

SUMMARY
Cortical pyramidal cells are generated locally, from pre-programmed progenitors, to form functionally distinct areas. By contrast, striatal projection neurons (SPNs) are generated remotely from a common source, undergo migration to form mosaics of striosomes and matrix, and become incorporated into functionally distinct sectors. Striatal circuits might thus have a unique logic of developmental organization, distinct from those of the neocortex. We explore this possibility in mice by mapping one set of SPNs, those in striosomes, with striatonigral projections to the dopamine-containing substantia nigra pars compacta (SNpc). Same-age SPNs exhibit topographic striatonigral projections, according to their resident sector. However, the different birth dates of resident SPNs within a given sector specify the destination of their axons within the SNpc. These findings highlight a logic intercalating birth date-dependent and birth date-independent factors in determining the trajectories of SPN axons and organizing specialized units of striatonigral circuitry that could influence behavioral expression and vulnerabilities to disease.
In Brief
Matsushima and Graybiel show that the birth dates of striatal projection neurons govern a center-surround organization in striosomes and the final destinations of their axons. These birth date-dependent controls seem to work in combination or in parallel with birth date-independent control of the final functional striatal sector in which they finally lie.
Graphical Abstract
INTRODUCTION
The brain is composed of myriads of elementary circuits that work uniquely yet coordinately. The prototypical, unique design of each circuit can be pre-programmed in progenitor cells from which the eventual circuit constituents are born (O’Leary et al., 2007) or can be post-mitotically bestowed after the constituent cells are generated from a common progenitor (Brown et al., 2011). Because the prototypes of elementary circuits are often subject to pre- and post-natal refinement via activity-dependent plasticity (Ben-Ari, 2002; Garaschuk et al., 2000; Hensch, 2005; Katz and Shatz, 1996; Klingler et al., 2019; Saint-Amant and Drapeau, 2000), it has been difficult to dissociate the “nature versus nurture” factors that specify the axonal projection target of each cell within a given circuit. This problem has hampered the identification of etiologies underlying disorders as well as mechanisms to organize normal neural circuits. Here, we tackled this question for the striatonigral circuit, as one of the most conserved circuits through vertebrate evolution (Reiner et al., 2004; Smeets et al., 2000; Yamamoto and Vernier, 2011), with expectation to have unique dependencies on development as organizational and operational principles in health and disease.
The developmental organization of neocortical circuits has been extensively studied. Pyramidal cells are known to be generated locally in the ventricular and subventricular zones of developing cortex, from ~E11.5 to ~E17.5 in mice (Molyneaux et al., 2007). The sequentially generated cells migrate outward to the pial surface along radial glial fibers, so that later-born cells migrate past the existing early-born cells to populate more-superficial layers (Angevine and Sidman, 1961; Rakic, 1974). As a consequence, the birth dates of pyramidal cells are highly correlated with their laminar position within each functional area. However, even with this strong correlation, birth dates of cells do not necessarily specify the projection target of their axons from the initial outgrowth (Hatanaka et al., 2016). Studies have now identified distinct transcriptomic profiles, for example, for two pyramidal subtypes, intermingled in a single layer (Tasic et al., 2018), that have distinct projection targets and functional roles in behavior (Economo et al., 2018). Although it remains controversial whether the fates of anatomical projections are progressively restricted in a lineage that shares a given birth date (Azim et al., 2009) or genetically predetermined in the heterogeneous progenitor pools dividing at the same time (Klingler et al., 2019), the birth date of neocortical pyramidal neurons appears not to be a definitive factor in the specification of their axonal projections.
By contrast, striatal projection neurons (SPNs), which compose the majority of striatal neurons, are not required to undergo extensive migrations but, instead, originate in a common germinal epithelium in the lateral ganglionic eminence (LGE), adjoining the striatal anlage, where they build the mature striatum (Turrero Garcıá and Harwell, 2017). SPNs are generated during a prolonged period from ~E10.5 to ~E18.5 in mice. SPNs born in the early phase of this time window become incorporated into a three-dimensional labyrinth of interconnected modules, called striosomes, whereas the later-born counterparts settle in the surrounding matrix (Graybiel and Hickey, 1982; Mason et al., 2005; Newman et al., 2015; Song and Harlan, 1994; van der Kooy and Fishell, 1987). This mosaic organization of the striatum is manifested by the differential expression of many molecular markers (Crittenden and Graybiel, 2011; Graybiel, 1990; Graybiel and Ragsdale, 1978) and is committed to differential input and output connections with the neocortex (Eblen and Graybiel, 1995; Hunnicutt et al., 2016; Ragsdale and Graybiel, 1981; Smith et al., 2016); thalamus (Berendse and Groenewegen, 1990; Fujiyama et al., 2019; Ragsdale and Graybiel, 1991; Smith et al., 2014); brainstem structures, including the substantia nigra (Fujiyama et al., 2011; Graybiel, 1984; Lévesque and Parent, 2005); and, indirectly via the pallidum, the lateral habenula (Hong et al., 2019; Rajakumar et al., 1993; Wallace et al., 2017). Advanced sequencing methods are beginning to identify distinct molecular profiles of striosomes (Gokce et al., 2016; Saunders et al., 2018). Differential vulnerability of the striosome and matrix compartments has been demonstrated in post-mortem analysis in Huntington’s disease and other disorders (Goto et al., 2005; Hedreen and Folstein, 1995; Tippett et al., 2007). However, the birth dates, the molecular profiles, and the connectivity patterns of each compartment at maturity are arranged across three-dimensional gradients that follow the anterior-posterior, medial-lateral, and dorsal-ventral axes of the striatum, greatly obscuring which factors assign a specific striatal circuit to the SPNs, the cells of origin of the great output pathways of the basal ganglia.
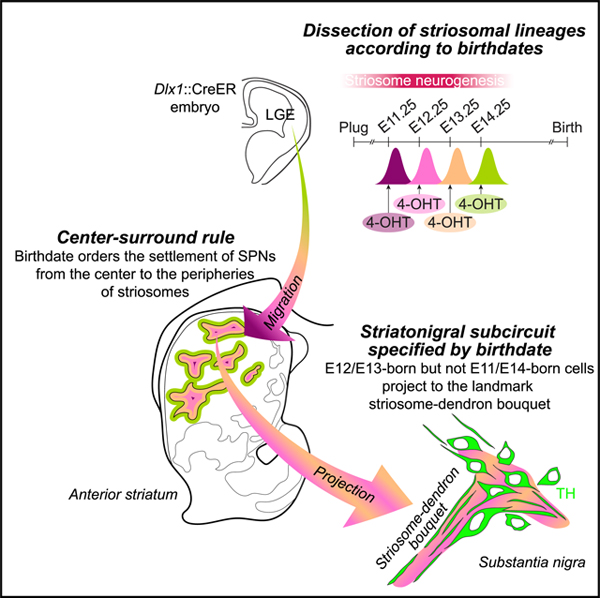
To isolate the role of birth date in the organization of the striatal circuits, we applied fast-kinetics 4-hydroxytamoxifen (4-OHT) (Ye et al., 2016) to Dlx1::CreER embryos (Feil et al., 1997; Kelly et al., 2018; Taniguchi et al., 2011). The fast kinetics allowed us to differentially label SPN sublineages born a single day apart. With that aid, we uncovered a sub-compartmental geography of SPN settlement, from the center (early-born) to surround (late-born) of the striosomes, as though forming a center-surround rule. Furthermore, we demonstrated the decisive role of birth date in routing striatonigral SPN axons to a specialized dopamine-containing nigral structure, a function beyond or separate from the topographic restriction imposed by an unspecified, birth date-independent factor, manifested as the functional striatal sector within which the SPNs settle. This combinatorial control of circuits could be critical for constructing behavioral repertoires, hardwired in the basal ganglia, i.e., one of the oldest circuits in the evolution (Puelles et al., 2000; Reiner et al., 1998a, 2004). This combinatorial code could also influence vulnerabilities of sub-compartments to genetic and epigenomic insult.
RESULTS
Neurogenesis of SPNs starts shortly after the formation of LGE at E9.5 in mice (Sousa and Fishell, 2010) and continues until birth (Graybiel and Hickey, 1982; Song and Harlan, 1994; van der Kooy and Fishell, 1987). Among the many other genes involved in striatal neurogenesis, a homeobox gene Dlx1/2 provides one of the best genomic loci to capture temporally specific stages of SPN development. Namely, Dlx1 is upregulated in newborn SPNs just after the terminal mitosis (Kelly et al., 2018), followed by its downregulation during their differentiation into mature SPNs (Liu et al., 1997; Yun et al., 2002). The inducible Dlx1::CreER driver (Taniguchi et al., 2011) has proven to be a powerful tool to leave a permanent marker of DNA recombination in specific lineages of SPNs (Kelly et al., 2018), allowing post hoc assessment of the distribution of their cell bodies and neurite arborizations long after the initial induction. For a fine-grain dissection of SPNs depending on their birth dates, we applied the fast-acting formulation of 4-OHT, previously shown in adults to be cleared within hours in vivo (Ye et al., 2016). Cells marked by this method must have the history that CreER was expressed under the control of the Dlx1 promoter, i.e., around the time of SPN neurogenesis, and at the same time was bound by 4-OHT (Indra et al., 1999), i.e., around the time of its administration, so that CreER could be liberated from cytosolic traps to enter the nucleus (Figure 1A). When crossed with Ai14, nuclear Cre excises the stop sequence upstream of tdTomato, resulting in the permanent expression of the reporter in the cell. Here, we used the reporter expression at maturity as an indicator of birth dates of SPNs. For example, we name SPNs as E12.25-born SPNs when they were labeled by 4-OHT administered at E12.25.
Figure 1. Sharpened Birth-Dating with 4-OHT.
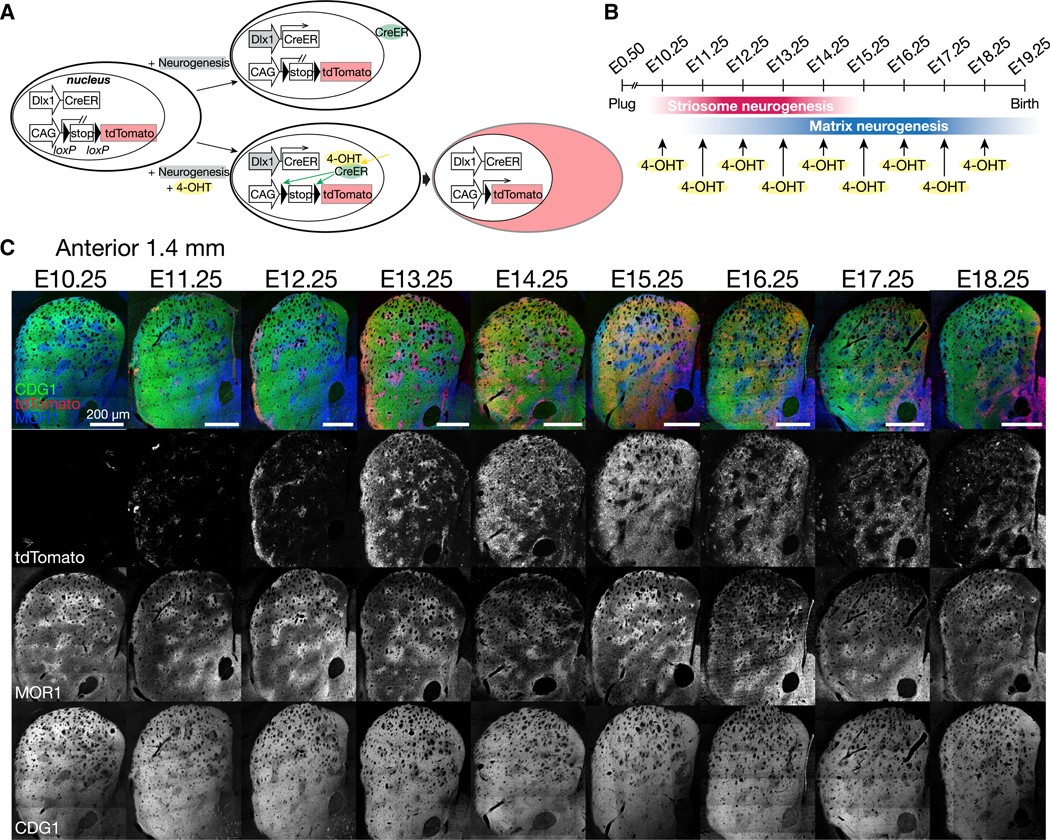
(A) In Dlx1::CreER/Ai14 reporter mice, cells are permanently labeled by tdTomato only if 4-OHT has been administered when the Dlx1 promoter is active, i.e., the neurogenetic phase of striatal progenitors.
(B) 4-OHT was administered at successive developmental times, separated by a single day.
(C) Striatum of 4-OHT pulse-labeled mice was assessed at maturity. Coronal sections in upper panels were stained for tdTomato (red), CalDAG-GEFI (CDG1, green), and MOR1 (blue). Lower panels show individual channels in grayscale.
The terminology can be justified by the estimated time course of DNA recombination. The absolute requirement is the existence of the 4-OHT/CreER complex in the nucleus, under the control of the Dlx1 promoter, CreER expression level, intracellular 4-OHT concentration, nuclear translocation of 4-OHT/CreER complex, and exclusion of CreER from the nucleus after the detachment of 4-OHT. Cytosolic CreER is inert because of the lack of access to DNA. Previous literature allows us to estimate the upper and lower limits of the temporal precision. Guenthner et al. (2013) showed that 4-OHT administered 6 h before or after light stimulation to the visually deprived Fos::CreER mice could hardly, if ever, induce DNA recombination. This indicates that, even if cytosolic CreER remained after the termination of the Fos promoter activation, DNA recombination could not be induced by 4-OHT administered with a 6h delay or lead time. In addition, they used 4-OHT dissolved in oil, but not in an aqueous solution as we did, so their pharmacokinetics were likely slower than ours. Thus, we can estimate 6 h as the upper limit of temporal separation between 4-OHT administration and the trigger of the promoter activation upstream of CreER (i.e., neuronal activation for Fos and terminal mitosis for Dlx1). As the lower limit, Ye et al. (2016) showed that 4-OHT reached a maximum concentration in murine brains within 2 h after intraperitoneal injection. Our goal was to dissociate SPNs born 24 h apart (Figure 1B). Thus, none of these limits or the inherent ambiguity in the identification of the time of fertilization (less than a half day) affects our interpretation of the results.
