

Abstract
Free fatty acids (FA) are a vital component of cells and are critical to cellular structure and function, so much so that alterations in FA are often associated with cell malfunction and disease. Analysis of FA from biological samples can be achieved by mass spectrometry (MS), but these analyses are often not capable of distinguishing the fine structural alterations within FA isomers and often limited to global profiling of lipids without spatial resolution. Here, we present the use of ultraviolet photodissociation (UVPD) for the characterization of double bond positional isomers of charge inverted dication·FA complexes and the subsequent implementation of this method for online desorption electrospray ionization (DESI) MS imaging of FA isomers from human tissue sections. This method allows relative quantification of FA isomers from heterogeneous biological tissue sections, yielding spatially resolved information about alterations in double bond isomers within these samples. Applying this method to the analysis of the monounsaturated FA 18:1 within breast cancer subtypes uncovered a correlation between double bond positional isomer abundance and the hormone receptor status of the tissue sample, an important factor in the prognosis and treatment of breast cancer patients. This result further validates similar studies that suggest FA synthase activity and FA isomer abundances are significantly altered within breast cancer tissue.
Graphical Abstarct
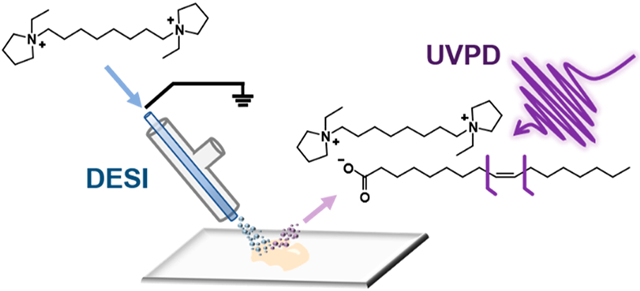
Free fatty acids (FA) have a variety of roles within eukaryotic cells that are vital to cellular structure and functions. Often termed the “building blocks” of lipids, FAs play a crucial role in the formation of cell membranes and often contribute to their biophysical properties. Acyl chain length, degree of unsaturation, and double bond position of the FA chains of lipids dictate membrane fluidity, which can often affect biological processes occurring within or near cells.1 FAs are also a significant source of energy for cells as they can be completely oxidized into CO2 by beta-oxidation, in turn producing energy-rich adenosine triphosphates to power cellular processes.2 Cells can obtain FAs in two ways: scavenging from exogenous sources or by de novo synthesis through FA synthase. Typically, healthy cells rely predominantly on the former to source their required FAs. However, cancerous cells have been shown to produce FAs endogenously in order to supply the molecules required for rapid cell growth.3 Increased levels of FA synthase have been observed in breast,4,5 ovarian,6 and prostate7 cancers, resulting in increased abundance of FAs within the tissues and blood of these patients. Additionally, FA synthase overexpression has been associated with more aggressive forms of cancer. Kuhajda et al., for example, found that overexpression of the functional FA synthase OA-519 was associated with an aggressive form of breast cancer and increased de novo FA synthesis.8 Further, cancer cells are known to display alterations in FA desaturation enzymes, particularly stearoyl-CoA desaturases, suggesting potential differences in the extent and position of desaturations in FA in diseased cells.9,10 Due to these difference between healthy and cancerous cells, there has been a strong interest in understanding the alterations in FA composition caused by cancer as these differences may be exploited for determination of disease biomarkers and possible therapeutic targets.11,12
Mass spectrometry (MS) provides a versatile tool for analysis of FAs from biological samples. Gas chromatography (GC) coupled to electron ionization (EI)-MS has been routinely used as a robust method for evaluation of FA composition in biological extracts, providing both the sum FA composition of the lipids and quantitative analysis.13,14 Yet, the mass spectra obtained with GC-EI-MS often fail to differentiate FAs with structural variations, such as different chain branching moieties and double bond locations.13,14 Liquid chromatography (LC) coupled to electrospray ionization (ESI) and shotgun analysis with ESI are alternative approaches used for soft ionization and sensitive detection of the deprotonated [FA-H]− anions when performed in the negative ion mode.13-15 Although LC-ESI-MS analysis reveals the FA sum composition, tandem mass spectrometry (MS/MS) analyses are needed to elucidate FA structure. Dissociation of FAs in the negative ion mode via traditional low energy collisional activation, however, predominantly produces uninformative neutral losses of water and CO2, precluding the assignment of structural features such as double bond location.13-15 In order to circumvent this limitation, double bond derivatization, charge-switching strategies, and alternative ion activation methods have been implemented to induce structurally informative dissociation. For example, the Paterno-Büchi reaction16-19 or epoxidation with meta-chloroperoxybenzoic acid (m-CPBA)20,21 have been used to derivatize double bonds prior to collisional activation, resulting in cleavage of the derivatized unsaturation to produce diagnostic product ions that reveal double bond positions in FAs. In a different approach, ion activation of cationized FAs produced through charge-fixing modifications of the carboxylate moiety promotes dissociation of the acyl chain and MS/MS spectra that are dependent on double bond position.22-27 Metal adduction,26,27 fixed-charge headgroup derivatization,26,27 and gas-phase ion/ion reactions22,23 are among the charge-switching strategies implemented to localize FA double-bond positions. Moreover, combining charge-switching strategies with activation techniques such as electron induced dissociation (EID)28 and ozone-induced dissociation (OzID)29 has been demonstrated to enhance FA detection and double-bond localization.
