Brechbiel M, Bernardo M, Choyke PL, J. Magn. Reson. Imaging 2007, 26, 235 10.1002/jmri.20991. [DOI] [PubMed] [Google Scholar]
- [39].Egeland TA et al. , Radiat. Res 2008, 169, 689 10.1667/RR1311.1. [DOI] [PubMed] [Google Scholar]
- [40].Cho H et al. , Neoplasia 2009,11, 247-, 242 p following 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Hoffmann U, Brix G, Knopp MV, Hess T, Lorenz WJ, Magn. Reson. Med 1995, 33, 506. [DOI] [PubMed] [Google Scholar]
- [42].Leach MO et al. , Eur. Radiol 2012, 22, 1451 10.1007/s00330-012-2446-x. [DOI] [PubMed] [Google Scholar]
- [43].Ingrisch M, Sourbron S, J. Pharmacokinet. Pharmacodyn 2013, 40, 281 10.1007/s10928-013-939315-3. [DOI] [PubMed] [Google Scholar]
- [44].Ramamonjisoa N, Ellen A, Front. Oncol 2017, 7, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ackerstaff E et al. , 25th Annual Meeting of the International Society for Magnetic Resonance in Medicine, International Society for Magnetic Resonance in Medicine, Honolulu, HI, 2017, p. 307. [Google Scholar]
- [46].Ackerstaff E et al. 24th Scientific Meeting and Exhibition of the International Society for Magnetic Resonance in Medicine #2782 (2016). Singapore: The Proceedings of the International Society of Magnetic Resonance in Medicine. [Google Scholar]
- [47].Chang YC et al. , Sci. Rep 2017, 7, 9746 10.1038/s41598-017-09932-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Han S, Stoyanova R, Lee H, Carlin SD, Koutcher JA, Cho HJ, Ackerstaff E, Magn. Reson. Med 2018, 79, 1736 10.1002/mrm.26822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Stoyanova R, Huang K, Sandler K, Cho HJ, Carlin S, Zanzonico PB, Koutcher JA, Ackerstaff E, Trans Oncol 2012, 5, 437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Jolly LA, Novitskiy S, Owens P, Massoll N, Cheng N, Fang W, Moses HL, Franco AT, Cancer Res. 2016, 76, 1804 10.1158/0008-5472.CAN-15-2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Kirkpatrick ND, Andreou S, Hoying JB, Utzinger U, Am. J. Physiol. Heart Circ. Physiol 2007, 292, H3198 10.1152/ajpheart.01234.2006. [DOI] [PubMed] [Google Scholar]
- [52].Maller O, Hansen KC, Lyons TR, Acerbi I, Weaver VM, Prekeris R, Tan AC, Schedin P, J. Cell Sci 2013, 126, 4108 10.1242/jcs.121590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Bleiweiss I, Pathology of Breast Cancer. UpToDate (Ed: Post TW), Waltham, MA, 2020. [Google Scholar]
- [54].Elmore JG, Longton GM, Carney PA, Geller BM, Onega T, Tosteson ANA, Nelson HD, Pepe MS, Allison KH, Schnitt SJ, O’Malley FP, Weaver DL, JAMA 2015, 313, 1122 10.1001/jama.2015.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Mendonça AM, Cardoso F, Sousa AV, Campilho A, International Conference on Image Analysis and Recognition (ICIAR) (Eds: Campilho A, Kamel M), Springer, Berlin, Heidelberg, 2012. [Google Scholar]
- [56].Kumar RP et al. , 6th European Conference of the International Federation for Medical and Biological Engineering (IFMBE) (Eds: Lackovic I, Vasic D), Springer, Cham, 2014. [Google Scholar]
- [57].Calvo D, Ortega M, Penedo MG, Rouco J, Comput. Methods Programs Biomed 2011,103, 28. [DOI] [PubMed] [Google Scholar]
- [58].Huang A, Nielson GM, Razdan A, Farin GE, Baluch DP, Capco DG, IEEE Trans. Vis. Comput. Graph 2005, 12, 93. [DOI] [PubMed] [Google Scholar]
- [59].Movasaghi Z, Rehman S, Rehman IU, Appl. Spectrosc. Rev 2007, 42, 493. [Google Scholar]
- [60].He J, Zhang F, Rachel Tay LW, Boroda S, Nian W, Levental KR, Levental I, Harris TE, Chang JT, du G, FASEB J. 2017, 31, 2893 10.1096/fj.201601353R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Luo X, Cheng C, Tan Z, Li N, Tang M, Yang L, Cao Y, Mol. Cancer 2017, 16, 76 10.1186/s12943-017-0646-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Beloribi-Djefaflia S, Vasseur S, Guillaumond F, Oncogene 2016, 5, e189 10.1038/oncsis.2015.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Ackerstaff E, Glunde K, Bhujwalla ZM, J. Cell. Biochem 2003, 90, 525 10.1002/jcb.10659. [DOI] [PubMed] [Google Scholar]