Indeed, as we administered 4-OHT to Dlx1::CreER/Ai14 embryos at successive developmental times from E10.25 to E18.25, separated only by a single day (Figure 1B), each time resulted in a unique pattern of tdTomato in relation to the compartments (Figure 1C). With that fine resolution, we examined the developmental organization manifested as a patterned distribution of SPNs born in close succession during embryogenesis.
Center-Surround Rule Uncovered by Sharpened Birth-Dating with 4-OHT
Early (E11.25)-born cells settled near the centers of striosomes (second column in Figure 2A), followed by the later (E12~E13)-born cells filling the striosomal profiles up to, and even beyond, their borders. Then, yet later, E14.25-born cells dispersed into the surrounding matrix (from the third to fifth columns in Figure 2A). These observations suggested a “center-surround” rule: the earliest-born cells become located at the center of the striosomes, as though they seeded the cluster formation, whereas later-born cells settled in the outskirts of the striosomes and ultimately in the surrounding matrix.
Figure 2. Center-Surround Rule in the Anterior Striatum.
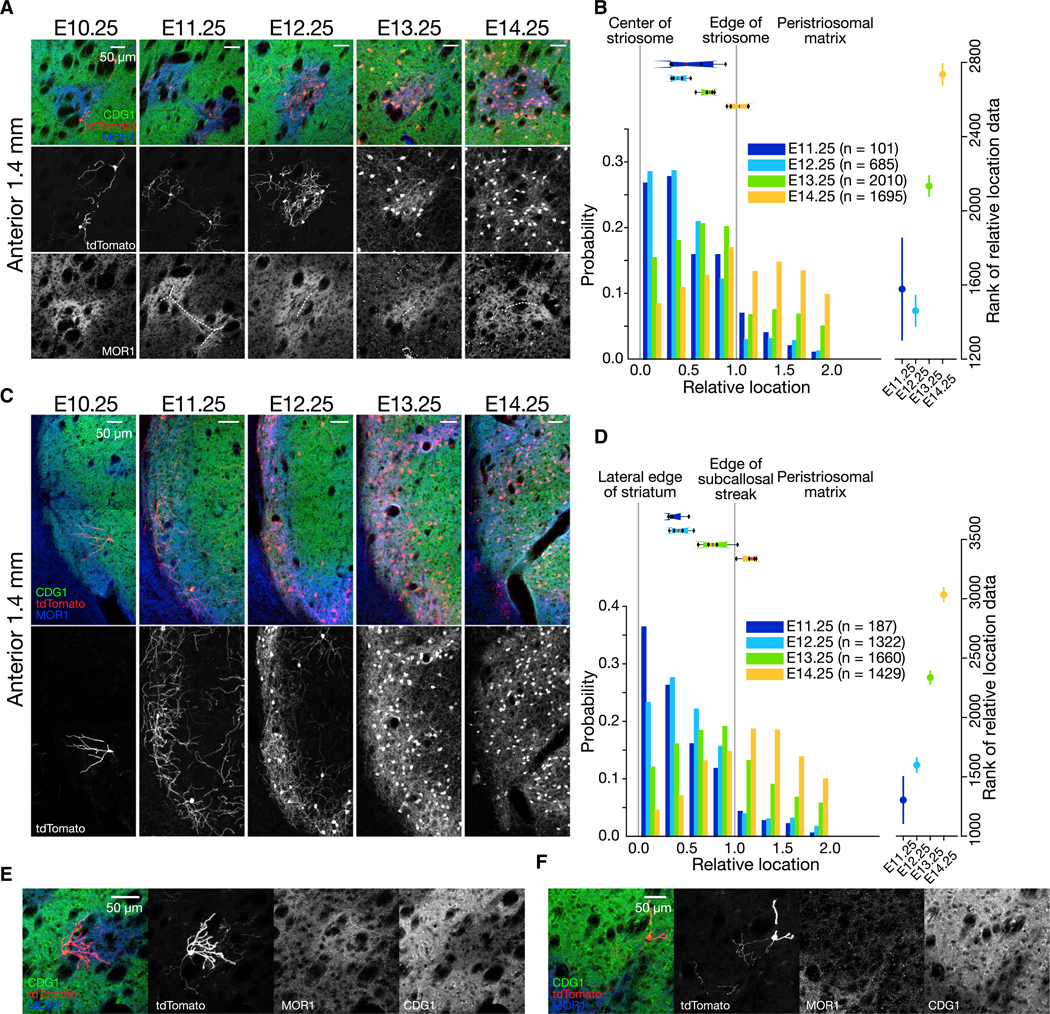
(A) Circular striosomes in the anterior striatum at the same level shown in Figure 1. Time of 4-OHT administration is indicated above. Striatal sections were stained for tdTomato (red), CDG1 (green), and MOR1 (blue). Lower panels show tdTomato (middle) or MOR1 (bottom) in grayscale. White, dotted lines indicate manually annotated centers of striosomes.
(B) Birth date-ordered settlement of SPNs from the center to the peripheries of striosomes. Left: Y axis shows the probability of cells falling into the corresponding bin among the all counted cells for a given birth date indicated by distinct colors. Boxplots above show the distribution of median values for individual animals by the 25th, 50th, and 75th percentiles. Right: Mean and 95% confidence intervals of data ranks for each birth date cohort. No overlap of confidence intervals indicates the significant differences detected by post hoc pairwise comparison (p < 0.05), following Kruskal-Wallis test. Number of mice = 4. Number of slices analyzed per mouse = 3.44 ± 0.63. Number of cells sampled per mouse = 25.25 ± 3.95 (E11.25), 171.3 ± 142.7 (E12.25), 502.5 ± 175.7 (E13.25), and 423.8 ± 167.5 (E14.25); means ± SD.
(C) Subcallosal streaks in the anterior striatum. Lower panels show tdTomato expression in grayscale.
(D) Birth date-ordered settlement of SPNs around the subcallosal streak, shown in the same format as in (B). Number of mice = 4. Number of slices analyzed permouse = 3.44 ± 0.51. Number of cells sampled per mouse = 46.8 ± 13.9 (E11.25), 330.5 ± 223.74 (E12.25), 415.0 ± 44.0 (E13.25), and 357.3 ± 158.7 (E14.25).
(E) Representative E10.25-born cell at striosome-matrix border with dendrites arborizing in a striosome and with axon extending toward the matrix.
(F) Representative E10.25-born cell at striosome-matrix border with dendrites arborizing in the matrix and with axon extending toward a striosome.
See also Table S1.
We confirmed that pattern by taking geometric measurements and by performing statistical tests (Figure 2B). First, we registered the location of each tdTomato-positive cell in relation to the center and the border of the striosome identified by muopioid receptor 1 (MOR1) immunostaining. Cells at the center were assigned to 0, and those at the border were assigned to 1. Each cohort of SPNs born at a specific developmental time, from E11.25 to E14.25, exhibited a distinct probability distribution of settling into the ranges of relative location (i.e., shown as a histogram in Figure 2B, with a bin size of 0.25). The birth date-dependent shift of the distributions was statistically significant, both when each cell was regarded as a single datum (right panel in Figure 2B with 95% confidence intervals, Kruskal-Wallis test, p < 1 × 10−10, see Table S1 for post hoc pairwise comparisons) and when each mouse was regarded as a single datum (p < 0.05, see boxplot above the histogram in Figure 2B for variability across animals). This was also true when we excluded cells outside the striosomes (i.e., relative location > 1.0, Kruskal-Wallis test, p < 1 × 10−10 for cell), but not statistically for data per mouse (p = 0.0667). These data strongly support the existence of a birth date-dictated center-surround rule for the striosomal system, which is smoothly transitioned into the surrounding matrix.
A similar rule appeared to hold for the organization of the socalled subcallosal streak, the band of MOR1-positive striosomal cells along the lateral edge of the caudoputamen, except that its anisotropy, rather than the isotropic organization of “circular” striosomes, was the organizing geometry (Figures 2C and 2D). The birth date-dependent wave of settlement was statistically confirmed (Figure 2D; Kruskal-Wallis test, p < 1 × 10−10 for cells, p < 0.01 for mice), even when we excluded matrix cells from the analysis (Kruskal-Wallis test, p < 1 × 10−10 for cells, p < 0.01 for mice).
In this study, E10.25-born cells represent the minority of cells. Some of these cells had dendrites ramifying within striosomes but with their axon extending toward the matrix (Figures 2C and 2E), whereas others had dendrites extending into the matrix but axons extending toward striosomes (Figure 2F).
Despite Anteroposterior Gradients, the Center-Surround Rule Holds in the Posterior Striatum
SPNs in the posterior striatum are known to be born earlier than those in the anterior striatum (Kelly et al., 2018; Newman et al., 2015), so that the composition of the striosomes and the matrix should be shifted in terms of birth dates. Consistent with that expectation, from anterior to posterior, the SPNs spreading out from the confines of striosomes were born at E14.25 at 1.4 mm anterior to the bregma (fifth column in Figure 2A), but at E13.25 at 0.3 mm anterior to the bregma (forth column in Figure 3A). The birth date-dependent wave of settlement was statistically confirmed (Figure 3B; Kruskal-Wallis test, p < 1 × 10−10 for cells, p < 0.01 for mice), even when excluding matrix cells (Kruskal-Wallis test, p < 1 × 10−10 for cells, p < 0.01 for mice). Thus, although the center-surround rule was commonly observed, irrespective of the location along the anterior-posterior axis of the striatum, the exact birth dates of the component SPNs respected the gradient and shifted accordingly. Across the gradient, the three-dimensional architecture constructed by the same-age cells (e.g., E13.25) seems to form a cometlike structure with the leading anterior point entering into the center of the striosomes, followed by the trailing halo surrounding the striosomes, which might reflect their migration paths during development.
Figure 3. Despite Anteroposterior Gradients, Center-Surround Rule Holds in the Posterior Parts of Striatum.
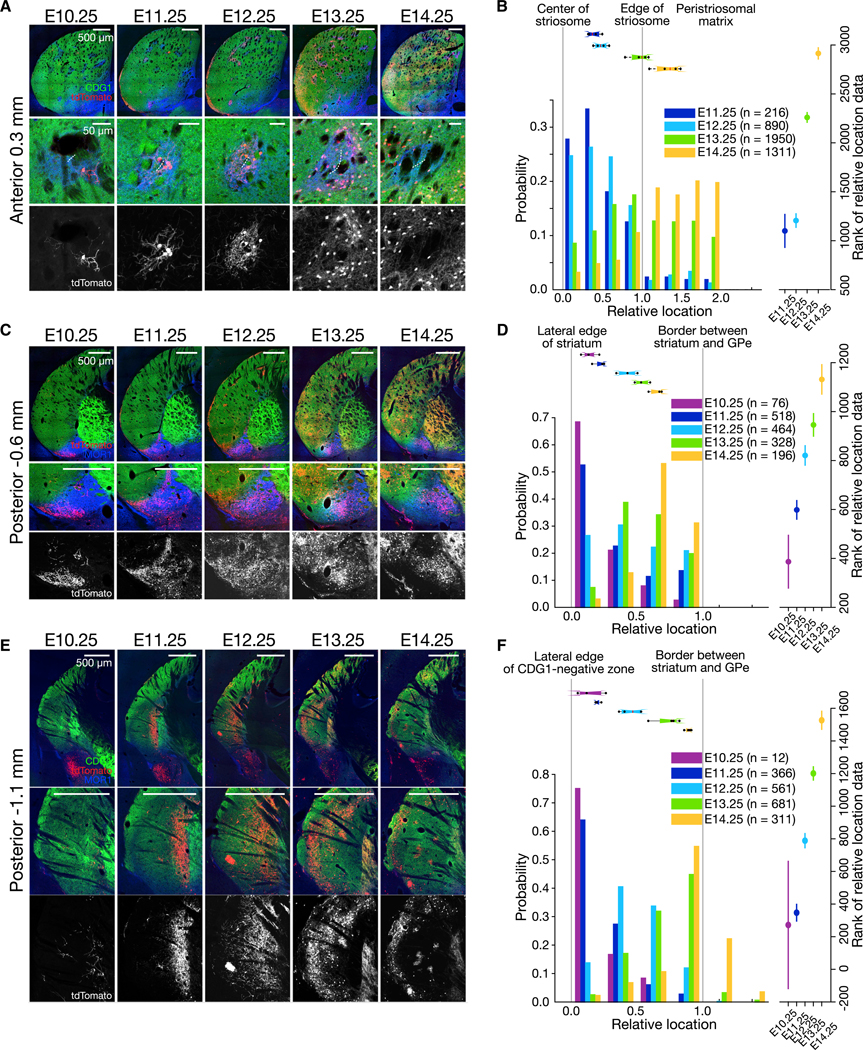
(A) Coronal sections at anterior 0.3 mm (top). The color code is the same as in Figure 1. Lower panels show circular striosomes. White, dotted lines in the middle panels indicate manually annotated centers of striosomes.
(B) Birth date-ordered settlement of SPNs around striosomes. Geometrical registration and figure format are the same as in Figure 2B. Number of mice = 4. Number of slices analyzed per mouse = 3.69 ± 0.48. Number of cells sampled per mouse = 54.0 ± 8.83 (E11.25), 222.5 ± 48.11 (E12.25), 487.5 ± 167.5 (E13.25), and 327.8 ± 41.08 (E14.25).
(C) Coronal sections for the caudal striatum with ventral MOR1-positive zone, in which E10.25-born cells form a cluster.
(D) Birth date-ordered settlement of SPNs in the ventral MOR1-positive zone. Number of mice = 4. Number of slices analyzed per mouse = 1.50 ± 0.51. Number of cells sampled per mouse = 19.0 ± 10.49 (E10.25), 129.5 ± 37.17 (E11.25), 116.0 ± 50.00 (E12.25), 82.00 ± 39.50 (E13.25), and 49.00 ± 10.80 (E14.25).
(E) Coronal sections of the tail of caudate nucleus where CDG1 expression is low.
(F) Birth date-ordered settlement of SPNs in the caudate tail. Number of mice = 4. Number of slices analyzed per mouse = 1.45 ± 0.51. Number of cells sampled per mouse = 3.00 ± 2.58 (E10.25), 91.50 ± 29.73 (E11.25), 140.3 ± 66.00 (E12.25), 170.25 ± 48.45 (E13.25), and 77.75 ± 9.50 (E14.25).