While shotgun and chromatographic analyses of biological extracts allow sensitive and in-depth investigation of the metabolic alterations in biological samples, they do not provide insight into the distribution or spatial localization of FAs or lipids in tissue. Human cancer tissues, for example, often present complex histology including viable tumor regions within necrotic tissue, healthy stroma, calcifications, connective tissue, and regions of inflammation. As each of these cell populations undergoes unique metabolic processes, tissue homogenization precludes precise identification of the molecular alterations occurring within tissue regions that are enriched with a specific cell type. Alternatively, MS imaging techniques allow acquisition of spatially accurate molecular information with the spatial resolution needed to correlate molecular and histologic information directly from tissues sections. Desorption electrospray ionization (DESI) MS, for example, has been extensively used to investigate the metabolic alterations occurring in diseased tissues, especially lipids.30,31 DESI-MS imaging has been used to identify alterations in free FA in a variety of human tissues including cancer.31,32 To improve detection of FA directly from tissues, the inclusion of silver ions and dicationic compounds (DCs) in the DESI solvent system or the matrix assisted lased desorption/ionization (MALDI) matrix has been used to enable detection of negatively charged lipids in the positive ion mode.15,33,34 DESI and other MS imaging techniques have been integrated with double bond elucidation methods to investigate lipid isomerism within tissue sections. For example, MALDI has been integrated with gas-phase OzID and Paterno–Büchi derivatization for the imaging of double bond and sn-positional isomers of phosphatidylcholines (PC), glycerophospholipids (GP), and glycosphingolipids in mouse brain tissues.35-37 Tang et al. integrated online Paternò–Büchi derivatization with a liquid microjunction surface sampling probe (LMJ-SSP) to perform in situ isomeric ratio analysis of free FA and PC lipids in healthy and disease mouse tissue sections.16 Kuo et al. utilized offline m-CPBA derivatization coupled to DESI-MS imaging to resolve FA isomers within kidney and metastatic lung tissue sections from murine cancer models.21 Both works by Kuo et al. and Tang et al. showed significant alterations in the double bond position of FA 18:1, either free or conjugated to GP, within the murine cancerous tissue, suggesting dysregulation of FA synthase and desaturase.
Ultraviolet photodissociation (UVPD) is an alternative ion activation method that provides detailed structural characterization of molecules in MS analyses. In particular, 193 nm UVPD has previously been implemented in shotgun and HPLC workflows to resolve and quantify phospho- and sphingo- lipid isomers, including double-bond positional isomers via the production of diagnostic product ions with a distinctive mass difference of 24 Da, resulting from cleavage of carbon–carbon bonds adjacent to double bonds.38-41 266 nm UVPD has also been explored in ESI experiments to characterize fixed-charge derivatized FAs through radical directed dissociation.42 Structural characterization of a series of unsaturated FAs adducted to alkali metal cations has been recently demonstrated using 193 nm UVPD from mixtures of standards and extracts of colorectal cancer cell lines.43 Our group recently integrated 193 nm UVPD with DESI on an Orbitrap platform for MS imaging of GP isomers in biological tissue sections.44 Using this approach, isomeric double bond distribution was observed in subregions of human tissues with different histologic compositions. For example, the 11Δ positional isomer of PC 18:1_18:1 was found at higher relative abundances in normal lymph node tissue when compared to the abundance within an adjacent thyroid metastasis, suggesting differential uptake or synthesis of FAs between these tissue types. In the present study, we used 193 nm UVPD coupled to reactive DESI to evaluate double-bond positions in charge-inverted free FAs directly from biological tissue sections and uncovered differences in free FA isomer compositions between tissue subtypes.
MATERIALS AND METHODS
ESI and DESI UVPD MS Analyses.
Mass spectra were collected on a Thermo Scientific Orbitrap Fusion Lumos mass spectrometer (San Jose, CA) modified to perform UVPD in the dual linear ion traps with a 193 nm Coherent Excistar excimer laser (Santa Cruz, CA) as previously described.45 Dicationic compund (DC)-bound FA complexes were generated via ESI in the positive ion mode by doping 1,8-ethyl DC (5 μM) to the FA methanolic solution. For DESI-MS analysis, a 2D Omni Spay DESI imaging platform (Prosolia Inc. Indianapolis, IN) was coupled to an Orbitrap Fusion Lumos mass spectrometer (Thermo Scientific, San Jose, CA) using an adaptor (Prosolia, Indianapolis, IN) and a lab-built extended capillary, as described previously.44 The maximum injection time was set to 1800 ms to obtain optimal signal-to-noise of fragment peaks. The DESI solvent system utilized was methanol:acetone 80:20 with 15 μM 1,8-ethyl DC at a flow rate of 7 μL/min, yielding a pixel size of the MS image of 200 μm. The FAs and DC·FAs were manually selected and isolated in the quadrupole and subjected to 193 nm UVPD in the high-pressure ion trap with 20 laser pulses at 4 mJ per pulse. Further experimental details, including chemicals used, the synthesis and evaluation of the DCs, and preparation of tissue samples are included in the Supporting Information. DESI-UVPD-MS imaging data used in this manuscript has been uploaded to Dataverse repository and can be accessed at 10.7910/DVN/EDUJFW.
Data Analysis.
Nonparametric t tests of the data were performed by calculating the mean log ratio of the diagnostic ions for each sample, followed by calculation of the mean for the tissue subsets to be analyzed (metastatic vs nonmetastatic, estrogen receptor (ER) positive vs ER negative, and progesterone (PR) positive vs PR negative). We then permuted the sample log means between the two groups and calculated the hypothetical distribution of the permutated sample sets (repetitions = 5000). The p-values were calculated by dividing the number of times the absolute mean difference of the observed value was smaller than the absolute value of the permuted mean by the number of repetitions. As three hypotheses were tested using the same data, a Bonferroni correction was applied resulting in a new significance cutoff value of α = 0.0167. Further information regarding image processing and data extraction are including in the Supporting Information.
Nomenclature.
Lipid shorthand, as described by Liebsich et al., was adopted.46 Please see the SI for further information regarding this nomenclature and its adaptation for this manuscript.
RESULTS AND DISCUSSION
Double Bond Localization within Fatty Acids by Dication Complexation and UVPD.
We first explored the utility of 193 nm UVPD in cleaving and characterizing double bonds within deprotonated FAs in the negative ion mode. Activating unsaturated phospho- and sphingo-lipids with 193 nm UVPD reproducibly produces fragment ions originating from cleavage of both C–C bonds adjacent to a double bond.38-40 The resulting pair of product ions, proposed to originate through a cis 1,2 elimination reaction,39 display a diagnostic mass difference of 24 Da that allows the facile localization of double bonds within acyl chains. Application of this activation method to free FAs was explored and shown to result in low abundance fragment ions (Figure S4) that paralleled the dissociation pathways of phospho- and sphingolipids. For example, UVPD of isomeric FA 18:1(9Z) and FA 18:1(11Z) resulted in distinct pairs of low abundance (average 0.030% of TIC for FA 18:1(9Z) and 0.040% of TIC for FA18:1(11Z), n = 3 mass spectra) diagnostic products differing by 24 Da (m/z 143.11/167.11 and m/z 171.14/195.14 for 9Z and 11Z, respectively) that localize the double bond in each case. However, 193 nm UVPD of polyunsaturated FAs yielded uninformative neutral losses, as in the case of linoleic acid (FA 18:2(9Z,12Z), Figure S5A,B) or uninformative dissociation of C–C bonds, as seen for arachidonic acid (FA 20:4(5Z,8Z,11Z,14Z), Figure S5C,D), precluding the ability to assign double bond positions. Noting the low or nonexistent efficiency for double bond diagnostic products, alternate approaches were explored to enhance structurally informative UVPD fragmentation of free FAs.