- [64].Mecham RP, Curr. Protoc. Cell Biol Chapter 10, Unit 10 2012, 11, 10.1.1–10.1.16. 10.1002/0471143030.cb1001s57. [DOI] [PubMed] [Google Scholar]
- [65].Gasior-Glogowska M, Komorowska M, Hanuza J, Ptak M, Kobielarz M, Acta Bioeng. Biomech 2010, 12, 55. [PubMed] [Google Scholar]
- [66].Nguyen TT, Gobinet C, Feru J, Brassart-Pasco S, Manfait M, Piot O, Spectrosc. Int. J 2012, 27, 421 10.1155/2012/686183. [DOI] [Google Scholar]
- [67].Fang M, Yuan J, Peng C, Li Y, Tumour Biol. 2014, 35, 2871 10.1007/s13277-013-1511-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Li H, Adaln T, Wang W, Emge D, Cichocki A, J. VLSI Signal Process. 2007, 48, 83 10.1007/s11265-006-0039-0. [DOI] [Google Scholar]
- [69].Lee DD, Seung HS, Nature 1999, 401, 788 10.1038/44565. [DOI] [PubMed] [Google Scholar]
- [70].Wu B, Smith J, Zhang L, Gao X, Alfano RR, Proc SPIE. 2018, 10489, 104890X. [Google Scholar]
- [71].Liao D, Luo Y, Markowitz D, Xiang R, Reisfeld RA, PLoS One 2009, 4, e7965 10.1371/journal.pone.0007965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Gangadhara S, Barrett-Lee P, Nicholson RI, Hiscox S, Future Oncol. 2012, 8, 1427 10.2217/fon.12.134. [DOI] [PubMed] [Google Scholar]
- [73].Morandi A, Chiarugi P, J. Mol. Med 2014, 92, 117 10.1007/s00109-014-1124-7. [DOI] [PubMed] [Google Scholar]
- [74].Mao Y, Keller ET, Garfield DH, Shen K, Wang J, Cancer Metastasis Rev. 2013, 32, 303 10.1007/s10555-012-9415-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Surmacki J, Brozek-Pluska B, Kordek R, Abramczyk H, Analyst 2015, 140, 2121. [DOI] [PubMed] [Google Scholar]
- [76].Zhou Y, Liu C-H, Wu B, Yu X, Cheng G, Zhu K, Wang K, Zhang C, Zhao M, Zong R, Zhang L, Shi L, Alfano RR, J. Biomed. Opt 2019, 24, 095001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Rattigan YI, Patel BB, Ackerstaff E, Sukenick G, Koutcher JA, Glod JW, Banerjee D, Exp. Cell Res 2012, 318, 326 10.1016/j.yexcr.2011.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Patel BB, Ackerstaff E, Serganova IS, Kerrigan JE, Blasberg RG, Koutcher JA, Banerjee D, Exp. Cell Res 2017, 352, 20 10.1016/j.yexcr.2017.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Tao K, Fang M, Alroy J, Sahagian GG, BMC Cancer 2008, 8, 228 10.1186/1471-2407-8-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Aslakson CJ, Miller FR, Cancer Res. 1992, 52, 1399 10.1002/0471142735.im2002s39. [DOI] [PubMed] [Google Scholar]
- [81].Simoes RV, Serganova IS, Kruchevsky N, Leftin A, Shestov AA, Thaler HT, Sukenick G, Locasale JW, Blasberg RG, Koutcher JA, Ackerstaff E, Neoplasia 2015, 17, 671 10.1016/j.neo.2015.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Tomayko MM, Reynolds CP, Cancer Chemother. Pharmacol 1989, 24, 148. [DOI] [PubMed] [Google Scholar]
- [83].Lou Y, McDonald PC, Oloumi A, Chia S, Ostlund C, Ahmadi A, Kyle A, auf dem Keller U, Leung S, Huntsman D, Clarke A, Sutherland BW, Waterhouse D, Bally M, Roskelley B, Overall CM, Minchinton A, Pacchiano F, Carta F, Scozzafava A, Touisni N, Winum JY, Supuran CT, Dedhar S, Cancer Res. 2011, 71, 3364 10.1158/0008-5472.CAN-10-4261. [DOI] [PubMed] [Google Scholar]
- [84].Huang Z, McWilliams A, Lam S, English J, McLean D, Lui H, Zeng H, Int. J. Oncol 2003, 23, 649. [PubMed] [Google Scholar]
- [85].Shim MG, Wilson BC, Photochem. Photobiol 1996, 63, 662. [DOI] [PubMed] [Google Scholar]
- [86].Dirk-Jan K Hessian Based Frangi Vesselness Filter (2010). Hessian based Frangi Vesselness filter, MATLAB Central File Exchange https://www.mathworks.com/matlabcentral/fileexchange/24409-hessian-based-frangi-vesselness-filter. [Google Scholar]
- [87].Pu Y, Wang W, Alfano RR, Proc. SPIE 2016, 9703, 97031M. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.