See also Table S1.
At the level of −0.6 mm posterior to the bregma, we again saw birth date-dependent settlement in the ventral MOR1-positive zone (Figures 3C and 3D). The settlement from the lateral edge of striatum to the border of the external segment of the globus pallidus was statistically confirmed (Figure 3D; Kruskal-Wallis test, p < 1 × 10−10 for cells, p < 0.01 for mice). As we assessed the laminar organization at the tail of caudate nucleus (Gangarossa et al., 2013; Miyamoto et al., 2018) (−1.1 mm posterior to the bregma; Figures 3E and 3F), we found that the birth dates could account for the lamination (Figure 3F; Kruskal-Wallis test, p < 1 × 10−10 for cells, p < 0.01 for mice).
The precision of 4-OHT birth-dating provided critical snapshots of the sequential settlement of SPNs. They point to the logic of striatal development initiated from the earliest seeding of the clustering, proceeding to the maturation of the striosomal system, followed by the dispersed formation of the matrix. The general center-surround rule is implemented from the anterior to the posterior parts of the striatum, as ordered settlements of SPNs born in succession within a time-window, which shift according to the spatial gradient but unequivocally beginning at one of the striosomal birth dates.
Birth Date-Dependent Gradients in Construction of the Striatonigral Circuit
In the neocortex, the birth dates of the pyramidal cells are highly correlated with the layers in which they will settle, but birth dates do not definitively predict targets of axonal projection (Economo et al., 2018; Hatanaka et al., 2016; Tasic et al., 2018). Analogously, our findings for the striatum illustrate that the birth dates of SPNs are highly correlated with the sub-compartment of settling, namely, from center to border and from peripheries of striosomes to the outlying matrix. These experiments, however, leave open the question of whether, and if so, how the axonal projections of the SPNs, which directly relates to their function, depend on their birthdates. To address this question, we focused on one of the most conserved circuits of the vertebrate brain, the striatonigral circuit, especially projections onto the striosome-dendron bouquets (intertwined striosomal afferent and the descending dopamine-containing dendrites from the ventral tier of the SNpc; see Figure 4A, right) and the posteriorly adjoining posterior cell cluster (PCC; tyrosine hydroxylase [TH]positive cells clustered deep in the medial substantia nigra pars reticulata [SNpr]; see Figure 4B, right).
Figure 4. Birth Date-Dependent Gradients in Construction of the Striatonigral Circuit.
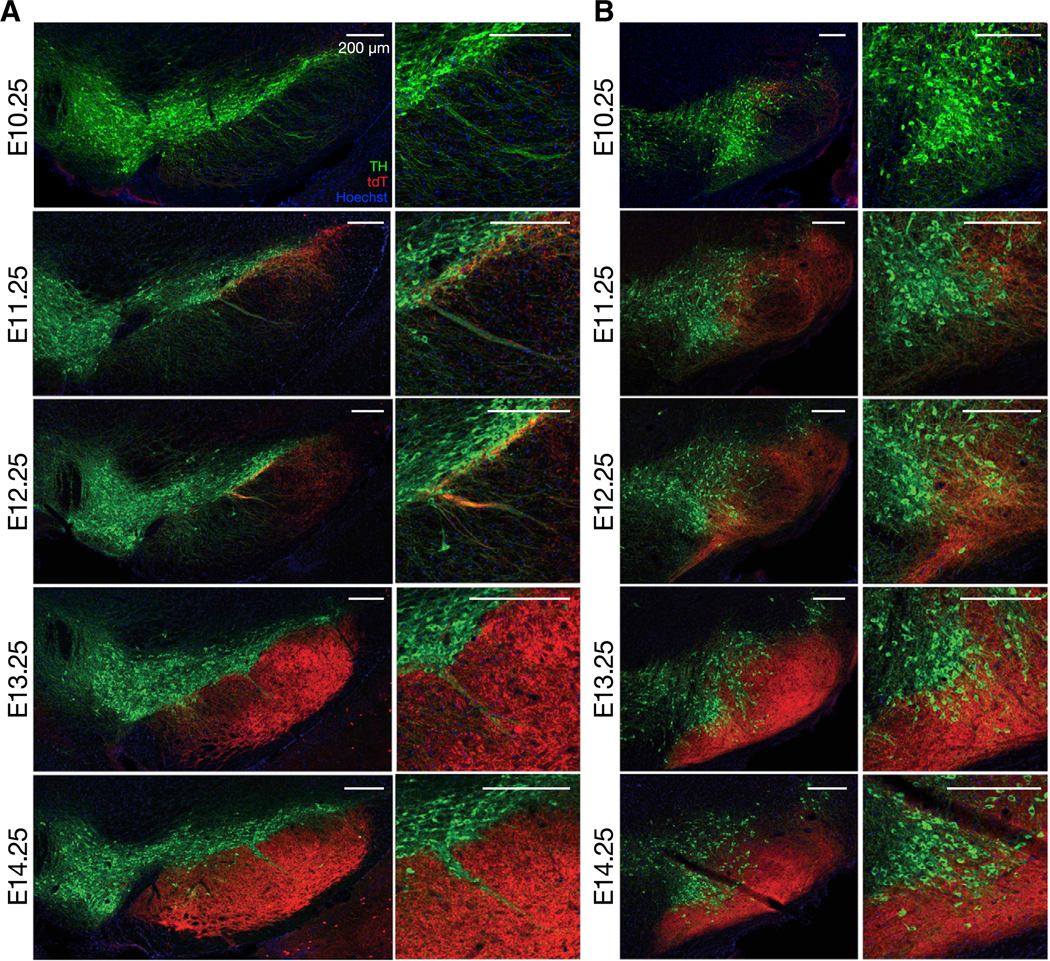
(A) Nigral sections of adult Dlx1::CreER/Ai14 mice administered with 4-OHT at embryonic time points indicated left. Left: coronal sections of anterior SNpc, in which the central striosome-dendron bouquet is formed, stained for tdTomato (red), tyrosine hydroxylase (TH, green), and Hoechst (blue). Right: magnification of striosome-dendron bouquet.
(B) Same as in (A) but in posterior SNpc with the posterior cell cluster, magnified in the right panels.
Most SPNs innervating the landmark bouquet (third column in Figure 4A) and the PCC (third column in Figure 4B) were born near the peak of the striosomal neurogenesis, E12.25. The axons of SPNs born 1 day earlier (i.e., E11.25) reached up to the ventral tier of the lateral SNpc but were largely precluded from the striosome-dendron bouquets and the PCC (second columns in Figures 4A and 4B, respectively). Those of SPNs born 1 day later (i.e., E13.25) innervated mainly the SNpr, with relatively few, presumably late-born striosomal SPN axons, intertwining themselves with the bouquets and the PCC (forth columns in Figures 4A and 4B, respectively). After that time window (e.g., E14.25), SPNs largely avoided the landmark structures (fifth columns in Figures 4A and 4B).
This detailed tracking of projection targets of striatonigral axons suggested, but did not prove, that the birth dates of SPNs determined (1) to which dopamine-containing or nondopamine-containing nigral cells they projected, and (2) to which nigral region, from the lateral to medial, they projected. These two features, however, still were confounded with one another in the Dlx1::CreER/Ai14 model because both the striosomal cells that prefer to project to dopamine-containing neurons (McGregor et al., 2019) and the matrix cells that mainly project to the non-dopaminergic SNpr neurons can express tdTomato because of their common birthdate (e.g., E13.25-born cells settled in matrix at −0.6 mm posterior to the bregma as shown in Figure 3C, but in striosomes, at 1.4 mm anterior to the bregma as shown in Figure 1C). Thus, as far as using Dlx1::CreER/Ai14, we could not determine whether tdTomato-positive striatonigral fibers were from the striosomal versus matrix cells or from the anterior versus posterior parts of the striatum. We, therefore, turned to a Dlx1::CreER/LSL-Flpo mouse line in combination with localized intrastriatal injection of a Flp-dependent adeno-associated virus (AAV), so that the fluorescent reporter was expressed exclusively in cells that went through the two steps: born at the time of 4-OHT administration to express Flp, and infected by Flp-dependent AAV injected locally. In this way, we could independently vary either the birth date of the SPNs, by the time of 4-OHT administration, or the striatal sectors into which they had settled at maturity, by the site of AAV injection.
Topographic Restriction of the Striatonigral Projection According to the Resident Sector in the Striatum
First, we mapped striatonigral inputs arising from SPNs in each of three different striatal sectors in which they had settled, holding birth dates constant. For that purpose, we injected a Flp-dependent reporter AAV that had minimal reporter expression in Flp-negative mice (Figure 7E) into the striatum of Dlx1:: CreER/LSL-Flpo mice pulse-labeled by 4-OHT at a single embryonic time point, E12.25, near the midpoint of striosomal neurogenesis. This strategy allowed us to use the central striosome-dendron bouquet as the landmark innervated by E12.25-born SPNs (Figure 4). Critically, this strategy confined the reporter expression to striatal SPNs born at the time of 4-OHT administration (i.e., E12.25); and, at the same time, distributed within the striatal sector in which the AAV was injected into a given mouse (Figure 5A).
Figure 7. Birth Date-Dependent Routing of Axons from a Single Sector of Striatum to the Central Striosome-Dendron Bouquet.
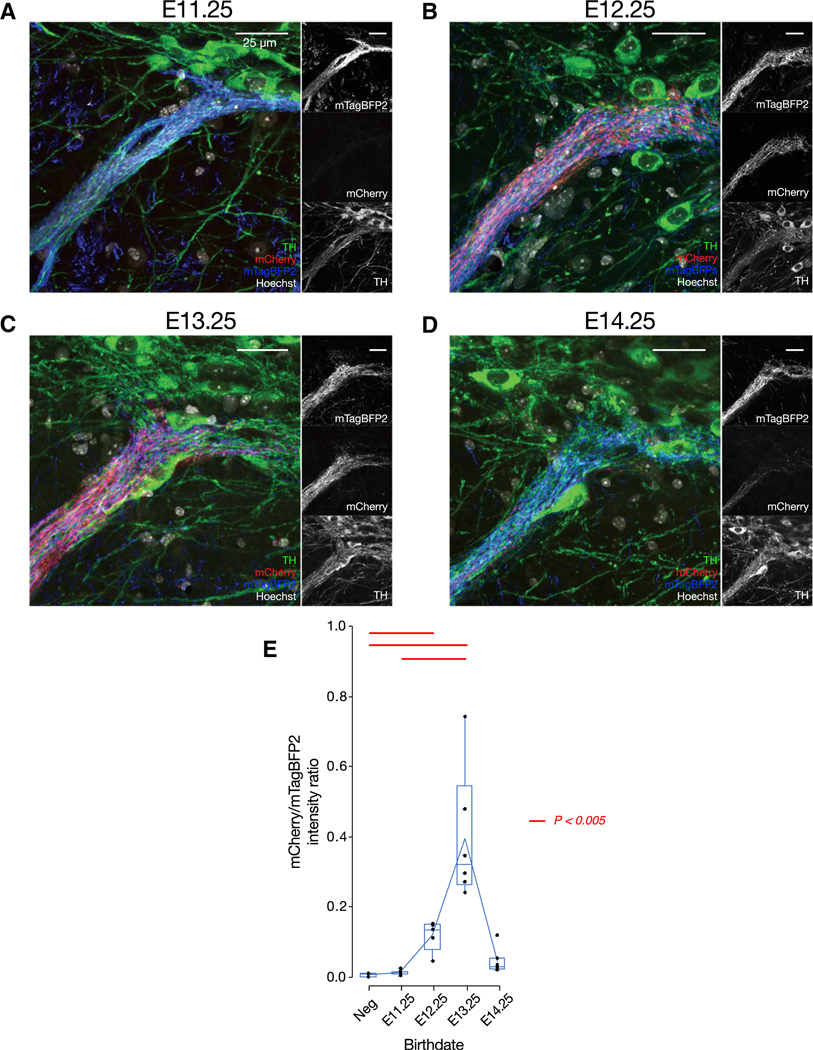
(A–D) The central striosome-dendron bouquet for E11.25 (A), E12.25 (B), E13.25 (C), and E14.25 (D), shown in Figure 6C.
(E) Statistical verification of birth date-dependent routing of SPN axons into the central striosome-dendron bouquet, by the intensity ratio of birth date-dependent mCherry to infection marker mTagBFP2 fluorescence. Single data point corresponds to a single mouse. Red horizontal lines above each pair of boxes indicate significant differences (p < 0.005, two-tailed t test with Bonferroni correction). Number of mice = 3 (Dlx1-negative [Neg]), 7 (E11.25) (A), 5 (E12.25) (B), 6 (E13.25) (C), and 7 (E14.25) (D). Upper and lower edges of boxes indicate, respectively, 75th and 25th percentiles of the data. Whiskers indicate the maximum and minimum data values excluding outliers. Outliers were defined as values distant from the edge of boxes by more than 1.5 times of interquartile range.
Figure 5. Topographic Restriction of the Striatonigral Projection According to the Resident Sector in the Striatum.