Charge remote fragmentation (CRF) processes have been shown to produce informative products for both mono- and poly-unsaturated FAs.47,48 CRF dissociation pathways may be induced for FAs using charge inversion strategies.22-27,47,48 Specially designed DCs, for example, have been shown to readily adduct and charge-invert free FAs from negative to positive ion mode without compromising sensitivity when implemented in a DESI workflow.15,49 In this approach, the anionic carboxylate group present on the FA structure binds electrostatically with the DC possessing a fixed 2+ charge, generating a DC·FA complex with a net +1 charge. Although collisional activation of noncovalent DC·lipid complexes is dominated by neutral losses from the dication group,15 193 nm UVPD has been shown to yield product ions that retain noncovalent interactions. In essence, the fast, high energy deposition caused by UV photoabsorption allows cleavage of covalent bonds on a time scale faster than disassembly of all noncovalent interactions. Thus, we hypothesized that 193 nm UVPD would yield CRF products that would retain DC – lipid interactions50 and allow structural characterization of FAs complexed with DC.
We explored the utility of DC complexation for the structural analysis of monounsaturated FAs by examining the dissociation patterns for DC·FA 18:1 complexes. Doping DC into FA 18:1(9Z) in solution resulted in an abundant complex of m/z 591.58 (Figure S6), corresponding to [M-H+DC]+. Higher-energy collisional dissociation (HCD) of the complex (Figure S7) resulted in fragment ions from the charge carrying DC (m/z 182.19, 210.22, 281.29, and 492.48) and no fragment ions from the FA portion of the complex, aligning with the charge-mediated nature of collisional dissociation pathways. On the other hand, activation of the complex of m/z 591.58 via 193 nm UVPD resulted in a rich mass spectrum composed of fragment ions originating from both the FA and DC structures (Figure 1A,B). Notably, nearly all C–C bonds in the FA chain were cleaved, with the most abundant fragments in the series (m/z 453.44 and 477.44) featuring a diagnostic mass difference of 24 Da that originates from the cleavage of the C–C bonds adjacent to the double bond. Detection of the diagnostic ions allowed confident identification and localization of the unsaturation at C9═C10. Although a series of fragment ions differing in mass by 14 Da (m/z 369–437) mirrors CRF fragmentation,47,48 the Δ14 Da series is incomplete for products originating from C–C bonds methyl-terminal to the double bond, a result of multiple UVPD fragmentation pathways generating both terminally saturated and unsaturated products. Similar results were achieved upon 193 nm UVPD of the double bond positional isomer FA 18:1(11Z) complexed to DC (Figure 1C,D). The Δ14 Da series is analogous to that observed by Narreddula et. al for the structural characterization of derivatized FAs.51 Fragments originating from the DC were also observed (m/z 182.19, 210.22, 281.29, and 492.48), with analogous C–C cleavage products arising from the FA structure. The most abundant (average relative abundance of 0.57% and 0.63% for 9Zp and 9Zd, respectively, for n = 3 mass spectra) products originated from cleavage of carbon–carbon bonds adjacent to the double bond (m/z 481.47 and 505.47), which allowed confident identification and localization of the unsaturation as C11═C12. Thus, 193 nm UVPD of DC·FA complexes results in informative product ions with a diagnostic mass difference of 24 Da that can be readily identified. Importantly, UVPD of the DC complexed FAs also yielded increased efficiency in the formation of the diagnostic ions (0.54% of the total ion current for DC·FA 18:1(9Z)) over the counterparts generated by UVPD of the deprotonated FAs in the negative ion mode (0.03% of the total ion current). The effects of cis/trans double bond geometry on UVPD spectra were not explored here. Photon absorption induces immediate isomerization of alkenes,52 and thus, dissociation via UVPD is expected to be indiscriminate toward double bond geometry, as suggested by Ryan et al.39
Figure 1.

193 nm UVPD (20 pulses, 4 mJ) spectra for FA 18:1 double bond isomers complexed to 1,8-ethyl DC (m/z 591.58). (A) UVPD spectrum for DC·FA 18:1(9Z) complex and (B) corresponding fragment map. (C) UVPD spectrum for DC·FA 18:1(11Z) complex and (D) corresponding fragment map. Pairs of diagnostic ions that localize the double bond are highlighted in red font. Insets display expanded regions with FA dissociation products. Selected precursor ions are designated with a star.
DESI-MS Imaging and UVPD for Double Bond Identification within Fatty Acids in Tissue Sections.
A primary motivation for development of the DC complexation-UVPD strategy is the potential for adapting it for double bond characterization of FAs during DESI-MS imaging without requiring special sample pretreatment. Methods proposed for double bond elucidation utilizing chemical derivatization of the double bond typically require seconds to minutes of reaction time which would be difficult to implement within the millisecond reaction time frame in DESI-MS experiments.16,21,35 On the other hand, reactive DESI-MS analysis of a mouse brain tissue sections with the DC in the spray solvent, as outlined by Lostun et al., enables formation of the DC·FA complexes with a high relative abundance (Figure S8).15 We thus explored reactive DESI-MS imaging of a tissue section in tandem with UVPD of the DC·FA complexes to acquire spatially correlated information regarding the distribution of FA isomers within heterogeneous biological tissues.