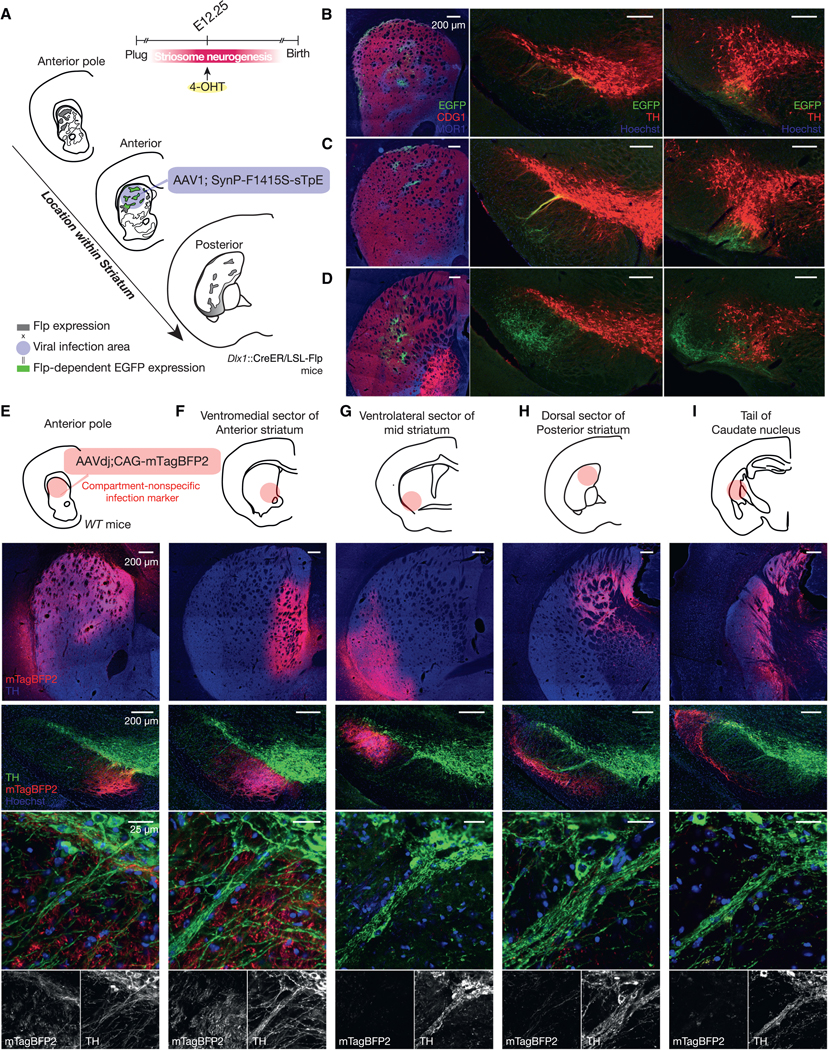
(A) Anterograde tracing of striatonigral axons from same-age SPNs settled in different functional sectors.
(B) Striatonigral projections of E12.25-born SPNs settled in the anterior pole of striatum. Left: striatal injection site stained for MOR1 (blue), CDG1 (red), and EGFP(green). Middle and right: anterior (middle) and posterior (right) SN stained for TH (red), GFP (green), and Hoechst (blue). Number of mice = 3.
(C and D) Same as in (B) but for E12.25-born SPNs settled in the anterior (C) and posterior (D) striatum. Number of mice = 3.
(E–I) Compartment-nonspecific infection marker was injected into the anterior pole (E), ventromedial/limbic sector of anterior striatum (F), ventrolateral sector of mid striatum (G), dorsal sector of posterior striatum (H), or tail of caudate nucleus (I). Upper panels: injection sites stained for mTagBFP2 (red) and TH (blue). Lower panels: nigral sections of the same mice stained for TH (green), mTagBFP2 (red), and Hoechst (blue). None of them route axons of resident SPNs to the central striosome-dendron bouquet. Number of mice = 2.6 ± 0.55.
The striatal region where the E12.25-born SPNs settled clearly controlled the topographic pattern of their striatonigral projection fields, as judged here by the landmark central bouquet. E12.25-born SPNs at the anterior pole of the striatum innervated the small bouquet near the anterior tip of the SNpc (Figure 5B). The E12.25-born SPNs in the anterodorsal striatum targeted the central, most conspicuous striosome-dendron bouquet (middle panel in Figure 5C). The E12.25-born cells in the posterior striatum innervated mainly the SNpr (Figure 5D). These findings demonstrate that the striatonigral projections of SPNs are topographically defined according to their eventual striatal sector of settling, even if they are born at approximately the same time during development.
To determine whether the source of a striatonigral projection to the landmark central bouquet was restricted to the anterodorsal sector so far identified, we next injected five different sectors of the striatum with an AAV-expressing mTagBFP2 in a compartment-nonspecific manner under the CAG promoter (Figures 5E–5I). None of the labeled SPNs in the anterior pole (Figure 5E), ventromedial/limbic sector of anterior striatum (Figure 5F), ventrolateral sector of mid striatum (Figure 5G), dorsal sector of posterior striatum (Figure 5H), or tail of caudate nucleus (Figure 5I) sent their axons to the landmark central bouquet. Only axon terminals from those in the anterodorsal sector were detectable at the resolution of our microscopy. These results indicate that the SPNs settling in this anterodorsal striatal sector, including at least those born at E12.25, are the main, if not the sole, source of striatal inputs to the landmark bouquet. We note that the topographic factor shown here is birth date independent because we compared the projection of sameage, E12.25-born SPNs that settled in different striatal sectors.
Birth Date-Dependent Routing of Axons from a Single Sector of the Striatum to the Central Striosome-Dendron Bouquet
Striatonigral projections, especially those from striosomes in the dorsal striatum, empower the striatum to shut down the activity of dopamine-containing nigral neurons (Evans et al., 2017, 2019; McGregor et al., 2019). We, therefore, next asked whether a specific birth date, as a measure of a developmental program, authorizes the SPNs settled in a particular striatal region–here, focusing on the anterodorsal striatum–to have privileged potential to modulate dopamine-containing cells forming the striosome-dendron bouquet and PCC systems. To address this, we focused on the single sector and varied the birth dates of the SPNs involved in the circuits.
We injected Flp-dependent AAVs into the same, anterodorsal sector of Dlx1::CreER/LSL-Flpo mice pulse-labeled at one of four striosomal birth dates: E11.25, E12.25, E13.25, and E14.25 (Figures 6A and 6B). To have a valid comparison across animals, we co-injected an infection marker AAV expressing mTagBFP2 to verify whether the injection sites were comparable across the mice and, also, to be sure that the injections hit the sensorimotor sector housing SPNs projecting to the landmark central bouquet. As intended, we found mTagBFP2 was consistently expressed in the anterodorsal sector across the mice (upper row in Figure 6B), whereas the mCherry (fused to hM3D) expression, which was Flp dependent and thus birth date dependent, varied, depending on the time of 4-OHT administration corresponding to the patterns shown in Figures 1 and 2 (lower row in Figure 6B). In the nigral sections from the same mice (Figure 6C), birth date-nonspecific, mTagBFP2-positive fibers labeled the landmark central bouquet as well as the SNpr proper, demonstrating that we did succeed in injecting the anterodorsal sector in every group of mice, directing striatonigral axon terminals of resident SPNs around the bouquet and adjoining SNpr (Figure 5).
Figure 6. Birth Date-Dependent Projection of SPNs Settled in the Same Functional Sector.
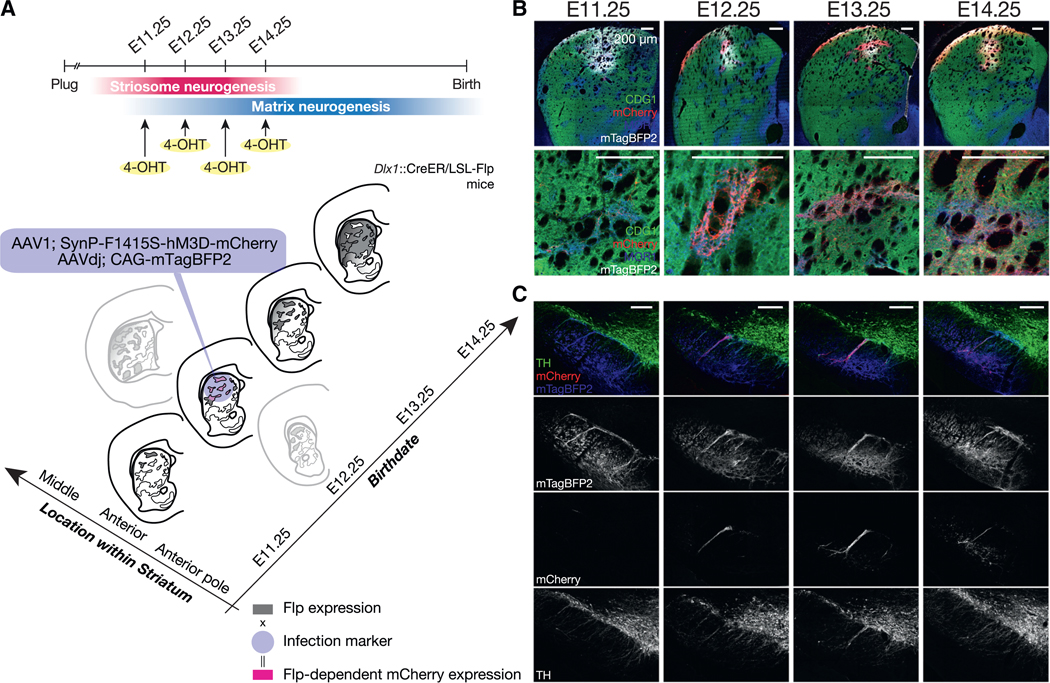
(A) Anterograde tracing of striatonigral axons of different-age SPNs settled in the same anterodorsal sector.
(B) Infection marker (mTagBFP2) was expressed in the anterodorsal sector consistently across mice. Top: striatal injection sites stained for MOR1 (blue), CDG1 (green), mCherry (red), and mTagBFP2 (gray). Bottom: enlarged images including striosomes.
(C) Coronal nigral sections stained for mTagBFP2 (blue in the first row and in grayscale in the second row), birth date-dependent mCherry (red in the first row and in grayscale in the third row), and TH (green in the first row, and in grayscale in the bottom row) at the level of the central striosome-dendron bouquet.
We next asked, as a test case for selectivity of striatonigral targeting, whether the particular birth dates of the SPNs coexisting in the same sensorimotor sector further confine the SPN axonal projections exactly to the landmark striosome-dendron bouquet (Figure 7). We found this to be the case. The axons of E11.25-born SPNs, labeled with mCherry, were hardly detectable in the control mTagBFP2-positive fiber bundles intertwined within the central bouquet (Figure 7A). Statistically, the projection strength, quantified by the intensity ratio of mCherry/mTagBFP2 in the central bouquet, was not significantly different from that in the Dlx1::CreER-negative control mice (Figure 7E). In sharp contrast, the mCherry-positive axons from E12.25-(Figure 7B) and E13.25-born (Figure 7C) SPNs were clearly colocalized with the mTagBFP2-expressing fiber bundles within TH-positive dendrons, forming synaptic varicosities. The mCherry/mTagBFP2 ratio at the central bouquet was significantly higher than the negative control (Figure 7E). Finally, the axons from E14.25-born SPNs had receded from the central bouquet (Figure 7D) and shifted ventrally toward the other mass of mTagBFP2 fibers observed in the ventral SNpr (Figure 6C). The strength of the E14.25 projection to the landmark bouquet was not significantly different from that of the negative control or from those of the E11.25, E12.25, and E13.25 samples (Figure 7E).
Taken together, these findings demonstrate that SPNs within the anterodorsal sector of the striatum born between E12.25 to E13.25, but not SPNs born at E11.25 or at E14.25, participated in building the specialized striatonigral circuit forming the striosome-dendron bouquets (Crittenden et al., 2016; Jimenez-Castellanos and Graybiel, 1989; Lévesque and Parent, 2005).
DISCUSSION
Our findings demonstrate that the striatonigral pathway arising from striosomal SPNs is restricted according to the precise times of births, and yet, that restriction is downstream of, or separately formulated from, the topographic organization of this pathway imposed by the functional sector within which the SPNs lie. This combinatorial set of birth date-dependent and -independent controls confers degrees of freedom in the striatal regulation over the activity of dopamine-containing neurons in the SNpc, known, in turn, to influence aspects of movement, motivation, and mood. Viewed in that light, our findings raise the possibility that this combinatorial control could be critical in searching for the mechanisms underlying vulnerabilities of striatonigral systems to systemic insults and for targeted therapeutics against neuronal disorders. For example, in both Huntington’s disease and Parkinson’s disease, there are clear topographic gradients of cellular and afferent fiber loss. In addition, there is differential vulnerability of striosomes along spatial axes of the striatum (Goto et al., 2005; Kuo and Liu, 2017; Tippett et al., 2007). These vulnerable cells can be precisely separated and genetically targeted for therapeutics because recent, single-nucleus RNA sequencing (RNA-seq) data indicate that the striosomal system is not only recognizable but also is composed of heterogeneous subpopulations (Gokce et al., 2016; Saunders et al., 2018). Given the strong influence that nigro-striato-nigral loops confers on behavior, from motor to motivational control, our findings also speak to the fundamental issue of the developmental and evolutionary restrictions on the behavioral repertoires characterizing a given species (Edinger, 1908).
Birth Date Specification and Its Relation to the Evolutionary Organization of Forebrain Circuits
In the evolution of amniotes over ~300 million years, the basal ganglia have maintained many features, from connectivity to molecular and developmental profiles (Grillner and Robertson, 2016; Puelles et al., 2000; Reiner et al., 1998a, 2004), in contrast to the radical modifications of the pallium and cerebral cortex (Briscoe and Ragsdale, 2018; Dugas-Ford and Ragsdale, 2015). Genetically, the homeobox gene Dlx1/2 defines the birthplace of striatal cells in birds and mammals. Anatomically, the SNpc, as a cluster of dopamine-containing neurons in the midbrain, is conserved across amniotes, located next to the γ-aminobutyric acid (GABAergic) SNpr, and bidirectionally connected with the striatum-containing part of the basal ganglia (Medina and Reiner, 1995; Reiner et al., 2004; Smeets et al., 2000). However, further distant from amniote, the existence of SNpc is still controversial. Species belonging to Agnatha (e.g., lamprey) and Actinopterygii (e.g., zebrafish), for example, appear to possess a homologous striatal structure but no midbrain dopamine-containing neurons (Yamamoto and Vernier, 2011). Apart from the difficulty in defining homologous circuits in distant species (Strausfeld and Hirth, 2013; Striedter, 2005), the developmental organization that we demonstrate here in Dlx1::CreER model mice might reflect canonical principles that, at least, the stem amniote adopted in the face of selection pressures.
Concentric Structure as an Organizing Geometry
Concentric structure, over and beyond the lamination, could be one of the schemas that biological systems tend to introduce for efficient arrangement of multiple lineages involving distinct circuits. In the murine striatum, we have found evidence for such a center-surround rule in organizing the settlement of SPNs. In birds, multiple functional centers are also arranged into core and shell regions that are composed by molecularly, hodologicaly, and functionally distinct lineages (Achiro et al., 2017; Johnson et al., 1995; Reiner et al., 2004), which might be ordered according to the times of birth. In contrast to the neocortex (He et al., 2015), however, we have no clue, as of now, about the mechanisms contributing to the ordered settlement of SPNs within striosomes. Possibilities are that these could be the product of birth date-dependent migration behavior (Hagimoto et al., 2017), chemoattractant and repellant interactions (Passante et al., 2008; Tai et al., 2013), anchoring of endfeet of radial glial progenitors to blood vessels (Tan et al., 2016; Vasudevan et al., 2008), the phase of a cell cycle (McConnell and Kaznowski, 1991), and/or innervation by dopamine-containing fibers during development (Hu et al., 2004; Newman et al., 2015; Specht et al., 1981; Voorn et al., 1988). Identification of these underlying mechanisms will be valuable in understanding not only the development of mammalian striatal systems but also whether the organization is the result of a parallel or convergent evolution yielding multiple specialized, yet separately orchestrated, circuits.
Origin of Functional Topography of Striatal Circuits
Our data provide clear evidence that the birth dates of SPNs control the multiplicity and layouts of striatonigral circuits. In parallel, we have demonstrated the existence of a birth date-independent control, which, at least in the adulthood, is manifested as the striatal sectors in which SPNs settled (Figure 5). The circuit topography might be initiated as genetically distinct subregions within the LGE (Tucker et al., 2008), refined by pruning postnatally (Friel et al., 2013; Hagihara et al., 2015; Hensch, 2005; Katz and Shatz, 1996), and maintained by the spatial gradient of genetic profiles (Märtin et al., 2019). Given that nigrostriatal projections are also organized topographically (Poulin et al., 2018), there may also exist a mechanism to align striatonigral and nigrostriatal projections to form closed and/or open loops, similar to corticostriatal circuits multiplexed for striosomes and sub-compartments in the matrix (matrisomes) (Alexander et al., 1986; Flaherty and Graybiel, 1993, 1994).
Heterogeneities within Striosomes at Maturity
We demonstrate that the SPNs within a given striatal compartment (i.e., striosome or matrix) can be developmentally distinct in terms of time of birth, and yet, on the other hand, that the SPNs settling in those different compartments can share common, compartment-dependent birth dates. These developmental profiles may account for aspects of the heterogeneity reported within the striosomal system at a sub-compartmental scale. The striosomal border regions (i.e., annular compartment) (Brimblecombe and Cragg, 2015; Faull et al., 1989) are vulnerable in patients with Huntington’s disease who have mood symptoms (Hedreen and Folstein, 1995; Tippett et al., 2007) and are prominent in the mature as well as the perinatal human brain (Graybiel and Ragsdale, 1980). They are enriched with local interneurons as well as late-born, rim-forming SPNs (Cowan et al., 1990; Graybiel et al., 1986; Kelly et al., 2018; Kubota and Kawaguchi, 1993; Rushlow et al., 1996). Among the few E10.25-born cells observed in this study, we found some striosomal SPNs extending its dendrites and axons across compartments (Bolam et al., 1988), in contrast to the processes of most later-born SPNs, which rarely cross the borders (Banghart et al., 2015; Bolam et al., 1988; Walker et al., 1993). Still to be determined is the relationship of birth dates and the expression patterns of D1and D2 receptors that correlate with striatonigral and striatopallidal SPNs (Gangarossa et al., 2013), MOR1 and d-opioid receptor expression (Banghart et al., 2015) and, more generally, the degree to which birth dates are decisive in the arrangement of heterogeneous cell-types defined molecularly and/or hodologically. The relationships among those attributes, as well as different metabolic demands of SPNs, may critically control their differential vulnerability to systemic insults in neurologic or neuropsychiatric disorders (Gagnon et al., 2018; Goto et al., 2005; Hayrapetyan et al., 2014; Kuo and Liu, 2017; Saka and Graybiel, 2003; Tippett et al., 2007).
Striosome-Dendron Bouquets as Possible Building Blocks to Influence Behavior
Striosome-dendron bouquets, by their anatomical character, stand in a position to exert powerful control over dopamine-containing neurons. Their unusual structure, with the intertwined bundles of ventrally extended, dopamine-containing dendrons and striosomal axons, might also have a long evolutional history. In birds, in which striatonigral and striatopallidal neurons are segregated into medial and lateral striatum, respectively (Karten and Dubbeldam, 1973; Reiner et al., 2004), striatonigral axon terminals were found to project dopaminergic dendrites extending into the adjacent SNpr (Reiner et al., 1998b). In primates, in correlation with the enlargement of the striatum and substantia nigra, there are larger numbers of descending dopamine-containing dendrites originating from multiple foci in the ventral tier of SNpc. It is not clear that these structures identified in other species correspond to the striosome-dendron bouquets discovered in mice. Our findings, however, suggest that at least in mice, the striosome-dendron bouquets are formed under the dual control of birth date (i.e., time) and the resident sector (i.e., space) of the SPNs. As the striatonigral system evolved to be larger and required longer embryonic periods to generate its constituent cells, it is possible that specialized circuits, including those crystallized as striosome-dendron bouquets, were added to the system one after another in proportion to the number of combinations of birth date and the destined striatal sectors of migration. The circuit elaboration, in turn, could have granted individual organisms survival-tested repertoires of behaviors, built upon the finely tuned tone of the dopamine for the appropriate movements (Albin et al., 1989; DeLong, 1990; Howe and Dombeck, 2016; Klaus et al., 2017), vigor (da Silva et al., 2018; Panigrahi et al., 2015; Salamone et al., 2018), alertness (Bromberg-Martin et al., 2010; Menegas et al., 2018), and motivation (Kawagoe et al., 1998; Richard et al., 2013; Schultz, 2006).
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ann M. Graybiel (graybiel@mit.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
Original/source data for Figures 2, 3, and 7 in the paper is available [i.e., Mendeley Data, https://doi.org/10.17632/pst2tzmpc4.3].
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
We used Dlx1-CreER mice (Taniguchi et al., 2011, MGI Cat# 5298042, RRID:MGI:5298042) crossed with either Ai14 (Madisen et al., 2010; IMSR Cat# JAX:007908, RRID:IMSR_JAX:007908) or LSL-Flpo (He el al., 2015, IMSR Cat# JAX:028584, RRID:IMSR_ JAX:028584) mice for fate-mapping of SPNs (Kelly et al., 2018). All mouse colonies were maintained in accordance with husbandry protocols approved by the Committee on Animal Care at MIT. Mice were housed under a standard 12-hr light/dark cycle with free access to food and water. We used both male and female adult mice, at least 3 weeks old and no older than 4 months of age at perfusion. Typically, we injected viruses at 2 months of age. All experimental procedures were approved by the Committee on Animal Care at MIT and were performed in accordance with the U.S. National Research Council Guide for the Care and Use of Laboratory Animals.
Timed Mating
Dlx1-CreER(het);Ai14(homo) or Dlx1-CreER(het); LSL-Flpo(homo) male breeders in FVB background, in which problematic alleles (i.e., Disc1del, Pde6brd1, mt-Atp8m1) were corrected by crossing with B6J mice (IMSR Cat# JAX:000664, RRID:IMSR_JAX:000664), were single housed at least for 1 week before the initiation of timed mating. Female breeders either in the same FVB background (IMSR Cat# JAX:001800, RRID:IMSR_JAX:001800) or Swiss webster (Taconic Biosciences, IMSR Cat# TAC:sw, RRID:IMSR_TAC:sw) were put into the cage with a male mouse after 5 pm during the light cycle. Presence of plug is checked in the next morning before 10 am, and if it exists, the female breeder is separately single-housed and designated as E0.5.
METHOD DETAILS
4-OHT Administration
We used aqueous solution of 4-OHT. Fifty mg of 4-OHT (Sigma-Aldrich, Cat# H6278–50) was dissolved, with or without 25 mg of progesterone (Sigma-Aldrich, Cat# P3972–5G), in 1250 μl DMSO and stored at −20°C, at 50 μl aliquots (40 mg/ml). On the day of experiment, each aliquot without progesterone was diluted by 950 μl of 2% Tween 80/saline to administer at the dose of 4-OHT 20 mg/kg. To administer at the dose of 4-OHT 15 mg/kg with progesterone 7.5 mg/kg, 37.5 μl of aliquots were added to 962.5 μl of 2% Tween 80/saline. The aqueous solution was subcutaneously injected at the volume of 0.01 ml/g body weight, either at E10.25 (in the morning of 10th day after the plug), E11.25, E12.25, E13.25, E14.25, E15.25, E16.25, E17.25, or E18.25.
AAVs and Plasmids
AAV1; SynP-F14F15S-splitTVA-EGFP (3.0 E12 gc/ml) and AAVdj; CAG-mTagBFP2 (3.25 E12 gc/ml) are kind gifts from Dr. Ian R. Wickersham. F14F15S is a short version (as denoted by S), i.e., lacking 13-bp tandem repeat element and a following single base pair, of Flp-dependent ‘FLEX’ (Atasoy et al., 2008) construct which consists of pairs of orthogonal FRT sites, F14 and F15 (Chatterjee et al., 2018; Turan et al., 2011). The design and sequence of splitTVA-EGFP is the same as described previously (Kohara et al., 2014). Plasmid of SynP-F14F15S-hM3D-mCherry was generated in house by sub-cloning hM3D-mCherry (Krashes et al., 2011; RRID:Addgene_44361, kindly provided by Bryan Roth) into the SynP-F14F15S vector provided by Dr. Ian R. Wickersham. The plasmid was packaged into AAV1 by Vigene Biosciences (2.49 E13 gc/ml).
Stereotaxic Virus Injections
Viruses were injected stereotactically (Stoelting Co.), through pulled glass micropipettes (Model P-2000, Sutter Instrument Co.), by applying pulses of air (Pneumatic PicoPump, PV800, WPI) triggered by stimulator (SEN-3301, Nihon Kohden). Typically, viruses were injected with 25 ms-long pulses spaced by 500 ms, at the speed of 1 nl/pulse. Mice were anesthetized and maintained with isoflurane during surgery. All AAV vectors were stocked at −80°C until use and were injected into mice within less than two freeze-thaw cycles.
For the experiments shown in Figures 5B–5D, we injected 210 nL of AAV1; SynP-F14F15S-splitTVA-EGFP either into the right anterior pole of the striatum (A/P 1.74 mm, M/L +1.35 mm, D/V −2.24 mm), anterior striatum (A/P 0.90 mm, M/L +1.90 mm, D/V −2.00 mm), or posterior striatum (A/P 0.06 mm, M/L +2.40 mm, D/V −2.81 mm). Then, mice were transcardially perfused after the survival time of 4 weeks. For the experiments shown in Figures 5E–5H, we injected AAVdj; CAG-mTagBFP2 either into the anterior pole of the striatum (A/P 1.74 mm, M/L +1.10 mm, D/V −2.65 mm, 210 nl), ventromedial sector of anterior striatum (A/P 0.90 mm, M/ L +1.31 mm, D/V –3.27 mm, 350 nl), dorsal sector of posterior striatum (A/P 0.06 mm, M/L +2.40 mm, D/V −2.90 mm, 350 nl), or tail of caudate nucleus (A/P −1.29 mm, M/L +3.35 mm, D/V −2.80 mm, 350 nl). Then, mice were transcardially perfused after the survival time of 1 week. For the experiments shown in Figures 6 and 7, we mixed the following three reagents with ratio of 1:2:9.4 in volume: AAVdj; CAG-tagBFP2, AAV1; SynP-F14F15S-hM3D-mCherry, and phosphate-buffered saline (PBS). Then, 210 nL of the mixture was injected into anterior striatum (A/P 1.32, M/L +1.63 mm, D/V −2.12 mm). Then, mice were transcardially perfused after the survival time of 3 weeks.
Sample Preparation for Histological Analysis
Mice were deeply anesthetized and then transcardially perfused with PBS followed by 4% paraformaldehyde in a 0.16 M phosphate buffer (pH7.4). Brains were post-fixed in the same fixative for < 12 hr, replaced in a 30% sucrose solution with PBS, embedded in OCT compound (Tissue-Tech) and frozen in dry ice-cold isopentane. In order to avoid any damage to samples, we froze samples within 5 days from perfusion. Samples were stored at −80°C until use.
Frozen specimen was cut with a cryostat into 20- or 25-μm-thick sections, fixed for a given experiment. After washed in PBS, the sections were blocked with 1% BSA / 5% normal goat serum / 0.3% Triton X-100 / PBS for 1 hr at room temperature, when primary antibodies to be used did not include one raised in goat. Otherwise, we used 1% BSA / 5% donkey serum / 0.3% Triton X-100 / PBS for the blocking. Then, the sections were incubated with primary antibody diluted in the same blocking buffer at 4°C on a shaker for 2 overnights.
The antibodies that we used were rabbit polyclonal anti-CalDAG-GEF I (Crittenden et al., 2010: 1:4000), rat monoclonal anti-mCherry (1:1000, Thermo Fisher Scientific Cat# M11217, RRID:AB_2536611), goat polyclonal anti-MOR1 (1:500, Santa Cruz Biotechnology Cat# sc-7488, RRID:AB_2156522), rabbit polyclonal anti-TH (1:1000, Abcam Cat# ab112, RRID:AB_297840), chicken polyclonal anti-mCherry (1:2000, Abcam Cat# ab205402, RRID:AB_2722769), mouse monoclonal anti-mu-crystallin (1:500, Santa Cruz Biotechnology Cat# sc-376687, RRID:AB_11150103), chicken polyclonal anti-GFP (1:2000, Abcam Cat# ab13970, RRID:AB_300798), mouse monoclonal anti-TH (1:4000, ImmunoStar Cat# 22941, RRID:AB_572268), sheep polyclonal anti-TH (1:1000, Abcam Cat# ab113, RRID:AB_297905), and rabbit polyclonal anti-tRFP (1:4000, Abcam Cat# ab13970, RRID:AB_300798).
After washing with PBS three times, tissue sections were incubated with the highly cross-absorbed secondary antibody that was conjugated to Alexa Fluor 488, DyLight 550, Alexa Fluor 546, Alexa Fluor 555 or Alexa Fluor 647 and was raised in goat or donkey, the same species to the blocking buffer, at the concentration of 4 μg/ml each. Finally, sections were stained with Hoechst 33342 (Thermo Fisher Scientific, Cat# H3570), further washed with PBS three times, then mounted on glass slides using Krystalon Mounting Medium (Sigma-Aldrich, Cat# 64969–71). All staining was performed on freshly prepared cryosections without any halts in the process.