DESI-MS was coupled to an Orbitrap Fusion Lumos mass spectrometer equipped with a 193 nm excimer laser to perform UVPD-MS imaging of a human ovarian tumor tissue sample containing a region of high grade serous ovarian carcinoma adjacent to a region of normal ovarian tissue. The molecular profile of the ovarian tissue acquired in the full m/z 500–1000 range showed high abundance of FA species complexed with DC (Figure 2A, Table S3), including various saturated, mono- and polyunsaturated FA species normally detected from biological tissues using DESI-MS imaging in the negative ion mode of biological tissues. It is important to note that glycerophospholipid hydrolysis products could contribute to the FA levels detected in DESI-MS analysis, despite best experimental efforts in handling tissues prior to and during DESI-MS analysis.53 DC complexed to other GP species, primarily glycerophosphoethanolamines, were also observed in the mass spectra, but UVPD of these species were not explored in this study. The most abundant complex of m/z 591.581 identified as DC·FA 18:1 was targeted for UVPD. Figure 2B shows an average UVPD mass spectrum of the m/z 591.581 complex acquired during DESI-MS imaging across the tissue section, resulting in two pairs of diagnostic products characteristic of a mixture of 9Δ and 11Δ FA 18:1 isomers. The relative abundance of the 9Δ ions of m/z 453.44 and m/z 477.44 was approximately 1 order of magnitude higher than those from the 11Δ ions of m/z 481.47 and m/z 505.47 across the tissue section. To investigate the spatial distribution of the isomers more closely, ion images of the precursor ion, the proximal isomeric fragments (9Δp and 11Δp), and the 9Δp:1lΔp ratio were generated (Figure 2C). All images showed consistent signal intensity across the tissue section, including the normal and tumor ovarian tissue regions, with the normal region having a log mean ratio of 0.779 ± 0.123 and the cancer region having a log mean ratio of 0.832 ± 0.164. This suggests that any alterations in the isomeric distribution of FA 18:1 between tumor and normal ovarian tissues from this patient are minimal and, if present, are less prominent than the random fluctuations in signal intensity associated with DESI-UVPD MS analysis (17.8% RSD for precursor ion signal and an average 27.9% RSD for Δ9 and Δ11 fragment ions).
Figure 2.

DESI-UVPD MS imaging of the distribution of the FA 18:1 double bond isomers within an ovarian tumor tissue. (A) Full MS profile of ovarian tissue analyzed by DESI-MS in the positive ion mode with the DC reagent added to the solvent. Peaks labeled in red are FA·DC complexes and are identified in Table S3. (B) The DESI-UVPD spectrum shows two pairs of diagnostic ions resulting from the fragmentation of the 9Δ and 11Δ positional isomers of FA 18:1. (C) The ion images from the precursor ion (m/z 591.58), the proximal fragment peaks for each isomer (9Δp at m/z 453.44 and 11Δp at m/z 481.47), and the 9Δp:11Δp ratio, illustrating that there is no observable difference in the isomer distribution when comparing the tumor and normal ovary tissue regions for this patient.
UVPD-MS Analysis of Polyunsaturated Fatty Acid Isomers.
Although UVPD of polyunsaturated FA anions was ineffective (Figure S5), charge inversion via DC complexation promoted rich UVPD mass spectra suited for double bond localization. Along with cleavage of most C–C bonds in the FA, UVPD of the DC·FA 18:2(9Z,12Z) complex resulted in diagnostic product ion pairs of m/z 453.44/477.44 and m/z 493.47/517.47 for the 9Z and 12Z double bonds, respectively (Figure S9). Additional products from the cleavage of the C–C bonds (m/z 451.42, 478.45, 479.46, 491.46, 492.46, and 519.49) were detected, as shown in the expansion of the mass range from m/z 450–520 (Figure S10). Similar products have been previously observed upon UVPD of phospholipids and have been proposed to arise from photoinduced excitation of the C═C bonds to diradical states, leading to competing dissociation pathways involving hydrogen atom migration and radical products.40 In addition, cleavage of C–C bonds allylic to the double bond (resulting in m/z 531.49 and m/z 437.41) yielded products more abundant than the expected diagnostic products, differing from UVPD of monounsaturated FAs. Due to the consequent complexity of the mass spectra, we evaluated the propensity to misidentify double bond locations in polyunsaturated FAs by searching for the theoretical diagnostic product ions for the ω-3 (FA 18:2(12Z,15Z)) isomer. The isomer FA 18:2(12Z,15Z) would theoretically produce diagnostic products of m/z 495.49/519.49 for the 12Z C═C, and m/z 535.52/559.52 for the 15Z C═C. When examining the UVPD mass spectra for DC-18:2(9Z,12Z), only an ion matching the m/z value of one diagnostic product for each double bond isomer was observed (m/z 519.49 and m/z 559.52). Thus, misidentification would not occur owing to the lack of a complete diagnostic pair needed for confident identification of each double bond. Similarly, when searching for theoretical product ions that would result from the dissociation of a different isomer, DC-18:2(6Z,9Z), only one of the theoretical products (m/z 451.43) was detected in the UVPD spectrum of DC·FA 18:2(9Z,12Z), mitigating the concern about mischaracterization. Thus, despite mass spectral complexity, double bonds in polyunsaturated fatty acids may be reliably localized via detection of diagnostic fragment pairs with a mass difference of 24 Da.
Applying DESI-UVPD MS imaging to analyze DC·FA 18:2 (m/z 589.56) directly from ovarian tissue confirmed that polyunsaturated FAs can also be characterized from biological samples. As there is diversity in the abundance of ω-9, ω-6, and ω-3 FAs in the human diet that may be detected in tissue samples, investigation of the potential presence of polyunsaturated FA isomers was performed. DESI-UVPD-MS imaging of DC·FA 18:2 consistently displayed the pair of double bond diagnostic products that would arise for FA 18:2 (9Δ,12Δ), suggesting a homogeneous presence of this FA within the sample (Figure S11). Only one fragment ion, at most, was observed per double bond that would correspond to the hypothetical diagnostic products for FA 18:2 (12Δ,15Δ) and FA 18:2 (6Δ,9Δ), as shown in Table S4, thus providing no positive confirmatory evidence for these isomers in the samples.
Remarkably, although mass spectral complexity increases for FAs featuring an increased degree of unsaturation, double-bond assignment through identification of the Δ24 fragment pairs remains possible. 193 nm UVPD of the DC·arachidonic acid (FA 20:4(5Z,8Z,11Z,14Z)) complex resulted in cleavage of all C–C bonds in the acyl chain, including those adjacent to the double bond, producing four pairs of diagnostic fragments with a mass difference of 24 Da (m/z 397.38/421.38 for 5Z; m/z 437.41/461.41 for 8Z; m/z 477.44/501.44 for 11Z; m/z 517.47/541.47 for 14Z; Figure S12). Multiple fragment ions were attributed to cleavage of each C–C bond adjacent to a double bond, as shown in the expansion of the m/z 390–545 range (Figure S13). DESI-UVPD-MS imaging of FA 20:4 from ovarian tissue also yielded complex mass spectra, yet, all double bond diagnostic pairs of fragments for FA 20:4- (5Δ,8Δ,11Δ,14Δ), the most prominent isomer of 20:4 in nature, were observed in the mass spectra and the ion images, as shown in Figure S14. As seen for the imaging of DC·FA 18:2, possible diagnostic fragment ions for the ω-3 FA 20:4 isomer were detected within the mass spectra (Table S5) but only one diagnostic ion corresponding to the cleavage of each double bond position was detected, therefore preventing confident identification of the presence of this isomer. Although dissociation of the 20:4 and 18:2 FAs did not demonstrate distinction of polyunsaturated double bond isomers in human tissue, characterization of polyunsaturated FAs at the double bond level was nonetheless demonstrated. An additional strategy for ensuring interfering fragment ions from coisolated species during DESI-MS imaging experiments are not mistaken for diagnostic fragments is discussed in the SI.
Relative Quantitation of Double Bond Isomers with Direct Infusion and DESI-MS Imaging.
Previous UVPD analyses of unsaturated FAs within GPs demonstrated a linear relationship between the abundances of the diagnostic ions and the relative concentration of double bond isomers.38,44 To explore if DC complexation followed by UVPD would similarly allow relative quantification of FA isomers, mixtures containing various molar ratios of the monounsaturated FA 18:1 (9Z) and FA 18:1 (11Z) were analyzed by direct infusion ESI. UVPD mass spectra were recorded for each isomer mixture to correlate the relative concentration of the isomers with the abundances of their diagnostic fragment ions. In previous studies, the abundances of diagnostic ions corresponding to each isomer were summed together in order to increase sensitivity. However, UVPD of DC·FA 18:1 (9Z) resulted in an ion of m/z 505.47 that is isomeric with the distal fragment of FA 18:1 (11Z) (Figure S16A,B). The added abundance from the confounding product disrupts the linear relationship between the concentration ratio of the isomers and signal abundance of the diagnostic ions (Figure S16C), and is also responsible for the different Δp/ Δd ratios observed for the FA 18:1(11) (Figure 1C) standard and FA 18:1(11) in ovarian tumor tissue (Figure 2B). To circumvent this challenge, a calibration curve was built excluding the distal fragments from both isomers, which led to excellent linearity of the response curve (R2 = 0.9998) (Figure S17) thus suggesting that this method can be used to quantify the relative concentrations of isomers from monounsaturated FAs in mixtures. Moreover, the slope of the regression line approaches unity (0.991) indicating both a lack of preferential ionization and dissociation of either isomer, as well as a lack of preferential dissociation based on double bond position. These features should enable the accurate relative quantitation of double bond positional isomers without constructing a calibration curve or in the absence of standards. Due to the complexity of the spectra, quantitation of polyunsaturated fatty acids was not explored.
To evaluate if DESI-MS also allowed relative quantitation of the FA isomers, imaging of the standard mixtures was performed in tandem with UVPD of the FA 18:1 precursor ion (Figure 3A). Ion images from the precursor ion show a consistent intensity across all sample spots at a similar absolute intensity value (NL ≈ 1E5 during UVPD) to what was detected from the ovarian tissue sections (NL ≈ 8E4–2E5 during UVPD) allowing extrapolation of calibration results to biological tissue sections. Plotting ion intensities for the 9Z and 11Z proximal diagnostic ions (m/z 453.44 and m/z 481.47, respectively) showed a clear gradient of intensities associated with altered concentration ratios. Plotting absolute concentration of the isomers against the average ion signal intensity of the respective proximal diagnostic peak for all spots (Figure S18) illustrated a linear relationship for both isomers (9Zp R2 = 0.9953, 11Zp R2 = 0.9965). The calculated limits of detection for the isomers were 0.568 and 0.492 μM for the 9Z and 11Z diagnostic fragments, respectively, indicating that spots featuring the lowest concentrations of the isomers (0.5 μM) were not reliable. A DESI-MS imaging calibration curve was then constructed based on the ratio of isomer concentrations within the spots and the ratio of their resulting diagnostic fragment peak intensities for every pixel, excluding the 19:1 and 1:19 9Z:11Z ratios (Figure 3B). The resulting curve showed a similar linear response to the one constructed via direct infusion (Figure S17) but with larger standard deviations and a decreased slope (m = 0.892 compared to m = 0.991 for direct infusion). These differences can be primarily attributed to lower overall signal intensity of diagnostic peaks within the DESI-MS data, which is ~5% of the signal intensity achieved by ESI. Additionally, as no averaging was used in order to more closely resemble DESI-MS imaging data where each pixel represents one mass spectrum, the standard deviation of the 9Z:11Z ratio within each spot is higher than what was obtained by direct infusion. Nevertheless, the linearity of the ratio concentration suggests DESI-UVPD of FA-DC complexes can be used for relative quantification of double bond isomers, as demonstrated in the next section.
Figure 3.

(A) DESI-UVPD MS ion images of DC·FA 18:1 varying FA 18:1(9Z)/FA 18:1(11Z) rations. (B) Plot of the concentration ratios of FA 18:1(9Z):FA 18:1(11Z) versus the ratio of the intensity of 9Z:11Z proximal diagnostic ions. Each concentration ratio point is comprised of the average 9Zp:11Zp ratio where each technical replicate is one pixel from the spot shown in A, approximately 100 pixels per spot. The inset shows an expanded view of the low concentration ratio range (0–1.0).
Investigation of Double Bond Localization in Human Breast Cancer Subtypes.