QUANTIFICATION AND STATISTICAL ANALYSIS
Image Acquisition and Analysis
Confocal images were obtained using an LSM710 (Carl Zeiss). For images shown in Figures 1, 2, and 3, we took tiled images using Plan-Apochromat 20x/0.8, using ZEN (ZEN Digital Imaging for Light Microscopy, RRID:SCR_013672) software, at the resolution of 0.42 μm and 16 bit/pixel. Images were analyzed by Fiji (Fiji, RRID:SCR_002285). In order to count the cells with respect to compartmental border, subcallosal streak and striosomes were first identified and demarcated manually as CDG1-negative and MOR1-positive zones. Next, the center of the compartments was manually defined as point(s) equidistant from the nearest points on the boundary (see Figures 2A and 3A for representative examples). So, for a completely symmetrical circle, the center point will be a single point. For asymmetrical boundaries, the center points are a set of points, i.e., line, which constitutes an equidistant set of nearest two points on the boundary. Note that for these first two steps, tdTomato channel was not overlaid, so that the boundary or center identification was solely dependent on matrix (CDG1) and striosome (MOR1) markers. Then, tdTomato-positive somata were identified semi-automatically by in-house programmed macros utilizing ImageJ plugins as Kuwahara filter, repeated morphological operations, and particle analysis plugin, and these operations were supervised and verified by eye. Finally, the distances between the soma and the border (distanceborder) and between the soma and the center (distancecenter) were measured by Exact Euclidean Distance Transform (3D). Measured data were saved as spreadsheets and fed into MATLAB (MATLAB, RRID:SCR_001622) for further statistical analysis across samples. For each cell, we calculated the relative location in reference to the center and border of a striosome, and assigned 0 to cells at the center and 1 to those at the border. More specifically, when a cell was located within a striosome, the relative location assigned to the cell was distancecenter / (distancecenter + distanceborder). When a cell was located outside of a striosome, the relative location assigned to the cell was distancecenter / (distancecenter distanceborder). In this way, each cell’s distance to the center of the nearest striosome was indexed in a unit of diameter of the striosome. We excluded cells located too far away from striosomes (relative location > 2). We used built-in MATLAB function (‘kruskalwallis’) to test the significance of differences in distribution between cells born at different birthdates. Exact values of biological (i.e., mice) and technical (i.e., sections/sampled cells per mouse) replicates can be found in the section ‘Sample Sizes’ below.
For images shown in Figure 4, we took tiled images using Plan-Apochromat 20×/0.8, at the resolution of 0.21 μm and 16 bit/pixel. For striatal images shown in Figures 5B–5D, we took tiled images using Plan-Apochromat 20×/0.8 or EC Plan-Neofluor 10×/0.30 M27. For nigral images shown in Figures 5B–5D, we took tiled images using EC Plan-Neofluor 10×/0.30 M27 at the resolution of 0.83 and 16 bit/pixel. For striatal images shown in Figures 5E–5H, we took tiled images using EC Plan-Neofluor 10×/0.30 M27 at the resolution of 0.83 μm and 16 bit/pixel. For nigral images shown in Figures 5E–5H, we took tiled images using Plan-Apochromat 20×/0.8 at the resolution of 0.42 mm and 16 bit/pixel, or high resolution images using Plan-Apochromat 63×/1.40 Oil DIC M27 at the resolution of 0.09 μm and 16 bit/pixel. For striatal images shown in Figure 6B, we took tiled images using EC Plan-Neofluor 10×/0.30 M27 at the resolution of 0.83 μm and 16 bit/pixel (upper) or Plan-Apochromat 20×/0.8 at the resolution of 0.42 μm and 16 bit/pixel. For nigral images shown in Figure 6C, we took tiled images using Plan-Apochromat 20×/0.8, at the resolution of 0.21 μm and 16 bit/pixel. For images shown in Figure 7, we took z stacked images using Plan-Apochromat 63×/01.40 Oil DIC M27, 0.46 mm z-steps (twice of optical section), across ~5–7 μm wide depth at the resolution of 0.07 μm and 16 bit/pixel. For presentation in Figures 7A–7D, we selected 4 sections with maximal intensity and made maximum intensity projection using ImageJ. For quantification in Figure 7E, the region corresponding to striosome-dendron bouquets was manually demarcated, and average intensity in the demarcated region was measured for the infection marker (i.e., mTagBFP2) and mCherry signals. As the background fluorescence, we also measured the average intensity in the region without mTagBFP2-positive fibers, again for each of the two channels, and subtracted from the average intensity measured for the central bouquet. Finally, we calculated the ratio of the two intensities, i.e., mCherry/mTagBFP2, as the index of projection strength. When multiple bouquets, at the middle of SNpc, were found in a single mouse, we averaged the ratio for the multiple bouquets to obtain a single point per mouse.
We set the optical section as 1 airy unit of the channel with longest wavelength (e.g., Alexa 647).
Exclusion of another Possibility to Explain Specific Innervation to the Central Bouquet
The abundance of projections to the central bouquet could not be accounted for by the abundance of the striosomal area occupied in the sectors. We quantified the percentage of MOR1-positive areas (presumed striosomes) within areas of the corresponding regions analyzed. We found the following percentages (mean ± std, biological replicate = 7): anterodorsal sector, 32.9 ± 6.2%; anterior pole, 46.2 ± 8.3%; ventromedial sector, 31.2 ± 5.0%; and ventrolateral sector, 17.3 ± 5.4%. For example, the anterodorsal sector had a smaller striosomal area than the anterior pole (two-tailed t test with Bonferroni correction, p < 0.0083), but evidently had a greater projection to the central bouquet. And although the anterodorsal sector had a comparable total striosomal area as the ventromedial sector, the ventromedial sector had only small, if any, projections to the central bouquet.
Sample Sizes and Statistical Test Used
Exact values of biological (i.e., mice) and technical (i.e., sections/sampled cells per mouse) replicates are shown in figure legends as mean ± std. For Figures 2 and 3, Kruskal-Wallis test (using MATLAB function ‘kruskalwallis’) was used to detect significant differences in the distributions of relative locations across the groups of cells born at different time points, or those of median value per mice administered by 4-OHT at different time points. We chose a non-parametric test because we would like to address the meaningful order of data across the groups, and due to the unbalanced number of samples across groups (i.e., number of cells per group) or the relatively small number of samples (i.e., number of mice per group). Outputs of the Kruskal-Wallis test were fed into the post hoc pairwise comparison function (using MATLAB function ‘multcompare’), and the significance of pairwise comparison between the data ranks (p < 0.05) are shown as a lack of overlap of the 95% confidence intervals in the right panel.
For the data shown in Figures 5E–5I, we prepared 2 mice, one injection site per mouse, for the anterograde tracings from dorsal sector of posterior striatum (Figure 5H), and tail of caudate nucleus (Figure 5I), taken into account of the redundancy of these two experiments targeting the far posterior to the anterodorsal sector.
For the data shown in Figure 7, we used two-tailed T test (‘TTEST’ in Microsoft Excel, with the variables tails = 2, type = 3) in the main text and figure, rather than non-parametric (e.g., Wilcoxon-Mann-Whitney) test, since 1) the data were normally distributed except for the E14.25-group (tested by MATLAB function ‘jbtest’, Dlx1-neg; p = 0.1985, E11.25; p = 0.5000, E12.25; p = 0.0926, E13.25; p = 0.0739, E14.25; p = 0.0085), and 2) we aimed to test the difference in the parametric value (i.e., projection strength). Previous work indicates that, for certain sets of data, non-parametric tests (e.g., Wilcoxon-Mann-Whitney test) can erroneously reject the null hypothesis with higher probability than the parametric t test (Fagerland, 2012; Zimmerman, 1998). We therefore have employed t test evaluations. Indeed, we found more significant difference using Wilcoxon-Mann-Whitney test (MATLAB function ‘ranksum’) for the following comparisons: p = 0.0025 for E11 versus E12, p = 0.0012 for E11 versus E13, p = 0.0023 for E11 versus E14, p = 0.0043 for E12 versus E13, and p = 0.0012 for E13 versus E14.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-CalDAG-GEF I | Crittenden et al., 2010 | N/A |
| Rat monoclonal anti-mCherry | Thermo Fisher Scientific | Cat# M11217, RRID:AB_2536611 |
| Goat polyclonal anti-MOR1 | Santa Cruz Biotechnology | Cat# sc-7488, RRID:AB_2156522 |
| Rabbit polyclonal anti-TH | Abcam | Cat# ab112, RRID:AB_297840 |
| Chicken polyclonal anti-mCherry | Abcam | Cat# ab205402, RRID:AB_2722769 |
| Mouse monoclonal anti-mu-crystallin | Santa Cruz Biotechnology | Cat# sc-376687, RRID:AB11150103 |
| Chicken polyclonal anti-GFP | Abcam | Cat# ab13970, RRID:AB_300798 |
| Mouse monoclonal anti-TH | ImmunoStar | Cat# 22941, RRID:AB_572268 |
| Sheep polyclonal anti-TH | Abcam | Cat# ab113, RRID:AB_297905 |
| Rabbit polyclonal anti-tRFP | Abcam | Cat# ab13970, RRID:AB_300798 |
| Donkey anti rabbit Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-21206, RRID:AB_2535792 |
| Donkey anti rat DyLight 550 | Thermo Fisher Scientific | Cat# SA5–10027, RRID:AB_2556607 |
| Donkey anti goat Alexa Fluor 647 | Thermo Fisher Scientific | Cat# A-21447, RRID:AB_2535864 |
| Goat anti rabbit Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-11034, RRID:AB_2576217 |
| Goat anti chicken Alexa Fluor 555 | Abcam | Cat# ab150174 |
| Goat anti mouse Alexa Fluor 647 | Thermo Fisher Scientific | Cat# A-21236, RRID:AB_2535805 |
| Donkey anti chicken Alexa Fluor 488 | Abcam | Abcam Cat# ab63507, RRID:AB_1139472 |
| Donkey anti mouse Alexa Fluor 546 | Thermo Fisher Scientific | Cat# A10036, RRID:AB_2534012 |
| Donkey anti sheep Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-11015, RRID:AB_2534082 |
| Donkey anti rabbit Alexa Fluor 647 | Thermo Fisher Scientific | Cat# A-31573, RRID:AB_2536183 |
| Bacterial and Virus Strains | ||
| AAV1; SynP-F14F15S-splitTVA-EGFP | Laboratory of Dr. Ian Wickersham | N/A |
| AAVdj; CAG-mTagBFP2 | Laboratory of Dr. Ian Wickersham | N/A |
| AAV1; SynP-F14F15S-hM3D-mCherry | This paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 4-OHT | Sigma-Aldrich | Cat# H6278–50 |
| Progesterone | Sigma-Aldrich | Cat# P3972–5G |
| Deposited Data | ||
| Raw data | This paper | https://doi.org/10.17632/pst2tzmpc4.3 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Dlx1tm1(cre/ERT2)Zjh/Dlx1+ | Taniguchi et al., 2011 | MGI Cat# 5298042, RRID:MGI:5298042 |
| Mouse: B6;129S6-Gt(ROSA) 26Sortm14(CAG-tdTomato)Hze/J | Madisen et al., 2010 | IMSR Cat# JAX:007908, RRID:IMSR_JAX:007908 |
| Mouse: B6;129S4-Gt(ROSA) 26Sortm5(CAG-flpo)Zjh/J | He et al., 2015 | IMSR Cat# JAX:028584, RRID:IMSR_JAX:028584 |
| Mouse: C57BL/6J | The Jackson Laboratory | IMSR Cat# JAX:000664, RRID:IMSR_JAX:000664 |
| Mouse: FVB/NJ | The Jackson Laboratory | IMSR Cat# JAX:001800, RRID:IMSR_JAX:001800 |
| Mouse: Tac:SW | Taconic Biosciences | IMSR Cat# TAC:sw, RRID:IMSR_TAC:sw |
| Oligonucleotides | ||
| Recombinant DNA | ||
| hM3D-mCherry | Krashes et al., 2011 | RRID:Addgene_44361 |
| pAAV- SynP-F14F15S-hM3D-mCherry | This paper | N/A |
| Software and Algorithms | ||
| ZEN | ZEN Digital Imaging for Light Microscopy | RRID:SCR_013672 |
| Fiji | Fiji | RRID:SCR_002285 |
| MATLAB | MATLAB | RRID:SCR_001622 |
| Microsoft Excel | Microsoft | RRID:SCR_016137 |
Highlights.