Dysregulation of FA synthase resulting in overexpression of free FA has been associated with a variety of cancers, including breast carcinomas.3,54 A few MS studies on mouse and human breast tissues have investigated the altered FA 18:1 isomer distribution within normal and cancerous breast tissue. For example, Xia and co-workers found significant differences in the 9Δ:11Δ ratio in FA 18:1 containing glycerophospholipids between normal and cancerous human breast tissues as well as alterations in the free FA 18:1 9Δ:11Δ ratio from mouse breast tissue.16,55 However, further evidence has suggested that FA metabolism is altered with respect to other factors within breast cancer, including subtype, receptor status, and lymphatic system invasion.4,56 Therefore, we aimed to use DESI-UVPD-MS to evaluate the FA 18:1 isomer distribution within breast carcinoma tissues related to various molecular and clinical features, including ER status, PR status, and metastatic status (invasion seen in sentinel lymph nodes).
A sample set of 18 breast tumors with detailed diagnosis and molecular subtyping information was used to classify samples into groups (Table S6). Note that ER and PR status were not available for four samples, yielding a smaller sample set for those analyses. DESI-UVPD-MS imaging was performed while targeting the DC·FA 18:1 complex. Samples were subsequently H&E stained and evaluated by a pathologist to determine areas of breast tumor. Breast tissue is notoriously heterogeneous, displaying normal, cancerous, necrotic, and fat tissue within each section, but the use of DESI-MS imaging allows extraction of mass spectra from tissue regions with predominant tumor content to ensure unambiguous correlation between molecular information and histologic diagnosis. The log 9Δ:11Δ ratio was calculated for each pixel and separated by three distinguishing features of the sample: metastatic vs nonmetastatic to sentinel lymph nodes, ER+ vs ER− and PR+ vs PR−. Per-pixel density plots for each of these evaluations are shown in Figure 4A-C. The means for each sample distribution and the mean difference for each evaluated parameter were calculated. All three sample sets showed separation in mean difference between the two groups (−0.018 for metastatic vs nonmetastatic, 0.109 for ER+ vs ER− and 0.211 for PR+ vs PR−), but both the metastatic and ER status density plots contained shoulders that suggested incomplete separation of the groups. Nonparametric permutations of the samples were performed for each parameter to generate a random sampling of the possible mean difference values for these sample sets and determine the significance of the observed mean differences. Histograms of the randomly generated mean differences with the observed difference value shown as a red dotted line for each sample set are provided in Figure 4D-F. Interestingly, PR status was the only mean difference that was statistically significant (α = 0.0187) from the generated distribution with a p value of 0.003, showing an increased relative abundance of the Δ11 isomer within PR+ samples compared to PR− samples. The ion images of the 9Δp:1lΔp ratio from 6 representative samples, 3 PR+ and 3 PR−, allows for visual confirmation of this difference (Figure 4G), with PR+ samples exhibiting a decreased 9Δp:1lΔp ratio and thus an increased relative abundance of the 11Δ isomer. For example, the two images within the center for Figure 4G have drastically different 9Δp:1lΔp ratios, with the PR− sample having an average ratio of 7.3 while the PR+ sample had an average ratio of 2.2. This alteration in the 9Δp:1lΔp isomer ratios in PR+ samples is consistent with previous studies correlating hormone receptor status with dysregulation in FA synthesis and desaturation.5,10 All DESI-UVPD images from PR+ and PR− samples are provided in Figure S19. These results suggest that FA isomer distribution is not only related to molecular alterations between normal and cancerous breast tissues, as suggested in studies performed with other MS methods, but may also correlate with aberrations in molecular processes that occur in breast cancer subtypes as determined by receptor status. Further studies are being performed to validate these trends and expand our sample set to include a larger sample cohort to adjust for patient-to-patient variability and diverse sample populations. While traditional MS imaging has shown promise in distinguishing ER/PR status,57-59 the addition of isomer data obtained via DESI-UVPD-MS could provide additional insights into the metabolite and lipid alterations detected in the full MS data and biological alterations that may be causing these changes.
Figure 4.

Density plots of the log of the 9Δp:11Δp diagnostic peak intensities for (A) metastatic (B) ER and (C) PR statuses. Histograms from the nonparametric permutation of the mean difference of the 9Δp:11Δp diagnostic ion abundances and the observed mean difference (red line) for the (D) metastatic, (E) ER, and (F) PR groupings. (G) DESI-UVPD MS 9Δp:11Δp ratio ion images of three PR− and three PR+ breast cancer samples, showing an increased ratio for the PR− samples. The areas outlined in black on the H&E stained tissue sections are the areas of pure tumor tissue that were extracted for data analysis, while the other area comprise of various tissue types including stroma, fibrosis, and necrosis as well as other cell types such as lymphocytes and inflammatory cells.
CONCLUSIONS
Mounting evidence correlating subtle structural variations in lipids with disease state demands development of methods capable of both distinguishing isomeric biomolecules and identifying the biosynthetic dysregulation of the molecules in question. To this end, UVPD was coupled with reactive DESI to enhance structural characterization of FA in an imaging workflow. Synergistic charge inversion via reactive DESI augmented the capabilities of UVPD for FA isomer distinction, while permitting the distribution of structural isomers to be spatially resolved without time-consuming off-line chemical derivatization. Using this method for the analysis of human breast tumor tissues suggested that alterations in the FA (18:1) isomer abundance may be correlated to other clinically relevant factors in breast cancer such as PR status, an outcome that merits further investigation. To better understand the role other FAs may play in disease progression and patient outcome, future studies are planned to explore the relationship between FA synthase and FA isomer abundance ratios, and the importance of these factors within breast cancer molecular subtypes and other molecular receptors. Note that the method described here has limitations that should be considered including the lower sensitivity of DESI when compared to ESI, the lack of double bond geometry assessment, and the limited structural characterization achieved for polyunsaturated FA isomers. In regards to sensitivity, alternate strategies such as a laser capture microdissection-LCMS workflow could be implemented in conjunction with UVPD to further explore the tissue dependent isomer abundances detected in this study, as well as allow double bond localization of low abundance polyunsaturated FA isomers that were not definitely identified in this study. Separations methods coupled to MS have also been described to investigate double bond geometry, although experimentally challenging.13,14 Optimization of DESI-MS imaging and UVPD experimental conditions, and derivatization with other DC species will be pursued to expand the analysis beyond FA(18:1) explored here. Nevertheless, our study demonstrated that DESI-UVPD-MS allows for spatially resolved FA isomer information without significant deviations from a traditional DESI-MS workflow, presenting a useful tool for the exploration of isomeric alterations within tissue samples.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by The Welch Foundation (Grants F-1895 LSE and F-1155 JSB), the National Institutes of Health (Grants R00CA190783 to L.S.E. and R01GM103655 to J.S.B.). The authors would like to thank Logan Wilder from Prof. Richard Crooks’ research lab for advising on the synthesis of the dicationic compounds. Advice on the choice of statistical method was provided by Spencer Woody and Yangxinyu Xie. The authors would also like to thank Kyana Garza for her breast cancer expertise and John Lin for composing the original code to compile the molecular images. We are grateful to Dr. Jinsong Liu (MD Anderson Cancer Center) and Dr. Chandandeep Nagi (Baylor College of Medicine) for the pathological evaluation of our samples. A subset of the tissue samples was provided by the CHTN which is funded by the National Cancer Institute.
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.0c00970.
Supplementary methods and references, sample demographics, evaluation of multiple DCs, polyunsaturated FA fragmentation, and images (PDF)
NOTE ADDED AFTER ASAP PUBLICATION
This paper was originally published ASAP on June 1, 2020. Reference 43 was added to support a new sentence in the Introduction. The corrected version was reposted on June 4, 2020.
Contributor Information
Clara L. Feider, Department of Chemistry, The University of Texas at Austin, Austin, Texas 78712, United States.
Luis A. Macias, Department of Chemistry, The University of Texas at Austin, Austin, Texas 78712, United States.
Jennifer S. Brodbelt, Department of Chemistry, The University of Texas at Austin, Austin, Texas 78712, United States.
Livia S. Eberlin, Department of Chemistry, The University of Texas at Austin, Austin, Texas 78712, United States.
REFERENCES
- (1).Helmreich EJ M. Biophys. Chem 2002, 100 (1), 519–534. [DOI] [PubMed] [Google Scholar]
- (2).de Carvalho CCCR; Caramujo MJ Molecules 2018, 23(10), 2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Kuhajda FP Nutrition 2000, 16 (3), 202–208. [DOI] [PubMed] [Google Scholar]
- (4).Alo PL; Visca P; Marci A; Mangoni A; Botti C; Di Tondo U Cancer 1996, 77 (3), 474–482. [DOI] [PubMed] [Google Scholar]
- (5).Alò PL; Visca P; Trombetta G; Mangoni A; Lenti L; Monaco S; Botti C; Serpieri DE; Di Tondo U Tumori 1999, 85 (1), 35–40. [DOI] [PubMed] [Google Scholar]
- (6).Pizer ES; Lax SF; Kuhajda FP; Pasternack GR; Kurman RJ Cancer 1998, 83 (3), 528–537. [PubMed] [Google Scholar]
- (7).Epstein JI; Carmichael M; Partin AW Urology 1995, 45 (1), 81–86. [DOI] [PubMed] [Google Scholar]
- (8).Kuhajda FP; Jenner K; Wood FD; Hennigar RA; Jacobs LB; Dick JD; Pasternack GR Proc. Natl. Acad. Sci. U. S. A 1994, 91 (14), 6379–6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Peck B; Schulze A FEBS J. 2016, 283 (15), 2767–2778. [DOI] [PubMed] [Google Scholar]
- (10).Holder AM; Gonzalez-Angulo AM; Chen H; Akcakanat A; Do K-A; Fraser Symmans W; Pusztai L; Hortobagyi GN; Mills GB; Meric-Bernstam F Breast Cancer Res. Treat 2013, 137 (1), 319–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Kridel SJ; Lowther WT; Pemble CW Expert Opin. Invest. Drugs 2007, 16 (11), 1817–1829. [DOI] [PubMed] [Google Scholar]
- (12).Menendez JA; Lupu R Expert Opin. Ther. Targets 2017, 21 (11) 1001–1016. [DOI] [PubMed] [Google Scholar]
- (13).Murphy RC; Axelsen PH Mass Spectrom. Rev 2011, 30 (4), 579–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Mitchell TW; Pham H; Thomas MC; Blanksby SJJ Chromatogr. B: Anal. Technol. Biomed. Life Sci 2009, 877 (26), 2722–2735. [DOI] [PubMed] [Google Scholar]
- (15).Lostun D; Perez CJ; Licence P; Barrett DA; Ifa DR Anal. Chem 2015, 87 (6), 3286–3293. [DOI] [PubMed] [Google Scholar]
- (16).Tang F; Guo C; Ma X; Zhang J; Su Y; Tian R; Shi R; Xia Y; Wang X; Ouyang Z Anal. Chem 2018, 90 (9), 5612–5619. [DOI] [PubMed] [Google Scholar]
- (17).Ma X; Zhao X; Li J; Zhang W; Cheng J-X; Ouyang Z; Xia Y Anal. Chem 2016, 88 (18), 8931–8935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Murphy RC; Okuno T; Johnson CA; Barkley RM Anal. Chem 2017, 89 (16), 8545–8553. [DOI] [PubMed] [Google Scholar]
- (19).Xie X; Xia Y Anal. Chem 2019, 91 (11), 7173–7180. [DOI] [PubMed] [Google Scholar]
- (20).Feng Y; Chen B; Yu Q; Li L Anal. Chem 2019, 91 (3), 1791–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Kuo T-H; Chung H-H; Chang H-Y; Lin C-W; Wang M-Y; Shen T-L; Hsu C-C Anal. Chem 2019, 91 (18), 11905–11915. [DOI] [PubMed] [Google Scholar]
- (22).Randolph CE; Foreman DJ; Blanksby SJ; McLuckey SA Anal. Chem 2019, 91 (14), 9032–9040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Randolph CE; Foreman DJ; Betancourt SK; Blanksby SJ; McLuckey SA Anal. Chem 2018, 90 (21), 12861–12869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Afonso C; Riu A; Xu Y; Fournier F; Tabet J-CJ Mass Spectrom. 2005, 40 (3), 342–349. [DOI] [PubMed] [Google Scholar]
- (25).Hsu F-F; Turk JJ Am. Soc. Mass Spectrom 1999, 10 (7), 600–612. [DOI] [PubMed] [Google Scholar]
- (26).Wang M; Han RH; Han X Anal. Chem 2013, 85 (19), 9312–9320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Yang K; Dilthey BG; Gross RW Anal. Chem 2013, 85 (20), 9742–9750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Yoo HJ; Håkansson K Anal. Chem 2010, 82 (16), 6940–6946. [DOI] [PubMed] [Google Scholar]