Fast kinetics of 4-OHT birth dating uncovers a network of striatonigral circuits
Striatonigral circuit elements are built serially during striosomal neurogenesis
Striatonigral circuits of same-age SPNs are ordered by striatal sector of settling
Axon terminal destinations of these SPNs are ordered by their differing birth dates
ACKNOWLEDGMENTS
We thank Dr. Josh Z. Huang (Cold Spring Harbor) for his original gift of the Dlx1-CreER line, now maintained in our laboratory, Dr. Ian Wickersham and Heather Anne Sullivan for generously providing viruses, as well as Graybiel laboratory members for their inputs and comments. This work was supported by the CHDI Foundation (A-5552), the Nancy Lurie Marks Family Foundation, NIH/NIMH (R01 MH060379), the Saks Kavanaugh Foundation, and a JSPS fellowship (20160378 to A.M.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107778.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Achiro JM, Shen J, and Bottjer SW (2017). Neural activity in cortico-basal ganglia circuits of juvenile songbirds encodes performance during goaldirected learning. eLife 6, E26973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albin RL, Young AB, and Penney JB (1989). The functional anatomy of basal ganglia disorders. Trends Neurosci. 12, 366–375. [DOI] [PubMed] [Google Scholar]
- Alexander GE, DeLong MR, and Strick PL (1986). Parallel organization of functionally segregated circuits linking basal ganglia and cortex. Annu. Rev. Neurosci. 9, 357–381. [DOI] [PubMed] [Google Scholar]
- Angevine JB Jr., and Sidman RL (1961). Autoradiographic study of cell migration during histogenesis of cerebral cortex in the mouse. Nature 192, 766–768. [DOI] [PubMed] [Google Scholar]
- Atasoy D, Aponte Y, Su HH, and Sternson SM (2008). A FLEX switch targets Channelrhodopsin-2 to multiple cell types for imaging and long-range circuit mapping. J. Neurosci. 28, 7025–7030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azim E, Shnider SJ, Cederquist GY, Sohur US, and Macklis JD (2009). Lmo4 and Clim1 progressively delineate cortical projection neuron subtypes during development. Cereb. Cortex 19 (Suppl 1), i62–i69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banghart MR, Neufeld SQ, Wong NC, and Sabatini BL (2015). Enkephalin disinhibits mu opioid receptor-rich striatal patches via delta opioid receptors. Neuron 88, 1227–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y (2002). Excitatory actions of gaba during development: the nature of the nurture. Nat. Rev. Neurosci. 3, 728–739. [DOI] [PubMed] [Google Scholar]
- Berendse HW, and Groenewegen HJ (1990). Organization of the thalamostriatal projections in the rat, with special emphasis on the ventral striatum. J. Comp. Neurol. 299, 187–228. [DOI] [PubMed] [Google Scholar]
- Bolam JP, Izzo PN, and Graybiel AM (1988). Cellular substrate of the histochemically defined striosome/matrix system of the caudate nucleus: a combined Golgi and immunocytochemical study in cat and ferret. Neuroscience 24, 853–875. [DOI] [PubMed] [Google Scholar]
- Brimblecombe KR, and Cragg SJ (2015). Substance P weights striatal dopamine transmission differently within the striosome-matrix axis. J. Neurosci. 35, 9017–9023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briscoe SD, and Ragsdale CW (2018). Homology, neocortex, and the evolution of developmental mechanisms. Science 362, 190–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg-Martin ES, Matsumoto M, and Hikosaka O (2010). Dopamine in motivational control: rewarding, aversive, and alerting. Neuron 68, 815–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KN, Chen S, Han Z, Lu CH, Tan X, Zhang XJ, Ding L, LopezCruz A, Saur D, Anderson SA, et al. (2011). Clonal production and organization of inhibitory interneurons in the neocortex. Science 334, 480–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee S, Sullivan HA, MacLennan BJ, Xu R, Hou Y, Lavin TK, Lea NE, Michalski JE, Babcock KR, Dietrich S, et al. (2018). Nontoxic, double-deletion-mutant rabies viral vectors for retrograde targeting of projection neurons. Nat. Neurosci. 21, 638–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan RL, Wilson CJ, Emson PC, and Heizmann CW (1990). Parvalbumin-containing GABAergic interneurons in the rat neostriatum. J. Comp. Neurol. 302, 197–205. [DOI] [PubMed] [Google Scholar]
- Crittenden JR, and Graybiel AM (2011). Basal Ganglia disorders associated with imbalances in the striatal striosome and matrix compartments. Front. Neuroanat. 5, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crittenden JR, Dunn DE, Merali FI, Woodman B, Yim M, Borkowska AE, Frosch MP, Bates GP, Housman DE, Lo DC, and Graybiel AM (2010). CalDAG-GEFI down-regulation in the striatum as a neuroprotective change in Huntington’s disease. Hum. Mol. Genet. 19, 1756–1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crittenden JR, Tillberg PW, Riad MH, Shima Y, Gerfen CR, Curry J, Housman DE, Nelson SB, Boyden ES, and Graybiel AM (2016). Striosome-dendron bouquets highlight a unique striatonigral circuit targeting dopamine-containing neurons. Proc. Natl. Acad. Sci. USA 113, 11318–11323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva JA, Tecuapetla F, Paixão V, and Costa RM (2018). Dopamine neuron activity before action initiation gates and invigorates future movements. Nature 554, 244–248. [DOI] [PubMed] [Google Scholar]
- DeLong MR (1990). Primate models of movement disorders of basal ganglia origin. Trends Neurosci. 13, 281–285. [DOI] [PubMed] [Google Scholar]
- Dugas-Ford J, and Ragsdale CW (2015). Levels of homology and the problem of neocortex. Annu. Rev. Neurosci. 38, 351–368. [DOI] [PubMed] [Google Scholar]
- Eblen F, and Graybiel AM (1995). Highly restricted origin of prefrontal cortical inputs to striosomes in the macaque monkey. J. Neurosci. 15, 5999–6013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Economo MN, Viswanathan S, Tasic B, Bas E, Winnubst J, Menon V, Graybuck LT, Nguyen TN, Smith KA, Yao Z, et al. (2018). Distinct descending motor cortex pathways and their roles in movement. Nature 563, 79–84. [DOI] [PubMed] [Google Scholar]
- Edinger L (1908). The relations of comparative anatomy to comparative psychology. J. Comp. Neurol. Psychol. 18, 437–457. [Google Scholar]
- Evans RC, Zhu M, and Khaliq ZM (2017). Dopamine inhibition differentially controls excitability of substantia nigra dopamine neuron subpopulations through T-type calcium channels. J. Neurosci. 37, 3704–3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans R, Twedell E, Zhu M, Ascencio J, Zhang R, and Khaliq Z (2019). Functional dissection of basal ganglia inhibitory input onto SNc dopaminergic neurons. bioRxiv. 10.1101/856617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagerland MW (2012). t-tests, non-parametric tests, and large studies–a paradox of statistical practice? BMC Med. Res. Methodol. 12, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faull RL, Dragunow M, and Villiger JW (1989). The distribution of neurotensin receptors and acetylcholinesterase in the human caudate nucleus: evidence for the existence of a third neurochemical compartment. Brain Res. 488, 381–386. [DOI] [PubMed] [Google Scholar]
- Feil R, Wagner J, Metzger D, and Chambon P (1997). Regulation of Cre recombinase activity by mutated estrogen receptor ligand-binding domains. Biochem. Biophys. Res. Commun. 237, 752–757. [DOI] [PubMed] [Google Scholar]
- Flaherty AW, and Graybiel AM (1993). Two input systems for body representations in the primate striatal matrix: experimental evidence in the squirrel monkey. J. Neurosci. 13, 1120–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty AW, and Graybiel AM (1994). Input-output organization of the sensorimotor striatum in the squirrel monkey. J. Neurosci. 14, 599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friel KM, Chakrabarty S, and Martin JH (2013). Pathophysiological mechanisms of impaired limb use and repair strategies for motor systems after unilateral injury of the developing brain. Dev. Med. Child Neurol. 55 (Suppl 4), 27–31. [DOI] [PubMed] [Google Scholar]
- Fujiyama F, Sohn J, Nakano T, Furuta T, Nakamura KC, Matsuda W, and Kaneko T (2011). Exclusive and common targets of neostriatofugal projections of rat striosome neurons: a single neuron-tracing study using a viral vector. Eur. J. Neurosci. 33, 668–677. [DOI] [PubMed] [Google Scholar]
- Fujiyama F, Unzai T, and Karube F (2019). Thalamostriatal projections and striosome-matrix compartments. Neurochem. Int. 125, 67–73. [DOI] [PubMed] [Google Scholar]
- Gagnon D, Eid L, Coudé D, Whissel C, Di Paolo T, Parent A, and Parent M (2018). Evidence for sprouting of dopamine and serotonin axons in the pallidum of parkinsonian monkeys. Front. Neuroanat. 12, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangarossa G, Espallergues J, Mailly P, De Bundel D, de Kerchove d’Exaerde A, Hervé D, Girault JA, Valjent E, and Krieger P (2013). Spatial distribution of D1R- and D2R-expressing medium-sized spiny neurons differs along the rostro-caudal axis of the mouse dorsal striatum. Front. Neural Circuits 7, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garaschuk O, Linn J, Eilers J, and Konnerth A (2000). Large-scale oscillatory calcium waves in the immature cortex. Nat. Neurosci. 3, 452–459. [DOI] [PubMed] [Google Scholar]
- Gokce O, Stanley GM, Treutlein B, Neff NF, Camp JG, Malenka RC, Rothwell PE, Fuccillo MV, Südhof TC, and Quake SR (2016). Cellular taxonomy of the mouse striatum as revealed by single-cell RNA-Seq. Cell Rep. 16, 1126–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto S, Lee LV, Munoz EL, Tooyama I, Tamiya G, Makino S, Ando S, Dantes MB, Yamada K, Matsumoto S, et al. (2005). Functional anatomy of the basal ganglia in X-linked recessive dystonia-parkinsonism. Ann. Neurol. 58, 7–17. [DOI] [PubMed] [Google Scholar]
- Graybiel AM (1984). Correspondence between the dopamine islands and striosomes of the mammalian striatum. Neuroscience 13, 1157–1187. [DOI] [PubMed] [Google Scholar]
- Graybiel AM (1990). Neurotransmitters and neuromodulators in the basal ganglia. Trends Neurosci. 13, 244–254. [DOI] [PubMed] [Google Scholar]
- Graybiel AM, and Hickey TL (1982). Chemospecificity of ontogenetic units in the striatum: demonstration by combining [3H]thymidine neuronography and histochemical staining. Proc. Natl. Acad. Sci. USA 79, 198–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graybiel AM, and Ragsdale CW Jr. (1978). Histochemically distinct compartments in the striatum of human, monkeys, and cat demonstrated by acetylthiocholinesterase staining. Proc. Natl. Acad. Sci. USA 75, 5723–5726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graybiel AM, and Ragsdale CW Jr. (1980). Clumping of acetylcholinesterase activity in the developing striatum of the human fetus and young infant. Proc. Natl. Acad. Sci. USA 77, 1214–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graybiel AM, Baughman RW, and Eckenstein F (1986). Cholinergic neuropil of the striatum observes striosomal boundaries. Nature 323, 625–627. [DOI] [PubMed] [Google Scholar]
- Grillner S, and Robertson B (2016). The basal ganglia over 500 million years. Curr. Biol. 26, R1088–R1100. [DOI] [PubMed] [Google Scholar]
- Guenthner CJ, Miyamichi K, Yang HH, Heller HC, and Luo L (2013). Permanent genetic access to transiently active neurons via TRAP: targeted recombination in active populations. Neuron 78, 773–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagihara KM, Murakami T, Yoshida T, Tagawa Y, and Ohki K (2015). Neuronal activity is not required for the initial formation and maturation of visual selectivity. Nat. Neurosci. 18, 1780–1788. [DOI] [PubMed] [Google Scholar]
- Hagimoto K, Takami S, Murakami F, and Tanabe Y (2017). Distinct migratory behaviors of striosome and matrix cells underlying the mosaic formation in the developing striatum. J. Comp. Neurol. 525, 794–817. [DOI] [PubMed] [Google Scholar]
- Hatanaka Y, Namikawa T, Yamauchi K, and Kawaguchi Y (2016). Cortical divergent projections in mice originate from two sequentially generated, distinct populations of excitatory cortical neurons with different initial axonal outgrowth characteristics. Cereb. Cortex 26, 2257–2270. [DOI] [PubMed] [Google Scholar]
- Hayrapetyan V, Castro S, Sukharnikova T, Yu C, Cao X, Jiang YH, and Yin HH (2014). Region-specific impairments in striatal synaptic transmission and impaired instrumental learning in a mouse model of Angelman syndrome. Eur. J. Neurosci. 39, 1018–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Li Z, Ge S, Yu YC, and Shi SH (2015). Inside-out radial migration facilitates lineage-dependent neocortical microcircuit assembly. Neuron 86, 1159–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedreen JC, and Folstein SE (1995). Early loss of neostriatal striosome neurons in Huntington’s disease. J. Neuropathol. Exp. Neurol. 54, 105–120. [DOI] [PubMed] [Google Scholar]
- Hensch TK (2005). Critical period plasticity in local cortical circuits. Nat. Rev. Neurosci. 6, 877–888. [DOI] [PubMed] [Google Scholar]
- Hong S, Amemori S, Chung E, Gibson DJ, Amemori KI, and Graybiel AM (2019). Predominant striatal input to the lateral habenula in macaques comes from striosomes. Curr. Biol. 29, 51–61.e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe MW, and Dombeck DA (2016). Rapid signalling in distinct dopaminergic axons during locomotion and reward. Nature 535, 505–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Cooper M, Crockett DP, and Zhou R (2004). Differentiation of the midbrain dopaminergic pathways during mouse development. J. Comp. Neurol. 476, 301–311. [DOI] [PubMed] [Google Scholar]
- Hunnicutt BJ, Jongbloets BC, Birdsong WT, Gertz KJ, Zhong H, and Mao T (2016). A comprehensive excitatory input map of the striatum reveals novel functional organization. eLife 5, e19103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indra AK, Warot X, Brocard J, Bornert JM, Xiao JH, Chambon P, and Metzger D (1999). Temporally-controlled site-specific mutagenesis in the basal layer of the epidermis: comparison of the recombinase activity of the tamoxifen-inducible Cre-ER(T) and Cre-ER(T2) recombinases. Nucleic Acids Res. 27, 4324–4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Castellanos J, and Graybiel AM (1989). Evidence that histochemically distinct zones of the primate substantia nigra pars compacta are related to patterned distributions of nigrostriatal projection neurons and striatonigral fibers. Exp. Brain Res. 74, 227–238. [DOI] [PubMed] [Google Scholar]
- Johnson F, Sablan MM, and Bottjer SW (1995). Topographic organization of a forebrain pathway involved with vocal learning in zebra finches. J. Comp. Neurol. 358, 260–278. [DOI] [PubMed] [Google Scholar]