- (29).Poad BLJ; Marshall DL; Harazim E; Gupta R; Narreddula VR; Young RSE; Duchoslav E; Campbell JL; Broadbent JA; Cvačka J; Mitchell TW; Blanksby SJ J. Am. Soc. Mass Spectrom 2019, 30 (10), 2135–2143. [DOI] [PubMed] [Google Scholar]
- (30).Sun C; Li T; Song X; Huang L; Zang Q; Xu J; Bi N; Jiao G; Hao Y; Chen Y; Zhang R; Luo Z; Li X; Wang L; Wang Z; Song Y; He J; Abliz Z Proc. Natl. Acad. Sci. U. S. A 2019, 116 (1), 52–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Calligaris D; Caragacianu D; Liu X; Norton I; Thompson CJ; Richardson AL; Golshan M; Easterling ML; Santagata S; Dillon DA; Jolesz FA; Agar NY R. Proc. Natl. Acad. Sci. U. S. A 2014, 111 (42), 15184–15189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Guo S; Wang Y; Zhou D; Li Z Sci. Rep 2015, 4, 5959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Jackson AU; Shum T; Sokol E; Dill A; Cooks RG Anal. Bioanal. Chem 2011, 399 (1), 367–376. [DOI] [PubMed] [Google Scholar]
- (34).Yang E; Fournelle F; Chaurand PJ Mass Spectrom. 2020, 55 (4), e4428. [DOI] [PubMed] [Google Scholar]
- (35).Bednařík A; Bölsker S; Soltwisch J; Dreisewerd K Angew. Chem., Int. Ed 2018, 57 (37), 12092–12096. [DOI] [PubMed] [Google Scholar]
- (36).Wäldchen F; Spengler B; Heiles SJ Am. Chem. Soc 2019, 141 (30), 11816–11820. [DOI] [PubMed] [Google Scholar]
- (37).Paine MRL; Poad BLJ; Eijkel GB; Marshall DL; Blanksby SJ; Heeren RMA; Ellis SR Angew. Chem., Int. Ed 2018, 57 (33), 10530–10534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Klein DR; Brodbelt JS Anal. Chem 2017, 89 (3), 1516–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Ryan E; Nguyen CQN; Shiea C; Reid GE J. Am. Soc. Mass Spectrom 2017, 28 (7), 1406–1419. [DOI] [PubMed] [Google Scholar]
- (40).Williams PE; Klein DR; Greer SM; Brodbelt JS J. Am. Chem. Soc 2017, 139 (44), 15681–15690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Becher S; Esch P; Heiles S Anal. Chem 2018, 90 (19), 11486–11494. [DOI] [PubMed] [Google Scholar]
- (42).Narreddula VR; Boase NR; Ailuri R; Marshall DL; Poad BLJ; Kelso MJ; Trevitt AJ; Mitchell TW; Blanksby SJ Anal. Chem 2019, 91 (15), 9901–9909. [DOI] [PubMed] [Google Scholar]
- (43).Fang M; Rustam Y; Palmieri M; Sieber OM; Reid GE Anal Bioanal Chem 2020, 412, 2339–2351. [DOI] [PubMed] [Google Scholar]
- (44).Klein DR; Feider CL; Garza KY; Lin JQ; Eberlin LS; Brodbelt JS Anal. Chem 2018, 90 (17), 10100–10104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Klein DR; Holden DD; Brodbelt JS Anal. Chem 2016, 88, 1044–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Liebisch G; Vizcaío JA; Köfeler H; Trötzmüller M; Griffiths WJ; Schmitz G; Spener F; Wakelam MJO J. Lipid Res. 2013, 54 (6), 1523–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Cheng C; Gross ML Mass Spectrom. Rev 2000, 19 (6), 398–420. [DOI] [PubMed] [Google Scholar]
- (48).Gross ML Int. J. Mass Spectrom 2000, 200 (1), 611–624. [Google Scholar]
- (49).Rao W; Mitchell D; Licence P; Barrett DA Rapid Commun. Mass Spectrom 2014, 28 (6), 616–624. [DOI] [PubMed] [Google Scholar]
- (50).Brodbelt JS Chem. Soc. Rev 2014, 43 (8), 2757–2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Narreddula VR; Boase NR; Ailuri R; Marshall DL; Poad BLJ; Kelso MJ; Trevitt AJ; Mitchell TW; Blanksby SJ Anal. Chem 2019, 91 (15), 9901–9909. [DOI] [PubMed] [Google Scholar]
- (52).Kropp P Pure Appl. Chem 1970, 24, 585–598. [Google Scholar]
- (53).Dill A; Eberlin L; Costa A; Ifa D; Cooks R Anal. Bioanal. Chem 2011, 401, 1949–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Currie E; Schulze A; Zechner R; Walther TC; Farese RV Cell Metab. 2013, 18 (2), 153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Zhang W; Zhang D; Chen Q; Wu J; Ouyang Z; Xia Y Nat. Commun 2019, 10 (1), 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Monaco ME Oncotarget 2017, 8 (17), 29487–29500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Porcari AM; Zhang J; Garza KY; Rodrigues-Peres RM; Lin JQ; Young JH; Tibshirani R; Nagi C; Paiva GR; Carter SA; Sarian LO; Eberlin MN; Eberlin LS Anal. Chem 2018, 90 (19), 11324–11332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Guenther S; Muirhead LJ; Speller AVM; Golf O; Strittmatter N; Ramakrishnan R; Goldin RD; Jones E; Veselkov K; Nicholson J; Darzi A; Takats Z Cancer Res. 2015, 75 (9), 1828–1837. [DOI] [PubMed] [Google Scholar]
- (59).Kang HS; Lee SC; Park YS; Jeon YE; Lee JH; Jung S-Y; Park IH; Jang SH; Park HM; Yoo CW; Park SH; Han SY; Kim KP; Kim YH; Ro J; Kim HK BMC Cancer 2011, 11, 465–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.