- Karten HJ, and Dubbeldam JL (1973). The organization and projections of the paleostriatal complex in the pigeon (Columba livia). J. Comp. Neurol. 148, 61–89. [DOI] [PubMed] [Google Scholar]
- Katz LC, and Shatz CJ (1996). Synaptic activity and the construction of cortical circuits. Science 274, 1133–1138. [DOI] [PubMed] [Google Scholar]
- Kawagoe R, Takikawa Y, and Hikosaka O (1998). Expectation of reward modulates cognitive signals in the basal ganglia. Nat. Neurosci. 1, 411–416. [DOI] [PubMed] [Google Scholar]
- Kelly SM, Raudales R, He M, Lee JH, Kim Y, Gibb LG, Wu P, Matho K, Osten P, Graybiel AM, and Huang ZJ (2018). Radial glial lineage progression and differential intermediate progenitor amplification underlie striatal compartments and circuit organization. Neuron 99, 345–361.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaus A, Martins GJ, Paixao VB, Zhou P, Paninski L, and Costa RM (2017). The spatiotemporal organization of the striatum encodes action space. Neuron 95, 1171–1180.e1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingler E, De la Rossa A, Fièvre S, Devaraju K, Abe P, and Jabaudon D (2019). A translaminar genetic logic for the circuit identity of intracortically projecting neurons. Curr. Biol. 29, 332–339.e5. [DOI] [PubMed] [Google Scholar]
- Kohara K, Pignatelli M, Rivest AJ, Jung HY, Kitamura T, Suh J, Frank D, Kajikawa K, Mise N, Obata Y, et al. (2014). Cell type-specific genetic and optogenetic tools reveal hippocampal CA2 circuits. Nat. Neurosci. 17, 269–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krashes MJ, Koda S, Ye C, Rogan SC, Adams AC, Cusher DS, Maratos-Flier E, Roth BL, and Lowell BB (2011). Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J. Clin. Invest. 121, 1424–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota Y, and Kawaguchi Y (1993). Spatial distributions of chemically identified intrinsic neurons in relation to patch and matrix compartments of rat neostriatum. J. Comp. Neurol. 332, 499–513. [DOI] [PubMed] [Google Scholar]
- Kuo HY, and Liu FC (2017). Valproic acid induces aberrant development of striatal compartments and corticostriatal pathways in a mouse model of autism spectrum disorder. FASEB J. 31, 4458–4471. [DOI] [PubMed] [Google Scholar]
- Lévesque M, and Parent A (2005). The striatofugal fiber system in primates: a reevaluation of its organization based on single-axon tracing studies. Proc. Natl. Acad. Sci. USA 102, 11888–11893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JK, Ghattas I, Liu S, Chen S, and Rubenstein JL (1997). Dlx genes encode DNA-binding proteins that are expressed in an overlapping and sequential pattern during basal ganglia differentiation. Dev. Dyn. 210, 498–512. [DOI] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Märtin A, Calvigioni D, Tzortzi O, Fuzik J, Wä rnberg, E., and Meletis, K. (2019). A Spatiomolecular map of the striatum. Cell Rep. 29, 4320–4333.e5. [DOI] [PubMed] [Google Scholar]
- Mason HA, Rakowiecki SM, Raftopoulou M, Nery S, Huang Y, Gridley T, and Fishell G (2005). Notch signaling coordinates the patterning of striatal compartments. Development 132, 4247–4258. [DOI] [PubMed] [Google Scholar]
- McConnell SK, and Kaznowski CE (1991). Cell cycle dependence of laminar determination in developing neocortex. Science 254, 282–285. [DOI] [PubMed] [Google Scholar]
- McGregor MM, McKinsey GL, Girasole AE, Bair-Marshall CJ, Rubenstein JLR, and Nelson AB (2019). Functionally distinct connectivity of developmentally targeted striosome neurons. Cell Rep. 29, 1419–1428.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina L, and Reiner A (1995). Neurotransmitter organization and connectivity of the basal ganglia in vertebrates: implications for the evolution of basal ganglia. Brain Behav. Evol. 46, 235–258. [DOI] [PubMed] [Google Scholar]
- Menegas W, Akiti K, Amo R, Uchida N, and Watabe-Uchida M (2018). Dopamine neurons projecting to the posterior striatum reinforce avoidance of threatening stimuli. Nat. Neurosci. 21, 1421–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto Y, Katayama S, Shigematsu N, Nishi A, and Fukuda T (2018). Striosome-based map of the mouse striatum that is conformable to both cortical afferent topography and uneven distributions of dopamine D1 and D2 receptor-expressing cells. Brain Struct. Funct. 223, 4275–4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molyneaux BJ, Arlotta P, Menezes JR, and Macklis JD (2007). Neuronal subtype specification in the cerebral cortex. Nat. Rev. Neurosci. 8, 427–437. [DOI] [PubMed] [Google Scholar]
- Newman H, Liu FC, and Graybiel AM (2015). Dynamic ordering of early generated striatal cells destined to form the striosomal compartment of the striatum. J. Comp. Neurol. 523, 943–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary DD, Chou SJ, and Sahara S (2007). Area patterning of the mammalian cortex. Neuron 56, 252–269. [DOI] [PubMed] [Google Scholar]
- Panigrahi B, Martin KA, Li Y, Graves AR, Vollmer A, Olson L, Mensh BD, Karpova AY, and Dudman JT (2015). Dopamine is required for the neural representation and control of movement vigor. Cell 162, 1418–1430. [DOI] [PubMed] [Google Scholar]
- Passante L, Gaspard N, Degraeve M, Frise n, J., Kullander K, De Maertelaer V, and Vanderhaeghen P (2008). Temporal regulation of ephrin/Eph signalling is required for the spatial patterning of the mammalian striatum. Development 135, 3281–3290. [DOI] [PubMed] [Google Scholar]
- Poulin JF, Caronia G, Hofer C, Cui Q, Helm B, Ramakrishnan C, Chan CS, Dombeck DA, Deisseroth K, and Awatramani R (2018). Mapping projections of molecularly defined dopamine neuron subtypes using intersectional genetic approaches. Nat. Neurosci. 21, 1260–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puelles L, Kuwana E, Puelles E, Bulfone A, Shimamura K, Keleher J, Smiga S, and Rubenstein JL (2000). Pallial and subpallial derivatives in the embryonic chick and mouse telencephalon, traced by the expression of the genes Dlx-2, Emx-1, Nkx-2.1, Pax-6, and Tbr-1. J. Comp. Neurol. 424, 409–438. [DOI] [PubMed] [Google Scholar]
- Ragsdale CW Jr., and Graybiel AM (1981). The fronto-striatal projection in the cat and monkey and its relationship to inhomogeneities established by acetylcholinesterase histochemistry. Brain Res. 208, 259–266. [DOI] [PubMed] [Google Scholar]
- Ragsdale CW Jr., and Graybiel AM (1991). Compartmental organization of the thalamostriatal connection in the cat. J. Comp. Neurol. 311, 134–167. [DOI] [PubMed] [Google Scholar]
- Rajakumar N, Elisevich K, and Flumerfelt BA (1993). Compartmental origin of the striato-entopeduncular projection in the rat. J. Comp. Neurol. 331, 286–296. [DOI] [PubMed] [Google Scholar]
- Rakic P (1974). Neurons in rhesus monkey visual cortex: systematic relation between time of origin and eventual disposition. Science 183, 425–427. [DOI] [PubMed] [Google Scholar]
- Reiner A, Medina L, and Veenman CL (1998a). Structural and functional evolution of the basal ganglia in vertebrates. Brain Res. Brain Res. Rev. 28, 235–285. [DOI] [PubMed] [Google Scholar]
- Reiner A, Perera M, Paullus R, and Medina L (1998b). Immunohistochemical localization of DARPP32 in striatal projection neurons and striatal interneurons in pigeons. J. Chem. Neuroanat. 16, 17–33. [DOI] [PubMed] [Google Scholar]
- Reiner A, Perkel DJ, Bruce LL, Butler AB, Csillag A, Kuenzel W, Medina L, Paxinos G, Shimizu T, Striedter G, et al. ; Avian Brain Nomenclature Forum (2004). Revised nomenclature for avian telencephalon and some related brainstem nuclei. J. Comp. Neurol. 473, 377–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard JM, Castro DC, Difeliceantonio AG, Robinson MJ, and Berridge KC (2013). Mapping brain circuits of reward and motivation: in the footsteps of Ann Kelley. Neurosci. Biobehav. Rev. 37 (9 Pt A), 1919–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rushlow W, Naus CC, and Flumerfelt BA (1996). Somatostatin and the patch/matrix compartments of the rat caudate-putamen. J. Comp. Neurol. 364, 184–190. [DOI] [PubMed] [Google Scholar]
- Saint-Amant L, and Drapeau P (2000). Motoneuron activity patterns related to the earliest behavior of the zebrafish embryo. J. Neurosci. 20, 3964–3972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saka E, and Graybiel AM (2003). Pathophysiology of Tourette’s syndrome: striatal pathways revisited. Brain Dev. 25 (Suppl 1), S15–S19. [DOI] [PubMed] [Google Scholar]
- Salamone JD, Correa M, Yang JH, Rotolo R, and Presby R (2018). Dopamine, effort-based choice, and behavioral economics: basic and translational research. Front. Behav. Neurosci. 12, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders A, Macosko EZ, Wysoker A, Goldman M, Krienen FM, de Rivera H, Bien E, Baum M, Bortolin L, Wang S, et al. (2018). Molecular diversity and specializations among the cells of the adult mouse brain. Cell 174, 1015–1030.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz W (2006). Behavioral theories and the neurophysiology of reward. Annu. Rev. Psychol. 57, 87–115. [DOI] [PubMed] [Google Scholar]
- Smeets WJ, Marín O, and González A (2000). Evolution of the basal ganglia: new perspectives through a comparative approach. J. Anat. 196, 501–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith Y, Galvan A, Ellender TJ, Doig N, Villalba RM, Huerta-Ocampo I, Wichmann T, and Bolam JP (2014). The thalamostriatal system in normal and diseased states. Front. Syst. Neurosci. 8, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JB, Klug JR, Ross DL, Howard CD, Hollon NG, Ko VI, Hoffman H, Callaway EM, Gerfen CR, and Jin X (2016). Genetic-based dissection unveils the inputs and outputs of striatal patch and matrix compartments. Neuron 91, 1069–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song DD, and Harlan RE (1994). Genesis and migration patterns of neurons forming the patch and matrix compartments of the rat striatum. Brain Res. Dev. Brain Res. 83, 233–245. [DOI] [PubMed] [Google Scholar]
- Sousa VH, and Fishell G (2010). Sonic hedgehog functions through dynamic changes in temporal competence in the developing forebrain. Curr. Opin. Genet. Dev. 20, 391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Specht LA, Pickel VM, Joh TH, and Reis DJ (1981). Fine structure of the nigrostriatal anlage in fetal rat brain by immunocytochemical localization of tyrosine hydroxylase. Brain Res. 218, 49–65. [DOI] [PubMed] [Google Scholar]
- Strausfeld NJ, and Hirth F (2013). Deep homology of arthropod central complex and vertebrate basal ganglia. Science 340, 157–161. [DOI] [PubMed] [Google Scholar]
- Striedter GF (2005). Principles of Brain Evolution (Sinauer Associates). [Google Scholar]
- Tai AX, Cassidy RM, and Kromer LF (2013). EphA7 expression identifies a unique neuronal compartment in the rat striatum. J. Comp. Neurol. 521, 2663–2679. [DOI] [PubMed] [Google Scholar]
- Tan X, Liu WA, Zhang XJ, Shi W, Ren SQ, Li Z, Brown KN, and Shi SH (2016). Vascular influence on ventral telencephalic progenitors and neocortical interneuron production. Dev. Cell 36, 624–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi H, He M, Wu P, Kim S, Paik R, Sugino K, Kvitsiani D, Fu Y, Lu J, Lin Y, et al. (2011). A resource of Cre driver lines for genetic targeting of GABAergic neurons in cerebral cortex. Neuron 71, 995–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasic B, Yao Z, Graybuck LT, Smith KA, Nguyen TN, Bertagnolli D, Goldy J, Garren E, Economo MN, Viswanathan S, et al. (2018). Shared and distinct transcriptomic cell types across neocortical areas. Nature 563, 72–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tippett LJ, Waldvogel HJ, Thomas SJ, Hogg VM, van Roon-Mom W, Synek BJ, Graybiel AM, and Faull RL (2007). Striosomes and mood dysfunction in Huntington’s disease. Brain 130, 206–221. [DOI] [PubMed] [Google Scholar]
- Tucker ES, Segall S, Gopalakrishna D, Wu Y, Vernon M, Polleux F, and Lamantia AS (2008). Molecular specification and patterning of progenitor cells in the lateral and medial ganglionic eminences. J. Neurosci. 28, 9504–9518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turan S, Galla M, Ernst E, Qiao J, Voelkel C, Schiedlmeier B, Zehe C, and Bode J (2011). Recombinase-mediated cassette exchange (RMCE): traditional concepts and current challenges. J. Mol. Biol. 407, 193–221. [DOI] [PubMed] [Google Scholar]
- Turrero García M, and Harwell CC (2017). Radial glia in the ventral telencephalon. FEBS Lett. 591, 3942–3959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Kooy D, and Fishell G (1987). Neuronal birthdate underlies the development of striatal compartments. Brain Res. 401, 155–161. [DOI] [PubMed] [Google Scholar]
- Vasudevan A, Long JE, Crandall JE, Rubenstein JL, and Bhide PG (2008). Compartment-specific transcription factors orchestrate angiogenesis gradients in the embryonic brain. Nat. Neurosci. 11, 429–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voorn P, Kalsbeek A, Jorritsma-Byham B, and Groenewegen HJ (1988). The pre- and postnatal development of the dopaminergic cell groups in the ventral mesencephalon and the dopaminergic innervation of the striatum of the rat. Neuroscience 25, 857–887. [DOI] [PubMed] [Google Scholar]
- Walker RH, Arbuthnott GW, Baughman RW, and Graybiel AM (1993). Dendritic domains of medium spiny neurons in the primate striatum: relationships to striosomal borders. J. Comp. Neurol. 337, 614–628. [DOI] [PubMed] [Google Scholar]
- Wallace ML, Saunders A, Huang KW, Philson AC, Goldman M, Macosko EZ, McCarroll SA, and Sabatini BL (2017). Genetically distinct parallel pathways in the entopeduncular nucleus for limbic and sensorimotor output of the basal ganglia. Neuron 94, 138–152 e135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, and Vernier P (2011). The evolution of dopamine systems in chordates. Front. Neuroanat. 5, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye L, Allen WE, Thompson KR, Tian Q, Hsueh B, Ramakrishnan C, Wang AC, Jennings JH, Adhikari A, Halpern CH, et al. (2016). Wiring and molecular features of prefrontal ensembles representing distinct experiences. Cell 165, 1776–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun K, Fischman S, Johnson J, Hrabe de Angelis M, Weinmaster G, and Rubenstein JL (2002). Modulation of the notch signaling by Mash1 and Dlx1/2 regulates sequential specification and differentiation of progenitor cell types in the subcortical telencephalon. Development 129, 5029–5040. [DOI] [PubMed] [Google Scholar]
- Zimmerman DW (1998). Invalidation of parametric and nonparametric statistical tests by concurrent violation of two assumptions. J. Exp. Educ. 67, 55–68. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original/source data for Figures 2, 3, and 7 in the paper is available [i.e., Mendeley Data, https://doi.org/10.17632/pst2tzmpc4.3].